
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 14 February 2023
Sec. Adrenal Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1015773
This article is part of the Research Topic Recent advances in diagnosis and treatment of congenital adrenal hyperplasia due to 21-hydroxylase deficiency View all 12 articles
 Zhiyuan Zhao1†
Zhiyuan Zhao1† Yinjie Gao1†
Yinjie Gao1† Lin Lu1
Lin Lu1 Anli Tong1
Anli Tong1 Shi Chen1
Shi Chen1 Wei Zhang1Xiaoxia Zhang1Bang Sun1Xueyan Wu1
Wei Zhang1Xiaoxia Zhang1Bang Sun1Xueyan Wu1 Jiangfeng Mao1Xi Wang1
Jiangfeng Mao1Xi Wang1 Min Nie1,2*
Min Nie1,2*Objective: To analyze the relationship between genotype and phenotype in 21-Hydroxylase deficiency patients harboring P31L variant and the underlying mechanism.
Methods: A total of 29 Chinese patients with 21-OHD harboring P31L variant were recruited, and the detailed clinical features of the patients were extracted and analyzed retrospectively. The TA clone combined with sequencing of the region containing the promotor and exon1 of CYP21A2 was performed to determine whether the variants in promotor and P31L aligned in cis. We further compared the clinical characteristics of 21-OHD patients between the promoter variant group and no promoter variant group.
Results: Among the 29 patients diagnosed with 21-OHD harboring P31L variant, the incidence of classical simple virilizing form was 62.1%. Thirteen patients owned promoter variants (1 homozygote and 12 heterozygote) and all exhibited SV form. The promoter variants and the P31L variant were located in the same mutant allele as validated by TA cloning and sequencing. There were statistically significant differences in clinical phenotype and 17-OHP level between the patients with and without promoter region variations (P<0.05).
Conclusion: There exists high incidence (57.4%) of SV form among the 21-OHD patients harboring P31L variant, and the underlying mechanism is partially due to both the promoter variants and P31L aligning in cis on one allele. Further sequencing of promoter region will provide important hints for the explanation of phenotype in patients harboring P31L.
21-hydroxylase deficiency (21-OHD), ranks among one of the most frequent inborn errors of the adrenal endocrine metabolism following an autosomal recessive trait (1). It is characterized by the impairment of cortisol synthesis with or without aldosterone deficiency, and increased androgen synthesis (1, 2). Based on the clinical manifestations, 21-OHD can be classified into three types: the classical salt-wasting (SW) form (approximately 75% of the classic 21-OHD), classical simple virilizing (SV) form (about 25% of the classic 21-OHD) and nonclassical form (NC-21OHD). The SW form is characterized by life-threatening adrenal crises in the neonatal period accompanied with hyperandrogenemia causing sexual ambiguity in affected females (3). The classical SV form usually exhibited precocious puberty combining with accelerated linear growth velocity, the affected females present virilization of external genitalia (i.e., clitoromegaly) or urogenital sinus in the early postnatal period (4). The NC-21OHD may be asymptomatic or clinically mild in the early stages and tend to display signs or symptoms of androgen excess until preadolescent, adolescent or young adult period, characterized by hirsutism, acne, menstrual disorders, subfertility and recurrent miscarriage (5–8).
The original cause of 21-OHD can be ascribed to the decrease or abrogation of P450C21 enzyme activity, which is encoded by the CYP21A2 gene. CYP21A2 is located on chromosome 6p21.3 adjacent to a nonfunctional pseudogene CYP21A1P. The CYP21A2 and CYP21A1P genes show a high homology, with a nucleotide identity of 98% in their exon and 96% in their intron sequences (9–11). Approximately 95% of CYP21A2 pathogenic variants are CYP21A1P-derived or large deletions due to non-homologous recombination events in meiotic (3, 12–14).
The clinical phenotype of 21-OHD is usually well correlated with the residual enzyme activity of mutant P450C21 resulted from CYP21A2 variant (1, 15). Variants leading to 0 to 1% enzymatic activity remaining of mutant P450C21 typically correspond to classical SW 21-OHD, such as 30KB deletions, L308Ffs*6(F308+T), R357W, E6 Cluster (I237D/V238E/M240K), 8bp deletion(E3Δ8bp, c.332_339del GAGACTAC, p.G111Vfs∗21), Q319* and c.293-13A/C>G(i2g). Variants resulting in nearly 1% to 2% enzyme activity retaining of mutant P450C21 frequently cause the classical SV 21-OHD, as demonstrated by I173N. Mutant P450C21 preserving 20% to 60% enzyme activity (e.g.,P31L, V282L and P454S) usually bring about the NC-21OHD (16). About 65–70% of patients with 21-OHD are compound heterozygous, while the clinical phenotype is generally considered to be determined by the less severely affected allele (4, 14).
However, some discrepancies between genotype/phenotype correlation had been found in 21-OHD patients. For example, P31L mutant P450C21 usually lead to NC-21OHD phenotype due to its retention of more than 50% residual enzyme activity (17), while the patients with P31L variant might present SV phenotype (8, 18). Some investigators showed that it could be the result of the occurrence of both promoter region variants and P31L in the same allele (19). In this study, we mainly focused on the relationship between genotype and phenotype in 21-OHD patients harboring P31L variant and its mechanism to augment our understanding of 21-OHD.
A total of 29 Chinese patients with 21-OHD harboring P31L variant identified by genetic testing were recruited for this study, who presented to Peking Union Medical College Hospital (Beijing) between 2003 and 2021.
The study was approved by the ethics committee of Peking Union Medical College Hospital (No.JS-2111).
This was a retrospective study. Detailed medical data pertaining to age, sex, previous medical history (clinical diagnosis and history of vulvar surgery), clinical presentations (hirsutism, acne, menstrual abnormalities, clitoromegaly or labial fusion and precocious pubarche), laboratory data including cortisol(F), testosterone(T), 17α-hydroxyprogesterone (17- OHP) and plasma adrenocorticotropic hormone (ACTH) at baseline (without treatment) or after discontinuation of treatment were extracted and analyzed. The variants of CYP21A2 were identified.
Plasma adrenocorticotropic hormone (ACTH) and serum cortisol(F) at 8:00 AM were measured by chemiluminescence immunoassay (Advia Centaur XP, Bayer). Serum testosterone (T) was measured with chemiluminescence (ACS:180; Automatic Chemiluminescence Systems, Bayer). 17α-hydroxyprogesterone (17-OHP) concentrations was determined by radioimmunoassay (Active 17α-OHP Progesterone DSL-5000, DSL). The intra and inter assay coefficients of variation were 5.6% and 6.6% for T, 6.7% and 8.2% for ACTH,5.3% and 5.7% for serum cortisol, 3.9% and 5.6% for 17-OHP, respectively.
Genomic deoxyribonucleic acid (DNA) from the peripheral blood leukocytes were obtained from all patients using a standard procedure (Omega Blood DNA Midi Kit, Omega Bio-Tek, USA). Multiplex Ligation-dependent probe amplification (MLPA) and PCR combined with sequencing were employed to detect the variants in the region between 700 bp upstream from the start codon ATG of CYP21A2 gene and the entire CYP21A2. The specific primer sequences and PCR amplification methods were described previously (20). The sequencing results were compared with the reference sequence NM_000500.9 of the CYP21A2 through the NCBI website to determine the variants. MLPA (P050-C1 CAH Kit, MRC Holland) was performed according to the manufacturer’s instructions.
The subjects harboring variants both in promoter region and P31L were further sequenced. The fragments containing 5’ UTR and exon1 of CYP21A2 were cloned into pMD19T vector by TA cloning kit (Takara, Bio, Inc.) according to the instruction to determine whether the variants in promoter region were aligned with P31L in cis.
All statistical analyses were performed using SPSS (version 26, SPSS Inc., IBM). Shapiro-Wilk test was performed to determine whether the continuous variables conform to normal distribution. The normally distributed quantitative variables were represented as mean ± standard deviation ( ± s), and the non-normally quantitative distributed variables were expressed as median (upper and lower quartiles) [M (Q1, Q3)]. Comparison between two groups was performed using independent t-test or Mann–Whitney U test, as appropriate. Categorical variables were expressed as cases (n) and percentages and were compared by the Chi-square test or Fisher exact test. P value less than 0.05 was considered statistically significant.
Among the 29 patients diagnosed with 21-OHD harboring P31L variant, 18 patients presented classical SV form, and the remaining 11 patients manifested as nonclassical form. The patients in the SV group were younger than NC group (18.72 ± 6.96 years v.s. 25.55 ± 6.49years, respectively) and with higher serum 17-OHP (58.95 ± 26.20ng/ml v.s. 19.91 ± 16.94ng/ml, respectively). No significant differences were found in other laboratory results (Table 1).
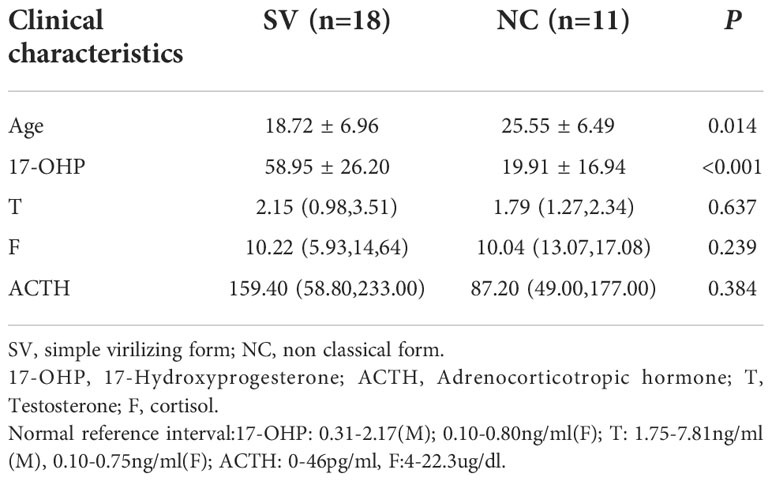
Table 1 Comparison of clinical characteristics of 21-OHD patients harboring P31L between SV and NC group.
A total of 13 patients have the variants in promoter region, and 12 patients were compound heterozygote, while one patient was homozygote (Table 2). TA cloning and sequencing showed that the promoter variants and the P31L variant were located in the same allele, and the detailed promoter variants are listed as follows: -4C→T,-103A→G,-110T→C,-113G→A,-126C→T,-198InsT,-201C→T,-212T→C,-284T→G,-286A→G,-297A→C,-298T→C,-310G→C,-448InsA. The above variants are consistent with the corresponding locus in the CYP21A1P gene (Supplementary Figure S1).

Table 2 Genotypes and phenotypes of 29 patients with 21-OHD harboring P31L.
The 29 patients were divided into promoter variant (PV) group and no promoter variant(NPV) group according to whether the promoter variations exist or not.17-OHP level were higher in the PV group than the NPV group. The clinical phenotype of 21-OHD patients in the PV group were quite different from the NPV group (P<0.05). All the 13 patients in the PV group were SV form, and 9 of them had undergone clitoroplasty. While most (11/16,68.8%) 21-OHD patients in the NPV group showed NC-21OHD form and the 5 other patients SV form. No statistical difference was noted in the other laboratory results and residual enzyme activity of the variant on the other allele (other than P31L located allele) (Table 3).

Table 3 Comparison of clinical characteristics of 21-OHD patients with both promoter variation and P31L variation versus P31L variation alone.
This study investigated the relation of genotype and phenotype and the occurrence of promoter variants among 21-OHD patients harboring P31L variant in a single medical center. Our findings demonstrated that the incidence of SV was 57.4% in 21-OHD patients harboring P31L variant, of which 84.6% patients were caused by promoter variants besides P31L on the same allele of CYP21A2.
In recessive disorders, the clinical phenotype of 21-OHD is usually determined by the activity of the milder variant in compound heterozygotes. According to the in vitro studies, residual enzyme activity of the P31L mutant usually leads to a relatively mild NC-21OHD (21, 22). In this study, the ratio of the classical SV to the NC-21OHD female patients harboring P31L variants was 18:11. Another study from Argentina found that the ratio of the SV form to the NC-21OHD patients with P31L variants was 1:1 (23). Other studies have found that in different 21-OHD patient series harboring P31L variants, the incidence of SV clinical phenotype ranged from 20% to 100% (4, 22, 24–29). As described in previous literature (30), comparing with other variants causing NC-21OHD, the patients affected by P31L tended to have more severe clinical phenotype and even exhibit the SV form. Our study showed the similar results, which strongly indicates that there also existed the genotype/phenotype discrepancies among the Chinese 21-OHD patients harboring P31L. For 21-OHD patients harboring P31L variants, additional attention should be paid to whether more severe clinical phenotype exists or not, so as to provide important basis for precise clinical intervention.
In this study, we found that all the 21-OHD patients with promoter variants in cis with the P31L variant presented SV phenotype. These results suggest that, in addition to the impairment of P450C21 protein activity by the P31L variant, promoter variants may further affect the function of the enzyme, and some evidence supported it. It was reported that c.-113G>A variant of CYP21A2 could reduce the basal transcriptional activity to 20% of CYP21A2 (31), and the c.-126C>T could decrease the transcriptional activity of CYP21A2 to 52% (32). The transcriptional activity of variants c.-126C > T, c.-113G > A, c.-110T > C and c.-103T > C in the promoter is reduced to 20% of the wild type and correlated with the SV 21-OHD (20, 33, 34). Our TA clone sequencing results demonstrated that CYP21A2 promoter variants within c.-500bp to c.-1bp upstream of the ATG occurred in all thirteen patients, including c.-448InsA, c.-310G>C, c.-298T>C, c.-297A>C, c.-286A>G, c.-284T>G, c.-212T>C, c.-201C>T, c.-198InsT, c.-126C>T, c.-113G>A, c.-110T>C, c.-103A>G, c.-4C>T, along with c.91C>T (P31L), are consistent with the corresponding locus of CYP21A1P, implying that the gene conversion from the CYP21A1P to the CYP21A2. For 21-OHD patients tested out P31L variant, further sequencing of genetic locus in the promoter region are needed to obtain the comprehensive and complete molecular diagnosis results, thus providing important basis for subsequent precise clinical treatment and reliable genetic risk assessment.
In our study, the promoter region variation might account for 72.2% of all the classical SV form 21-OHD patients harboring P31L, which is similar to the previous results (19, 23, 34) (Table 4), and their general occurrence of promoter variant in SV 21-OHD patients harboring P31L were 84.6%. Our study demonstrated that 5 patients in the NPV group also showed the clinical phenotype of SV form. This implied that other reasons (besides promoter variants) could account for the more severe clinical phenotype of 21-OHD patients haboring P31L. The following three mechanisms were reported. First, CYP3A7 gene and its transcriptional regulator constitutive androstane receptor (CAR) might be involved in fetal virilization in female 21-OHD. Specifically, the CAR variant could account for a higher degree of external genitalia virilization (35). Second, CAG repetition in the exon 1 of androgen receptor gene matters and SV 21-OHD patients tended to have fewer CAG repeats (36). Third, the alternative pathway of androgen biosynthesis during embryonic development and corresponding proteins (SRD5A1, AKR1C1/3, HSD17B6, etc.) may be involved in fetal virilization in females, through which the 17-OHP accumulated in the 21-OHD may be converted to the androgen and thus aggravate female virilization (37).

Table 4 The summary of incidence of SV phenotype and promoter variants in 21-OHD patients harboring P31L.
There are some limitations to this research. This was a retrospective study, hence, the medical information, including the neonatal virilization of external genitalia, might have been affected by recall bias. This might have resulted in an underestimation of the incidence of SV form among the 21-OHD patients harboring P31L variant.
There exists high incidence (57.4%) of SV form among the 21-OHD patients harboring P31L variant, and the underlying mechanism is partially due to both the promoter variants and P31L aligning in cis on one allele. Further sequencing of promoter region will provide important hints for the explanation of phenotype in patients harboring P31L.
The original contributions presented in the study are included in the article/supplementary files, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by ethics committee of Peking Union Medical College Hospital (No.JS-2111). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
MN conceived the project, designed the experiments. LL, AT, SC, XiW, JM and XuW collected the clinical data, YG, WZ, BS and ZZ performed the experiments. MN and ZZ analyzed the data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS, Grant No.2021-I2M-1-003), National High Level Hospital Clinical Research Funding (Grant No.2022-PUMCH-D-002), National Natural Science Foundation of China (Grant No.81971375), Natural Science Foundation of Beijing (Grant No. 7212080 and No. 7202151).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.1015773/full#supplementary-material
1. Merke DP, Auchus RJ. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med (2020) 383(13):1248–61. doi: 10.1056/NEJMra1909786
2. El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet (2017) 390(10108):2194–210. doi: 10.1016/s0140-6736(17)31431-9
3. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An endocrine society clinical practice guideline. J Clin Endocrinol Metab (2018) 103(11):4043–88. doi: 10.1210/jc.2018-01865
4. New MI, Abraham M, Gonzalez B, Dumic M, Razzaghy-Azar M, Chitayat D, et al. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci U S A (2013) 110(7):2611–6. doi: 10.1073/pnas.1300057110
5. Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou A-M, et al. The spectrum of clinical, hormonal and molecular findings in 280 individuals with nonclassical congenital adrenal hyperplasia caused by mutations of the CYP21A2 gene. Clin Endocrinol (Oxf) (2015) 82(4):543–9. doi: 10.1111/cen.12543
6. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, Tardy V, Billaud L, Laborde K, et al. Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. J Clin Endocrinol Metab (2009) 94(5):1570–8. doi: 10.1210/jc.2008-1582
7. Nordenström A, Falhammar H. MANAGEMENT OF ENDOCRINE DISEASE: Diagnosis and management of the patient with non-classic CAH due to 21-hydroxylase deficiency. Eur J Endocrinol (2019) 180(3):R127–r145. doi: 10.1530/eje-18-0712
8. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: Clinical presentation, diagnosis, treatment, and outcome. Endocrine (2015) 50(1):32–50. doi: 10.1007/s12020-015-0656-0
9. Yang Z, Mendoza AR, Welch TR, Zipf WB, Yu CY. Modular variations of the human major histocompatibility complex class III genes for serine/threonine kinase RP, complement component C4, steroid 21-hydroxylase CYP21, and tenascin TNX (the RCCX module). A mechanism for gene deletions and disease associations. J Biol Chem (1999) 274(17):12147–56. doi: 10.1074/jbc.274.17.12147
10. White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci U S A (1986) 83(14):5111–5. doi: 10.1073/pnas.83.14.5111
11. Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: A pseudogene and a genuine gene. Proc Natl Acad Sci U S A (1986) 83(9):2841–5. doi: 10.1073/pnas.83.9.2841
12. Chen JM, Cooper DN, Chuzhanova N, Férec C, Patrinos GP. Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet (2007) 8(10):762–75. doi: 10.1038/nrg2193
13. Parsa AA, New MI. Steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia. J Steroid Biochem Mol Biol (2017) 165(Pt A):2–11. doi: 10.1016/j.jsbmb.2016.06.015
14. Concolino P, Costella A. Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency: A comprehensive focus on 233 pathogenic variants of CYP21A2 gene. Mol Diagn Ther (2018) 22(3):261–80. doi: 10.1007/s40291-018-0319-y
15. Riedl S, Röhl FW, Bonfig W, Brämswig J, Richter-Unruh A, Fricke-Otto S, et al. Genotype/phenotype correlations in 538 congenital adrenal hyperplasia patients from Germany and Austria: discordances in milder genotypes and in screened versus prescreening patients. Endocr Connect (2019) 8(2):86–94. doi: 10.1530/ec-18-0281
16. Hannah-Shmouni F, Chen W, Merke DP. Genetics of congenital adrenal hyperplasia. Endocrinol Metab Clin North Am (2017) 46(2):435–58. doi: 10.1016/j.ecl.2017.01.008
17. Hannah-Shmouni F, Morissette R, Sinaii N, Elman M, Prezant TR, Chen WY, et al. Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US ashkenazi jews and caucasians. Genet Med (2017) 19(11):1276–9. doi: 10.1038/gim.2017.46
18. New MI. Extensive clinical experience: Nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab (2006) 91(11):4205–14. doi: 10.1210/jc.2006-1645
19. Dolzan V, Stopar-Obreza M, Zerjav-Tansek M, Breskvar K, Krzisnik C, Battelino T. Mutational spectrum of congenital adrenal hyperplasia in Slovenian patients: a novel Ala15Thr mutation and Pro30Leu within a larger gene conversion associated with a severe form of the disease. Eur J Endocrinol (2003) 149(2):137–44. doi: 10.1530/eje.0.1490137
20. Zhang HJ, Yang J, Zhang MN, Zhang W, Liu JM, Wang WQ, et al. Variations in the promoter of CYP21A2 gene identified in a Chinese patient with simple virilizing form of 21-hydroxylase deficiency. Clin Endocrinol (Oxf) (2009) 70(2):201–7. doi: 10.1111/j.1365-2265.2008.03356.x
21. Neves Cruz J, da Costa KS, de Carvalho TAA, de Alencar NAN. Measuring the structural impact of mutations on cytochrome P450 21A2, the major steroid 21-hydroxylase related to congenital adrenal hyperplasia. J Biomol Struct Dyn (2020) 38(5):1425–34. doi: 10.1080/07391102.2019.1607560
22. Tusie-Luna MT, Speiser PW, Dumic M, New MI, White PC. A mutation (Pro-30 to leu) in CYP21 represents a potential nonclassic steroid 21-hydroxylase deficiency allele. Mol Endocrinol (1991) 5(5):685–92. doi: 10.1210/mend-5-5-685
23. Marino R, Ramirez P, Galeano J, Garrido NP, Rocco C, Ciaccio M, et al. Steroid 21-hydroxylase gene mutational spectrum in 454 argentinean patients: genotype-phenotype correlation in a large cohort of patients with congenital adrenal hyperplasia. Clin Endocrinol (Oxf) (2011) 75(4):427–35. doi: 10.1111/j.1365-2265.2011.04123.x
24. Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency? comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J Clin Endocrinol Metab (2000) 85(3):1059–65. doi: 10.1210/jcem.85.3.6441
25. Kocova M, Anastasovska V, Bitovska I. The impact of CYP21A2 (P30L/I172N) genotype on female fertility in one family. Eur J Med Res (2019) 24(1):21. doi: 10.1186/s40001-019-0379-4
26. Zhang B, Lu L, Lu Z. Molecular diagnosis of Chinese patients with 21-hydroxylase deficiency and analysis of genotype-phenotype correlations. J Int Med Res (2017) 45(2):481–92. doi: 10.1177/0300060516685204
27. Gurgov S, Bernabé KJ, Stites J, Cunniff CM, Lin-Su K, Felsen D, et al. Linking the degree of virilization in females with congenital adrenal hyperplasia to genotype. Ann N Y Acad Sci (2017) 1402(1):56–63. doi: 10.1111/nyas.13370
28. Anastasovska V, Milenković T, Kocova M. Direct molecular diagnosis of CYP21A2 point mutations in Macedonian and Serbian patients with 21-hydroxylase deficiency. J Med Biochem (2015) 34(1):52–7. doi: 10.2478/jomb-2014-0048
29. Dracopoulou-Vabouli M, Maniati-Christidi M, Dacou-Voutetakis C. The spectrum of molecular defects of the CYP21 gene in the Hellenic population: variable concordance between genotype and phenotype in the different forms of congenital adrenal hyperplasia. J Clin Endocrinol Metab (2001) 86(6):2845–8. doi: 10.1210/jcem.86.6.7574
30. Kocova M, Anastasovska V, Falhammar H. Clinical outcomes and characteristics of P30L mutations in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine (2020) 69(2):262–77. doi: 10.1007/s12020-020-02323-3
31. Chin KK, Chang SF. The -104G nucleotide of the human CYP21 gene is important for CYP21 transcription activity and protein interaction. Nucleic Acids Res (1998) 26(8):1959–64. doi: 10.1093/nar/26.8.1959
32. Araújo RS, Mendonca BB, Barbosa AS, Lin CJ, Marcondes JAM, Billerbeck AEC, et al. Microconversion between CYP21A2 and CYP21A1P promoter regions causes the nonclassical form of 21-hydroxylase deficiency. J Clin Endocrinol Metab (2007) 92(10):4028–34. doi: 10.1210/jc.2006-2163
33. Chang SF, Chung BC. Difference in transcriptional activity of two homologous CYP21A genes. Mol Endocrinol (1995) 9(10):1330–6. doi: 10.1210/mend.9.10.8544841
34. Araujo RS, Billerbeck AE, Madureira G, Mendonca BB, Bachega TA. Substitutions in the CYP21A2 promoter explain the simple-virilizing form of 21-hydroxylase deficiency in patients harbouring a P30L mutation. Clin Endocrinol (Oxf) (2005) 62(2):132–6. doi: 10.1111/j.1365-2265.2005.02184.x
35. Kaupert LC, Lemos-Marini SH, De Mello MP, Moreira RP, Brito VN, Jorge AAL, et al. The effect of fetal androgen metabolism-related gene variants on external genitalia virilization in congenital adrenal hyperplasia. Clin Genet Nov (2013) 84(5):482–8. doi: 10.1111/cge.12016
36. Moura-Massari VO, Cunha FS, Gomes LG, Gomes DBD, Marcondes JAM, Madureira G, et al. The presence of clitoromegaly in the nonclassical form of 21-hydroxylase deficiency could be partially modulated by the CAG polymorphic tract of the androgen receptor gene. PloS One (2016) 11(2):e0148548. doi: 10.1371/journal.pone.0148548
Keywords: classical simple virilizing, promoter variation, 21- hydroxylase deficiency, P31L, congenital adrenal hyperplasia
Citation: Zhao Z, Gao Y, Lu L, Tong A, Chen S, Zhang W, Zhang X, Sun B, Wu X, Mao J, Wang X and Nie M (2023) The underlying cause of the simple virilizing phenotype in patients with 21-hydroxylase deficiency harboring P31L variant. Front. Endocrinol. 13:1015773. doi: 10.3389/fendo.2022.1015773
Received: 10 August 2022; Accepted: 11 November 2022;
Published: 14 February 2023.
Edited by:
Henrik Falhammar, Karolinska Institutet (KI), SwedenReviewed by:
Tony Yuen, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2023 Zhao, Gao, Lu, Tong, Chen, Zhang, Zhang, Sun, Wu, Mao, Wang and Nie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Nie, bm1fcHVtY2hAYWxpeXVuLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.