 Paulina Villaseca
Paulina Villaseca Pedro Cisternas
Pedro Cisternas Nibaldo C. Inestrosa
Nibaldo C. Inestrosa- 1Centro de Excelencia en Biomedicina de Magallanes (CEBIMA), Universidad de Magallanes, Punta Arenas, Chile
- 2Instituto de Ciencias de la Salud, Universidad de O´Higgins, Rancagua, Chile
- 3Centro de Envejecimiento y Regeneración (CARE UC), Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, Santiago, Chile
Late onset Alzheimer´s disease (AD) is a neurodegenerative disease with gender differences in its onset and progression, being the prevalence predominant in women and at an earlier age than in men. The pathophysiology of the menopausal condition has been associated to this dementia, playing major roles regarding both endocrine and glucose metabolism changes, amongst other mechanisms. In the current review we address the role of estrogen deficiency in the processes involved in the development of AD, including amyloid precursor protein (APP) processing to form senile plaques, Tau phosphorylation forming neurofibrillary tangles, Wnt signaling and AD neuropathology, the role of glucose brain metabolism, Wnt signaling and glucose transport in the brain, and our research contribution to these topics.
Introduction
The endocrine status that comes with menopause, with the cessation of ovarian estrogens and progesterone synthesis and increase of gonadotropins (FSH and LH), brings a loss of neuroprotector mechanisms that could explain cognitive decline (1) and the risk of developing AD later in life more prevalent in women (2). Estradiol (E2), the main estrogen acting during women´s reproductive years, is well known to exert neuroprotection through various mechanisms (3–5). Even more, neuron derived E2 regulates synaptic plasticity and memory (6, 7).
On the other hand, it is also well known that Wnt signaling plays important roles in various systemic physiological processes: cell differentiation, polarity and migration (8, 9). At the central nervous system (CNS) as well, Wnt signaling has been described in various cell types including radial glia, oligodendrocytes, microglia, astrocytes, and neurons (10–12), and regulates neural patterning, stem cells proliferation and neurogenesis (9, 13, 14). Back in 2000, we proposed that Wnt signaling might play a key role in AD neuroprotection (15). We have since studied the function of this signaling pathway in the CNS and its importance in AD, with the objective to explore pharmacological strategies that could strike the burden of this disease. In humans, 19 Wnt ligands and 10 Frizzled (Fz) receptors are recognized (16). Wnt proteins are post-translationally palmitoylated to be secreted and to be bound to Fz receptor (17). Wnt ligands play a key role in the formation and function of synapses through lifetime. Wnt7a, a canonical ligand, stimulates dendritic spine morphogenesis inducing the postsynaptic density-95 (PSD-95) protein expression (18).
Dysfunction of Wnt signaling has been described in aging and menopause, both associated with hippocampal memory impairments (9, 19). Our studies on hippocampal cultured neurons, as well as in transgenic mice models of AD, indicated that Wnt signaling protects against the amyloid β (αβ) peptide neurotoxicity. Wnt signaling is also involved in Tau phosphorylation by its enzyme glycogen synthase kinase-3β (GSK-3β) and, finally, the activation of Wnt signaling was shown to be involved in learning and memory (20–22).
One of the mechanisms through which E2 protects from brain tissue ischemia is by interfering the induction of dickkopf-1 (Dkk1) (23), an antagonist of the Wnt signaling pathway (24). Studies with hippocampal neurons in culture showed that Dkk1 is required for amyloid-mediated synaptic loss (25). Dkk1, though crucially expressed during neurodevelopment, its elevation in adult life has shown to be a principal mediator of neurodegeneration (9, 19).
Another mechanism related to a decrease in brain function of AD patients is the decrease in glucose consumption. Our research group described that Wnt activators increase both brain glucose consumption and cognitive performance in transgenic AD mice (26).
A Wnt activator Andrographolide (ANDRO), a labdane diterpene, was found to reduce Aβ burden, astrogliosis, interleukin-6 and oxidative stress (27), all these mechanisms present in AD. Our studies are consistent with the idea that the activation of Wnt signaling might delay the onset of AD (9).
In this review, we describe studies on the role of Wnt signaling on the neuroprotective activity of E2, and on energy metabolism and glucose transport in neurons, in search of a method that could delay or avoid the development of this progressive dementia with no effective treatment available.
Menopause and development of AD
A particularly interesting aspect is the role of sex steroids on neuroprotection (28). In fact, postmenopausal women are most frequently affected by sporadic AD in a ratio 3:1, differently from the incidence in the infrequent hereditary illness occurring in younger patients in a ratio 1:1 (29). Estrogen deficiency occurring in spontaneous or surgical menopause has been associated to cognitive decline; in this context, several studies have shown that early E2 replacement exerts neuroprotective effects (7, 30).
Since AD is the most prevalent dementia and incurable disease, understanding its molecular pathogenesis could lead to the development of a specific therapeutic strategy, here we will describe effects of E2 on Aβ peptide production and Tau phosphorylation.
E2 affects the production of Aβ peptide by blocking the amyloid precursor protein processing
The APP is the precursor of a 40-42 amino acid peptide called amyloid-β-peptide (Aβ), that forms the central core of senile plaques (31). APP is cleaved by distinctive enzymatic activities, denominated secretases: α-secretase was the first described and cuts the Aβ peptide between the 15 and 17 amino acids and thus yields a large soluble amino-terminal fragment of APP known as the soluble non-amyloidogenic APP (sAPP), that does not form senile plaques. The second enzyme, named β-secretase (BACE1), cuts the Aβ peptide at its N-terminal domain, generating a cell-associated amyloidogenic carboxy-terminal fragment which is processed by a third enzymatic complex enzyme called γ-secretase, which produces a soluble Aβ by cutting the APP in the middle of the neuronal membrane, generating an aggregating amyloidogenic Aβ peptide that forms the senile plaques (31).
One of the first indications that E2 was protective against AD was obtained when E2 was found to stimulate the processing of APP, at the level of the α-secretase enzyme, preventing the generation of Aβ, generating a soluble non-amyloidogenic APP (32); therefore, E2 blocks the amyloid plaques formation (32), a key neuropathological lesion in AD (31). In addition, brain E2 deficiency accelerates amyloid plaque formation, as shown in an AD animal model (33). Finally, E2 also decreases the formation of amyloid fibrils as well as the formation of Aβ oligomers in vitro (34). Table 1 indicates some of the E2 effects on Aβ peptide production.

Table 1 Summary of Neuroprotective actions of E2.
Wnt signaling activity and Aβ peptide production
Recent studies in mouse AD models indicate that the activation of the Wnt signaling pathway decreases the Aβ peptide levels, due to a low β-secretase (BACE1) availability, since Wnt signaling activation repressed BACE1 transcription at the nuclear level (40, 41). These results indicate that the activation of Wnt signaling by acting in a different secretase than E2, also decreases the amount of Aβ peptide and diminishes the amyloid plaques formation (9, 41). These studies also indicated that reducing the β-cleavage of APP may protect against the appearance of AD in the animal models. Interestingly, molecular genetics studies in Iceland (42) identified coding variants in the APP that were tested for an association with AD, after studying whole-genome sequencing of 1,795 subjects. Variant A673T, corresponding to a single nucleotide alanine-to-threonine substitution adjacent to the β-secretase (BACE1) site in APP, was markedly more common in the elderly control than in the AD subjects; this is consistent with an ≈ 40% reduction in the Aβ, formation observed in vitro (42).
E2 affects Tau phosphorylation
Intraneuronal communication occurs through microtubules and the associated Tau proteins that stabilize microtubule structure and function. In AD, kinases phosphorylate Tau proteins, detaching them from the microtubules; these “speedways” lose structure and function, and synaptic vesicles cannot be driven to the synaptic region, and twisted filaments are aggregated in tangles – the neurofibrillary tangles. In fact, Tau is phosphorylated by several kinases, including GSK-3β (43). In general, GSK-3β can phosphorylate around 42 Tau sites, of which at least 29 are found phosphorylated in human AD brains (44).
In this context, it is interesting to mention that E2 prevents neural Tau hyperphosphorylation; moreover, E2 increased Tau dephosphorylation as measured by using a Tau-1 antibody which identifies a site of Tau (proline) which is non-phosphorylated. In addition, E2 prevented okadaic acid-induced hyperphosphorylation of Tau in both proline- and non-proline sites, and this effect was blocked by an anti-E2 antibody (45). In this study, the Estradiol Receptor (ER) appeared to be responsible of these effects probably via the Akt metabolic pathway (35). Finally, in transgenic mice expressing the wild-type human Tau, it was shown that E2 inhibited the Aβ-mediated conformation of Tau called paired helical filaments (PHF) through both antioxidant activity and regulation of the miRNA-218 (28, 46). Table 1 indicates some effects of E2 on Tau protein.
Wnt signaling activity and Tau hyperphosphorylation
β-Catenin is a key protein of Wnt signaling which is regulated by GSK-3β, studies in ovariectomized rats sacrificed 1h after E2 treatment, showed that β-catenin and GSK-3β are co-immunoprecipitated with the (ERα) in the hippocampus. This observation is consistent with the hypothesis that a multi-complex is formed by ERα, β-catenin and GSK-3β, that inhibited GSK-3β activity and thus regulating Tau phosphorylation, avoiding its hyperphosphorylation through Wnt and estradiol action (44, 47).
AD mutations in presenilin-1 also promote GSK-3β activity and Tau phosphorylation (48). Overexpression of GSK-3β in the adult mouse brain leads to a decrease in β-catenin and an increase in Tau phosphorylation (45). Also, hippocampal infusion of Dkk1, a Wnt antagonist of the Canonical Wnt signaling triggers PHF1 Tau phosphorylation in rats (46).
Energy metabolism and metabolic syndrome
The excess of energy from carbohydrates and fats in inadequate diet habits, leads to progressive metabolic disorders at any age. In the perimenopausal years, when E2 synthesis becomes variable and fluctuating, and FSH increases, the risk of the appearance of the metabolic syndrome increases significantly; when E2 decline is established with menopause, the metabolic syndrome is clearly higher than in premenopausal years (49). All components of metabolic syndrome become more conspicuous with menopause: higher blood glucose, lower HDL-cholesterol, higher blood triglycerides, higher blood pressure, and increase in visceral adiposity (larger waist circumference) (50).
In postmenopausal women, treatment with estrogens improves all components of the metabolic syndrome, as well as insulin resistance parameters (51). Estrogens have a transcendent role on metabolism by modulating directly whole-body energy management, controlling glucose availability and facilitating insulin secretion, and modulating energy partition by favoring lipids as the main substrate for energy when they are more available than carbohydrates, shifting from lipid storage to their oxidation as substrate. As well, E2 maintains energy balance by influencing energy intake and energy expenditure, regulating body weight homeostasis. The E2 roles on energy balance are mediated by the (ERα), which is abundant in the hypothalamus. Estrogen deficiency in menopause and the consequent loss of ERα activity, determine a decrease in energy expenditure, increased food intake and increased adiposity (52–54). In animal experiments, treatment with E2 decreases obesity (55), decreases hepatic steatosis, and limits fat deposition (54, 56).
Estrogens, as well, decrease lipogenesis and inhibit adipogenesis through ERα activation (57). The role of E2 on lipid metabolism is coordinated with effects on carbohydrate metabolism, also through ERα, by lowering insulin resistance and fat storage (58, 59). The insulin-sensitizing actions of E2 work through improving insulin-mediated glucose uptake, insulin signaling, and glucose transport in adipose. muscular and brain tissue. E2 also controls the metabolic sensor in the hypothalamus, protecting the brain from hypoglycemia (60). Otherwise, E2 potentiates the oxidative capacity of mitochondria and E2 deprivation induces mitochondrial dysfunction and insulin resistance, mechanisms involved in the development of alterations in cognitive function (61). Thus, the metabolic changes associated to menopause can play a key role in triggering AD, in addition to the direct mechanisms favoring the Aβ peptide production of amyloid plaques and the neurofibrillary tangles described above.
The role of glucose metabolism in the brain and its regulation by Wnt signaling is discussed now.
Glucose metabolism in the brain
The adult brain represents only 2% of the total body mass but is responsible for the utilization of almost 25% of total ATP produced by the body (62–64). Glucose is the principal energy source of the brain (65, 66). The uptake of glucose occurs via glucose transporters (GLUTs): these are 14 isoforms, and several GLUTs are expressed in the brain. At cellular levels the specific isoform expressed depends on the brain region, i.e., in the cortex, astrocytes express exclusively GLUT1 while neurons express mainly GLUT3 (67–69). In the hippocampus the expression of mRNA of GLUT4 mainly occurs in neurons; this is an interesting observation because GLUT4 is regulated by insulin and AMP-activated protein kinase (AMPK) pathways (70, 71). In different AD models, a significant decrease in the expression of GLUT4 has been described, suggesting a role for this transporter in AD pathogenesis (72).
After glucose uptake by the cells, this molecule can be used by several pathways including glycolysis, the Krebs cycle and oxidative phosphorylation, the pentose phosphate pathway (PPP) and glycogen synthesis (73–75). The use of glucose by glycolysis, together with the Krebs cycle are the major source of ATP to neurons (76–78). The PPP is required to obtain reducing equivalents in the form of nicotinamide adenine dinucleotide phosphate (NADP(H+)), which in turn is required for neuronal defense against oxidative stress since it is necessary for the recycling of antioxidant molecules, such as glutathione and ascorbate (74, 79). The glucose utilization by brain cells plays a central role in the physiology of the brain and a decrease in this metabolism has been related with almost all the neurodegenerative diseases, including AD (62).
Glucose metabolism in AD
A decrease in glucose utilization has been described in several brain regions of AD patients, mainly in brain zones related functions such as memory/learning, including the hippocampus and cortex (62, 72, 80). At the molecular level, the lower glucose utilization in AD patients has been associated with alterations, including decreased expression of GLUT1, -3 and -4 in the cortex and hippocampus, insulin resistance in the brain, mitochondrial dysfunction, deregulation of the Krebs cycle, and oxidative phosphorylation, which are triggered by the loss of key enzymes (81–83). On the other hand, stimulating glucose metabolism in AD patients through the administration of insulin or GLP-1, significantly improves the cognitive function, supporting the close relationship between the deregulation of cerebral glucose metabolism and the cognitive failures described in AD (84, 85). In the context of menopause, hot flashes could be a neurovascular compensatory response to brain hypometabolism, to increase blood flow and glucose in the brain, as has been shown in perimenopausal women (86).
Wnt signaling and glucose metabolism
Wnt ligands have been related with the regulation of glucose metabolism. Animal models and human studies have suggested that some components of Wnt signaling increase the risk for the development of diabetes and age-related dementia (9, 87).
The increase in Ca+2 levels by the Wnt signaling pathway significantly affects glucose metabolism in neurons and astrocytes; thus, Wnt/Ca2+ signaling may represent a newly identified mode for the regulation of glucose metabolism in neurons (88–90). Previously, we showed that acute treatment with Wnt5a stimulates glucose uptake in neurons, in a time-dependent manner, this was correlated with an increase in both hexokinase activity and the glycolytic rate. Furthermore, we observed an increase in the activity of glucose-6-phosphate dehydrogenase and PPP. The effects of Wnt5a were dependent on the generation of nitric oxide (NO) downstream of Wnt5a signaling (62, 91). These results support that the activation of non-Canonical Wnt signaling pathway regulates cellular glucose metabolism in neurons in a NO-dependent manner (74, 92).
When we treated the neurons with a canonical Wnt ligand, like Wnt3a, we observed an increase in glucose uptake in neurons without changing the expression or localization of GLUT3, the main GLUT transporter in these cells. Furthermore, we described that the acute treatment with Wnt3a stimulates the activation of Akt, the activity of hexokinase and the glycolytic rate. These effects of Wnt3a were dependent on activation of the Akt pathway and was independent of both the transcription of Wnt target genes and synaptic effects (93).
In addition to Wnt ligands, other molecules such as ANDRO, obtained from Andrographis paniculata, activates the Wnt signaling pathway by inhibiting GSK-3β and it was shown to protect neurons (94, 95). ANDRO also increases the uptake of glucose in vivo and in vitro promoting in both conditions a recovery of the brain glucose metabolism and cognitive performance (9, 26, 96). Furthermore, we also demonstrated in a transgenic mice model of AD that the administration of ANDRO in pre-symptomatic stages can delay the appearance of several markers of AD, promoting a general rescue of the brain metabolic parameters (9, 62, 97).
These studies support that Wnt signaling promote glucose metabolism in neurons, stimulating the ATP production in these cells to satisfy the energy demands of neurons (62, 91, 93).
Globally, estrogens play a role on neurodegeneration interrelated with Wnt signaling and glucose brain metabolism as described in Figure 1, a scheme comparing the estrogenic adult female with the postmenopauseal estrogen-deprived brain.
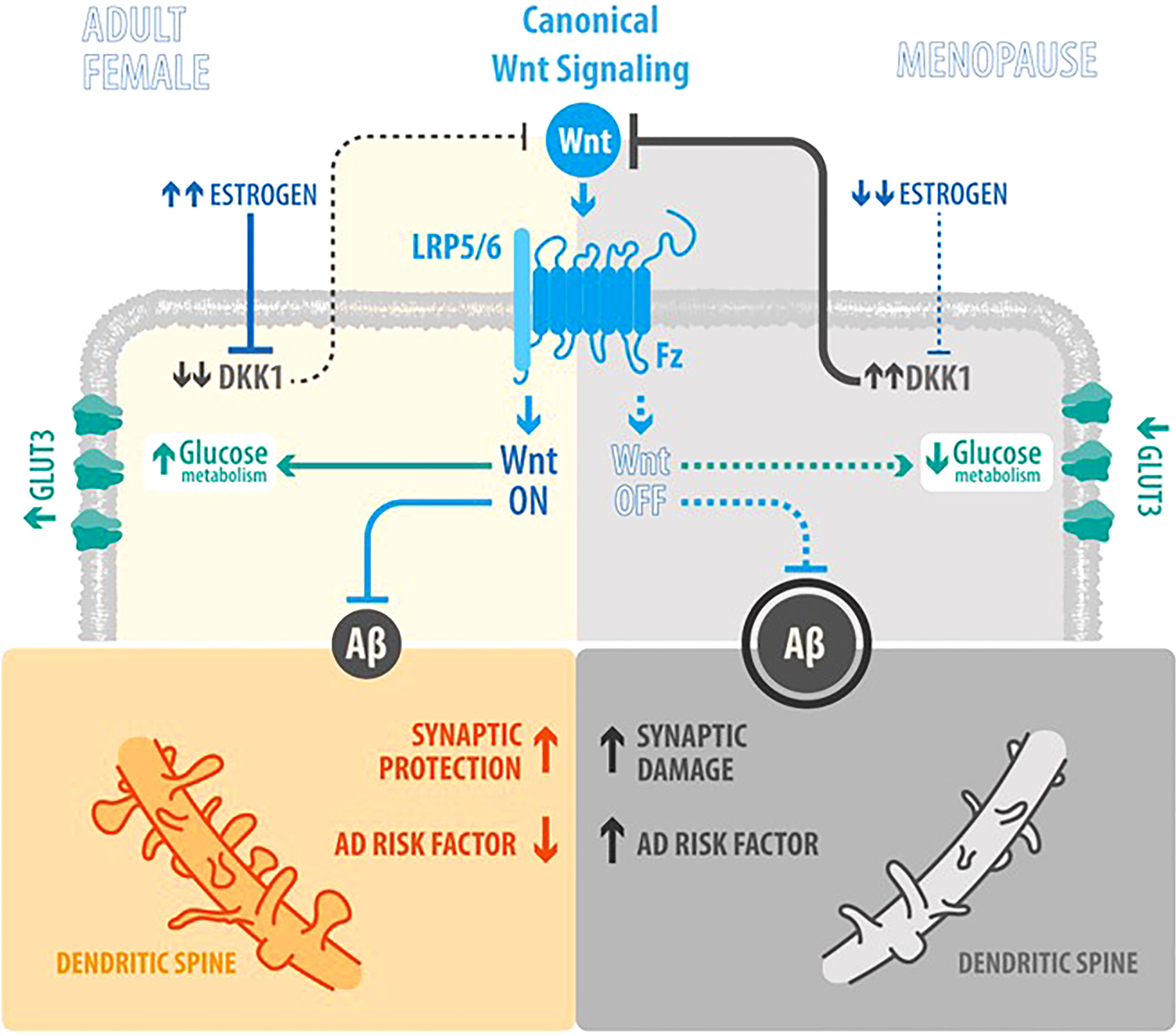
Figure 1 How factors are interrelated and their role in AD (Estrogen, Wnt signaling, glucose metabolism and neurodegeneration). Scheme integrating the actions of Wnt signaling, Estrogen levels, Aβ peptide synthesis and brain glucose uptake in the Adult Female as opposed to the Menopause status: In the Adult Female, estrogen (E2) inhibits the synthesis of Dkk1 (a physiological antagonist of Wnt canonical signaling) (23), thus promoting the activation of Wnt canonical signaling and leading to a decrease of Aβ peptide synthesis, an increase in synaptic protection and development of dendritic spines; in parallel, also leading to increased brain glucose uptake. In Menopause, the decrease of E2 induces an increase of Dkk1 and the consequent decrease in Wnt signaling activity, that lead to an increase and aggregation of Aβ peptide and neuronal damage, as the loss of dendritic spines by the accumulation of plaques; in parallel, the decrease in Wnt signaling activity, decreases brain glucose metabolism.
A strength of this review is the involvement of our group in a lifetime research on this topic, beginning in the early 1998 (3, 4) and ongoing research (9).
Conclusion
Our aim to integrate the actions of different factors: Wnt signaling, E2 levels, synthesis of Aβ peptide, Tau phosphorylation and brain glucose uptake have been detailed in this contribution, and are intimately entwined for neuroprotection in the female reproductive years. These mechanims are lost with menopause leading to a cascade of events towards neurodegeneration in the postmenopausal years.
Further research is required for a better understanding of the cellular and physio-pathological mechanisms related to the endocrine and metabolic changes that occur with menopause, globally and in the brain. This could lead to discover triggering factors that initiate late onset neurodegeneration and that facilitate its development. Thence, science could be nearer to intervene and strike on this disease burden in an aging population worldwide.
Author contributions
PV and NI designed the review. PV, PC and NI wrote the article and edited the article. NI prepared Table 1. PC prepared Figure 1 with graphic designer Felipe Serrano. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Basal Centre of Excellence in Aging and Regeneration AFB170005 and ACE210009 from ANID (Agencia Nacional de Investigación y Desarrollo of Chile) to NCI.
Acknowledgments
We thank to Illustrative Science (www.illustrative-science.com), for the Illustrated Graphic of Figure 1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Reuben R, Karkaby L, McNamee C, Phillips NA, Einstein G. Menopause, and cognitive complaints: are ovarian hormones linked with subjective cognitive decline? Climacteric (2021) 24(4):321–32. doi: 10.1080/13697137.2021.1892627
2. Jessen F, Wiese B, Bachmann C, Eifflaender-Gorfer S, Haller F, Kölsch H, et al. German Study on aging, cognition and dementia in primary care patients study group. prediction of dementia by subjective memory impairment:effects of severity and temporal association with cognitive impairment. Arch Gen Psychiatry (2010) 67(4):414–22. doi: 10.1001/archgenpsychiatry.2010.30
3. Inestrosa NC, Marzolo MP, Bonnefont AB. Cellular and molecular basis of estrogen´s neuroprotection. potential relevance for alzheimer´s disease. Mol Neurobiol (1998) 17(1-3):73–86. doi: 10.1007/BF02802025
4. Bonnefont AB, Muñoz FJ, Inestrosa NC. Estrogen protects neuronal cells from the cytotoxicity induced by acetylcholinesterase-amyloid complexes. FEBS Lett (1998) 441(2):220–4. doi: 10.1016/s0014-5793(98)01552-x
5. Cheng YJ, Lin CH, Lane HY. From menopause to neurodegeneration-molecular basis and potential therapy. Int J Mol Sci (2021) 22(16):8654. doi: 10.3390/ijms22168654
6. Brann DW, Lu Y, Wang J, Sareddy GR, Pratap UP, Zhang Q, et al. Neuron-derived estrogen-a key neuromodulator in synaptic function and memory. Int J Mol Sci (2021) 22(24):13242. doi: 10.3390/ijms222413242
7. Brann DW, Lu Y, Wang J, Zhang Q, Thakkar R, Sareddy GR, et al. Brain-derived estrogen and neural function. Neurosci Biobehav Rev (2022) 132:793–817. doi: 10.1016/j.neubiorev.2021.11.014
8. Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell (2017) 169(6):985–99. doi: 10.1016/j.cell.2017.05.016
9. Inestrosa NC, Tapia-Rojas C, Cerpa W, Cisternas P, Zolezzi JM. WNT signaling is a key player in alzheimer's disease. Handb Exp Pharmacol (2021) 269:357–82. doi: 10.1007/164_2021_532
10. Briona LK, Poulain FE, Mosimann C, Dorsky RI. Wnt/b-catenin signaling is required for radial glial neurogenesis following spinal cord injury. Dev Biol (2015) 403(1):15–21. doi: 10.1016/j.ybio.2015.03.025
11. Marchetti B. Wnt/β-catenin signaling pathway governs a full program for dopaminergic neuron survival, neurorescue and regeneration in the MPTP mouse model of parkinson´s diseases. Int J Mol Sci (2018) 19(12):3743. doi: 10.3390/ijms19123743
12. Wexler EM, Paucer A, Kornblum HI, Palmer TD, Geschwind DH. Endogenous wnt signaling maintains neural progenitor cell potency. Stem Cells (2009) 27(5):1130–40. doi: 10.1002/stem.36
13. Budnik V, Salinas PC. Wnt signaling during synaptic development and plasticity. Curr Opin Neurobiol (2011) 21:151–9. doi: 10.1016/j.conb.2010.12.002
14. Inestrosa NC, Arenas E. Emerging roles of wnts in the adult nervous system. Nat Rev Neurosci (2010) 11:77–86. doi: 10.1038/nrn2755
15. De Ferrari GV, Inestrosa NC. Wnt signaling function in alzheimer´s disease. Brain Res Rev (2000) 33(1):1–12. doi: 10.1016/s0165-0173(00)00021-7
16. Oliva CA, Montecinos-Oliva C, Inestrosa NC. Wnt signaling in the central nervous System:New insights in health and disease. Prog Mol Biol Transl Sci (2018) 153:81–130. doi: 10.1016/bs.pmbts.2017.11.018
17. Torres VI, Godoy JA, Inestrosa NC. Modulating wnt signaling at the root:Porcupine and wnt acylation. Pharmacol Ther (2019) 198:34–45. doi: 10.1016/j.pharmthera.2019.02.009
18. Ramos-Fernández E, Tapia-Rojas C, Ramírez VT, Inestrosa NC. Wnt-7a stimulates dendritic spine morphogenesis and PSD-95 expression through canonical signaling. Mol Neurobiol (2019) 56(3):1870–82. doi: 10.1007/s12035-018-1162-1
19. Inestrosa NC, Tapia-Rojas C, Lindsay CB, Zolezzi JM. Wnt signaling pathway dysregulation in the aging Brain:Lessons from the octodon degus. Front Cell Dev Biol (2020) 8:734. doi: 10.3389/fcell.2020.00734
20. Toledo EM, Inestrosa NC. Activation of wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of alzheimer's disease. Mol Psychiatry (2010) 15(3):272–85, 228. doi: 10.1038/mp.2009.72
21. Tapia-Rojas C, Inestrosa NC. Loss of canonical wnt signaling is involved in the pathogenesis of alzheimer's disease. Neural Regener Res (2018) 13(10):1705–10. doi: 10.4103/1673-5374.238606
22. Palomer E, Buechler J, Salinas PC. Wnt signaling deregulation in the aging and alzheimer's brain. Front Cell Neurosci (2019) 13:227. doi: 10.3389/fncel.2019.00227
23. Scott EL, Brann DW. Estrogen regulation of Dkk1 and wnt/β-catenin signaling in neurodegenerative disease. Brain Res (2013) 1514:63–74. doi: 10.1016/j.brainres.2012.12.015
24. Caricasole A, Copani A, Caraci F, Aronica E, Rozemuller AJ, Caruso A, et al. Induction of dickkopf-1, a negative modulator of the wnt pathway, is associated with neuronal degeneration in alzheimer´s brain. J Neurosci (2004) 24:6021–7. doi: 10.1523/JNEUROSCI.1381-04.2004
25. Purro SA, Dickins EM, Salinas PC. The secreted wnt antagonist dickkopf-1 is required for amyloid β-mediated synaptic loss. J Neurosci (2012) 32(10):3492–8. doi: 10.1523/JNEUROSCI.4562-11.2012
26. Cisternas P, Zolezzi J, Martinez M, Torres VI, Wong GW, Inestrosa NC. Wnt-induced activation of glucose metabolism mediates the in vivo neuroprotective roles of wnt signaling in Alzheimer disease, J. Neurochem (2018) 149(1):54–72. doi: 10.1111/jnc.14608
27. Lindsay CB, Zolezzi JM, Rivera DS, Cisternas P, Bozinovic F, Inestrosa NC. Andrographolide reduces neuroinflammation and oxidative stress in aged octodon degus. Mol Neurobiol (2020) 57(2):1131–45. doi: 10.1007/s12035-019-01784-6
28. Tamagno E, Guglielmotto M. Estrogens still represent an attractive therapeutic approach for alzheimer's disease. Neural Regener Res (2022) 17(1):93–4. doi: 10.4103/1673-5374.314295
29. Pike CJ. Sex and the development of alzheimer's disease. J Neurosci Res (2017) 95(1-2):671–80. doi: 10.1002/jnr.23827
30. Merlo S, Spampinato SF, Sortino MA. Estrogen and alzheimer's disease:Still an attractive topic despite disappointment from early clinical results. Eur J Pharmacol (2017) 817:51–8. doi: 10.1016/j.ejphar.2017.05.059
31. Selkoe DJ. Cell biology of the amyloid β-protein precursor and the mechanism of alzheimer's disease. Annu Rev Cell Biol (1994) 10:373–403. doi: 10.1146/annurev.cb.10.110194.002105
32. Jaffe AB, Toran-Allerand CD, Greengard P, Gandy SE. Estrogen regulates metabolism of Alzheimer amyloid β precursor protein. J Biol Chem (1994) 269(18):13065–8. doi: 10.1016/S0021-9258(17)36796-0
33. Yue X, Lu M, Lancaster T, Cao P, Honda S, Staufenbiel M, et al. Brain estrogen deficiency accelerates aβ plaque formation in an alzheimer's disease animal model. Proc Natl Acad Sci U S A (2005) 102(52):19198–203. doi: 10.1073/pnas.0505203102
34. Morinaga A, Ono K, Takasaki J, Ikeda T, Hirohata M, Yamada M. Effects of sex hormones on alzheimer's disease-associated β-amyloid oligomer formation in vitro. Exp Neurol (2011) 228(2):298–302. doi: 10.1016/j.expneurol.2011.01.011
35. Alvarez-de-la-Rosa M, Silva I, Nilsen J, Perez MM, Garcia-Segura LM, Avila J, et al. Estradiol prevents neural tau hyperphosphorylation characteristic of alzheimer´s disease. Ann N Y Acad Sci (2005) 1052:210–24. doi: 10.1196/annals.1347.016
36. Guglielmotto M, Manassero G, Vasciaveo V, Venezia M, Tabaton M, Tamagno E. Estrogens inhibit amyloid-β-Mediated paired helical filament-like conformation of tau through antioxi.dant activity and miRNA 218 regulation in hTau mice. J Alzheimers Dis (2020) 77(3):1339–51. doi: 10.3233/JAD-200707
37. Yao M, Nguyen TV, Pike CJ. Estrogen regulates bcl-w and bim expression:role in protection against beta-amyloid peptide-induced neuronal death. J Neurosci (2007) 27(6):1422–33. doi: 10.1523/JNEUROSCI.2382-06.2007
38. Merlo S, Sortino MA. Estrogen activates matrix metalloproteinases-2 and -9 to increase β amyloid degradation. Mol Cell Neurosci (2012) 49(4):423–9. doi: 10.1016/j.mcn.2012.02.005
39. Morinaga A, Hirohata M, Ono K, Yamada M. Estrogen has anti-amyloidogenic effects on alzheimer's β-amyloid fibrils. vitro Biochem Biophys Res Commun (2007) 359(3):697–702. doi: 10.1016/j.bbrc.2007.05.158
40. Parr C, Mirzaei N, Christian M, Sastre M. Activation of the wnt/β-catenin pathway represses the transcription of the β-amyloid precursor protein cleaving enzyme (BACE1) via binding of T-cell factor-4 to BACE1 promoter. FASEB J (2015) 29(2):623–35. doi: 10.1096/fj.14-253211
41. Tapia-Rojas C, Burgos PV, Inestrosa NC. Inhibition of wnt signaling induces amyloidogenic processing of amyloid precursor protein and the production and aggregation of amyloid-β (Aβ)42 peptides. J Neurochem (2016) 139(6):1175–91. doi: 10.1111/jnc.13873
42. Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in APP protects against alzheimer's disease and age-related cognitive decline. Nature (2012) 488(7409):96–9. doi: 10.1038/nature11283
43. Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med (2012) 2(7):a006247. doi: 10.1101/cshperspect.a006247
44. Muñoz-Mayorga D, Guerra-Araiza C, Torner L, Morales T. Tau phosphorylation in female Neurodegeneration:Role of estrogens, progesterone, and prolactin. Front Endocrinol (Lausanne) (2018) 9:133. doi: 10.3389/fendo.2018.00133
45. Lucas JJ, Hernández F, Gómez-Ramos P, Morán MA, Hen R, Avila J. Decreased nuclear β-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J (2001) 20(1-2):27–39. doi: 10.1093/emboj/20.1.27
46. Scali C, Caraci F, Gianfriddo M, Diodato E, Roncarati R, Pollio G, et al. Inhibition of wnt signaling, modulation of tau phosphorylation and induction of neuronal cell death by DKK1. Neurobiol Dis (2006) 24(2):254–65. doi: 10.1016/j.nbd.2006.06.016
47. Azcoitia I, Arevalo MA, De Nicola AF, Garcia-Segura LM. Neuroprotective actions of estradiol revisited. Trends Endocrinol Metab (2011) 22(12):467–73. doi: 10.1016/j.tem.2011.08.002
48. Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, et al. Presenilin 1 associates with glycogen synthase kinase-3β and its substrate tau. Proc Natl Acad Sci U S A (1998) 95(16):9637–41. doi: 10.1073/pnas.95.16.9637
49. Davis SR, Castelo-Branco C, Chedraui P, Lumsden MA, Nappi RE, Shah D, et al. Understanding weight gain at menopause. Climacteric (2012) 15(5):419–29. doi: 10.3109/13697137.2012.707385
50. Janssen I, Powell LH, Crawford S, Lasley B, Sutton-Tyrrell K. Menopause and the metabolic syndrome:the study of women's health across the nation. Arch Intern Med (2008) 168(14):1568–75. doi: 10.1001/archinte.168.14.1568
51. Salpeter SR, Walsh JM, Ormiston TM, Greyber E, Buckley NS, Salpeter EE. Meta-analysis:effect of hormone-replacement therapy on components of the metabolic syndrome in postmenopausal women. Diabetes Obes Metab (2006) 8(5):538–54. doi: 10.1111/j.1463-1326.2005.00545.x
52. Xu Y, Nedungadi TP, Zhu L, Sobhani N, Irani BG, Davis KE, et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab (2011) 14:453–65. doi: 10.1016/j.cmet.2011.08.009
53. Musatov X, Chen W, Pfaff DW, Mobbs CV, Yang XJ, Clegg DJ, et al. Silencing of estrogen receptor α in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci USA (2007) 104(7):2501–6. doi: 10.1073/pnas.0610787104
54. Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev (2013) 34(3):309–38. doi: 10.1210/er.2012-1055
55. Ting WJ, Huang CY, Jiang CH, Lin YM, Chung LC, Shen CY, et al. Treatment with 17β-estradiol reduced body weight and the risk of cardiovascular disease in a high-fat diet-induced animal model of obesity. Int J Mol Sci (2017) 18:629. doi: 10.3390/ijms18030629
56. Litwak SA, Wilson JL, Chen W, Garcia-Rudaz C, Khaksari M, Cowley MA, et al. Estradiol prevents fat accumulation and overcomes leptin resistance in female high-fat diet mice. Endocrinology (2014) 155:4447–60. doi: 10.1210/en.2014-1342
57. Ahluwalia A, Hoa N, Ge L, Blumberg B, Levin ER. Mechanisms by which membrane and nuclear ER α inhibit adipogenesis in cells isolated from female mice. Endocrinology (2020) 161(11):bqaa175. doi: 10.1210/endocr/bqaa175
58. Handgraaf S, Riant E, Fabre A, Waget A, Burcelin R, Lière P, et al. Prevention of obesity and insulin resistance by estrogens requires ERα activation function-2 (ERαAF-2), whereas ERαAF-1 is dispensable. Diabetes (2013) 62:4098–108. doi: 10.2337/db13-0282
59. Jelenik T, Roden M. How estrogens prevent from lipid-induced insulin resistance. Endocrinology (2013) 154:989–92. doi: 10.1210/en.2013-1112
60. Kang S, Park YM, Kwon DJ, Chung YJ, Namkung J, Han K, et al. Reproductive life span and severe hypoglycemia risk in postmenopausal women with type 2 diabetes mellitus. Diabetes Metab J (2022) 46:578–91. doi: 10.4093/dmj.2021.0135
61. Pratchayasakul W, Sa-Nguanmoo P, Sivasinprasasn S, Pintana H, Tawinvisan R, Sripetchwandee J, et al. Obesity accelerates cognitive decline by aggravating mitochondrial dysfunction, insulin resistance and synaptic dysfunction under estrogen deprived conditions. Horm Behav (2015) 72:68–77. doi: 10.1016/j.yhbeh.2015.04.023
62. Cisternas P, Inestrosa NC. Brain glucose metabolism:Role of wnt signaling in the metabolic impairment in alzheimer's disease. Neurosci Biobehav Rev (2017) 80:316–28. doi: 10.1016/j.neubiorev.2017.06.004
63. Siesjo B. Brain energy metabolism and catecholaminergic activity in hypoxia, hypercapnia and ischemia. J Neural Transm Suppl (1978) 14:17–22. doi: 10.2337/db12-0365db12-0365
64. Sokoloff L. Relation between physiological function and energy metabolism in the central nervous system. J Neurochem (1977) 29(1):13–26. doi: 10.1111/j.1471-4159.1977.tb03919.x
65. Vannucci RC, Yager JY, Vannucci SJ. Cerebral glucose and energy utilization during the evolution of hypoxic-ischemic brain damage in the immature rat. J Cereb Blood Flow Metab (1994) 14(2):279–88. doi: 10.1038/jcbfm.1994.35
66. Vannucci SJ. Developmental expression of GLUT1 and GLUT3 glucose transporters in rat brain. J Neurochem (1994) 62(1):240–6. doi: 10.1046/j.1471-4159.1994.62010240.x
67. Caprile T, Salazar K, Astuya A, Cisternas P, Silva-Alvarez C, Montecinos H, et al. The na+-dependent l-ascorbic acid transporter SVCT2 expressed in brainstem cells, neurons, and neuroblastoma cells is inhibited by flavonoids. J Neurochem (2009) 108:563–77. doi: 10.1111/j.1471-4159.2008.05788.x
68. Cisternas P, Silva-Alvarez C, Martínez F, Fernandez E, Ferrada L, Oyarce K, et al. The oxidized form of vitamin c, dehyd, oascorbic acid, regulates neuronal energy metabolism. J Neurochem (2014) 129:663–71. doi: 10.1111/jnc.12663
69. Vannucci SJ, Reinhart R, Maher F, Bondy CA, Lee WH, Vannucci RC, et al. Alterations in GLUT1 and GLUT3 glucose transporter gene expression following unilateral hypoxia-ischemia in the immature rat brain. Brain Res Dev Brain Res (1998) 107:255–64. doi: 10.1016/s0165-3806(98)00021-2
70. Fernando RN, Albiston AL. Chai SY the insulin-regulated aminopeptidase IRAP is colocalised with GLUT4 in the mouse hippocampus–potential role in modulation of glucose uptake in neurones? Eur J Neurosci (2008) 28:588–98. doi: 10.1111/j.1460-9568.2008.06347.x
71. Grillo CA, Piroli GG, Hendry RM, Reagan LP. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res (2009) 1296:35–45. doi: 10.1016/j.brainres.2009.08.005
72. Chen Z, Zhong C. Decoding alzheimer’s disease from perturbed cerebral glucose metabolism:implications for diagnostic and therapeutic strategies. Prog Neurobiol (2013) 108:21–43. doi: 10.1016/j.pneurobio.2013.06.004S0301-0082(13)00053-1
73. Bolaños JP, Almeida A, Moncada S. Glycolysis:a bioenergetic or a survival pathway? Trends Biochem Sci (2010) 35:145–9. doi: 10.1016/j.tibs.2009.10.006S0968-0004(09)00207-2
74. Bolaños JP, Almeida A. The pentose-phosphate pathway in neuronal survival against nitrosative stress. IUBMB Life (2010) 62:14–8. doi: 10.1002/iub.280
75. Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol (2009) 11:747–52. doi: 10.1038/ncb1881
76. Brekke E, Morken TS, Sonnewald U. Glucose metabolism and astrocyte-neuron interactions in the neonatal brain. Neurochem Int (2015) 82:33–41. doi: 10.1016/j.neuint.2015.02.002
77. Carey M, Kehlenbrink S, Hawkins M. Evidence for central regulation of glucose metabolism. J Biol Chem (2013) 288:34981–8. doi: 10.1074/jbc.R113.506782R113.506782
78. Kann O, Kovacs R. Mitochondria and neuronal activity. AJP Cell Physiol (2006) 292:C641–57. doi: 10.1152/ajpcell.00222.2006
79. Dringen R. Glutathione metabolism and oxidative stress in neurodegeneration. Eur J Biochem (2000) 267:4903, ejb1651. doi: 10.1046/j.1432-1327.2000.01651.x
80. Doraiswamy PM, Sperling RA, Coleman RE, Johnson KA, Reiman EM, Davis MD, et al. Amyloid-β assessed by florbetapir f 18 PET and 18-month cognitive decline:a multicenter study. Neurology (2012) 79:1636–44. doi: 10.1212/WNL.0b013e3182661f74
81. Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain:mechanistic implications. Ann Neurol (2005) 57:695–703. doi: 10.1002/ana.20474
82. Harr SD, Simonian NA, Hyman BT. Functional alterations in alzheimer's disease:decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J Neuropathol Exp Neurol (1995) 54(1):38–41. doi: 10.1097/00005072-199501000-00005
83. Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with alzheimer’s disease. Ann Neurol (1994) 35:546–51. doi: 10.1002/ana.410350507
84. Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment:a pilot clinical trial. Arch Neurol (2012) 69:29–38. doi: 10.1001/archneurol.2011.233archneurol.2011.233
85. Parthsarathy V, Holscher C. Chronic treatment with the GLP1 analogue liraglutide increases cell proliferation and differentiation into neurons in an AD mouse model. PloS One (2013) 8:e58784. doi: 10.1371/journal.pone.0058784PONE-D-12-21092
86. Mosconi L, Berti V, Quinn C, McHugh P, Petrongolo G, Osorio RS, et al. Perimenopause and emergence of an alzheimer's bioenergetic phenotype in brain and periphery. PloS One (2017) 12(10):e0185926. doi: 10.1371/journal.pone.0185926
87. Rios JA, Cisternas P, Arrese M, Barja S, Inestrosa NC. Is alzheimer’s disease related to metabolic syndrome? a wnt signaling conundrum. Prog Neurobiol (2014) 121:125–46. doi: 10.1016/j.pneurobio.2014.07.004
88. Gibson GE. Interactions of oxidative stress with cellular calcium dynamics and glucose metabolism in alzheimer’s disease. Free Radic Biol Med (2002) 32:1061–70. doi: 10.1016/S0891-5849(02)00802-X
89. Kristian T. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium (2004) 36:221–33. doi: 10.1016/j.ceca.2004.02.016S0143416004000600
90. Silva-Alvarez C, Arrázola MS, Godoy JA, Ordenes D, Inestrosa NC. Canonical wnt signaling protects hippocampal neurons from aβ oligomers:role of non-canonical wnt-5a/Ca(2+) in mitochondrial dynamics. Front Cell Neurosci (2013) 7:97. doi: 10.3389/fncel.2013.00097
91. Cisternas P, Salazar P, Silva-Álvarez C, Barros LF, Inestrosa NC. Wnt5a increases the glycolytic rate and the activity of the pentose phosphate pathway in cortical neurons. Neural Plast (2016) 2016:983948. doi: 10.1155/2016/9839348
92. Bolaños JP, Almeida A. Modulation of astroglial energy metabolism by nitric oxide. Antioxid Redox Signal (2006) 8:955–65. doi: 10.1089/ars.2006.8.955
93. Cisternas P, Salazar P, Silva-Álvarez C, Barros LF, Inestrosa NC. Activation of wnt signaling in cortical neurons enhances glucose utilization through glycolysis. J Biol Chem (2016) 291(50):25950–64. doi: 10.1074/jbc.M116.735373
94. Serrano FG, Tapia-Rojas C, Carvajal FJ, Hancke J, Cerpa W, Inestrosa NC. Andrographolide reduces cognitive impairment in young and mature AβPPswe/PS-1 mice. Mol Neurodegener (2014) . 9:61. doi: 10.1186/1750-1326-9-61
95. Tapia-Rojas C, Schüller A, Lindsay CB, Ureta RC, Mejias-Reyes C, Hancke J, et al. Andrographolide activates the canonical wnt signalling pathway by a mechanism that implicates the non-ATP competitive inhibition of GSK-3β:autoregulation of GSK-3β in vivo. Biochem J (2015) 466:415–30. doi: 10.1042/BJ20140207
96. Gherardelli C, Cisternas P, Gutiérrez J, Martinez M, Inestrosa NC. Andrographolide restores glucose uptake in rat hippocampal neurons. J Neurochem (2021) 157(4):1222–33. doi: 10.1111/jnc.15229
Keywords: wnt signaling, menopause, estrogen, glucose brain metabolism, alzheimer´s disease, tau phosphorylation, APP - amyloid precursor protein
Citation: Villaseca P, Cisternas P and Inestrosa NC (2022) Menopause and development of Alzheimer’s disease: Roles of neural glucose metabolism and Wnt signaling. Front. Endocrinol. 13:1021796. doi: 10.3389/fendo.2022.1021796
Received: 17 August 2022; Accepted: 28 September 2022;
Published: 19 October 2022.
Edited by:
Gaetano Santulli, Albert Einstein College of Medicine, United StatesReviewed by:
Benjamín Florán, Instituto Politécnico Nacional de México (CINVESTAV), MexicoCopyright © 2022 Villaseca, Cisternas and Inestrosa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nibaldo C. Inestrosa, bmluZXN0cm9zYUBiaW8ucHVjLmNs