
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 10 January 2023
Sec. Reproduction
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1013894
This article is part of the Research Topic Systemic Regulation of Organ Homeostasis and Implications of Hormones and Immunity, Volume II View all 17 articles
 Xiaopan Chen1,2*†
Xiaopan Chen1,2*† Linjie Chen3†
Linjie Chen3† Yang Wang2,4†Chongyi Shu1Yier Zhou1Ruifang Wu1
Yang Wang2,4†Chongyi Shu1Yier Zhou1Ruifang Wu1 Bihui Jin1Leixiang Yang2
Bihui Jin1Leixiang Yang2 Junhui Sun5
Junhui Sun5 Ming Qi6
Ming Qi6 Jing Shu1,4*
Jing Shu1,4*Primary ovarian insufficiency (POI) is among the foremost causes of women infertility due to premature partial or total loss of ovarian function. Resistant ovary syndrome (ROS) is a subtype of POI manifested as normal ovarian reserve but insensitive to gonadotropin stimulation. Inactivating variants of follicle-stimulating hormone receptor (FSHR), a class A G-protein coupled receptor, have been associated with POI and are inherited via an autosomal recessive pattern. In this study, we investigated the genetic causes of a primary infertility patient manifested as POI with ROS, and elucidated the structural and functional impact of variants of uncertain significance. Next-generation sequencing (NGS) combined with Sanger sequencing revealed novel compound heterozygous FSHR variants: c.1384G>C/p.Ala462Pro and c.1862C>T/p.Ala621Val, inherited from her father and mother, respectively. The two altered amino acid sequences, localized in the third and seventh transmembrane helix of FSHR, were predicted as deleterious by in silico prediction. In vitro experiments revealed that the p.Ala462Pro variant resulted in barely detectable levels of intracellular signaling both in cAMP-dependent CRE-reporter activity and ERK activation and displayed a severely reduced plasma membrane receptor expression. In contrast, the p.Ala621Val variant resulted in partial loss of receptor activation without disruption of cell surface expression. In conclusion, two unreported inactivating FSHR variants potentially responsible for POI with ROS were first identified. This study expands the current phenotypic and genotypic spectrum of POI.
Primary ovarian insufficiency (POI) is an etiologically and clinically heterogeneous condition caused by the early loss or complete absence of ovarian activity (1, 2). POI affects roughly 1%–2% of females <40 years old (3), making it among the foremost causes of women infertility. Resistant ovary syndrome (ROS) is a special form of POI characterized by the presence of normal ovarian reserve and resistance to gonadotropin (GN) stimulation (4–6). The etiopathogenesis of POI is multifactorial, including genetic alterations, autoimmune and metabolic disorders, viral infections, and environmental or iatrogenic factors (7). Genetic factors represent the most commonly identified causes; however, the current understanding of its hereditary basis is incomplete (8, 9).
The follicle-stimulating hormone receptor (FSHR) is expressed in granulosa cells from the primary follicle stage onwards (10, 11) and rigidly controls follicle development in response to cyclic pituitary FSH discharge (11, 12). FSHR belongs to a highly conserved subfamily of G protein-coupled receptors with a remarkably long amino-terminal extracellular domain (ECD), an intracellular carboxyl-terminal tail (C-tail), and a typical structural architecture comprising seven transmembrane helixes (TMH) interconnected by three extracellular loops (ECL) and three intracellular loops (ICL) (13, 14). Upon FSH binding, the receptor adopts a conformational change and subsequently couples to Gαs protein, which in turn activates the adenylyl cyclase, resulting in increased cAMP signaling (15, 16). Variations in any of these domains by alterations in primary DNA sequences may potentially lead to receptor dysfunction and eventually to disease (17).
Genetic variants that inactivate protein-coding genes, collectively known as loss-of-function variants, are often single-nucleotide variants that disrupt the structure and function of the protein. FSHR inactivating variants, one of the rare causes of POI, are inherited recessively and require homozygous or compound heterozygous status to develop the clinical phenotype (18). After the first POI-causative FSHR variant-p.Ala189Val in the ECD was reported (19), diverse inactivating variants have been identified in different FSHR domains, including the ECD (20–29); the second, fourth, and sixth TMH (26, 29–34); the first, second, and third ECL (22, 28, 35, 36); second and third ICL (21, 37); and the C-tail (27). However, pathogenic inactivating variants in the third and seventh TMH of FSHR have not been reported so far in patients with POI.
Numerous evidence indicates that the clinical manifestations of FSHR variants are not uniform, ranging from primary amenorrhea with puberty disorders to secondary amenorrhea, oligomenorrhea, and premature menopause, depending on the specific domain and type of variant and the degree of inactivation. Thus, expanding the POI-causing variant spectrum of FSHR is crucial to understand the etiopathogenesis of POI and for better clinical diagnosis that involves patients and their offspring. Herein, we reported two novel FSHR variants (c.1862C>T/p.Ala621Val and c.1384G>C/p.Ala462Pro) in a compound heterozygous state in a Chinese infertile woman who manifests hypergonadotropic hypogonadism (HH) with oligomenorrhea, presence of normal ovarian reserve, and resistance to exogenous GNs: three clinical characteristics of POI with ROS. Using in vitro functional experiments, we characterized the molecular features underlying this disease phenotype, thereby providing new insights into POI with ROS. To our knowledge, this is the first study identifying pathogenic variants in the TMH3 and TMH7 of FSHR as causative variants for POI.
The study procedures were reviewed and approved by the Institutional Ethics Committee of Zhejiang provincial People’s Hospital (Approval number: 2019KY205). Written informed consent was obtained from all participants. All genetic materials were handled in accordance with the National Regulation on Human Genetic Resources.
A 29-year-old Chinese female was referred to our outpatient center for 5 years of primary infertility. The female patient reached menarche at age 15 and had experienced irregular menstrual cycles (15 - 180 days) in a progressively prolonged manner. At the age of 21 years, she consulted a local hospital for irregular periods, and after sex hormonal evaluation, she was diagnosed with ovarian dysfunction and began to intermittently take exogenous hormone replacement therapy to induce artificial menstrual cycles. After the age of 21 years, her menstrual cycle never appeared again without hormone therapy. A dominant follicle was detected during an occasional ultrasound examination when she was 26-year-old. She was otherwise healthy and had an unexceptional past clinical history. No family history of consanguinity, reproductive anomaly or infertility was documented.
Upon examination, the patient’s height and weight were found to be 160 cm and 62.5 kg, respectively. Physical examination of secondary sex characteristics revealed normal breast development, normal appearance of female external genitalia, and normal armpit and pubic hair. Hormonal assays were carried out in the presence of low doses of estrogen (Estradiol Valerate Tablets 1-2mg/d, Delpharm Lille S.A.S., Lys-lez-Lannoy, France). The repeated basal hormonal evaluations in our department revealed elevated serum FSH (35.98 and 37.73 IU/L) and luteinizing hormone (LH) (21.23 and 21 IU/L) (Table 1). The patient’s thyroid stimulating hormone (TSH) level was normal (2.34 mIU/L), as was her prolactin (PRL) (10.03 ng/mL) (Table 1). Despite the increase in GNs, the patient’s anti-Müllerian hormone (AMH) level was 2.89 ng/mL (Table 1), suggesting a normal ovarian reserve. Transvaginal ultrasonography examination displayed a normal-sized uterus and normal-sized bilateral ovaries, consisting of 3–5 small antral follicles 3–5 mm in diameter (Supplemental Figure). The patient had no autoimmune condition, including absence of anti-nuclear and anti-cardiolipin antibodies. Immunoglobulin and natural killer cell levels were within normal ranges. Chromosome analysis and Fragile X DNA test were normal. No other relevant clinical features were observed.

Table 1 Clinical characteristics of the proband.
After failing to observe follicle development in the natural cycle, an initial attempt of controlled ovarian stimulation (COS) was performed by a daily combination of 50 mg clomiphene and 150 IU human menopausal gonadotropins (hMG). However, the stimulation was cancelled after 8 days of treatment with a total dose of 1,200 IU FSH, as no ovarian follicle development was observed (peak serum estradiol level, 39 pg/mL). To exclude the possibility that the ovarian unresponsiveness to exogenous GNs was due to stimulation insufficiency, another COS cycle with increased daily hMG dose and prolonged stimulation was conducted. Despite increasing the daily dose of exogenous GNs up to 375 IU per day, growth of immature follicles remained unresponsive even after 15 days of stimulation with total FSH dose of 4,350 IU. Thus, the patient was clinically diagnosed with ROS according to previously described criteria (6, 38).
The FSH, LH, estrogen (E2), PRL, and TSH concentrations were tested by a set of commercial enzyme immunoassay kits (7K72-78 & 2P13-40, Abbott, Chicago, USA). The ovarian reserve was measured by an AMH detection kit (C86002, YHLO, Shenzhen, China). Monitoring of follicle growth and development was conducted by transvaginal ultrasonography (Hitachi, Tokyo, Japan).
Whole-exome sequencing was conducted on DNA samples isolated from the patient’s peripheral blood leukocytes, according to the manufacturer’s manual (Agilent Technologies, Santa Clara, USA; Illumina, San Diego, USA). In brief, genomic DNA was fragmented, barcoded, purified, and hybridized with capture probes during library preparation for massively parallel sequencing. The captured sequences were then enriched with streptavidin-conjugated paramagnetic beads and further amplified before being subjected to NGS. Clusters were generated and sequenced on the Illumina NextSeq 550 system using the Illumina Nextseq High Output Kit (20024908) to obtain approximately 90 million reads per sample (2 × 151 cycles). After quality assessment, the resulting paired-end reads were mapped to the GRCh37 assembly (hg19) of the human genome. All called variants were annotated based on public databases, including the 1KGB (39), ExAC (39), gnomAD (40), dbSNP (41), OMIM (42), ClinVar (43), and LOVD (44). Major focus of the analysis was on the 4 relevant disease-causing genes (FSHB, FSHR, LHB and LHCGR). Variant interpretation was based on the gene clinical and pathogenic relevance according to ACMG guidelines (45).
Validation of NGS results was conducted by Sanger sequencing. The primers used to amplify target region of the human FSHR gene were designed as follows: 5′-CAAACTGGGGCAGGCTGTGATG-3′ and 5′-CTTGCATTTCATAGCAGCCACAC-3′. All the PCRs were carried out at 50 μl reaction volume using 100 ng genomic DNA on an ABI GeneAmp 9700 PCR system (Applied Biosystems, Bedford, USA), and the amplified fragments were subsequently extracted from the agarose gel, purified, and directly sequenced by Big Dye Direct Cycle Sequencing (4458688, ThermoFisher Scientific, Waltham, USA) on an ABI 3730 Genetic Analyzer (Applied Biosystems, Bedford, USA). The nucleotide sequences were blasted with the published sequence of human FSHR gene (http://www.ncbi.nlm.nih.gov).
Protein topology prediction of FSHR variants was conducted using the TMHMM architecture (46). Variant pathogenicity prediction was performed using three online tools: PolyPhen-2 (47), SIFT (48), and MutationTaster (49). Evolutionary conservation analysis was performed based on multiple-alignment of the amino acids across multispecies from the HomoloGene database.
The wild-type (WT) FSHR plasmid was constructed by subcloning human FSHR cDNA from pCR-BluntII-TOPO-FSHR (P19532, Miaolingbio, Wuhan, China). The obtained polymerase chain reaction products were then cloned into pcDNA3.1 and pEGFP-N1 with the Seamless Cloning Kit (D7010, Beyotime, Shanghai, China). Point mutations were performed by using the QuickMutation Kit (Beyotime, Shanghai, China). The sequences of primers used for plasmid construction were provided in Supplemental Table. The pCRE-luciferase vector was kindly provided by Dr. Naiming Zhou (50). All vectors were thoroughly sequenced to confirm sequence integrity.
The HEK293 cell line was routinely cultured in Dulbecco’s modified Eagle’s medium (Gibco, Waltham, USA) containing 10% heat-inactivated fetal bovine serum (SV30087, Hyclone, Logan, China) as described previously (51). Transfection of plasmid constructs into cells was conducted using lipofectamine 3000 reagent (L3000-015, Invitrogen, Carlsbad, USA). Variants were transfected alone (3 μg) to mimic the homozygous state, or together in equimolar concentration (1.5 μg each) to mimic the compound heterozygous state.
Cells pre-seeded in a 96-well plate were co-transfected with either an expression vector (pWT-cDNA or variants) and reporter vector (pCRE-Luc). At 48 h after transfection, cells were incubated with the DMEM containing various concentrations of recombinant FSH (S20150007, Jinsai, Changchun, China) for 4 h at 37°C. cAMP-dependent CRE-luciferase activity was measured using the firefly luciferase assay system (RG042S, Beyotime, Shanghai, China) with a multiple function microplate reader (Tecan, Männedorf, Switzerland). Briefly, the culture medium in the plate was removed, and 50 μL per well reporter lysis buffer was added immediately. The plate was then placed on a horizontal shaker for complete lysis. The cell lysate was collected and centrifuged in 10,000 × g for 5 min. Then, 20 μl supernatant was mixed with 100 μl luciferase substrate and incubated for 5 min. Finally, the mixture was transferred into a 96-well plate, and luminance was read on Tecan luminometer 1 s/per well at the 470 nM. Efficacy and potency were determined by a three-parameter nonlinear logistic regression using GraphPad Prism 7.0 (Graph Pad Software, San Diego, USA).
Cells transiently expressing wild-type receptor or variants were stimulated with the FSHR agonist (S20150007, Jinsai, Changchun, China) for the indicated durations. Drug incubation was terminated by washing the cells with ice-cold phosphate buffered saline followed by the addition of lysis buffer containing complete protease inhibitor (P1010, Beyotime, Shanghai, China) and phosphatase inhibitors cocktail (P1082, Beyotime, Shanghai, China). Equal protein amounts from cell lysates were electrophoresed on a 10% SDS-polyacrylamide gel, and then transferred to a PVDF membrane and incubated with rabbit monoclonal anti-phospho-ERK1/2 antibody (4370, Cell Signaling, Danvers, USA) followed by stripping and reprobing with anti-total ERK antibody (4695, Cell Signaling, Danvers, USA) according to manufacturers’ protocols. Chemiluminescence was detected using an ECL substrate (FD8000, Fdbio science, Hangzhou, China) with ChemiDoc Touch Imaging System (Bio-Rad, Hercules). All immunoblots were semi-quantified using the Adobe Photoshop CC software, and ERK1/2 activation was calculated as the level of phosphor-ERK1/2 normalized by the total-ERK.
Cells pre-seeded in a coverslip-covered 12-well plate were transiently transfected with pWT-EGFP, pA462P-EGFP, or pA621V-EGFP. At 48 h after transfection, the coverslip seeded with transfected cells was invertedly placed on a microscope slide and observed immediately under a confocal laser-scanning microscope (Leica, Wetzlar, Germany). The transfection efficiency was determined by counting number of cells that express fluorescence divided by the total number of observed cells. Quantification of the FSHR level on cell membrane by their mean fluorescent signal intensity was measured using ImageJ (NIH, Bethesda, USA); moreover, the plugin of MorphoLibJ in ImageJ was used to create segmentation borders. Furthermore, a region of interest area was created for a final measure.
Statistical analysis of in vitro experimentation was performed with GraphPad Prism software using data derived from ≥3 different biological replicates. Collected data were examined for normality of distribution using Shapiro–Wilk normality test. Comparisons between groups were carried out using one-way analysis of variance followed by Tukey’s multiple comparisons tests for post-hoc analysis. Differences between groups were considered statistically significant at P < 0.05.
Bioinformatics analysis following NGS revealed two missense variants. The first variant was a transition of guanine to cytosine at nucleotide position 1384 in Exon 10 producing an Alanine to Proline replacement at position 462 (FSHR_ex10 c.1384G>C/p.Ala462Pro). The second variant was a transition of cytosine to thymine at nucleotide position 1862 in Exon 10 of FSHR gene resulting in an amino acid substitution of Alanine to Valine at position 621 (FSHR_ex10 c.1862C>T/p.Ala621Val). Sanger sequencing confirmed the suspected variants and showed that the patient’s parents were heterozygous carriers; the father and mother being a heterozygous carrier of c.1384G>C/p.Ala462Pro and c.1862C>T/p.Ala621Val variants, respectively (Figures 1A-C). Both variants were neither found in public population databases (1KGB, ExAC, gnomAD, and dbSNP) nor in disease databases (OMIM, ClinVar, and LOVD) (Table 2). Multiple sequence alignments were performed for FSHR proteins form different species; both A462 and A621 were highly evolutionarily conserved over multiple species (Figure 1D and Table 2). Protein topology prediction of the variants revealed that p.Ala462Pro and p.Ala621Val were localized in the third and seventh TMH of FSHR, respectively (Figure 1E). Furthermore, the two variants were predicted to be “Probably damaging” by PolyPhen-2, “Disease causing” by MutationTaster, and “Intolerated” by SIFT, indicating potential pathogenic effects of these two variants (Table 2). Thus, both p.Ala621Val and p.Ala462Pro were ranked as variants of uncertain significance according to ACMG pathogenicity assessing criteria (Table 2).
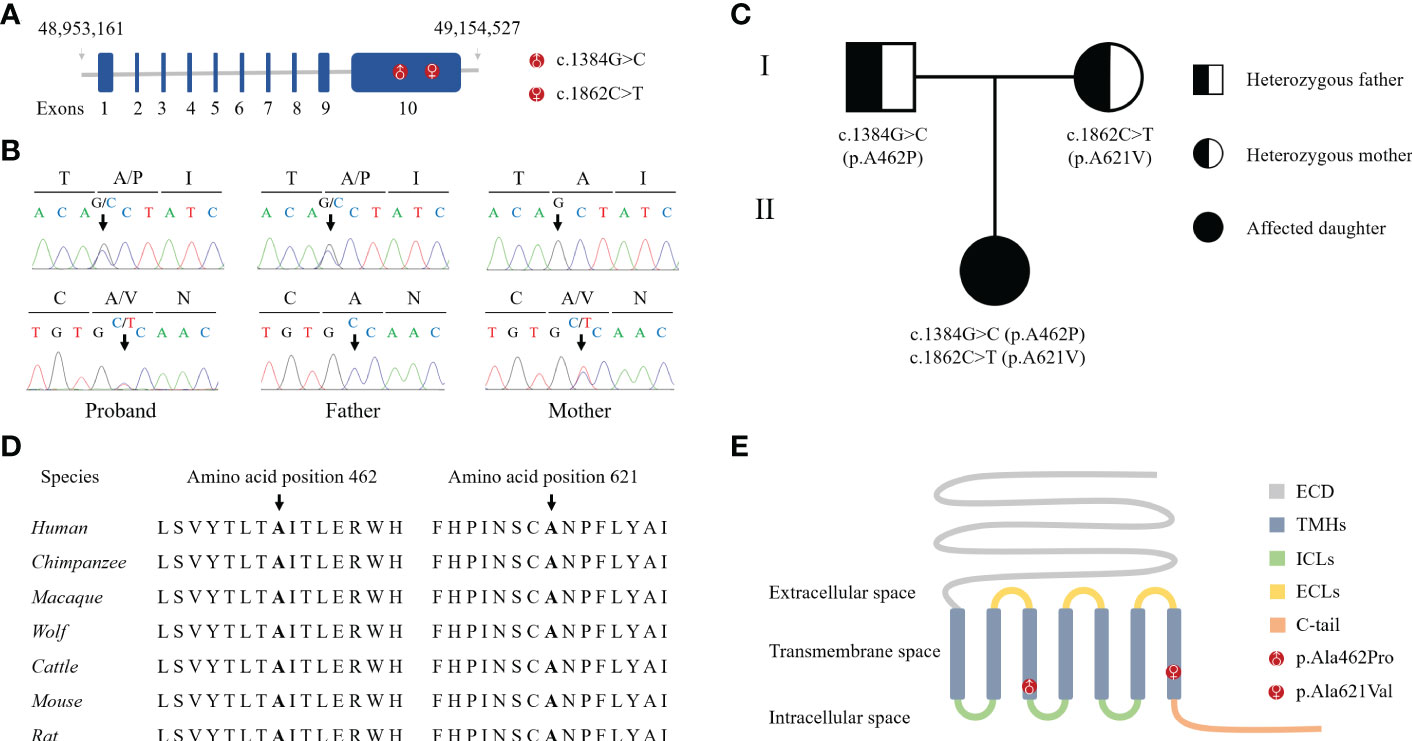
Figure 1 Identification of novel compound heterozygous variants in FSHR. (A) Schematic representation of the hFSHR gene and localization of newly identified variants. Size of exons is drawn to scale. (B) Validation of NGS by Sanger sequencing. (C) Pedigree of the family examined in the present study. (D) Multiple amino acid sequences alignment of FSHR target sites and their two flanks across different species. (E) Schematic representation of hFSHR and localization of newly identified variants.
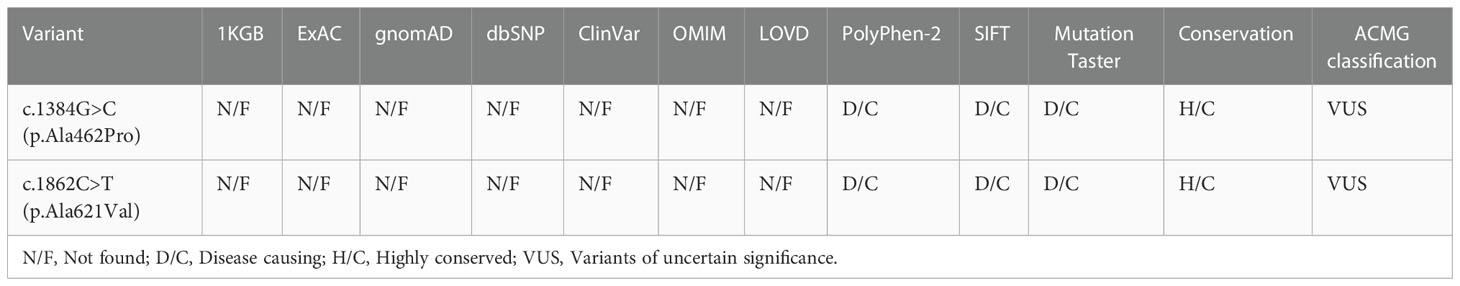
Table 2 Bioinformatic prediction of the FSHR variants.
Examination of FSHR-mediated cAMP-dependent CRE-promoter transcription activity was conducted by co-transfection of HEK293 cells with the variants and CRE-luciferase reporter. In the presence of FSH, a dose-dependent increase in luciferase activity (EC50 = 11.98 IU) was recorded in cells harboring WT FSHR (Figure 2A). However, both FSH efficacy and potency to elicit cAMP-dependent CRE-reporter activity were significantly impaired in cells transfected with p.A621V variant as demonstrated by decreased maximal stimulation (59.72% of WT) and increased EC50 (81 IU) (Figure 2A). The most deteriorating impact in cAMP-dependent CRE-promoter activity was observed in cells expressing the p.A462P variant, whose luciferase activity was barely detectable (maximal stimulation = 4.24% of WT FSHR; EC50 = 32.3 IU) even in the presence 10,000 IU FSH, the highest concentration in our study (Figure 2A). When a blend of A462P and A621V was transfected together in equimolar concentration to mimic the compound heterozygous state found in the proband, there was a significant reduction either in the potency (EC50 = 62.16 IU) or in the efficacy (maximal stimulation = 44.95% of WT) as compared to those observed with the wild-type receptor.
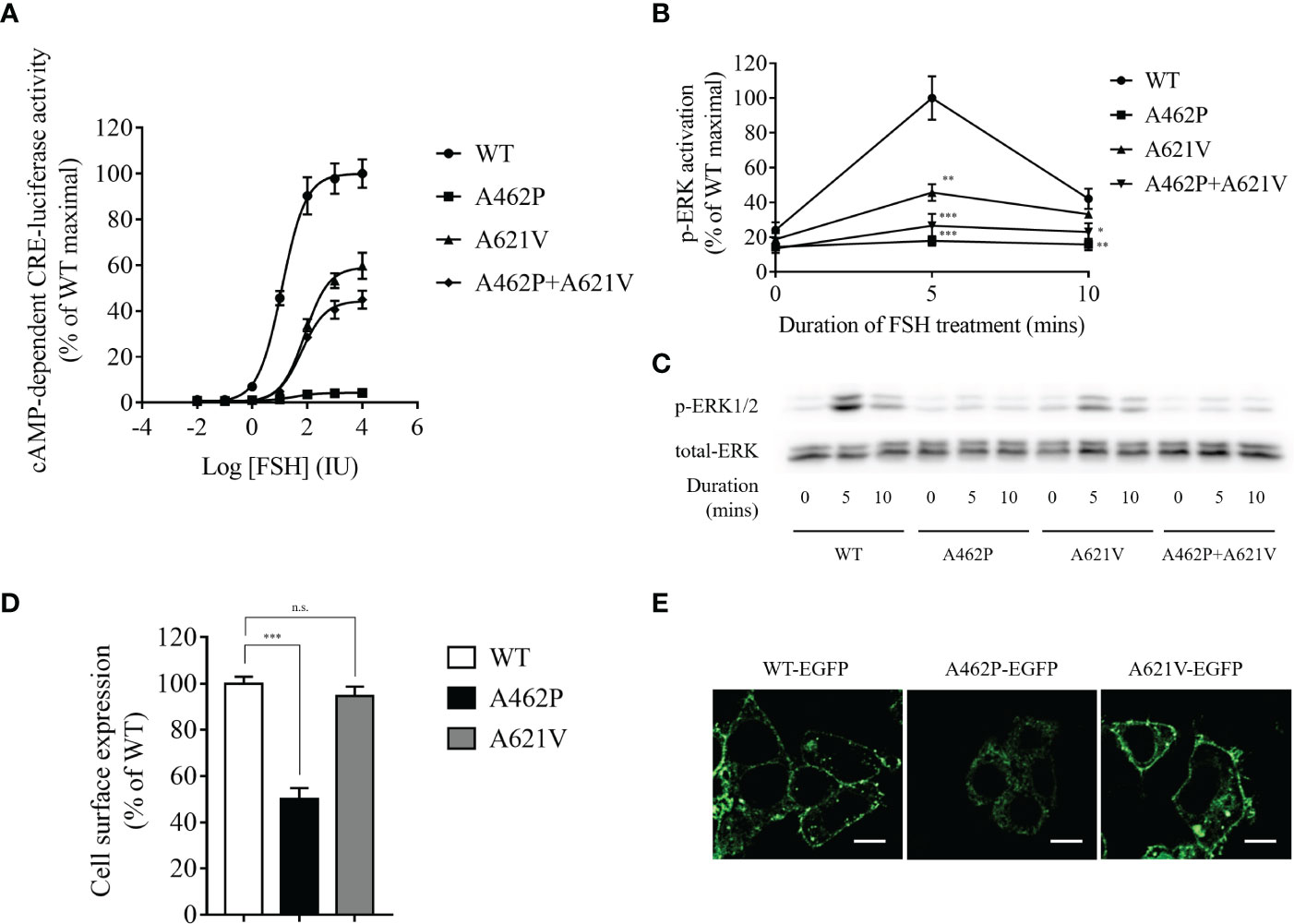
Figure 2 Functional evaluation of variants. (A) Dose-dependent curve of cAMP-dependent CRE-luciferase activity for FSHR variants upon FSH treatment. Variant transfected HEK293 cells were incubated with various FSH concentrations for 4 h. Data are expressed as percentage of WT maximal stimulation and represent the mean ± SEM of six independent experiments. (B) Time course of FSH-stimulated phosphorylation of ERK1/2 in cells expressing variants. Data are expressed as percentage of WT maximal stimulation and represent the mean ± SEM of three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. WT FSHR. (C) Representative images of immunoblots using antibody against phosphorylated ERK1/2. Transfected cells were incubated with 500 IU FSH for the indicated time. (D) Bar plots showing quantitative fluorescence microscopy analysis of cell surface expression for all variants. Data are expressed as percentage of WT FSHR and represent the mean ± SEM of six independent experiments. *** P < 0.001 vs. WT FSHR, n.s. not significant. (E) Representative fluorescence microscopy images of the receptor cellular localization. Bar = 20 µm.
To further explore whether ERK activation is altered by the variants, we performed Western immunoblot using a monoclonal antibody against phosphorylated ERK1/2. As shown in Figures 2B, C, FSHR-related ERK activation was time-dependent with a maximal activation at 5 min and a subsequent reduction to 42.14% of maximal levels at 10 min in WT receptor transfected HEK293 cells after stimulation with 500 IU FSH. We found that receptors homozygous for either variant exhibited significantly reduced ERK activation compared to the WT receptor (Figures 2B, C). The homozygous A462P variant receptor almost completely abolished ERK activation at both treatment periods, whereas homozygous A621V variant receptors carried 45.79% and 33.21% of maximal ERK activation at 5 and 10 min, respectively. When both A462P and A621V variants were expressed together to mimic the compound heterozygous state of the patient, the magnitude of ERK activation in response to 5 and 10 min of FSH stimulation was severely impaired compared with the WT FSHR (Figures 2B, C).
Examination of the cell surface expression of p.A462P and p.A621V was conducted by transfection of HEK293 cells with these variants fused with EGFP. As shown in Figures 2D, E, the p.A621V variant and WT receptor exhibited comparable cell surface expression levels, but the p.A462P variant lost 50% of cell surface expression and was trapped intracellularly, suggesting that the nearly total loss of function in this variant might be due to reduced membrane receptor expression.
In this study, we investigated a case of primary infertility in a woman previously diagnosed with ovarian dysfunction who in fact had POI with ROS; we identified two novel missense variants in the FSHR gene, c.1384G>C/p.Ala462Pro and c.1862C>T/p.Ala621Val, inherited from her father and mother, respectively. These two variants were predicted to be disease causing by bioinformatics analysis. In vitro functional studies confirmed that these two variants are both pathogenic inactivating variants
FSHR variants are functionally distinct with respect to their ligand binding, membrane expression, cAMP production, and other intracellular signaling characteristics (17, 52). The complexity of these variants is further amplified by the fact that causative variant of POI can be either homozygous or compound heterozygous (19, 21, 30, 53, 54). As a consequence, in a single FSHR gene, genetic alteration can result in various clinical manifestations, leading to the broad phenotypic spectrum of conditions arising from these variants. Thus, correlation of novel FSHR variants with a new case of POI characterized by endogenous HH, normal ovarian reserve, and hypo-responsiveness to hyper-physiological exogenous GN stimulation testifies to the phenotypic and etiological complexity of POI.
Patients with partially inactivating FSHR variants have displayed a range of clinical manifestations such as primary amenorrhea, secondary amenorrhea, and oligomenorrhea, which is in contrast to completely inactivating FSHR variants that cause fully absence of sexual development (55). The patient in our study displayed normal puberty and secondary sex characteristics, and presented with secondary amenorrhea and normal-sized bilateral ovaries, in which developing follicles were detected by ultrasonography. This phenotype is notably differed from patients with severe variants (19, 36, 56). The term ROS has been used to describe this special form of POI, which is caused by FSH resistance rather than follicular depletion (21, 22, 26, 27, 33, 54). Although most ROS patients presented with developing antral follicles (2-8 mm) on ultrasonography and follicular arrest at the early antral stage on ovarian biopsies, further maturation of follicle is usually blocked in these patients (21, 22, 24–27). The patient’s follicles seemed incapable of proceeding through the later stages of follicular maturation, but likely produced adequate sex hormones at puberty to drive development of comparable secondary sex characteristics which accords with other studies with less severe variants (21, 22, 26, 27). The above surmise was supported by the normal AMH level detected in our case, which is consistent with previous studies that ROS patients have age-matched AMH values while patients with common POI have low or undetectable levels of AMH (25–27, 31, 54). These observations suggest that POI caused by inactivating FSHR variants could have wide-range effects on clinical manifestations, with disturbance severity likely varying with the severity of inactivation and the resultant stage of follicular block.
The FSHR protein is encoded by the FSHR gene positioned at chromosome 2p21 (57). The two variants herein are both positioned at exon 10, which is the largest exon, mainly responsible for encoding the non-ECD domains. The p.A462P and p.A621V variants are positioned at the TMH3 and TMH7 of the receptor, respectively, where no mutation had formerly been observed in patients. Our in vitro functional study demonstrated that the p.A462P variant causes the intracellular retention of the receptor, leading to impaired FSHR cell membrane expression. Several previous studies have reported the association of lower membrane expression of FSHR variants with impaired intracellular signaling of the receptor (19, 21, 22, 25, 29, 31, 54, 56). In our study, the lower membrane expression of the p.A462P variant was linked with an approximately 96% reduction in the maximal response elicited by the FSH. However, FSHR cell surface localization was not affected by the p.A621V variant, but its intracellular signaling was significantly impaired. Consistent with these results, three previously identified inactivating variants of FSHR showing intact cell surface expression displayed a dramatic reduction in cAMP signaling (21, 22, 27). Partially functional impaired FSHR caused by variants affects the further development of follicular growth (21, 28, 31, 55), indicating that the partial functional impairment of FSHR mediated by these two novel variants might explain the follicles advanced to the small antral phase and normal AMH levels found in this patient.
Even after receiving a maximal daily FSH dose (375 IU), the patient’s ovaries still remained unresponsive, which was consistent with several previous studies where partial or complete inactivating FSHR variants could not respond to intense and continual FSH stimulation (21, 22, 27, 28, 34, 36, 54, 58). It is worth noting that only one study has reported successful FSH treatment for ovarian stimulation in patient with ROS (24). The patient with successful treatment carried one heterozygous variant and two heterozygous SNPs. These clinical findings demonstrate that the differences in the ovarian responses to FSH stimulation noted between the successful and failed cases likely depend on the FSHR genotype. Patients who failed to react to follicular stimulation following an in vitro fertilization cycle could consider in vitro maturation (IVM) of the oocytes isolated from the blocked follicles. Successful IVM of oocytes was found in a primary infertility patient who carried compound heterozygous FSHR variants and had no response to FSH stimulation (54). Recently, Benammar et al. also reported a successful case of IVM live birth in a woman with primary infertility and ovarian resistance to FSH cause by compound heterozygous FSHR variants (58).
The lack of ovarian response to excessive FSH stimulation seems contradictory with the residual intracellular signaling observed in vitro. However, although in vitro functional data can be used as reference for in vivo scenarios, the selection of functional assay model may affect in vitro FSHR functionality (28). Moreover, receptor expression levels resulting from in vitro overexpression are far higher than in in vivo physiological environments, indicating that the real intracellular signaling of these variants in response to FSH stimulation would be probably poor in vivo. This observation suggests that in the presence of adequate FSH stimulation, partially functional but impaired FSHR might be critical for follicular growth.
In conclusion, we uncovered two novel FSHR variants, c.1384G>C/p.A462P and c.1862C>T/p.A621V, in a primary infertility patient affected by POI with ROS. To the best of our knowledge, these variants represent the first pathogenic inactivating variants found in the third and seventh TMH of FSHR. This study offers novel insights into the molecular basis of POI, expands the current knowledge of genotype–phenotype correlations and genetic variant spectrum of POI, and promotes the clinical diagnosis of patients with POI.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA687912; https://www.ncbi.nlm.nih.gov/, SCV001478347; https://www.ncbi.nlm.nih.gov/, SCV001478348.
The studies involving human participants were reviewed and approved by The Institutional Ethics Committee of Zhejiang provincial People’ Hospital. The patients/participants provided their written informed consent to participate in this study.
XC was responsible for the experimental design, critical data analysis, interpretation, article writing and editing, and participated in conducting experimental procedures and coordination. LC, YW, CS, YZ, and LY conducted the main experimental procedures, and participated in experimental protocol design, data analysis and interpretation. BJ and RW contributed to the clinical data collection and interpretation. JHS and MQ contributed to the genetic counseling and data interpretation. JS conceived the study, participated in experimental design and coordination, and was responsible for the project supervision and final approval of the article. All authors contributed to the article and approved the submission version.
This work was supported by grants from National Natural Science Foundation of China (81701460 to XC), General Research Program for Medicine and Health of Zhejiang Province (2020KY414 to JS, 2019KY021 to RW, and 2020KY448 to XC), Natural Science Foundation of Zhejiang Province (LY20H160046 to LY, LQ19H040005 to BJ), and Key Laboratory of Reproductive Genetics (Zhejiang University), Ministry of Education, P. R. China (ZDFY2020-RG-0006 to XC).
We would like to thank the patient and her parents for their participation in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.1013894/full#supplementary-material
1. Nelson LM. Clinical practice. primary ovarian insufficiency. N Engl J Med (2009) 360(6):606–14. doi: 10.1056/NEJMcp0808697
2. Welt CK. Primary ovarian insufficiency: A more accurate term for premature ovarian failure. Clin Endocrinol (Oxf) (2008) 68(4):499–509. doi: 10.1111/j.1365-2265.2007.03073.x
3. Coulam CB, Adamson SC, Annegers JF. Incidence of premature ovarian failure. Obstet Gynecol (1986) 67(4):604–6.
4. Jones GS, De Moraes-Ruehsen M. A new syndrome of amenorrhae in association with hypergonadotropism and apparently normal ovarian follicular apparatus. Am J Obstet Gynecol (1969) 104(4):597–600. doi: 10.1016/S0002-9378(16)34255-7
5. Koninckx PR, Brosens IA. The "gonadotropin-resistant ovary" syndrome as a cause of secondary amenorrhea and infertility. Fertil Steril (1977) 28(9):926–31. doi: 10.1016/S0015-0282(16)42792-5
6. Shangold MM, Turksoy RN, Bashford RA, Hammond CB. Pregnancy following the "insensitive ovary syndrome". Fertil Steril (1977) 28(11):1179–81. doi: 10.1016/S0015-0282(16)42914-6
7. Huhtaniemi I, Hovatta O, La Marca A, Livera G, Monniaux D, Persani L, et al. Advances in the molecular pathophysiology, genetics, and treatment of primary ovarian insufficiency. Trends Endocrinol Metab (2018) 29(6):400–19. doi: 10.1016/j.tem.2018.03.010
8. Qin Y, Jiao X, Simpson JL, Chen ZJ. Genetics of primary ovarian insufficiency: new developments and opportunities. Hum Reprod Update (2015) 21(6):787–808. doi: 10.1093/humupd/dmv036
9. Jaillard S, Bell K, Akloul L, Walton K, McElreavy K, Stocker WA, et al. New insights into the genetic basis of premature ovarian insufficiency: Novel causative variants and candidate genes revealed by genomic sequencing. Maturitas (2020) 141:9–19. doi: 10.1016/j.maturitas.2020.06.004
10. Oktay K, Briggs D, Gosden RG. Ontogeny of follicle-stimulating hormone receptor gene expression in isolated human ovarian follicles. J Clin Endocrinol Metab (1997) 82(11):3748–51. doi: 10.1210/jc.82.11.3748
11. Simoni M, Gromoll J, Nieschlag E. The follicle-stimulating hormone receptor: Biochemistry, molecular biology, physiology, and pathophysiology. Endocr Rev (1997) 18(6):739–73. doi: 10.1210/edrv.18.6.0320
12. Richards JS, Pangas SA. The ovary: Basic biology and clinical implications. J Clin Invest (2010) 120(4):963–72. doi: 10.1172/JCI41350
13. Jiang X, Liu H, Chen X, Chen PH, Fischer D, Sriraman V, et al. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc Natl Acad Sci USA (2012) 109(31):12491–6. doi: 10.1073/pnas.1206643109
14. Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol (2003) 63(6):1256–72. doi: 10.1124/mol.63.6.1256
15. Jiang X, Dias JA, He X. Structural biology of glycoprotein hormones and their receptors: Insights to signaling. Mol Cell Endocrinol (2014) 382(1):424–51. doi: 10.1016/j.mce.2013.08.021
16. Casarini L, Crepieux P. Molecular mechanisms of action of FSH. Front Endocrinol (Lausanne) (2019) 10:305. doi: 10.3389/fendo.2019.00305
17. Ulloa-Aguirre A, Zarinan T, Jardon-Valadez E, Gutierrez-Sagal R, Dias JA. Structure-function relationships of the follicle-stimulating hormone receptor. Front Endocrinol (Lausanne) (2018) 9:707. doi: 10.3389/fendo.2018.00707
18. Desai SS, Roy BS, Mahale SD. Mutations and polymorphisms in FSH receptor: functional implications in human reproduction. Reproduction (2013) 146(6):R235–48. doi: 10.1530/REP-13-0351
19. Aittomaki K, Lucena JL, Pakarinen P, Sistonen P, Tapanainen J, Gromoll J, et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell (1995) 82(6):959–68. doi: 10.1016/0092-8674(95)90275-9
20. Gromoll J, Simoni M, Nordhoff V, Behre HM, De Geyter C, Nieschlag E. Functional and clinical consequences of mutations in the FSH receptor. Mol Cell Endocrinol (1996) 125(1-2):177–82. doi: 10.1016/S0303-7207(96)03949-4
21. Beau I, Touraine P, Meduri G, Gougeon A, Desroches A, Matuchansky C, et al. A novel phenotype related to partial loss of function mutations of the follicle stimulating hormone receptor. J Clin Invest (1998) 102(7):1352–9. doi: 10.1172/JCI3795
22. Touraine P, Beau I, Gougeon A, Meduri G, Desroches A, Pichard C, et al. New natural inactivating mutations of the follicle-stimulating hormone receptor: Correlations between receptor function and phenotype. Mol Endocrinol (1999) 13(11):1844–54. doi: 10.1210/mend.13.11.0370
23. Allen LA, Achermann JC, Pakarinen P, Kotlar TJ, Huhtaniemi IT, Jameson JL, et al. A novel loss of function mutation in exon 10 of the FSH receptor gene causing hypergonadotrophic hypogonadism: Clinical and molecular characteristics. Hum Reprod (2003) 18(2):251–6. doi: 10.1093/humrep/deg046
24. Nakamura Y, Maekawa R, Yamagata Y, Tamura I, Sugino N. A novel mutation in exon8 of the follicle-stimulating hormone receptor in a woman with primary amenorrhea. Gynecol Endocrinol (2008) 24(12):708–12. doi: 10.1080/09513590802454927
25. Liu H, Xu X, Han T, Yan L, Cheng L, Qin Y, et al. A novel homozygous mutation in the FSHR gene is causative for primary ovarian insufficiency. Fertil Steril (2017) 108(6):1050–5 e2. doi: 10.1016/j.fertnstert.2017.09.010
26. He WB, Du J, Yang XW, Li W, Tang WL, Dai C, et al. Novel inactivating mutations in the FSH receptor cause premature ovarian insufficiency with resistant ovary syndrome. Reprod BioMed Online (2019) 38(3):397–406. doi: 10.1016/j.rbmo.2018.11.011
27. Khor S, Lyu Q, Kuang Y, Lu X. Novel FSHR variants causing female resistant ovary syndrome. Mol Genet Genomic Med (2020) 8(2):e1082. doi: 10.1002/mgg3.1082
28. Sassi A, Désir J, Janssens Véronique, Marangoni M, Daneels D, Gheldof A, et al. Novel inactivating follicle-stimulating hormone receptor (FSHR) mutations in a patient with premature ovarian insufficiency identified by next generation sequencing gene panel analysis. F&S Rep (2020) 1(3):193–201. doi: 10.1016/j.xfre.2020.08.008
29. Liu H, Guo T, Gong Z, Yu Y, Zhang Y, Zhao S, et al. Novel FSHR mutations in han Chinese women with sporadic premature ovarian insufficiency. Mol Cell Endocrinol (2019) 492:110446. doi: 10.1016/j.mce.2019.05.005
30. Doherty E, Pakarinen P, Tiitinen A, Kiilavuori A, Huhtaniemi I, Forrest S, et al. A novel mutation in the FSH receptor inhibiting signal transduction and causing primary ovarian failure. J Clin Endocrinol Metab (2002) 87(3):1151–5. doi: 10.1210/jcem.87.3.8319
31. Bramble MS, Goldstein EH, Lipson A, Ngun T, Eskin A, Gosschalk JE, et al. A novel follicle-stimulating hormone receptor mutation causing primary ovarian failure: A fertility application of whole exome sequencing. Hum Reprod (2016) 31(4):905–14. doi: 10.1093/humrep/dew025
32. Katari S, Wood-Trageser MA, Jiang H, Kalynchuk E, Muzumdar R, Yatsenko SA, et al. Novel inactivating mutation of the FSH receptor in two siblings of Indian origin with premature ovarian failure. J Clin Endocrinol Metab (2015) 100(6):2154–7. doi: 10.1210/jc.2015-1401
33. Kuechler A, Hauffa BP, Koninger A, Kleinau G, Albrecht B, Horsthemke B, et al. An unbalanced translocation unmasks a recessive mutation in the follicle-stimulating hormone receptor (FSHR) gene and causes FSH resistance. Eur J Hum Genet (2010) 18(6):656–61. doi: 10.1038/ejhg.2009.244
34. Zarinan T, Mayorga J, Jardon-Valadez E, Gutierrez-Sagal R, Maravillas-Montero JL, Mejia-Dominguez NR, et al. A novel mutation in the FSH receptor (I423T) affecting receptor activation and leading to primary ovarian failure. J Clin Endocrinol Metab (2021) 106(2):e534–e50. doi: 10.1210/clinem/dgaa782
35. Franca MM, Lerario AM, Funari MFA, Nishi MY, Narcizo AM, de Mello MP, et al. A novel homozygous missense FSHR variant associated with hypergonadotropic hypogonadism in two siblings from a Brazilian family. Sex Dev (2017) 11(3):137–42. doi: 10.1159/000477193
36. Meduri G, Touraine P, Beau I, Lahuna O, Desroches A, Vacher-Lavenu MC, et al. Delayed puberty and primary amenorrhea associated with a novel mutation of the human follicle-stimulating hormone receptor: Clinical, histological, and molecular studies. J Clin Endocrinol Metab (2003) 88(8):3491–8. doi: 10.1210/jc.2003-030217
37. Woad KJ, Prendergast D, Winship IM, Shelling AN. FSH receptor gene variants are rarely associated with premature ovarian failure. Reprod BioMed Online (2013) 26(4):396–9. doi: 10.1016/j.rbmo.2013.01.004
38. European Society for Human R, Embryology Guideline Group on POI, Webber L, Davies M, Anderson R, Bartlett J, et al. ESHRE guideline: Management of women with premature ovarian insufficiency. Hum Reprod (2016) 31(5):926–37. doi: 10.1093/humrep/dew027
39. Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature (2015) 526(7571):68–74. doi: 10.1038/nature15393
40. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature (2020) 581(7809):434–43. doi: 10.1038/s41586-020-2308-7
41. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res (2001) 29(1):308–11. doi: 10.1093/nar/29.1.308
42. Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online mendelian inheritance in man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res (2015) 43(Database issue):D789–98. doi: 10.1093/nar/gku1205
43. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res (2014) 42(Database issue):D980–5. doi: 10.1093/nar/gkt1113
44. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: The next generation in gene variant databases. Hum Mutat (2011) 32(5):557–63. doi: 10.1002/humu.21438
45. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
46. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol (2001) 305(3):567–80. doi: 10.1006/jmbi.2000.4315
47. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods (2010) 7(4):248–9. doi: 10.1038/nmeth0410-248
48. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res (2003) 31(13):3812–4. doi: 10.1093/nar/gkg509
49. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods (2014) 11(4):361–2. doi: 10.1038/nmeth.2890
50. Zhu C, Huang H, Hua R, Li G, Yang D, Luo J, et al. Molecular and functional characterization of adipokinetic hormone receptor and its peptide ligands in bombyx mori. FEBS Lett (2009) 583(9):1463–8. doi: 10.1016/j.febslet.2009.03.060
51. Chen XP, Yang W, Fan Y, Luo JS, Hong K, Wang Z, et al. Structural determinants in the second intracellular loop of the human cannabinoid CB1 receptor mediate selective coupling to g(s) and g(i). Br J Pharmacol (2010) 161(8):1817–34. doi: 10.1111/j.1476-5381.2010.01006.x
52. Ulloa-Aguirre A, Reiter E, Crepieux P. FSH receptor signaling: Complexity of interactions and signal diversity. Endocrinology (2018) 159(8):3020–35. doi: 10.1210/en.2018-00452
53. Latronico AC, Arnhold IJ. Gonadotropin resistance. Endocr Dev (2013) 24:25–32. doi: 10.1159/000342496
54. Flageole C, Toufaily C, Bernard DJ, Ates S, Blais V, Chenier S, et al. Successful in vitro maturation of oocytes in a woman with gonadotropin-resistant ovary syndrome associated with a novel combination of FSH receptor gene variants: A case report. J Assist Reprod Genet (2019) 36(3):425–32. doi: 10.1007/s10815-018-1394-z
55. Oduwole OO, Huhtaniemi IT, Misrahi M. The roles of luteinizing hormone, follicle-stimulating hormone and testosterone in spermatogenesis and folliculogenesis revisited. Int J Mol Sci (2021) 22(23):12735. doi: 10.3390/ijms222312735
56. Desai SS, Achrekar SK, Sahasrabuddhe KA, Meharji PK, Desai SK, Mangoli VS, et al. Functional characterization of two naturally occurring mutations (Val514Ala and Ala575Val) in follicle-stimulating hormone receptor. J Clin Endocrinol Metab (2015) 100(4):E638–45. doi: 10.1210/jc.2014-3662
57. Gromoll J, Pekel E, Nieschlag E. The structure and organization of the human follicle-stimulating hormone receptor (FSHR) gene. Genomics (1996) 35(2):308–11. doi: 10.1006/geno.1996.0361
Keywords: primary ovarian insufficiency, resistant ovary syndrome, follicle-stimulating hormone receptor, compound heterozygous variant, transmembrane helix
Citation: Chen X, Chen L, Wang Y, Shu C, Zhou Y, Wu R, Jin B, Yang L, Sun J, Qi M and Shu J (2023) Identification and characterization of novel compound heterozygous variants in FSHR causing primary ovarian insufficiency with resistant ovary syndrome. Front. Endocrinol. 13:1013894. doi: 10.3389/fendo.2022.1013894
Received: 08 August 2022; Accepted: 20 December 2022;
Published: 10 January 2023.
Edited by:
Bianca Bianco, Faculdade de Medicina do ABC, BrazilReviewed by:
Alfredo Ulloa-Aguirre, National Autonomous University of Mexico, MexicoCopyright © 2023 Chen, Chen, Wang, Shu, Zhou, Wu, Jin, Yang, Sun, Qi and Shu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Shu, U2h1amluZ0BobWMuZWR1LmNu; Xiaopan Chen, Q2hlbnhpYW9wYW5AaG1jLmVkdS5jbg==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.