
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 18 November 2022
Sec. Pediatric Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1030398
This article is part of the Research Topic Endocrine Aspects of Noonan Syndrome and Related Syndromes View all 6 articles
 M. A. Siano1
M. A. Siano1 R. Pivonello2
R. Pivonello2 M. Salerno3
M. Salerno3 M. Falco1
M. Falco1 C. Mauro1D. De Brasi4
C. Mauro1D. De Brasi4 A. Klain4
A. Klain4 S. Sestito5
S. Sestito5 A. De Luca6V. Pinna6
A. De Luca6V. Pinna6 C. Simeoli2
C. Simeoli2 D. Concolino5Ciro Gabriele Mainolfi7T. Mannarino7P. Strisciuglio3
D. Concolino5Ciro Gabriele Mainolfi7T. Mannarino7P. Strisciuglio3 M. Tartaglia8
M. Tartaglia8 D. Melis1,3*
D. Melis1,3*Background and Objectives: Endocrine complications have been described in patients affected by RASopathies but no systematic assessment has been reported. In this study, we investigate the prevalence of endocrine disorders in a consecutive unselected cohort of patients with RASopathies.
Study Design: 72 patients with a genetically confirmed RASopathy (Noonan syndrome [NS], N=53; 29 LEOPARD syndrome [LS], N=2; cardiofaciocutaneous syndrome [CFCS], N=14; subjects showing co-occurring pathogenic variants in PTPN11 and NF1, N=3) and an age- and sex-matched healthy controls were included in the study. Endocrine system involvement was investigated by assessing the thyroid function, pubertal development, auxological parameters, adrenal function and bone metabolism.
Results: Short stature was detected in 40% and 64% of the NS and CFCS subcohorts, respectively. Patients showed lower Z-scores at DXA than controls (p<0.05) when considering the entire case load and both NS and CFCS groups. Vitamin D and Calcitonin levels were significantly lower (p< 0.01), Parathormone levels significantly higher (p<0.05) in patients compared to the control group (p<0.05). Patients with lower BMD showed reduced physical activity and joint pain. Finally, anti-TPO antibody levels were significantly higher in patients than in controls when considering the entire case load and both NS and CFCS groups.
Conclusions: The collected data demonstrate a high prevalence of thyroid autoimmunity, confirming an increased risk to develop autoimmune disorders both in NS and CFCS. Reduced BMD, probably associated to reduced physical activity and inflammatory cytokines, also occurs. These findings are expected to have implications for the follow-up and prevention of osteopenia/osteoporosis in both NS and CFCS.
RASopathies are a clinically defined group of genetic syndromes affecting development that are caused by germline mutations in genes encoding signal transducers or regulators of the RAS/mitogen activated protein kinase (MAPK) pathway (1, 2). This family of disorders include Noonan syndrome (NS), LEOPARD syndrome (also known as Noonan syndrome with multiple lentigines, LS), cardiofaciocutaneous syndrome (CFCS), Costello syndrome (CS), Mazzanti syndrome (also known as Noonan syndrome-like disorder with loose anagen hair, NS/LAH), neurofibromatosis type I (NFNS), Legius syndrome and an increasing number of other clinically related conditions (3–5). Most of these disorders share craniofacial dysmorphisms, a wide spectrum of congenital cardiac defects, hypertrophic cardiomyopathy, postnatal reduced growth, cutaneous and musculoskeletal abnormalities, and variable neurocognitive impairment and predisposition to certain cancers (3, 5). While short stature is a well-recognized feature in these patients, endocrine system involvement has rarely been systematically assessed. Possible endocrine complications of NS, such as thyroid disease, gonadal functional impairment and abnormal bone metabolism, have recently been reported (6, 7).
In affected male and female patients prenatal growth is generally normal as well as length at birth. Growth failure often appears during the first year of life (8–10). Mean height is usually at lower limit for age during childhood, and then it usually further declines because of delayed puberty and an attenuated pubertal growth spurt. Bone maturity is usually delayed. Genotype–phenotype correlations have been reported indicating that NS patients with pathogenic PTPN11 and RAF1 variants show more severely impaired growth than those with other genotypes (e.g., pathogenic variants in SOS1) (11). The different molecular role of the individual signal transducers and modulators implicated in these disorders on intracellular signaling possibly underlies this and other genotype-phenotype correlations (12–14).
In NS patients, normal-to-elevated serum GH levels associated with low serum IGF1 levels are found, suggesting occurrence of GH insensitivity at the postreceptor level (15, 16). In children with NS/LAH, short stature is commonly associated to proven GH deficiency (17, 18). In vivo and in vitro studies have shown that upregulated RAS/MAPK signaling is associated with impaired IGF1 production. Moreover, pharmacological inhibition of this pathway also improved growth in other NS mouse models (19–21). However, a direct effect of RAS/MAPK functional upregulation has also been demonstrated at the level of the growth plate and longitudinal bone growth. Indeed, recent studies have revealed an important physiological role for SHP2 in growth plate development (22), and hyperactive SHP2 and BRAF function has been shown to impair chondrocyte differentiation during endochondral bone growth (23–25).
In both sexes, the onset of puberty is usually delayed in NS patients and is associated with a decreased pubertal growth spurt (26–28). Fertility does not seem to be affected in women with NS, while gonadal dysfunction with defective spermatogenesis has been reported in males (29–31), which has been related to cryptorchidism, occurring in up to 80% of NS males (27, 32–34). However, defect in spermatogenesis have been described in male patients with and without cryptorchidism (29, 30, 35), and recent studies documented occurrence of Sertoli cell-specific primary testicular insufficiency (36). Consistently, a critical role of SHP2 in the maintenance of Sertoli cell function has been demonstrated (37). Thyroid dysfunction has been described in many patients with RASopathies, including overt and subliclinal hypothyroidism. Moreover, several studies reported occurrence of thyroid autoantibodies in patients with NS (27, 38–42).
Endocrine complications of NS include possible pathology in bone metabolism. Values of bone mineral density assessed by different techniques (dual-energy radiograph absorptiometry or phalangeal quantitative ultrasound) are decreased in NS, NF1 and CS (43, 44–51), with increased risk of osteoporosis.
Recent studies confirmed that 25(OH)vitD and BMD parameters are reduced in CS, and vitamin D supplementation is not sufficient to restore proper BMD values (52). The impairment of bone metabolism could be the result of up-regulation osteoclast development and function associated with a decreased activity of osteoblasts (22, 53). Indeed, increased levels of bone resorption have been reported in NS patients (54).
The aim of the present study was to perform a systematic study of the involvement of the endocrine system in patients with RASopathies, to establish the timing of follow-up and prevention strategies.
The current study is a prospective case-control study evaluating the complete baseline endocrine profile, as expression of the hypothalamus-pituitary function, in patients with RASopathies and control subjects. Clinical features suggestive for involvement of any specific endocrine system were recorded. In particular the presence of short stature, delayed growth pattern, delayed or precocious puberty, poor or excessive weight gain, recurrent infections, fatigue, bone and/or joint pain, muscle aches and/or weakness, recurrent infections, and increased tendency to fractures were recorded.
Physical activity and sun exposure were also recorded during the enrolment visit in order to study their contribution on bone impairment.
The clinical and biochemical data were correlated with molecular data including involved gene and specific mutation.
Seventy-two patients affected by RASopathies were enrolled in the study. Subjects were followed at the Pediatric Genetic Section of the University Federico II of Naples (N = 50), Department of Medicine, Surgery and Dentistry “Scuola Medica Salernitana”, Salerno (N = 8), Department of Clinical and Experimental Medicine of the University Magna Graecia of Catanzaro (N = 7) and Santobono Hospital in Naples (N = 7).
The protocol was discussed with each patient (or legal tutor) and signed informed consent was obtained. The whole cohort (44 males, 28 female) included the following RASopathies: NS (N = 53), CFCS (N = 14), LS (N = 2). Three subjects showed co-occurring pathogenic variants in PTPN11 and NF1. The mean age at enrolment was 8.7 years, (range: 2 to 26 years). A control group was also enrolled including 50 apparently healthy subjects (30 males, 20 females) with mean age 9.0 years (range: 0-26). The enrolment was carried out according to the following inclusion criteria: (i) molecularly confirmed clinical diagnosis of RASopathy, (ii) informed consent expression to participate to the study.
Within the NS subcohort, patients were heterozygous for pathogenic variants in PTPN11 (36/53, 67.9%), SOS1 (9/53, 16.9%), RIT1 (4/53, 7.5%), RAF1 (2/53, 3.7%), and LZTR1 (2/53, 3.7%). Within the CFCS subcohort, patients were heterozygous for pathogenic variants in BRAF (11/14, 78.57%), MAP2K1 (1/14, 7.14%), MAP2K2 (1/14, 7.14%), and KRAS (1/14, 7.14%) (Table 1).
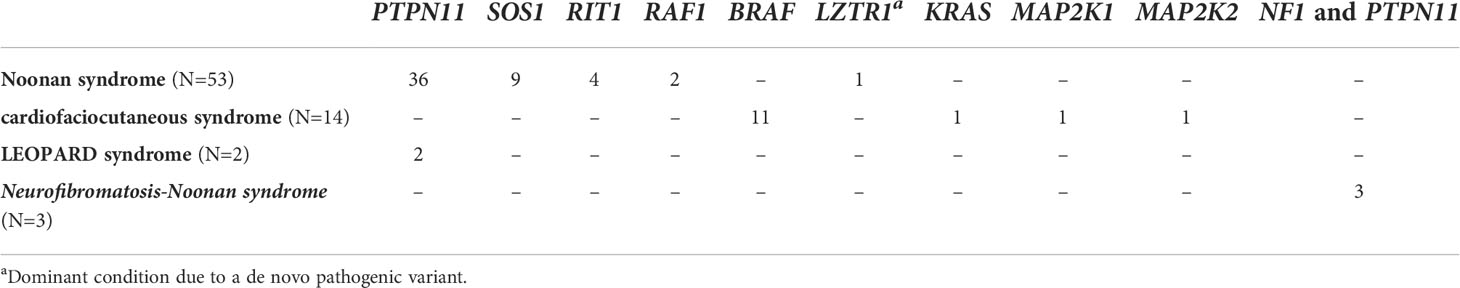
Table 1 The study cohort. The different RASopathies are subdivided taking into account the involved disease gene(s).
The diagnosis of NS vs CFCS was performed on the basis of both clinical and molecular data. All the patients carrying BRAF mutation received a clinical diagnosis of CFCS.
All patients underwent clinical assessment, including auxological parameters, and patients/families were also contacted to collect available clinical information. Indeed clinical data were available from medical records over the past 20 years. In the medical record a diagnosis of lymphoblastic leukemia was performed in a patient when she was 7; at the time of the study entry the patient was 16. Among NS patients, 8 showed cardiomyopathy. In our patient group, renal abnormalities were reported in the medical record, including double kidney district (1 patient), kidney cyst (1 patient) and pyelectasis (2 patients).
We evaluated height and weight, with their percentile and SD scores, BMI, with its percentile and SD scores, growth velocity, and pubertal stage. Short stature was defined when height was at least 2 SD less than the mean for chronologic age. In adults, a BMI between 25 and 30 was considered overweight, whereas, a BMI of 30 was considered obesity. In children, the 85th and 95th percentiles of BMI for age and sex were considered the thresholds for overweight and obesity, respectively. Delayed puberty was defined differently in accordance with sex. In girls, delayed puberty was defined as lack of breast development by 13 years, lack of pubic hair by 14 years, lack of menarche by 16 years, or a the documentation of a period >5 years between thelarche and menarche. In boys, delayed puberty was defined as lack of testicular enlargement by 14 years, lack of pubic hair by 15 years, or the documentation of a period >5 years to complete genital enlargement. Skeletal maturation was evaluated by measuring skeletal age through the examination of a radiograph of the left hand and wrist, according to the method of Greulich and Pyle. Bone mineralization was investigated by dual-energy X-ray absorptiometry (DXA). The presence of bone/joint pain, muscle aches/muscle weakness, recurrent fatigue and increased tendency to fractures was also recorded. Clinical features suggestive for periodontitis and recurrent infections (especially respiratory tract) as well as mood alterations were also investigated. In order to assess the potential contribution of “environmental” factors, on both Calcium metabolism and BMD, physical activity and sun exposure were recorded during the enrolment visit.
Physical activity was evaluated by administering the International Physical Activity Questionnaire (IPAQ) (55). For each patient the time spent experiencing different kind of activities during the previous week was calculated: vigorous-intensity activity (hard physical effort and the patient breathe harder than normal), moderate-intensity activity (moderate physical effort, walking) and the time spent sitting.
Sun exposure was considered low with at least two of the following criteria: no arm and skin exposure during summer months; no sunbathing or holiday in sunny places; no working outdoors. Otherwise, it was deemed sufficient.
The complete baseline endocrine profile, as expression of the hypothalamus-pituitary function was evaluated in patients with RASopathy and control subjects.
The somatotropic axis was evaluated by analyzing basal serum growth hormone (GH) and insulin-like growth factor 1 (IGF1) via chemiluminescence immune assay (CLIA, Diasorin, Liaison XL). In patients with low IGF1, GH stimulation tests (arginine and, if necessary, clonidine) were performed.
The thyrotropic axis function was assessed by analyzing serum thyroid-stimulating hormone (TSH), free triiodothyronine (fT3), free thyroxine (fT4), anti-Tg and anti-TPO (Siemens, Advia Centaur Immunoassay Systems). Anti-r-TSH autoantibodies were assayed by ELISA (TSH Receptor Autoantibody Human ELISA 2. Generation, Biovendor).
The adrenocorticotropic axis function was investigated by analyzing plasma adrenocorticotropic hormone (ACTH), serum cortisol, androstenedione, dehydroepiandrosterone sulphate (DHEA-S), all measured by using automated immunoassays (Siemens, Immulite 2000 XPi). The gonadotropic axis function was investigated by analyzing serum follicle-stimulating hormone (FSH), luteinizing hormone (LH), 17β-estradiol, testosterone levels, by automated immunoassays (Siemens, Immulite 2000 XPi). 17hydroxyprogesteron (17OHP) blood concentration was determined by CLIA (Pantec, IDS ISYS).
Bone metabolism was studied evaluating parathyroid hormone (PTH), calcitonin (56), 25OH vitamin D levels, using immunoassay with chemiluminescence immune assays (Diasorin, Liaison XL). Considering the vitamin D levels, deficiency was defined by 25OHD levels below or equal to 20 ng/ml, while insufficiency was considered by 25OHD levels between 21 and 29 mg/ml (57). Drug intake and fracture history were included in the medical history. Bone mineralization was investigated by dual-energy X-ray absorptiometry (DXA). Osteopenia was defined as the presence of BMD between -1.5 and -2 SD while Osteoporosis was defined as BMD below -2SD.
In a group of 10 patients and 10 controls available for further blood sampling, screening of a panel of inflammatory molecules was performed including PDGF, IL-1b, IL-1ra, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12(p70), IL-13, IL-15, IL-17, Eotaxin, FGF basic, G-CSF, GM-CSF, IFN-g, IP-10, MCP-1(MCAF), MIP-1a, MIP-1b, RANTES, TNF-a, VEGF. The results of these investigations have been previously reported (42).
Each numerical variable was expressed as mean +/- SD. Statistical analysis was performed using SPSS package. Differences in studied parameters between patients and controls were analysed using the t test for unpaired data corrected for Fisher exact test. To investigate the presence of an association between severity of phenotype and either DNA mutation or specific gene involved, χ2 test was performed.
Differences with P <0.05 were considered to be significant in all instances.
Within the NS and CFCS subcohorts, short stature was detected in 21/53 (39.6%) and 9/14 (64.3%) patients, respectively. No patient showed obesity or poor weight gain. Pubertal delay was reported in 1/35 NS males and 1/18 NS females. No patient had history of fractures. The presence of bone/joint pain was recorded in 8 NS patients and 1 CFC patient. Recurrent fatigue was recorded in 6 NS patients. No patient showed clinical features suggestive for periodontitis, recurrent infections as well as mood alterations were investigated.
Physical activity was significantly reduced in patients than in controls. Particularly both vigorous-intensity activity (0.24 ± 0.5 hours vs 10.25 ± 3, p<0.001), moderate-intensity activity (2.81 ± 0.2 vs 6.98 ± 2.5, p=0.012) and walking (2.96 ± 3.6 vs 7.2 ± 6.9, p=0.02) appeared significantly reduced. The time spent sitting every day during the previous week was higher in patients than in controls (62.6 ± 46.2 vs 32.8 ± 7, p=0.007).
Sun exposure was low in 21/46 (45.6%) patients and in 10/50 (20%) control subjects.
Two patients, one CFCS and one NS patients showed GH deficiency (GH <8 ng/ml at both arginine and clonidine stimulation test). Both patients had short stature (<-2DS), pathological growth rate and delayed bone age. They have been treated with GH with improvement of growth pattern. No patient who underwent GH treatment showed sign of heart damage or cardiomyopathy at echocardiography. In the remaining patients both baseline GH and IGF1 were in the normal range. No correlation was detected between molecular data and GH deficiency or short stature.
Thyroid function was normal in all tested patients and we did not find any clinical signs and symptoms of hypo-hyperthyroidism. No significant difference was recorded between the CFCS and NS groups and controls. Compared to the control population, anti-TPO autoantibody levels were significantly higher both in the whole cohort and in the NS and CFCS subgroup (Tables 2A–C).
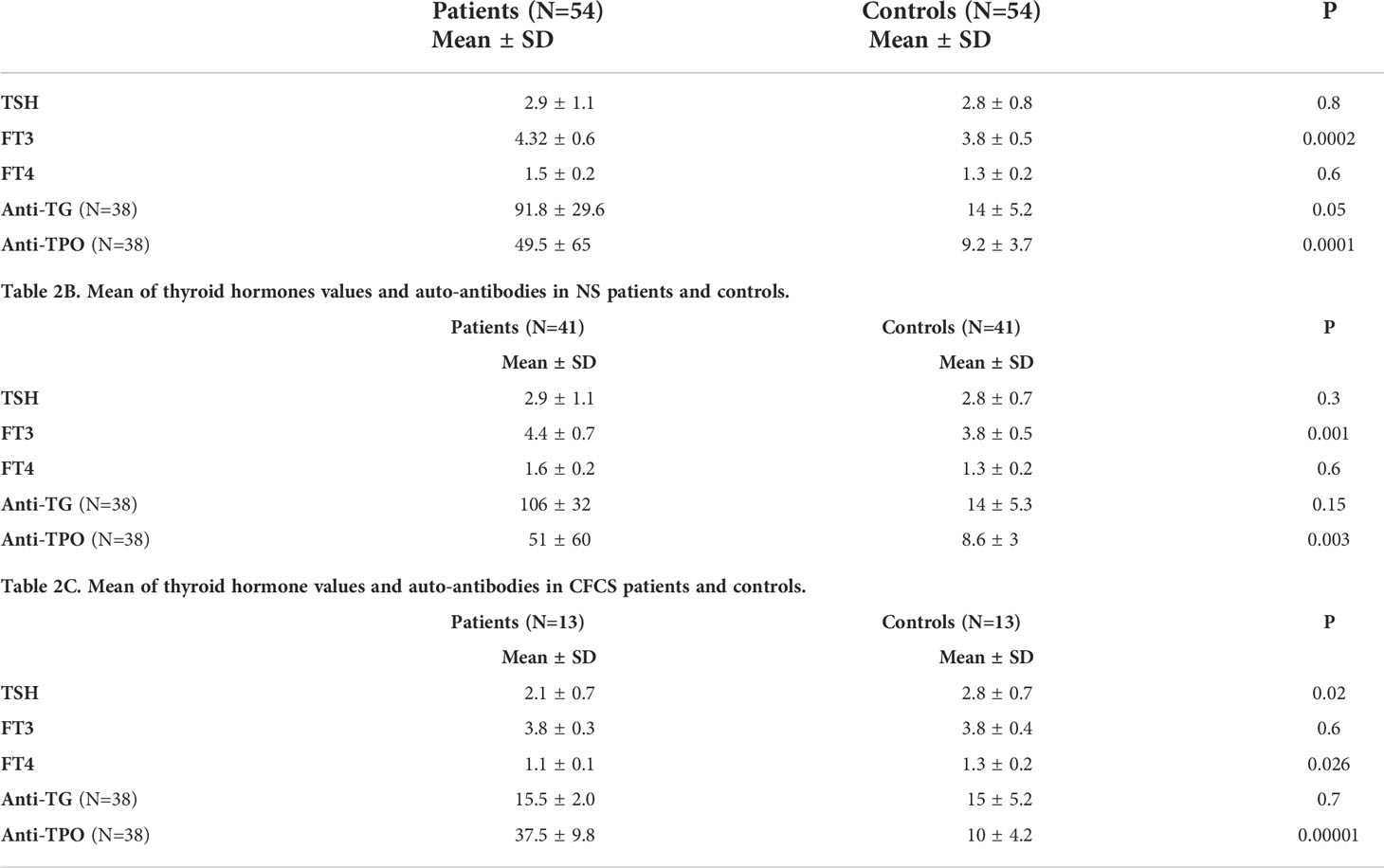
Table 2A Mean of thyroid hormones values and auto-antibodies in all patients and controls.
Cryptorchidism was found in 21/44 males (47.7%). Others genital malformations were rare. The age of the enrolled patients did not allow to systematically assess gonadal function. No significant differences were observed between patients and controls, when the gonadotropic axis function was tested (Tables 3A–C).
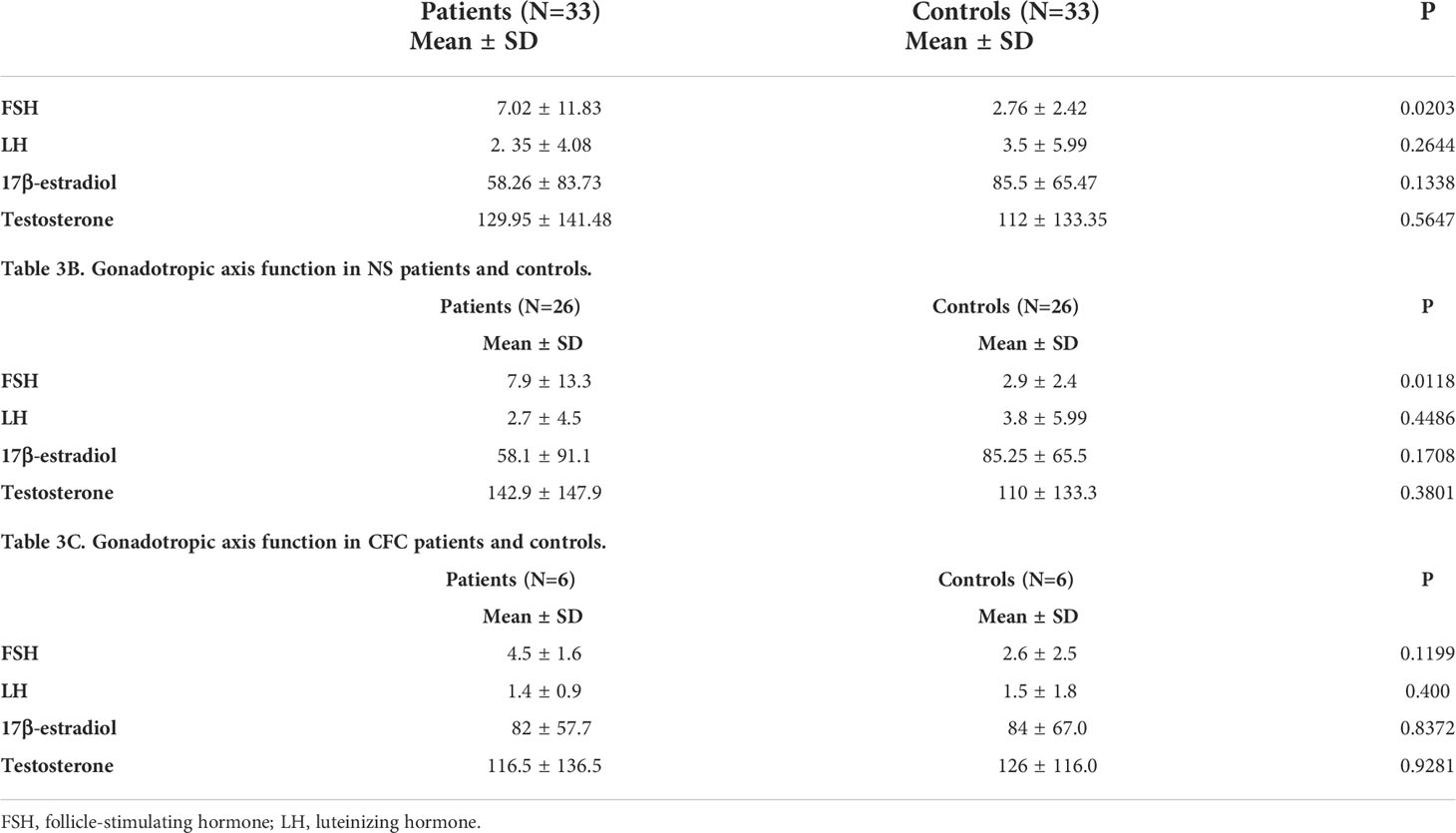
Table 3A Gonadotropic axis function in all patients and controls.
The adrenocorticotropic axis function was normal. No significant differences were observed between patients and controls, when we compared plasma ACTH, serum cortisol, androstenedione, 17OHP and DHEA-S.
None of the enrolled subjects were taking corticosteroids, or any other medication affecting bone metabolism. None of our patients had history of fractures. The presence of bone/joint pain was recorded in 8 NS patients and 1 CFCS patient. In these NS patients BMD was significantly reduced when compared to patients with no pain (-2.49 ± 0.8 vs -1.1 ± 0.4; p=0.03). Recurrent fatigue was recorded in 6 NS patients. No patient showed clinical features suggestive for periodontitis, recurrent infections as well as mood alterations were investigated.
Biochemical evaluation for bone turnover was performed. Biochemical parameters revealed calcium and phosphorous levels in the normal range in all tested patients. Compared to the age-and sex matched control population, however, vitamin D serum levels were significantly lower in patients and in the NS and CFCS subgroups (Tables 4A–C).

Table 4A Calcium and phosphorus metabolism in all patients and controls.
PTH serum levels were significantly higher in patients than in controls while calcitonin serum levels were significantly lower. Patients showed lower Z-scores at DXA than controls (Table 4A). BMD z-scores correlated with physical activity. The time spent sitting every day inversely correlated with BMD z-score levels (r= -0.638, p=0.008). No correlation between BMD data and short stature and/or sun exposure was recorded. It is noteworthy that 4 patients with low BMD showed high cytokines levels (data not shown). No correlation was detected between molecular data and BMD was recorded.
Endocrine system involvement, such as thyroid disease, gonads impairment and abnormal bone metabolism, have been reported in NS (6). The present study provide a general picture of the endocrine system involvement in patients with RASopathies. In particular, this report offers an endocrine system profiling of an unselected CFCS cohort.
As expected, the collected data underlined a high prevalence of short stature both in NS and in CFCS group (58, 59). The reduced growth in both disorders likely results from the dysregulation of the RAS-MAPK signaling pathways at different levels, being related to GH insensitivity at the postreceptor level (15, 16), and to a direct effect at the growth plate (22, 24). While GH deficiency was demonstrated in 1 NS patient and 1 CFCS patient, none of our patients showed a GH insensitivity. No correlation between genotype and short stature was observed.
Thyroid dysfunction has been described in patients with RASopathies, including overt and subclinal hypothyroidism. Moreover, several studies reported occurrence of thyroid autoantibodies in patients with NS (27, 38–42). Our findings outline the high prevalence of thyroid autoimmunity in both NS and CFCS. These data confirm our previous report indicating a high risk to develop autoimmune disease in these groups of patients. Of note, the presence of thyroid autoantibodies was associated to normal thyroid function, in line with previously reported studies (27, 34, 38–41). We hypothesize that these antibodies can precede the clinical symptoms of the disease by years, and could be used for diagnostic and prognostic purposes (42).
Although osteopenia/osteoporosis have anecdotally been reported in NS, pathophysiology and correlations with underlying genetic defects are poorly understood. Reduced BMD in presence of normal bone mineralization marker has previously been reported in NS (44). Low 25-OH vitamin D concentration has been documented in NF1 patients (60, 61) and circumstantially in other RASopathies. However, no clear correlation between vitamin D levels and BMD was reported (45). In the current study, analysis of bone metabolism documented a lower BMD in patients (including both NS and CFCS subgroups) compared to controls. Vitamin D serum levels were significantly reduced, PTH levels significantly increased and calcitonin serum levels reduced in patients than in controls when the entire case load was considered. The observation of decreased levels of calcitonin, marker of osteoblastic activity and bone neo-formation in these patients might be considered as a compensatory mechanism against the increased resorption, which is in turn pointed out by high levels of PTH.
Both NS and CFCS patients also showed lower vitamin D serum levels than controls. However no correlation between vitamin D levels and BMD was demonstrated, suggesting that other factors might cause bone damage in patients affected by RASopathy. The impairment of bone metabolism could be the result of up-regulation of osteoclast development and function (22, 53) with a decreased activity of osteoblasts by the action of SHP2. Increased levels of a bone resorption marker (degraded cross links of mature collagen excreted in the urine) have been reported in NS patients (54), confirming an important role of osteoclasts. Several studies underline that the Ras-MAPK signal transduction pathway is important in bone homeostasis (54, 62, 63), suggesting that increased signaling through this cascade impacts bone remodeling (64, 65). Our results support the hypothesis that metabolic bone disease due to variation in osteoclast vs osteoblast activity as a consequence of a dysregulation of the RAS MAPK signaling pathway is at the basis of impaired bone metabolism (43). However, additional factors that could contribute to the increased bone resorption markers (e.g., inactivity, hypotonia, and poor motor function) should be considered. Consistently, recent study confirms that a combination of different factors, including reduced sun exposure, possibly associated with reduced serum vitamin D levels, and poor physical activity, concur to the impaired bone status in NF1 patients (61).
In this respect it is noteworthy that we demonstrated a correlation between physical activity and BMD data. We also underline that patients with lower BMD show high levels of inflammatory cytokines and high prevalence of joint pain. On the basis of the current data we suggest to investigate bone metabolism and BMD in patients complaining joint pain.
Endocrine imbalance may also be involved in bone abnormalities. The anabolic effect of IGF1 on the bone is well known, regulating bone growth and enhancing osteoblast proliferation. Recently, it has been shown that IGF1 promotes osteoblastic activity activating mTOR pathway. IGF1 affects bone status also acting in a paracrine way, in response to mechanical load, such as during physical activity. It might be hypothesized that low IGF1 levels due to GH resistance might contribute to reduced BMD in both NS and CFCS patients. Furthermore, reduced physical activity could also contribute to the reduction of the DXA z-score in patients as the expression of IGF-1 is stimulated in response to mechanical load.
On the basis of the collected data, we suggest that that the combination of several factor including the dysregulation of the RAS-MAPK pathway, the reduced physical activity, the presence of inflammatory cytokines and the impaired IGF1 activity might contribute to the impaired bone metabolism in patients with RASopathy.
We recommended routine monitoring of bone homeostasis to prevent bone deterioration and possible fractures in both NS and CFCS patients. Regular physical activity should also be encouraged in patients affected by RASopathy when allowed by heart involvement.
The main limitation of this study is the relatively small number of enrolled patients. Further studies are necessary to investigate the effects of other factors influencing BMD in patients. Longitudinal studies are needed to evaluate the progression of endocrine involvement in patients with NS and CFCS.
Endocrine complications have been described in patients with RASopathies, though an evaluation of the endocrine system status has not systematically been performed. The collected data demonstrate a high prevalence of thyroid autoimmunity, confirming an increased risk to develop autoimmune disorders both in NS and CFCS. Reduced BMD, probably associated to reduced physical activity and inflammatory cytokines, also occurs. These findings are expected to have implications for the follow-up and prevention of osteopenia/osteoporosis in both NS and CFCS. In order to recommend systematic evaluation of all endocrinological aspects in patients with asymptomatic RASopathy, continuous monitoring will be required and other studies are necessary to confirm our results.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Comitato Etico Campania Sud. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Conceptualization, DM, MAS, RP. Methodology, AL, VP, CM, TM. software MAS, AL, VP, CGM, TM. Validation, DM, CS, AL, VP, CGM, TM. Formal analysis MAS, CS, MF, AL, VP, CGM, TM. Investigation CS, CM, AK, SS. Data curation, DM, RP, MS, MF, CM, DB, AK, SS, DC. Writing—original draft preparation, MAS, MF, CM. writing—review and editing. DM, TM, RP, DC, PS. Visualization, DM. supervision, DM, MT, PS, RP. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol. (2010) 1(1):2–26. doi: 10.1159/000276766
2. Tidyman WE, Rauen KA. Expansion of the RASopathies. Curr Genet Med Rep (2016) 4(3):57–64. doi: 10.1007/s40142-016-0100-7
3. Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab (2011) 25(1):161–79. doi: 10.1016/j.beem.2010.09.002
4. Motta M, Pannone L, Pantaleoni F, Bocchinfuso G, Radio FC, Cecchetti S, et al. Enhanced MAPK1 function causes a neurodevelopmental disorder within the RASopathy clinical spectrum. Am J Hum Genet (2020) 107(3):499–513. doi: 10.1016/j.ajhg.2020.06.018
5. Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet (2013) 14:355–69. doi: 10.1146/annurev-genom-091212-153523
6. Venugopal V, Romero CJ. Endocrine complications of noonan syndrome beyond short stature. PediatrEndocrinol. Rev (2019) 16(Suppl 2):465–70. doi: 10.17458/per.vol16.2019.vr.endocrinecomplicationsnoonan
7. Stevenson DA, Schwarz EL, Carey JC, Viskochil DH, Hanson H, Bauer S, et al. Bone resorption in syndromes of the Ras/MAPK pathway. Clin Genet (2011) 80(6):566–73. doi: 10.1111/j.1399-0004.2010.01619.x
8. Da Silva FM, Jorge AA, Malaquias A, da Costa Pereira A, Yamamoto GL, Kim CA, et al. Nutritional aspects of noonan syndrome and noonan-related disorders. Am J Med Genet A (2016) 170:1525–31. doi: 10.1002/ajmg.a.37639
9. Leoni C, Onesimo R, Giorgio V, Diamanti A, Giorgio D, Martini L, et al. Understanding growth failure in Costello syndrome: increased resting energy expenditure. J Pediatr (2016) 170:322–4. doi: 10.1016/j.jpeds.2015.11.076
10. Yart A, Edouard T. Noonan syndrome: an update on growth and development. CurrOpinEndocrinol. Diabetes Obes (2018) 25(1):67–73. doi: 10.1097/MED.0000000000000380
11. Tartaglia M, Martinelli S, Stella L, et al. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet (2006) 78(2):279–90. doi: 10.1086/499925
12. Leoni C, Giorgio V, Onesimo R, Kuczynska E, Zampino G. Impact of Costello syndrome on growth patterns. Am J Med Genet A. (2020) 182(11):2797–9. doi: 10.1002/ajmg.a.61812
13. Cessans C, Ehlinger V, Arnaud C, Yart A, Capri Y, Barat P, et al. Growth patterns of patients with noonan syndrome: correlation with age and genotype. Eur J Endocrinol (2016) 174:641–50. doi: 10.1530/EJE-15-0922
14. Malaquias AC, Brasil AS, Pereira AC, Arnhold IJ, Mendonca BB, Bertola DR, et al. Growth standards of patients with noonan and noonan-like syndromes with mutations in the RAS/MAPK pathway. Am J Med Genet A (2012) 158A:2700–6. doi: 10.1002/ajmg.a.35519
15. Binder G, Neuer K, Ranke MB, Wittekindt NE. PTPN11 mutations are associated with mild growth hormone resistance in individuals with noonan syndrome. J ClinEndocrinolMetab. (2005) 90:5377–81. doi: 10.1210/jc.2005-0995
16. Limal JM, Parfait B, Cabrol S, Bonnet D, Leheup B, Lyonnet S, et al. Noonan syndrome: relationships between genotype, growth, and growth factors. J Clin Endocrinol Metab (2006) 91(1):300–6. doi: 10.1210/jc.2005-0983
17. Cordeddu V, Di Schiavi E, Pennacchio LA, Ma'ayan A, Sarkozy A, Fodale V, et al. Mutation of SHOC2 promotes aberrant protein n-myristoylation and causes noonan-like syndrome with loose anagen hair. NatGenet. (2009) 41(9):1022–6. doi: 10.1038/ng.425
18. Mazzanti L, Tamburrino F, Scarano E, Perri A, Vestrucci B, Guidetti M, et al. GH therapy and first final height data in noonan-like syndrome with loose anagen hair (Mazzanti syndrome). Am J Med Genet A (2013) 161A(11):2756–61. doi: 10.1002/ajmg.a.36255
19. De Rocca Serra-Ne´de´lec A, Edouard T, Treguer K, Tajan M, Araki T, Dance M, et al. Noonan syndromecausing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc Natl Acad Sci USA (2012) 109:4257–62. doi: 10.1073/pnas.1119803109
20. Nakamura T, Gulick J, Pratt R, Robbins J. Noonan syndrome is associated with enhanced pERK activity, the repression of which can prevent craniofacial malformations. ProcNatlAcadSci. U.S.A. (2009) 106:15436–41. doi: 10.1073/pnas.0903302106
21. Wu X, Simpson J, Hong JH, Kim KH, Thavarajah NK, Backx PH, et al. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of noonan syndrome associated with the Raf1(L613V) mutation. J Clin Invest. (2011) 121(3):1009–25. doi: 10.1172/JCI44929
22. Bauler TJ, Kamiya N, Lapinski PE, Langewisch E, Mishina Y, Wilkinson JE, et al. Development of severe skeletal defects in induced SHP-2-deficient adult mice: a model of skeletal malformation in humans with SHP-2 mutations. Dis Model Mech (2011) 4(2):228–39. doi: 10.1242/dmm.006130
23. Tajan M, Pernin-Grandjean J, Beton N, Gennero I, Capilla F, Neel BG, et al. Noonan syndrome-causing SHP2 mutants impair ERK-dependent chondrocyte differentiation during endochondral bone growth. Hum Mol Genet (2018) 27(13):2276–89. doi: 10.1093/hmg/ddy133
24. Edouard T, Combier JP, Nédélec A, Bel-Vialar S, Métrich M, Conte-Auriol F, et al. Functional effects of PTPN11 (SHP2) mutations causing LEOPARD syndrome on epidermal growth factor-induced phosphoinositide 3-kinase/AKT/glycogen synthase kinase 3beta signaling. Mol Cell Biol (2010) 30(10):2498–507. doi: 10.1128/MCB.00646-09
25. Inoue SI, Morozumi N, Yoshikiyo K, Maeda H, Aoki Y. C-type natriuretic peptide improves growth retardation in a mouse model of cardio-facio-cutaneous syndrome. Hum Mol Genet (2019) 28(1):74–83. doi: 10.1093/hmg/ddy333
26. Ranke MB, Heidemann P, Knupfer C, Enders H, Schmaltz AA, Bierich JR. Noonan syndrome: growth andclinical manifestations in 144 cases. Eur J Pediatr (1988) 148:220–7. doi: 10.1007/BF00441408
27. Sharland M, Burch M, McKenna WM, Paton MA. A clinical study of noonan syndrome. Arch Dis Child (1992) 67:178–83. doi: 10.1136/adc.67.2.178
28. Shaw AC, Kalidas K, Crosby AH, Jeffery S, Patton MA. The natural history of noonan syndrome: a long-term follow-up study. Arch Dis Child. (2007) 92(2):128–32. doi: 10.1136/adc.2006.104547
29. Ankarberg-Lindgren C, Westphal O, Dahlgren J. Testicular size development and reproductive hormones in boys and adult males with noonan syndrome: a longitudinal study. Eur J Endocrinol (2011) 165(1):137–44. doi: 10.1530/EJE-11-0092
30. Marcus KA, Sweep CG, van der Burgt I, Noordam C. Impaired sertoli cell function in males diagnosed with noonan syndrome. J Pediatr Endocrinol Metab. (2008) 21:1079–84. doi: 10.1515/jpem.2008.21.11.1079
31. Theintz G, Savage MO. Growth and pubertal development in five boys with noonan's syndrome. Arch Dis Child. (1982) 57(1):13–7.
32. Okuyama A, Nishimoto N, Yoshioka T, Namiki M, Itatani H, Takaha M, et al. Gonadal findings in cryptorchid boys with noonan’s phenotype. EurUrol (1981) 7(5):274–7. doi: 10.1159/000473239
33. Sasagawa I, Nakada T, Kubota Y, Sawamura T, Tateno T, Ishigooka M. Gonadal function and testicular histology in noonan’s syndrome with bilateral cryptorchidism. Arch Androl. (1994) 32:135–440. doi: 10.3109/01485019408987778
34. Elsawi MM, Pryor JP, Klufio G, Barnes C, Patton MA. Genital tract function in men with noonan syndrome. J Med Genet (1994) 31:468–70. doi: 10.1136/jmg.31.6.468
35. Trsinar B, Muravec UR. Fertility potential after unilateral and bilateral orchidopexy for cryptorchidism. World J Urol. (2009) 27(4):513–9. doi: 10.1007/s00345-009-0406-0
36. Moniez S, Pienkowski C, Lepage B, Hamdi S, Daudin M, Oliver I, et al. Noonan syndrome males display sertoli cell-specific primary testicular insufficiency. Eur J Endocrinol (2018) 179(6):409–18. doi: 10.1530/EJE-18-0582
37. Hu X, Tang Z, Li Y, Liu W, Zhang S, Wang B, et al. Deletion of the tyrosine phosphatase Shp2 in sertoli cells causes infertility in mice. Sci Rep (2015) 5:12982. doi: 10.1038/srep12982
38. Vesterhus P, Aarskog D. Noonan's syndrome and autoimmune thyroiditis. J Pediatr (1973) 83(2):237–40. doi: 10.1016/s0022-3476(73)80482-2
39. Amoroso A, Garzia P, Vadacca M, Galluzzo S, Del Porto F, Mitterhofer AP, et al. The unusual association of three autoimmune diseases in a patient with noonan syndrome. J Adolesc Health (2003) 32(1):94–7. doi: 10.1016/s1054-139x(02)00364-6
40. Svensson J, Carlsson A, Ericsson UB, Westphal O, Ivarsson SA. Noonan’s syndrome and autoimmune diseases. J Pediatr Endocrinol Metab (2003) 16:217–8. doi: 10.1515/jpem.2003.16.2.217
41. Quaio CR, Carvalho JF, da Silva CA, Bueno C, Brasil AS, Pereira AC, et al. Autoimmune disease and multiple autoantibodies in 42 patients with RASopathies. Am J Med Genet A (2012) 158A(5):1077–82. doi: 10.1002/ajmg.a.35290
42. Siano MA, Marchetti V, Pagano S, Di Candia F, Alessio M, De Brasi D, et al. Risk of autoimmune diseases in patients with RASopathies: systematicstudy of humoral and cellularimmunity. Orphanet. J Rare Dis (2021) 16(1):410. doi: 10.1186/s13023-021-02050-6
43. Baldassarre G, Mussa A, Carli D, Molinatto C, Ferrero GB. Constitutional bone impairment in noonan syndrome. Am J Med Genet A. (2017) 173(3):692–8. doi: 10.1002/ajmg.a.38086
44. Choudhry KS, Grover M, Tran AA, O'Brian Smith E, Ellis KJ, Lee BH. Decreased bone mineralization in children with noonan syndrome: another consequence of dysregulated RAS MAPKinase pathway? MolGenetMetab (2012) 106:237–40. doi: 10.1016/j.ymgme.2012.04.003
45. Leoni C, Stevenson DA, Martini L, De Sanctis R, Mascolo G, Pantaleoni F, et al. Decreased bone mineraldensity in Costello syndrome. Mol Genet Metab (2014) 111(1):41–5. doi: 10.1016/j.ymgme.2013.08.007
46. Brunetti-Pierri N, Doty SB, Hicks J, Phan K, Mendoza-Londono R, Blazo M, et al. Generalized metabolic bone disease in neurofibromatosis type I. Mol Genet Metab (2008) 94:105–11. doi: 10.1016/j.ymgme.2007.12.004
47. Dulai S, Briody J, Schindeler A, North KN, Cowell CT, Little DG. Decreased bone mineral density in neurofibromatosis type 1: results from a pediatric cohort. J Pediatr Orthop. (2007) 27:472–5. doi: 10.1097/01.bpb.0000271310.87997.ae
48. Illés T, Halmai V, de Jonge T, Dubousset J. Decreased bone mineral density in neurofibromatosis-1 patients with spinal deformities. Osteoporos. Int (2001) 12:823–7. doi: 10.1007/s001980170032
49. Kuorilehto T, Pöyhönen M, Bloigu R, Heikkinen J, Väänänen K, Peltonen J. Decreased bone mineral density and content in neurofibromatosis type 1: lowest local values are located in the load-carrying parts of the body. Osteoporos. Int (2005) 16:928–36. doi: 10.1007/s00198-004-1801-4
50. Lodish MB, Dagalakis U, Sinaii N, Bornstein E, Kim A, Lokie KB, et al. Bone mineral density in children and young adults with neurofibromatosis type 1. Endocr Relat Cancer. (2012) 19(6):817–25. doi: 10.1530/ERC-12-0293
51. Stevenson DA, Moyer-Mileur LJ, Murray M, Slater H, Sheng X, Carey JC, et al. Bone mineral density in children and adolescents with neurofibromatosis type 1. J Pediatr (2007) 150(1):83–8. doi: 10.1016/j.jpeds.2006.10.048
52. Leoni C, Viscogliosi G, Tartaglia M, Aoki Y, Zampino G. Multidisciplinary management of Costello syndrome: Current perspectives. J Multidiscip Healthc. (2022) 15:1277–96. doi: 10.2147/JMDH.S291757
53. Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q, et al. PTPN11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature. (2013) 499(7459):491–5. doi: 10.1038/nature12396
54. Stevenson DA, Viskochil DH, Carey JC, Sheng X, Murray M, Moyer-Mileur L, et al. Pediatric 25-hydroxyvitamin d concentrations in neurofibromatosis type 1. J Pediatr Endocrinol Metab (2011) 24(3-4):169–74. doi: 10.1515/jpem.2011.092
55. Lee PH, Macfarlane DJ, Lam TH. Stewart SMInt validity of the international physical activity questionnaire short form (IPAQ-SF): a systematic review. J Behav Nutr Phys Act (2011) 8:115. doi: 10.1186/1479-5868-8-115
56. Castagna MG, Fugazzola L, Maino F, Covelli D, Memmo S, Sestini F, et al. Reference range of serum calcitonin in pediatric population. J Clin Endocrinol Metab. (2015) 100(5):1780–4. doi: 10.1210/jc.2014-4508
57. Cianferotti L, Brandi ML Guidance for the diagnosis, prevention and therapy of osteoporosis in Italy. Clin Cases Miner. Bone Metab (2012) 9:170–8.
58. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. (2013) 381(9863):333–42. doi: 10.1016/S0140-6736(12)61023-X
59. Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, et al. The cardiofaciocutaneous syndrome. J Med Genet (2006) 43(11):833–42. doi: 10.1136/jmg.2006.042796
60. Lammert M, Friedman JM, Roth HJ, Friedrich RE, Kluwe L, Atkins D, et al. Vitamin d deficiency associated with number of neurofibromas in neurofibromatosis 1. J Med Genet (2006) 43(10):810–3. doi: 10.1136/jmg.2006.041095
61. Ferrara UP, Tortora C, Rosano C, Assunto A, Rossi A, Pagano S, et al. Bone metabolism in patients with type 1 neurofibromatosis: Key role of sun exposure and physical activity. Sci Rep (2022) 12(1):4368. doi: 10.1038/s41598-022-07855-4
62. Stevenson DA, Schwarz EL, Viskochil DH, Moyer-Mileur LJ, Murray M, Firth SD, et al. Evidence of increased bone resorption in neurofibromatosis type 1 using urinary pyridinium crosslink analysis. Pediatr Res (2008) 63(6):697–701. doi: 10.1203/PDR.0b013e31816fee45
63. Yu X, Chen S, Potter OL, Murthy SM, Li J, Pulcini JM, et al. Neurofibromin and its inactivation of ras are prerequisites for osteoblast functioning. Bone. (2005) 36(5):793–802. doi: 10.1016/j.bone.2005.01.022
64. Yang FC, Chen S, Robling AG, Yu X, Nebesio TD, Yan J, et al. Hyperactivation of p21ras and PI3K cooperate to alter murine and human neurofibromatosis type 1-haploinsufficient osteoclast functions. J Clin Invest. (2006) 116(11):2880–91. doi: 10.1172/JCI29092
Keywords: RASopathies, bone mineral density, vitamin D, autoimmunity, CFC
Citation: Siano MA, Pivonello R, Salerno M, Falco M, Mauro C, De Brasi D, Klain A, Sestito S, De Luca A, Pinna V, Simeoli C, Concolino D, Mainolfi CG, Mannarino T, Strisciuglio P, Tartaglia M and Melis D (2022) Endocrine system involvement in patients with RASopathies: A case series. Front. Endocrinol. 13:1030398. doi: 10.3389/fendo.2022.1030398
Received: 28 August 2022; Accepted: 26 October 2022;
Published: 18 November 2022.
Edited by:
Vandana Jain, All India Institute of Medical Sciences, IndiaReviewed by:
Sahar Mansour, St George’s University Hospitals NHS Foundation Trust, United KingdomCopyright © 2022 Siano, Pivonello, Salerno, Falco, Mauro, De Brasi, Klain, Sestito, De Luca, Pinna, Simeoli, Concolino, Mainolfi, Mannarino, Strisciuglio, Tartaglia and Melis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: D. Melis, ZG1lbGlzQHVuaXNhLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.