
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 19 October 2022
Sec. Bone Research
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1032614
This article is part of the Research Topic Influence of the Immune System on Bone Metabolism View all 7 articles
 Xinli Hu1,2†
Xinli Hu1,2† Zheng Wang1,2†
Zheng Wang1,2† Chao Kong1,2Yu Wang1,2Weiguo Zhu1,2Wei Wang1,2Yongjin Li1,2Wei Wang1,2*
Chao Kong1,2Yu Wang1,2Weiguo Zhu1,2Wei Wang1,2Yongjin Li1,2Wei Wang1,2* Shibao Lu1,2*
Shibao Lu1,2*Multiple causes may contribute to osteoporosis, characterized by a loss in bone mass and density as a consequence of the degradation of bone microstructure and a resultant rise in bone fragility. Recently, increasing attention has been paid to the role of necroptosis in the development of osteoporosis. Necroptosis is orchestrated by a set of proteins known as receptor-interacting protein kinase (RIPK)1, RIPK3, and mixed lineage kinase domain-like protein (MLKL). A necrosome is formed by MLKL, RIPK1, RIPK3, and RIPK3-RIPK3. A dissociated MLKL forms pores in the plasma membrane and eventually leads to necroptosis after translocating from the necrosome. In this review, we discuss a detailed understanding of necroptosis and its associated processes, a better understanding of its interactions with osteoclasts, osteoblasts, and osteocytes, and the associations between necroptosis and diabetic osteoporosis, steroid-induced osteoporosis, and postmenopausal osteoporosis. In addition, a variety of experimental medicines capable of modulating crucial necroptosis processes are highlighted. It’s important to note that this is the first review paper to consolidate current data on the role of necroptosis in osteoporosis, and it offers fresh hope for the future treatment of this disease.
Micro-architecture of bone tissue degeneration and reduced bone mass define osteoporosis (OP), a systemic disease (1). Osteoporosis affects over 200 million individuals worldwide, and the number continues to rise yearly. In particular, hip fractures, vertebral fractures, and distal forearm fractures are at an increased risk due to increased bone fragility (2). The most common cause of fracture in elderly individuals is osteoporosis, which accounts for around 9.0 million fractures annually (3). A considerable amount of pain and suffering is involved in fragility fractures, as well as disability, death, and societal costs associated with these injuries. The bone is constantly resorbed and reformed according to a process called remodeling, controlled by two major cell types: osteoclasts and osteoblasts (4). Osteoporosis (OP) is caused by an imbalance in bone metabolism. Osteoporosis occurs when bone resorption outpaces bone creation, which may result from normal aging or other pathological diseases (5). Currently, the treatment strategies for OP mainly use drug therapy, including calcitonin, bisphosphonates and fluorides (6, 7). These drugs may relief some of the bone loss and clinical symptoms. However, their long-term clinical efficacy is limited. Because they have poor tolerance, significant side effects and high cost (8, 9). All these reasons making the development of new, safe and effective OP treatment is imperative.
Programmed cell death with a necrotic appearance is called necroptosis and occurs in several biological processes, including inflammation, the immunological response and metabolic disorders (10).,RIPK1 (receptor-interacting protein kinase 1), RIPK3 (receptor-interacting protein kinase 3), and MLKL (mixed-lineage kinase domain-like pseudokinase) play an important role in this process (11). Studies have shown that death receptors such as tumor necrosis factor receptor 1 (TNFR1), Fas ligand receptor (FasR), TNF-related apoptosis-inducing ligand receptor (TRAIL-R), interferons (IFNs), intracellular RNA and DNA sensors, and other mediators are effective in inducing necroptosis (12, 13). Among these signaling, TNFR1 signaling is best characterized and believed to be extensively involved in necroptosis (14).
The association between necroptosis and osteoporosis has increased in recent studies. We draw a Figure 1 to illustrate the momentous events for necroptosis and its research in osteoporosis. One of the most common types of osteoporosis in women is postmenopausal osteoporosis (PMOP), which arises after the cessation of estrogen (E2) in women. Ovarian ovariectomized rats in several studies had higher levels of tumor necrosis factor-alpha (TNF-α) when estrogen was deficient (15, 16). Moreover, TNF-α is an essential cytokine that activates TNFR1 and induces necroptosis (17). Necroptosis hence may have a significant role in PMOP. In addition, rats with glucocorticoid-induced osteoporosis who were administered necroptosis inhibitor (Nec-1) had improved bone production (18). These findings may indicate that necroptosis is intimately associated with osteoporosis. Here, we summarize the basic pathological features of necroptosis, the relationship between necroptosis and osteoclasts and osteoblasts as well as the relationship between necroptosis and osteoporosis. We also illustrate how necroptosis influences diabetic osteoporosis (DOP), glucocorticoid-induced osteoporosis (GIOP), and postmenopausal osteoporosis (PMOP).
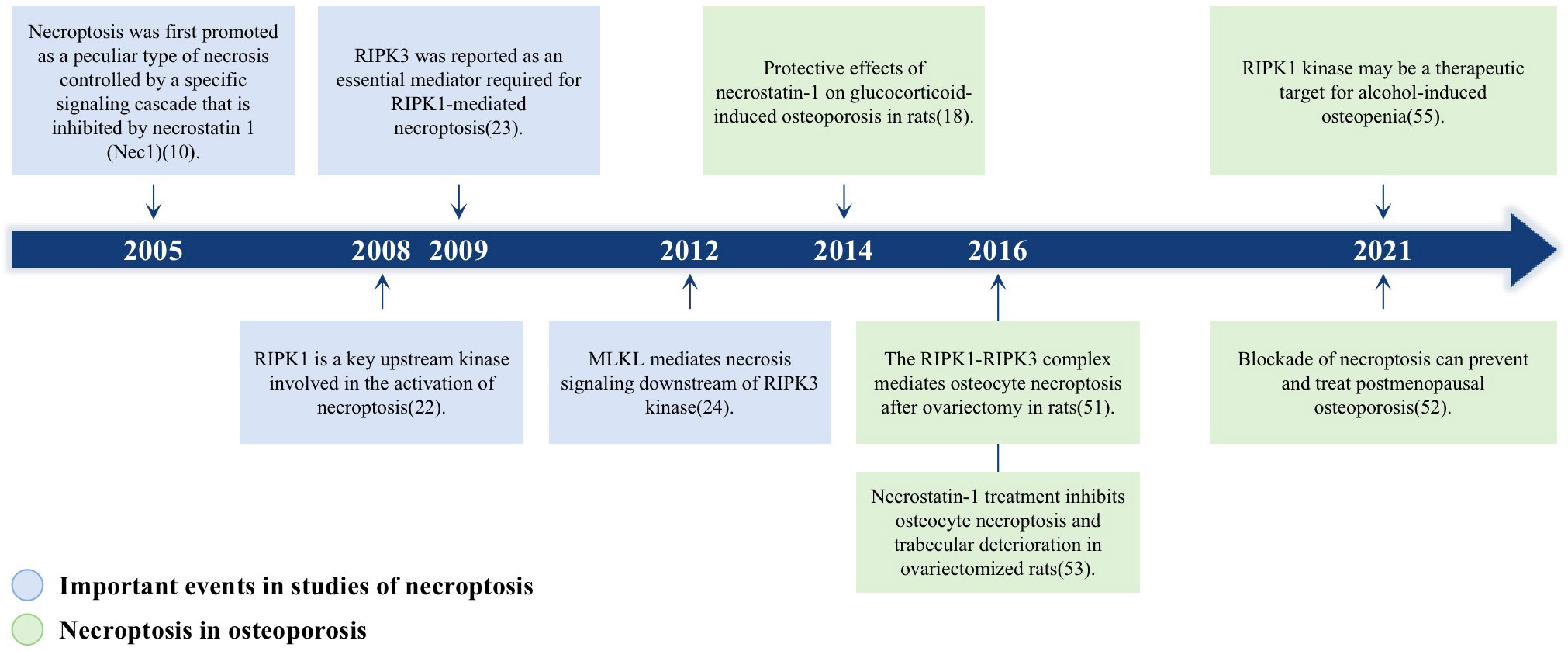
Figure 1 A timeline of the momentous events for necroptosis and its role in osteoporosis. Blue boxes highlight the major findings in the discovery of necroptosis signaling pathway; Green boxes represent the researches about the role of necroptosis in osteoporosis.
According to the morphological and biochemical changes of cell death. The Nomenclature Committee on Cell Death (NCCD) roughly classifies cell death into three categories: apoptosis, autophagy and necrosis (19, 20). Apoptosis is the first type of cell death, characterized by small cells, cell membrane blebbing, nuclear fragmentation and chromatin concentration (21). Two apoptosis pathways have been found in organisms, including the intrinsic and the extrinsic apoptotic pathway. Its biochemical characteristics and mechanism are defined as cascading by the activation of caspases-8 or 9 and subsequent activation of caspases-3/6/7 (22). The second type of cell death is autophagy, which is characterized by the accumulation of double membrane covered vacuoles covering cytoplasm or cytoplasmic organelles, and the redistribution of light chain 3 (LC3) to autophagy membranes (19). Autophagy-related (ATG) families, mammalian target of rapamycin (mTOR), LC3-I, LC3-II and p62 are involved in autophagy (23). Necrosis, as the third type of cell death, is characterized by the rupture of the plasma membrane and release of cell contents (24). The release of damage associated molecular model (DAMP) and cell contents provides feedforward signals to enhance programmed necrosis in other cells (25). The distinctive features among these different death modes are listed in Table 1. Most studies of osteoporosis focus on apoptosis and autophagy, because necrosis has long been considered irreversible (26, 27). However, recently, more and more evidence has revealed a special form of necrosis, called necrotic apoptosis, involving osteoporosis (28, 29). First, A distinct signaling cascade blocked by necrostatin-1(Nec1) governs necroptosis, described in 2005 by Degterev and colleagues (10). The term necroptosis was first used in 2018, in reference to a type of programmed necrosis not triggered by cysteine-aspartic proteases (caspases) and governed mainly by two receptor interacting protein kinases: RIPK1 and RIPK3 (19). Necroptosis is a newly found mechanism of intentional cell death that has a distinct morphology from apoptosis. It is characterized by plasma membrane rupture, cell content leaking, and organelle enlargement (30, 31). Necroptosis is not reliant on caspase, but is regulated by various cellular signal transduction proteins, such as RIPK1, RIPK3, and MLKL (32–34). In research studies, several cell mediators have been demonstrated to trigger the necroptosis pathway, such as members of the tumor necrosis factor (TNF) family, Fas ligands, interferon, and RNA or DNA sensors (35–37). Death receptor ligands, particularly TNF and FAS, are the most well-studied inducers of necroptosis (19). Once TNF interacts with TNFR1, TNFR1 recruits TRADD, TRAF2/5, cIAP1/2, LUBAC, and RIPK1 to form the so-called complex I, which activates NF-kB and its transcription of pro-survival and pro-inflammatory genes (37). Deubiquitination of RIPK1 by cylindromatosis (CYLD) and ubiquitin- editing enzyme A20(A20) can result in RIPK1 dissociating from complex I; then, the remaining complex recruits TRADD, FADD and caspase-8 and forms complex IIa (composed of TRADD, Fas-associated death domain (FADD), RIPK1 and caspase-8), which activates apoptosis (38, 39). At the same time, deubiquitination also result in complex I transformed into complex IIb to induce necroptosis, which consists of RIPK1, RIPK3, Fas-associated death domain protein (FADD), and caspase-8 (38, 40). It means that the deubiquitination of complex I transforms into complex II (IIa and IIb). Two distinct types of complex II can be distinguished based on their composition and the activity of their proteins.
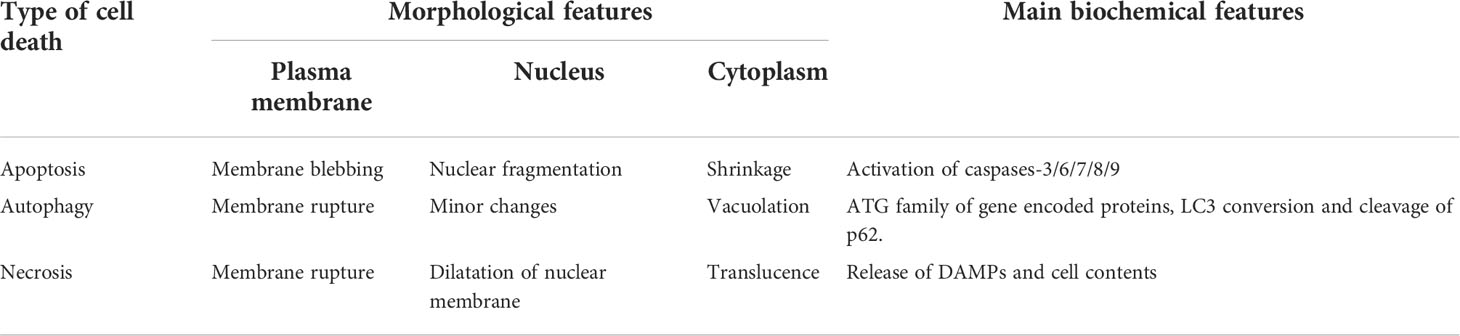
Table 1 The distinctive morphological and main biochemical features of apoptosis, autophagy, and necrosis.
To summize, complex I is pro-survival, complex IIa is pro-apoptotic, and complex IIb is pro-necroptotic (19). The type of complex II (a or b) and the state of caspase 8 (activated or inactivated) determine the transition to apoptosis or necroptosis (41). RIPK3, considered one of the key regulators of necroptosis, is required for necroptosis (42). A necrosome is formed when complex IIb recruits substantial quantities of RIPK3 in the presence of high levels of this protein (43). RIPK1 and RIPK3 are degraded by caspase-8 in the necrosome, preventing necroptosis (37). When zVAD inhibits caspase-8, RIPK1 recruits RIPK3 and phosphorylates RIPK3 in response (33). A phosphorylated RIPK3 recruits MLKL and phosphorylates MLKL, and then MLKL occurs oligomerization and translocation into the plasma membrane. Exactly how MLKL causes necrosis is still a mystery, although there are two competing hypotheses at the moment: It’s possible that MLKL serves as a plasma membrane platform for the recruitment of Ca2+ or Na+ ion channels (44, 45), or that MLKL forms part of a complex that creates a pore in the membrane (46). In order to better understand the process of necroptosis, we draw a Figure 2 to show the process.
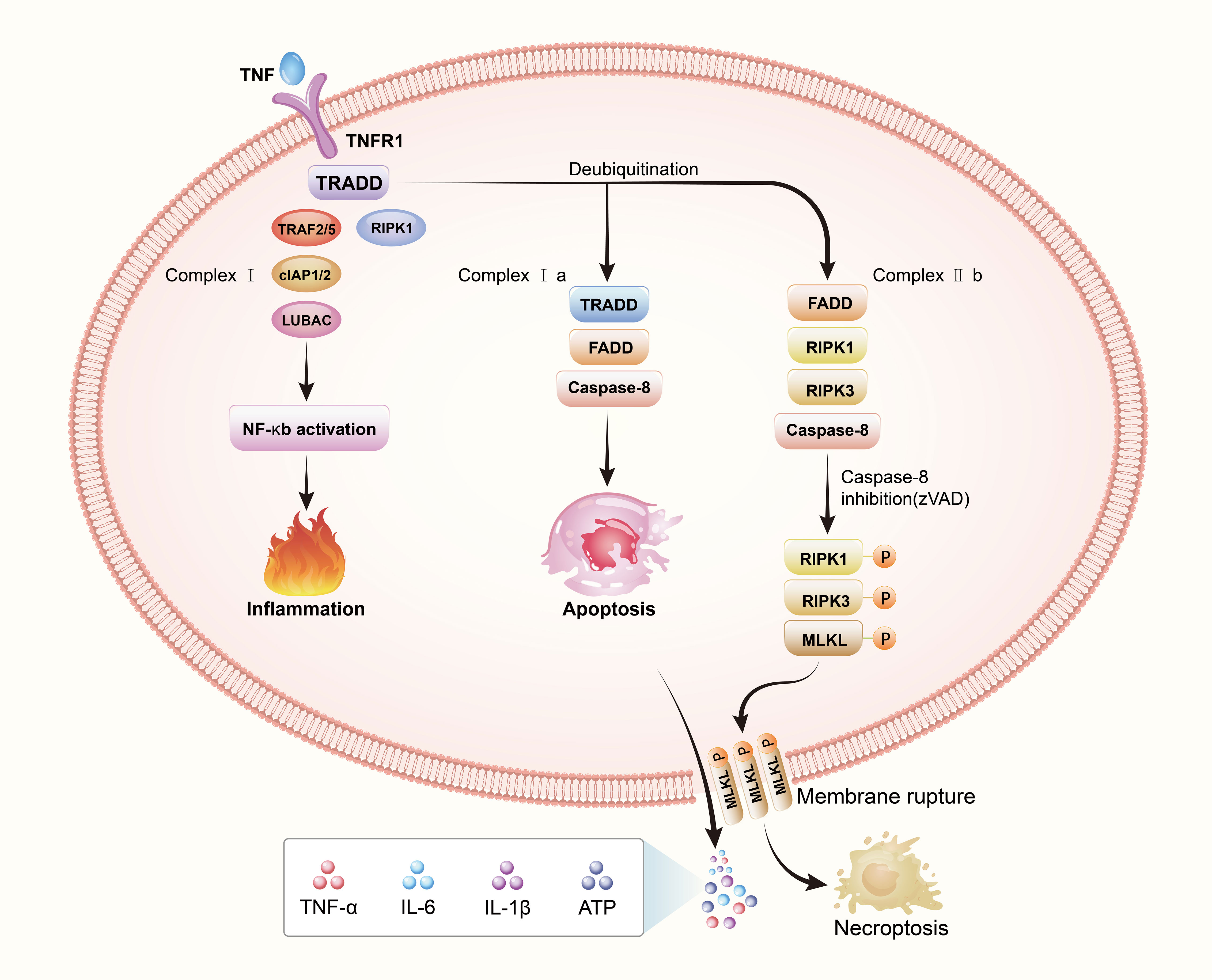
Figure 2 Necroptosis: a regulatory cell death. The death receptor signaling pathway is responsible for necroptosis This image depicts the interaction between tumor necrosis factor (TNF) and TNF receptor 1(TNFR1). When the binding of tumor necrosis factor-α(TNFα) to TNF receptor 1 (TNFR1), TNFR1 ligation triggers the assembly of complex I composed of TRADD, RIPK1, TRAF2/5, cIAP1/2 and LUBAC. Cell survival, apoptosis, and necroptosis are all possible consequences of complex I formation, which are regulated by different downstream cytosolic signaling complexes. Cell survival is boosted by ubiquitination of RIPK1, which activates the NF-κB pathway and promotes cell proliferation. Under a certain circumstance, deubiquitination of RIPK1 can result in the complex recruiting TRADD, FADD and caspase-8 forming complex IIa which activates apoptosis, or recruiting FADD, RIPK3, and caspase-8 forming complex IIb, which activates necroptosis. By blocking caspase-8, the binding of activated RIPK1 and RIPK3 leads to RIPK3 activation, which then phosphorylates and activates a pseudokinase called MLKL. MLKL undergoes oligomerization and translocates to and permeabilizes the plasma membrane to induce necroptosis.
A bone cell consists of osteocytes, osteoclasts, and osteoblasts. Adult skeletons are composed primarily of osteoclasts. A large percentage of bone cells are comprised of osteocytes (90-95%), while a smaller percentage is comprised of osteoclasts and osteoblasts (5-10%) (47). A bone mesenchymal stem cell (BMSC) differentiates into osteoblasts and osteocytes (48). Osteocytes become encased in their surrounding recently-mineralized bone matrix and then the new bone is formed (49, 50). The bone remodeling process and the incidence of osteoporosis are influenced by osteoocytes, osteoclasts, and osteoblasts (51). Maintaining a healthy balance between bone formation and destruction, as well as a continuous cycle of bone remodeling, are essential for maintaining the integrity of bone tissue. Osteoclasts mainly play the role of bone resorption, whereas osteoblasts mainly play the role of bone reconstruction, including the formation, mineralization, and secretion of osteocytes. They mutually restrict and balance the metabolism of bone tissues (52, 53). When an imbalance appears between the bone resorptive and bone-forming, this imbalance leads to loss of bone mass and strength, resulting in osteoporosis (54, 55). Obsession with the link between necroptosis and osteoporosis has grown during the past two decades. Necroptosis, which occurs in bone cells, such as osteocytes, osteoclasts and osteoblasts, has been linked to bone loss.
Approximately 90% to 95% of the bone’s mature cells are osteocytes (56, 57). Osteocytes play a vital function in transmitting diverse signals to the interfaces between osteoclasts and osteoblasts (58), demonstrating that osteocytes may engage in the regulation of bone resorption and creation (58). In addition, mounting evidence is that the continuing loss of osteocytes due to cell death significantly contributes to osteoporosis (59, 60). It was discovered in 2016 by Cui et al. that ovariectomy (OVX) rats suffering from estrogen deficiency had higher rates of osteocyte apoptosis and necroptosis (29). Nec-1 (a particular inhibitor of receptor-interacting protein 1) was shown to prevent significantly necroptotic death of osteocytes, thereby slowing down the loss of bone mass in the animals. However, it has not been shown to a considerable degree that necroptosis and apoptosis are responsible for the loss of osteocytes, however. That’s why the study of necroptosis and apoptotic cell death in rats with osteoporosis deficiency has been extended by He et al. (60). Apoptosis and necroptosis were found to contribute to osteocyte loss in OVX-induced osteoporosis, with necroptosis having a more significant influence than apoptosis on osteocyte loss. There is a strong correlation between necroptosis and osteoporosis development based on these findings.
One of the most significant aspects of bone regeneration is osteoblasts, which play a crucial role in a bone matrix formation, secretion, and mineralization (61). Increased necroptosis of osteoblasts and reduced bone formation and osteogenesis were seen in a model of excessive ethyl alcohol consumption in mice, as shown by Guo et al. in their study. A key inhibitor of RIPK1 kinase in the necroptosis pathway, necrostatin-1 (Nec-1), inhibits RIPK1/RIPK3/MLKL signaling, and thus might inhibit alcohol-induced osteopenia through decreased activation of osteoblast necroptosis (28). Furthermore, Shi et al. discovered concurrently identical events implicating necroptosis in the cell death of mouse osteoblasts (62).
In the bone-resorbing pit between the osteoclasts and the bone, The Osteoclasts secrete lysosomal enzymes to degrade the mineralized bone matrix and perform the function of bone resorption (63). The osteoclastogenesis process depends on transforming growth factor (TGF-)-activated kinase 1 (TAK1). Loss of TAK1 potentiates spontaneous necroptosis in osteoclasts. It suggests necroptosis occur in osteoclasts (64). Inhibiting RIPK1 ubiquitination occurs using Smac-mimetics (SM), which activates RIPK1 and causes necroptosis. After application of the SM, human osteoclasts showed less stability and viability, and induced TNF-dependent cell death in osteoclasts precursors (pre-osteoclasts) (65). It shows that necroptosis inhibits human osteoclastogenesis, and that the regulatory mechanism may provide interesting avenues for lowering bone deterioration. Mullinet et al. proved, from the standpoint of genetic gene variations, that RIPK3 is one of the genes identified in the research as associated with osteoporosis (66). In RIPK3-deficient mice, the degree of diaphyseal trabecularisation was observed to be drastically reduced. Analysis of bone sections stained with TRAP indicated a substantial increase in the overall number of osteoclasts in the bones of RIPK3-deficient animals. An excess of osteoclasts might result from the absence of RIPK3, which is involved in osteoclast necroptosis. Loss of necroptosis may lead to an excess of osteoclasts. Ultimately contribute to significant bone loss.Following the aforementioned research, we assumed that necroptosis of osteoclasts would reduce the occurrence of bone resorption and increase bone formation. In contrast, necroptosis of osteoblasts and osteocytes would lead to reduced bone and bone formation.
As a result of osteoporosis, bone mass declines, along with its microarchitecture, making the bones more fragile and more likely to break (67). OP is classified as primary, secondary, and idiopathic. Senile osteoporosis (SOP) and postmenopausal osteoporosis (PMOP) are the most common forms of OP. Age-related bone loss is the hallmark of SOP. In general, it refers to osteoporosis after 70 years of age and is caused by specific biological aging of the skeletal system (68). Another primary osteoporosis is PMOP, mainly due to estrogen deficiency. The most common secondary osteoporosis is diabetic osteoporosis (DOP) and glucocorticoid-induced osteoporosis (GIOP) (69). The necroptosis in osteocytes, osteoblasts and osteoclasts and the imbalance of bone metabolism are involved in the pathophysiological process of primary and secondary osteoporosis. In order to better understand the relationship between necroptosis and osteoporosis, we draw a Figure 3 to show their relationship.
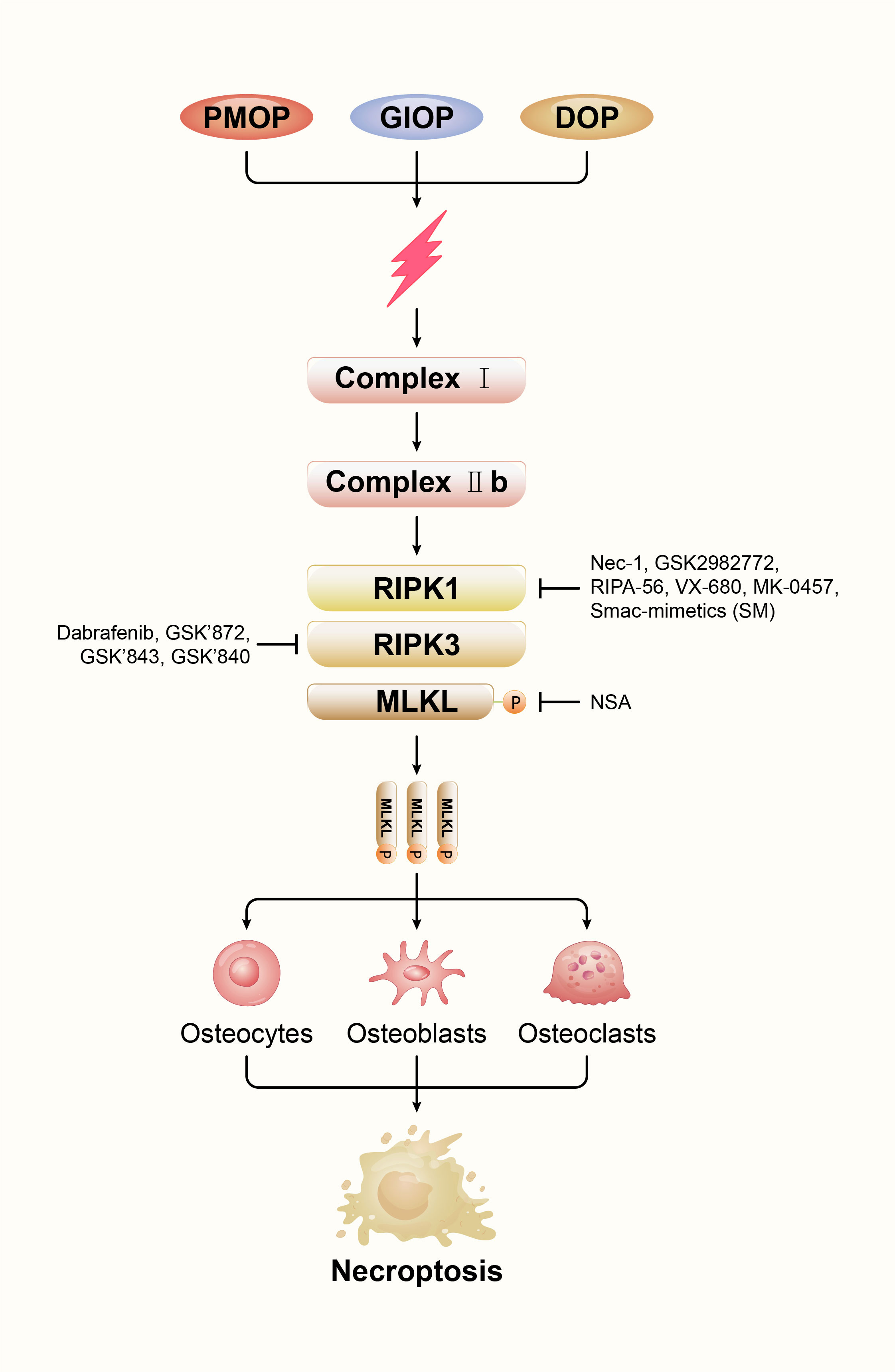
Figure 3 Effects of various therapeutic inhibitors of necroptosis on components of necrosomes.
One of the most common types of osteoporosis in women is PMOP, which arises after the cessation of estrogen (E2) in women. Customarily, the twin processes of bone resorption and creation are closely connected to preserving bone homeostasis (70). With PMOP, postmenopausal women have a certain degree of bone loss, resulting in a shift toward higher bone resorption and reduced bone growth (71). Bone cell apoptosis and necroptosis have been substantially linked to PMOP (51, 66). In addition to apoptosis, OVX-induced estrogen deficiency in the rat femur enhanced necroptosis in osteocytes, according to Cui et al. Osteocyte necroptosis is shown to be a substantial contributor to estrogen deficiency-induced bone loss in the OVX rat model (59). As far, as the roles of necroptosis and apoptosis in osteocyte loss remain largely a scientific mystery. When estrogen shortage is present, necroptosis and apoptosis play a significant role in the gradual loss of osteocytes (60). Their findings suggest that OVX-induced osteoporosis is caused by necroptosis, which may have a greater impact on osteoclast loss than apoptosis, and apoptosis, which is thought to have a less impact on osteoclast loss. Nec-1 therapy also significantly reduces necroptosis in OVX mice, therefore preventing osteoporosis development, according to Cui et al. Because of this, scientists believe that the death process known as necroptosis might be a unique method of treating osteoporosis (29).
GIOP has become the most common form of secondary OP in recent years, and it is commonplace in adolescents and young adults (72). The suppression of bone growth by GCs has been regarded as a crucial factor in this occurrence, the fact that the specific processes behind GIOP are still unclear. A previous study reported that apoptosis is an essential mechanism in the GIOP process (73). However, the modest amount of apoptotic osteoblasts cannot explain the elevated bone fragility observed in GC-treated patients (74). It might suggest that GIOP involves other cell death pathways in osteocytes and osteoblasts. For the first time, Feng et al. are examining the expression of RIPK1 and RIPK3 in GIOP rats after glucocorticoid (dexamethasone) injection to induce osteoporosis pharmacologically (18). Nec-1 protect GIOP rats is then tested utilizing biochemical markers and histomorphometric indices. When given necroptosis inhibitor Nec-1, rats suffering from glucocorticoids produces more bones because of the necroptosis inhibitor Nec-1, the researchers found. This evidence has indicated that necroptosis play an important role in the progression of GIOP. Thus, more studies are required to better understand the processes of necroptosis in GIOP.
By 2045, 578 million and 700 million persons are expected to live with diabetes, respectively. Diabetes is expected to affect one in every eleven adults in 2019, or 463 million (75). The condition of DOP occurs when there is a decrease in bone mineral density (BMD) and a change in bone microstructure, as well as an increase in bone fragility due to diabetes. A significant financial burden is imposed on patients suffering from DOP, which significantly diminishes their quality of life (76). On a yearly basis, DOP rates rise as diabetes prevalence increases around the world. There is evidence from clinical studies that between 50 and 65% of people with diabetes have lower bone mineral density and are at a higher risk of fractures, while roughly 35% have osteoporosis (77). The exact processes of DOP are still unclear. Loss of osteocyte viability has been proposed as a major pathogenic component in DOP (78). While it is yet unknown how osteocytes occurs cell death, Some studies show that apoptosis, autophagy, and ferroptosis are significant mechanisms of osteocyte death in DOP (78–80). However, whether necroptosis is involved in DOP has not been experimentally confirmed. Among other complications of diabetes, such as diabetic nephropathy, diabetic retinopathy, and diabetic cardiomyopathy, many studies have reported that necroptosis has an important role (81–83). Therefore, we speculate that necroptosis also has a role in DOP. This will be an interesting direction for future research.
A specific group of protein molecules mediates necroptosis. Therefore, inhibitors have been developed by researchers to target these key proteins. We described the effects of various inhibitors on necroptosis, with the aim of identifying new and therapeutic agents for necroptosis (Figure 3). These findings of necroptosis inhibitors will provide potential treatment strategies for osteoporosis.
Degterev et al. in 2005 demonstrated a particularly potent small-molecule necrosis inhibitor called Nec-1 (10). The mechanism of action of Nec-1 is its binding to the hydrophobic pocket of the carboxyl and amino lobes of the RIPK1 kinase domain (32, 84). Cui et al. investigated that Nec-1 can prevent necroptotic osteocytes, mediated by RIPK1, and inhibit osteoporosis progression in OVX rats (29). In addition to Nec-1, GSK2982772, compound 56 (RIPA-56) and VX-680 and MK-0457 were confirmed as emerging inhibitors of RIPK1 (85–87). These drugs have been used in other diseases and have potential for the treatment of pathology caused or aggravated by necroptotic cell death. However, its application in osteoporosis still needs to be further explored. Collectively, RIPK1 kinase may serve as a new target for the development of therapeutic drugs for osteoporosis.
RIPK3 is a critical regulator of necroptosis. The B-Raf inhibitor dabrafenib was demonstrated to be a potent inhibitor of RIPK3 and is used in the treatment of melanoma (88). The mechanism of dabrafenib is to disrupt the interaction between RIPK3 and MLKL (89). In mouse models of ischemic brain injury, Dabrafenib may attenuate TNF-α-induced necroptotic pathway (90). However, whether dabrafenib can inhibit necroptosis and delay the progression of osteoporosis remains to be determined. In addition, GSK’872, GSK’843 and GSK’840 were identified as a class of specific RIPK3 inhibitors of necroptosis by Mocarski and colleagues (91). The application of these drugs to osteoporosis remains to be explored.
The phosphorylation of MLKL has a significant role in regulating necrosis. However, few effective inhibitors have yet been identified. Hildebrand et al. reported a small molecule (unnamed) that binds the MLKL pseudokinase domain and retards membrane translocation to inhibit necroptotic signalling (92). In addition, Necrosulfonamide (NSA) inhibits MLKL-Mediated necrosis by blocking its N-Terminal CC domain function of MLKL (34). Considering that MLKL is an essential protein for the execution of necroptosis, it may be a potential ideal target for inhibiting necroptosis.
Deficiencies in bone density and microstructure are hallmarks of osteoporosis, a systemic metabolic bone disease (93). Osteoporosis and its debilitating effects continue to pose difficulties for clinicians. Therefore, understanding the molecular mechanisms of necroptosis and osteoporosis can provide substantial insights into the field of bone metabolism.
Necroptosis and osteoporosis are discussed in this article, specifically PMOP, GIOP, and DOP. Although tremendous progress has been made in the research of necroptosis, several challenges need to be resolved. Necroptosis research is still in its infancy, and its precise mechanism and associated signalling pathways are yet unknown. To create successful treatments, it is necessary to investigate these newly identified processes, their interconnected signalling networks, and molecular targets. An effective method of treating osteoporosis is to use Nec-1, an inhibitor of necroptosis that has been demonstrated to be a specific inhibitor of necroptosis (18, 29). Studies on the pharmacologic inhibition of necroptosis in osteoporosis is limited. Therefore, it’s necessary to found other potential drugs to depress necroptosis in osteoporosis. In addition, the role of necroptosis has been confirmed in castrated mice and glucocorticoid mice. However, in diabetic mice, it has not yet been confirmed, so it is necessary to confirm the relationship between osteoporosis and necroptosis in diabetic mice.
Currently, there is a lot of interest in finding out more about the processes that underlie necroptosis and osteoporosis thanks to these promising new paths in study. We have a few ideas for further research: (1).Bone reconstruction is orchestrated by osteoblast, osteocyte and osteoclast mutually (94). The interaction and crosstalk among osteoclasts, osteoblasts, and osteocytes should be considered in the treatment of osteoporosis, especially when osteoclasts, osteoblasts, and osteocytes undergo necroptosis. (2) One research has systematically confirmed that the necroptosis pathway is involved in the iron overload-induced death of osteoblastic cells for the first time. ROS as its key molecule (95). This suggests a link between necroptosis and ferroptosis. Moreover, cell death includes autophagy, pyroptosis, ferroptosis, necroptosis, and lysosomal cell death. So are these types of cell deaths having a connection with osteoporosis? Are there interactions and crosstalk between these cell deaths in osteoporosis? Thus, further researches are needed to be comprehensively investigated the correlation among autophagy, necroptosis, ferroptosis, pyroptosis and lysosomal cell death in osteoporosis. (3).it is essential to elucidate which type of programmed cell death plays the dominant role in osteoporosis
Necroptosis is a regulated programmed cell death involved in developing and progressing many diseases. Osteoporosis is attracting more and more attention because of its high morbidity. An overview of necroptosis and its relevance to osteoporosis is provided in this review. A better understanding of necroptosis may generate new ideas for bone metabolism research. Osteoporosis medications now on the market target osteoclasts, which are responsible for bone resorption. These medications can slow the growth of osteoporosis, but they also have some unwanted side effects. Necroptosis seems to have a therapeutic impact on osteoporosis by influencing osteoclasts, osteoblasts, and osteocytes, according to this promising new study. It is predicted to be a novel treatment for osteoporosis prevention and treatment with fewer adverse effects as a result of necroptosis management. Despite necroptosis being a unique kind of cell death, additional research is needed to uncover new targets for controlling necroptosis in the therapy of osteoporotic bone loss, which might lead to more effective and better therapeutic alternatives.
XH and ZW searched and reviewed literature, drafted manuscript and revision; CK and YW discussed and revised the manuscript; WZ and WW (8th author) and YL provided critical comments; WW (8th author) and SL designed and formulated the review theme, revised and finalized the manuscript.
This study was supported by the National Key Research and Development Program of China (No.2020YFC2004900), National Natural Science Foundation of China (H0608), Natural Science Foundation of China (No.81672201, 81871794) and Beijing Hospitals Authority’Ascent Plan (No.DFL20190802).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Genant HK, Cooper C, Poor G, Reid I, Ehrlich G, Kanis J, et al. Interim report and recommendations of the world health organization task-force for osteoporosis. Osteoporos Int (1999) 10(4):259–64. doi: 10.1007/s001980050224
2. Tao Z, Wang J, Wen K, Yao R, Da W, Zhou S, et al. Pyroptosis in osteoblasts: A novel hypothesis underlying the pathogenesis of osteoporosis. Front Endocrinol (Lausanne) (2020) 11:548812. doi: 10.3389/fendo.2020.548812
3. Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int (2006) 17(12):1726–33. doi: 10.1007/s00198-006-0172-4
4. Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem (2010) 285(33):25103–8. doi: 10.1074/jbc.R109.041087
5. Borciani G, Montalbano G, Baldini N, Cerqueni G, Vitale-Brovarone C. And ciapetti G Co-culture systems of osteoblasts and osteoclasts: Simulating in vitro bone remodeling in regenerative approaches. Acta Biomater (2020) 108:22–45. doi: 10.1016/j.actbio.2020.03.043
6. Olszynski WP, Shawn Davison K, Adachi JD, Brown JP, Cummings SR, Hanley DA, et al. Osteoporosis in men: epidemiology, diagnosis, prevention, and treatment. Clin Ther (2004) 26(1):15–28. doi: 10.1016/s0149-2918(04)90002-1
7. Tella SH, Gallagher JC. Prevention and treatment of postmenopausal osteoporosis. J Steroid Biochem Mol Biol (2014) 142:155–70. doi: 10.1016/j.jsbmb.2013.09.008
8. Aihara S, Yamada S, Oka H, Kamimura T, Nakano T, Tsuruya K, et al. Hypercalcemia and acute kidney injury induced by eldecalcitol in patients with osteoporosis: a case series of 32 patients at a single facility. Ren Fail (2019) 41(1):88–97. doi: 10.1080/0886022x.2019.1578667
9. Geusens P, Oates M, Miyauchi A, Adachi JD, Lazaretti-Castro M, Ebeling PR, et al. The effect of 1 year of romosozumab on the incidence of clinical vertebral fractures in postmenopausal women with osteoporosis: Results from the FRAME study. JBMR Plus (2019) 3(10):e10211. doi: 10.1002/jbm4.10211
10. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol (2005) 1(2):112–9. doi: 10.1038/nchembio711
11. Shan B, Pan H, Najafov A, Yuan J. Necroptosis in development and diseases. Genes Dev (2018) 32(5-6):327–40. doi: 10.1101/gad.312561.118
12. Pasparakis M. Vandenabeele p necroptosis and its role in inflammation. Nature (2015) 517(7534):311–20. doi: 10.1038/nature14191
13. Moquin D, Chan FK. The molecular regulation of programmed necrotic cell injury. Trends Biochem Sci (2010) 35(8):434–41. doi: 10.1016/j.tibs.2010.03.001
14. Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta (2013) 1833(12):3448–59. doi: 10.1016/j.bbamcr.2013.06.001
15. Pacifici R. T Cells and post menopausal osteoporosis in murine models. Arthritis Res Ther (2007) 9(2):102. doi: 10.1186/ar2126
16. Tyagi AM, Srivastava K, Sharan K, Yadav D, Maurya R, Singh D. Daidzein prevents the increase in CD4+CD28null T cells and b lymphopoesis in ovariectomized mice: a key mechanism for anti-osteoclastogenic effect. PloS One (2011) 6(6):e21216. doi: 10.1371/journal.pone.0021216
17. Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol (2010) 22(2):263–8. doi: 10.1016/j.ceb.2009.12.003
18. Feng M, Zhang R, Gong F, Yang P, Fan L, Ni J, et al. Protective effects of necrostatin-1 on glucocorticoid-induced osteoporosis in rats. J Steroid Biochem Mol Biol (2014) 144:455–62. doi: 10.1016/j.jsbmb.2014.09.005
19. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
20. Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, et al. Classification of cell death: recommendations of the nomenclature committee on cell death. Cell Death Differ (2005) 12 Suppl 2:1463–7. doi: 10.1038/sj.cdd.4401724
21. Metzstein MM, Stanfield GM, Horvitz HR. Genetics of programmed cell death in c. elegans: past, present and future. Trends Genet (1998) 14(10):410–6. doi: 10.1016/s0168-9525(98)01573-x
22. Delhalle S, Duvoix A, Schnekenburger M, Morceau F, Dicato M, Diederich M. An introduction to the molecular mechanisms of apoptosis. Ann N Y Acad Sci (2003) 1010:1–8. doi: 10.1196/annals.1299.001
23. Bergmann A. Autophagy and cell death: no longer at odds. Cell (2007) 131(6):1032–4. doi: 10.1016/j.cell.2007.11.027
24. Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev (2004) 18(11):1272–82. doi: 10.1101/gad.1199904
25. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ (2009) 16(1):3–11. doi: 10.1038/cdd.2008.150
26. Wang L, Liu S, Zhao Y, Liu D, Liu Y, Chen C, et al. Osteoblast-induced osteoclast apoptosis by fas ligand/FAS pathway is required for maintenance of bone mass. Cell Death Differ (2015) 22(10):1654–64. doi: 10.1038/cdd.2015.14
27. Song S, Guo Y, Yang Y, Fu D. Advances in pathogenesis and therapeutic strategies for osteoporosis. Pharmacol Ther (2022) 237:108168. doi: 10.1016/j.pharmthera.2022.108168
28. Guo M, Huang YL, Wu Q, Chai L, Jiang ZZ, Zeng Y, et al. Chronic ethanol consumption induces osteopenia via activation of osteoblast necroptosis. Oxid Med Cell Longev (2021) 2021:3027954. doi: 10.1155/2021/3027954
29. Cui H, Zhu Y, Yang Q, Zhao W, Zhang S, Zhou A, et al. Necrostatin-1 treatment inhibits osteocyte necroptosis and trabecular deterioration in ovariectomized rats. Sci Rep (2016) 6:33803. doi: 10.1038/srep33803
30. Galluzzi L, Kepp O, Chan FK, Kroemer. Necroptosis: Mechanisms and relevance to disease. Annu Rev Pathol (2017) 12:103–30:G. doi: 10.1146/annurev-pathol-052016-100247
31. Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol (2014) 35:24–32. doi: 10.1016/j.semcdb.2014.02.006
32. Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol (2008) 4(5):313–21. doi: 10.1038/nchembio.83
33. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science (2009) 325(5938):332–6. doi: 10.1126/science.1172308
34. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031
35. Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe (2010) 7(4):302–13. doi: 10.1016/j.chom.2010.03.006
36. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol (2000) 1(6):489–95. doi: 10.1038/82732
37. Zhang Y, Chen X, Gueydan C, Han J. Plasma membrane changes during programmed cell deaths. Cell Res (2018) 28(1):9–21. doi: 10.1038/cr.2017.133
38. Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ (2013) 20(10):1381–92. doi: 10.1038/cdd.2013.94
39. Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell (2008) 133(4):693–703. doi: 10.1016/j.cell.2008.03.036
40. Legarda-Addison D, Hase H, O'Donnell MA, Ting AT. NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell Death Differ (2009) 16(9):1279–88. doi: 10.1038/cdd.2009.41
41. Jaco I, Annibaldi A, Lalaoui N, Wilson R, Tenev T, Laurien L, et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol Cell (2017) 66(5):698–710.e5. doi: 10.1016/j.molcel.2017.05.003
42. Moujalled DM, Cook WD, Okamoto T, Murphy J, Lawlor KE, Vince JE, et al. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis (2013) 4(1):e465. doi: 10.1038/cddis.2012.201
43. Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol (2015) 16(7):689–97. doi: 10.1038/ni.3206
44. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol (2014) 16(1):55–65. doi: 10.1038/ncb2883
45. Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res (2014) 24(1):105–21. doi: 10.1038/cr.2013.171
46. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell (2014) 54(1):133–46. doi: 10.1016/j.molcel.2014.03.003
47. Bonewald LF. Osteocytes as dynamic multifunctional cells. Ann N Y Acad Sci (2007) 1116:281–90. doi: 10.1196/annals.1402.018
49. Franz-Odendaal TA, Hall BK, Witten PE. Buried alive: how osteoblasts become osteocytes. Dev Dyn (2006) 235(1):176–90. doi: 10.1002/dvdy.20603
50. Dallas SL, Prideaux M, Bonewald LF. The osteocyte: an endocrine cell ... and more. Endocr Rev (2013) 34(5):658–90. doi: 10.1210/er.2012-1026
51. Han Y, You X, Xing W, Zhang Z, Zou W. Paracrine and endocrine actions of bone-the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res (2018) 6:16. doi: 10.1038/s41413-018-0019-6
52. Zhao H, Ito Y, Chappel J, Andrews NW, Teitelbaum SL, Ross FP. Synaptotagmin VII regulates bone remodeling by modulating osteoclast and osteoblast secretion. Dev Cell (2008) 14(6):914–25. doi: 10.1016/j.devcel.2008.03.022
53. Kołodziejska B, Stępień N, Kolmas J. The influence of strontium on bone tissue metabolism and its application in osteoporosis treatment. Int J Mol Sci (2021) 22(12):6564. doi: 10.3390/ijms22126564
54. Compston JE, McClung MR, Leslie WD. Osteoporosis. Lancet (2019) 393(10169):364–76. doi: 10.1016/s0140-6736(18)32112-3
55. Yamamoto Y, Chiba T, Dohmae S, Higashi K, Nakazawa A. Osteoporosis medication after fracture in older adults: an administrative data analysis. Osteoporos Int (2021) 32(6):1245–6. doi: 10.1007/s00198-021-05973-9
56. Milovanovic P, Zimmermann EA, Hahn M, Djonic D, Püschel K, Djuric M, et al. Osteocytic canalicular networks: morphological implications for altered mechanosensitivity. ACS Nano (2013) 7(9):7542–51. doi: 10.1021/nn401360u
57. Mattinzoli D, Messa P, Corbelli A, Ikehata M, Mondini A, Zennaro C, et al. Application of retinoic acid to obtain osteocytes cultures from primary mouse osteoblasts. J Vis Exp (2014) 13(87):51465. doi: 10.3791/51465
58. Heino TJ, Hentunen TA, Väänänen HK. Conditioned medium from osteocytes stimulates the proliferation of bone marrow mesenchymal stem cells and their differentiation into osteoblasts. Exp Cell Res (2004) 294(2):458–68. doi: 10.1016/j.yexcr.2003.11.016
59. Cui H, Zhu Y, Jiang D. The RIP1-RIP3 complex mediates osteocyte necroptosis after ovariectomy in rats. PloS One (2016) 11(3):e0150805. doi: 10.1371/journal.pone.0150805
60. He B, Zhu Y, Cui H, Sun B, Su T, Wen P. Comparison of necroptosis with apoptosis for OVX-induced osteoporosis. Front Mol Biosci (2021) 8:790613. doi: 10.3389/fmolb.2021.790613
61. Florencio-Silva R, Sasso GR, Sasso-Cerri E, Simões MJ, Cerri PS. Biology of bone tissue: Structure, function, and factors that influence bone cells. BioMed Res Int (2015) 2015:421746. doi: 10.1155/2015/421746
62. Shi G, Jia P, Chen H, Bao L, Feng F, Tang H. Necroptosis occurs in osteoblasts during tumor necrosis factor-α stimulation and caspase-8 inhibition. Braz J Med Biol Res (2018) 52(1):e7844. doi: 10.1590/1414-431x20187844
63. Teitelbaum SL. Bone resorption by osteoclasts. Science (2000) 289(5484):1504–8. doi: 10.1126/science.289.5484.1504
64. Lamothe B, Lai Y, Xie M, Schneider MD, Darnay BG. TAK1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis. Mol Cell Biol (2013) 33(3):582–95. doi: 10.1128/mcb.01225-12
65. Moen IN, Westhrin M, Håland E, Haug M, Nonstad U, Klaharn M, et al. Smac-mimetics reduce numbers and viability of human osteoclasts. Cell Death Discovery (2021) 7(1):36. doi: 10.1038/s41420-021-00415-1
66. Mullin BH, Tickner J, Zhu K, Kenny J, Mullin S, Brown SJ, et al. Characterisation of genetic regulatory effects for osteoporosis risk variants in human osteoclasts. Genome Biol (2020) 21(1):80. doi: 10.1186/s13059-020-01997-2
67. Lane NE. Epidemiology, etiology, and diagnosis of osteoporosis. Am J Obstet Gynecol (2006) 194(2 Suppl):S3–11. doi: 10.1016/j.ajog.2005.08.047
68. Guo Y, Jia X, Cui Y, Song Y, Wang S, Geng Y, et al. Sirt3-mediated mitophagy regulates AGEs-induced BMSCs senescence and senile osteoporosis. Redox Biol (2021) 41:101915. doi: 10.1016/j.redox.2021.101915
69. Gao Z, Chen Z, Xiong Z, Liu X. Ferroptosis - a new target of osteoporosis. Exp Gerontol (2022) 165:111836. doi: 10.1016/j.exger.2022.111836
70. Eastell R. Treatment of postmenopausal osteoporosis. N Engl J Med (1998) 338(11):736–46. doi: 10.1056/nejm199803123381107
71. Syed FA, Hoey KA. Integrative physiology of the aging bone: insights from animal and cellular models. Ann N Y Acad Sci (2010) 1211:95–106. doi: 10.1111/j.1749-6632.2010.05813.x
72. Briot K, Roux C. Glucocorticoid-induced osteoporosis. RMD Open (2015) 1(1):e000014. doi: 10.1136/rmdopen-2014-000014
73. Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. potential mechanisms of their deleterious effects on bone. J Clin Invest (1998) 102(2):274–82. doi: 10.1172/jci2799
74. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Use of oral corticosteroids and risk of fractures. J Bone Miner Res (2000) 15(6):993–1000. doi: 10.1359/jbmr.2000.15.6.993
75. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the international diabetes federation diabetes atlas, 9(th) edition. Diabetes Res Clin Pract (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
76. Qi S, He J, Han H, Zheng H, Jiang H, Hu CY, et al. Anthocyanin-rich extract from black rice (Oryza sativa l. japonica) ameliorates diabetic osteoporosis in rats. Food Funct (2019) 10(9):5350–60. doi: 10.1039/c9fo00681h
77. Ma R, Zhu R, Wang L, Guo Y, Liu C, Liu H, et al. Diabetic osteoporosis: A review of its traditional Chinese medicinal use and clinical and preclinical research. Evid Based Complement Alternat Med (2016) 2016:3218313. doi: 10.1155/2016/3218313
78. Yang Y, Lin Y, Wang M, Yuan K, Wang Q, Mu P, et al. Targeting ferroptosis suppresses osteocyte glucolipotoxicity and alleviates diabetic osteoporosis. Bone Res (2022) 10(1):26. doi: 10.1038/s41413-022-00198-w
79. Wang N, Xu P, Wu R, Wang X, Wang Y, Shou D, et al. Timosaponin BII improved osteoporosis caused by hyperglycemia through promoting autophagy of osteoblasts via suppressing the mTOR/NFκB signaling pathway. Free Radic Biol Med (2021) 171:112–23. doi: 10.1016/j.freeradbiomed.2021.05.014
80. Zhao W, Zhang WL, Yang B, Sun J, Yang MW. NIPA2 regulates osteoblast function via its effect on apoptosis pathways in type 2 diabetes osteoporosis. Biochem Biophys Res Commun (2019) 513(4):883–90. doi: 10.1016/j.bbrc.2019.04.030
81. Chung H, Lee SW, Hyun M, Kim SY, Cho HG, Lee ES, et al. Curcumin blocks high glucose-induced podocyte injury via RIPK3-dependent pathway. Front Cell Dev Biol (2022) 10:800574. doi: 10.3389/fcell.2022.800574
82. Xu X, Lan X, Fu S, Zhang Q, Gui F, Jin Q, et al. Dickkopf-1 exerts protective effects by inhibiting PANoptosis and retinal neovascularization in diabetic retinopathy. Biochem Biophys Res Commun (2022) 617(Pt 2):69–76. doi: 10.1016/j.bbrc.2022.05.001
83. Gong W, Zhang S, Chen Y, Shen J, Zheng Y, Liu X, et al. Protective role of hydrogen sulfide against diabetic cardiomyopathy via alleviating necroptosis. Free Radic Biol Med (2022) 181:29–42. doi: 10.1016/j.freeradbiomed.2022.01.028
84. Xie T, Peng W, Yan C, Wu J, Gong X. Shi y structural insights into RIP3-mediated necroptotic signaling. Cell Rep (2013) 5(1):70–8. doi: 10.1016/j.celrep.2013.08.044
85. Harris PA, Berger SB, Jeong JU, Nagilla R, Bandyopadhyay D, Campobasso N, et al. Discovery of a first-in-Class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J Med Chem (2017) 60(4):1247–61. doi: 10.1021/acs.jmedchem.6b01751
86. Ren Y, Su Y, Sun L, He S, Meng L, Liao D, et al. Discovery of a highly potent, selective, and metabolically stable inhibitor of receptor-interacting protein 1 (RIP1) for the treatment of systemic inflammatory response syndrome. J Med Chem (2017) 60(3):972–86. doi: 10.1021/acs.jmedchem.6b01196
87. Martens S, Goossens V, Devisscher L, Hofmans S, Claeys P, Vuylsteke M, et al. RIPK1-dependent cell death: a novel target of the aurora kinase inhibitor tozasertib (VX-680). Cell Death Dis (2018) 9(2):211. doi: 10.1038/s41419-017-0245-7
88. Alcalá AM, Flaherty KT. BRAF inhibitors for the treatment of metastatic melanoma: clinical trials and mechanisms of resistance. Clin Cancer Res (2012) 18(1):33–9. doi: 10.1158/1078-0432.Ccr-11-0997
89. Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su Y, et al. The b-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis (2014) 5(6):e1278. doi: 10.1038/cddis.2014.241
90. Cruz SA, Qin Z, Stewart AFR, Chen HH. Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regener Res (2018) 13(2):252–6. doi: 10.4103/1673-5374.226394
91. Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell (2014) 56(4):481–95. doi: 10.1016/j.molcel.2014.10.021
92. Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U.S.A. (2014) 111(42):15072–7. doi: 10.1073/pnas.1408987111
93. Srivastava M, Deal C. Osteoporosis in elderly: prevention and treatment. Clin Geriatr Med (2002) 18(3):529–55. doi: 10.1016/s0749-0690(02)00022-8
94. Xiong Y, Chen L, Yan C, Zhou W, Yu T, Sun Y, et al. M2 macrophagy-derived exosomal miRNA-5106 induces bone mesenchymal stem cells towards osteoblastic fate by targeting salt-inducible kinase 2 and 3. J Nanobiotechnology (2020) 18(1):66. doi: 10.1186/s12951-020-00622-5
Keywords: osteoporosis, necroptosis, cell death, osteoclasts, osteoblasts
Citation: Hu X, Wang Z, Kong C, Wang Y, Zhu W, Wang W, Li Y, Wang W and Lu S (2022) Necroptosis: A new target for prevention of osteoporosis. Front. Endocrinol. 13:1032614. doi: 10.3389/fendo.2022.1032614
Received: 31 August 2022; Accepted: 03 October 2022;
Published: 19 October 2022.
Edited by:
Giacomina Brunetti, University of Bari Aldo Moro, ItalyReviewed by:
Xiao Lv, Huazhong University of Science and Technology, ChinaCopyright © 2022 Hu, Wang, Kong, Wang, Zhu, Wang, Li, Wang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Wang, d2FuZ3dlaTM3QGJ1YWEuZWR1LmNu; Shibao Lu, c3BpbmVsdUAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.