 Sara Arefhosseini
Sara Arefhosseini Mehrangiz Ebrahimi-Mameghani
Mehrangiz Ebrahimi-Mameghani Farzad Najafipour
Farzad Najafipour Helda Tutunchi
Helda Tutunchi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Endocrinol. , 07 November 2022
Sec. Translational and Clinical Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1032361
This article is part of the Research Topic Non-Alcoholic Fatty Liver Disease Across Endocrine Diseases: From Pathophysiology to Novel Therapeutic Approaches View all 5 articles
Nonalcoholic fatty liver disease (NAFLD) has emerged as the most frequent chronic liver disease globally. NAFLD is strongly associated with metabolic syndrome and it has been recently suggested that to rename NAFLD as metabolic dysfunction-associated fatty liver disease (MAFLD). NAFLD has been studied in different endocrine axes and accumulating body of clinical and experimental studies have suggested that NAFLD is associated with polycystic ovarian syndrome (PCOS), hypopituitarism, growth hormone deficiency (GHD), hypogonadism and other endocrine disorders. In fact, endocrine dysfunction may be considered as the major contributor for the development, progression, and severity of NAFLD. In the present comprehensive review, we discussed the epidemiological and clinical evidence on the epidemiology, pathophysiology, and management of NAFLD in endocrine disorders, with an emphasis on the effects of sex-specific hormones/conditions as well as molecular basis of NAFLD development in these endocrine diseases.
The liver, a dynamic endocrine organ, displays a crosstalk with extra-hepatic organs in order to mediate numerous metabolic, endocrine and hormonal signaling pathways (1). Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease and is associated with a wide spectrum of hepatic disorders ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), cirrhosis and hepatocellular carcinoma (HCC) (2). It is estimated that NAFLD may affect almost one third of the general population by 2030 (3). The “Multiple-Hit” theory, based on the synergic role of genetic and epigenetic factors including insulin resistance (IR), inflammation, oxidative stress (OS) and gut dysbiosis has been suggested for disease pathogenesis (4). Based on the metabolic roots of NAFLD, it has been recently suggested that to rename NAFLD as metabolic dysfunction-associated fatty liver disease (MAFLD) (5). Moreover, considering NAFLD as the “barometer of metabolic health” highlights the importance of metabolic disorders and associated endocrine dysfunction during disease progression (1, 6). NAFLD has been studied in different endocrine axes and to our knowledge, endocrinopathies are potent factors contributing to the incidence, progression and severity of NAFLD (7). Recently, accumulating body of clinical and experimental studies have suggested the association of NAFLD with polycystic ovarian syndrome (PCOS), hypopituitarism, growth hormone deficiency (GHD), hypogonadism and other endocrine disorders. Despite the extensive research that has been carried out, the exact inter-relationship between NAFLD and these conditions are not fully understood. In this review, we aimed to make an overview and summarize the epidemiologic, pathophysiologic and molecular approaches of recently published data on the association between NAFLD and endocrine disorders with an emphasize on the effects of sex-specific hormones/conditions.
NAFLD-associated endocrinopathies, may be clinically displayed as other pathological conditions such as sex hormone imbalance. PCOS-a multifaceted disorder with a spectrum of clinical presentations- is known as the most prevalent endocrine disease in women of childbearing age (8). PCOS is a potent risk factor for anovulatory infertility with a prevalence of 5-20% worldwide based on the used diagnostic criteria (9). More precisely, disease prevalence has been reported 6-10% based on the classic definition, while, the number reaches to 18-20% by considering the Rotterdam criteria due to comprising meddler phenotypes in the absence of hyperandrogenism (HA) (10). Current diagnostic characteristics of PCOS include chronic ovulatory dysfunction, clinical and/or biochemical HA and polycystic ovarian morphology confirmed through ultrasonography (8). PCOS is diagnosed when at least two of the mentioned criteria are present after excluding other endocrine conditions (8). PCOS is a multifactorial disorder with a combination of metabolic, endocrinological, and genetic disturbances (8). Regarding the Rotterdam criteria, PCOS is classified into three phenotypes including “classical”, “ovulatory” and “normo-androgenic” with different risks for the development of metabolic disorders (11). The women in “classical” subgroup exhibit the highest risk of IR and metabolic syndrome (Mets) in comparison to others (11). In this context, PCOS is known as a multi-system disease due to its concomitance with other disorders such as Mets, type 2 diabetes mellitus (T2DM), cardiovascular- and cerebrovascular –diseases (10). Indeed, obesity is an additional finding in almost 40% of PCOS patients, while it has been eliminated from disease diagnostic features (11). Obesity, in turn, may also lead to IR with subsequent hyperinsulinemia as an innate feature (11). Moreover, a number of studies imply that women with PCOS experience higher IR compared with healthy subjects, independent of body mass index (BMI) status (12). Given that up to 70% of patients with PCOS are diagnosed with IR, it is considered as one of the focal pathogenic pathways linking PCOS to other metabolic and endocrine disorders (13). Taken together, IR, obesity, metabolic disorder and androgen excess commonly trigger the risk of NAFLD among these patients at the baseline or over the time (14).
To date, NAFLD is a growing global health problem with a great concern for the health-care system, highlighting the need for exploring high-risk populations and managing related outcomes. NAFLD and PCOS are not only co-exist conditions, but also show a synergic burden based on their common pathological pathways (15). Moreover, the emerging rapid increase in the prevalence of NAFLD and PCOS shows the importance of conducting related epidemiological studies. The role of PCOS in NAFLD progression seems to remain unclear. To the best of our knowledge, while a great number of studies have documented the significant increased prevalence of NAFLD (generally in ultrasound-proven subjects) in patients with PCOS, other results remained inconclusive (16). For the assessment, PCOS was diagnosed through National Institutes of Health (NIH) 1990 definition, Rotterdam 2003 criteria, or Androgen Excess Society criteria (17). Furthermore, fatty liver was detected via assessing serum liver enzymes, ultrasonography, transient elastography, magnetic resonance spectroscopy (MRS) and liver biopsy. Brown et al. (18) first hypothesized a relation between PCOS and NAFLD in a case report in a 24-years old woman with PCOS and IR who was assessed for NAFLD via liver biopsy. Regarding to the literature review, four previous meta-analyses have investigated the interaction between NAFLD and PCOS. Ramezani-Binabaj et al. (19) first performed a meta-analysis of 7 studies in 2014 and reported the significant higher prevalence of NAFLD in women with PCOS as well as introducing PCOS as a probable risk factor for NAFLD progression. Due to the limitation of included studies, Rocha et al. (20) with regard to the collected data from 17 studies of 2734 cases vs. 2561 age and BMI-matched controls, confirmed previous results beside assessing the levels of serum androgen. In this meta-analysis, the higher prevalence of NAFLD among PCOS patients was attributed to increased free androgen index (FAI), serum total testosterone (TT), BMI and IR (20). Moreover, serum androgens were known as independent risk factors for NAFLD in PCOS patients by considering the finding that normo-androgen-PCOS women had normal liver function indices in comparison to PCOS women with HA (21). Subsequently, another meta-analysis in 2018 was conducted on 17 observational studies to clarify whether PCOS acts as an independent trigger for NAFLD or their co-existence is related to their mutual risk factors (22). Results were in accordance with former studies. Furthermore, after stratification for BMI and geographic origin, the marked higher risk of NAFLD was reported in PCOS women with HA (22). Wu et al. (22) shed a light into the possible correlation between androgen levels and NAFLD in an independent manner of IR. The most recent updated meta-analysis in 2021 with a larger sample size was carried out by Shengir et al. (15) including 23 studies with 7148 participants (4164 PCOS women vs 2984 controls) in agreement with previous meta-analyses. In all, aforementioned studies suggest that while liver biopsy is the gold-standard diagnostic method, the majority of studies have used ultrasonography for assessing NAFLD due to its reproducibility, low cost and noninvasiveness as well as satisfactory sensitivity and specificity for epidemiologic studies. Nevertheless, a small group of studies have used liver enzymes, biopsy and other diagnostic methods. As mentioned by Rocha et al. (20), the results seem to indicate an increased risk of NAFLD in PCOS suggesting that the heterogeneity in study designs and screening tools did not affect the overall result.
Based on the previous reports, contributing factors in NAFLD progression among PCOS women include IR, HA and disturbed metabolic markers, meanwhile, the current study depicts BMI as the main cofactor (15). A number of observational studies assessed the correlation between higher serum liver enzymes and PCOS (23–27). In this context, Schwimmer et al. (25) reported the higher risk of elevated serum alanine transaminase (ALT) in PCOS subjects after adjusting for confounders (30% and 15% increased risk for > 35 U/L and > 60 U/L cut-off values, respectively). The correlation between PCOS and increased serum aminotransferases was also observed in adolescent PCOS patients with a 15.4% prevalence (28). Two main factors, namely age and TT, have been reported to accelerate the progression of liver steatosis in adolescents (29). Taken together, current evidence demonstrates a positive correlation between increased aminotransferase levels and PCOS, although reported variations in prevalence may be related to study characteristics and performed diagnostic criteria (13). Besides, ALT levels have been attributed to lower insulin sensitivity in PCOS women (30). Other studies investigated the epi-phenomenon by using ultrasound data for detecting NAFLD (19, 23, 31–33). Gambarin-Gelwan et al. (12) demonstrated a 55% prevalence of NAFLD among PCOS women via ultrasonography. Interestingly 39% of the NAFLD proven subjects were lean. However, other studies did not report any significant correlation between ALT levels and PCOS in lean subgroup (34, 35). According to the lower prevalence of PCOS in lean women, the correlation between NAFLD and PCOS is still doubted in lean subjects (36). Compensatory, case- control studies also were performed to assess the issue. A study by Cerda et al. (23) showed higher prevalence of ultrasound confirmed-NAFLD (41.5% vs 19.4%, respectively) and elevated ALT levels (39% and 3.2%, respectively) in patients with PCOS in comparison to healthy controls. Moreover, ultrasound findings have illustrated increased thickness of mesenteric fat in patients with NAFLD and PCOS, suggesting an independent risk factor in these patients (33). Latter investigations used transient elastography as another non-invasive tool for the assessment and conducted cross-sectional studies in South Asia and Mexico reported similar results that represent the significant higher risk (37, 38). MRS was also used to detect intra-hepatic fat content and showed similar findings (21). Furthermore, other studies considered liver biopsy as the gold standard for the detection of steatosis (24, 39). Setji et al. (24) for the first time investigated histological aspects of biopsy-proven liver fibrosis and PCOS co-incidence. Based on their results, 15% of the PCOS patients experienced higher aminotransferase levels. Moreover, all of the patients with permanent increased ALT levels showed liver fibrosis signs through liver biopsy (24). Hence, increased risk of advanced NASH and liver fibrosis is anticipated in PCOS (40). Two further cross-sectional studies conducted in tertiary gastroenterology centers confirmed the presence of PCOS in 51%-71% of biopsy-proven NASH patients (39, 41). Limited number of studies have also evaluated liver steatosis/fibrosis-related indices in PCOS populations. In this regard, Polyzos et al. (42) revealed a positive correlation between fatty liver index (FLI), lipid accumulation products (LAP), hepatic steatosis index (HSI) and AST to platelet ratio (APRI) with PCOS (27, 42). A most recent large scale cross-sectional study from US national database, containing data from 50,785,354 women including 77,415 PCOS patients demonstrated a 4.3- fold higher prevalence of NAFLD in PCOS subjects, after adjusting for confounders (43). They also considered HA as the potential link in this epi-phenomenon (43). In this line, a great number of studies were performed to assess the predictors of NAFLD among PCOS patients. Collectively, the predictors include obesity, Mets, IR and HA. To the best of our knowledge a majority of studies have suggested androgen excess as the main risk factor for co-existence of PCOS and NAFLD (37, 38, 44, 45). The “classical phenotype” of PCOS exhibited a marked higher prevalence of NAFLD in comparison to other phenotypes (84.3% vs 41.1%, respectively) (38). Hence, HA has been considered as a putative risk factor solely (21, 31, 37, 46, 47) or along with increased BMI (37) or IR (45). In this regard, a large scale retrospective cohort study was conducted by Kumarendran et al. (48) and the raised incidence of NAFLD among PCOS women was attributed to HA, BMI and dysglycemia after assessing 63,000 PCOS women vs 121,000 controls. The latest study by Won et al. (44) involved 586 PCOS subjects and highlighted HA and Mets as the main related risk factors for the incidence of NAFLD in these patients. Although meta-analyses confirmed mentioned data and introduced serum androgens as independent predictors of NAFLD in PCOS women, there are a few number of findings making this issue controversial (20). Two studies did not observe any marked association between sex hormone-binding globulin (SHBG), FAI, TT and 17-hydroxyprogesterone levels with NAFLD (49). Overall, some studies have reported the correlation between FAI, SHBG and TT with NAFLD (26, 31, 35, 41, 50, 51) while other studies have failed to confirm these associations (12, 35, 49, 52–54). BMI, Mets, metabolic disturbances (e.g. higher serum triglyceride (TG), LAP) are among other important factors linking two diseases (15, 53). In addition, limited studies have suggested a number of other mutual risk factors including exacerbating inflammatory status and menstrual/reproductive markers, in an overweight-obesity-independent manner (47). The results of studies on risk factors for development of NAFLD in PCOS patients are summarized in Table 1.
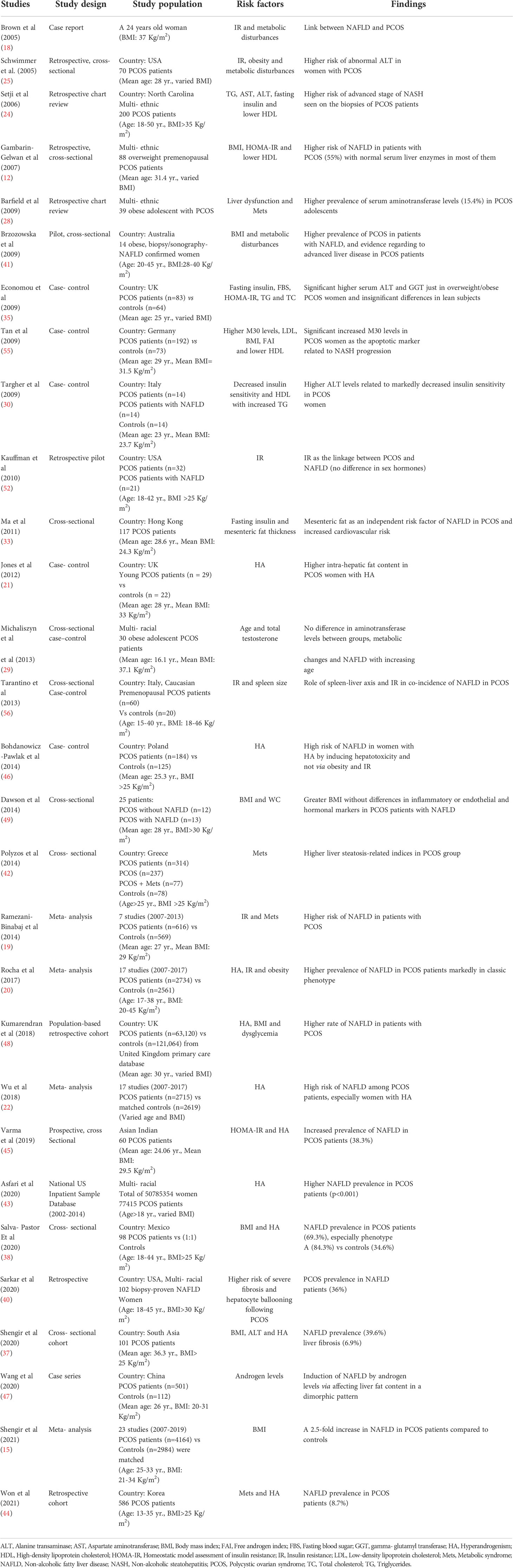
Table 1 Studies investigating the risk factors for the development of NAFLD in PCOS patients.
In the light of the controversy among current epidemiological findings, a number of studies suggest the correlation between NAFLD and PCOS, while others do not (15). Meanwhile, to date, there are four met-analysis studies that document higher risk of NAFLD in PCOS women via their common risk factors including obesity (19), BMI (15), IR (19), HA (20) and metabolic factors (15). Hence, it seems that a multifactorial pathological mechanism- as the complex of genetic and acquired factors- contributes to this co-existence (16). A vicious cycle of genetic variations, adipocyte- dysfunction, HA, systemic IR and chronic inflammation contribute to the synergistic catastrophic consequence of aforementioned diseases (16). Evidence seems to be insufficient for determining whether the association between NAFLD and PCOS is resulted from their common risk factors or PCOS acts as an independent risk factor for NAFLD. Different mechanisms linking PCOS and NAFLD are summarized in Figures 1 and 2.
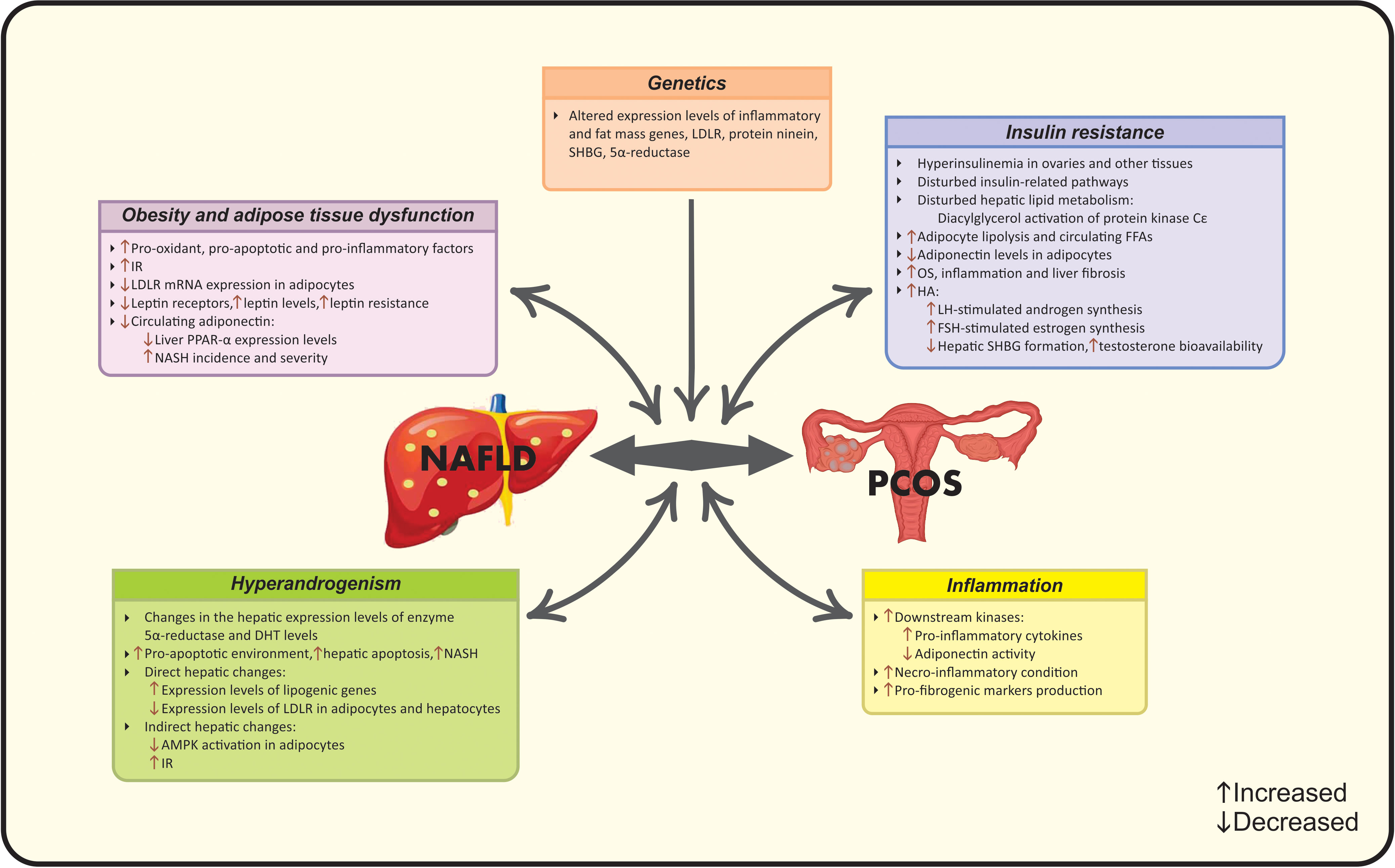
Figure 1 Established pathophysiological mechanisms linking PCOS and NAFLD. AMPK, Adenosine monophosphate-activated protein kinase; DHT, Dihydrotestosterone; FFA, Free fatty acid; FSH, follicle-stimulating hormone; HA, Hyperandrogenism; IR, Insulin resistance; LDLR, Low-density lipoprotein receptor; LH, Luteinizing hormone; NAFLD, Nonalcoholic fatty liver disease; NASH, Non-alcoholic steatohepatitis; OS, Oxidative stress; PCOS, Polycystic ovarian syndrome; PPARα, Peroxisome proliferator-activated receptor-α; SHBG, Sex hormone-binding globulin.
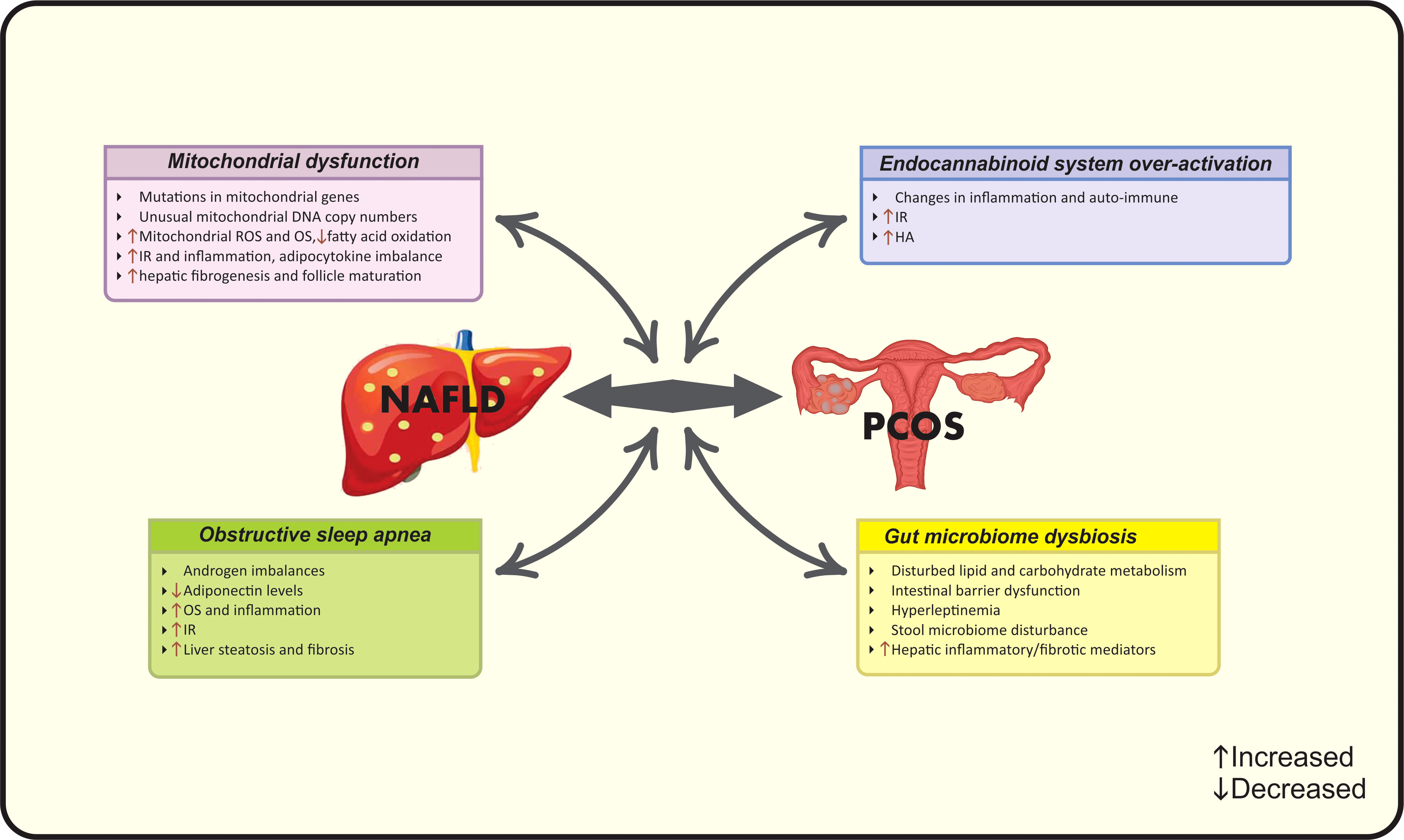
Figure 2 Possible new pathophysiological mechanisms linking PCOS and NAFLD. HA, Hyperandrogenism; IR, Insulin resistance; NAFLD, Nonalcoholic fatty liver disease; OS, Oxidative stress; PCOS, Polycystic ovarian syndrome; ROS, Reactive oxygen species.
Abdominal obesity is another common finding in both NAFLD and PCOS patients and at the same time is associated with other related mutual risk factors (17). The term “hypertrophic obesity” used for PCOS women refers to the larger adipocytes (shown as its 25% increased diameter compared with healthy obese controls) as well as higher amounts of visceral fat, markedly in mesenteric and intraperitoneal sites (57). Villa et al. (57) reported that although this specific pattern of fat distribution in PCOS patients is not a putative factor in disease progression, the adipose tissue dysfunction is linked to several metabolic abnormalities. Several observational studies have introduced obesity as a common mechanism linking NAFLD and PCOS (58, 59). Noteworthy, the impaired visceral adipose tissue becomes a target for systemic alterations and pathologic mediators such as pro-oxidant, pro-apoptotic and pro-inflammatory factors and thereby significant metabolic/hormonal disorders emerge as the co-existence of endocrine-related diseases such as PCOS and NAFLD (16). IR, HA and apoptosis are the main contributors. In addition to IR, which is a characteristic feature of hypertrophic adipocytes, HA induces IR in visceral adipocytes and deteriorates abdominal adiposity in PCOS women (57). Another androgen-dependent change refers to increased serum M30 levels-as an apoptosis biomarker- along with decreased low-density lipoprotein receptor (LDLR) mRNA expression in adipocytes of PCOS women with NAFLD compared with NAFLD patients that sheds a light into the importance of adipocyte dysfunction (59).
Based on the clinical evidence, IR is present in 80% of patients with NAFLD and 50-80% of patients with PCOS plus NAFLD (60). IR plays an important role in the development of NAFLD and PCOS (60). The severity of both diseases is associated with higher IR (30). IR may be a consequence of obesity or lowered glucose uptake in muscles followed by increased hepatic de novo lipogenesis (10). The accumulation of hepatic lipotoxic products reduces mitochondrial fatty acid oxidation and manifests as IR in NAFLD (61). IR, in turn, is linked with a wide spectrum of adverse conditions such as OS, inflammation, adipocytokine imbalance and liver fibrosis (56, 61). IR is also associated with increased adipocyte lipolysis and higher levels of free fatty acids in circulation (38). In the case of PCOS, impaired insulin signaling, due to the post-receptor defects in insulin signal transduction in adipocytes and muscles, leads to the additional hyperinsulinemic condition in other tissues such as the ovaries (61). Finally, these changes emerge as the IR-phenotype in women with PCOS which make changes in androgenesis (10). Insulin acts as a co-gonadotropin to increase luteinizing hormone (LH)-stimulated androgen synthesis and follicle-stimulating hormone (FSH)-stimulated estrogen synthesis as well as to decrease hepatic formation of SHBG which is the key binding protein for testosterone and prolongs its metabolic clearance, leading to enhanced testosterone bioavailability (13). This androgen-regulating aspect of insulin is amplified during the hyperinsulinemic state of PCOS, especially in theca cells (61). Hence, insulin can regulate the generation, bioavailability and clearance of androgens and make androgen excess state (13). Androgen excess in turn may lead to IR by lowering adiponectin levels in adipocytes that is followed by enhanced visceral adiposity and disturbed insulin action (62).
PCOS is already known as an outstanding “heyperandrogenic syndrome”, encompassing a wide range of changes in endocrine and metabolic markers (13). The majority of studies have investigated the androgen changes in PCOS along with NAFLD. It is considered that androgen levels contribute to the development of NAFLD in a sex-dependent manner i.e. hypogonadism in men and hyperandrogenism in women (63). In the concept of “hepato-ovarian axis”, liver is known as the origin and the target of hormonal changes at the same time. Two pathways are involved; hepatic involvement in peripheral sex-steroid transformation beside the hepatic-surface expression of androgen-estrogen receptors (64). The presence of persist IR state along with lowered HA after the ovarian ablation surgery sheds a light into the issue that IR and HA are the main causes of the co-existence of NAFLD and PCOS, but there is a gap in the priority between them (25, 45). As mentioned before, IR results in HA-phenotype. Wang et al. (47) also confirmed that HA in turn contributes to the development of NAFLD. These mechanisms of action are via direct and/or indirect hepatic changes; direct effect of androgen excess leads to hepatic fat accumulation, while, indirect effects include (a) suppressing adenosine monophosphate-activated protein kinase (AMPK) activation in adipocytes that results in higher lipogenesis and increased visceral adiposity and (b) exerting alterations in insulin sensitivity or both mechanisms together (22, 65). Changes in insulin sensitivity have been observed as HA-induced-IR via the suppression of insulin signaling transduction (66). Aforementioned changes induce disturbances in lipid metabolism and emerge as adipose tissue dysfunction, collectively named as “androgen-mediated adipose lipotoxicity”, linking PCOS to NAFLD (67). HA leads to changes in the expression levels of genes involved in lipid metabolism represent in liver and adipocytes, thereby making PCOS subjects more susceptible to NAFLD (59). Kumarendan et al. (48) recently observed a correlation between higher free testosterone levels (>3.0 nmol/L) and increased prevalence of NAFLD in PCOS women. In this regard, the hepatocytes express the enzyme 5α-reductase, which is involved in the converting circulating testosterone to dihydrotestosterone (DHT) and, on the other hand, modulates insulin pathway, liver steatosis and fibrosis (68). Androgens also induce a pro-apoptotic environment in PCOS patients (59, 69). Tan et al. (55) demonstrated higher serum levels of “M30” for assessing caspase 3-cleaved fragment of cytokeratin 18 (CK18)- the apoptotic biomarkers- in PCOS women and its role in the prediction of NASH. Another study showing higher M30 levels in patients with PCOS and NAFLD (proved by biopsy), confirmed previous data and highlighted the increased risk of hepatic apoptosis and NASH in women with PCOS (59). These findings highlighted the independent role of androgen levels in NAFLD regardless of IR or obesity (31). In this context, a recent meta-analysis has confirmed that PCOS patients with HA exhibit higher incidence of NAFLD after adjusting for confounders, while patients without HA show similar features to healthy subjects (22). Interestingly, there are a few number of studies in contrast with these findings, providing the non-significant difference in NAFLD prevalence among PCOS patients with or without HA (12, 35, 49, 52–54, 70).
Low-grade inflammatory state not only can be identified as an additional link between two pathological entities but also plays a pivotal role in the progression of them (16). Inflammatory mediators can lead to IR and thereby, facilitate the progression of NAFLD in a multi-step manner (16). Tumor necrosis factor-alpha (TNF-α) is an important pro-inflammatory biomarker involved in this epiphenomenon. It is reported that patients with IR exhibit higher circulating TNF-α levels in an obesity-independent manner. Moreover, increased expression levels of androgen receptors (ARs) up-regulates the expression of TNF-α (58, 71). Increased TNFα induces IR and triggers downstream kinases, leading to higher cytokine formation along with impaired adiponectin activity (71). Taken together, low-grade inflammation plays a fundamental role in IR, abdominal obesity and NAFLD development (14). More severe forms of NAFLD are more susceptible to PCOS, due to the necro-inflammatory state that leads to the overproduction of multiple pro-inflammatory and profibrogenic markers (16).
Genetic polymorphisms are responsible for observed inter-ethnic differences in the prevalence of NAFLD in PCOS patients. Genome-wide association studies (GWAS) aimed to identify the molecular and genetic pathways involved in pathological conditions (72). The first GWAS identifies susceptibility loci for PCOS on chromosome 2p16.3, 2p21 and 9q33.3 (72). Moreover, polymorphisms in pro-inflammatory cytokine genes (interleukins) are detectable in PCOS (73). GWAS on NAFLD documented a missense mutation in patatin-like phospholipase domain-containing (PNPLA) 3 gene that completely overshadows NAFLD spectrum (74). Bohdanowicz-pawlak et al. (46) did not document any association between polymorphisms rs328 and rs268 of the lipoprotein lipase gene and NAFLD occurrence in women with PCOS. Previous studies have identified alterations in the expression levels of genes involved in both NAFLD and PCOS including LDLR, protein ninein, fat mass gene, SHBG, 5-alpha reductase along with inflammatory genes (59, 60).
Newly, a body of evidence indicates the role of mitochondrial dysfunction in NAFLD-PCOS connection. Patients with PCOS have shown mutations in mitochondrial genes and unusual mitochondrial DNA copy numbers (75). Moreover, this condition has been observed in PCOS patients with normal ranges of BMI (76). Indeed, IR as well as hepatic fat accumulation results in mitochondrial dysfunction and reduced fatty acid oxidation (61). It is considered that increased production of mitochondrial reactive oxygen species (ROS) plays a key role in producing liver damage and initiating hepatic fibrogenesis (77). Mitochondrial dysfunction induces OS that subsequently aggravates lipid metabolism, IR, inflammation, adipocytokine imbalance, and follicle maturation leading to the coexistence of PCOS and NAFLD (61, 75).
“Gut-liver axis” has a predominant role in the liver health. The intestinal factors such as dysbiosis and intestinal barrier dysfunction along with hepatic factors such as inflammatory/fibrotic mediators and hyperleptinemia lead to NAFLD progression through disturbed lipid and carbohydrate metabolism (78). On the other hand, changes in the gut microbiome also play a role in PCOS progression and are explained by the disturbances in stool microbiome associated with IR, leading to reproductive and metabolic dysfunction of PCOS women (79). Collectively, gut microbiome can be considered as a newly diagnosed link between PCOS and NAFLD.
To date, over-activation of the endocannabinoid system (ECS) is also involved in both diseases. ECS- a complex cellular signaling network- modulates a wide range of human functions and regulates inflammation, auto-immune and energy homeostasis (80). ECS is linked with non-receptor mediated pathways including the induction of MAP kinases and PI3 kinases beside G protein-independent pathways (81). The ECS includes cannabinoid receptors (CB1 and CB2) and their lipidic ligands and over-activated ECS is involved in different pathological conditions such as liver disease (82). ECS over-activation exerts catastrophic effects on insulin signaling and leads to IR, increased hepatic lipogenesis, inflammation, steatosis and subsequent NAFLD (80). CB1 polymorphism leads to the development of NAFLD and PCOS by progressing HA state (83).
A commonly observed condition in women with PCOS and NAFLD is obstructive sleep apnea (OSA). Subjects with OSA have a 2.6- fold increased risk of NAFLD and fibrosis progression, while this number increases to 7.6-fold in patients with PCOS and OSA (84). OSA is linked with androgen imbalances and IR through the role of chronic intermittent hypoxia that leads to decreased levels of adiponectin, increased OS and inflammatory state, which ultimately manifests as liver steatosis (85). Regarding to the role of androgens in OSA, PCOS women with higher free testosterone levels are more prone to mentioned conditions (84).
As discussed below, the role of abnormalities in IR pathway during NAFLD and PCOS has been confirmed previously. Accumulating evidence illustrated the disturbances in the initial part of glycogen metabolism and/or insulin- signaling cascades shown in IR-phenotype of PCOS (86). Insulin stimulates P450c17 activity through phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways, consisting from subgroup mediators such as MAP kinase-activated protein kinase-3, mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK/ERK) and MAP kinase-activated protein kinase-4/c-Jun N-terminal kinase (87). Hence, not only the direct effects of insulin on androgens happen, but also disturbances in insulin-related pathways (PI3K, MAPK and MEK/ERK) are involved in androgen excess pattern in the IR- related- PCOS and NAFLD (88). Moreover, a controversy has been observed according to the differences in the underlying mechanisms of lean PCOS subjects compared to overweight/obese ones. It is suggested that in overweight/obese women with PCOS, IR state is due to the impairments in the proximal part of insulin signaling pathway in skeletal muscles, whereas in lean subjects, adiponectin affects skeletal muscle through AMPK mediators (89). IR state-found in either NAFLD or PCOS- may be also derived from the diacylglycerol activation of protein kinase Cϵ that is stimulated via disturbed hepatic lipid metabolism during diseases (90). IR promotes the hyperandrogenic aspect of PCOS. Androgen excess is also involved in liver lipotoxicity, as suggested by the impact of higher TT levels on increasing the expression levels of lipogenic genes as well as hepatic de novo lipogenesis in hepatic cells of women (67). More precisely, androgen excess reduces the expression levels of LDLR mRNA in adipocytes and hepatocytes of PCOS women (50, 59). These changes may result in extended half-life of very low-density lipoprotein cholesterol (VLDL-C), LDL-C, increased hepatic fat accumulation and eventually liver steatosis (50, 59). Oppositely, estrogen inhibits stellate cell activation and fibrogenesis. Women with PCOS and NAFLD showed lower estradiol levels compared with PCOS ones (91). Yang et al. (92) reported the increased risk of severe hepatic fibrosis in men when compared with premenopausal women. Moreover, the severity of NAFLD correlates with estrogen reduction (93). These findings may be explained by the protective role of estrogen against fibrogenesis.
Although there is limited evidence with regard to the role of adipokines in the mentioned co-existence, it is hypothesized that decreased number of leptin receptors may result in leptin resistance and increased leptin levels (94). Conversely, lowered circulating adiponectin is documented in PCOS women and NAFLD patients, respectively (95, 96). Meanwhile, Baranova et al. (60) did not find any significant variations according to adiponectin levels in NAFLD patients with or without PCOS. The underlying mechanism is based on the positive correlation between serum adiponectin levels and liver peroxisome proliferator-activated receptor-α (PPAR-α) mRNA expression level that exerts an inverse relation with NASH incidence and severity (97).
As mentioned, women with PCOS and NAFLD have an elevated fat mass, increased prevalence of hypertrophic obesity, androgen imbalance, lipid disorders, IR, inflammation, OS, HTN, Mets and cardiovascular disorders. The main finding that differentiates the characteristics of PCOS plus NAFLD women from NAFLD patients might be related to HA, as PCOS women with HA experience a higher risk of NAFLD in comparison to PCOS patients without HA, even after adjusting for other potential confounders that have been mentioned above (20).
Applying powerful diagnostic approaches for NAFLD among PCOS women seems necessary due to; firstly, preventing patients from more severe forms of liver disease by considering the higher risk of NASH progression among them and secondly, considering the benefits of early intervention in eliminating NAFLD and related hepatic/metabolic complications (26, 39). NAFLD has been reported to be present in up to 39% of lean PCOS subjects, it is suggested that PCOS patients should undergo NAFLD screening regardless of BMI status or HA (12). A number of diagnostic strategies have been implemented for NAFLD, however the optimal approach is still unclear. Ultrasonography as the first-line screening approach as well as transient elastography (Fibro scan) can be combined with non-invasive liver markers such as serum liver enzyme levels, NAFLD fibrosis score or Fibrosis-4 score to identify NAFLD in PCOS women (98). The combination of ultrasound and serum aminotransferases is commonly used for detection, while liver enzymes cannot be sufficient solely (99). Liver biopsy and MRS are not used in routine practice (99). In addition, PCOS patients should be screened for hepatic and cardio-metabolic disorders. In this context, early diagnosis and managing a useful lifestyle intervention seems necessary (13). The first-line therapeutic strategy for PCOS patients with NAFLD should focus on lifestyle modification including calorie-restricted diet plus regular physical activity resulting in a weigh loss of 5-10% of initial body weight (11, 17, 100). A second-line strategy may also focus on lifestyle modification plus pharmacological therapy (14). Aforementioned interventions aim to improve biochemical, histological and clinical features of both conditions simultaneously (17, 100). Pharmacotherapy is an additional therapeutic approach combined with first-line interventions in a proportion of cases (13). In this context, metformin might be the drug of choice for patients with PCOS and NAFLD (100). It is reported that metformin acts through inhibiting hepatic gluconeogenesis, relieving IR, reducing BMI, improving serum liver enzymes and ovulatory menstrual cycles, but has no effect on liver histology (13, 101). Although PPAR agonists, specially pioglitazone, have shown beneficial effects on IR, elevating adiponectin in circulation, lowering serum ALT, TG and macrophage activation (102, 103). Moreover, the prevalence of NAFLD was reduced in approximately 68% of patients after treatment with liraglutide as a member of glucagon-like peptide-1 (GLP-1) receptor agonists (104). The mechanism of action may be related to increased hepatic insulin sensitivity as well as reduced lipogenesis and liver enzymes (105). Noticeably, evidence regarding the effects of anti-androgenic drugs such as oral contraceptive pills (OCP) on liver markers is still unclear (106). Farnesoid X receptor (FXR) agonists such as obeticholic acid act by reducing bile acid synthesis, liver enzymes, BMI and improving liver steatosis/fibrosis (105). Indeed, FGF-21 inhibitors, FGF-19 analogs and antiobesity agents like aramchol, orlistat or gut microbiome modulators are other therapeutic options for NAFLD treatment that ameliorate liver damage, insulin sensitivity, body weight and the activation of Kupffer cells (105). In the case of gut microbiome modulators, reduced delivery of lipopolysaccharides (LPS) from gut to liver results in treatment (105). Pioneering studies have confirmed the effects of anti-oxidant, anti-inflammatory and immunomodulator agents on PCOS and NAFLD. Emricasan and pentoxifylline have been used as anti-TNF-α medications that alleviate liver enzymes and fibrosis (105). However, amlexanox and cenicriviroc have been examined trough their immune-modulatory activities resulting in down-regulated liver inflammatory genes, steatosis markers as well as IR (105). Omega-3 fatty acids and vitamin E have shown favorable effects on OS- mediated injury, hepatocyte ballooning and lobular inflammation that eventually improve liver steatosis, but further studies are needed to evaluate their clinical effectiveness (107, 108). Other pharmacologic agents e.g. statins, aramchol, anti-fibrotics and probiotics could be used as suggested approaches for NAFLD treatment in PCOS women (10, 17). Suggested pharmacological treatments for management of NAFLD in PCOS are summarized in Figure 3. In addition to the aforementioned pharmacotherapies, further investigations are needed to identify the best approach in this context based on the pathological interactions and subsequent complications in both diseases.
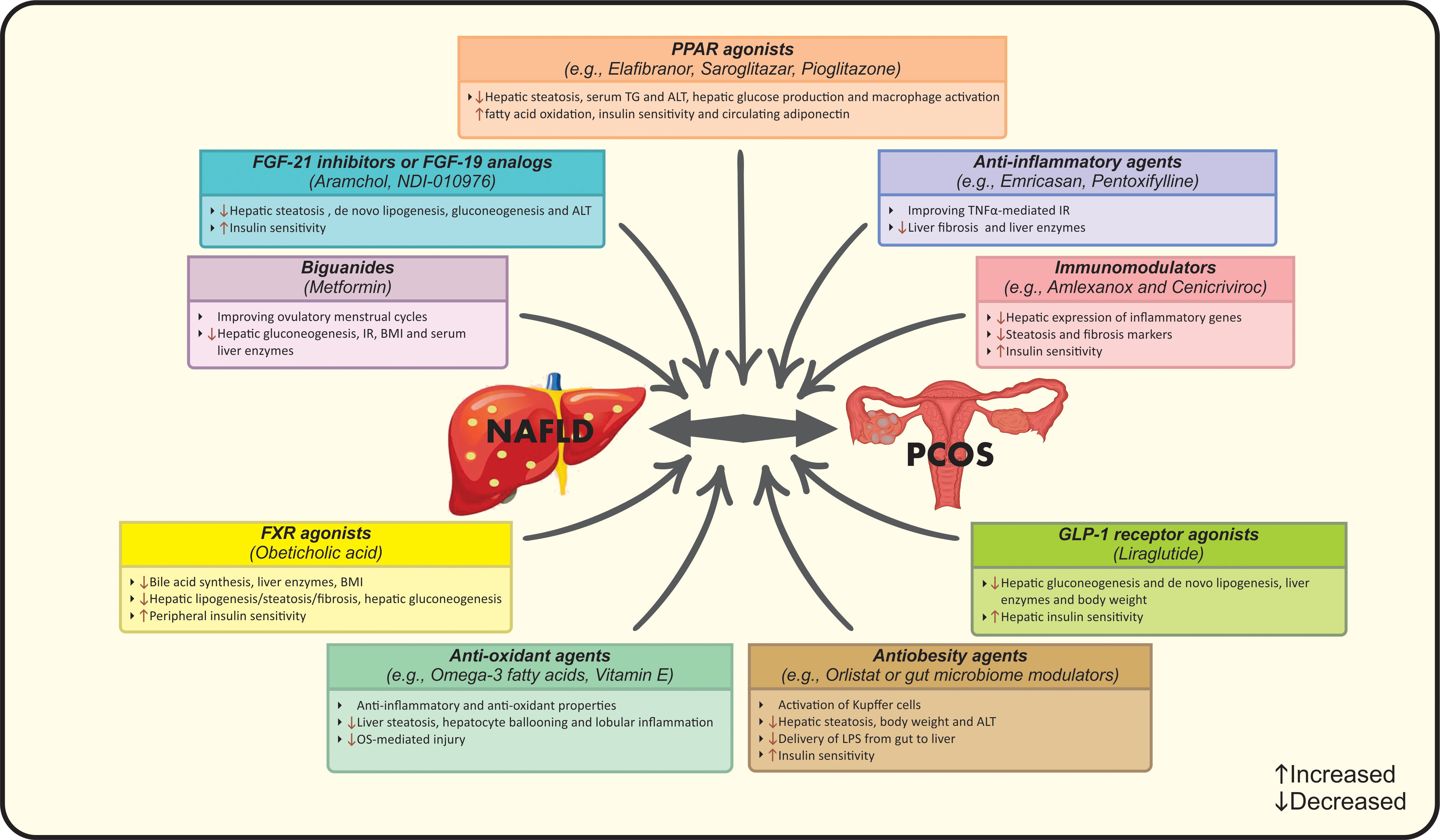
Figure 3 Suggested pharmacological treatments for management of NAFLD in PCOS. ALT, Alanine transaminase; BMI, Body mass index; FGF, Fibroblast growth factor; FXR, Farnesoid X receptor; GLP-1, Glucagon-like peptide 1; IR, Insulin resistance; LPS, Lipopolysaccharides; NAFLD, Nonalcoholic fatty liver disease; OS, Oxidative stress; PCOS, Polycystic ovarian syndrome; PPAR, Peroxisome proliferator-activated receptor; TG, Triglycerides; TNFα, Tumor necrosis factor-alpha.
Further studies are needed to investigate other comorbidities associated with NAFLD in PCOS women including cardiometabolic risk factors, gut-liver axis dysfunction, gut microbiota and cardiovascular disease due to the increased risk of mentioned conditions in patients with either disease. Furthermore, larger well- designed cohort studies including periodic assessment of liver function based on biochemical, histological and hormone markers are necessary to explore the current epi-phenomenon with an emphasis on FAI and SHBG. By considering the limitations of previous meta-analysis studies (small sample size, limitations in search strategy and underlying risk factors) conducting more detailed compensatory meta-analysis studies seems desirable. The mechanisms underlying mitochondrial dysfunction during either disease or both together must be noticed. Indeed, improvement of mitochondrial function and endocannabinoid system can be studied as new therapeutic targets. Finally, the most effective, accurate, and evidence-based approaches for the treatment of NAFLD in PCOS women remain unknown, multi-disciplinary research seems to shed light on this issue. NAFLD and PCOS are known as the hepatic and ovarian manifestations of Mets, respectively. In conclusion, the review of literature suggests the significant higher risk of PCOS in NAFLD. The underlying responsible mechanistic pathways are not completely explored. Further research is needed to evaluate the current issue from the “hepato-ovarian axis” to epidemiological, clinical and pharmacological aspects.
The total or partial loss of pituitary gland function caused by pituitary or hypothalamic disorders is defined as hypopituitarism (109). Pituitary gland contributes to the secretion of predominant hormones targeting various organs, including growth hormone (GH), thyroid-stimulating hormone (TSH), gonadotropins and prolactin (110). Hypopituitarism is classified into “primary” and “secondary” subgroups that refer to the internal pituitary gland disorders and other related disorders (hypothalamic and central nervous system), respectively (111). The global prevalence of hypopituitarism ranges from 290 to 455 cases per million and the current incidence is 42.1 cases per million (112). Despite the relative scarcity of hypopituitarism, it is reported that Mets and NAFLD are common conditions that have been observed in these patients (110). More precisely, subjects with hypopituitarism have a potential risk for NAFLD due to the interplay of low GH levels and hypogonadism in the pathogenesis of NAFLD (113). Leptin resistance is another common condition in NAFLD and hypopituitarism and leptin levels correlate with the severity of liver fibrosis in surgical-hypopituitarism patients (114). The main pathophysiological mechanisms linking hypopituitarism to NAFLD are summarized in Figure 4.
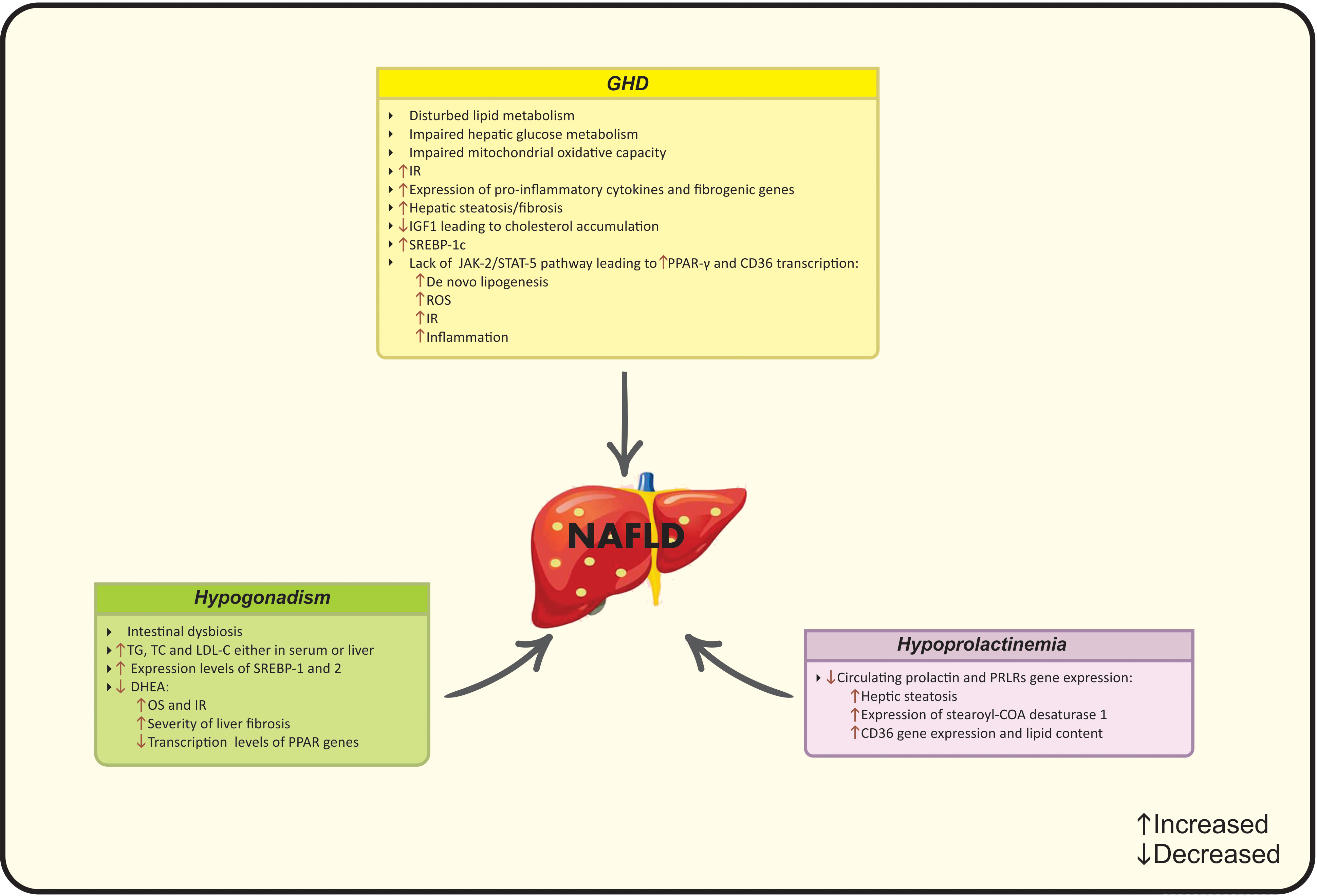
Figure 4 Possible mechanisms of hypopituitarism on NAFLD development. CD36, Cluster of differentiation 36; COA, Coenzyme A; DHEA, Dehydroepiandrosterone; IGF-1, Insulin-like growth factor-1, IR, Insulin resistance; JAK2, Janus kinase 2; LDL-C, Low-density lipoprotein cholesterol; NAFLD, Nonalcoholic fatty liver disease; OS, Oxidative stress; PCOS, Polycystic ovarian syndrome; PPAR, Peroxisome proliferator-activated receptor; PRLR; Prolactin receptor; ROS, Reactive oxygen species; SREBP, Sterol regulatory element- binding protein; STAT-5, Signal transducer and the activation of activator of transcription 5; TC, Total cholesterol; TG, Triglycerides.
GH and insulin-like growth factor-1 (IGF-1) are mainly mediated by “hypothalamic-pituitary somatotropic axis” and exert several physiological effects, ranging from promoting cell division/proliferation and growth development to macro-nutrients metabolism in adipocytes, liver and skeletal muscle through direct or indirect-complex mechanisms (115, 116). GH also stimulates protein synthesis and lipolysis, hepatic gluconeogenesis and glycogenolysis beside eliminating peripheral insulin sensitivity (117). GHD -a rare clinical syndrome- is resulted from genetic or structural disorders and occurs in both children and adults (110). GHD is defined by insufficient GH secretion from the pituitary gland and is associated with disturbed glucose and lipid metabolism, hypertension and cardiac dysfunction (113, 118).
A number of epidemiological studies have demonstrated that GHD patients exhibit higher risk of NAFLD progression, and a negative correlation between GH levels and the severity of NAFLD has been documented. Meanwhile, some studies do not report same results due to the heterogeneity in the characteristics of study participants and sample sizes. The epidemiological studies that have assessed GH and related risk factors in NAFLD patients have been summarized in Table 2. Of note, a retrospective study reported a 2% approximate prevalence of NAFLD in patients with hypothalamic/pituitary dysfunctions (120). Additionally, Ichikawa et al. (119) mentioned that patients with GHD exhibit a higher risk for the development of NAFLD, compared to other patients with pituitary dysfunction and the possible association between GHD and NAFLD was suggested based on the “Mets-like phenotype” of adults with GHD (121). Subsequently, cumulative evidence confirmed the aforementioned association in several studies by assessing transaminases (135), ultrasound (122, 123, 127, 132) and biopsy findings (121, 126, 128). Nishizawa et al. (126) reported a 6.4-fold increased prevalence of NAFLD and NASH-related histological findings in subjects with GHD in comparison to matched controls. It should be noted that hypothalamic/pituitary disorders, especially GHD are the risk factors for the secondary forms of NAFLD that are not reversible even after lifestyle modifications (125, 136).
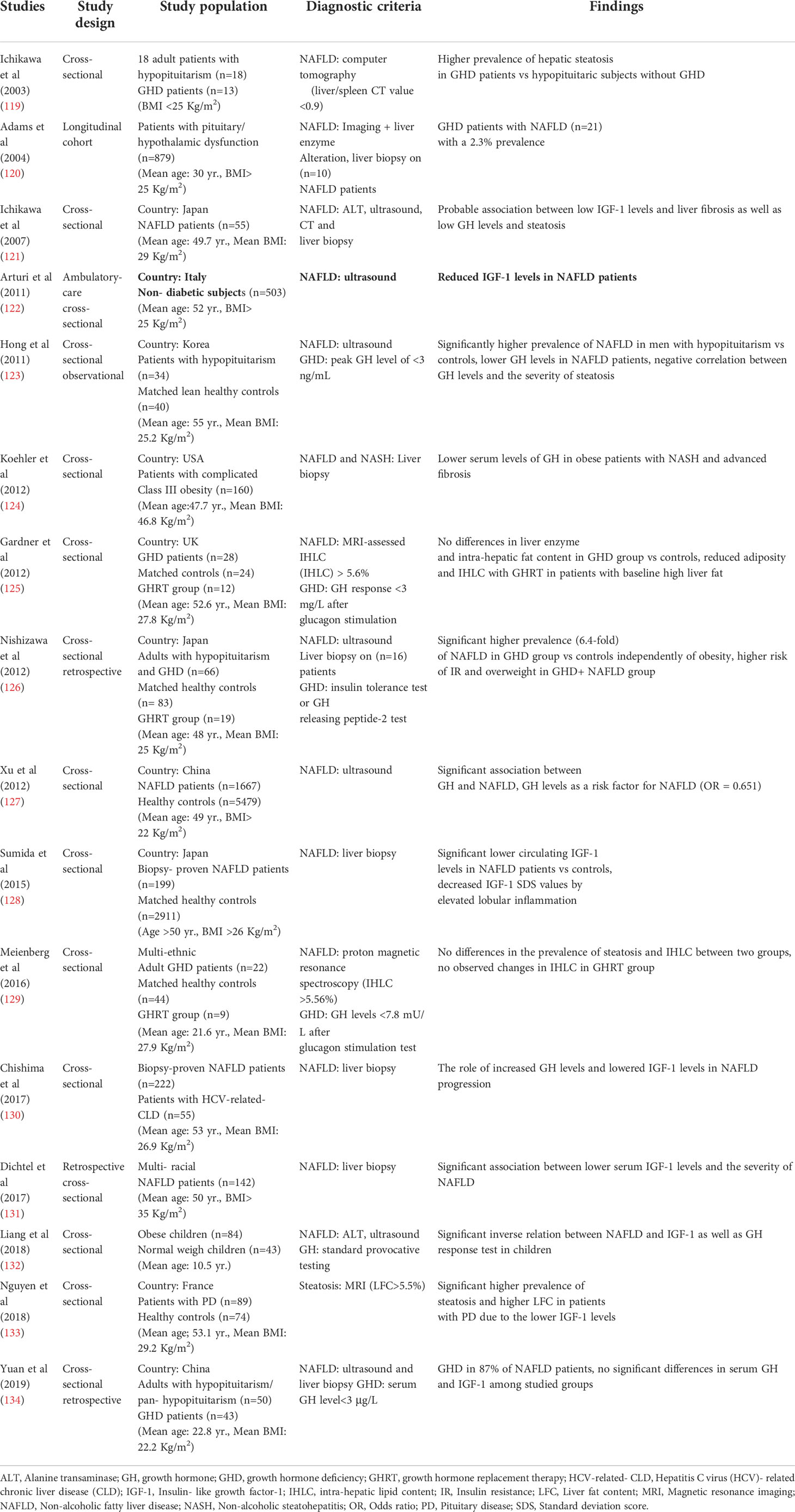
Table 2 Studies investigating the risk factors for the development of NAFLD in GHD patients.
Recently, Xu et al. (127) conducted a large cross-sectional study reporting a significant relation between GH levels and the risk factors of NAFLD (OR: 0.651). Lower GH and IGF-1 levels are correlated with the severity of NAFLD (122, 127, 128, 130–133, 137). In this regard, mRNA levels of hepatic IGF-1 obviously decreases in NASH patients in comparison to patients with liver steatosis and predicts the degrees of hepatic inflammation (138). Moreover, the current body of evidence suggests that NAFLD/NASH patients, even with advanced liver fibrosis, have low serum GH levels (123, 124, 128, 132). Hence, NAFLD might be known as a hepatic complication of GDH. Despite the agreement among the majority of studies, Yuan et al. (134) have not found any differences in GH and IGF-1 levels between NAFLD patients and healthy controls. Furthermore, another study reported similar prevalence of NAFLD through magnetic resonance imaging (MRI)-assessment between GHD patients and healthy controls (129).
GHD is followed by a wide range of metabolic consequences that are linked to the onset of NAFLD, including visceral obesity, IR and dyslipidemia (139). Beside the direct effects of GH, it could act indirectly through IGF-1 production mainly by the liver (117). IGF-1 amplifies GH actions after binding to specific proteins and receptors (117). In this regard, GH/IGF-1 axis is involved in the progression of NAFLD and predicts liver steatosis/fibrosis. Moreover, based on the evidence from animal studies, lower IGF-1 levels are associated with lower protection against IR, inflammation and liver steatosis, while, treatment with IGF-1 improves liver enzymes, steatosis and fibrosis (115). Impaired hepatic GH signaling is immediately followed by liver steatosis and NASH that highlights the role of GH in the liver in an independent manner from metabolic dysfunction (113).
There is an inverse interaction between GH and insulin signaling via the stimulation of hepatic gluconeogenesis and glycogenolysis. However, GHD is along with IR and impaired hepatic glucose metabolism due to the changes in lipid flux (140).
GHD up-regulates the expression levels of adipocytes and pro-inflammatory cytokines that leads to increased hepatic and visceral fat accumulation and IR (139). Elevated expression of inflammatory/fibrogenic genes including TNFα and chemokine (C-C motif) ligand 3 (CCL3) are observed in animals with liver GH receptor deletion (141). This systemic low-grade inflammation also stimulates the progression of NAFLD in GHD patients. IGF-1 acts as an anti-inflammatory and anti-oxidant agent by suppressing ROS generation and promoting hepatic mitochondrial function (126). Moreover, GH replacement improves inflammatory state in human (14).
Current evidence suggest the protective role of GH against OS by regulating mitochondrial oxidative capacity. OS is a key pathologic hit in the progression of NAFLD. As discussed, lower IGF-1 levels increase OS and induce NAFLD that suggests IGF-1 as an anti-oxidant factor (142). Recent studies have investigated an association between IGF-1 and sirtuin-4 (Sirt4) levels (143). Sirt4- a mitochondrial NAD-dependent ADP ribosyltransferase- negatively regulates the oxidative capacity and lowers levels of free fatty acid and ROS (143).
Melatonin is another contributing factor involved in the epi-phenomenon. Melatonin as a pleiotropic hormone act as a ROS-scavenger and exerts ant-oxidative activity (144). During NAFLD, melatonin improves oxidative state and liver histology (144). Notably, melatonin could modulate the secretion of GH and exert beneficial effects on GHD and NAFLD in the same time (145).
The correlation of GH and IGF-1 with NAFLD has been reported in epidemiological studies. IGF-1 mediates lipid metabolism via several intracellular pathways. IGF-1 up-regulates the expression of ATP-binding cassette transporter A1 (ABCA1) and prevents cholesterol accumulation (146). Taken together, based on the changes in liver homeostasis, function and structure due to impaired GH/IGF-1 axis, treatment with IGF-1 induces the senescence of the liver stellate cells and improves hepatic fibrosis (147). Additionally, the involvement of GH/IGF-1 axis in lipid metabolism is also observed through regulating Janus kinase 2 (JAK-2) and signal transducer and the activation of activator of transcription 5 (STAT-5) (148). GHD is along with severe hepatic steatosis, fibrosis, adipogenesis, and IR due to the lack of aforementioned genes (139). GH stimulates JAK2-STAT5 activation, promotes lipolysis and insulin sensitivity and attenuates lipogenesis (149). The activation of STAT-5b may inhibit the expression of downstream genes. In this context, GH inhibits PPAR-γ and Cluster of differentiation 36 (CD36) transcription as the chief regulators of free fatty acid uptake and attenuates de novo lipogenesis (150). PPAR-γ/CD36 are lipid activating factors involved in the ROS production in NAFLD (151). PPAR-γ stimulates the expression of genes and production of enzymes related to lipid metabolism and CD36 facilitates lipid endocytosis (152, 153). Higher CD36 production aggravates IR and inflammation. GH reduces the production of CD36, and the deletion of CD36 increases leptin secretion and suppresses lipid absorption (153). Sterol regulatory element- binding protein-1c (SREBP-1c) is a transcription factor involved in both conditions that regulates hepatic adipogenesis and induces lipid storage (154). Suppressed expression of SREBP-1c eliminates the risk of NAFLD, while, SREBP-1c is up-regulated in GHD diseases (139). GH administration interacts with this condition and improves lipid status as well as body weight (155).
Several tests are implemented in clinical practice with the aim of GHD diagnosis including insulin tolerance test and GH-releasing hormone (GHRH)-arginine test (14). Although there is not any definite identified cut-off value for GHD, ranges of <5.1 g/L for the insulin tolerance test and 4.1 g/L for the GHRH-arginine test suggest the state of GHD (143). GH replacement therapy has been used in a number of studies exerting improvements in transaminase levels, inflammation, mitochondrial function, lipid profile, liver steatosis/fibrosis and weight reduction (14, 113). GH replacement therapy reduces serum TNF-α, OS and other NAFLD-related conditions after six-month intervention (143). Moreover, recombinant human GH has been applied in adolescents with NAFLD (156). Collectively, despite the mentioned favorable effects, the efficacy of GH-replacement therapy (GHRT) in GHD patients with NAFLD is still controversial. Combining lifestyle intervention to specific therapy seems to be more effective for the treatment, but not sufficient for the prevention of NAFLD in these patients (14).
In summary, NAFLD screening is needed in all patients with GHD. Despite the controversy among recent finding, treatment with GH or IGF-1 might be a useful strategy in NAFLD patients with GHD. However, subsequent well-designed studies can evaluate the efficacy of aforementioned therapeutic strategies.
As shown before, metabolic disturbances are parallel to hormonal abnormalities, triggering each other. According to the “sex hormone-liver axis”, male hypogonadism and female hyperandrogenism are recently known as the independent risk factors for the development of NAFLD (157). The diminished function/activity of the reproductive organs followed by reduced sex hormone concentrations in either sex and regardless of its pathological causes is defined as hypogonadism and is classified into primary and secondary subtypes. Secondary hypogonadism is related to the reduced hypothalamus/pituitary gland function in producing sex hormones (158). The basic diagnostic criteria include the combination of clinical and biochemical signs of low sex hormones beside changes in physical health. Moreover, considering the co-existence of hypogonadism along with other endocrinopathies, such as hypopituitarism may be helpful (159). NAFLD is a common observed condition in these patients and shares a bi-directional connection with hypogonadism.
To the best of our knowledge, a great number of studies have investigated the bi-directional relationship between hypogonadism and NAFLD (160–162). Women with hypogonadism and hypoestrogenism exhibit higher prevalence of liver enzyme levels, NAFLD and advanced liver fibrosis (91, 92, 160, 163–168). Table 3 summarizes the results of studies investigating the potent association between hypogonadism and NAFLD in men and women. As discussed, increased duration of estrogen deficiency has been reported to be associated with an increased risk of hepatic fibrosis (160). Mckenzie et al. (165) reported that hormone replacement therapy (HRT) alleviates serum liver enzymes in diabetic post-menopausal women.
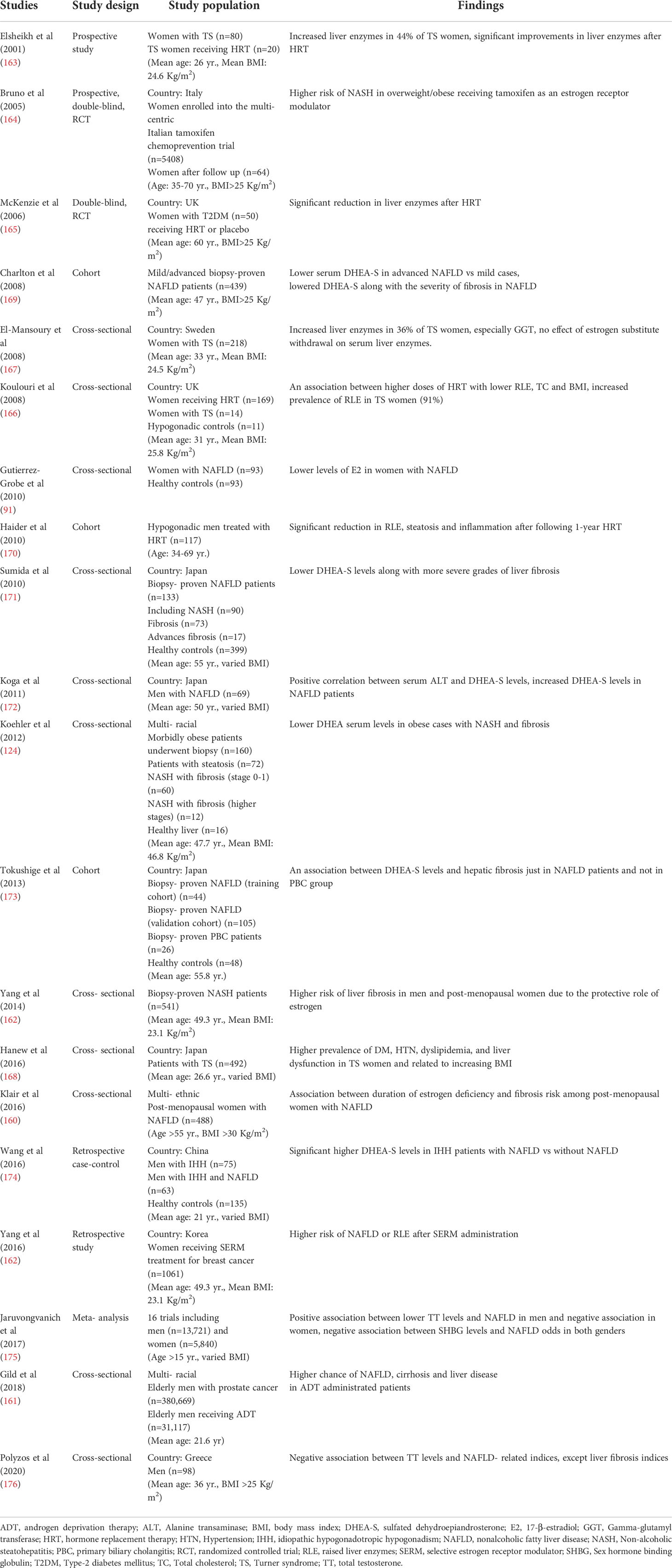
Table 3 Studies investigating the risk of NAFLD in Hypogonadism.
Serum TT levels are associated with central fat accumulation, IR and Mets (139). Seo et al. (177) in a cross-sectional study on 1944 men, reported lower TT levels in NAFLD patients in comparison to controls. Indeed, other studies confirmed these findings (170, 178, 179). Haider et al. (170) reported the favorable effects of testosterone replacement therapy on liver function of male NAFLD subjects. Additionally, the higher risk of NAFLD after androgen deprivation therapy in men with prostate cancer may be eliminated through hormone replacement therapy (HRT) (161). Jaruvongvanich et al. (175) performed a meta-analysis on 16 observational studies (13,721 men) and documented that TT levels are lower in NAFLD men in comparison to controls. Based on the sex-dependent relation, TT levels have inverse-association with NAFLD in men and positive-association in women, while, higher SHBG levels are followed by lower NAFLD odds in either gender (175).
In this regard, the same association has been observed in children and adolescent, as Mueller et al. (180) in a cross-sectional multiethnic study reported that boys with increased TT had lower steatosis/fibrosis in histological findings, while girls with higher TT experienced more severe forms of NAFLD and approved the sex-dependent association of TT levels with NAFLD even in pediatric population. Polyzos et al. (176) observed a negative-relationship between TT levels and steatosis-related indices, but not with fibrosis indices.
In addition, some studies have investigated the relationship between lower dehydroepiandrosterone (DHEA) levels and liver-related complications in NASH patients and have suggested DHEA as a marker for assessing the risk of NASH with advanced fibrosis (14).
The exact underlying mechanism in the incidence of “disturbed sex hormone- liver axis” state remains unclear, while deregulated pancreatic β-cell, adipocyte and hepatocyte function and changes in energy homeostasis could be considered as indirect changes during hormonal imbalances (181). Sex hormone deficiency is associated with disruption of homeostasis of lipid and glucose metabolism, triggering the molecular pathways leading to NAFLD (14). Although, detecting the exact molecular mediators is very complex, a number of common underlying pathogenic pathways are involved.
Decreases in sex steroids are observed in women during menopause (158). Men and post-menopause women experience higher risk of Mets and NAFLD compared with pre-menopause women (182). This higher risk reaches to approximately two-fold higher risk of NAFLD in post-menopausal women (139). The underlying mechanism is the effect of reduced estrogen levels on increasing hepatic fat accumulation, dyslipidemia and the incidence of NAFLD (158). Estrogen also improves anti-oxidant state and protects women from liver injury. Furthermore, increased testosterone and decreased SHBG levels are associated with NAFLD in post- menopause women (183, 184).
Hypogonadism in females is commonly resulted from Turner syndrome-defined by premature ovarian failure and subsequent hypoestrogenism- that eventually leads to elevated hepatic enzymes, NAFLD and liver injury (185). In addition, female rats have shown development of NAFLD due to the lowered fibroblast growth factor (FGF)-21 after ovariectomy (186).
Low testosterone levels are observed in male hypogonadism. Low testosterone levels are correlated with IR in a bi-directional manner followed by abdominal/visceral fat deposition (187). In turn, adipocytes generate leptin and pro-inflammatory factors that inhibit the production of testosterone. Taken together, epidemiologic studies mention that lower testosterone levels are correlated with the prevalence of NAFLD (178, 179).
Sex steroid deficiency is mainly involved in the pathogenesis of NAFLD as estrogen and testosterone play role in liver steatosis. Moreover, a number of other pathologic changes also occur during hypogonadal state.
Estrogen regulates energy and lipid homeostasis through its classic nuclear estrogen receptors (ERs) in the liver, exerts non-nuclear activities and inhibits liver fibrosis (64, 143). Estrogen reduces hepatic lipid deposition and promotes fatty acid oxidation via inhibiting PPAR-α signaling pathway (139). Indeed, increases the expression levels of insulin receptor gene. Therefor, hypoestrogenism triggers IR and disturbed glucose tolerance (188). Lower estradiol levels are also correlated with hepatic fat accumulation (189). Moreover, hypoestrogenism is associated with increased macrophage infiltration and higher expression of pro-inflammatory genes in the liver (190).
The major circulating androgens-testosterone and dihydrotestosterone (DHT)-affect hepatic metabolism. Testosterone modulates gene expression by translocating to the nucleus after binding to the hepatic ARs in both male and females (158). The highest expression levels of ARs is attributed to males and subjects in reproductive years (64). The polymorphic CAG repeat sequence of AR gene affects fat accumulation (191, 192). Of note, testosterone improves pancreatic function, suppresses TG uptake, adipose accumulation and inhibits adipocyte differentiation (193). Testosterone levels negatively correlate with visceral fat mass and the incidence of obesity and obesity-related conditions including Mets, T2DM and NAFLD in men with hypogonadism (194, 195).
Collectively, observational studies suggest that testosterone levels are associated with NAFLD in a sex- dependent manner, as higher serum TT levels lead to steatosis in women, while may be even protective in men. More precisely, TT levels in men and post-menopause women are negatively correlated with the incidence of NAFLD, whereas the correlation seems positive in PCOS women (157, 175). In the case of SHBG, diminished SHBG levels are associated with NAFLD in either sex (183).
DHEA deficiency is commonly observed in pan-hypopituitarism condition and amplifies the rapid progression of NASH to cirrhosis. DHEA is a steroid hormone with high abundance levels and modulates homeostasis, mediates ROS scavengers and OS, reduces IR, and increases the transcription levels of PPAR genes (169, 196). Several studies have illustrated the association between diminished serum DHEA levels and the higher severity of liver fibrosis in biopsy-proven NASH patients (124, 169, 171–174, 177). In this regard, the settled studies have been summarized in Table 3.
Hypogonadism triggers a wide range of impairments in cardiovascular risk factors. The pattern of fat distribution as well as regional adiposity is dependent on sex hormones and hypogonadism which causes obesity, dyslipidemia, hypertension and dysglycemia and resulting in the higher progression of NAFLD (14). Based on former studies, animals in hypogonadal state experience increased TG, total cholesterol and LDL-C either in serum or liver and show elevated expression levels of SREBP-1 and 2 that eventually enhance liver steatosis (197).
Based on the evidence from animal studies, intestinal dysbiosis links hypogonadism to the development of NAFLD. Changes in Lactobacillus numbers as well as Firmicutes/Bacteroidetes ratio could be resulted from androgen loss in castrated male mice (198).
Patients with hypogonadism-induced NAFLD should regularly be visited by both endocrinologist and hepatologist and undergo liver related assessments after excluding other steatogenic causes to prevent liver damage (199). The general management includes a combination of lifestyle modifications, dietary restrictions as well as regular physical activity (200). Specific treatment plans should be sex-dependent, i.e. female hypogonadism is treated by estrogen replacement, while in men with hypogonadism testosterone is the drug of choice (14, 201).
Further studies evaluating the potential impact of HRT in patients with hypogonadism and NAFLD are needed. As mentioned before, hypogonadism is linked to NAFLD in a bi-directional manner and these patients should be screened routinely.
Anterior-pituitary is responsible for the production of prolactin- a multifunctional lactotroph polypeptide hormone (202). Hypopituitarism is usually along with reduced levels of prolactin and expression levels of prolactin receptors (PRLRs) gene (139). Prolactin is involved in growth, immunity, food intake and metabolic health via modulating glucose and lipid metabolisms as well as stimulating adipogenesis (113). Prolactin is also involved in the biosynthesis of monounsaturated fatty acids by down-regulating the expression of stearoyl-CoA desaturase1 (SCD1) (203). Moreover, it acts through its specific hepatic receptors and down-regulates CD36, resulting in reduced hepatic TG accumulation, lowered lipid content and protection against hepatic steatosis (203, 204). Luque et al. (205) highlighted the probable role of prolactin in de novo lipogenesis and subsequent NAFLD progression. Zhang et al. (204) in a study on 456 NAFLD subjects reported lower circulating prolactin levels in NAFLD group in comparison to controls. Prolactin levels were lower in patients with more severe hepatic steatosis (139). In conclusion, although regarding evidence seems to be insufficient and further in vivo and in vitro studies are needed, chronic lower prolactin levels may exert catastrophic effects on liver health.
Taken together, the current body of evidence highlights the inter-relationship between NAFLD and a wide range of endocrinopathies and suggests the need of NAFLD screening, simultaneously by hepatologists and endocrinologists. Further well-designed studies are needed to discover the exact underlying molecular contact-point of aforementioned diseases as well as the possible new screening/treatment strategies for NAFLD patients with endocrine disorders.
SA and HT contributed to data extraction and manuscript drafting. ME-M and FN contributed to the conception of the article as well as to the final revision of the manuscript. All authors read and approved the final version of the manuscript (grant No 71003).
This study was supported by the Research Vice-Chancellor and Endocrine Research Center of Tabriz University of Medical Sciences (grant No 71003).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nobili V, Svegliati-Baroni G, Alisi A, Miele L, Valenti L, Vajro P. A 360-degree overview of paediatric NAFLD: recent insights. J Hepatol (2013) 58(6):1218–29. doi: 10.1016/j.jhep.2012.12.003
2. Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, et al. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American association for the study of liver diseases, American college of gastroenterology, and the American gastroenterological association. Hepatology (2012) 55(6):2005–23. doi: 10.1002/hep.25762
3. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology (2018) 67(1):123–33. doi: 10.1002/hep.29466
4. Marchisello S, Di Pino A, Scicali R, Urbano F, Piro S, Purrello F, et al. Pathophysiological, molecular and therapeutic issues of nonalcoholic fatty liver disease: an overview. Int J Mol Sci (2019) 20(8):1948. doi: 10.3390/ijms20081948
5. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol (2020) 73(1):202–9. doi: 10.1016/j.jhep.2020.03.039
6. Tutunchi H, Saghafi-Asl M, Asghari-Jafarabadi M, Ostadrahimi A. The relationship between severity of liver steatosis and metabolic parameters in a sample of Iranian adults. BMC Res Notes (2020) 13(1):218. doi: 10.1186/s13104-020-05059-5
7. Hazlehurst JM, Tomlinson JW. Mechanisms in endocrinology: Non-alcoholic fatty liver disease in common endocrine disorders. Eur J Endocrinol (2013) 169(2):R27–37. doi: 10.1530/EJE-13-0296
8. Elasam AN, Ahmed MA, Ahmed A, Sharif ME, Abusham A, Hassan B, et al. The prevalence and phenotypic manifestations of polycystic ovary syndrome (PCOS) among infertile Sudanese women: a cross-sectional study. BMC Women's Health (2022) 22(1):1–7. doi: 10.1186/s12905-022-01762-6
9. Ntumy M, Maya E, Lizneva D, Adanu R, Azziz R. The pressing need for standardization in epidemiologic studies of PCOS across the globe. Gynecological Endocrinol (2019) 35(1):1–3.
10. Spremović Rađenović S, Pupovac M, Andjić M, Bila J, Srećković S, Gudović A, et al. Prevalence, risk factors, and pathophysiology of nonalcoholic fatty liver disease (NAFLD) in women with polycystic ovary syndrome (PCOS). Biomedicines (2022) 10(1):131. doi: 10.3390/biomedicines10010131
11. Sirmans SM, Pate KA. Epidemiology, diagnosis, and management of polycystic ovary syndrome. Clin Epidemiol (2014) 6:1. doi: 10.2147/CLEP.S37559
12. Gambarin–Gelwan M, Kinkhabwala SV, Schiano TD, Bodian C, Yeh HC, Futterweit W. Prevalence of nonalcoholic fatty liver disease in women with polycystic ovary syndrome. Clin Gastroenterol Hepatol (2007) 5(4):496–501. doi: 10.1016/j.cgh.2006.10.010
13. Vassilatou E. Nonalcoholic fatty liver disease and polycystic ovary syndrome. World J Gastroenterol (2014) 20(26):8351. doi: 10.3748/wjg.v20.i26.8351
14. Lonardo A, Mantovani A, Lugari S, Targher G. NAFLD in some common endocrine diseases: prevalence, pathophysiology, and principles of diagnosis and management. Int J Mol Sci (2019) 20(11):2841. doi: 10.3390/ijms20112841
15. Shengir M, Chen T, Guadagno E, Ramanakumar AV, Ghali P, Deschenes M, et al. Non-alcoholic fatty liver disease in premenopausal women with polycystic ovary syndrome: A systematic review and meta-analysis. JGH Open (2021) 5(4):434–45. doi: 10.1002/jgh3.12512
16. Targher G, Rossini M, Lonardo A. Evidence that non-alcoholic fatty liver disease and polycystic ovary syndrome are associated by necessity rather than chance: a novel hepato-ovarian axis? Endocrine (2016) 51(2):211–21. doi: 10.1007/s12020-015-0640-8
17. Kelley CE, Brown AJ, Diehl AM, Setji TL. Review of nonalcoholic fatty liver disease in women with polycystic ovary syndrome. World J Gastroenterol (2014) 20(39):14172. doi: 10.3748/wjg.v20.i39.14172
18. Brown AJ, Tendler DA, McMurray RG, Setji TL. Polycystic ovary syndrome and severe nonalcoholic steatohepatitis: beneficial effect of modest weight loss and exercise on liver biopsy findings. Endocri Pract (2005) 11(5):319–24. doi: 10.4158/EP.11.5.319
19. Ramezani-Binabaj M, Motalebi M, Karimi-Sari H, Rezaee-Zavareh MS, Alavian SM. Are women with polycystic ovarian syndrome at a high risk of non-alcoholic fatty liver disease; a meta-analysis. Hepatitis Monthly (2014) 14(11):e23235. doi: 10.5812/hepatmon.23235
20. Rocha A, Faria L, Guimarães T, Moreira G, Cândido A, Couto C, et al. Non-alcoholic fatty liver disease in women with polycystic ovary syndrome: systematic review and meta-analysis. J Endocrinol Invest (2017) 40(12):1279–88. doi: 10.1007/s40618-017-0708-9
21. Jones H, Sprung VS, Pugh CJ, Daousi C, Irwin A, Aziz N, et al. Polycystic ovary syndrome with hyperandrogenism is characterized by an increased risk of hepatic steatosis compared to nonhyperandrogenic PCOS phenotypes and healthy controls, independent of obesity and insulin resistance. J Clin Endocrinol Metab (2012) 97(10):3709–16. doi: 10.1210/jc.2012-1382
22. Wu J, Yao X-Y, Shi R-X, Liu S-F, Wang X-Y. A potential link between polycystic ovary syndrome and non-alcoholic fatty liver disease: an update meta-analysis. Reprod Health (2018) 15(1):1–9. doi: 10.1186/s12978-018-0519-2
23. Cerda C, Pérez-Ayuso RM, Riquelme A, Soza A, Villaseca P, Sir-Petermann T, et al. Nonalcoholic fatty liver disease in women with polycystic ovary syndrome. J Hepatol (2007) 47(3):412–7. doi: 10.1016/j.jhep.2007.04.012
24. Setji TL, Holland ND, Sanders LL, Pereira KC, Diehl AM, Brown AJ. Nonalcoholic steatohepatitis and nonalcoholic fatty liver disease in young women with polycystic ovary syndrome. J Clin Endocrinol Metab (2006) 91(5):1741–7. doi: 10.1210/jc.2005-2774
25. Schwimmer JB, Khorram O, Chiu V, Schwimmer WB. Abnormal aminotransferase activity in women with polycystic ovary syndrome. Fertil Steril (2005) 83(2):494–7. doi: 10.1016/j.fertnstert.2004.08.020
26. Vassilatou E, Lafoyianni S, Vryonidou A, Ioannidis D, Kosma L, Katsoulis K, et al. Increased androgen bioavailability is associated with non-alcoholic fatty liver disease in women with polycystic ovary syndrome. Hum Reprod (2010) 25(1):212–20. doi: 10.1093/humrep/dep380
27. Lerchbaum E, Gruber H-J, Schwetz V, Giuliani A, Möller R, Pieber TR, et al. Fatty liver index in polycystic ovary syndrome. Eur J Endocrinol (2011) 165(6):935–43. doi: 10.1530/EJE-11-0614
28. Barfield E, Liu Y-H, Kessler M, Pawelczak M, David R, Shah B. The prevalence of abnormal liver enzymes and metabolic syndrome in obese adolescent females with polycystic ovary syndrome. J Pediatr Adolesc Gynecol (2009) 22(5):318–22. doi: 10.1016/j.jpag.2009.03.003
29. Michaliszyn SF, Lee S, Tfayli H, Arslanian S. Polycystic ovary syndrome and nonalcoholic fatty liver in obese adolescents: association with metabolic risk profile. Fertil Steril (2013) 100(6):1745–51. doi: 10.1016/j.fertnstert.2013.08.015
30. Targher G, Solagna E, Tosi F, Castello R, Spiazzi G, Zoppini G, et al. Abnormal serum alanine aminotransferase levels are associated with impaired insulin sensitivity in young women with polycystic ovary syndrome. J Endocrinol Invest (2009) 32(8):695–700. doi: 10.1007/BF03345743
31. Kim J, Kim D, Yim J, Kang J, Han K, Kim S, et al. Polycystic ovary syndrome with hyperandrogenism as a risk factor for non-obese non-alcoholic fatty liver disease. Aliment Pharmacol Ther (2017) 45(11):1403–12. doi: 10.1111/apt.14058
32. Romanowski MD, Parolin MB, Freitas AC, Piazza MJ, Basso J, Urbanetz AA. Prevalence of non-alcoholic fatty liver disease in women with polycystic ovary syndrome and its correlation with metabolic syndrome. Arquivos Gastroenterol (2015) 52:117–23. doi: 10.1590/S0004-28032015000200008
33. Ma R, Liu K, Lam P, Cheung L, Tam W, Ko G, et al. Sonographic measurement of mesenteric fat predicts presence of fatty liver among subjects with polycystic ovary syndrome. J Clin Endocrinol Metab (2011) 96(3):799–807. doi: 10.1210/jc.2010-1608
34. Markou A, Androulakis II, Mourmouris C, Tsikkini A, Samara C, Sougioultzis S, et al. Hepatic steatosis in young lean insulin resistant women with polycystic ovary syndrome. Fertil Steril (2010) 93(4):1220–6. doi: 10.1016/j.fertnstert.2008.12.008
35. Economou F, Xyrafis X, Livadas S, Androulakis II, Argyrakopoulou G, Christakou CD, et al. In overweight/obese but not in normal-weight women, polycystic ovary syndrome is associated with elevated liver enzymes compared to controls. Hormones (2009) 8(3):199–206. doi: 10.14310/horm.2002.1236
36. March WA, Moore VM, Willson KJ, Phillips DI, Norman RJ, Davies MJ. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum Reprod (2010) 25(2):544–51. doi: 10.1093/humrep/dep399
37. Shengir M, Krishnamurthy S, Ghali P, Deschenes M, Wong P, Chen T, et al. Prevalence and predictors of nonalcoholic fatty liver disease in south Asian women with polycystic ovary syndrome. World J Gastroenterol (2020) 26(44):7046. doi: 10.3748/wjg.v26.i44.7046
38. Salva-Pastor N, López-Sánchez GN, Chávez-Tapia NC, Audifred-Salomón JR, Niebla-Cárdenas D, Topete-Estrada R, et al. Polycystic ovary syndrome with feasible equivalence to overweight as a risk factor for non-alcoholic fatty liver disease development and severity in Mexican population. Ann Hepatol (2020) 19(3):251–7. doi: 10.1016/j.aohep.2020.01.004
39. Hossain N, Stepanova M, Afendy A, Nader F, Younossi Y, Rafiq N, et al. Non-alcoholic steatohepatitis (NASH) in patients with polycystic ovarian syndrome (PCOS). Scand J Gastroenterol (2011) 46(4):479–84. doi: 10.3109/00365521.2010.539251
40. Sarkar M, Terrault N, Chan W, Cedars MI, Huddleston HG, Duwaerts CC, et al. Polycystic ovary syndrome (PCOS) is associated with NASH severity and advanced fibrosis. Liver Int (2020) 40(2):355–9. doi: 10.1111/liv.14279
41. Brzozowska MM, Ostapowicz G, Weltman MD. An association between non-alcoholic fatty liver disease and polycystic ovarian syndrome. J Gastroenterol Hepatol (2009) 24(2):243–7. doi: 10.1111/j.1440-1746.2008.05740.x
42. Polyzos SA, Goulis DG, Kountouras J, Mintziori G, Chatzis P, Papadakis E, et al. Non-alcoholic fatty liver disease in women with polycystic ovary syndrome: assessment of non-invasive indices predicting hepatic steatosis and fibrosis. Hormones (2014) 13(4):519–31. doi: 10.14310/horm.2002.1493
43. Asfari MM, Sarmini MT, Baidoun F, Al-Khadra Y, Ezaizi Y, Dasarathy S, et al. Association of non-alcoholic fatty liver disease and polycystic ovarian syndrome. BMJ Open Gastroenterol (2020) 7(1):e000352. doi: 10.1136/bmjgast-2019-000352
44. Won YB, Seo SK, Yun BH, Cho S, Choi YS, Lee BS. Non-alcoholic fatty liver disease in polycystic ovary syndrome women. Sci Rep (2021) 11(1):1–11. doi: 10.1038/s41598-021-86697-y
45. Varma SH, Tirupati S, Pradeep T, Sarathi V, Kumar D. Insulin resistance and hyperandrogenemia independently predict nonalcoholic fatty liver disease in women with polycystic ovary syndrome. Diabetes Metab Syndrome: Clin Res Rev (2019) 13(2):1065–9. doi: 10.1016/j.dsx.2018.12.020
46. Bohdanowicz-Pawlak A, Lenarcik-Kabza A, Brona A, Kuliczkowska-Płaksej J, Łaczmański Ł, Zaleska-Dorobisz U, et al. Non-alcoholic fatty liver disease in women with polycystic ovary syndrome–clinical and metabolic aspects and lipoprotein lipase gene polymorphism. Endokrynol Polska (2014) 65(6):416–21. doi: 10.5603/EP.2014.0058
47. Wang Y, Guo X, Xu W, Cai J, Zhang Y, Wu C, et al. Role of androgen in liver fat content in women: metabolically advantageous or disadvantageous? Endocri Pract (2020) 26(9):1003–16. doi: 10.4158/EP-2019-0407
48. Kumarendran B, O’Reilly MW, Manolopoulos KN, Toulis KA, Gokhale KM, Sitch AJ, et al. Polycystic ovary syndrome, androgen excess, and the risk of nonalcoholic fatty liver disease in women: a longitudinal study based on a united kingdom primary care database. PloS Med (2018) 15(3):e1002542. doi: 10.1371/journal.pmed.1002542
49. Dawson AJ, Sathyapalan T, Smithson JA, Vince RV, Coady AM, Ajjan R, et al. A comparison of cardiovascular risk indices in patients with polycystic ovary syndrome with and without coexisting nonalcoholic fatty liver disease. Clin Endocrinol (2014) 80(6):843–9. doi: 10.1111/cen.12258
50. Cai J, Wu C, Zhang Y, Wang Y, Xu W, Lin T, et al. High-free androgen index is associated with increased risk of non-alcoholic fatty liver disease in women with polycystic ovary syndrome, independent of obesity and insulin resistance. Int J Obes (2017) 41(9):1341–7. doi: 10.1038/ijo.2017.116
51. Chen M-J, Chiu H-M, Chen C-L, Yang W-S, Yang Y-S, Ho H-N. Hyperandrogenemia is independently associated with elevated alanine aminotransferase activity in young women with polycystic ovary syndrome. J Clin Endocrinol Metab (2010) 95(7):3332–41. doi: 10.1210/jc.2009-2698
52. Kauffman RP, Baker TE, Baker V, Kauffman MM, Castracane VD. Endocrine factors associated with non-alcoholic fatty liver disease in women with polycystic ovary syndrome: do androgens play a role? Gynecol Endocrinol (2010) 26(1):39–46. doi: 10.3109/09513590903184084
53. Macut D, Tziomalos K, Božić-Antić I, Bjekić-Macut J, Katsikis I, Papadakis E, et al. Non-alcoholic fatty liver disease is associated with insulin resistance and lipid accumulation product in women with polycystic ovary syndrome. Hum Reprod (2016) 31(6):1347–53. doi: 10.1093/humrep/dew076
54. Cussons AJ, Watts GF, Mori TA, Stuckey BG. Omega-3 fatty acid supplementation decreases liver fat content in polycystic ovary syndrome: a randomized controlled trial employing proton magnetic resonance spectroscopy. J Clin Endocrinol Metab (2009) 94(10):3842–8. doi: 10.1210/jc.2009-0870
55. Tan S, Bechmann LP, Benson S, Dietz T, Eichner S, Hahn S, et al. Apoptotic markers indicate nonalcoholic steatohepatitis in polycystic ovary syndrome. J Clin Endocrinol Metab (2010) 95(1):343–8. doi: 10.1210/jc.2009-1834
56. Tarantino G, Di Somma C, Pizza G, Brancato V, Nedi V, Valentino R, et al. Polycystic ovary syndrome and hepatic steatosis: could low-grade chronic inflammation be mediated by the spleen? Eur J Inflamm (2013) 11(1):179–91. doi: 10.1177/1721727X1301100117
57. Villa J, Pratley RE. Adipose tissue dysfunction in polycystic ovary syndrome. Curr Diabetes Rep (2011) 11(3):179–84. doi: 10.1007/s11892-011-0189-8
58. Zeng X, Y-j X, Liu Y-T, Long S-L, Mo Z-C. Polycystic ovarian syndrome: correlation between hyperandrogenism, insulin resistance and obesity. Clinica Chimica Acta (2020) 502:214–21. doi: 10.1016/j.cca.2019.11.003
59. Baranova A, Tran TP, Afendy A, Wang L, Shamsaddini A, Mehta R, et al. Molecular signature of adipose tissue in patients with both non-alcoholic fatty liver disease (NAFLD) and polycystic ovarian syndrome (PCOS). J Trans Med (2013) 11(1):1–8. doi: 10.1186/1479-5876-11-133
60. Baranova A, Tran T, Birerdinc A, Younossi Z. Systematic review: association of polycystic ovary syndrome with metabolic syndrome and non-alcoholic fatty liver disease. Aliment Pharmacol Ther (2011) 33(7):801–14. doi: 10.1111/j.1365-2036.2011.04579.x
61. Gültepe İ, Başaranoğlu M, Süleymanoğlu Y, Başaranoğlu G, Beyazıt F. Ovaries are more vulnerable than hepatocytes for insulin resistance and hyperinsulinemia. Turk J Gastroenterol (2016) 27(1):62–7. doi: 10.5152/tjg.2015.150473
62. Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocri Rev (2012) 33(6):981–1030. doi: 10.1210/er.2011-1034
63. Grossmann M, Wierman ME, Angus P, Handelsman DJ. Reproductive endocrinology of nonalcoholic fatty liver disease. Endocri Rev (2019) 40(2):417–46. doi: 10.1210/er.2018-00158
64. Shen M, Shi H. Sex hormones and their receptors regulate liver energy homeostasis. Int J Endocrinol (2015) 2015:294278. doi: 10.1155/2015/294278
65. McInnes KJ, Corbould A, Simpson ER, Jones ME. Regulation of adenosine 5′, monophosphate-activated protein kinase and lipogenesis by androgens contributes to visceral obesity in an estrogen-deficient state. Endocrinology (2006) 147(12):5907–13. doi: 10.1210/en.2006-0879
66. Su C, Chen M, Xu W, Lin J. The impacts of testosterone on insulin sensitivity and chronic low-grade. Zhonghua yi xue za zhi (2017) 97(1):47–52. doi: 10.3760/cma.j.issn.0376-2491.2017.01.012
67. Schiffer L, Kempegowda P, Arlt W, O’Reilly MW. Mechanisms in endocrinology: the sexually dimorphic role of androgens in human metabolic disease. Eur J Endocrinol (2017) 177(3):R125–R43. doi: 10.1530/EJE-17-0124
68. Livingstone DE, Barat P, Di Rollo EM, Rees GA, Weldin BA, Rog-Zielinska EA, et al. 5α-reductase type 1 deficiency or inhibition predisposes to insulin resistance, hepatic steatosis, and liver fibrosis in rodents. Diabetes (2015) 64(2):447–58. doi: 10.2337/db14-0249
69. Dai R, Yan D, Li J, Chen S, Liu Y, Chen R, et al. Activation of PKR/eIF2α signaling cascade is associated with dihydrotestosterone-induced cell cycle arrest and apoptosis in human liver cells. J Cell Biochem (2012) 113(5):1800–8. doi: 10.1002/jcb.24051
70. Chitturi S, Abeygunasekera S, Farrell GC, Holmes-Walker J, Hui JM, Fung C, et al. NASH and insulin resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology (2002) 35(2):373–9. doi: 10.1053/jhep.2002.30692
71. González F, Sia CL, Bearson DM, Blair HE. Hyperandrogenism induces a proinflammatory TNFα response to glucose ingestion in a receptor-dependent fashion. J Clin Endocrinol Metab (2014) 99(5):E848–54. doi: 10.1210/jc.2013-4109
72. Chen Z-J, Zhao H, He L, Shi Y, Qin Y, Shi Y, et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16. 3, 2p21 and 9q33. 3. Nat Genet (2011) 43(1):55–9. doi: 10.1038/ng.732
73. Deligeoroglou E, Vrachnis N, Athanasopoulos N, Iliodromiti Z, Sifakis S, Iliodromiti S, et al. Mediators of chronic inflammation in polycystic ovarian syndrome. Gynecol Endocrinol (2012) 28(12):974–8. doi: 10.3109/09513590.2012.683082
74. Dongiovanni P, Donati B, Fares R, Lombardi R, Mancina RM, Romeo S, et al. PNPLA3 I148M polymorphism and progressive liver disease. World J Gastroenterol (2013) 19(41):6969. doi: 10.3748/wjg.v19.i41.6969
75. Zeng X, Huang Q, Long SL, Zhong Q, Mo Z. Mitochondrial dysfunction in polycystic ovary syndrome. DNA Cell Biol (2020) 39(8):1401–9. doi: 10.1089/dna.2019.5172
76. Cree-Green M, Rahat H, Newcomer BR, Bergman BC, Brown MS, Coe GV, et al. Insulin resistance, hyperinsulinemia and mitochondria dysfunction in non-obese girls with polycystic ovarian syndrome. J Endocri Soc (2017) 1(7):931–44. doi: 10.1210/js.2017-00192
77. Simões IC, Fontes A, Pinton P, Zischka H, Wieckowski MR. Mitochondria in non-alcoholic fatty liver disease. Int J Biochem Cell Biol (2018) 95:93–9. doi: 10.1016/j.biocel.2017.12.019
78. Kessoku T, Kobayashi T, Tanaka K, Yamamoto A, Takahashi K, Iwaki M, et al. The role of leaky gut in nonalcoholic fatty liver disease: a novel therapeutic target. Int J Mol Sci (2021) 22(15):8161. doi: 10.3390/ijms22158161
79. Lindheim L, Bashir M, Münzker J, Trummer C, Zachhuber V, Leber B, et al. Alterations in gut microbiome composition and barrier function are associated with reproductive and metabolic defects in women with polycystic ovary syndrome (PCOS): A pilot study. PloS One (2017) 12(1):e0168390. doi: 10.1371/journal.pone.0168390
80. Juan C-C, Chen K-H, Wang P-H, Hwang J-L, Seow K-M. Endocannabinoid system activation may be associated with insulin resistance in women with polycystic ovary syndrome. Fertil Steril (2015) 104(1):200–6. doi: 10.1016/j.fertnstert.2015.03.027
81. Guzmán M, Galve-Roperh I, Sánchez C. Ceramide: a new second messenger of cannabinoid action. Trends Pharmacol Sci (2001) 22(1):19–22. doi: 10.1016/S0165-6147(00)01586-8
82. Mallat A, Lotersztajn S. Endocannabinoids and their role in fatty liver disease. Digest Dis (2010) 28(1):261–6. doi: 10.1159/000282100
83. Kuliczkowska Plaksej J, Laczmanski L, Milewicz A, Lenarcik-Kabza A, Trzmiel-Bira A, Zaleska-Dorobisz U, et al. Cannabinoid receptor 1 gene polymorphisms and nonalcoholic fatty liver disease in women with polycystic ovary syndrome and in healthy controls. Int J Endocrinol (2014) 2014:232975. doi: 10.1155/2014/232975
84. Tock L, Carneiro G, Togeiro SM, Hachul H, Pereira AZ, Tufik S, et al. Obstructive sleep apnea predisposes to nonalcoholic fatty liver disease in patients with polycystic ovary syndrome. Endocri Pract (2014) 20(3):244–51. doi: 10.4158/EP12366.OR
85. Sookoian S, Pirola CJ. Obstructive sleep apnea is associated with fatty liver and abnormal liver enzymes: a meta-analysis. Obes Surg (2013) 23(11):1815–25. doi: 10.1007/s11695-013-0981-4
86. Glintborg D, Højlund K, Andersen NR, Hansen BF, Beck-Nielsen H, Wojtaszewski JF. Impaired insulin activation and dephosphorylation of glycogen synthase in skeletal muscle of women with polycystic ovary syndrome is reversed by pioglitazone treatment. J Clin Endocrinol Metab (2008) 93(9):3618–26. doi: 10.1210/jc.2008-0760
87. Munir I, Yen H-W, Geller DH, Torbati D, Bierden RM, Weitsman SR, et al. Insulin augmentation of 17α-hydroxylase activity is mediated by phosphatidyl inositol 3-kinase but not extracellular signal-regulated kinase-1/2 in human ovarian theca cells. Endocrinology (2004) 145(1):175–83. doi: 10.1210/en.2003-0329
88. Abbott DH, Dumesic DA, Levine JE. Hyperandrogenic origins of polycystic ovary syndrome–implications for pathophysiology and therapy. Expert Rev Endocrinol Metab (2019) 14(2):131–43. doi: 10.1080/17446651.2019.1576522
89. Hansen SL, Svendsen PF, Jeppesen JF, Hoeg LD, Andersen NR, Kristensen JM, et al. Molecular mechanisms in skeletal muscle underlying insulin resistance in women who are lean with polycystic ovary syndrome. J Clin Endocrinol Metab (2019) 104(5):1841–54. doi: 10.1210/jc.2018-01771
90. Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature (2014) 510(7503):84–91. doi: 10.1038/nature13478
91. Gutierrez-Grobe Y, Ponciano-Rodríguez G, Ramos MH, Uribe M, Méndez-Sánchez N. Prevalence of non alcoholic fatty liver disease in premenopausal, posmenopausal and polycystic ovary syndrome women. Role Estrogens. Ann Hepatol (2010) 9(4):402–9. doi: 10.1016/S1665-2681(19)31616-3
92. Yang JD, Abdelmalek MF, Pang H, Guy CD, Smith AD, Diehl AM, et al. Gender and menopause impact severity of fibrosis among patients with nonalcoholic steatohepatitis. Hepatology (2014) 59(4):1406–14. doi: 10.1002/hep.26761
93. Shimizu I, Kohno N, Tamaki K, Shono M, Huang H-W, He J-H, et al. Female hepatology: favorable role of estrogen in chronic liver disease with hepatitis b virus infection. World J Gastroenterol (2007) 13(32):4295. doi: 10.3748/wjg.v13.i32.4295
94. Hahn S, Haselhorst U, Quadbeck B, Tan S, Kimmig R, Mann K, et al. Decreased soluble leptin receptor levels in women with polycystic ovary syndrome. Eur J Endocrinol (2006) 154(2):287–94. doi: 10.1530/eje.1.02078
95. Yilmaz M, Bukan N, Demırcı H, Öztürk Ç, Kan E, Ayvaz G, et al. Serum resistin and adiponectin levels in women with polycystic ovary syndrome. Gynecol Endocrinol (2009) 25(4):246–52. doi: 10.1080/09513590802653833
96. Targher G, Bertolini L, Zenari L. Hypoadiponectinemia is closely associated with nonalcoholic hepatic steatosis in obese subjects. Diabetes Care (2004) 27(8):2085–6. doi: 10.2337/diacare.27.8.2085
97. Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J Hepatol (2015) 63(1):164–73. doi: 10.1016/j.jhep.2015.02.019
98. Byrne CD, Patel J, Scorletti E, Targher G. Tests for diagnosing and monitoring non-alcoholic fatty liver disease in adults. Bmj (2018) 362:k2734. doi: 10.1136/bmj.k2734
99. Papatheodoridi M, Cholongitas E. Diagnosis of non-alcoholic fatty liver disease (NAFLD): Current concepts. Curr Pharm Design (2018) 24(38):4574–86. doi: 10.2174/1381612825666190117102111
100. Legro RS, Arslanian SA, Ehrmann DA, Hoeger KM, Murad MH, Pasquali R, et al. Diagnosis and treatment of polycystic ovary syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2013) 98(12):4565–92. doi: 10.1210/jc.2013-2350
101. Gangale MF, Miele L, Lanzone A, Sagnella F, Martinez D, Tropea A, et al. Long-term metformin treatment is able to reduce the prevalence of metabolic syndrome and its hepatic involvement in young hyperinsulinaemic overweight patients with polycystic ovarian syndrome. Clin Endocrinol (2011) 75(4):520–7. doi: 10.1111/j.1365-2265.2011.04093.x
102. Tutunchi H, Ostadrahimi A, Saghafi-Asl M, Roshanravan N, Shakeri-Bavil A, Asghari-Jafarabadi M, et al. Expression of NF-κB, IL-6, and IL-10 genes, body composition, and hepatic fibrosis in obese patients with NAFLD-combined effects of oleoylethanolamide supplementation and calorie restriction: A triple-blind randomized controlled clinical trial. J Cell Physiol (2021) 236(1):417–26. doi: 10.1002/jcp.29870
104. Frøssing S, Nylander M, Chabanova E, Frystyk J, Holst JJ, Kistorp C, et al. Effect of liraglutide on ectopic fat in polycystic ovary syndrome: a randomized clinical trial. Diabetes Obes Metab (2018) 20(1):215–8. doi: 10.1111/dom.13053
105. Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut (2017) 66(1):180–90. doi: 10.1136/gutjnl-2016-312431
106. Liu S-H, Lazo M, Koteish A, Kao W, Shih M-H, Bonekamp S, et al. Oral contraceptive pill use is associated with reduced odds of nonalcoholic fatty liver disease in menstruating women: results from NHANES III. J Gastroenterol (2013) 48(10):1151–9. doi: 10.1007/s00535-012-0715-8
107. Parker HM, Johnson NA, Burdon CA, Cohn JS, O’Connor HT, George J. Omega-3 supplementation and non-alcoholic fatty liver disease: a systematic review and meta-analysis. J Hepatol (2012) 56(4):944–51. doi: 10.1016/j.jhep.2011.08.018
108. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin e, or placebo for nonalcoholic steatohepatitis. N Engl J Med (2010) 362(18):1675–85. doi: 10.1056/NEJMoa0907929
109. Yeliosof O, Gangat M. Diagnosis and management of hypopituitarism. Curr Opin Pediatr (2019) 31(4):531–6. doi: 10.1097/MOP.0000000000000779
110. Singeap A-M, Stanciu C, Huiban L, Muzica CM, Cuciureanu T, Girleanu I, et al. Association between nonalcoholic fatty liver disease and endocrinopathies: clinical implications. Can J Gastroenterol Hepatol (2021) 2021:6678142. doi: 10.1155/2021/6678142
111. Kim SY. Diagnosis and treatment of hypopituitarism. Endocrinol Metab (2015) 30(4):443–55. doi: 10.3803/EnM.2015.30.4.443
112. Jasim S, Alahdab F, Ahmed AT, Tamhane S, Prokop LJ, Nippoldt TB, et al. Mortality in adults with hypopituitarism: a systematic review and meta-analysis. Endocrine (2017) 56(1):33–42. doi: 10.1007/s12020-016-1159-3
113. Von-Hafe M, Borges-Canha M, Vale C, Leite AR, Sérgio Neves J, Carvalho D, et al. Nonalcoholic fatty liver disease and endocrine axes–a scoping review. Metabolites (2022) 12(4):298. doi: 10.3390/metabo12040298
114. Kodama K, Ichihara A, Seki Y, Ikarashi Y, Sagawa T, Kogiso T, et al. Characteristics of NAFLD based on hypopituitarism. Can J Gastroenterol Hepatol (2020) 2020:8814435. doi: 10.1155/2020/8814435
115. Takahashi Y. The role of growth hormone and insulin-like growth factor-I in the liver. Int J Mol Sci (2017) 18(7):1447. doi: 10.3390/ijms18071447
116. Press M. Growth hormone and metabolism. Diabetes/Metab Rev (1988) 4(4):391–414. doi: 10.1002/dmr.5610040406
117. Kargi AY, Merriam GR. Diagnosis and treatment of growth hormone deficiency in adults. Nat Rev Endocrinol (2013) 9(6):335–45. doi: 10.1038/nrendo.2013.77
118. Gazzaruso C, Gola M, Karamouzis I, Giubbini R, Giustina A. Cardiovascular risk in adult patients with growth hormone (GH) deficiency and following substitution with GH–an update. J Clin Endocrinol Metab (2014) 99(1):18–29. doi: 10.1210/jc.2013-2394
119. Ichikawa T, Hamasaki K, Ishikawa H, Ejima E, Eguchi K, Nakao K. Non-alcoholic steatohepatitis and hepatic steatosis in patients with adult onset growth hormone deficiency. Gut (2003) 52(6):914–. doi: 10.1136/gut.52.6.914
120. Adams LA, Feldstein A, Lindor KD, Angulo P. Nonalcoholic fatty liver disease among patients with hypothalamic and pituitary dysfunction. Hepatology (2004) 39(4):909–14. doi: 10.1002/hep.20140
121. Ichikawa T, Nakao K, Hamasaki K, Furukawa R, Tsuruta S, Ueda Y, et al. Role of growth hormone, insulin-like growth factor 1 and insulin-like growth factor-binding protein 3 in development of non-alcoholic fatty liver disease. Hepatol Int (2007) 1(2):287–94. doi: 10.1007/s12072-007-9007-4
122. Arturi F, Succurro E, Procopio C, Pedace E, Mannino GC, Lugarà M, et al. Nonalcoholic fatty liver disease is associated with low circulating levels of insulin-like growth factor-I. J Clin Endocrinol Metab (2011) 96(10):E1640–E4. doi: 10.1210/jc.2011-1227
123. Hong J, Kim J, Kim Y-E, Lee E. Metabolic parameters and nonalcoholic fatty liver disease in hypopituitary men. Hormone Metab Res (2011) 43(01):48–54. doi: 10.1055/s-0030-1265217
124. Koehler E, Swain J, Sanderson S, Krishnan A, Watt K, Charlton M. Growth hormone, dehydroepiandrosterone and adiponectin levels in non-alcoholic steatohepatitis: an endocrine signature for advanced fibrosis in obese patients. Liver Int (2012) 32(2):279–86. doi: 10.1111/j.1478-3231.2011.02637.x
125. Gardner CJ, Irwin A, Daousi C, McFarlane IA, Joseph F, Bell JD, et al. Hepatic steatosis, GH deficiency and the effects of GH replacement: a Liverpool magnetic resonance spectroscopy study. Eur J Endocrinol (2012) 166:993–1002. doi: 10.1530/EJE-12-0002
126. Nishizawa H, Iguchi G, Murawaki A, Fukuoka H, Hayashi Y, Kaji H, et al. Nonalcoholic fatty liver disease in adult hypopituitary patients with growth hormone deficiency and the impact of growth hormone replacement therapy. Eur J Endocrinol (2012) 167:67–74. doi: 10.1530/EJE-12-0252
127. Xu L, Xu C, Yu C, Miao M, Zhang X, Zhu Z, et al. Association between serum growth hormone levels and nonalcoholic fatty liver disease: A cross-sectional study. PLOS ONE (2012) 7(8):e44136. doi: 10.1371/journal.pone.0044136
128. Sumida Y, Yonei Y, Tanaka S, Mori K, Kanemasa K, Imai S, et al. Lower levels of insulin-like growth factor-1 standard deviation score are associated with histological severity of non-alcoholic fatty liver disease. Hepatol Res (2015) 45(7):771–81. doi: 10.1111/hepr.12408
129. Meienberg F, Yee M, Johnston D, Cox J, Robinson S, Bell JD, et al. Liver fat in adults with GH deficiency: comparison to matched controls and the effect of GH replacement. Clin Endocrinol (2016) 85(1):76–84. doi: 10.1111/cen.13042
130. Chishima S, Kogiso T, Matsushita N, Hashimoto E, Tokushige K. The relationship between the growth hormone/insulin-like growth factor system and the histological features of nonalcoholic fatty liver disease. Internal Med (2017) 56(5):473–80. doi: 10.2169/internalmedicine.56.7626
131. Dichtel LE, Corey KE, Misdraji J, Bredella MA, Schorr M, Osganian SA, et al. The association between IGF-1 levels and the histologic severity of nonalcoholic fatty liver disease. Clin Trans Gastroenterol (2017) 8(1):e217. doi: 10.1038/ctg.2016.72
132. Liang S, Yu Z, Song X, Wang Y, Li M, Xue J. Reduced growth hormone secretion is associated with nonalcoholic fatty liver disease in obese children. Hormone Metab Res (2018) 50(03):250–6. doi: 10.1055/s-0043-124970
133. Nguyen A, Ricolfi F, Lemogne B, Aho S, Lemaire S, Bouillet B, et al. Liver fat content in people with pituitary diseases: influence of serum IGF1 levels. Hormone Metab Res (2018) 50(04):303–7. doi: 10.1055/s-0043-120673
134. Yuan X-X, Zhu H-J, Pan H, Chen S, Liu Z-Y, Li Y, et al. Clinical characteristics of non-alcoholic fatty liver disease in Chinese adult hypopituitary patients. World J Gastroenterol (2019) 25(14):1741. doi: 10.3748/wjg.v25.i14.1741
135. Matsumoto R, Fukuoka H, Iguchi G, Nishizawa H, Bando H, Suda K, et al. Long-term effects of growth hormone replacement therapy on liver function in adult patients with growth hormone deficiency. Growth Hormone IGF Res (2014) 24(5):174–9. doi: 10.1016/j.ghir.2014.07.002
136. Gilliland T, Dufour S, Shulman GI, Petersen KF, Emre SH. Resolution of non-alcoholic steatohepatitis after growth hormone replacement in a pediatric liver transplant patient with panhypopituitarism. Pediatr Transplant (2016) 20(8):1157–63. doi: 10.1111/petr.12819
137. Lonardo A, Loria P, Leonardi F, Ganazzi D, Carulli N. Growth hormone plasma levels in nonalcoholic fatty liver disease. Am J Gastroenterol (2002) 97(4):1071. doi: 10.1111/j.1572-0241.2002.05641.x
138. Rufinatscha K, Ress C, Folie S, Haas S, Salzmann K, Moser P, et al. Metabolic effects of reduced growth hormone action in fatty liver disease. Hepatol Int (2018) 12(5):474–81. doi: 10.1007/s12072-018-9893-7
139. Zhang X, Tian H, Li Y. Pathophysiological mechanism between hypopituitarism and nonalcoholic fatty liver disease. iLiver (2022) 1(1):65–71. doi: 10.1016/j.iliver.2022.02.004
140. Vijayakumar A, Novosyadlyy R, Wu Y, Yakar S, LeRoith D. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Hormone IGF Res (2010) 20(1):1–7. doi: 10.1016/j.ghir.2009.09.002
141. Fan Y, Fang X, Tajima A, Geng X, Ranganathan S, Dong H, et al. Evolution of hepatic steatosis to fibrosis and adenoma formation in liver-specific growth hormone receptor knockout mice. Front Endocrinol (2014) 5:218. doi: 10.3389/fendo.2014.00218
142. Kokoszko A, Dąbrowski J, Lewiński A, Karbownik-Lewińska M. Protective effects of GH and IGF-I against iron-induced lipid peroxidation in vivo. Exp Toxicol Pathol (2008) 60(6):453–8. doi: 10.1016/j.etp.2008.04.012
143. Marino L, Jornayvaz FR. Endocrine causes of nonalcoholic fatty liver disease. World J Gastroenterol (2015) 21(39):11053. doi: 10.3748/wjg.v21.i39.11053
144. Zhou H, Du W, Li Y, Shi C, Hu N, Ma S, et al. Effects of melatonin on fatty liver disease: The role of NR 4A1/DNA-PK cs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res (2018) 64(1):e12450. doi: 10.1111/jpi.12450
145. Smythe G, Lazarus L. Growth hormone regulation by melatonin and serotonin. Nature (1973) 244(5413):230–1. doi: 10.1038/244230a0
146. Fukunaga K, Imachi H, Lyu J, Dong T, Sato S, Ibata T, et al. IGF1 suppresses cholesterol accumulation in the liver of growth hormone-deficient mice via the activation of ABCA1. Am J Physiol Endocrinol Metab (2018) 315(6):E1232–E41. doi: 10.1152/ajpendo.00134.2018
147. Sanz S, Pucilowska J, Liu S, Rodriguez-Ortigosa CM, Lund P, Brenner D, et al. Expression of insulin-like growth factor I by activated hepatic stellate cells reduces fibrogenesis and enhances regeneration after liver injury. Gut (2005) 54(1):134–41. doi: 10.1136/gut.2003.024505
148. Kim S-H, Park M-J. Effects of growth hormone on glucose metabolism and insulin resistance in human. Ann Pediatr Endocrinol Metab (2017) 22(3):145. doi: 10.6065/apem.2017.22.3.145
149. Qiu H, Yang J-K, Chen C. Influence of insulin on growth hormone secretion, level and growth hormone signalling. Sheng Li Xue Bao (2017) 69(5):541–56. doi: 10.13294/j.aps.2017.0062
150. Barclay JL, Nelson CN, Ishikawa M, Murray LA, Kerr LM, McPhee TR, et al. GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinology (2011) 152(1):181–92. doi: 10.1210/en.2010-0537
151. Skat-Rørdam J, Højland Ipsen D, Lykkesfeldt J, Tveden-Nyborg P. A role of peroxisome proliferator-activated receptor γ in non-alcoholic fatty liver disease. Basic Clin Pharmacol Toxicol (2019) 124(5):528–37. doi: 10.1111/bcpt.13190
152. Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr (2014) 34:281. doi: 10.1146/annurev-nutr-071812-161220
153. Cai L, Wang Z, Ji A, Meyer JM, van der Westhuyzen DR. Scavenger receptor CD36 expression contributes to adipose tissue inflammation and cell death in diet-induced obesity. PloS One (2012) 7(5):e36785. doi: 10.1371/journal.pone.0036785
154. Kotzka J, Knebel B, Avci H, Jacob S, Nitzgen U, Jockenhovel F, et al. Phosphorylation of sterol regulatory element-binding protein (SREBP)-1a links growth hormone action to lipid metabolism in hepatocytes. Atherosclerosis (2010) 213(1):156–65. doi: 10.1016/j.atherosclerosis.2010.08.046
155. Smith TM, Gilliland K, Clawson GA, Thiboutot D. IGF-1 induces SREBP-1 expression and lipogenesis in SEB-1 sebocytes via activation of the phosphoinositide 3-kinase/Akt pathway. J Invest Dermatol (2008) 128(5):1286–93. doi: 10.1038/sj.jid.5701155
156. Pan CS, Weiss JJ, Fourman LT, Buckless C, Branch KL, Lee H, et al. Effect of recombinant human growth hormone on liver fat content in young adults with nonalcoholic fatty liver disease. Clin Endocrinol (2021) 94(2):183–92. doi: 10.1111/cen.14344
157. Yim JY, Kim J, Kim D, Ahmed A. Serum testosterone and non-alcoholic fatty liver disease in men and women in the US. Liver Int (2018) 38(11):2051–9. doi: 10.1111/liv.13735
158. Mintziori G, Poulakos P, Tsametis C, Goulis DG. Hypogonadism and non-alcoholic fatty liver disease. Minerva Endocrinol (2016) 42(2):145–50. doi: 10.23736/S0391-1977.16.02570-0
159. Al-Sharefi A, Quinton R. Current national and international guidelines for the management of male hypogonadism: Helping clinicians to navigate variation in diagnostic criteria and treatment recommendations. Endocrinol Metab (2020) 35(3):526. doi: 10.3803/EnM.2020.760
160. Klair JS, Yang JD, Abdelmalek MF, Guy CD, Gill RM, Yates K, et al. A longer duration of estrogen deficiency increases fibrosis risk among postmenopausal women with nonalcoholic fatty liver disease. Hepatology (2016) 64(1):85–91. doi: 10.1002/hep.28514
161. Gild P, Cole AP, Krasnova A, Dickerman BA, von Landenberg N, Sun M, et al. Liver disease in men undergoing androgen deprivation therapy for prostate cancer. J Urol (2018) 200(3):573–81. doi: 10.1016/j.juro.2018.03.135
162. Yang Y-J, Kim KM, An JH, Lee DB, Shim JH, Lim Y-S, et al. Clinical significance of fatty liver disease induced by tamoxifen and toremifene in breast cancer patients. Breast (2016) 28:67–72. doi: 10.1016/j.breast.2016.04.017
163. Elsheikh M, Hodgson H, Wass J, Conway G. Hormone replacement therapy may improve hepatic function in women with turner's syndrome. Clin Endocrinol (2001) 55(2):227–31. doi: 10.1046/j.1365-2265.2001.01321.x
164. Bruno S, Maisonneuve P, Castellana P, Rotmensz N, Rossi S, Maggioni M, et al. Incidence and risk factors for non-alcoholic steatohepatitis: Prospective study of 5408 women enrolled in Italian tamoxifen chemoprevention trial. Bmj (2005) 330(7497):932. doi: 10.1136/bmj.38391.663287.E0
165. McKenzie J, Fisher BM, Jaap AJ, Stanley A, Paterson K, Sattar N. Effects of HRT on liver enzyme levels in women with type 2 diabetes: A randomized placebo-controlled trial. Clin Endocrinol (2006) 65(1):40–4. doi: 10.1111/j.1365-2265.2006.02543.x
166. Koulouri O, Ostberg J, Conway GS. Liver dysfunction in turner syndrome: Prevalence, natural history and effect of exogenous estrogen. Obstet Gynecol Survey (2008) 63(11):706–7. doi: 10.1097/01.ogx.0000334736.56501.ec
167. El-Mansoury M, Berntorp K, Bryman I, Hanson C, Innala E, Karlsson A, et al. Elevated liver enzymes in turner syndrome during a 5-year follow-up study. Clin Endocrinol (2008) 68(3):485–90. doi: 10.1111/j.1365-2265.2007.03166.x
168. Hanew K, Tanaka T, Horikawa R, Hasegawa T, Fujita K, Yokoya S. Women with turner syndrome are at high risk of lifestyle-related disease–from questionnaire surveys by the foundation for growth science in Japan. Endocri J (2016) 63(5):449–56. doi: 10.1507/endocrj.EJ15-0288
169. Charlton M, Angulo P, Chalasani N, Merriman R, Viker K, Charatcharoenwitthaya P, et al. Low circulating levels of dehydroepiandrosterone in histologically advanced nonalcoholic fatty liver disease. Hepatology (2008) 47(2):484–92. doi: 10.1002/hep.22063
170. Haider A, Gooren L, Padungtod P, Saad F. Improvement of the metabolic syndrome and of non-alcoholic liver steatosis upon treatment of hypogonadal elderly men with parenteral testosterone undecanoate. Exp Clin Endocrinol Diabetes (2010) 118(03):167–71. doi: 10.1055/s-0029-1202774
171. Sumida Y, Yonei Y, Kanemasa K, Hara T, Inada Y, Sakai K, et al. Lower circulating levels of dehydroepiandrosterone, independent of insulin resistance, is an important determinant of severity of non-alcoholic steatohepatitis in Japanese patients. Hepatol Res (2010) 40(9):901–10. doi: 10.1111/j.1872-034X.2010.00704.x
172. Koga M, Saito H, Mukai M, Saibara T, Kasayama S. Serum dehydroepiandrosterone sulphate levels in patients with non-alcoholic fatty liver disease. Internal Med (2011) 50(16):1657–61. doi: 10.2169/internalmedicine.50.4682
173. Tokushige K, Hashimoto E, Kodama K, Tobari M, Matsushita N, Kogiso T, et al. Serum metabolomic profile and potential biomarkers for severity of fibrosis in nonalcoholic fatty liver disease. J Gastroenterol (2013) 48(12):1392–400. doi: 10.1007/s00535-013-0766-5
174. Wang W-B, She F, Xie L-F, Yan W-H, Ouyang J-Z, Wang B-A, et al. Evaluation of basal serum adrenocorticotropic hormone and cortisol levels and their relationship with nonalcoholic fatty liver disease in male patients with idiopathic hypogonadotropic hypogonadism. Chin Med J (2016) 129(10):1147–53. doi: 10.4103/0366-6999.181967
175. Jaruvongvanich V, Sanguankeo A, Riangwiwat T, Upala S. Testosterone, sex hormone-binding globulin and nonalcoholic fatty liver disease: a systematic review and meta-analysis. Ann Hepatol (2017) 16(3):382–94. doi: 10.5604/01.3001.0009.8593
176. Polyzos SA, Mousiolis A, Mintziori G, Goulis DG. Nonalcoholic fatty liver disease in males with low testosterone concentrations. Diabetes Metab Syndrome: Clin Res Rev (2020) 14(5):1571–7. doi: 10.1016/j.dsx.2020.07.049
177. Seo NK, Koo HS, Haam Jh, HY K, MJ K, KC P, et al. Prediction of prevalent but not incident non-alcoholic fatty liver disease by levels of serum testosterone. J Gastroenterol Hepatol (2015) 30(7):1211–6. doi: 10.1111/jgh.12935
178. Barbonetti A, Caterina Vassallo MR, Cotugno M, Felzani G, Francavilla S, Francavilla F. Low testosterone and non-alcoholic fatty liver disease: Evidence for their independent association in men with chronic spinal cord injury. J Spinal Cord Med (2016) 39(4):443–9. doi: 10.1179/2045772314Y.0000000288
179. Völzke H, Aumann N, Krebs A, Nauck M, Steveling A, Lerch MM, et al. Hepatic steatosis is associated with low serum testosterone and high serum DHEAS levels in men. Int J Androl (2010) 33(1):45–53. doi: 10.1111/j.1365-2605.2009.00953.x
180. Mueller NT, Liu T, Mitchel EB, Yates KP, Suzuki A, Behling C, et al. Sex hormone relations to histologic severity of pediatric nonalcoholic fatty liver disease. J Clin Endocrinol Metab (2020) 105(11):3496–504. doi: 10.1210/clinem/dgaa574
181. Yang M, Ma F, Guan M. Role of steroid hormones in the pathogenesis of nonalcoholic fatty liver disease. Metabolites (2021) 11(5):320. doi: 10.3390/metabo11050320
182. Kander MC, Cui Y, Liu Z. Gender difference in oxidative stress: a new look at the mechanisms for cardiovascular diseases. J Cell Mol Med (2017) 21(5):1024–32. doi: 10.1111/jcmm.13038
183. Lazo M, Zeb I, Nasir K, Tracy RP, Budoff MJ, Ouyang P, et al. Association between endogenous sex hormones and liver fat in a multiethnic study of atherosclerosis. Clin Gastroenterol Hepatol (2015) 13(9):1686–93.e2. doi: 10.1016/j.cgh.2014.12.033
184. Polyzos SA, Kountouras J, Tsatsoulis A, Zafeiriadou E, Katsiki E, Patsiaoura K, et al. Sex steroids and sex hormone-binding globulin in postmenopausal women with nonalcoholic fatty liver disease. Hormones (2013) 12(3):405–16. doi: 10.1007/BF03401306
185. Carvalho AB, Guerra-Junior G, Baptista MTM, Marques-de-Faria AP, de Lemos-Marini SH, Maciel-Guerra AT. Turner syndrome: a pediatric diagnosis frequently made by non-pediatricians. Jornal Pediatria (2010) 86:121–5. doi: 10.2223/JPED.1985
186. Chukijrungroat N, Khamphaya T, Weerachayaphorn J, Songserm T, Saengsirisuwan V. Hepatic FGF21 mediates sex differences in high-fat high-fructose diet-induced fatty liver. Am J Physiol Endocrinol Metab (2017) 313(2):E203–E12. doi: 10.1152/ajpendo.00076.2017
187. Kupelian V, Page ST, Araujo AB, Travison TG, Bremner WJ, McKinlay JB. Low sex hormone-binding globulin, total testosterone, and symptomatic androgen deficiency are associated with development of the metabolic syndrome in nonobese men. J Clin Endocrinol Metab (2006) 91(3):843–50. doi: 10.1210/jc.2005-1326
188. Suba Z, Kásler M. Interactions of insulin and estrogen in the regulation of cell proliferation and carcinogenesis. Orvosi Hetilap (2012) 153(4):125–36. doi: 10.1556/OH.2012.29287
189. Nemoto Y, Toda K, Ono M, Fujikawa-Adachi K, Saibara T, Onishi S, et al. Altered expression of fatty acid–metabolizing enzymes in aromatase-deficient mice. J Clin Invest (2000) 105(12):1819–25. doi: 10.1172/JCI9575
190. Kamada Y, Kiso S, Yoshida Y, Chatani N, Kizu T, Hamano M, et al. Estrogen deficiency worsens steatohepatitis in mice fed high-fat and high-cholesterol diet. Am J Physiol Gastrointest Liver Physiol (2011) 301(6):G1031–G43. doi: 10.1152/ajpgi.00211.2011
191. Stanworth R, Kapoor D, Channer K, Jones T. Androgen receptor CAG repeat polymorphism is associated with serum testosterone levels, obesity and serum leptin in men with type 2 diabetes. Eur J Endocrinol (2008) 159(6):739. doi: 10.1530/EJE-08-0266
192. Kelly D, Jones T. Testosterone and obesity. Obes Rev (2015) 16(7):581–606. doi: 10.1111/obr.12282
193. Singh R, Artaza JN, Taylor WE, Braga M, Yuan X, Gonzalez-Cadavid NF, et al. Testosterone inhibits adipogenic differentiation in 3T3-L1 cells: nuclear translocation of androgen receptor complex with β-catenin and T-cell factor 4 may bypass canonical wnt signaling to down-regulate adipogenic transcription factors. Endocrinology (2006) 147(1):141–54. doi: 10.1210/en.2004-1649
194. Kim S, Kwon H, Park J-H, Cho B, Kim D, Oh S-W, et al. A low level of serum total testosterone is independently associated with nonalcoholic fatty liver disease. BMC Gastroenterol (2012) 12(1):1–8. doi: 10.1186/1471-230X-12-69
195. Stellato RK, Feldman HA, Hamdy O, Horton ES, McKinlay JB. Testosterone, sex hormone-binding globulin, and the development of type 2 diabetes in middle-aged men: prospective results from the Massachusetts male aging study. Diabetes Care (2000) 23(4):490–4. doi: 10.2337/diacare.23.4.490
196. Peters JM, Zhou Y-C, Ram PA, Lee S, Gonzalez FJ, Waxman DJ. Peroxisome proliferator-activated receptor alpha required for gene induction by dehydroepiandrosterone-3 beta-sulfate. Mol Pharmacol (1996) 50(1):67–74.
197. Sakr HF, Hussein AM, Eid EA, AlKhateeb M. Possible mechanisms underlying fatty liver in a rat model of male hypogonadism: A protective role for testosterone. Steroids (2018) 135:21–30. doi: 10.1016/j.steroids.2018.04.004
198. Harada N, Hanaoka R, Hanada K, Izawa T, Inui H, Yamaji R. Hypogonadism alters cecal and fecal microbiota in male mice. Gut Microbes (2016) 7(6):533–9. doi: 10.1080/19490976.2016.1239680
199. Lonardo A, Nascimbeni F, Targher G, Bernardi M, Bonino F, Bugianesi E, et al. AISF position paper on nonalcoholic fatty liver disease (NAFLD): Updates and future directions. Digest Liver Dis (2017) 49(5):471–83. doi: 10.1016/j.dld.2017.01.147
200. Khera M, Broderick GA, Carson CC III, Dobs AS, Faraday MM, Goldstein I, et al. Adult-onset hypogonadism. Mayo Clin Proc (2016) 91(7):908–26. doi: 10.1016/j.mayocp.2016.04.022
201. Klein KO, Rosenfield RL, Santen RJ, Gawlik AM, Backeljauw PF, Gravholt CH, et al. Estrogen replacement in turner syndrome: literature review and practical considerations. J Clin Endocrinol Metab (2018) 103(5):1790–803. doi: 10.1210/jc.2017-02183
202. Bernard V, Young J, Binart N. Prolactin–a pleiotropic factor in health and disease. Nat Rev Endocrinol (2019) 15(6):356–65. doi: 10.1038/s41574-019-0194-6
203. Shao S, Yao Z, Lu J, Song Y, He Z, Yu C, et al. Ablation of prolactin receptor increases hepatic triglyceride accumulation. Biochem Biophys Res Commun (2018) 498(3):693–9. doi: 10.1016/j.bbrc.2018.03.048
204. Zhang P, Ge Z, Wang H, Feng W, Sun X, Chu X, et al. Prolactin improves hepatic steatosis via CD36 pathway. J Hepatol (2018) 68(6):1247–55. doi: 10.1016/j.jhep.2018.01.035
205. Luque GM, Lopez-Vicchi F, Ornstein AM, Brie B, De Winne C, Fiore E, et al. Chronic hyperprolactinemia evoked by disruption of lactotrope dopamine D2 receptors impacts on liver and adipocyte genes related to glucose and insulin balance. Am J Physiol Endocrinol Metab (2016) 311(6):E974–E88. doi: 10.1152/ajpendo.00200.2016

Keywords: growth hormone deficiency (GHD), nonalcoholic fatty liver disease (NAFLD), polycystic ovarian syndrome (PCOS), hypogonadism, hypopituitarism
Citation: Arefhosseini S, Ebrahimi-Mameghani M, Najafipour F and Tutunchi H (2022) Non-alcoholic fatty liver disease across endocrinopathies: Interaction with sex hormones. Front. Endocrinol. 13:1032361. doi: 10.3389/fendo.2022.1032361
Received: 30 August 2022; Accepted: 24 October 2022;
Published: 07 November 2022.
Edited by:
Guglielmo Beccuti, University of Turinn, ItalyReviewed by:
Alessandro Mantovani, University of Verona, ItalyCopyright © 2022 Arefhosseini, Ebrahimi-Mameghani, Najafipour and Tutunchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helda Tutunchi, SGVsZGEubnV0cml0aW9uQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.