 Ana Cristina García-Gaytán†
Ana Cristina García-Gaytán† Andy Hernández-Abrego
Andy Hernández-Abrego Mauricio Díaz-Muñoz
Mauricio Díaz-Muñoz Isabel Méndez
Isabel Méndez
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 15 November 2022
Sec. Cancer Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1029210
This article is part of the Research Topic Immunoendocrine System from Physiology to Pathology View all 6 articles
Glutamate is one of the most abundant amino acids in the blood. Besides its role as a neurotransmitter in the brain, it is a key substrate in several metabolic pathways and a primary messenger that acts through its receptors outside the central nervous system (CNS). The two main types of glutamate receptors, ionotropic and metabotropic, are well characterized in CNS and have been recently analyzed for their roles in non-neural organs. Glutamate receptor expression may be particularly important for tumor growth in organs with high concentrations of glutamate and might also influence the propensity of such tumors to set metastases in glutamate-rich organs, such as the liver. The study of glutamate transporters has also acquired relevance in the physiology and pathologies outside the CNS, especially in the field of cancer research. In this review, we address the recent findings about the expression of glutamatergic system components, such as receptors and transporters, their role in the physiology and pathology of cancer in non-neural organs, and their possible use as biomarkers and therapeutic targets.
Glutamatergic system components are located in neural and non-neural cells. Recent studies have demonstrated that ionotropic and metabotropic glutamate receptors, along with membrane glutamate transporters, regulate a broad variety of processes in the pathophysiology of non-neural diseases, especially those related to oncologic and inflammatory conditions, highlighting their potential use as biomarkers and/or therapeutic targets. This review aims to provide an overview of recent progress in the study of glutamatergic system components and their function in cancer of non-neural organs.
Metabolic networks are complex systems that make coordinated biochemical and energy transformations possible among and within a variety of cellular compartments. In this context, some molecules, including glutamate, play important nodal roles in strategic pathways involving the concerted interactions of dozens of different intermediates.
Glutamate (2-aminopentanedioic acid) is a non-essential α-amino acid that is recognized as one of the most abundant biomolecules in cellular systems. It displays properties according to the reactivity of molecule at the tautomeric 2 carbon and at the 4- and 5-C positions, making it serve as a metabolic intermediate. The 5-carbon skeleton of its structure allows the keto-enol tautomers and transient, joint-electron activation states at the 2,3- and 4,5-bounds in positions of enzymatic catalysis. Glutamate is present in prokaryotes and along the entire phylogenetic scale, and it can be formed in prebiotic conditions from simpler entities such as HCN, NH4+, and some aldehydes (1). As a relevant prebiotic reaction, glutamate can be oxidatively decarboxylated by sunlight photolysis towards succinate (2). Glutamate acts as a link between energy and nitrogen metabolism, and it is a key intermediate between anabolic and catabolic transformations. Glutamate functions as a 1) substrate for protein synthesis, 2) a component of the rigid β-sheet structure enriched in polyglutamate, 3) a precursor of ornithine and proline, 4) a part of the arginine cycle, 5) an inhibitor of the glutaminase reaction, 6) an energy source for some tissues (mucosa), 7) a substrate for vitamin k-dependent carboxylation of clotting factors (with high Ca2+ affinity), and 8) a precursor of γ-aminobutyrate (GABA; neurotransmitter and osmolyte) and glutathione (for review see (3)).
Nitrogen is a key element in the structure of biomolecules especially peptides and nucleic acids. To maintain nitrogen homeostasis in the brain and the whole organism, glutamate is required, functioning as a substrate and a product of several enzymes. For example, transaminases, a family of pyridoxal phosphate-dependent enzymes (aminotransferases), usually catalyze reversible reactions in which two types of conversion are possible: 1) most amino acids can transfer their amino group to α-ketoglutarate and converge in the formation of glutamate; 2) glutamate can donate its amino group to a variety of keto acids to favor the formation of the any corresponding amino acid (4). In an ATP-dependent reaction, glutamate and NH4+ are condensed to form glutamine by glutamine synthetase (GS). GS is an oligomeric enzyme expressed in the liver (exclusively in pericentral hepatocytes) and in the brain, primarily in astrocytes (5). During hyperammonemia, production of glutamine from glutamate is significantly favored. In another reaction, glutamate donates NH4+ by mitochondrial oxidative deamination in a reversible redox reaction catalyzed by glutamate dehydrogenase (GDH). In the NH4+-resulting reaction, α-ketoglutarate is formed and NAD+ is reduced to NADH. NADPH is oxidized in the condensation of α-ketoglutarate, derived from the Krebs cycle (6) and NH4+ to glutamate (7). Because of this biochemical equilibrium between glutamate and NH4+, in hyperammonemia conditions, such as those associated with hepatic or kidney disorders, glutamate metabolism acts as a pathogenic factor (8). Urea is the major nitrogenous excretory product in mammals; it is synthesized in the liver and is part of a key process to maintain nitrogen balance. NH4+ produced by oxidative deamination is a substrate of the urea cycle rate-limiting enzyme, carbamoyl phosphate synthetase I, and in an ATP-dependent reaction, it reacts with HCO3- to form carbamoyl phosphate (9). In the brain, the main pathway for glutamate and ammonia removal is by GS and by glutamate decarboxylase action to produce the inhibitory neurotransmitter GABA (10).
Glutamate is the most abundant amino acid in the brain with concentrations in the range of 10-100 mM within synaptosomal vesicles. Since glutamate is a major excitatory neurotransmitter, its concentration in the cerebral extracellular fluid must be kept at low micromole levels (0.5–5 μM) (11). A large proportion of cerebral glutamate is synthesized mainly via glutaminase reaction and by aminotransferases, and to a lesser extent by GDH reaction (12). The carbon skeleton of the glutamate molecule is formed within the mitochondria as a cataplerotic reaction based on the incorporation of an NH4+ molecule to α-ketoglutarate derived from the Krebs cycle. This pathway was also described in the liver but with less efficacy (6). GDH activity is favored towards the excision of glutamate mainly by enhancing its phosphorylation and reducing ADP-ribosylation and lysine acetylation. While NH4+ derived from this reaction is channeled to urea synthesis, α-ketoglutarate turns into gluconeogenic intermediates, mainly oxaloacetate, which is decarboxylated by PEPCK (phosphoenolpyruvate carboxykinase). In this way, both ATP-dependent metabolic pathways combine transit among organelles, as well as redox requirements, to accomplish nitrogen disposal and synthesis of energy molecules (13).
Glutamate also plays a role in the malate-aspartate shuttle, which is a link between cytosolic redox reactions with mitochondrial electron transport and oxidative phosphorylation to generate energy. Since mitochondria are impermeable to glycolytic NADH, the reducing equivalent is introduced into the mitochondria by a set of coupled redox reactions: 1) cytosolic malate dehydrogenase forms malate from oxaloacetate (in parallel, NADH is oxidized to NAD+); 2) malate enters the mitochondria via an antiporter in exchange for α-ketoglutarate; 3) mitochondrial malate dehydrogenase restitutes NADH that is available for electron transport chain (in parallel, oxaloacetate is formed); 4) glutamate allows the transamination of oxaloacetate to aspartate; 5) aspartate goes to the cytosol via an antiporter in exchange for cytosolic glutamate; 6) cytosolic transaminase regenerates oxaloacetate and glutamate to complete the cycle. Shortage of cytosolic glutamate may impair the function of the malate-aspartate shuttle (14).
Cancer is a collection of pathologies characterized by loss of genetic and metabolic control set-points. A set of hallmarks have been postulated as the principal feature that distinguishes cancerous cells from non-cancerous cells (15). Among the most accepted cancerogenic elements are metabolic rewiring, immune modulation, altered stress response, invasion and metastasis, vascularization, selective growth and proliferative advantage, and an inciting microenvironment (16).
Reprogrammed metabolism allows cancerous cells to sustain rapid proliferation. Two central pathways that permit biochemical adaptations of cancer metabolism are increased glycolysis and glutamine catabolism (17). These biochemical characteristics are part of a metabolic modulation that has incidence in the transit of metabolites towards and from the mitochondria. Despite oxygen availability, cancerous cells display intense glucose consumption and lactate production, known as the Warburg effect; this enhanced glycolytic activity promotes sufficient energy as ATP, coupled with glutamine metabolism, to support cellular sustainability. At the same time, substrates from the Krebs cycle, such as citrate, escape from the mitochondria to serve as precursors for the synthesis of fatty acids and generation of reducing power as NADPH (18).
Glutamine regulation is tightly controlled by metabolic intermediates that allosterically inhibit and activate GDH, which fuels the Krebs cycle by converting glutamine-derived glutamate to α-ketoglutarate. The result of this metabolic reprogramming is that mitochondria contribute with biosynthetic intermediates for membrane synthesis and DNA formation, allowing NADPH availability. At the same time, this organelle uptakes glutamine and glutamate carbon skeletons to sustain its energy generation. The coordinated response in oncogenic conditions between oxidative glycolysis and glutaminolysis involves synergistic regulation of several genes encoding metabolic enzymes, including lactate dehydrogenase isoforms A (LDHA) and B (LDHB), and mitochondrial glutamic pyruvate transaminase 2 (19).
In addition to its multiple metabolic roles, glutamate also forms part of a signaling system, acting as a specific ligand for an array of ionotropic and metabotropic receptors (20, 21).
Outside the neural environment, glutamate functions as a signaling molecule. Over the last 20 years, investigators have dedicated their research to discovering the presence of glutamatergic system components in non-neural organs. Several studies indicate an aberrant expression of glutamate receptors and/or transporters in the pathogenesis of cancer cells of non-neural organs. Diverse non-neural cancer cells have been shown to release elevated levels of glutamate, suggesting a putative autocrine loop of glutamatergic signaling (22, 23).
Glutamate mediates cellular responses via the interaction with receptors in the plasmatic membranes of target cells. Glutamate acts via two classes of receptors: ion channels named ionotropic glutamate receptors (iGluRs) and G protein-coupled receptors named metabotropic glutamate receptors (mGluRs).
iGluRs are glutamate-gated ion channels characterized based on the amino acid sequences, functional properties, and molecular structure, and are classified as NMDA (N-methyl-d-aspartate), AMPA (α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate), and kainate (Figure 1). iGluRs consist of a tetrameric or pentameric unit that forms an ion pore-forming segment (24, 25). In mammals, many different subunits form iGluRs: NMDA (GluN1, 2A-D, 3A-B encoded by Grin1, 2a-d, 3a-b), AMPA (GluA1-4 encoded by Gria1-4), and kainate (GluK1-5 encoded by Grik1-5) (to review the previous nomenclature, see the revision by Sprengel et al. (24)). These receptors are selective cation channels, allowing the passage of monovalent (Na+, K+) or divalent (Ca2+) cations (or both). iGluRs are regulated by postranscritional mechanisms like RNA splicing and editing, posttranslational modifications, like phosphorylation and ubiquitination, and trafficking to the plasma membrane (26, 27). In the CNS, these receptors have been widely characterized as mediators of synaptic transmission.
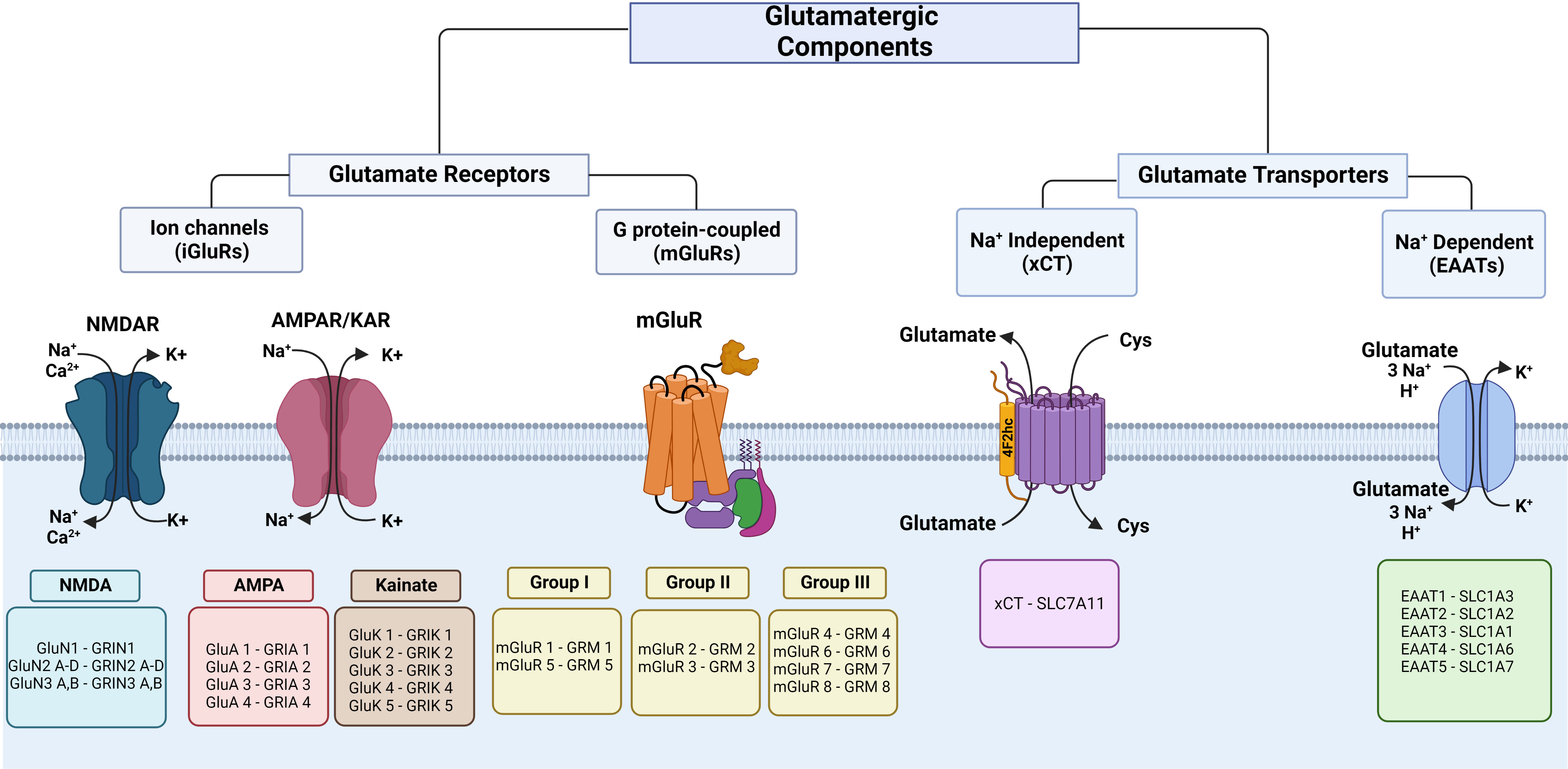
Figure 1 Classification of glutamate receptors and transporters. The left panel shows iGluR subunits and the respective coding genes. In the middle, the proteins and respective coding genes of mGluR families are shown. The right panel shows glutamate transporters of glutamate efflux and influx. Cys, cystine. This figure was created with BioRender.com.
NMDA receptors mediate the majority of excitatory neurotransmission in the central nervous system (CNS) and Mg2+ blocks them at rested potential (28). After glutamate binding to NMDA receptors, rapidly depolarize the postsynaptic membrane and initiate signaling pathways (28). These receptors are highly permeable for Ca2+ and voltage dependent, showing high single-channel conductance (29). Under physiological conditions, cell membrane depolarization is necessary from a resting potential of -70 to -30–0 mV induced by the entrance of Na+ to release Mg2+ blocking and allow the activation of the channel by glutamate. In neurons, the released Mg2+ contributes to CREB activation by NMDA receptor in an extracellular calcium-independent manner (30). AMPA receptors support the depolarization that results in NMDA receptor activation. GluN1 is the main subunit that forms all functional NMDA receptors in the CNS (31). Glycine and D-serine serves as a co-agonist of glutamate for NMDA receptor activation (32, 33). The Ca2+ influx by brain NMDA receptors activates signaling via phosphorylation, activation of calmodulin-dependent kinase II (CaMKII) and protein kinase C (PCK) related to synaptic plasticity.
AMPA receptors evoke excitatory postsynaptic potentials and mediate the fast signal transmission of glutamate and receptor turnover in the CNS (27, 34). They are also involved in synaptic plasticity, which forms the basis of learning and memory, and display rapid desensitization (34). Posttranscriptional modifications make AMPA receptor subtypes that form homo and heterotetrameric channels highly diverse (35, 36) A specialized set of auxiliary proteins interacts with these receptors providing functional regulation (37). AMPA receptors are permeable to Na+ and K+ and a minor population is mainly present in interneurons for Na+ and Ca2+ (38, 39). Glutamate released by a presynaptic neuron activates AMPA receptors and depolarizes postsynaptic membranes that allow a pre-activated state of NMDA receptors to be activated by a second glutamate release.
Kainate receptors are formed by a tetrameric unit conformed by the low affinity for the ligand subunits GluK1-GluK3 and/or the high agonist affinity for the ligand subunits GluK4-GluK5 (26). These receptors are selective for Na+ and Ca2+ (24) and are regulated by postranscritional mechanisms like RNA splicing and editing, posttranslational modifications, and trafficking to the plasma membrane (26). Kainate receptors mediate slower synaptic transmission and are expressed both at pre-synapse, which occurs in excitatory neurotransmission, and at post-synapse which regulates neurotransmitter release. The iGluR proteins and coding genes are shown in Figure 1.
mGluRs are seven transmembrane G-protein coupled receptors. In the CNS, they participate in increasing neuronal excitability and promote synaptic plasticity, which are key for learning and memory, through second messenger signaling pathways (40). These receptors are voltage-sensitive, and depolarization modifies the binding with G proteins and affinity for agonists (41, 42). mGluRs have been characterized into eight subtypes (mGluR1-8) encoded by GRM1-8 and classified into three groups (Figure 1) according to their pharmacology, sequence of amino acid homology, and signal transduction pathways (43). Group I (mGluR1 and mGluR5) is mostly localized post-synaptically, is normally stimulatory, and particularly, the subcellular location of mGluR1 in perisynaptic and extrasynaptic areas is associated with postsynaptic specialization of excitatory synapses (44), group II (mGluR2 and mGluR3) is found on post-synaptic and pre-synaptic cells and has a key function in reducing neurotransmitter release (45), and group III (mGluR4, mGluR6-8) is localized pre-synaptically and controls synaptic transmission (44). A variety of mGluRs that are expressed in different cell types play important roles in the immunology of the CNS, both in physiology and pathological processes associated with neurodegenerative disorders (44, 45). These receptors form homodimers, although they can eventually form heterodimers with other kinds of receptors, such as dopamine and adenosine receptors (46). Glutamate acts on mGluRs by activating gene expression and protein synthesis to exert a variety of regulatory effects through the recruitment of second messengers (47). Group I is coupled to Gq and activates phospholipase C, which produces inositol triphosphate and releases Ca2+ from intracellular stores in the endoplasmic reticulum, and diacylglycerol activates protein kinase C (47). Also, these receptors activate K+ channels and inhibit voltage-gated Ca2+ channels (48–50). Group II and group III are coupled to Gi and inhibit adenylate cyclase, and group III activates mitogen-activated protein kinases (MAPK) and PI-3 kinase (44, 47, 51). The mGluR proteins and coding genes are shown in Figure 1.
Several glutamate receptors are expressed in a variety of organs and cells outside the CNS (Figure 2). Expression of both iGluRs and mGluRs was reported in androgen-dependent and androgen-independent prostate cancer cell lines (52) in human skin fibroblasts and various human cancer non-neural cells lines; the expression level of these receptors was lower in these cell lines than in the human brain (53). Upregulation, and in some cases downregulation, of glutamate receptors has been associated with cancer. Therefore, gain or loss of function could be possible for tumor survival with an aggressive environment associated with oxidative stress and inflammatory conditions. In recent years, studies have demonstrated that glutamate receptors are expressed in various types of cancer in non-neural cells, and although their precise involvement is still uncertain, there is strong evidence that they participate in different aspects of cancer development, such as augmented proliferation and metastasis, and adaptive metabolic changes (125, 126).
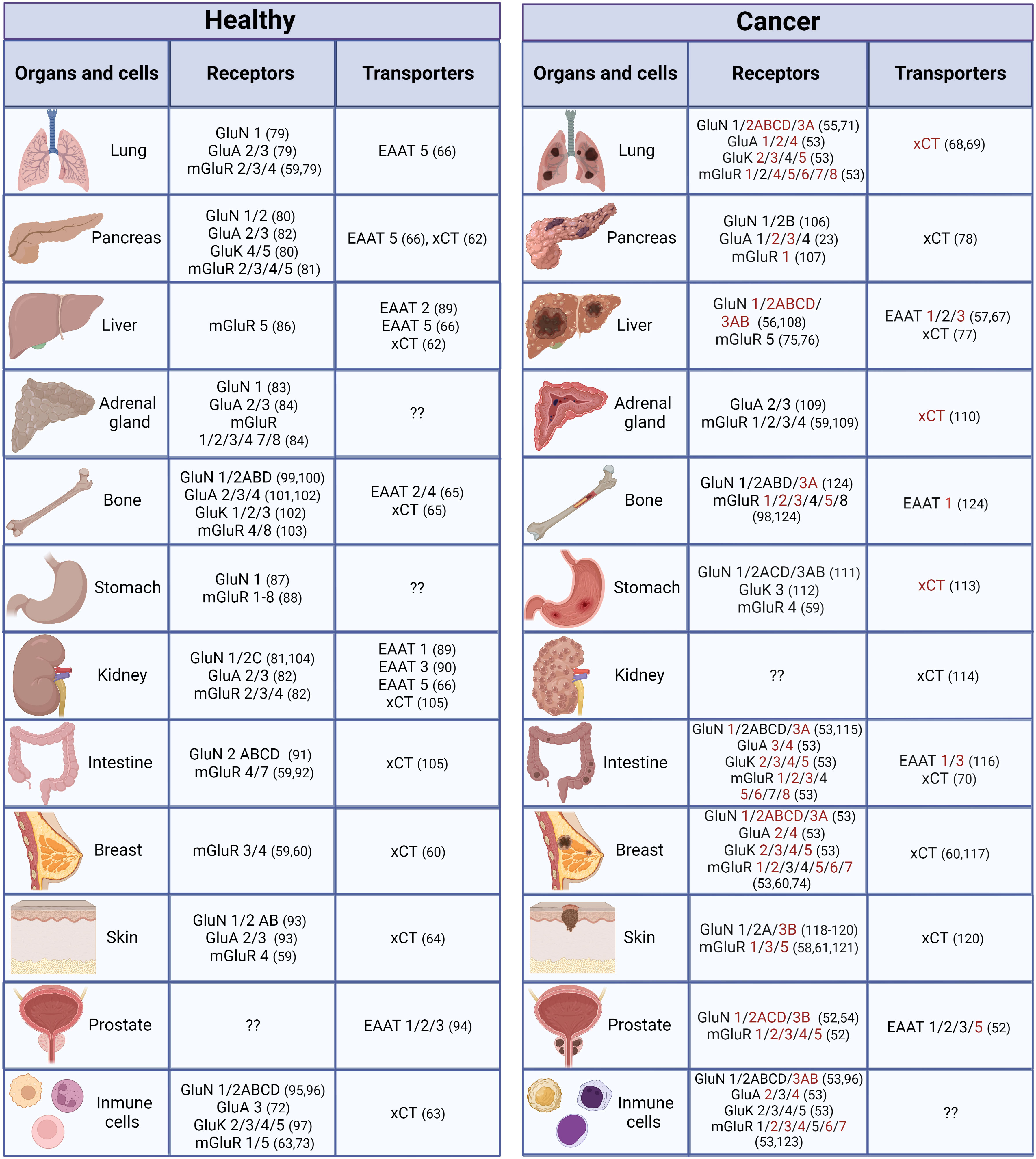
Figure 2 Distribution of glutamatergic system components in non-neural organs and cells under normal and cancer conditions. Components in red have only been reported under cancer conditions (they could be expressed de novo under cancer conditions or have not been studied under normal conditions). (23, 52–124).
Regarding iGluRs, both NMDAR and AMPA have been detected by immunostaining in colon adenocarcinoma, lung, and breast carcinoma cell lines and it was reported that they favor proliferation in a Ca2+ influx-dependent manner (127). In particular, NMDAR1 expression was found in tissue sections of prostate, breast, and colon cancers, and has been proven to be functional in tumor-derived cell lines stimulating cell proliferation (54). NMDAR1 staining was observed in 145 prostate cancer specimens: positive stromal staining was low in 30%, moderate in 26%, and high in 15%, and either stromal or epithelial staining was observed in 81% of them. Of 18 benign prostatic hyperplasia specimens, 12 were normal prostate specimens, and none had stromal or scarce epithelial NMDAR1 staining (54). NMDAR1 and NMDAR2 have been identified in diverse cancer cell lines derived from peripheral sites, such as human lung adenocarcinoma cells A549, human breast cancer cells MDA-MB-231, and rat prostate cancer cells MAT-LyLu, and their activation also stimulated proliferation (128). In skin, functional NMDAR1 has been found in normal keratinocytes, especially at areas of cell-to-cell contact, influencing proliferation, differentiation, and migration during epithelialization, and playing a role in contact-mediated inhibition of growth. In squamous cell carcinomas, NMDAR1 is missing, which is consistent with the lack of contact-mediated growing (129). Conversely, overexpression of NMDAR1 has been associated with metastasis, tumor size, cancer stage, and poor prognosis in oral squamous cell carcinoma (130). This inconsistency can be explained because in this last study, authors found positive NMDAR1 reactivity in 50 of 81 cases from a matched normal adjacent mucosa, and the level of NMDAR1 was significantly associated with prognosis-related factors. Otherwise, researchers identified NMDAR2B expression in gastric epithelial cells and decreased or absent expression in a diversity of human cancer gastric cell lines (131). Accordingly, transcriptional silencing of NMDAR2B due to gene hypermethylation was identified in human cancer gastric cells, as well as in a variety of esophageal squamous carcinoma cell lines (131, 132). In this context, NMDAR2B emerges as a tumor suppressor gene since transfection of NMDAR1-1a and NMDAR2B significantly decreased viability and induced cell death by apoptosis of esophageal squamous cell carcinoma cell line KYSE140 in the presence of NMDA (132). NMDAR2B transcriptional silencing by methylation was also found in non-small cell lung cancer, which correlates with some clinical features (55), whereas NMDAR1 and 2A-D expression was observed in hepatocellular carcinoma tissue from rat and mouse models (56). Interestingly, in this model NMDAR1 and NMDAR2B expression was identified since fibrosis, a precancerous stage of liver disease, correlated with demethylation in the promoter regions (56).
Hypoxia is a common feature of the microenvironment of solid tumors that induces epithelial-mesenchymal transition and angiogenesis resulting in cell migration and metastasis. A hypoxic environment enhances AMPA receptor expression and signaling. In hepatocarcinoma cells, but not in breast cancer cells, GRIA2 and GRIA3 (GluA 2 and 3) mRNA expression is upregulated by hypoxia through HIF-1α and HIF-2α, which contribute to cell proliferation and growth of tumor xenografts (57). AMPA receptors have been demonstrated to promote proliferation and invasion in pancreatic cancer cells. GluA1 subunit levels were increased in pancreatic cancer precursor lesions, whereas GluA1, 2, and 4 subunit expression was scarce in pancreatic ductal adenocarcinoma accompanied by increased glutamate levels (23). Interestingly, in ductal carcinoma cells SU.86.86, activation of GluA receptors by a selective agonist induced invasion and migration, and inhibition of GluA by a selective antagonist decreased these parameters that were associated with oncogenic signaling via MAPK and K-Ras, as well as with changes in the presence of matrix metalloproteinase-2 (23).
An inflammatory environment could lead to the dysregulation of glutamate receptors, altering functions such as learning, spatial memory discriminatory or other information acquisition processes in the CNS (133). In this context, NF-κB is a proinflammatory transcription factor involved in carcinogenesis (134, 135). Some NMDA and AMPA receptor promoters contain NF-κB binding sites and are therefore sensitive to an inflammatory environment that might affect the gene transcription and/or remodeling of chromatin through epigenetic modification as has been observed in neural cells (136).
Regarding mGluR expression outside the CNS, numerous studies have demonstrated the presence of these receptors in the periphery. Twenty-five years ago, functional mGluRs in non-neural cells were found in rat hepatocytes in culture (137). Since then, studies have consistently found that glutamatergic receptors are involved in the development of inflammatory-related pathologies, especially cancer. A melanoma mouse model revealed the presence of mGluRs in the cancer biology (138). Additionally, in human melanoma biopsies and cell lines, but not in benign tissues and melanocytes, expression of GRM1 (mGluR1) was detected (138). Mutations in mGluR3 have also been identified in melanoma, which were associated with increased anchorage-independent growth and migration (58). mGluR overexpression is associated with pathological clinical parameters and poor disease-free survival in cancer. For example, mGluR4 overexpression is related to a poor prognosis in colorectal carcinoma (59). Expression of mGluR4 was detected in 54% of 241 samples of colorectal carcinoma and, interestingly, 5% of them showed cytoplasmic expression; the association with clinical features was positive with recurrence and poor disease-free survival. Additionally, mGluR4 overexpression seems to mediate 5-fluorouracil resistance in colon cancer cells and its activation by the agonist L-AP 4 promotes cell survival (139). It seems that the presence of mGluR4 is detrimental for patients with colon cancer. mGluR3 is upregulated in most human colonic adenocarcinomas and colon cancer cell lines. Its reduction by knockdown expression decreased cell survival in vitro, whereas in vivo, it inhibited tumor growth by enhancing TGFβ tumor suppressor function (140). In breast cancer, mGluR3 promotes invasiveness through activation by autocrine action of glutamate generated by glutaminolysis exported to the extracellular milieu via the xCT transporter (60). In our hands, overexpression of mGluR3 was observed in the progression of cirrhosis to hepatocarcinoma development in rats and in HepG2 cell line, suggesting a role in the pathophysiology of the liver cancer (A.C. García-Gaytán, A. Hernández-Abrego, D. De Ita-Pérez, E. de los Ríos-Arellano, I. Turrubiate, E. Gámez, M. Díaz-Muñoz, I. Méndez, unpublished data).
mGluR1 can be an oncogene in epithelial cells and critical in the genesis of melanoma. Transfection of GRM1 induces transformation of mouse kidney cells that exhibit enhanced cell proliferation in vitro and significant tumorigenicity in vivo involving MAPK and AKT signaling, both crucial for tumorigenesis (141). High expression of mGluR1 was observed in primary or metastatic prostate cancer tissues, while in luminal acinar epithelial cells of normal or hyperplastic glands the expression was weak or absent (142). Moreover, mGluR1 is expressed with high prevalence in most breast cancer subgroups, such as invasive breast cancer with positivity in 60% of the ductal tumors and 47% in lobular tumors and is an unfavorable prognostic marker (143, 144). mGluR1 expression was associated with a lower tumor grade and with estrogen receptor-positive and progesterone receptor-positive tumors, suggesting that hormone stimulation regulates mGluR1 expression (144). Analysis of the progression from normal epithelium to triple-negative breast cancer demonstrated that mGluR1 overexpression induces transformation to a malignant phenotype that was reverted by silencing the GRM1 gene (143). Moreover, the expression of mGluR1 lead to the transformation of hyperplasic triple-negative breast cancer cells in human breast cancer cell xenografts in athymic nude mice (143). In addition to overexpression, mutations and single nucleotide polymorphisms or SNPs of GRM1 have been identified in multiple cancers, such as prostate (145), colorectal, and lung (146), that led to alterations in receptor activation, intracellular localization, and downstream signaling. mGluR5 expression induced melanoma in transgenic mice under a melanocyte-specific promoter, showing an aggressive phenotype with early onset (61). Hence, some of the mGluRs could be considered as prognostic markers and possible therapeutic targets for certain types of cancer.
As the major mediator of excitatory signals in the nervous system of mammals (147), tight regulation of extracellular glutamate concentrations at both synaptic and extra-synaptic locations is critical to ensure correct neurotransmission and prevent neurotoxicity events. In the CNS, glutamate is stored, and subsequently released from the presynaptic terminal to neighboring neurons by exocytosis, with successful replenishment by specialized recycling synaptic vesicles known as vesicular glutamate transporters (VGLUTs) (148). In non-neural organs, expression of some VGLUTs has been found in intestinal cells, pancreas, and osteoclasts; these VGLUTs contribute to glutamate release and exert autocrine and paracrine actions (149–151). VGLUT expression in liver, stomach, testis, and platelets suggests that they have numerous functions in the physiology of these sites (152–154). There is evidence of VGLUT expression in carcinoma stem cells that correlates with enhanced release of glutamate and cell proliferation via activation of NMDA receptors (155); therefore, VGLUT functions could also be implicated in cancer development.
In non-neural tissues, plasma membrane transporters act as non-vesicular routes of glutamate release or uptake. Non-vesicular releasing is the main source of extracellular glutamate in the brain, such as gap junction hemichannels (156) and heteromeric sodium-independent amino acid transporters, particularly system xCT (157).
System xCT (Sxc-) is a cystine/glutamate Na+-dependent antiporter that mediates the exchange of extracellular L-cystine and intracellular L-glutamate at a 1:1 ratio across the cellular plasma membrane, that is involved in neural protection against cerebral degeneration or damage (158–160). This system is a member of the heteromeric amino acid transporter family (glycoprotein‐associated amino acid exchangers) consisting of a heterodimer formed by the transporter subunit or light chain xCT (SLC7A11) and a regulatory subunit or heavy chain 4F2hc (4F2 cell-surface antigen heavy chain, acronyms: CD98, rBAT, FRP1, slc3 family SLC3A2) covalently linked through a disulfide bond to xCT (161). The xCT subunit is a hydrophobic, non‐glycosylated protein that has transport activity (161). 4F2hc acts as a chaperone for several amino acid transporters which serves to regulate trafficking to the plasma membrane maintaining xCT protein stability. xCT activity supports the non-vesicular release of glutamate and is critical for oxidative protection through reduced glutathione (GSH) production and provides intracellular L-cystine that is rapidly reduced in the cytoplasm to L-cysteine, a precursor for the enzymatic synthesis of GSH (162) (Figure 3).
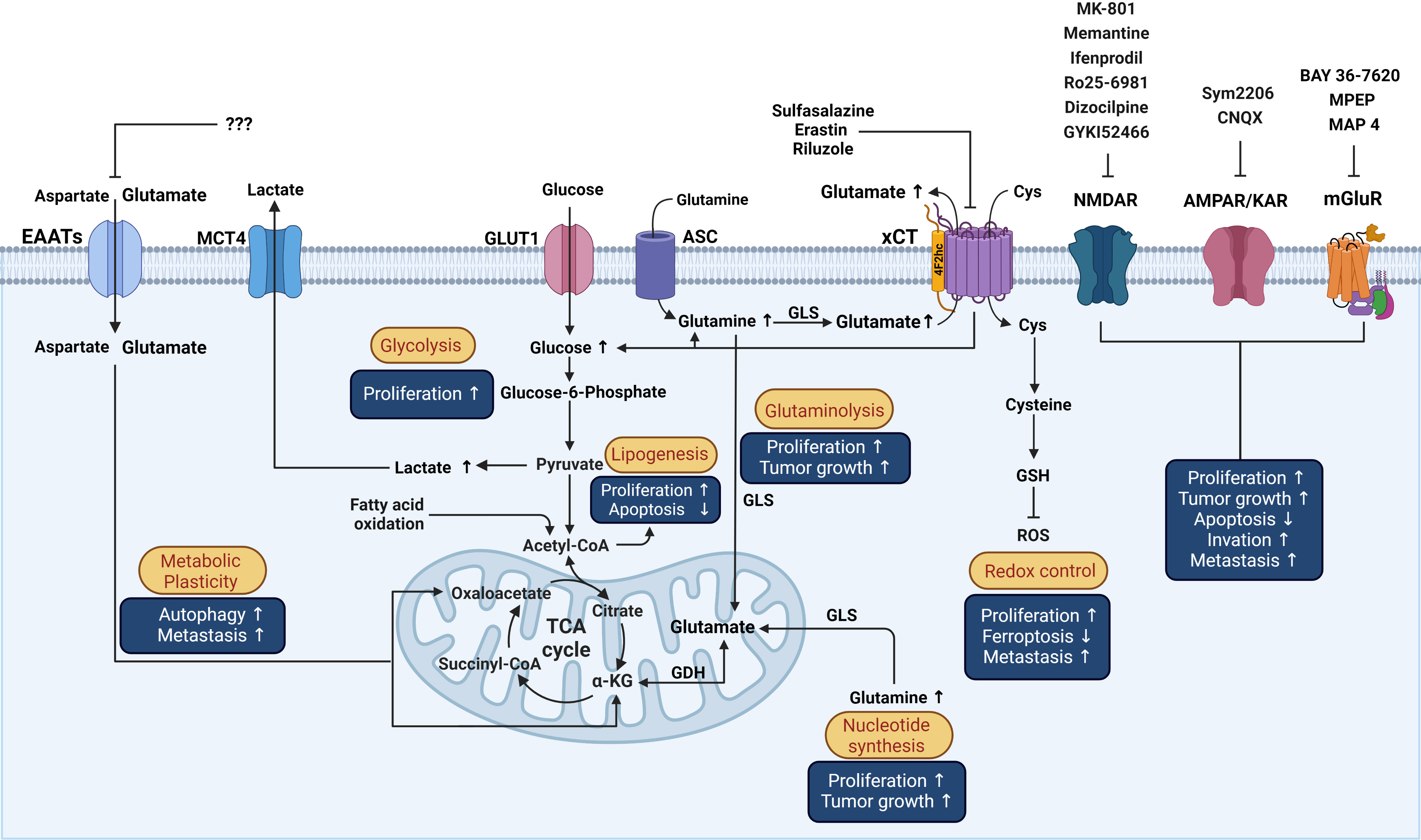
Figure 3 Schematic representation of the influence of glutamatergic system components on reprogrammed metabolism in cancer cells and the effects over processes associated with cancer. Possible actions of known drugs that affect glutamatergic components over different mechanisms involved in the pathophysiology of cancer that are described in the text are shown. Cys, cystine; GSH, Glutathione; GLS, Glutaminase; ASC, Alanine/serine/cysteine transporter; GDH, Glutamate dehydrogenase; α-KG, alpha-ketoglutarate; ROS, reactive oxygen species; TCA, Tricarboxylic acid cycle; GLUT1, Glucose transporter 1; MCT4, Monocarboxylate transporter 4. This figure was created with BioRender.com.
xCT is present in neurons and glia, especially in microglia and astrocytes, in the CNS, is the largest source of extracellular glutamate in the brain, and is dysregulated in neurological conditions (163, 164) and in various organs and cell types outside the CNS, such as liver, pancreas, skin, bone, dendritic cells, mammary epithelial cells, and fibroblasts, among others (60, 62–65, 158) (Figure 2). Interestingly, in the cancer stage, xCT is upregulated in some of those sites (e.g., breast cancer cells) (60) or expressed de novo (Figure 2). Several mechanisms are involved in the upregulation of xCT expression by amino acid deprivation, xenobiotics and, particularly, oxidative stress (165, 166). The production of reactive oxygen species (ROS) provokes cellular responses such as transcriptional activation of genes encoding proteins that participate in the mechanisms of protection against oxidative stress by xCT. Antioxidant response element (ARE) cis-acting sequences are present in the promotor regions of xCT that responds to phosphorylated nuclear factor erythroid‐2‐related factor (Nrf2) and increases its transcription (165, 167). Likewise, a proinflammatory environment increases the efflux of L-glutamate and induces the increase in L-cystine uptake through xCT in neurons co-cultured with astrocytes, leading to hypoxic neuronal injury (168). Both oxidative stress and inflammation promote oncogenesis. Sustained oxidative stress can induce chronic inflammation through the activation of several transcription factors, including NF-κB, p53, HIF-1α, and Nrf2, involved in the distinctive characteristics of cancer such as cell proliferation, migration, and survival (169).
Extracellular glutamate is not metabolized by any enzyme (170). Therefore, glutamate is able to interact with receptors until it is removed into the cell by specialized transporters located on the plasmatic membrane of pre- and post-synaptic neurons and astrocytes that play an essential role in the control of extracellular glutamate concentrations, with a high level of cooperation. Outside the CNS, glutamate reuptake is carried out by transporters that mobilize glutamate from the extracellular space.
Glutamate uptake is accomplished in the CNS by excitatory amino acid transporters (EAATs) that are present in the pre- and postsynaptic cell surface of neurons and astrocytes, where glutamate is preferentially recycled (163). EAATs belong to the solute carrier (slc) family 1 and transport L-glutamate and DL-aspartate with similar affinity. Five human EAATs have been identified: EAAT1-5: glutamate−aspartate transporter (GLAST/EAAT1), glutamate transporter (GLT/EAAT2), excitatory amino acid carrier (EAAC/EAAT3), and excitatory amino acid transporters 4 and 5 (EAAT 4 and EAAT5) (163) (Figure 1). EAAT1-3 are high-capacity glutamate transporters that play an important role in the regulation of the vulnerability to excitotoxicity modulating accumulation of glutamate, its receptors activation, and its metabolic homeostasis (171, 172). In addition, EAATs also serve as anion channels. EAATs catalyze coupled transport of 1H+ and 3Na+ with one molecule of glutamate and counter-transport 1K+ (Figure 1) (147, 173). Some EAATs operate as chloride channels in response to the glutamate transporter function (174). EAATs assemble as homo- or heterotrimers; each subunit exerts transport actions independently of the others (175) and form subpopulations of transporters with different affinities for glutamate (162). Their expression is under different mechanisms of regulation including transcriptional, epigenetic, and posttranslational control, that may influence on the trafficking and availability in the plasma membrane (162). Altered expression has been observed in pathologies such as Alzheimer disease that could indicate alterations in glutamate recycling (176). The mTOR signaling pathway is often activated in cancer (177) and is a potent regulator of EAATs (162). In peripheral organs, various EAATs express at the mRNA and protein levels in diverse sites, such as liver, kidney, and pancreas, among others (Figure 2) (66, 67).
During tumorigenesis, metabolic flexibility comprises reprogramming of catabolic and anabolic pathways to contend with their demands for energy, growth, proliferation, and detoxification, which involve the handling of several substrates related to aberrant glucose, lipid, and glutathione-glutamine-glutamate metabolism (178, 179). Metabolic reprogramming in cancer also increases oxidative stress; therefore, cancer cells tend to upregulate their antioxidant defenses in the form of GSH to maintain the appropriate redox balance. System xCT is instrumental in metabolic reprogramming, as it increases glucose consumption and lactate production, as well as glutamine dependency of cancer cells (68) (Figure 3). The oncogene RAS influences the transactivation of xCT promoter, enhancing GSH synthesis as part of the protection mechanism to favor oncogenic transformation (69). Thus, system xCT overexpression frequently found in an array of malignant tumors promotes survival features at the expense of increased GSH synthesis, such as protection from lipid peroxidation and oxidative stress-induced cell death (180, 181), cell proliferation, invasion, tumor growth, and chemoresistance (68, 70, 182–184). Conversely, there are cancer cells with a low expression level of xCT, such as chronic lymphocytic leukemia, which uses the capacity of stromal cells to synthesize GSH for survival and drug resistance to phenethyl isothiocyanate (185).
In contrast to the well-established pro-survival effects of xCT overexpression, xCT in renal cancer cells enhances cancer dependence on glucose and renders cancer cells more sensitive to cell death induced by glucose starvation (186). In fact, glucose starvation induces xCT overexpression through its major inductor Nrf2 transcription factor. It appears that xCT overexpression in these conditions serves as a ROS scavenger and protects the cells from glucose starvation. The bimodal role of xCT in protecting and promoting cell death in cancer make it challenging to elaborate therapeutic strategies that alleviate the symptoms caused by cancer. In this regard, it is important to consider the context of the disease, namely the oncogenic nature of the cancerous cell.
Regarding the role of the EAATs in the physiopathology of cancer, a differential expression of the EAAT family members has been observed in human cancers (187). An aberrant expression of several members of this family correlates with immune infiltration, tumor grade, and cancer stage, and nodal metastasis status in lung adenocarcinoma has a promising prognostic value and is a potential target for therapy in this cancer (187). In an analysis from The Cancer Genome Atlas, lung cancer patients with high expression of EAAT3 exhibited poor survival rate compared to patients with low EAAT3 expression, thus supporting the notion that EAAT3 is important in lung tumor progression (188). Overexpression of EAAT1, EAAT2, and EAAT3 contributes to cancer progression in non-neural solid tumors, such as gastric and lung cancers (188–190). EAAT1 and EAAT2 have been identified in adenocarcinomas from intestine, stomach, and genital tract and, depending on the cell confluence related to touching among cells, they re-locate from the plasma membrane to the nucleus, such that they no longer promote glutamate clearance when the cells are not in touch (191). While normal cells preserve membrane localization of EAATs, their mislocalization in malignant cells together with up-regulation of xCT lead to aberrant handling of glutamate that induces high levels of glutamate in the extracellular space, as seen in gliomas (192) and in breast cancer, where endocrine therapy-resistant cells promote autophagy and glutamate import via EAAT2 (60, 193). Interestingly, the relocation of EAAT1 and EAAT2 does not occur in astrocytoma; therefore, the regulation and effects of glutamate transporters should not necessarily be the same in all tumor types.
Several cancers are treated efficiently with endocrine therapy. However, it has been observed that a significant group of patients with breast cancer relapsed with the endocrine therapy-resistant disease. This pattern is associated with an enhanced influx of aspartate and glutamate via EAAT2 that serves as fuel for metabolic pathways and promotes autophagy and metastasis (193). Moreover, higher expression of EAAT2 is linked to changes in proliferation induced by treatments with an antagonist of estrogen receptors (fulvestrant) or an aromatase inhibitor (letrozole). Hence, the increased activity of EAAT2 promotes resistance to therapy that reinforces the overexpression of EAAT2 in a feedback loop. The challenge would be to end the vicious circle as a therapeutic strategy. Altogether these findings support a role for glutamate transporter EAATs in therapy-resistant cancer.
Hypoxia inducible factors (HIFs) mediate the transcription of multiple genes that allow cells to adapt to hypoxic environments, increase in cancer with a poor prognosis, and participate in various stages of carcinogenesis and cancer progression (194). In cells derived from hepatocarcinoma and renal carcinoma, but not from breast cancer, EAAT1 and EAAT3 expression is upregulated under hypoxic conditions through transactivation of gene expression by HIF-1α and HIF-2α (57). Hypoxia favors glutamate signaling through upregulation of AMPA receptors and EAATs. Since glutamate transport by EAATs could be reversible in the outward direction in neurons under certain conditions (e.g., when ratio [Na+]i/[K+]e decreases) (195), it seems that something similar occurs in hepatocarcinoma. This supposition needs further investigation.
Another member of the EAAT family, EAAT1, has been identified as a contributor to tumor initiation and progression, as well as asparaginase resistance in prostate cancer and breast cancer cells (196). Additionally, EAAT1 overexpression promotes cancer growth and correlates with a poor prognosis in a xenograft tumor growth model of gastric cancer (190). EAAT1 has been proposed as a potential biomarker for the pathogenesis and progression of chondrosarcoma (197).
Recent evidence suggests increasingly important actions of EAATs in various hallmarks of cancer development, including cell proliferation, reprogramming of energy metabolism, and drug resistance. Therefore, EAATs could be considered as another therapeutic target in searching for a more specific cancer treatment.
Progress in the management of cancer including chemotherapy, radiotherapy, and immunotherapy has been substantial (198). However, along with resistance to therapy, there is still the challenge of developing personalized therapy with fewer side effects. For many years, combined modality therapies have been developed for complete tumor eradication (198, 199). In this context, glutamate system components might be good candidates for concomitant therapy approaches with known treatments to avoid resistance, diminish toxicity, and offer individualized cancer treatment.
Malignancy frequently induces pain sense in peripheral organs that are innervated by afferent fibers. Several organs such as colon and liver produce and release glutamate, which mediates the activation of locality-specific nociceptive neurons (200). Another source of glutamate might be peripheral innervation like that of the epidermis (201). Pharmacological approaches have proposed antagonists of mGluRs and NMDA receptors in afferents that innervate peripheral organs, such as skin (202), colon (203), and bone (204), as therapeutic targets for the treatment of pain, since they have obtained analgesic effects. There is evidence that glutamate levels might increase in the serum or plasma of patients with cancer (205, 206); thus, they have been suggested as a biomarker of diagnosis or prognosis. Significantly higher serum glutamate levels correlated with aggressiveness in prostate cancer patients (142). Also, elevated glutamate levels were found in the serum of patients with hepatocellular carcinoma compared to those of patients with cirrhosis and healthy controls (207). Higher glutamate concentrations in the extracellular space could bind and signal through receptors to produce biological effects on the physiopathology of non-neural cells, independent of nervous system fibers. In addition to the high levels of circulating glutamate, the expression of other components of the glutamatergic system could serve as biomarkers and/or therapeutic targets for improving currently approved treatments.
In patients with oral squamous cell carcinoma, NMDAR1 is a likely prognostic biomarker and therapeutic target for cancer, as NMDAR1-positive tumors are associated with poorer survival compared to NMDAR1-negative tumors (130). Differential expression of NMDAR1 has been detected in stromal and epithelial cancer prostate tissues while a lack or scarce expression was observed in normal or hyperplastic prostate specimens (54), suggesting that NMDAR1 expression could be a biomarker for prostate cancer. In small-cell lung cancer cell lines, NMDAR1 and NMDAR2 promoted cell proliferation, and positive immunostaining for NMDAR1 was found in human tumor samples (71). Treatment of these small-cell lung cancer cells with the NMDAR1 antagonists dizocilpine (MK-801) and memantine impaired cell viability, whereas treatment with the NMDAR2 antagonists ifenprodil and Ro25-6981inhibited cell growth (71). Memantine has been proven to inhibit the growth of human prostate cancer cell lines (54). Moreover, dizocilpine prevented proliferative actions induced by epidermal, insulin, and basic fibroblast growth factors by upregulating the tumor suppressor protein p21 in lung adenocarcinoma cells and prolonged the survival in mice that were injected with human lung carcinoma cells into the peritoneal cavity (128).
Studies have shown that using antagonists of distinct iGluRs is effective because they exert anti-proliferative actions, inhibit migration, and alter the morphology of tumor cells, and that they have synergistic effects when used with chemotherapy agents, such as cisplatin or cyclophosphamide (127). The effects depend on the type of cancer and iGluR. For example, in colon adenocarcinoma and breast and lung carcinoma, cells are highly sensitive to the NMDAR antagonist dizocilpine and to the GluA 2/3 antagonist GYKI52466 (127). Treatments with iGluR antagonists resulted in modified membrane protrusions and thus a decreased motility and invasive phenotype of the thyroid carcinoma cells FTC238 (127). In addition, oncogenic signaling via MAPK and K-Ras was reduced by the non-competitive AMPA receptor antagonist SYM2206, and the invasiveness and migration were inhibited in the ductal pancreatic cell line Su86.86 (23). These data support the assessment of drugs that target iGluRs for potential uses in anticancer therapy.
On the other hand, DNA methylation pattern has emerged as a promising molecular biomarker for cancer detection, prognosis, and therapeutic response (208). Hypermethylation in promoter regions of cancer-associated genes can lead to blocked transcription (209). Accordingly, hypermethylation of NMDR2B detected in primary human esophageal squamous cell carcinoma and in primary gastric cancer tissues was associated with suppression of tumor features such as proliferation and colony formation (131, 132). Restoration by NMDAR2B transfection and activation in esophageal squamous cell carcinoma induced the suppressor phenotype eliciting cell death by apoptosis (132). Otherwise, increased expression of NMDAR1 and 2B was identified in hepatocellular carcinoma. This expression appeared from a precancerous stage of liver disease correlating with demethylation in CpG islands in the promoter regions in rat and mouse models of hepatocellular carcinoma (56). NMDAR2B emerges as a tumor suppressor gene since transfection of NMDAR1-1a and NMDAR2B significantly decreases viability and induces cell death by apoptosis of esophageal squamous cell carcinoma cell line KYSE140 in the presence of NMDA (132). Hence, the expression level of NMDARs and the methylation or demethylation of NMDAR in promoter regions might provide biomarkers to assess therapy response in these types of cancer.
In the immune system, expression of a plethora of iGluRs and mGluRs in human T cells has been reported in resting and activated cells and under normal and cancer conditions (210, 211). Both types of glutamate receptors have been shown to contribute to diverse functions. For example, iGluRs expression in T cells induces adhesion and chemotaxis (72), mediated by Ca2+ signaling, whereas mGluR promotes survival protecting from T cell activation-induced cell death (73). Additional evidence supports that glutamate released by dendritic cells through xCT antiporter acts in a paracrine way on mGluR1 to activate T cells (63). Considering that cancer cells avoid the immune response, activation of T cells via glutamate receptors could provide another effective T cell-mediated immune response against the tumor. Additionally, iGluRs and mGluRs expressed in non-solid tumors, T-cell leukemia, and T-cell lymphoma promote cancerous T cells by augmenting their extravasation and the expression of matrix metalloproteinases and evoking iCa2+ increase and concomitant expression of genes that regulate the cell cycle, respectively (211). Blocking AMPA receptors with antagonists reverted the effect over increased secretion of matrix metalloproteinases and limited tumor growth, suggesting that glutamate receptor inactivation could be another possible therapeutic strategy against cancer (212).
Regarding mGluRs as therapeutic targets, some drugs have been suggested as possible treatments for solid cancers. mGluR1 is visualized as a prognostic marker and a potential treatment target in some types of cancer. In triple-negative breast cancer cells, both a selective mGluR1 antagonist and silencing with shRNA targeting the gene GRM1 inhibited proliferation associated with apoptosis (143, 213). Interestingly, MDA-MB-231 triple-negative breast cancer cells and normal AB589 mammary epithelial cells express GRM1; however, treatment of cells with the mGluR1 antagonist BAY 36-7620 had a significant effect on cell growth only in breast cancer cells (213). In vivo research using MDA-MB-231 xenograft-bearing mice found that both the mGluR1 antagonist and riluzole were effective in reducing tumor volume (213). Riluzole is an oral drug with low toxicity that could be a therapeutic candidate for cancer therapy. This drug has been shown to inhibit the proliferation of human melanoma cell lines that express mGluR1 (22). Moreover, treatment of human melanoma cell xenografts in immunodeficient nude mice with riluzole inhibited tumor growth by 50% (22). A phase II trial of riluzole in melanoma seems to be promising (214).
mGluR1 has been suggested as a pro-angiogenic factor and a mediator of tumor progression in breast cancer (74). In this regard, primary endothelial cells and endothelial cell lines were shown to express high levels of mGluR1, and both riluzole and the non-competitive antagonist BAY36-7620 significantly inhibited cell proliferation in vitro and angiogenesis in subcutaneous implants in vivo (74). These data suggest that the inactivation of mGluR1 may represent a promising therapy for cancer related to angiogenesis. Although there was varying levels of mGluR5 in human oral squamous cell carcinoma tissues from patients, a significant positive association with overall survival was found (215). Moreover, the mGluR5 activation with the agonist DHPG increased tumor cell migration, invasion, and adhesion in the HSC3cell line, effects that were reverted by the antagonist MPEP (215). Inactivation of mGluR5 with the selective antagonist MPEP suppressed oncogenic actions; for instance, it induced inhibition of cell growth, migration, and invasion via ERK phosphorylation and induced apoptosis in the cell line HepG2. It also inhibited metalloproteinases 2 and 9, as well as tumor growth and metastasis in a xenograft model of hepatocellular carcinoma (75). mGluR5 expression is higher in hepatocellular carcinoma and chemotherapy agents caused cell death by decreasing mGluR5 in this cancerous system and increasing mGluR5 in normal hepatic cells. Moreover, inhibition of mGluR5 was shown to enhance the potency of chemotherapeutic agents in hepatocellular carcinoma cell lines and attenuate chemotoxicity over DNA damage by downregulating Ca2+-dependent MAPK signaling in the normal liver (76). Hence, mGluR5 seems to be a prognostic marker and a possible therapeutic target in oral squamous cell carcinoma and hepatocarcinoma.
Therefore, the pharmacology of glutamate receptor activity may be therapeutically useful for cancer treatment.
Overexpression of xCT correlates with tumor invasion, short survival, and a poor prognosis in patients and animal models of hepatocarcinoma (77, 216), acute myeloid leukemia (217), non-small cell lung cancer (68), prostate cancer (218), and colorectal cancer (219). Therefore, xCT has been considered as a possible biomarker of cancer progression and recurrence, and also as a therapeutic target for various types of cancer (77, 220). Moreover, xCT overexpression renders cancer cells, such as ovarian cancer cells, tongue squamous cell carcinoma cells, lung cancer cells, and gastric cancer cells, more resistant to chemotherapy with cisplatin (221–224).
Chemoresistance is still a challenge in cancer therapy. Chemotherapy-resistant cancer cells rely on cystine import for ROS resistance via the xCT. In addition to being a biomarker, xCT has been proposed as a therapeutic target for drug-resistant cancer types, such as triple-negative breast cancer, pancreatic cancer, and hepatocarcinoma, that depend on xCT for survival (78, 220, 225, 226). Patients with hepatocellular carcinoma with high xCT mRNA expression showed poorer overall and disease-free survival than those who lacked xCT mRNA (77). Therefore, xCT could be used as a biomarker for disease prognosis.
Multiple studies have demonstrated that cancer stem cells can promote tumorigenesis, tumor growth, and chemoresistance (227–229). Cluster of differentiation 44 (CD44) is an adhesion molecule in cancer stem cells that does not seem to be involved in stem cell properties and interacts with xCT. CD44 upregulates and stabilizes the membrane insertion and activity of xCT by interacting with the complex xCT-4F2hc (224, 230) to regulate the intracellular level of GSH, thereby controlling the defense against ROS generation. In gastric cancer, ablation of a CD44 variant (CD44v) diminishes membrane xCT and suppresses tumor growth accompanied by a greater expression of phospho-p38 MAPK and upregulation of p21 in the cancer cells that adopt a more differentiated and less proliferative state (230).
It has been demonstrated that xCT deficiency or inhibition suppresses cell proliferation and tumor growth and induces cell death by apoptosis or cell death mediated by accumulation of lipid ROS or lipid peroxidation, known as ferroptosis (231–234). Moreover, xCT inhibition potentiates the cytotoxic effects of gemcitabine and cisplatin in pancreatic ductal adenocarcinoma cells (234).
Inhibitors of xCT have been observed with therapeutic potential for the suppression of tumors. Sulfasalazine, a specific inhibitor of xCT and FDA-approved drug, is a potent suppressor of tumors that inhibits growth, invasion, and metastasis as has been revealed by the attenuation or suppression of lymphoma growth (231), metastasis of melanocytes (235), proliferation, colony formation, metastasis and invasion of gastric cancer cells (236), and stemness and metastasis of colorectal cancer (237). Further, sulfasalazine overcomes ionizing radiation and chemoresistance to anti-cancer therapies in hepatocarcinoma (183), improves cisplatin chemosensitivity in cholangiocarcinoma (238), and enhances the sensitivity to cell death by ferroptosis in paclitaxel-resistant uterine serous carcinoma cells (239). Repeated cisplatin treatment-induced cisplatin-resistant cells present high expression of xCT. Using different strategies, such as xCT inhibitors (sulfasalazine or erastin) or xCT siRNA, could sensitize human gastric cancer cells to cisplatin (240). Likewise, sulfasalazine suppresses CD44-dependent tumor growth promoting p38 in gastric cancer cells (230). In fact, hepatocarcinoma treated with hepatic arterial infusion chemoembolization therapy with cisplatin expresses a high level of CD44v9, suggesting that it is involved in the resistance to treatment (241). Interestingly, treatment with sulfasalazine was effective in combination with cisplatin in the hepatocarcinoma CD44v9-positive cell line HAK-1A (241). Sulfasalazine has been shown as a good candidate for anti-cancer therapy; however, its specificity is limited since its effects depend not only on the type of cancer but on other features of cancer cells of the same type of cancer. For example, in lung cancer, inhibition of proliferation in the cell lines A549 and H520 in response to sulfasalazine was not observed in the H1869 cell line (68). Inhibition of xCT alone is not sufficient since cysteine is still imported by additional transporters as the ASC system; thus, enzymatic treatment to eliminate extracellular cysteine has been proposed for prostate cancer treatment (242). Accordingly, it is still necessary to investigate the extent to which sulfasalazine, sorafenib, erastin analogs or any other xCT inhibitor could be harnessed for cancer therapy (70, 243). Erastin and sorafenib are antitumor agents that induce ferroptosis cell death by inhibiting xCT (244, 245). Even though their benefits as monotherapy are limited in survival because of primary and acquired resistance (246–248), they are able to enhance the sensitivity of chemotherapy and radiotherapy and function significantly better together with another antineoplastic agent (244, 249). Cancer immunotherapy is based on antigen-specific T cell function and is a potent toll against cancer with the advantage that it is better tolerated than common chemotherapy drugs (250). Immunotherapy functions well by itself for some types of cancers, and for others it works better together with other treatments. Combined treatment using immunotherapy results in a significant increase in durable responses, which suggest a viable strategy for improving anticancer immunotherapy (184). There is evidence that xCT deficiency does not impair T cell proliferation and antitumor immunity in vivo (184). Since xCT inhibition is dispensable for T cell proliferation, combined therapy of xCT inhibition with immunotherapy is a promising therapeutic strategy for cancer. Another strategy using vaccination of anti-xCT has been tested with good results on improving sensitivity to chemotherapy by doxorubicin in tumor growth and metastasis of breast cancer (251). Therefore, xCT-targeted therapy would impede ROS defense and compromise cell survival and may be ancillary, or even synergic, for other conventional treatments, such as chemotherapy or immunotherapy in refractory cancer cells but may be more susceptible to xCT-target drugs (252).
Glutamate transporters influence each other since the overexpression of some affects others. In non-small cell lung cancer, EAAT3 overexpression facilitates recycled extracellular glutamate into the cells by enhancing xCT activity and the consequent GSH biosynthesis to favor tumor growth and a malignant phenotype (188). Thus, increased extracellular glutamate by EAAT3 inhibition would restrict cystine uptake by xCT and promote cell death as a result of the blocking of GSH synthesis.
Regarding the role of EAATs in cancer therapy, it has been shown that the EAATs mediate therapeutic resistance related to altered tumor metabolic profiles. Cancer treatment schemes with enzymes that catabolize amino acids to deplete them have been successfully used in solid tumors with auxotrophy for arginine or asparagine with high clinical efficacy (253, 254). The limitation of this treatment is the intolerant toxicity (255) and therapy resistance, because cancer cells remain proliferative despite asparaginase treatment, the mechanism of which little is known. In this regard, SLC1A3 (EAAT1) has been identified to contribute to asparaginase resistance in prostate and breast cancer in which combined treatment with asparaginase and EAAT1 chemical inhibition restrains cancer cell proliferation and tumor progression (196). In response to this combination, metabolism is altered in prostate and breast cancer cells, including NAD+/NADH inhibition and lactate depletion which are important indicators of energy and redox status. It is clear that EAAT1 participates in enzyme treatment resistance. On the other hand, an RNA sequence analysis showed that SLC1A6 (EAAT4) was upregulated in nasopharyngeal carcinoma cell lines resistant to radiation and with low sensitivity to cisplatin (256). In addition, radiation upregulates EAAT4 expression in radioresistant cell lines that correlates with poor prognosis in patients with nasopharyngeal carcinoma (256).
In the complex context of cancer, reorganization of genome fragments results in new aberrant chimeric genes as a product of the fusion of the fragments, found less frequently in solid tumors (257). However, in primary gastric tumors and cell lines of gastric cancer and in primary colorectal cancer, the fusion of coding region of SLC1A2 (EAAT2) with a region of a probable promoter of CD44 results in a truncated but overexpressed and functional protein EAAT2 (189, 258). This truncated EAAT2 contributes to augmented glutamate concentrations that regulate the growth and development of gastric cancer (189). Furthermore, CD44-SLC1A2 gene silencing sensitizes gastric cancer cells to chemotherapy with cisplatin (189) which provides an additional target for therapy intervention.
Although more evidence is necessary for establishing the role of EAATs in the development and progression of cancer, here is another potential target for cancer therapy that includes selective inhibition of EAATs.
The use of drugs targeting glutamate receptors and/or transporters in combination with approved therapies could offer a more specific scheme of treatment with fewer adverse effects.
The identification and validation of biomarkers in early stages of cancer, as well as more specific therapeutic targets, is still a challenge for cancer research. The heterogenous nature of cancer makes it difficult to provide a prompt diagnosis and find a specific therapy to improve the prognosis of patients with cancer. Pharmacological approaches using receptor agonists or antagonists and transporter inhibitors, or molecular biology approaches, such as knock out, knock in, or silencing of gene expression, have been used to demonstrate that the glutamatergic system has an important role in oncogenesis. Glutamatergic system components are differentially expressed in non-neural organs in cancer and healthy organs, suggesting that this system supports cancer progression. In this regard, glutamate agonists or antagonists, glutamate transporter inhibitors, and molecular strategies could be used as powerful tools to interfere the mechanism of action of glutamatergic system components in order to treat malignant tumors. A variety of cancers depend on xCT overexpression for survival. Blocking of xCT could avoid the progression of a variety of cancer hallmarks by inhibiting protection against ROS overproduction and/or impeding its glutamate exporting activity and the subsequent binding and activation of glutamate receptors (Figure 3). For example, breast cancer has been well studied in which glutamate overproduction by enhanced glutaminase is released via the system xCT to activate mGluR3 signaling to increase invasiveness, and xCT inhibition with sulfasalazine decreases breast tumor growth. Besides, concomitant use of these drugs with other approved treatments may be helpful to eliminate tumors on multiple fronts. Several studies have highlighted the potential of combined treatment approaches as more effective therapeutic strategies than a single treatment for cancer. Hence, designing novel combinations of drugs that trigger glutamatergic system components, taking advantage of those with fewer side effects that do not penetrate the blood-brain barrier seems to be feasible. The efficacy and safety of combining drugs that include glutamatergic system components for clinical use requires further research that considers the expression pattern of the elements of the glutamatergic system (Figure 4).
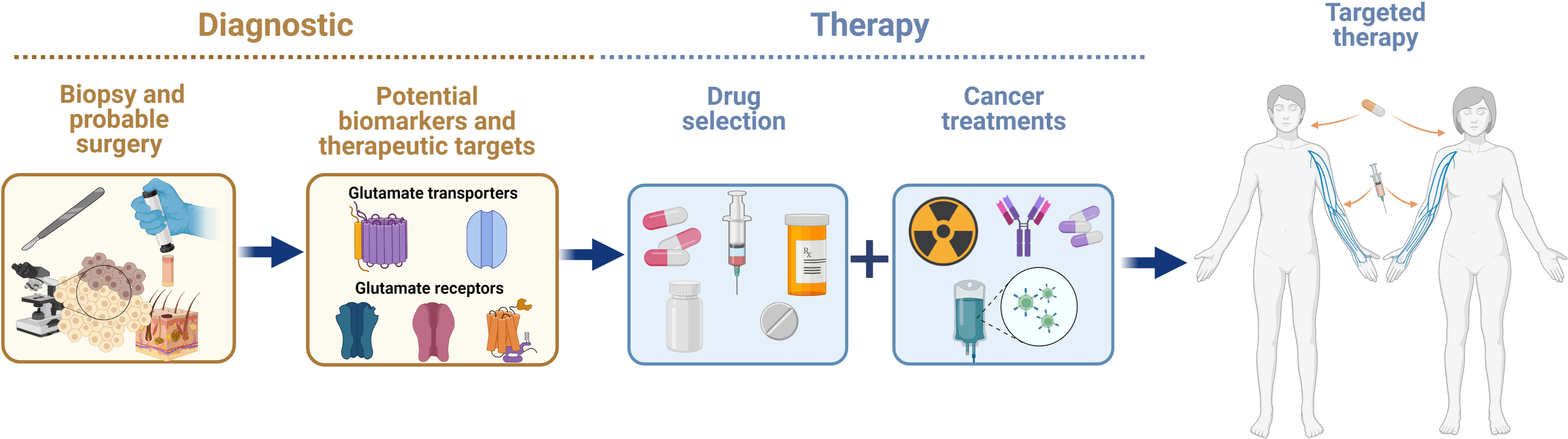
Figure 4 Advances in knowledge on the expression and actions of glutamatergic system components in cancer in non-neural organs have allowed researchers to consider them as potential biomarkers and therapeutic targets for alleviating the effects of cancer therapy.
IM provided critical advice during the writing. MD-M. critically revised the content. AH-A performed the creative design of the figures. All authors contributed to the writing of the article and approved the submitted version.
This work was supported by CONACyT, México (239250 for IM), PAPIIT-DGAPA, UNAM México (IN206418 and IN222821 for IM) (IN202121 for MD-M).
We thank Jessica Norris for critically editing the manuscript. AG-G is a Doctoral student from the Programa de Posgrado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM). AH-A is a Master student from Programa de Posgrado en Ciencias Bioquímicas, Universidad Nacional Autónoma de México (UNAM). Both students received fellowships from Mexico’s National Council of Science and Technology (CONACyT) (AH-A, scholarship 1084428).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Koga T, Naraoka H. A new family of extraterrestrial amino acids in the Murchison meteorite. Sci Rep (2017) 71:636. doi: 10.1038/s41598-017-00693-9
2. Waddell TG, Miller TJ. Chemical evolution of the citric acid cycle: Sunlight photolysis of the amino acids glutamate and aspartate. Orig Life Evol Biosph (1991) 21(4):219–23. doi: 10.1007/BF01809857
3. Young VR, Ajami AM. Glutamate: An amino acid of particular distinction. J Nutr (2000) 130(4S Suppl):892S–900S. doi: 10.1093/jn/130.4.892S
4. Kvamme E. Synthesis of glutamate and its regulation. Prog Brain Res (1998) 116:73–85. doi: 10.1016/s0079-6123(08)60431-8
5. van Straaten HW, He Y, van Duist MM, Labruyere WT, Vermeulen JL, van Dijk PJ, et al. Cellular concentrations of glutamine synthetase in murine organs. Biochem Cell Biol (2006) 84(2):215–31. doi: 10.1139/o05-170
6. Magnusson I, Schumann WC, Bartsch GE, Chandramouli V, Kumaran K, Wahren J, et al. Noninvasive tracing of Krebs cycle metabolism in liver. J Biol Chem (1991) 266(11):6975–84. doi: 10.1016/S0021-9258(20)89598-2
7. Spanaki C, Kotzamani D, Plaitakis A. Widening spectrum of cellular and subcellular expression of human Glud1 and Glud2 glutamate dehydrogenases suggests novel functions. Neurochem Res (2017) 42(1):92–107. doi: 10.1007/s11064-016-1986-x
8. Royes LF, Gabbi P, Ribeiro LR, Della-Pace ID, Rodrigues FS, de Oliveira Ferreira AP, et al. A neuronal disruption in redox homeostasis elicited by ammonia alters the Glycine/Glutamate (Gaba) cycle and contributes to mma-induced excitability. Amino Acids (2016) 48(6):1373–89. doi: 10.1007/s00726-015-2164-1
9. Brosnan ME, Brosnan JT. Hepatic glutamate metabolism: A tale of 2 hepatocytes. Am J Clin Nutr (2009) 90(3):857S–61S. doi: 10.3945/ajcn.2009.27462Z
10. Schousboe A, Scafidi S, Bak LK, Waagepetersen HS, McKenna MC. Glutamate metabolism in the brain focusing on astrocytes. Adv Neurobiol (2014) 11:13–30. doi: 10.1007/978-3-319-08894-5_2
11. Lajtha A. Basic neurochemistry: Molecular, cellular. Neurochem Res (1999) 247:973. doi: 10.1023/A:1020966216216
12. Cooper AJ, Jeitner TM. Central role of glutamate metabolism in the maintenance of nitrogen homeostasis in normal and hyperammonemic brain. Biomolecules (2016) 6(2): 1–33. doi: 10.3390/biom6020016
13. Brosnan JT. Glutamate, at the interface between amino acid and carbohydrate metabolism. J Nutr (2000) 130(4S Suppl):988S–90S. doi: 10.1093/jn/130.4.988S
14. Hertz L, Kala G. Energy metabolism in brain cells: Effects of elevated ammonia concentrations. Metab Brain Dis (2007) 22(3-4):199–218. doi: 10.1007/s11011-007-9068-z
16. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discovery (2022) 12(1):31–46. doi: 10.1158/2159-8290.Cd-21-1059
17. Stine ZE, Dang CV. Stress eating and tuning out: Cancer cells re-wire metabolism to counter stress. Crit Rev Biochem Mol Biol (2013) 48(6):609–19. doi: 10.3109/10409238.2013.844093
18. Icard P, Wu Z, Fournel L, Coquerel A, Lincet H, Alifano M. Atp citrate lyase: A central metabolic enzyme in cancer. Cancer Lett (2020) 471:125–34. doi: 10.1016/j.canlet.2019.12.010
19. Smith B, Schafer XL, Ambeskovic A, Spencer CM, Land H, Munger J. Addiction to coupling of the warburg effect with glutamine catabolism in cancer cells. Cell Rep (2016) 17(3):821–36. doi: 10.1016/j.celrep.2016.09.045
20. Cooper AJ. 13n as a tracer for studying glutamate metabolism. Neurochem Int (2011) 59(4):456–64. doi: 10.1016/j.neuint.2010.11.011
21. Willard SS, Koochekpour S. Glutamate, glutamate receptors, and downstream signaling pathways. Int J Biol Sci (2013) 9(9):948–59. doi: 10.7150/ijbs.6426
22. Namkoong J, Shin SS, Lee HJ, Marín YE, Wall BA, Goydos JS, et al. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res (2007) 67(5):2298–305. doi: 10.1158/0008-5472.Can-06-3665
23. Herner A, Sauliunaite D, Michalski CW, Erkan M, Oliveira TD, Abiatari I, et al. Glutamate increases pancreatic cancer cell invasion and migration Via ampa receptor activation and kras-mapk signaling. Int J Cancer (2011) 129(10):2349–59. doi: 10.1002/ijc.25898
24. Sprengel R. Ionotropic glutamate receptors. In: Pfaff DW, editor. Neuroscience in the 21st century: From basic to clinical. New York, NY: Springer New York (2013). p. 59–80.
25. Hawkins LM, Chazot PL, Stephenson FA. Biochemical evidence for the Co-association of three n-Methyl-D-Aspartate (Nmda) R2 subunits in recombinant nmda receptors*. J Biol Chem (1999) 274(38):27211–18. doi: 10.1074/jbc.274.38.27211
26. Dhingra S, Yadav J, Kumar J. Structure, function, and regulation of the kainate receptor. Subcell Biochem (2022) 99:317–50. doi: 10.1007/978-3-031-00793-4_10
27. Sumi T, Harada K. Mechanism underlying hippocampal long-term potentiation and depression based on competition between endocytosis and exocytosis of ampa receptors. Sci Rep (2020) 10(1):14711. doi: 10.1038/s41598-020-71528-3
28. Twomey EC, Sobolevsky AI. Structural mechanisms of gating in ionotropic glutamate receptors. Biochemistry (2018) 57(3):267–76. doi: 10.1021/acs.biochem.7b00891
29. MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. Nmda-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature (1986) 321(6069):519–22. doi: 10.1038/321519a0
30. Hou H, Wang L, Fu T, Papasergi M, Yule DI, Xia H. Magnesium acts as a second messenger in the regulation of nmda receptor-mediated creb signaling in neurons. Mol Neurobiol (2020) 57(6):2539–50. doi: 10.1007/s12035-020-01871-z
31. Clarke RJ, Glasgow NG, Johnson JW. Mechanistic and structural determinants of nmda receptor voltage-dependent gating and slow Mg2+ unblock. J Neurosci (2013) 33(9):4140–50. doi: 10.1523/jneurosci.3712-12.2013
32. Clements JD, Westbrook GL. Activation kinetics reveal the number of glutamate and glycine binding sites on the n-Methyl-D-Aspartate receptor. Neuron (1991) 7(4):605–13. doi: 10.1016/0896-6273(91)90373-8
33. Oliet SH, Mothet JP. Regulation of n-Methyl-D-Aspartate receptors by astrocytic d-serine. Neuroscience (2009) 158(1):275–83. doi: 10.1016/j.neuroscience.2008.01.071
34. Zanetti L, Regoni M, Ratti E, Valtorta F, Sassone J. Presynaptic ampa receptors in health and disease. Cells (2021) 10(9):1–21. doi: 10.3390/cells10092260
35. Monyer H, Seeburg PH, Wisden W. Glutamate-operated channels: Developmentally early and mature forms arise by alternative splicing. Neuron (1991) 6(5):799–810. doi: 10.1016/0896-6273(91)90176-Z
36. Sommer B, Keinänen K, Verdoorn TA, Wisden W, Burnashev N, Herb A, et al. Flip and flop: A cell-specific functional switch in glutamate-operated channels of the cns. Science (1990) 249(4976):1580–5. doi: 10.1126/science.1699275
37. Kamalova A, Nakagawa T. Ampa receptor structure and auxiliary subunits. J Physiol (2021) 599(2):453–69. doi: 10.1113/jp278701
38. Szabo A, Somogyi J, Cauli B, Lambolez B, Somogyi P, Lamsa KP. Calcium-permeable ampa receptors provide a common mechanism for ltp in glutamatergic synapses of distinct hippocampal interneuron types. J Neurosci (2012) 32(19):6511–16. doi: 10.1523/jneurosci.0206-12.2012
39. He X, Wang Y, Zhou G, Yang J, Li J, Li T, et al. A critical role for Γcamkii in decoding nmda signaling to regulate ampa receptors in putative inhibitory interneurons. Neurosci Bull (2022) 38(8):916–26. doi: 10.1007/s12264-022-00840-x
40. Gerber U, Gee CE, Benquet P. Metabotropic glutamate receptors: Intracellular signaling pathways. Curr Opin Pharmacol (2007) 7(1):56–61. doi: 10.1016/j.coph.2006.08.008
41. Ohana L, Barchad O, Parnas I, Parnas H. The metabotropic glutamate G-Protein-Coupled receptors Mglur3 and Mglur1a are voltage-sensitive*. J Biol Chem (2006) 281(34):24204–15. doi: 10.1074/jbc.M513447200
42. De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem (1980) 255(15):7108–17. doi: 10.1016/S0021-9258(20)79672-9
43. Niswender CM, Conn PJ. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol (2010) 501:295–322. doi: 10.1146/annurev.pharmtox.011008.145533
44. Crupi R, Impellizzeri D, Cuzzocrea S. Role of metabotropic glutamate receptors in neurological disorders. Front Molec Neurosci (2019) 12:20. doi: 10.3389/fnmol.2019.00020
45. Li SH, Abd-Elrahman KS, Ferguson SSG. Targeting Mglur2/3 for treatment of neurodegenerative and neuropsychiatric diseases. Pharmacol Ther (2022) 239:108275. doi: 10.1016/j.pharmthera.2022.108275
46. Fuxe K, Marcellino D, Rivera A, Diaz-Cabiale Z, Filip M, Gago B, et al. Receptor-receptor interactions within receptor mosaics. impact on neuropsychopharmacology. Brain Res Rev (2008) 58(2):415–52. doi: 10.1016/j.brainresrev.2007.11.007
47. Eddy K, Eddin MN, Fateeva A, Pompili SVB, Shah R, Doshi S, et al. Implications of a neuronal receptor family, metabotropic glutamate receptors, in cancer development and progression. Cells (2022) 11(18):1–21. doi: 10.3390/cells11182857
48. Chavis P, Shinozaki H, Bockaert J, Fagni L. The metabotropic glutamate receptor types 2/3 inhibit l-type calcium channels Via a pertussis toxin-sensitive G-protein in cultured cerebellar granule cells. J Neurosci (1994) 14(11 Pt 2):7067–76. doi: 10.1523/jneurosci.14-11-07067.1994
49. Choi S, Lovinger DM. Metabotropic glutamate receptor modulation of voltage-gated Ca2+ channels involves multiple receptor subtypes in cortical neurons. J Neurosci (1996) 16(1):36–45. doi: 10.1523/JNEUROSCI.16-01-00036.1996
50. Saugstad JA, Segerson TP, Westbrook GL. Metabotropic glutamate receptors activate G-Protein-Coupled inwardly rectifying potassium channels in xenopus oocytes. J Neurosci (1996) 16(19):5979–85. doi: 10.1523/JNEUROSCI.16-19-05979.1996
51. Iacovelli L, Bruno V, Salvatore L, Melchiorri D, Gradini R, Caricasole A, et al. Native group-iii metabotropic glutamate receptors are coupled to the mitogen-activated protein Kinase/Phosphatidylinositol-3-Kinase pathways. J Neurochem (2002) 82(2):216–23. doi: 10.1046/j.1471-4159.2002.00929.x
52. Pissimissis N, Papageorgiou E, Lembessis P, Armakolas A, Koutsilieris M. The glutamatergic system expression in human pc-3 and lncap prostate cancer cells. Anticancer Res (2009) 29(1):371–7.
53. Stepulak A, Luksch H, Gebhardt C, Uckermann O, Marzahn J, Sifringer M, et al. Expression of glutamate receptor subunits in human cancers. Histochem Cell Biol (2009) 132(4):435–45. doi: 10.1007/s00418-009-0613-1
54. Abdul M, Hoosein N. N-Methyl-D-Aspartate receptor in human prostate cancer. J Membr Biol (2005) 205(3):125–8. doi: 10.1007/s00232-005-0777-0
55. Tamura H, Suzuki M, Moriya Y, Hoshino H, Okamoto T, Yoshida S, et al. Aberrant methylation of n-Methyl-D-Aspartate receptor type 2b (Nmdar2b) in non-small cell carcinoma. BMC Cancer (2011) 11:220. doi: 10.1186/1471-2407-11-220
56. Stepanov YV, Golovynska I, Dziubenko NV, Kuznietsova HM, Petriv N, Skrypkina I, et al. Nmda receptor expression during cell transformation process at early stages of liver cancer in rodent models. Am J Physiol Gastrointest Liver Physiol (2022) 322(1):G142–g53. doi: 10.1152/ajpgi.00060.2021
57. Hu H, Takano N, Xiang L, Gilkes DM, Luo W, Semenza GL. Hypoxia-inducible factors enhance glutamate signaling in cancer cells. Oncotarget (2014) 5(19):8853–68. doi: 10.18632/oncotarget.2593
58. Prickett TD, Wei X, Cardenas-Navia I, Teer JK, Lin JC, Walia V, et al. Exon capture analysis of G protein-coupled receptors identifies activating mutations in Grm3 in melanoma. Nat Genet (2011) 43(11):1119–26. doi: 10.1038/ng.950
59. Chang HJ, Yoo BC, Lim SB, Jeong SY, Kim WH, Park JG. Metabotropic glutamate receptor 4 expression in colorectal carcinoma and its prognostic significance. Clin Cancer Res (2005) 11(9):3288–95. doi: 10.1158/1078-0432.Ccr-04-1912
60. Dornier E, Rabas N, Mitchell L, Novo D, Dhayade S, Marco S, et al. Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat Commun (2017) 8:14. doi: 10.1038/s41467-017-02101-2
61. Choi KY, Chang K, Pickel JM, Badger JD 2nd, Roche KW. Expression of the metabotropic glutamate receptor 5 (Mglur5) induces melanoma in transgenic mice. Proc Natl Acad Sci U.S.A. (2011) 108(37):15219–24. doi: 10.1073/pnas.1107304108
62. Cheng MF, Huang YY, Ho BY, Kuo TC, Hsin LW, Shiue CY, et al. Prospective comparison of (4s)-4-(3-(18)F-Fluoropropyl)-L-Glutamate versus (18)F-fluorodeoxyglucose Pet/Ct for detecting metastases from pancreatic ductal adenocarcinoma: A proof-of-Concept study. Eur J Nucl Med Mol Imaging (2019) 46(4):810–20. doi: 10.1007/s00259-018-4251-5
63. Pacheco R, Oliva H, Martinez-Navío JM, Climent N, Ciruela F, Gatell JM, et al. Glutamate released by dendritic cells as a novel modulator of T cell activation. J Immunol (2006) 17710:6695–704. doi: 10.4049/jimmunol.177.10.6695
64. Galván I, Inácio Â, Dañino M, Corbí-Llopis R, Monserrat MT, Bernabeu-Wittel J. High Slc7a11 expression in normal skin of melanoma patients. Cancer Epidemiol (2019) 62:101582. doi: 10.1016/j.canep.2019.101582
65. Hinoi E, Takarada T, Uno K, Inoue M, Murafuji Y, Yoneda Y. Glutamate suppresses osteoclastogenesis through the Cystine/Glutamate antiporter. Am J Pathol (2007) 170(4):1277–90. doi: 10.2353/ajpath.2007.061039
66. Lee A, Anderson AR, Stevens M, Beasley S, Barnett NL, Pow DV. Excitatory amino acid transporter 5 is widely expressed in peripheral tissues. Eur J Histochem (2013) 57(1):e11–1. doi: 10.4081/ejh.2013.e11
67. Najimi M, Stéphenne X, Sempoux C, Sokal E. Regulation of hepatic eaat-2 glutamate transporter expression in human liver cholestasis. World J Gastroenterol (2014) 20(6):1554–64. doi: 10.3748/wjg.v20.i6.1554
68. Ji X, Qian J, Rahman SMJ, Siska PJ, Zou Y, Harris BK, et al. Xct (Slc7a11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene (2018) 37(36):5007–19. doi: 10.1038/s41388-018-0307-z
69. Lim JKM, Delaidelli A, Minaker SW, Zhang HF, Colovic M, Yang H, et al. Cystine/Glutamate antiporter xct (Slc7a11) facilitates oncogenic ras transformation by preserving intracellular redox balance. Proc Natl Acad Sci U.S.A. (2019) 116(19):9433–42. doi: 10.1073/pnas.1821323116
70. Tang B, Zhu J, Liu F, Ding J, Wang Y, Fang S, et al. Xct contributes to colorectal cancer tumorigenesis through upregulation of the melk oncogene and activation of the Akt/Mtor cascade. Cell Death Dis (2022) 13(4):373. doi: 10.1038/s41419-022-04827-4
71. North WG, Gao G, Jensen A, Memoli VA, Du J. Nmda receptors are expressed by small-cell lung cancer and are potential targets for effective treatment. Clin Pharmacol (2010) 2:31–40. doi: 10.2147/cpaa.S6262
72. Ganor Y, Besser M, Ben-Zakay N, Unger T, Levite M. Human T cells express a functional ionotropic glutamate receptor Glur3, and glutamate by itself triggers integrin-mediated adhesion to laminin and fibronectin and chemotactic migration. J Immunol (2003) 170(8):4362–72. doi: 10.4049/jimmunol.170.8.4362
73. Chiocchetti A, Miglio G, Mesturini R, Varsaldi F, Mocellin M, Orilieri E, et al. Group I mglu receptor stimulation inhibits activation-induced cell death of human T lymphocytes. Br J Pharmacol (2006) 148(6):760–8. doi: 10.1038/sj.bjp.0706746
74. Speyer CL, Hachem AH, Assi AA, Johnson JS, DeVries JA, Gorski DH. Metabotropic glutamate receptor-1 as a novel target for the antiangiogenic treatment of breast cancer. PloS One (2014) 9(3):e88830. doi: 10.1371/journal.pone.0088830
75. Wu YL, Wang NN, Gu L, Yang HM, Xia N, Zhang H. The suppressive effect of metabotropic glutamate receptor 5 (Mglu5) inhibition on hepatocarcinogenesis. Biochimie (2012) 94(11):2366–75. doi: 10.1016/j.biochi.2012.06.006
76. Yang H-M, Hou T-Z, Zhang Y-N, Zhao S-D, Wu Y-L, Zhang H. Blocked metabotropic glutamate receptor 5 enhances chemosensitivity in hepatocellular carcinoma and attenuates chemotoxicity in the normal liver by regulating DNA damage. Cancer Gene Ther (2022) 29:1487–501. doi: 10.1038/s41417-022-00465-2
77. Kinoshita H, Okabe H, Beppu T, Chikamoto A, Hayashi H, Imai K, et al. Cystine/Glutamic acid transporter is a novel marker for predicting poor survival in patients with hepatocellular carcinoma. Oncol Rep (2013) 29(2):685–9. doi: 10.3892/or.2012.2162
78. Lo M, Ling V, Wang YZ, Gout PW. The xc- Cystine/Glutamate antiporter: A mediator of pancreatic cancer growth with a role in drug resistance. Br J Cancer (2008) 99(3):464–72. doi: 10.1038/sj.bjc.6604485
79. Gill SS, Mueller RW, McGuire PF, Pulido OM. Potential target sites in peripheral tissues for excitatory neurotransmission and excitotoxicity. Toxicol Pathol (2000) 28(2):277–84. doi: 10.1177/019262330002800207
80. Inagaki N, Kuromi H, Gonoi T, Okamoto Y, Ishida H, Seino Y, et al. Expression and role of ionotropic glutamate receptors in pancreatic islet cells. FASEB J (1995) 9(8):686–91. doi: 10.1096/fasebj.9.8.7768362
81. Brice NL, Varadi A, Ashcroft SJH, Molnar E. Metabotropic glutamate and gabab receptors contribute to the modulation of glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia (2002) 45(2):242–52. doi: 10.1007/s00125-001-0750-0
82. Gill SS, Pulido OM. Glutamate receptors in peripheral tissues: Current knowledge, future research, and implications for toxicology. Toxicol Pathol (2001) 29(2):208–23. doi: 10.1080/019262301317052486
83. Schwendt M, Jezová D. Gene expression of nmda receptor subunits in rat adrenals under basal and stress conditions. J Physiol Pharmacol (2001) 52(4 Pt 2):719–27.
84. Liu H-P, Tay SS-W, Leong S-K. Localization of glutamate receptor subunits of the A-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionate (Ampa) type in the pancreas of newborn Guinea pigs hai-ping liu. Pancreas (1997) 14(4):360–68. doi: 10.1097/00006676-199705000-00006
85. Fan Y-N, Li C, Huang L, Chen L, Tang Z, Han G, et al. Characterization of group I metabotropic glutamate receptors in rat and human adrenal glands. Front Physiol (2020) 11:401. doi: 10.3389/fphys.2020.00401
86. Storto M, de Grazia U, Knöpfel T, Canonico PL, Copani A, Richelmi P, et al. Selective blockade of Mglu5 metabotropic glutamate receptors protects rat hepatocytes against hypoxic damage. Hepatology (2000) 31(3):649–55. doi: 10.1002/hep.510310315
87. Golovynska I, Beregova TV, Falalyeyeva TM, Stepanova LI, Golovynskyi S, Qu J, et al. Peripheral n-Methyl-D-Aspartate receptor localization and role in gastric acid secretion regulation: Immunofluorescence and pharmacological studies. Sci Rep (2018) 8(1):7445. doi: 10.1038/s41598-018-25753-6
88. Nakamura E, Hasumura M, San Gabriel A, Uneyama H, Torii K. New frontiers in gut nutrient sensor research: Luminal glutamate-sensing cells in rat gastric mucosa. J Pharmacol Sci (2010) 112(1):13–8. doi: 10.1254/jphs.09r16fm
89. Berger UV, Hediger MA. Distribution of the glutamate transporters glt-1 (Slc1a2) and glast (Slc1a3) in peripheral organs. Anat Embryol (Berl) (2006) 211(6):595–606. doi: 10.1007/s00429-006-0109-x
90. Hu QX, Ottestad-Hansen S, Holmseth S, Hassel B, Danbolt NC, Zhou Y. Expression of glutamate transporters in mouse liver, kidney, and intestine. J Histochem Cytochem (2018) 66(3):189–202. doi: 10.1369/0022155417749828
91. Zhang W, Zhao D, Qi Q, Long X, Li Y, Wang P, et al. Nr2b-containing nmda receptors contribute to diarrhea-predominant irritable bowel syndrome. Oncotarget (2018) 5:s456–s68.
92. Julio-Pieper M, Hyland NP, Bravo JA, Dinan TG, Cryan JF. A novel role for the metabotropic glutamate receptor-7: Modulation of faecal water content and colonic electrolyte transport in the mouse. Br J Pharmacol (2010) 160(2):367–75. doi: 10.1111/j.1476-5381.2010.00713.x
93. Genever PG, Maxfield SJ, Kennovin GD, Maltman J, Bowgen CJ, Raxworthy MJ, et al. Evidence for a novel glutamate-mediated signaling pathway in keratinocytes. J Invest Dermatol (1999) 1123:337–42. doi: 10.1046/j.1523-1747.1999.00509.x
94. Franklin RB, Zou J, Yu Z, Costello LC. Eaac1 is expressed in rat and human prostate epithelial cells; functions as a high-affinity l-aspartate transporter; and is regulated by prolactin and testosterone. BMC Biochem (2006) 7:10. doi: 10.1186/1471-2091-7-10
95. Mashkina AP, Tyulina OV, Solovyova TI, Kovalenko EI, Kanevski LM, Johnson P, et al. The excitotoxic effect of nmda on human lymphocyte immune function. Neurochem Int (2007) 51(6-7):356–60. doi: 10.1016/j.neuint.2007.04.009
96. Miglio G, Varsaldi F, Lombardi G. Human T lymphocytes express n-Methyl-D-Aspartate receptors functionally active in controlling T cell activation. Biochem Biophys Res Commun (2005) 338(4):1875–83. doi: 10.1016/j.bbrc.2005.10.164
97. Sturgill JL, Mathews J, Scherle P, Conrad DH. Glutamate signaling through the kainate receptor enhances human immunoglobulin production. J Neuroimmunol (2011) 233(1-2):80–9. doi: 10.1016/j.jneuroim.2010.11.014
98. Liao S, Ruiz Y, Gulzar H, Yelskaya Z, Ait Taouit L, Houssou M, et al. Osteosarcoma cell proliferation and survival requires Mglur5 receptor activity and is blocked by riluzole. PloS One (2017) 12(2):e0171256. doi: 10.1371/journal.pone.0171256
99. Patton AJ, Genever PG, Birch MA, Suva LJ, Skerry TM. Expression of an n-Methyl-D-Aspartate-Type receptor by human and rat osteoblasts and osteoclasts suggests a novel glutamate signaling pathway in bone. Bone (1998) 22(6):645–9. doi: 10.1016/s8756-3282(98)00061-1
100. Itzstein C, Cheynel H, Burt-Pichat B, Merle B, Espinosa L, Delmas PD, et al. Molecular identification of nmda glutamate receptors expressed in bone cells. J Cell Biochem (2001) 82(1):134–44. doi: 10.1002/jcb.1114
101. Hinoi E, Fujimori S, Takemori A, Kurabayashi H, Nakamura Y, Yoneda Y. Demonstration of expression of mrna for particular ampa and kainate receptor subunits in immature and mature cultured rat calvarial osteoblasts. Brain Res (2002) 943(1):112–6. doi: 10.1016/s0006-8993(02)02726-9
102. Szczesniak AM, Gilbert RW, Mukhida M, Anderson GI. Mechanical loading modulates glutamate receptor subunit expression in bone. Bone (2005) 37(1):63–73. doi: 10.1016/j.bone.2003.10.016
103. Hinoi E, Fujimori S, Nakamura Y, Yoneda Y. Group iii metabotropic glutamate receptors in rat cultured calvarial osteoblasts. Biochem Biophys Res Commun (2001) 281(2):341–6. doi: 10.1006/bbrc.2001.4355
104. Leung JC, Marphis T, Craver RD, Silverstein DM. Altered nmda receptor expression in renal toxicity: Protection with a receptor antagonist. Kidney Int (2004) 66(1):167–76. doi: 10.1111/j.1523-1755.2004.00718.x
105. Burdo J, Dargusch R, Schubert D. Distribution of the Cystine/Glutamate antiporter system X–c in the brain, kidney, and duodenum. J Histochem Cytochem (2006) 54(5):549–57. doi: 10.1369/jhc.5A6840.2006
106. North WG, Liu FL, Lin LZ, Tian RY, Akerman B. Nmda receptors are important regulators of pancreatic cancer and are potential targets for treatment. Clin Pharmacol-Adv Appl (2017) 9:79–85. doi: 10.2147/cpaa.s140057
107. Ye Y, Li Y, Wei Y, Xu Y, Wang R, Fu Z, et al. Anticancer effect of hottip regulates human pancreatic cancer Via the metabotropic glutamate receptor 1 pathway. Oncol Lett (2018) 16(2):1937–42. doi: 10.3892/ol.2018.8870
108. Yamaguchi F, Hirata Y, Akram H, Kamitori K, Dong Y, Sui L, et al. Foxo/Txnip pathway is involved in the suppression of hepatocellular carcinoma growth by glutamate antagonist mk-801. BMC Cancer (2013) 13:468. doi: 10.1186/1471-2407-13-468
109. Felizola SJA, Nakamura Y, Satoh F, Morimoto R, Kikuchi K, Nakamura T, et al. Glutamate receptors and the regulation of steroidogenesis in the human adrenal gland: The metabotropic pathway. Mol Cell Endocrinol (2014) 382(1):170–77. doi: 10.1016/j.mce.2013.09.025
110. Kim HM, Koo JS. Expression of glutamine metabolism-related and amino acid transporter proteins in adrenal cortical neoplasms and pheochromocytomas. Dis Markers (2021) 2021:8850990. doi: 10.1155/2021/8850990
111. Watanabe K, Kanno T, Oshima T, Miwa H, Tashiro C, Nishizaki T. The nmda receptor Nr2a subunit regulates proliferation of Mkn45 human gastric cancer cells. Biochem Biophys Res Commun (2008) 367(2):487–90. doi: 10.1016/j.bbrc.2007.12.167
112. Wu CS, Lu YJ, Li HP, Hsueh C, Lu CY, Leu YW, et al. Glutamate receptor, ionotropic, kainate 2 silencing by DNA hypermethylation possesses tumor suppressor function in gastric cancer. Int J Cancer (2010) 126(11):2542–52. doi: 10.1002/ijc.24958
113. Guan Z, Chen J, Li X, Dong N. Tanshinone IIA induces ferroptosis in gastric cancer cells through P53-mediated Slc7a11 down-regulation. Biosci Rep (2020) 40(8):1–11. doi: 10.1042/bsr20201807
114. Hu J, Gu W, Ma N, Fan X, Ci X. Leonurine alleviates ferroptosis in cisplatin-induced acute kidney injury by activating the Nrf2 signalling pathway. Br J Pharmacol (2022) 179(15):3991–4009. doi: 10.1111/bph.15834
115. Yan Z, Li P, Xue Y, Tian H, Zhou T, Zhang G. Glutamate receptor, ionotropic, N−Methyl D−Aspartate−Associated protein 1 promotes colorectal cancer cell proliferation and metastasis, and is negatively regulated by Mir−296−3p. Mol Med Rep (2021) 24(4):1–11. doi: 10.3892/mmr.2021.12339
116. Biolley C, Tretola M, Bee G, Jud C, Silacci P. Punicalagin increases glutamate absorption in differentiated caco-2 cells by a mechanism involving gene expression regulation of an Eaat3 transporter. Food Funct (2019) 10(9):5333–38. doi: 10.1039/c9fo00191c
117. Ungard RG, Linher-Melville K, Nashed MG, Sharma M, Wen J, Singh G. Xct knockdown in human breast cancer cells delays onset of cancer-induced bone pain. Mol Pain (2019) 15: 1–14. doi: 10.1177/1744806918822185
118. Kang M, Cho JH, Koo JK, Noh SU, Kim MY, Kang H, et al. The expression of nmda receptor 1 correlates with clinicopathological parameters in cutaneous squamous cell carcinoma. Ann Dermatol (2009) 21(4):382–8. doi: 10.5021/ad.2009.21.4.382
119. Prickett TD, Zerlanko BJ, Hill VK, Gartner JJ, Qutob N, Jiang J, et al. Somatic mutation of Grin2a in malignant melanoma results in loss of tumor suppressor activity Via aberrant nmdar complex formation. J Invest Dermatol (2014) 134(9):2390–98. doi: 10.1038/jid.2014.190
120. Hajdú T, Juhász T, Szűcs-Somogyi C, Rácz K, Zákány R. Nr1 and Nr3b composed intranuclear n-Methyl-D-Aspartate receptor complexes in human melanoma cells. Int J Mol Sci (2018) 19(7):1–14. doi: 10.3390/ijms19071929
121. Wen Y, Li JD, Koo J, Shin SS, Lin Y, Jeong BS, et al. Activation of the glutamate receptor Grm1 enhances angiogenic signaling to drive melanoma progression. Cancer Res (2014) 74(9):2499–509. doi: 10.1158/0008-5472.can-13-1531
122. Shin SS, Jeong BS, Wall BA, Li J, Shan NL, Wen Y, et al. Participation of xct in melanoma cell proliferation in vitro and tumorigenesis in vivo. Oncogenesis (2018) 7(11):86. doi: 10.1038/s41389-018-0098-7
123. Pacheco R, Ciruela F, Casadó V, Mallol J, Gallart T, Lluis C, et al. Group I metabotropic glutamate receptors mediate a dual role of glutamate in T cell activation. J Biol Chem (2004) 279(32):33352–8. doi: 10.1074/jbc.M401761200
124. Kalariti N, Lembessis P, Koutsilieris M. Characterization of the glutametergic system in mg-63 osteoblast-like osteosarcoma cells. Anticancer Res (2004) 24(6):3923–9.
125. Stepulak A, Rola R, Polberg K, Ikonomidou C. Glutamate and its receptors in cancer. J Neural Transm (2014) 121(8):933–44. doi: 10.1007/s00702-014-1182-6
126. Yi H, Talmon G, Wang J. Glutamate in cancers: From metabolism to signaling. J BioMed Res (2019) 34(4):260–70. doi: 10.7555/jbr.34.20190037
127. Rzeski W, Turski L, Ikonomidou C. Glutamate antagonists limit tumor growth. Proc Natl Acad Sci (2001) 98(11):6372–77. doi: 10.1073/pnas.091113598
128. Stepulak A, Sifringer M, Rzeski W, Endesfelder S, Gratopp A, Pohl EE, et al. Nmda antagonist inhibits the extracellular signal-regulated kinase pathway and suppresses cancer growth. Proc Natl Acad Sci (2005) 102(43):15605–10. doi: 10.1073/pnas.0507679102
129. Nahm WK, Philpot BD, Adams MM, Badiavas EV, Zhou LH, Butmarc J, et al. Significance of n-Methyl-D-Aspartate (Nmda) receptor-mediated signaling in human keratinocytes. J Cell Physiol (2004) 200(2):309–17. doi: 10.1002/jcp.20010
130. Choi SW, Park SY, Hong SP, Pai H, Choi JY, Kim SG. The expression of nmda receptor 1 is associated with clinicopathological parameters and prognosis in the oral squamous cell carcinoma. J Oral Pathol Med (2004) 33(9):533–7. doi: 10.1111/j.1600-0714.2004.00226.x
131. Liu J-W, Kim MS, Nagpal J, Yamashita K, Poeta L, Chang X, et al. Quantitative hypermethylation of Nmdar2b in human gastric cancer. Int J Cancer (2007) 121(9):1994–2000. doi: 10.1002/ijc.22934
132. Kim MS, Yamashita K, Baek JH, Park HL, Carvalho AL, Osada M, et al. N-Methyl-D-Aspartate receptor type 2b is epigenetically inactivated and exhibits tumor-suppressive activity in human esophageal cancer. Cancer Res (2006) 66(7):3409–18. doi: 10.1158/0008-5472.Can-05-1608
133. Limón ID, Angulo-Cruz I, Sánchez-Abdon L, Patricio-Martínez A. Disturbance of the glutamate-glutamine cycle, secondary to hepatic damage, compromises memory function. Front Neurosci (2021) 15:578922. doi: 10.3389/fnins.2021.578922
134. Xia L, Tan S, Zhou Y, Lin J, Wang H, Oyang L, et al. Role of the nfκb-signaling pathway in cancer. Onco Targets Ther (2018) 11:2063–73. doi: 10.2147/ott.S161109
135. Taniguchi K, Karin M. Nf-Kb, inflammation, immunity and cancer: Coming of age. Nat Rev Immunol (2018) 18(5):309–24. doi: 10.1038/nri.2017.142
136. Cohen S, Greenberg ME. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol (2008) 24:183–209. doi: 10.1146/annurev.cellbio.24.110707.175235
137. Sureda F, Copani A, Bruno V, Knöpfel T, Meltzger G, Nicoletti F. Metabotropic glutamate receptor agonists stimulate polyphosphoinositide hydrolysis in primary cultures of rat hepatocytes. Eur J Pharmacol (1997) 338(2):R1–2. doi: 10.1016/S0014-2999(97)81950-4
138. Pollock PM, Cohen-Solal K, Sood R, Namkoong J, Martino JJ, Koganti A, et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat Genet (2003) 34(1):108–12. doi: 10.1038/ng1148
139. Yoo BC, Jeon E, Hong SH, Shin YK, Chang HJ, Park JG. Metabotropic glutamate receptor 4-mediated 5-fluorouracil resistance in a human colon cancer cell line. Clin Cancer Res (2004) 10(12 Pt 1):4176–84. doi: 10.1158/1078-0432.Ccr-1114-03
140. Yi H, Geng L, Black A, Talmon G, Berim L, Wang J. The mir-487b-3p/Grm3/Tgf beta signaling axis is an important regulator of colon cancer tumorigenesis. Oncogene (2017) 36(24):3477–89. doi: 10.1038/onc.2016.499
141. Martino JJ, Wall BA, Mastrantoni E, Wilimczyk BJ, La Cava SN, Degenhardt K, et al. Metabotropic glutamate receptor 1 (Grm1) is an oncogene in epithelial cells. Oncogene (2013) 32(37):4366–76. doi: 10.1038/onc.2012.471
142. Koochekpour S, Majumdar S, Azabdaftari G, Attwood K, Scioneaux R, Subramani D, et al. Serum glutamate levels correlate with Gleason score and glutamate blockade decreases proliferation, migration, and invasion and induces apoptosis in prostate cancer cells. Clin Cancer Res (2012) 18(21):5888–901. doi: 10.1158/1078-0432.CCR-12-1308
143. Banda M, Speyer CL, Semma SN, Osuala KO, Kounalakis N, Torres Torres KE, et al. Metabotropic glutamate receptor-1 contributes to progression in triple negative breast cancer. PloS One (2014) 9(1):e81126. doi: 10.1371/journal.pone.0081126
144. Bastiaansen AEM, Timmermans AM, Smid M, van Deurzen CHM, Hulsenboom ESP, Prager-van der Smissen WJC, et al. Metabotropic glutamate receptor 1 is associated with unfavorable prognosis in er-negative and triple-negative breast cancer. Sci Rep (2020) 10(1):22292. doi: 10.1038/s41598-020-79248-4
145. Ali S, Shourideh M, Koochekpour S. Identification of novel Grm1 mutations and single nucleotide polymorphisms in prostate cancer cell lines and tissues. PloS One (2014) 9(7):e103204. doi: 10.1371/journal.pone.0103204
146. Esseltine JL, Willard MD, Wulur IH, Lajiness ME, Barber TD, Ferguson SS. Somatic mutations in Grm1 in cancer alter metabotropic glutamate receptor 1 intracellular localization and signaling. Mol Pharmacol (2013) 83(4):770–80. doi: 10.1124/mol.112.081695
147. Zhou Y, Danbolt NC. Glutamate as a neurotransmitter in the healthy brain. J Neural Transm (Vienna) (2014) 121(8):799–817. doi: 10.1007/s00702-014-1180-8
148. Hori T, Takamori S. Physiological perspectives on molecular mechanisms and regulation of vesicular glutamate transport: Lessons from calyx of held synapses. Front Cell Neurosci (2021) 15:811892. doi: 10.3389/fncel.2021.811892
149. Uehara S, Jung SK, Morimoto R, Arioka S, Miyaji T, Juge N, et al. Vesicular storage and secretion of l-glutamate from glucagon-like peptide 1-secreting clonal intestinal l cells. J Neurochem (2006) 96(2):550–60. doi: 10.1111/j.1471-4159.2005.03575.x
150. Morimoto R, Uehara S, Yatsushiro S, Juge N, Hua Z, Senoh S, et al. Secretion of l-glutamate from osteoclasts through transcytosis. EMBO J (2006) 25(18):4175–86. doi: 10.1038/sj.emboj.7601317
151. Cabrera O, Jacques-Silva MC, Speier S, Yang SN, Köhler M, Fachado A, et al. Glutamate is a positive autocrine signal for glucagon release. Cell Metab (2008) 7(6):545–54. doi: 10.1016/j.cmet.2008.03.004
152. Hayashi M, Morimoto R, Yamamoto A, Moriyama Y. Expression and localization of vesicular glutamate transporters in pancreatic islets, upper gastrointestinal tract, and testis. J Histochem Cytochem (2003) 51(10):1375–90. doi: 10.1177/002215540305101014
153. Tremolizzo L, DiFrancesco JC, Rodriguez-Menendez V, Sirtori E, Longoni M, Cassetti A, et al. Human platelets express the synaptic markers Vglut1 and 2 and release glutamate following aggregation. Neurosci Lett (2006) 404(3):262–5. doi: 10.1016/j.neulet.2006.06.015
154. Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, et al. A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci (2002) 22(13):5442–51. doi: 10.1523/jneurosci.22-13-05442.2002
155. Teng L, Lei H-M, Sun F, An S-M, Tang Y-B, Meng S, et al. Autocrine glutamatergic transmission for the regulation of embryonal carcinoma stem cells. Oncotarget (2016) 7(31):49552–64. doi: 10.18632/oncotarget.9973
156. Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J Neurosci (2003) 23(9):3588–96. doi: 10.1523/jneurosci.23-09-03588.2003
157. Featherstone DE, Shippy SA. Regulation of synaptic transmission by ambient extracellular glutamate. Neuroscientist (2008) 14(2):171–81. doi: 10.1177/1073858407308518
158. Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem (1986) 261(5):2256–63. doi: 10.1016/S0021-9258(17)35926-4
159. Bentea E, De Pauw L, Verbruggen L, Winfrey LC, Deneyer L, Moore C, et al. Aged xct-deficient mice are less susceptible for lactacystin-, but not 1-Methyl-4-Phenyl-1,2,3,6- tetrahydropyridine-, induced degeneration of the nigrostriatal pathway. Front Cell Neurosci (2021) 15:796635. doi: 10.3389/fncel.2021.796635
160. He Y, Hewett SJ. The Cystine/Glutamate antiporter, system X(C) (-), contributes to cortical infarction after moderate but not severe focal cerebral ischemia in mice. Front Cell Neurosci (2022) 16:821036. doi: 10.3389/fncel.2022.821036
161. Bridges RJ, Natale NR, Patel SA. System Xc? Cystine/Glutamate antiporter: An update on molecular pharmacology and roles within the cns. Br J Pharmacol (2012) 165(1):20–34. doi: 10.1111/j.1476-5381.2011.01480.x
162. Rodríguez-Campuzano AG, Ortega A. Glutamate transporters: Critical components of glutamatergic transmission. Neuropharmacology (2021) 192:108602. doi: 10.1016/j.neuropharm.2021.108602
163. Featherstone DE. Intercellular glutamate signaling in the nervous system and beyond. ACS Chem Neurosci (2010) 11:4–12. doi: 10.1021/cn900006n
164. Beckers P, Lara O, Belo do Nascimento I, Desmet N, Massie A, Hermans E. Validation of a system X(C) (-) functional assay in cultured astrocytes and nervous tissue samples. Front Cell Neurosci (2021) 15:815771. doi: 10.3389/fncel.2021.815771
165. Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem (2000) 275(21):16023–9. doi: 10.1074/jbc.275.21.16023
166. Sato H, Nomura S, Maebara K, Sato K, Tamba M, Bannai S. Transcriptional control of Cystine/Glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun (2004) 325(1):109–16. doi: 10.1016/j.bbrc.2004.10.009
167. Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, et al. Electrophile response element-mediated induction of the Cystine/Glutamate exchange transporter gene expression. J Biol Chem (2002) 277(47):44765–71. doi: 10.1074/jbc.M208704200
168. Fogal B, Li J, Lobner D, McCullough LD, Hewett SJ. System X(C)- activity and astrocytes are necessary for interleukin-1 beta-mediated hypoxic neuronal injury. J Neurosci (2007) 27(38):10094–105. doi: 10.1523/JNEUROSCI.2459-07.2007
169. Vaidya FU, Chhipa AS, Sagar N, Pathak C. Oxidative stress and inflammation can fuel cancer. In: Maurya PK, Dua K, editors. Role of oxidative stress in pathophysiology of diseases. Singapore: Springer Singapore (2020). p. 229–58.
170. Danbolt NC. Glutamate uptake. Prog Neurobiol (2001) 65(1):1–105. doi: 10.1016/S0301-0082(00)00067-8
171. Rimmele TS, Li S, Andersen JV, Westi EW, Rotenberg A, Wang J, et al. Neuronal loss of the glutamate transporter glt-1 promotes excitotoxic injury in the hippocampus. Front Cell Neurosci (2021) 15:788262. doi: 10.3389/fncel.2021.788262
172. Andersen JV, Markussen KH, Jakobsen E, Schousboe A, Waagepetersen HS, Rosenberg PA, et al. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology (2021) 196:108719. doi: 10.1016/j.neuropharm.2021.108719
173. Tanaka K. Functions of glutamate transporters in the brain. Neurosci Res (2000) 37(1):15–9. doi: 10.1016/s0168-0102(00)00104-8
174. Wu Q, Akhter A, Pant S, Cho E, Zhu JX, Garner A, et al. Ataxia-linked Slc1a3 mutations alter Eaat1 chloride channel activity and glial regulation of cns function. J Clin Invest (2022) 132(7):1–14. doi: 10.1172/JCI154891
175. Koch HP, Brown RL, Larsson HP. The glutamate-activated anion conductance in excitatory amino acid transporters is gated independently by the individual subunits. J Neurosci (2007) 27(11):2943–7. doi: 10.1523/jneurosci.0118-07.2007
176. Yeung JHY, Palpagama TH, Wood OWG, Turner C, Waldvogel HJ, Faull RLM, et al. Eaat2 expression in the hippocampus, subiculum, entorhinal cortex and superior temporal gyrus in alzheimer's disease. Front Cell Neurosci (2021) 15:702824. doi: 10.3389/fncel.2021.702824
177. Zou Z, Tao T, Li H, Zhu X. Mtor signaling pathway and mtor inhibitors in cancer: Progress and challenges. Cell Bioscience (2020) 101:31. doi: 10.1186/s13578-020-00396-1
178. Koglin N, Mueller A, Berndt M, Schmitt-Willich H, Toschi L, Stephens AW, et al. Specific pet imaging of xc- transporter activity using a (1)(8)F-labeled glutamate derivative reveals a dominant pathway in tumor metabolism. Clin Cancer Res (2011) 17(18):6000–11. doi: 10.1158/1078-0432.CCR-11-0687
179. Chen M, Lu S, Zheng H, Xu M, Song J, Yang W, et al. Identification of the potential metabolic pathways involved in the hepatic tumorigenesis of rat diethylnitrosamine-induced hepatocellular carcinoma Via (1)H nmr-based metabolomic analysis. BioMed Res Int (2019) 2019:9367082. doi: 10.1155/2019/9367082
180. Banjac A, Perisic T, Sato H, Seiler A, Bannai S, Weiss N, et al. The Cystine/Cysteine cycle: A redox cycle regulating susceptibility versus resistance to cell death. Oncogene (2008) 27(11):1618–28. doi: 10.1038/sj.onc.1210796
181. Koppula P, Zhuang L, Gan B. Cystine transporter Slc7a11/Xct in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell (2021) 12(8):599–620. doi: 10.1007/s13238-020-00789-5
182. Yang Y, Yee D. Igf-I regulates redox status in breast cancer cells by activating the amino acid transport molecule xc. Cancer Res (2014) 74(8):2295–305. doi: 10.1158/0008-5472.Can-13-1803
183. Song Y, Jang J, Shin T-H, Bae SM, Kim J-S, Kim KM, et al. Sulfasalazine attenuates evading anticancer response of Cd133-positive hepatocellular carcinoma cells. J Exp Clin Cancer Res (2017) 36(1):38. doi: 10.1186/s13046-017-0511-7
184. Arensman MD, Yang XS, Leahy DM, Toral-Barza L, Mileski M, Rosfjord EC, et al. Cystine-glutamate antiporter xct deficiency suppresses tumor growth while preserving antitumor immunity. Proc Natl Acad Sci U.S.A. (2019) 116(19):9533–42. doi: 10.1073/pnas.1814932116
185. Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol (2012) 14(3):276–86. doi: 10.1038/ncb2432
186. Koppula P, Zhang Y, Shi J, Li W, Gan B. The Glutamate/Cystine antiporter Slc7a11/Xct enhances cancer cell dependency on glucose by exporting glutamate. J Biol Chem (2017) 292(34):14240–49. doi: 10.1074/jbc.M117.798405
187. Zhong X, Yao L, Xu L, Ma Q, Huang G, Yang M, et al. Comprehensive analysis of potential correlation between solute carrier 1a (Slc1a) family and lung adenocarcinoma. Int J Gen Med (2022) 15:2101–17. doi: 10.2147/IJGM.S350986
188. Guo W, Li K, Sun B, Xu D, Tong L, Yin H, et al. Dysregulated glutamate transporter Slc1a1 propels cystine uptake Via xc– for glutathione synthesis in lung cancer. Cancer Res (2021) 81(3):552–66. doi: 10.1158/0008-5472.Can-20-0617
189. Tao J, Deng NT, Ramnarayanan K, Huang B, Oh HK, Leong SH, et al. Cd44-Slc1a2 gene fusions in gastric cancer. Sci Trans Med (2011) 3(77):77ra30–0. doi: 10.1126/scitranslmed.3001423
190. Xu L, Chen J, Jia L, Chen X, Awaleh Moumin F, Cai J. Slc1a3 promotes gastric cancer progression Via the Pi3k/Akt signalling pathway. J Cell Mol Med (2020) 24(24):14392–404. doi: 10.1111/jcmm.16060
191. Varini K, Benzaria A, Taieb N, Di Scala C, Azmi A, Graoudi S, et al. Mislocalization of the exitatory amino-acid transporters (Eaats) in human astrocytoma and non-astrocytoma cancer cells: Effect of the cell confluence. J BioMed Sci (2012) 19:10. doi: 10.1186/1423-0127-19-10
192. Robert SM, Sontheimer H. Glutamate transporters in the biology of malignant gliomas. Cell Mol Life Sci (2014) 71(10):1839–54. doi: 10.1007/s00018-013-1521-z
193. Bacci M, Lorito N, Ippolito L, Ramazzotti M, Luti S, Romagnoli S, et al. Reprogramming of amino acid transporters to support aspartate and glutamate dependency sustains endocrine resistance in breast cancer. Cell Rep (2019) 28(1):104–18.e8. doi: 10.1016/j.celrep.2019.06.010
194. Jun JC, Rathore A, Younas H, Gilkes D, Polotsky VY. Hypoxia-inducible factors and cancer. Curr Sleep Med Rep (2017) 3(1):1–10. doi: 10.1007/s40675-017-0062-7
195. Grewer C, Gameiro A, Zhang Z, Tao Z, Braams S, Rauen T. Glutamate forward and reverse transport: From molecular mechanism to transporter-mediated release after ischemia. IUBMB Life (2008) 60(9):609–19. doi: 10.1002/iub.98
196. Sun J, Nagel R, Zaal EA, Ugalde AP, Han R, Proost N, et al. Slc1a3 contributes to l-asparaginase resistance in solid tumors. EMBO J (2019) 38(21):e102147. doi: 10.15252/embj.2019102147
197. He L, Shi X, Liu Z, Ren X, Zhang C, Yang Z, et al. Roles of Eaat1, dhfr, and fetuin-a in the pathogenesis, progression and prognosis of chondrosarcoma. Onco Targets Ther (2019) 12:8411–20. doi: 10.2147/ott.S222426
198. Scott AM, Cebon J. Clinical promise of tumour immunology. Lancet (1997) 349:S19–22. doi: 10.1016/S0140-6736(97)90016-7
199. Vokes EE. Combined modality therapy of solid tumours. Lancet (1997) 349:S4–6. doi: 10.1016/S0140-6736(97)90011-8
200. Najjar SA, Davis BM, Albers KM. Epithelial-neuronal communication in the colon: Implications for visceral pain. Trends Neurosci (2020) 43(3):170–81. doi: 10.1016/j.tins.2019.12.007
201. Fagan BM, Cahusac PM. Evidence for glutamate receptor mediated transmission at mechanoreceptors in the skin. Neuroreport (2001) 12(2):341–7. doi: 10.1097/00001756-200102120-00032
202. Davidson S, Golden JP, Copits BA, Ray PR, Vogt SK, Valtcheva MV, et al. Group ii mglurs suppress hyperexcitability in mouse and human nociceptors. PAIN (2016) 157(9):2081–88. doi: 10.1097/j.pain.0000000000000621
203. Najjar SA, Davis BM, Albers KM. Epithelial–neuronal communication in the colon: Implications for visceral pain. Trends Neurosci (2020) 43(3):170–81. doi: 10.1016/j.tins.2019.12.007
204. Dai WL, Yan B, Jiang N, Wu JJ, Liu XF, Liu JH, et al. Simultaneous inhibition of nmda and Mglu1/5 receptors by levo-corydalmine in rat spinal cord attenuates bone cancer pain. Int J Cancer (2017) 141(4):805–15. doi: 10.1002/ijc.30780
205. Ollenschläger G, Karner J, Karner-Hanusch J, Jansen S, Schindler J, Roth E. Plasma glutamate–a prognostic marker of cancer and of other immunodeficiency syndromes? Scand J Clin Lab Invest (1989) 49(8):773–7. doi: 10.3109/00365518909091556
206. Dröge W, Eck HP, Betzler M, Schlag P, Drings P, Ebert W. Plasma glutamate concentration and lymphocyte activity. J Cancer Res Clin Oncol (1988) 114(2):124–8. doi: 10.1007/bf00417824
207. Gao R, Cheng J, Fan C, Shi X, Cao Y, Sun B, et al. Serum metabolomics to identify the liver disease-specific biomarkers for the progression of hepatitis to hepatocellular carcinoma. Sci Rep (2015) 5:18175. doi: 10.1038/srep18175
208. Sidransky D. Emerging molecular markers of cancer. Nat Rev Cancer (2002) 2(3):210–9. doi: 10.1038/nrc755
209. Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, et al. 5' cpg island methylation is associated with transcriptional silencing of the tumour suppressor P16/Cdkn2/Mts1 in human cancers. Nat Med (1995) 1(7):686–92. doi: 10.1038/nm0795-686
210. Ganor Y, Levite M. The neurotransmitter glutamate and human T cells: Glutamate receptors and glutamate-induced direct and potent effects on normal human T cells, cancerous human leukemia and lymphoma T cells, and autoimmune human T cells. J Neural Transm (2014) 121(8):983–1006. doi: 10.1007/s00702-014-1167-5
211. Ganor Y, Levite M. Glutamate in the immune system: Glutamate receptors in immune cells, potent effects, endogenous production and involvement in disease. In: Levite M, editor. Nerve-driven immunity: Neurotransmitters and neuropeptides in the immune system. Vienna: Springer Vienna (2012). p. 121–61.
212. Ganor Y, Grinberg I, Reis A, Cooper I, Goldstein RS, Levite M. Human T-leukemia and T-lymphoma express glutamate receptor ampa Glur3, and the neurotransmitter glutamate elevates the cancer-related matrix-metalloproteinases inducer Cd147/Emmprin, mmp-9 secretion and engraftment of T-leukemia in vivo. Leuk Lymphoma (2009) 50(6):985–97. doi: 10.1080/10428190902878448
213. Speyer CL, Smith JS, Banda M, DeVries JA, Mekani T, Gorski DH. Metabotropic glutamate receptor-1: A potential therapeutic target for the treatment of breast cancer. Breast Cancer Res Treat (2012) 132(2):565–73. doi: 10.1007/s10549-011-1624-x
214. Mehnert JM, Silk AW, Lee JH, Dudek L, Jeong BS, Li J, et al. A phase ii trial of riluzole, an antagonist of metabotropic glutamate receptor 1 (Grm1) signaling, in patients with advanced melanoma. Pigment Cell Melanoma Res (2018) 31(4):534–40. doi: 10.1111/pcmr.12694
215. Park SY, Lee SA, Han IH, Yoo BC, Lee SH, Park JY, et al. Clinical significance of metabotropic glutamate receptor 5 expression in oral squamous cell carcinoma. Oncol Rep (2007) 17(1):81–7. doi: 10.3892/or.17.1.81
216. Zhang L, Huang Y, Ling J, Zhuo W, Yu Z, Luo Y, et al. Overexpression of Slc7a11: A novel oncogene and an indicator of unfavorable prognosis for liver carcinoma. Future Oncol (2018) 14(10):927–36. doi: 10.2217/fon-2017-0540
217. Zhao X, Li Y, Wu H. A novel scoring system for acute myeloid leukemia risk assessment based on the expression levels of six genes. Int J Mol Med (2018) 42(3):1495–507. doi: 10.3892/ijmm.2018.3739
218. Zhong W, Weiss HL, Jayswal RD, Hensley PJ, Downes LM, St Clair DK, et al. Extracellular redox state shift: A novel approach to target prostate cancer invasion. Free Radic Biol Med (2018) 117:99–109. doi: 10.1016/j.freeradbiomed.2018.01.023
219. Sugano K, Maeda K, Ohtani H, Nagahara H, Shibutani M, Hirakawa K. Expression of xct as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res (2015) 35(2):677–82.
220. Magri J, Gasparetto A, Conti L, Calautti E, Cossu C, Ruiu R, et al. Tumor-associated antigen xct and mutant-P53 as molecular targets for new combinatorial antitumor strategies. Cells (2021) 10(1):108. doi: 10.3390/cells10010108
221. Okuno S, Sato H, Kuriyama-Matsumura K, Tamba M, Wang H, Sohda S, et al. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br J Cancer (2003) 88(6):951–56. doi: 10.1038/sj.bjc.6600786
222. Zhang P, Wang W, Wei Z, Xu LI, Yang X, Du Y. Xct expression modulates cisplatin resistance in Tca8113 tongue carcinoma cells. Oncol Lett (2016) 12(1):307–14. doi: 10.3892/ol.2016.4571
223. Luo Y, Xiang W, Liu Z, Yao L, Tang L, Tan W, et al. Functional role of the Slc7a11-As1/Xct axis in the development of gastric cancer cisplatin-resistance by a gsh-dependent mechanism. Free Radical Biol Med (2022) 184:53–65. doi: 10.1016/j.freeradbiomed.2022.03.026
224. Horibe S, Kawauchi S, Tanahashi T, Sasaki N, Mizuno S, Rikitake Y. Cd44v-dependent upregulation of xct is involved in the acquisition of cisplatin-resistance in human lung cancer A549 Cells. Biochem Biophys Res Commun (2018) 507(1-4):426–32. doi: 10.1016/j.bbrc.2018.11.055
225. Timmerman Luika A, Holton T, Yuneva M, Louie Raymond J, Padró M, Daemen A, et al. Glutamine sensitivity analysis identifies the xct antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell (2013) 24(4):450–65. doi: 10.1016/j.ccr.2013.08.020
226. Guo W, Zhao Y, Zhang Z, Tan N, Zhao F, Ge C, et al. Disruption of xct inhibits cell growth Via the Ros/Autophagy pathway in hepatocellular carcinoma. Cancer Lett (2011) 312(1):55–61. doi: 10.1016/j.canlet.2011.07.024
227. Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer (2003) 3(12):895–902. doi: 10.1038/nrc1232
228. Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. Cd133+ hcc cancer stem cells confer chemoresistance by preferential expression of the Akt/Pkb survival pathway. Oncogene (2008) 27(12):1749–58. doi: 10.1038/sj.onc.1210811
229. Zhang Q, Shi S, Yen Y, Brown J, Ta JQ, Le AD. A subpopulation of Cd133(+) cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett (2010) 289(2):151–60. doi: 10.1016/j.canlet.2009.08.010
230. Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, et al. Cd44 variant regulates redox status in cancer cells by stabilizing the xct subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell (2011) 19(3):387–400. doi: 10.1016/j.ccr.2011.01.038
231. Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the X(C)- cystine transporter: A new action for an old drug. Leukemia (2001) 15(10):1633–40. doi: 10.1038/sj.leu.2402238
232. Lan H, Gao Y, Zhao Z, Mei Z, Wang F. Ferroptosis: Redox imbalance and hematological tumorigenesis. Front Oncol (2022) 12:834681. doi: 10.3389/fonc.2022.834681
233. Liu J, Xia X, Huang P. Xct: A critical molecule that links cancer metabolism to redox signaling. Mol Ther (2020) 28(11):2358–66. doi: 10.1016/j.ymthe.2020.08.021
234. Daher B, Parks SK, Durivault J, Cormerais Y, Baidarjad H, Tambutte E, et al. Genetic ablation of the cystine transporter xct in pdac cells inhibits Mtorc1, growth, survival, and tumor formation Via nutrient and oxidative stresses. Cancer Res (2019) 79(15):3877–90. doi: 10.1158/0008-5472.Can-18-3855
235. Chen RS, Song YM, Zhou ZY, Tong T, Li Y, Fu M, et al. Disruption of xct inhibits cancer cell metastasis Via the caveolin-1/Beta-Catenin pathway. Oncogene (2009) 28(4):599–609. doi: 10.1038/onc.2008.414
236. Zhuang J, Liu X, Yang Y, Zhang Y, Guan G. Sulfasalazine, a potent suppressor of gastric cancer proliferation and metastasis by inhibition of xct: Conventional drug in new use. J Cell Mol Med (2021) 25(12):5372–80. doi: 10.1111/jcmm.16548
237. Leung W-H, Shih J-W, Chen J-S, Mokgautsi N, Wei P-L, Huang Y-J. Preclinical identification of Sulfasalazine&Rsquo;S therapeutic potential for suppressing colorectal cancer stemness and metastasis through targeting Kras/Mmp7/Cd44 signaling. Biomedicines (2022) 10(2):377. doi: 10.3390/biomedicines10020377
238. Thanee M, Padthaisong S, Suksawat M, Dokduang H, Phetcharaburanin J, Klanrit P, et al. Sulfasalazine modifies metabolic profiles and enhances cisplatin chemosensitivity on cholangiocarcinoma cells in in vitro and in vivo models. Cancer Metab (2021) 9(1):11. doi: 10.1186/s40170-021-00249-6
239. Sugiyama A, Ohta T, Obata M, Takahashi K, Seino M, Nagase S. Xct inhibitor sulfasalazine depletes Paclitaxel−Resistant tumor cells through ferroptosis in uterine serous carcinoma. Oncol Lett (2020) 20(3):2689–700. doi: 10.3892/ol.2020.11813
240. Wang S-F, Chen M-S, Chou Y-C, Ueng Y-F, Yin P-H, Yeh T-S, et al. Mitochondrial dysfunction enhances cisplatin resistance in human gastric cancer cells Via the ros-activated Gcn2-Eif2α-Atf4-Xct pathway. Oncotarget (2016) 7(45):74132–51. doi: 10.18632/oncotarget.12356
241. Wada F, Koga H, Akiba J, Niizeki T, Iwamoto H, Ikezono Y, et al. High expression of Cd44v9 and xct in chemoresistant hepatocellular carcinoma: Potential targets by sulfasalazine. Cancer Sci (2018) 109(9):2801–10. doi: 10.1111/cas.13728
242. Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, et al. Systemic depletion of l-Cyst(E)Ine with Cyst(E)Inase increases reactive oxygen species and suppresses tumor growth. Nat Med (2017) 23(1):120–27. doi: 10.1038/nm.4232
243. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
244. Zhao Y, Li Y, Zhang R, Wang F, Wang T, Jiao Y. The role of erastin in ferroptosis and its prospects in cancer therapy. Onco Targets Ther (2020) 13:5429–41. doi: 10.2147/OTT.S254995
245. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife (2014) 3:e02523. doi: 10.7554/eLife.02523
246. Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1g facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology (2016) 64(2):488–500. doi: 10.1002/hep.28574
247. Battaglia AM, Sacco A, Perrotta ID, Faniello MC, Scalise M, Torella D, et al. Iron administration overcomes resistance to erastin-mediated ferroptosis in ovarian cancer cells. Front Oncol (2022) 12:868351. doi: 10.3389/fonc.2022.868351
248. Fornari F, Giovannini C, Piscaglia F, Gramantieri L. Elucidating the molecular basis of sorafenib resistance in hcc: Current findings and future directions. J Hepatocell Carcinoma (2021) 8:741–57. doi: 10.2147/JHC.S285726
249. Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduction Targeted Ther (2020) 5(1):87. doi: 10.1038/s41392-020-0187-x
250. Nishijima TF, Shachar SS, Nyrop KA, Muss HB. Safety and tolerability of pd-1/Pd-L1 inhibitors compared with chemotherapy in patients with advanced cancer: A meta-analysis. Oncologist (2017) 22(4):470–79. doi: 10.1634/theoncologist.2016-0419
251. Lanzardo S, Conti L, Rooke R, Ruiu R, Accart N, Bolli E, et al. Immunotargeting of antigen xct attenuates stem-like cell behavior and metastatic progression in breast cancer. Cancer Res (2016) 76(1):62–72. doi: 10.1158/0008-5472.Can-15-1208
252. Bi J, Yang S, Li L, Dai Q, Borcherding N, Wagner BA, et al. Metadherin enhances vulnerability of cancer cells to ferroptosis. Cell Death Dis (2019) 10(10):682. doi: 10.1038/s41419-019-1897-2
253. Pui C-H, Campana D, Pei D, Bowman WP, Sandlund JT, Kaste SC, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. New Engl J Med (2009) 360(26):2730–41. doi: 10.1056/NEJMoa0900386
254. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell (2017) 168(4):657–69. doi: 10.1016/j.cell.2016.12.039
255. Hays JL, Kim G, Walker A, Annunziata CM, Lee JM, Squires J, et al. A phase ii clinical trial of polyethylene glycol-conjugated l-asparaginase in patients with advanced ovarian cancer: Early closure for safety. Mol Clin Oncol (2013) 1(3):565–69. doi: 10.3892/mco.2013.99
256. Hao W, Wu L, Cao L, Yu J, Ning L, Wang J, et al. Radioresistant nasopharyngeal carcinoma cells exhibited decreased cisplatin sensitivity by inducing Slc1a6 expression. Front Pharmacol (2021) 12:629264. doi: 10.3389/fphar.2021.629264
257. Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer (2007) 7(4):233–45. doi: 10.1038/nrc2091
Keywords: ionotropic glutamate receptor (iGluR), metabotropic glutamate receptor (mGluR), excitatory amino acid transporter (EAAT), glutamate/cystine antiporter (xCT), cancer, non-neural organs
Citation: García-Gaytán AC, Hernández-Abrego A, Díaz-Muñoz M and Méndez I (2022) Glutamatergic system components as potential biomarkers and therapeutic targets in cancer in non-neural organs. Front. Endocrinol. 13:1029210. doi: 10.3389/fendo.2022.1029210
Received: 26 August 2022; Accepted: 24 October 2022;
Published: 15 November 2022.
Edited by:
Ignacio Camacho-Arroyo, National Autonomous University of Mexico, MexicoReviewed by:
Arturo Ortega, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoCopyright © 2022 García-Gaytán, Hernández-Abrego, Díaz-Muñoz and Méndez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabel Méndez, aXNhYmVsY21AdW5hbS5teA==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.