 Mellisa Renteria
Mellisa Renteria Jun Zhang
Jun Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Endocrinol., 29 September 2022
Sec. Cancer Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1013892
This article is part of the Research TopicSteroid Hormone Receptors and Cell Cycle in Breast CancerView all 7 articles
Breast cancer is the most commonly diagnosed cancer worldwide and remains the second leading cause of cancer death in the United States (1, 2). One distinct characteristic of breast cancer is that breast cancer tumorigenesis is strongly influenced by one or more sex steroid hormones, defined as hormone-sensitive/dependent cancers. It has been well described that two major female sex steroids, estrogen (E) and progesterone (PRG), are highly associated with the growth of some types of breast cancers, which usually contain two major sex steroid receptors, estrogen receptors (ERs) and progesterone receptors (PRs). Breast cancer cells derived from these hormone-sensitive breast cancers can become activated and proliferate when they are stimulated with hormones. Based on this phenomenon, certain human reproductive cancers have been labeled hormone-related cancers, including breast, endometrium, ovarian, prostatic, and testicular cancers, which may share a unique mechanism of carcinogenesis associated with sex steroids (3).
Approximately 70% of advanced breast cancers are considered to be ‘hormone responsive’ due to their expression of ERs, PRs, or both (4, 5). However, triple-negative breast cancer (TNBC), one of the most aggressive forms of breast cancer, defined by the lack of expression of the estrogen receptor (ER), classic PRG receptors (nPRs), and epidermal growth factor receptor 2 (HER2) (6), can be considered as hormone non-responsive breast cancer. TNBC is a notoriously heterogeneous disease and yet the most poorly understood (7), exhibiting different histological and molecular subtypes with varying clinical outcomes. TNBC accounts for roughly 15% of all breast cancers and exhibits the most aggressive metastatic behavior (8) with limited targeted therapies (9). Ironically, recent data demonstrated that there is still quite a significant biological and/or clinical relevance of sex steroids in TNBCs and the action of these hormones is exerted through similar molecular mechanisms (10–14). This suggests a possible existence of previously undefined sex steroid receptor-mediated signaling pathways that retain the sensitivity and vulnerability of TNBCs to endocrine hormones (15), reemphasizing that perturbed expression of sex hormone receptors and abnormally long-exposure to sex steroids should be equally evaluated for more effective preventative strategies for breast cancers (16, 17). Efforts on this aspect have thus far identified several candidate steroid receptors, including androgen receptors (ARs), novel classic ERs (ER-βs), and G-protein-coupled ERs (GPERs), in the pathobiological actions of sex hormones in TNBCs. Endocrine therapies continue to be the anchored treatment for breast cancers, though the development of endocrine resistance has become a large obstacle for this therapeutic strategy, suggesting again that there are undefined mediators and signaling pathways associated with endocrine resistance in breast cancers (18, 19). It is important to mention that steroid hormones bind either nuclear receptors or membrane receptors. However, the most studied class of steroid hormone receptors are the nuclear receptors due to their early discoveries (20). Similarly, although the term “sex hormone” in hormone-dependent/-responsive/-sensitive breast cancers can be referred to either E or PRG (14), it usually refers only to E (21, 22). ERs can be classified into nuclear receptors (ERα and ERβ) (10) and membrane receptors (mERs) (23, 24). mERs include G protein-coupled ER1 (GPER)/G protein-coupled receptor 30 (GPR30) (11), ERx (a type of less known mERs) (25), and Gq-coupled membrane ER (Gq-mER) (26). Current hormonal therapies for breast cancer, also known as antiestrogen therapies, usually target various types of ERs and selective ER modulators (SERMs) (27, 28) by default, for the treatment and prevention of breast cancers (29, 30), demonstrating the limitation of current endocrine therapy. The effects of PRG and its corresponding receptors on breast cancers are less emphasized (31).
As a sex steroid, PRG is essential for normal breast development through its positive role in promoting proliferation of human mammary epithelial cells. Therefore, PRG and its derivatives have been long suspected to be culprits for the development of breast cancers (17, 31–35). However, the underlying mechanism of how PRG plays a role in breast cancers through either enhancing EMT (epithelial-mesenchymal transition) (25, 36–40), inhibiting EMT (41), or having no effect on EMT (39), remains to be determined. Like ERs, PRG receptors can be classified into classic nuclear receptors (nPRs) and non-classic mPRs (42–46). It was reported that PRG excerpts its cellular actions through signaling cascades that involve either nPRs, mPRs, or combined responses (47). PRG binds to nPRs as a transcription factor to evoke classic actions (48). Alternatively, PRG can also bind to and activate mPRs, which subsequently leads to activation of nPRs, leading to a hypothesized model where PRG-dependent mPRs contribute to later nPR-mediated PRG actions (49). Two groups of non-classic mPRs have been recently identified, known as membrane PRG receptor/Class II progestin and adipoQ receptors (mPRs/PAQRs) and the cytochrome-related sigma-2/PRG receptor membrane components (S2R/PRGMCs) (42, 43). mPRs are highly expressed in reproductive tissues and were reported to execute rapid, non-genomic actions through interactions with G-protein coupled receptors (42, 43, 50).
Despite its significance, the relationship between nPRs and mPRs have been minimally explored. Our group recently provided strong evidence that the CCM signaling complex (CSC) plays an essential role to bridge the crosstalk between nPRs, mPRs, and their shared ligands (progestins/anti-progestins), such as PRG, to establish and modulate this cascade among nPR positive (+) breast cancer cells. There has been supporting evidence that PRG promotes cellular proliferation (51) and inhibits apoptosis in human nPR(+) breast cancer T47D cells, suggesting PRG might not be the benign hormone for nPR(+) breast cancers and may rather be pro-oncogenic (52). It has been speculated that PRG, its cellular metabolites, and its derivatives (progestins) promote cellular proliferation through induced activation of MAPK signaling pathways in both nPR(+/-) breast cancer cells, which is independent of PRs and ERs (53). These results also suggest existence of both independent and coordinated relationships between nPR-/mPR-mediated PRG signaling involved in proliferative signaling from multiple experiments with varying approaches (32, 34, 45, 51, 54–57).
Since both types of PRG receptors and their modulators can be predominant targets for breast cancer therapy (58), as an antiprogestin (36, 59–63) [one of the well-known common contraceptives (60–65)], mifepristone (MIF, RU486, antiprogestin) has certainly earned its candidacy in the treatment of reproductive cancers (58, 66–68) such as breast, prostate, ovarian, and endometrial cancers, and has been previously studied in many clinical trials (59, 62, 63, 69). It was demonstrated that elevated levels of MIF can enhance the growth inhibition and induction of apoptosis triggered by high doses of PRG in nPR(+/-) cancer cells (70, 71). A clinically relevant dose of MIF significantly improved the treatment efficacy of chemotherapy regimens for human ovarian carcinoma cells (72). However, there have been many contradictory results reported regarding whether MIF has growth inhibition or stimulation for hormone-responsive breast cancer cells, as an anti-progestin (73). MIF can act as a potent antagonist of steroid hormone receptors such as nuclear PRG receptors (nPRs), glucocorticoid receptors (GRs), and androgen receptors (ARs) through directly binding as the ligand (74–78). The degree of nPRs, GRs, and ARs inhibition by MIF are variable, depending on dosages of MIF and specific cell types (79). Recent data demonstrated that only nPRs play protective roles on the stability of the CSC under the negative effects of mPR-specific PRG actions, while both GRs and ARs play no role in this CmPn signaling network (57). Among three types of antiprogestins, MIF is defined as a type-II antiprogestin, which can also act as an agonist in nPR(+) tissues in a cell-specific manner, based on selective modulation of PRs (SPRM) criteria (80). MIF has been well known to exert its antagonist action, in-trans, with three dimeric forms of nPR isoforms (AA, AB, and BB) to inhibit nPR activation at concentration that are substoichiometric with PRG (81, 82), however, it is also reported that MIF can affect the ratio of nPG-A/nPG-B isoforms at both the transcriptional and translational levels, depending on dosages of MIF and specific cell types (83). Since MIF can act as an agonist, and the patient survival outcome depends on the ratio of nPR antagonists versus agonists effects of MIF, the mechanism of how MIF is either nPR antagonists or agonists needs to be define. In an in-vitro experiment with nPR knockout T47D cells (T47D-Y), cellular expression transactivation was observed only in the T47D-Y cells ectopically expressed nPR-B isoform (T47D-YB) (84), suggesting that nPR-B isoform has a unique activation domain that may confer agonist-like properties in the presence of MIF (85). Recent findings demonstrated that the regulated expression of nPRA and nPRB is critical to the breast cells’ response to synthetic progestins, this altered PRG receptor expression may be an important factor in the malignant transformation of breast cells (86). Regardless of tissue of origin and hormone responsiveness, the anti-proliferative activity of MIF in cancer cells has been found to be independent of nPRs (87). Similarly, the cellular effects of MIF on proliferation were also reported (54, 88). It seems that the pro- or anti-proliferative activities of MIF are cell-specific (69) and might be determined by the applied dosage of MIF, as well as ratio of nPR isoforms (36). These results indicate the importance to uncover key mediators of MIF and underlying mechanisms of its dual roles (pro- or anti-tumor activity) in breast malignancies (87). The possible convergence of classic and non-classic PRG actions (nPRs/mPRs) and CSC signaling on their common cellular targets in nPR(+) cells is an attractive model by which PRG and MIF can fine tune this intricate balance among these signaling pathways. Our recent findings suggest that combined PRG and MIF treatments can enhance the inhibitory effects on protein expression of CSC, which is independent of nPR(+/-) status. This indicates that PRG and MIF can work synergistically to inhibit the protein stability of the CSC, only through mPRs, termed as mPR-specific PRG action (57, 89–92).
Altered expression patterns of nPRs, mPRs and CCM genes across clinical breast tumors and their associated prognostic effects were observed (93), suggesting combined involvement of all three signaling pathways in influencing the size and extent of the primary breast tumor (57, 90, 91). Our recent findings that upon treatment of PRG and MIF, which only excerpt their PRG actions through mPR-specific PRG actions, the expression patterns of three CCM proteins which form the CSC are altered in the nPR(+) breast cancer cells, T47D, strongly suggesting the involvement of the CSC in breast tumorigenesis (57, 90, 91). Reciprocally, mPRs and nPRs signaling cascades are coupled through CSC modulation (57). These results solidify a novel network among the CSC, classic, and non-classic PRG receptors, termed the CSC-mPRs-PRG-nPRs (CmPn) signaling network, which is dynamically modulated and fine-tuned with a series of feedback regulations under PRG actions.
Strong evidence for the existence of PRG-mPRs signaling cascade in both nPR(+/-) breast cancer cells have been previously proposed (41, 94–97), suggesting that PRG signaling in nPR(-) cell lines can be mediated solely through mPR-mediated signaling (termed as mPR-specific PRG actions) (41). In our recent reports, we further defined the novel CSC-mPRs-PRG (CmP) signaling network in nPR(-) breast cancer cells which overlaps with our previously defined CmPn (CSC-mPRs-PRG-nPRs) network in nPR(+) breast cancer cells (57). In the CmP signaling network, the CSC is able to stabilize mPRs under steroid actions in a forward fashion (CSC➔mPRs), indicating an essential role of the CSC in maintaining the stability of mPRs in nPR(-) breast cancer cells under the mPR-specific PRG actions. Overall, the CSC can stabilize the expression of mPR proteins in TNBC cells in concordance with our observations in nPR(+) breast cancer cells, indicating the consistent function of the CSC on the stability of mPRs under PRG actions (57). Since nPR-mediated signaling pathways usually have protective roles over the CSC, the intricate balance of the CmPn signaling network is more stable under mPR-specific PRG actions compared to the CmP network. Therefore, in contrast to our previously observed nPR(+) breast cancer and nPR(-) endothelial cell data (57, 89, 92), mPR-specific PRG actions have variable effects on both RNA and protein expression levels on the CSC in TNBCs, which can be utilized for subtype classifications (90, 91). In general, the relationships among the CSC, mPRs, PRG, nPRs, and their mediated signaling pathways within either the CmPn or the CmP signaling networks can be summarized (Figure 1). Extensive omics has been performed confirming alterations in key tumorigenesis signaling pathways, suggesting their association with the CmPn/CmP signaling network in nPR(+/-) breast cancers (57, 90, 91, 93, 98).
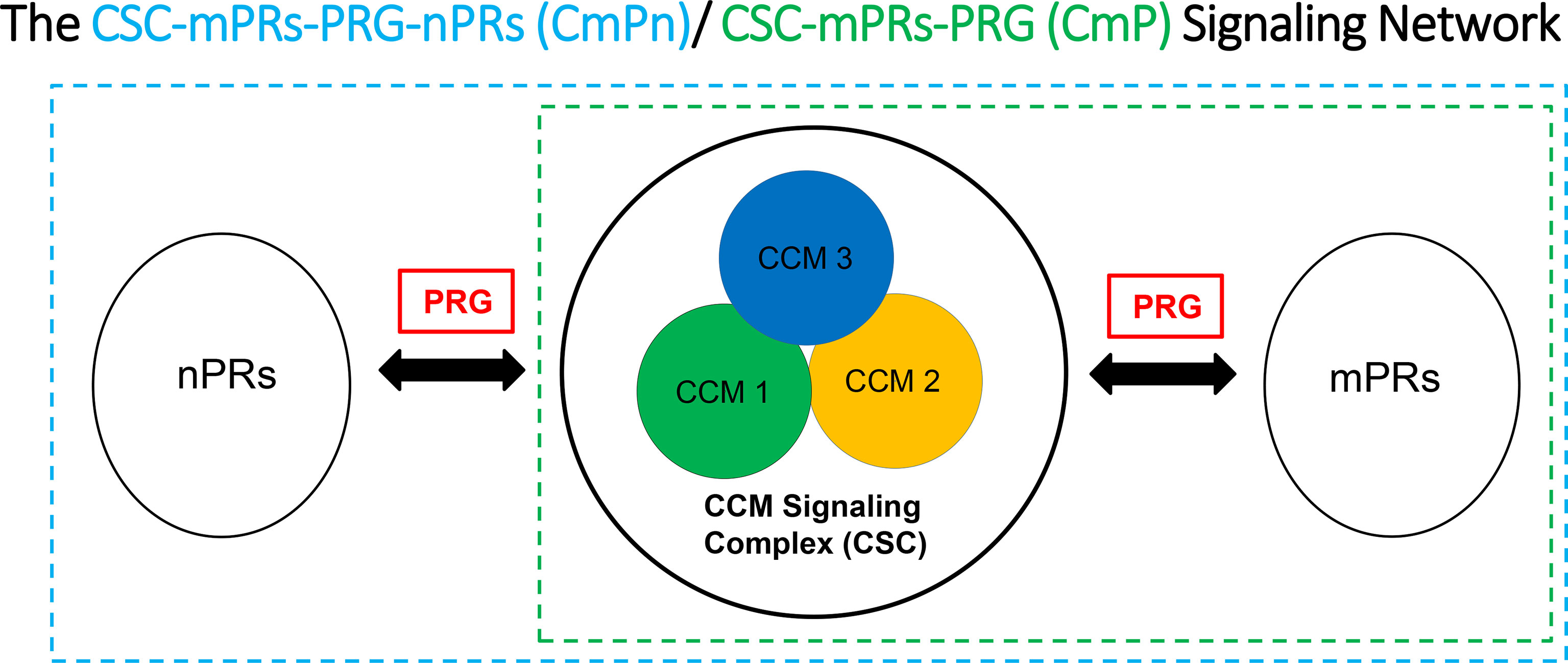
Figure 1 Signaling network for key CSC players and the effects of progesterone (PRG) on nPR and mPR. We suggest that the CSC functions as a channel modulator for the interactions of PRG between nPRs and mPRs.
The interplays between classic and non-classic PRG-mediated signaling have been long suspected (43, 44, 49, 99) and newly discovered evidence demonstrated that the CSC plays an essential role to bridge crosstalk among nPRs, mPRs, and their ligands to form the CmPn/CmP signaling networks in response to mPR-specific PRG actions among nPR(+/-) breast cancer cells (57, 90, 91, 93). The convergence of classic and non-classic PRG actions by the CSC on their common ligands and downstream cellular targets in breast cancer cells is an attractive model by which mPR-specific PRG actions play a central role for the stability of this system by fine-tuning the intricate balance within CmPn/CmP signaling network. Therefore, any activities that disrupt this intricate balance within the network, including patients under hormone replacement therapy (HRT), females taking hormonal contraceptives, or extended exposures (dietary and/or daily supplementation) to hormones during their reproductive ages, could result in perturbation of the CmPn/CmP signaling networks, with potential serious consequences of increased risks in breast cancers and compromised tumor therapy.
JZ proposed, designed and drafted manuscript. MR, OB involved in revision and comments. DJ, JA and MB involved in comments. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California cancer registry. Cancer. (2007) 109(9):1721–8. doi: 10.1002/cncr.22618
2. Baquet CR, Mishra SI, Commiskey P, Ellison GL, DeShields M. Breast cancer epidemiology in blacks and whites: Disparities in incidence, mortality, survival rates and histology. J Natl Med Assoc (2008) 100(5):480–8. doi: 10.1016/S0027-9684(15)31294-3
3. Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. (2000) 21(3):427–33. doi: 10.1093/carcin/21.3.427
4. Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol (2007) 608:1–22. doi: 10.1007/978-0-387-74039-3_1
5. Kohler BA, Sherman RL, Howlader N, Jemal A, Ryerson AB, Henry KA, et al. Annual report to the nation on the status of cancer, 1975-2011, featuring incidence of breast cancer subtypes by Race/Ethnicity, poverty, and state. J Natl Cancer Inst (2015) 107(6):djv048. doi: 10.1093/jnci/djv048
6. Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the Carolina breast cancer study. JAMA. (2006) 295(21):2492–502. doi: 10.1001/jama.295.21.2492
7. Yadav BS, Chanana P, Jhamb S. Biomarkers in triple negative breast cancer: A review. World J Clin Oncol (2015) 6(6):252–63. doi: 10.5306/wjco.v6.i6.252
8. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. (2000) 406(6797):747–52. doi: 10.1038/35021093
9. Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn HJ, et al. Strategies for subtypes–dealing with the diversity of breast cancer: Highlights of the st. gallen international expert consensus on the primary therapy of early breast cancer 2011. Ann Oncol (2011) 22(8):1736–47. doi: 10.1093/annonc/mdr304
10. Reese JM, Suman VJ, Subramaniam M, Wu X, Negron V, Gingery A, et al. ERbeta1: Characterization, prognosis, and evaluation of treatment strategies in ERalpha-positive and -negative breast cancer. BMC Cancer. (2014) 14:749. doi: 10.1186/1471-2407-14-749
11. Hernandez-Silva CD, Villegas-Pineda JC, Pereira-Suarez AL. Expression and role of the G protein-coupled estrogen receptor (GPR30/GPER) in the development and immune response in female reproductive cancers. Front Endocrinol (Lausanne). (2020) 11:544. doi: 10.3389/fendo.2020.00544
12. Sellitto A, D'Agostino Y, Alexandrova E, Lamberti J, Pecoraro G, Memoli D, et al. Insights into the role of estrogen receptor beta in triple-negative breast cancer. Cancers (Basel). (2020) 12(6):1477. doi: 10.3390/cancers12061477
13. Honma N, Matsuda Y, Mikami T. Carcinogenesis of triple-negative breast cancer and sex steroid hormones. Cancers (Basel). (2021) 13(11):2588. doi: 10.3390/cancers13112588
14. van Barele M, Heemskerk-Gerritsen BAM, Louwers YV, Vastbinder MB, Martens JWM, Hooning MJ, et al. Estrogens and progestogens in triple negative breast cancer: Do they harm? Cancers (Basel). (2021) 13(11):2506. doi: 10.3390/cancers13112506
15. Li K, Zong D, Sun J, Chen D, Ma M, Jia L. Rewiring of the endocrine network in triple-negative breast cancer. Front Oncol (2022) 12:830894. doi: 10.3389/fonc.2022.830894
16. Spicer DV, Pike MC. The prevention of breast cancer through reduced ovarian steroid exposure. Acta Oncol (1992) 31(2):167–74. doi: 10.3109/02841869209088898
17. Miyamoto H. Sex hormone receptor signals in human malignancies. Int J Mol Sci (2019) 20(11):2677. doi: 10.3390/ijms20112677
18. Murphy CG, Dickler MN. Endocrine resistance in hormone-responsive breast cancer: mechanisms and therapeutic strategies. Endocr Relat Cancer (2016) 23(8):R337–52. doi: 10.1530/ERC-16-0121
19. Hyder T, Marti JLG, Nasrazadani A, Brufsky AM. Statins and endocrine resistance in breast cancer. Cancer Drug Resist (2021) 4(2):356–64. doi: 10.20517/cdr.2020.112
20. Trevino LS, Gorelick DA. The interface of nuclear and membrane steroid signaling. Endocrinology (2021) 162(8):bqab107. doi: 10.1210/endocr/bqab107
21. Osborne CK, Bolan G, Monaco ME, Lippman ME. Hormone responsive human breast cancer in long-term tissue culture: effect of insulin. Proc Natl Acad Sci U S A (1976) 73(12):4536–40. doi: 10.1073/pnas.73.12.4536
22. Clarke R, Brunner N, Katzenellenbogen BS, Thompson EW, Norman MJ, Koppi C, et al. Progression of human breast cancer cells from hormone-dependent to hormone-independent growth both in vitro and in vivo. Proc Natl Acad Sci U S A (1989) 86(10):3649–53. doi: 10.1073/pnas.86.10.3649
23. Soltysik K, Czekaj P. Membrane estrogen receptors - is it an alternative way of estrogen action? J Physiol Pharmacol (2013) 64(2):129–42.
24. Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol (2019) 116:135–70. doi: 10.1016/bs.apcsb.2019.01.001
25. Kampa M, Notas G, Pelekanou V, Troullinaki M, Andrianaki M, Azariadis K, et al. Early membrane initiated transcriptional effects of estrogens in breast cancer cells: First pharmacological evidence for a novel membrane estrogen receptor element (ERx). Steroids. (2012) 77(10):959–67. doi: 10.1016/j.steroids.2012.02.011
26. Vail G, Roepke TA. Membrane-initiated estrogen signaling via gq-coupled GPCR in the central nervous system. Steroids. (2019) 142:77–83. doi: 10.1016/j.steroids.2018.01.010
27. Provenzano A, Kurian S, Abraham J. Overcoming endocrine resistance in breast cancer: role of the PI3K and the mTOR pathways. Expert Rev Anticancer Ther (2013) 13(2):143–7. doi: 10.1586/era.12.173
28. Fan W, Chang J, Fu P. Endocrine therapy resistance in breast cancer: current status, possible mechanisms and overcoming strategies. Future Med Chem (2015) 7(12):1511–9. doi: 10.4155/fmc.15.93
29. Journe F, Body JJ, Leclercq G, Laurent G. Hormone therapy for breast cancer, with an emphasis on the pure antiestrogen fulvestrant: mode of action, antitumor efficacy and effects on bone health. Expert Opin Drug Saf. (2008) 7(3):241–58. doi: 10.1517/14740338.7.3.241
30. Abdulkareem IH, Zurmi IB. Review of hormonal treatment of breast cancer. Niger J Clin Pract (2012) 15(1):9–14. doi: 10.4103/1119-3077.94088
31. Lange CA. Challenges to defining a role for progesterone in breast cancer. Steroids. (2008) 73(9-10):914–21. doi: 10.1016/j.steroids.2007.12.023
32. Lange CA, Richer JK, Horwitz KB. Hypothesis: Progesterone primes breast cancer cells for cross-talk with proliferative or antiproliferative signals. Mol Endocrinol (1999) 13(6):829–36. doi: 10.1210/mend.13.6.0290
33. Lange CA, Sartorius CA, Abdel-Hafiz H, Spillman MA, Horwitz KB, Jacobsen BM. Progesterone receptor action: translating studies in breast cancer models to clinical insights. Adv Exp Med Biol (2008) 630:94–111. doi: 10.1007/978-0-387-78818-0_7
34. Dressing GE, Lange CA. Integrated actions of progesterone receptor and cell cycle machinery regulate breast cancer cell proliferation. Steroids. (2009) 74(7):573–6. doi: 10.1016/j.steroids.2008.12.001
35. Trabert B, Sherman ME, Kannan N, Stanczyk FZ. Progesterone and breast cancer. Endocr Rev (2020) 41(2):320–44. doi: 10.1210/endrev/bnz001
36. Meyer ME, Pornon A, Ji JW, Bocquel MT, Chambon P, Gronemeyer H. Agonistic and antagonistic activities of RU486 on the functions of the human progesterone receptor. EMBO J (1990) 9(12):3923–32. doi: 10.1002/j.1460-2075.1990.tb07613.x
37. Conneely OM, Mulac-Jericevic B, Lydon JP. Progesterone-dependent regulation of female reproductive activity by two distinct progesterone receptor isoforms. Steroids. (2003) 68(10-13):771–8. doi: 10.1016/S0039-128X(03)00126-0
38. Kariagina A, Xie J, Langohr IM, Opreanu RC, Basson MD, Haslam SZ. Progesterone decreases levels of the adhesion protein e-cadherin and promotes invasiveness of steroid receptor positive breast cancers. Horm Cancer. (2013) 4(6):371–80. doi: 10.1007/s12672-013-0158-6
39. Kariagina A, Xie J, Langohr IM, Opreanu RC, Basson MD, Haslam SZ. Progesterone stimulates proliferation and promotes cytoplasmic localization of the cell cycle inhibitor p27 in steroid receptor positive breast cancers. Horm Cancer. (2013) 4(6):381–90. doi: 10.1007/s12672-013-0159-5
40. Rosati R, Oppat K, Huang Y, Kim S, Ratnam M. Clinical association of progesterone receptor isoform a with breast cancer metastasis consistent with its unique mechanistic role in preclinical models. BMC Cancer. (2020) 20(1):512. doi: 10.1186/s12885-020-07002-0
41. Zuo L, Li W, You S. Progesterone reverses the mesenchymal phenotypes of basal phenotype breast cancer cells via a membrane progesterone receptor mediated pathway. Breast Cancer Res (2010) 12(3):R34. doi: 10.1186/bcr2588
42. Zhu Y, Rice CD, Pang Y, Pace M, Thomas P. Cloning, expression, and characterization of a membrane progestin receptor and evidence it is an intermediary in meiotic maturation of fish oocytes. Proc Natl Acad Sci U S A (2003) 100(5):2231–6. doi: 10.1073/pnas.0336132100
43. Boonyaratanakornkit V, Edwards DP. Receptor mechanisms of rapid extranuclear signalling initiated by steroid hormones. Essays Biochem (2004) 40:105–20. doi: 10.1042/bse0400105
44. Boonyaratanakornkit V, McGowan E, Sherman L, Mancini MA, Cheskis BJ, Edwards DP. The role of extranuclear signaling actions of progesterone receptor in mediating progesterone regulation of gene expression and the cell cycle. Mol Endocrinol (2007) 21(2):359–75. doi: 10.1210/me.2006-0337
45. Carnevale RP, Proietti CJ, Salatino M, Urtreger A, Peluffo G, Edwards DP, et al. Progestin effects on breast cancer cell proliferation, proteases activation, and in vivo development of metastatic phenotype all depend on progesterone receptor capacity to activate cytoplasmic signaling pathways. Mol Endocrinol (2007) 21(6):1335–58. doi: 10.1210/me.2006-0304
46. Boonyaratanakornkit V, McGowan EM, Marquez-Garban DC, Burton LP, Hamilton N, Pateetin P, et al. Progesterone receptor signaling in the breast tumor microenvironment. Adv Exp Med Biol (2021) 1329:443–74. doi: 10.1007/978-3-030-73119-9_21
47. Mueck AO, Ruan X, Seeger H, Fehm T, Neubauer H. Genomic and non-genomic actions of progestogens in the breast. J Steroid Biochem Mol Biol (2014) 142:62–7. doi: 10.1016/j.jsbmb.2013.08.011
48. Giangrande PH, McDonnell DP. The a and b isoforms of the human progesterone receptor: Two functionally different transcription factors encoded by a single gene. Recent Prog Horm Res (1999) 54:291–313; discussion 13-4.
49. Boonyaratanakornkit V, Hamilton N, Marquez-Garban DC, Pateetin P, McGowan EM, Pietras RJ. Extranuclear signaling by sex steroid receptors and clinical implications in breast cancer. Mol Cell Endocrinol (2018) 466:51–72. doi: 10.1016/j.mce.2017.11.010
50. Pang Y, Dong J, Thomas P. Characterization, neurosteroid binding and brain distribution of human membrane progesterone receptors delta and {epsilon} (mPRdelta and mPR{epsilon}) and mPRdelta involvement in neurosteroid inhibition of apoptosis. Endocrinology. (2013) 154(1):283–95. doi: 10.1210/en.2012-1772
51. Proietti C, Salatino M, Rosemblit C, Carnevale R, Pecci A, Kornblihtt AR, et al. Progestins induce transcriptional activation of signal transducer and activator of transcription 3 (Stat3) via a jak- and src-dependent mechanism in breast cancer cells. Mol Cell Biol (2005) 25(12):4826–40. doi: 10.1128/MCB.25.12.4826-4840.2005
52. Moore MR, Spence JB, Kiningham KK, Dillon JL. Progestin inhibition of cell death in human breast cancer cell lines. J Steroid Biochem Mol Biol (2006) 98(4-5):218–27. doi: 10.1016/j.jsbmb.2005.09.008
53. Wiebe JP, Pawlak KJ, Kwok A. Mechanism of action of the breast cancer-promoter hormone, 5alpha-dihydroprogesterone (5alphaP), involves plasma membrane-associated receptors and MAPK activation. J Steroid Biochem Mol Biol (2016) 155(Pt A):166–76. doi: 10.1016/j.jsbmb.2015.10.021
54. Skildum A, Faivre E, Lange CA. Progesterone receptors induce cell cycle progression via activation of mitogen-activated protein kinases. Mol Endocrinol (2005) 19(2):327–39. doi: 10.1210/me.2004-0306
55. Diep CH, Ahrendt H, Lange CA. Progesterone induces progesterone receptor gene (PGR) expression via rapid activation of protein kinase pathways required for cooperative estrogen receptor alpha (ER) and progesterone receptor (PR) genomic action at ER/PR target genes. Steroids. (2016) 114:48–58. doi: 10.1016/j.steroids.2016.09.004
56. Salazar M, Lerma-Ortiz A, Hooks GM, Ashley AK, Ashley RL. Progestin-mediated activation of MAPK and AKT in nuclear progesterone receptor negative breast epithelial cells: The role of membrane progesterone receptors. Gene. (2016) 591(1):6–13. doi: 10.1016/j.gene.2016.06.044
57. Abou-Fadel J, Jiang X, Grajeda B, Padarti A, Ellis CC, Flores E, et al. CCM signaling complex (CSC) couples both classic and non-classic progesterone receptor signaling. Cell Commun Signal (2022) 20(1):120. doi: 10.1186/s12964-022-00926-z
58. Check JH, Check D. New insights as to why progesterone receptor modulators, such as mifepristone, seem to be more effective in treating cancers that are devoid of the classical nuclear progesterone receptor. Anticancer Res (2021) 41(12):5873–80. doi: 10.21873/anticanres.15407
59. Baird DT. Antigestogens. Br Med Bull (1993) 49(1):73–87. doi: 10.1093/oxfordjournals.bmb.a072607
60. Bygdeman M, Swahn ML, Gemzell-Danielsson K, Svalander P. Mode of action of RU 486. Ann Med (1993) 25(1):61–4. doi: 10.3109/07853899309147859
61. Spitz IM, Croxatto HB, Robbins A. Antiprogestins: mechanism of action and contraceptive potential. Annu Rev Pharmacol Toxicol (1996) 36:47–81. doi: 10.1146/annurev.pa.36.040196.000403
62. Cadepond F, Ulmann A, Baulieu EE. RU486 (mifepristone): mechanisms of action and clinical uses. Annu Rev Med (1997) 48:129–56. doi: 10.1146/annurev.med.48.1.129
63. Goldberg JR, Plescia MG, Anastasio GD. Mifepristone (RU 486): current knowledge and future prospects. Arch Fam Med (1998) 7(3):219–22. doi: 10.1001/archfami.7.3.219
64. Avrech OM, Golan A, Weinraub Z, Bukovsky I, Caspi E. Mifepristone (RU486) alone or in combination with a prostaglandin analogue for termination of early pregnancy: a review. Fertil Steril. (1991) 56(3):385–93. doi: 10.1016/S0015-0282(16)54527-0
65. Baulieu EE. RU 486 (mifepristone). a short overview of its mechanisms of action and clinical uses at the end of 1996. Ann N Y Acad Sci (1997) 828:47–58. doi: 10.1111/j.1749-6632.1997.tb48523.x
66. Check JH, Sansoucie L, Chern J, Amadi N, Srivastava M, Larece K. Evidence that progesterone receptor antagonists may help in the treatment of a variety of cancers by locally suppressing natural killer cell activity. Clin Exp Obstet Gynecol. (2007) 34(4):207–11.
67. Lee O, Choi MR, Christov K, Ivancic D, Khan SA. Progesterone receptor antagonism inhibits progestogen-related carcinogenesis and suppresses tumor cell proliferation. Cancer Lett (2016) 376(2):310–7. doi: 10.1016/j.canlet.2016.04.010
68. Check JH, Check DL. A hypothetical model suggesting some possible ways that the progesterone receptor may be involved in cancer proliferation. Int J Mol Sci (2021) 22(22):12351. doi: 10.3390/ijms222212351
69. Goyeneche AA, Seidel EE, Telleria CM. Growth inhibition induced by antiprogestins RU-38486, ORG-31710, and CDB-2914 in ovarian cancer cells involves inhibition of cyclin dependent kinase 2. Invest New Drugs (2012) 30(3):967–80. doi: 10.1007/s10637-011-9655-z
70. Moe BG, Vereide AB, Orbo A, Sager G. High concentrations of progesterone and mifepristone mutually reinforce cell cycle retardation and induction of apoptosis. Anticancer Res (2009) 29(4):1053–8.
71. Fjelldal R, Moe BT, Orbo A, Sager G. MCF-7 cell apoptosis and cell cycle arrest: non-genomic effects of progesterone and mifepristone (RU-486). Anticancer Res (2010) 30(12):4835–40.
72. Gamarra-Luques CD, Goyeneche AA, Hapon MB, Telleria CM. Mifepristone prevents repopulation of ovarian cancer cells escaping cisplatin-paclitaxel therapy. BMC Cancer. (2012) 12:200. doi: 10.1186/1471-2407-12-200
73. Iwasaki K, Underwood B, Herman M, Dinda S, Kodali S, Kloosterboer HJ, et al. Effects of antiprogestins on the rate of proliferation of breast cancer cells. Mol Cell Biochem (1999) 198(1-2):141–9. doi: 10.1023/A:1006945813508
74. Berrevoets CA, Umar A, Brinkmann AO. Antiandrogens: selective androgen receptor modulators. Mol Cell Endocrinol (2002) 198(1-2):97–103. doi: 10.1016/S0303-7207(02)00373-8
75. Song LN, Coghlan M, Gelmann EP. Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol Endocrinol (2004) 18(1):70–85. doi: 10.1210/me.2003-0189
76. Zhang J, Tsai FT, Geller DS. Differential interaction of RU486 with the progesterone and glucocorticoid receptors. J Mol Endocrinol (2006) 37(1):163–73. doi: 10.1677/jme.1.02089
77. Wilkinson JM, Hayes S, Thompson D, Whitney P, Bi K. Compound profiling using a panel of steroid hormone receptor cell-based assays. J Biomol Screen. (2008) 13(8):755–65. doi: 10.1177/1087057108322155
78. Sun Y, Fang M, Davies H, Hu Z. Mifepristone: a potential clinical agent based on its anti-progesterone and anti-glucocorticoid properties. Gynecol Endocrinol (2014) 30(3):169–73. doi: 10.3109/09513590.2013.856410
79. Horwitz KB. The molecular biology of RU486. is there a role for antiprogestins in the treatment of breast cancer? Endocr Rev (1992) 13(2):146–63. doi: 10.1210/edrv-13-2-146
80. Lanari C, Wargon V, Rojas P, Molinolo AA. Antiprogestins in breast cancer treatment: are we ready? Endocr Relat Cancer. (2012) 19(3):R35–50. doi: 10.1530/ERC-11-0378
81. Leonhardt SA, Altmann M, Edwards DP. Agonist and antagonists induce homodimerization and mixed ligand heterodimerization of human progesterone receptors in vivo by a mammalian two-hybrid assay. Mol Endocrinol (1998) 12(12):1914–30. doi: 10.1210/mend.12.12.0210
82. Leonhardt SA, Edwards DP. Mechanism of action of progesterone antagonists. Exp Biol Med (Maywood). (2002) 227(11):969–80. doi: 10.1177/153537020222701104
83. Rekawiecki R, Kowalik MK, Kotwica J. Onapristone (ZK299) and mifepristone (RU486) regulate the messenger RNA and protein expression levels of the progesterone receptor isoforms a and b in the bovine endometrium. Theriogenology. (2015) 84(3):348–57. doi: 10.1016/j.theriogenology.2015.03.024
84. Sartorius CA, Groshong SD, Miller LA, Powell RL, Tung L, Takimoto GS, et al. New T47D breast cancer cell lines for the independent study of progesterone b- and a-receptors: only antiprogestin-occupied b-receptors are switched to transcriptional agonists by cAMP. Cancer Res (1994) 54(14):3868–77.
85. Horwitz KB, Tung L, Takimoto GS. Novel mechanisms of antiprogestin action. J Steroid Biochem Mol Biol (1995) 53(1-6):9–17. doi: 10.1016/0960-0760(95)00035-X
86. Chen FP, Chien MH, Chen HY, Huang TS, Ng YT. Effects of estradiol and progestogens on human breast cells: regulation of sex steroid receptors. Taiwan J Obstet Gynecol. (2013) 52(3):365–73. doi: 10.1016/j.tjog.2012.09.038
87. Tieszen CR, Goyeneche AA, Brandhagen BN, Ortbahn CT, Telleria CM. Antiprogestin mifepristone inhibits the growth of cancer cells of reproductive and non-reproductive origin regardless of progesterone receptor expression. BMC Cancer. (2011) 11:207. doi: 10.1186/1471-2407-11-207
88. Bowden RT, Hissom JR, Moore MR. Growth stimulation of T47D human breast cancer cells by the anti-progestin RU486. Endocrinology. (1989) 124(5):2642–4. doi: 10.1210/endo-124-5-2642
89. Abou-Fadel J, Jiang X, Padarti A, Goswami D, Smith M, Grajeda B, et al. CCM signaling complex (CSC) is a master regulator governing homeostasis of progestins and their mediated signaling cascades. bioRxiv. (2020). doi: 10.1101/2020.06.10.145003
90. Abou-Fadel J, Bhalli M, Grajeda B, Zhang J. CmP signaling network leads to identification of prognostic biomarkers for triple-negative breast cancer in Caucasian women. Genet Test Mol Biomarkers. (2022) 26(4):198–219. doi: 10.1089/gtmb.2021.0221
91. Abou-Fadel J, Grajeda B, Jiang X, Cailing-De La OA, Flores E, Padarti A, et al. CmP signaling network unveils novel biomarkers for triple negative breast cancer in African American women. Cancer biomark (2022) 34(4):607–636. doi: 10.1101/2021.05.24.445510
92. Abou-Fadel J, Jiang X, Padarti A, Goswami DG, Smith M, Grajeda B, et al. mPR-specific actions influence maintenance of the Blood–Brain barrier (BBB). Int J Mol Sci (2022) 23(17):9684. doi: 10.3390/ijms23179684
93. Abou-Fadel J, Qu Y, Gonzalez E, Smith M, Zhang J. Emerging roles of CCM genes during tumorigenesis with potential application as novel biomarkers across major types of cancers. Oncol Rep (2020) 18:2020. doi: 10.3892/or.2020.7550
94. Karteris E, Zervou S, Pang Y, Dong J, Hillhouse EW, Randeva HS, et al. Progesterone signaling in human myometrium through two novel membrane G protein-coupled receptors: potential role in functional progesterone withdrawal at term. Mol Endocrinol (2006) 20(7):1519–34. doi: 10.1210/me.2005-0243
95. Dosiou C, Hamilton AE, Pang Y, Overgaard MT, Tulac S, Dong J, et al. Expression of membrane progesterone receptors on human T lymphocytes and jurkat cells and activation of G-proteins by progesterone. J Endocrinol (2008) 196(1):67–77. doi: 10.1677/JOE-07-0317
96. Sleiter N, Pang Y, Park C, Horton TH, Dong J, Thomas P, et al. (PRA) and PRB-independent effects of progesterone on gonadotropin-releasing hormone release. Endocrinology. (2009) 150(8):3833–44. doi: 10.1210/en.2008-0774
97. Pang Y, Thomas P. Progesterone signals through membrane progesterone receptors (mPRs) in MDA-MB-468 and mPR-transfected MDA-MB-231 breast cancer cells which lack full-length and n-terminally truncated isoforms of the nuclear progesterone receptor. Steroids. (2011) 76(9):921–8. doi: 10.1016/j.steroids.2011.01.008
98. Abou-Fadel J, Vasquez M, Grajeda B, Ellis C, Zhang J. Systems-wide analysis unravels the new roles of CCM signal complex (CSC). Heliyon. (2019) 5(12):e02899. doi: 10.1016/j.heliyon.2019.e02899
Keywords: breast cancer, CmPn signaling networks, CmP signaling networks, mPR-specific PRG actions, progesterone
Citation: Renteria M, Belkin O, Jang D, Aickareth J, Bhalli M and Zhang J (2022) CmPn signaling networks in the tumorigenesis of breast cancer. Front. Endocrinol. 13:1013892. doi: 10.3389/fendo.2022.1013892
Received: 08 August 2022; Accepted: 12 September 2022;
Published: 29 September 2022.
Edited by:
Victoria Fabris, CONICET Instituto de Biología y Medicina Experimental (IBYME), ArgentinaReviewed by:
Sebastian Giulianelli, CONICET Instituto de Biología de Organismos Marinos (IBIOMAR), ArgentinaCopyright © 2022 Renteria, Belkin, Jang, Aickareth, Bhalli and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Zhang, anVuLnpoYW5nMjAwMEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.