 Tarana Arman
Tarana Arman Peter S. Nelson
Peter S. Nelson- 1Division of Human Biology, Fred Hutchinson Cancer Center, Seattle, WA, United States
- 2Division of Clinical Research, Fred Hutchinson Cancer Center, Seattle, WA, United States
Prostate cancer is a common malignancy affecting men worldwide. While the vast majority of newly diagnosed prostate cancers are categorized as adenocarcinomas, a spectrum of uncommon tumor types occur including those with small cell and neuroendocrine cell features. Benign neuroendocrine cells exist in the normal prostate microenvironment, and these cells may give rise to primary neuroendocrine carcinomas. However, the more common development of neuroendocrine prostate cancer is observed after therapeutics designed to repress the signaling program regulated by the androgen receptor which is active in the majority of localized and metastatic adenocarcinomas. Neuroendocrine tumors are identified through immunohistochemical staining for common markers including chromogranin A/B, synaptophysin and neuron specific enolase (NSE). These markers are also common to neuroendocrine tumors that arise in other tissues and organs such as the gastrointestinal tract, pancreas, lung and skin. Notably, neuroendocrine prostate cancer shares biochemical features with nerve cells, particularly functions involving the secretion of a variety of peptides and proteins. These secreted factors have the potential to exert local paracrine effects, and distant endocrine effects that may modulate tumor progression, invasion, and resistance to therapy. This review discusses the spectrum of factors derived from neuroendocrine prostate cancers and their potential to influence the pathophysiology of localized and metastatic prostate cancer.
Introduction
Prostate cancer (PC) is the second most commonly diagnosed cancer worldwide among men (1). The American Cancer Society has estimated that ~270,000 men will be diagnosed with PC in 2022 in the United States and PC will cause in excess of 34,000 deaths (2). The vast majority of men diagnosed with PC present with localized disease and the histology of these tumors are primarily adenocarcinomas with distinctive architectures codified as Gleason patterns (3). The most characteristic feature displayed by the vast majority of localized and metastatic PCs involves the expression of the androgen receptor (AR) and a program of genes/proteins regulated by the AR including a group of secreted factors such as prostate specific antigen (PSA) (4). In addition to specifying prostate epithelial lineage and regulating metabolic and secretory functions, the AR serves as a key therapeutic target both for localized tumors and metastases (5, 6).
While the vast majority of PCs are adenocarcinomas with secretory epithelial features and an active AR program, PCs with a spectrum of other histological characteristics also occur. Among these are PCs with qualities of neuroendocrine (NE) cells (7). These prostate neuroendocrine carcinomas (NEPCs), representing <1% of all localized PCs, exhibit features found in benign NE cells and in NE carcinomas arising in other organs and tissues (8, 9). In the context of localized NEPC, the origin of these tumors has not been conclusively established as they may arise from resident benign NE cells or from stem-like, basal or luminal cells that usually serve as the cell of origin for typical adenocarcinoma (10–12). In the setting of metastatic PC (mPC), tumors with NE features are more common, ranging from 10-30% depending on the markers used for classification and the disease state with respect to the application of therapeutics that suppress AR signaling (13, 14). Preclinical models have demonstrated the occurrence of transdifferentiation whereby tumor cells with a typical epithelial phenotype and active AR program lose AR activity and gain NE characteristics during the development of resistance to AR repression (15–17). With the advent of more potent AR signaling inhibitors (ARSI) such as abiraterone and enzalutamide, the frequency of tumors with NE phenotypes is increasing (18). One feature of metastatic NEPC is the downregulation or complete absence of AR expression and AR signaling (18, 19). Notably, since a subset of these tumors harbor underlying genomic alterations commonly observed in AR-active PCs that serve to promote AR oncogenic functions such as TMPRSS2-ERG rearrangements and structural alterations in the AR locus – it is likely that these metastatic PCs arise through transdifferentiation processes that are usually repressed by an active AR program, and enhanced by the loss of key tumor suppressors that influence cell reprogramming such as TP53 and RB1 (13, 19–25).
While pure NEPC is evident in some tumor biopsies including a subset with small cell histology that is indistinguishable from small cell carcinomas arising in other organs such as the lung, other tumors show mixtures of ARPC and NEPC cells indicating a degree of intratumor heterogeneity (14, 26). Currently, neuroendocrine small cell carcinomas are primarily characterized by morphological features, lack of AR expression, and a higher expression of several canonical markers that reflects NE cell differentiation, e.g. the transcriptional factors (TFs) ASCL1, NEUROD1, INSM1, and NE function, for example, the secreted proteins synaptophysin (SYP), chromogranin A (CgA) and neuron specific enolase (NSE) (14, 27, 28).
The role of the TFs in NE differentiation has been an active area of investigation. ASCL1 plays a key role in promoting and maintaining NE features of luminal cell types by modulating chromatin dynamics, supporting lineage plasticity, and directly regulating the expression of secreted NE proteins (29). Similarly, NEUROD1 has been studied in the context of several aggressive neural/neuroendocrine carcinomas and are important for their survival, invasion, and metastasis (30). INSM1 is a zinc-finger transcriptional factor that functions as a transcriptional repressor, thus regulating cell cycle arrest and facilitating NE differentiation (31). On the other hand, endocrine and paracrine functions of NE secretory proteins, despite being some of the most commonly used NE markers have not been established. Most of these canonical markers are not specific to the prostate, but are rather expressed in a variety of tumors that belong to the diffuse neuroendocrine system (32).
Characteristics of neuroendocrine cells in the normal prostate and prostate carcinoma
The prostate is a complex secretory organ comprised of multiple cell types broadly partitioned into epithelium and stroma. The stroma includes predominant resident cell types of smooth muscle, fibroblasts, vascular endothelium and nerves, which are variably infiltrated with transitory inflammatory cell populations that include neutrophils, lymphocytes, and macrophages (33–35). The epithelial compartment is comprised of two primary cell types: basal and luminal/secretory cells, and a minor (<1%) population of neuroendocrine (NE) cells (7, 10, 12, 36) (Table 1). Increasingly sophisticated molecular profiling studies now subdivide these broadly classified types into subtypes with distinctive functions such as those with stem cell/self-renewal capabilities (35, 46, 47). The rare resident NE cells are not easily appreciated using standard H&E staining. They are better identified through immunohistochemistry (IHC) using common markers including chromogranin A (CgA), Synaptophysin (SYP), neuron-specific enolase (NSE), neural cell adhesion molecule (NCAM), Forkhead-box A2 (FOXA2) and CXC chemokine receptor 2 (CXCR2) (12, 48). NE cells are androgen-insensitive and postmitotic and have been shown to be preferentially situated around Ki-67 positive epithelial cells (49, 50), which are highly proliferative. Morphologically, there are two distinct populations of NE cells in the prostate: open, flask-shaped cells with long and slender extensions reaching the lumen, and closed cells without luminal extensions (51) (Figure 1). Although the distinct roles of these two NE sub-populations have not been addressed specifically, there is a distinction in their ability to interact with the prostate environment. Closed cells can only receive basal stimuli, whereas, open cells can also receive luminal stimuli (52). The functional role of NE cells in the mature prostate is not well-defined. Electron microscopic studies have shown that NE cells secrete a variety of products, including serotonin, histamine, chromogranin A and other related peptides, calcitonin, calcitonin gene-related peptide, katacalcin, neuropeptide Y, vasoactive intestinal peptide (VIP), bombesin/gastrin releasing peptide (GRP), somatostatin, alpha-human chorionic gonadotropin (aHCG), parathyroid hormone-related protein (PTHrP), thyroid stimulating hormone-like peptide, cholecystokinin, adrenomedullin and vascular endothelial growth factor (VEGF) (8, 53). The potential role of these factors on prostate cancer pathobiology has been detailed in Table 2, however, in normal physiology these secreted factors have growth promoting and angiogenic properties, justifying the proximity of NE cells near to the highly proliferative cells. The receptors for some of these products are detected in benign and neoplastic prostate epithelium. This suggests, the possible roles of NE cells in the regulation of growth and differentiation of the developing prostate and also regulation of secretory processes in the mature gland (8, 51, 53). Another postulated role of NE cells is regulation of sperm function, because many of the aforementioned secretory products are also detected in the seminal fluid (11, 53).
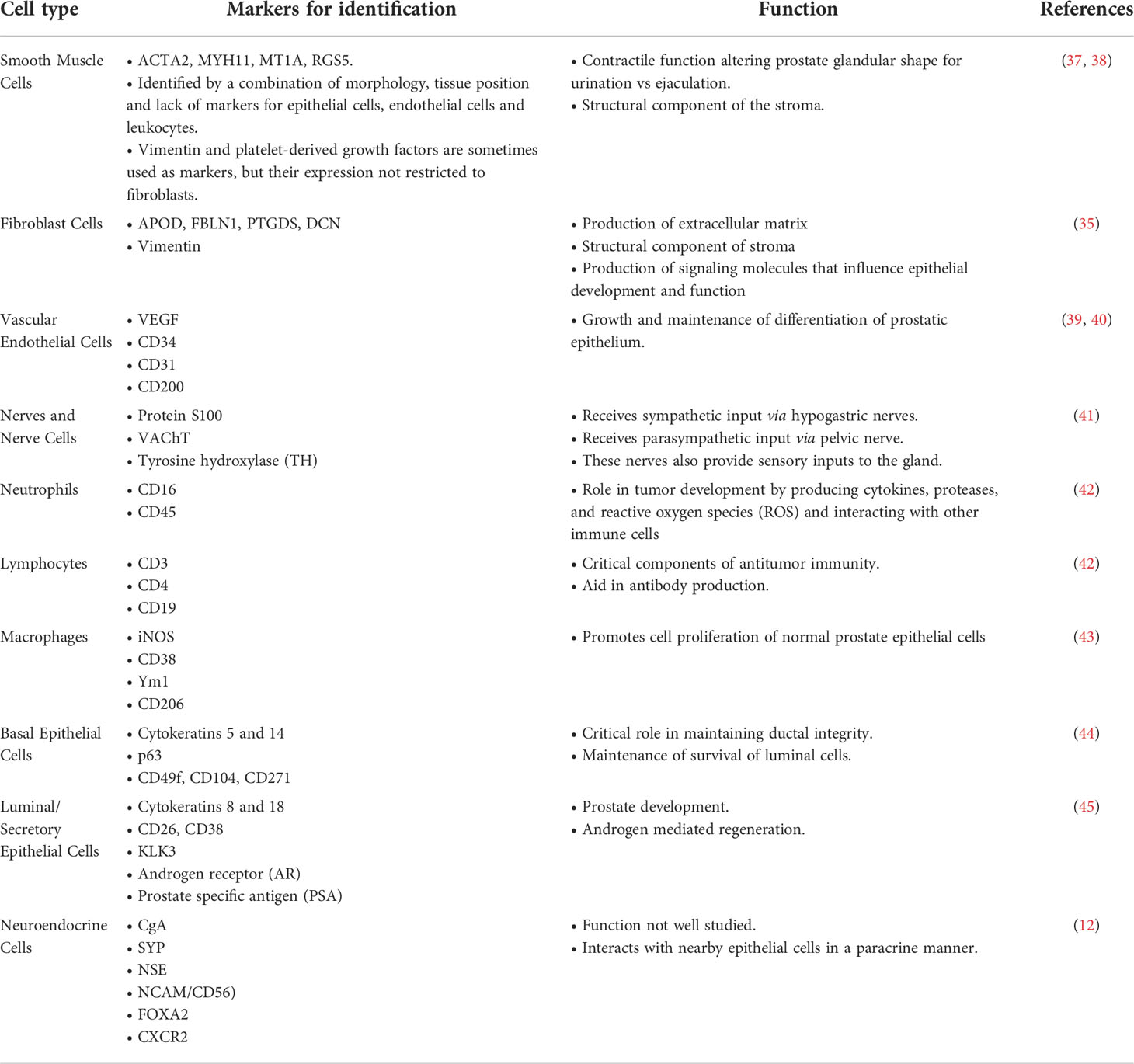
Table 1 Common cell types comprising the normal prostate.
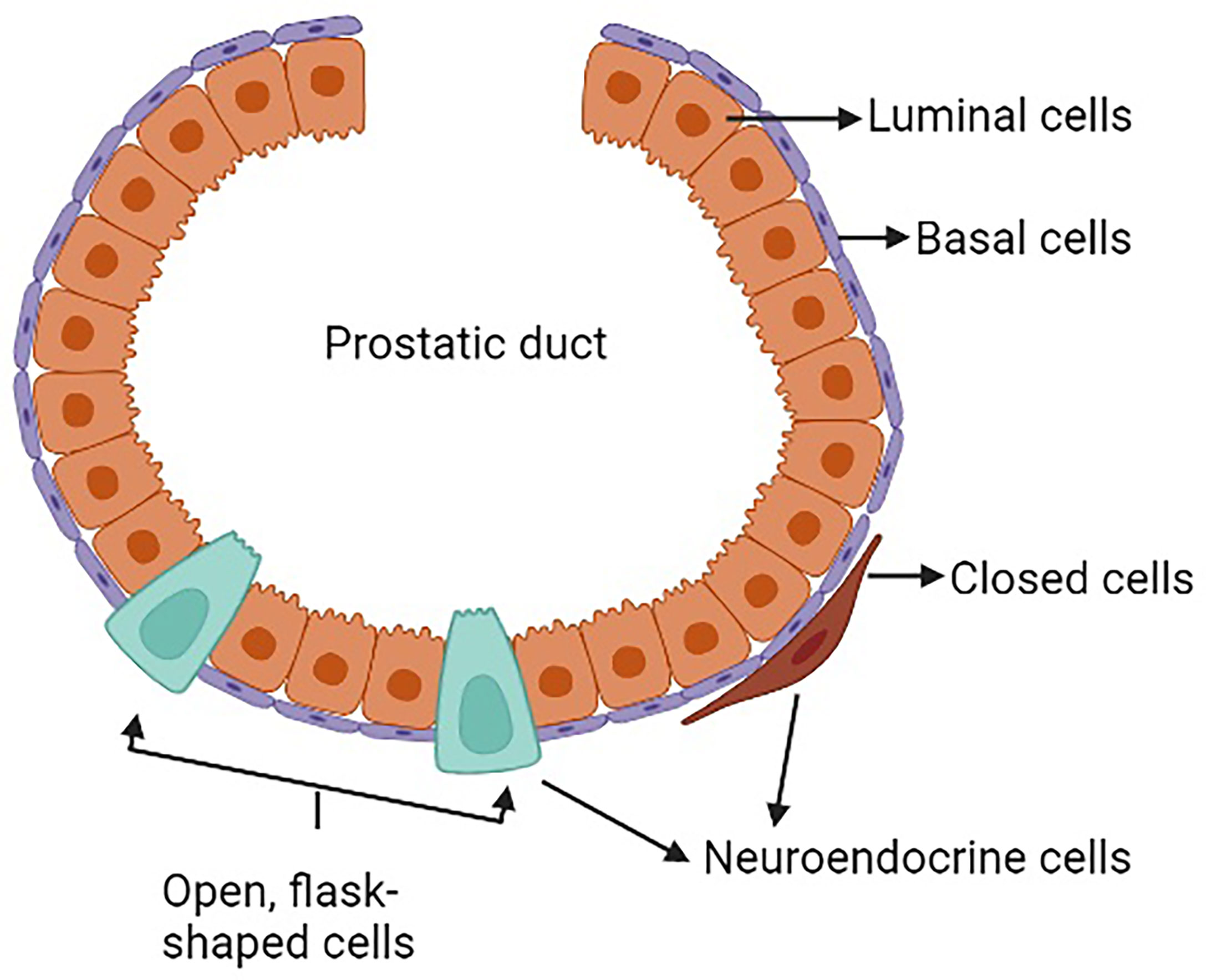
Figure 1 Neuroendocrine cells in a normal prostate.
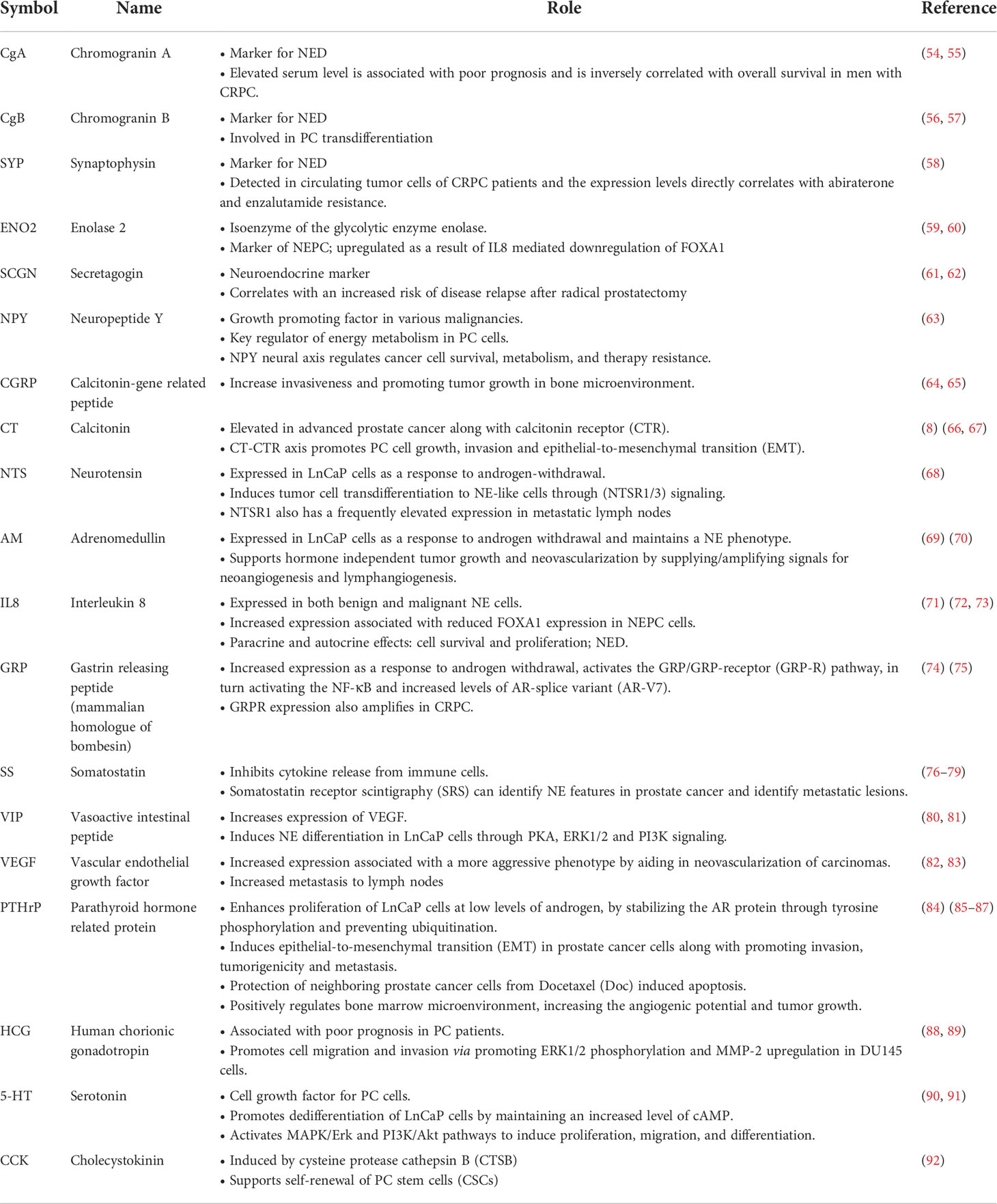
Table 2 Secreted factors from neuroendocrine cells and potential roles in PC pathobiology.
Neuroendocrine prostate cancer and neuroendocrine transdifferentiation
Detailed mechanisms influencing prostate carcinogenesis and tumor development have been previously reviewed (93, 94). Briefly, invasive prostate adenocarcinoma may develop directly from differentiated secretory epithelium, epithelium with stem-like characteristics, or from precursor lesions such as high grade prostatic intraepithelial neoplasia (HGPIN). Prostate adenocarcinoma exhibits characteristic features that include cytologic atypia with enlargement of nuclei and nucleoli, loss of the basal cell layer, branching morphogenesis, and ultimately loss of gland formation (93–95). While localized PC is generally treated by surgical removal or radiation therapy, metastatic PC requires systemic therapies – primarily drugs that repress AR activity. While most PCs resist AR targeting by maintaining or amplifying AR signaling, a subset of PC cells is capable of transdifferentiation – a process whereby a differentiated AR-active tumor cell with secretory luminal cell characteristics, change phenotypes with the resultant loss of AR/luminal cell features and the gain of NE attributes that may include alterations in morphology as well as the expression of NE transcription factors and secretory proteins indicative of differentiated NE cell types (25). This process termed neuroendocrine differentiation (NED) is an adaptive mechanism of PC cells to achieve therapy resistance as AR signaling is no longer operative or required for cell survival and proliferation (28). Determining the cellular mechanisms that initiate NED remains an active area of investigation, although studies have shown that loss of tumor suppressor proteins such as PTEN, RB1, TP53 increases the chances of tumors to acquire neuroendocrine like features (96, 97). However, these tumor suppressors appear to function as permissive rather than deterministic factors. While loss of AR activity and attendant enforcement of epithelial lineage is a key feature contributing to NED, the precise molecular switches responsible for gaining NE functions remain to be identified.
While a complete transition from ARPC to NEPC has been shown to occur in patients, detailed autopsy studies have shown that metastatic tumors may comprise heterogenous populations of ARPC and NEPC that co-exist. Inter- and intra-tumor heterogeneity with respect to tumor cells with ARPC and NEPC phenotypes is well-documented. Notably, other than representing a clear mechanism/pathway for bypassing AR-directed treatment, the role and influence of NE cells in PC pathology, particularly with respect to tumor progression and therapy resistance is not completely understood. This is relevant in view of the potential for NE-associated paracrine and endocrine factors to influence the behavior of non-NE cell types – either locally or distantly. In this context, a previous study reported that the NE cells promoted the growth of castration sensitive LnCaP cells, when grown as a xenograft in castrated mice (98). Further, NE cells were also shown to enhance the migration and metastasis of ARPC cells in the presence of androgen (99). Thus, NEPC cells may promote the continued survival of ARPC cells in an androgen deprived environment possibly through paracrine and endocrine mechanisms (discussed later).
Neuroendocrine carcinomas in non-prostate organs and tissues
Neuroendocrine tumors (NETs) are generally classified as neoplasms with both neural- and endocrine-like characteristics, and these malignancies often have the ability to store and secrete different peptides and neuroamines (100). Although rare, NETs can occur anywhere in the body. Some of the common sites of NET occurrence are the GI tract, lungs and pancreas. The definition of NE cells has changed over the years and in many instances their origins are still not clear. The generally accepted criteria for defining NE cells are: (1) production of a neurotransmitter, neuromodulator or neuropeptide hormone, (2) the presence of dense -core secretory granules from which hormones are released by exocytosis, and (3) the absence of axons and synapses (101). This section briefly discusses several of the more common types of NETs, though as noted above, NETs can arise in nearly every organ/tissue in the body and not all are described here, for example neuroblastoma which is a NE tumor type arising almost exclusively in children (102).
Gastric neuroendocrine tumor
These neoplasms are derived from enterochromaffin-like cells (ECL cells) of the gastric mucosa (103). Over the last several years, the incidence of gNETs is increasing, partly due to improved diagnostic techniques (104). gNETs can be clinically functioning (symptomatic) or silent (non-symptomatic) (105). Based on clinicopathological characteristics, and therapeutic and prognostic implications, gNETs are further subdivided into four types (Type I-Type IV) (104): Type I gNET comprise 70-80% of gNETs and are associated with autoimmune chronic atrophic gastritis (103, 106). These patients often suffer from hypergastrinemia (increased gastrin production by G cells) (103). Patients with type I tumors are usually asymptomatic, and the tumors are rarely metastatic (<2%) (106). However, these tumor cells strongly stain positive for NE markers: chromogranin A (CgA) and neuron specific enolase (NSE) (106), but have very low proliferation rates; Type II gNET represent~7% of gNETs and behave like type I tumors and are caused by gastrinomas. These tumors show an increased staining for CgA compared to the type I tumors and exhibit a higher metastatic potential (103); Type III gNET are aggressive with tissue invasion and metastasis and have a poor prognosis (106). The tumor cells also show a greater frequency of staining for the proliferation marker Ki-67, but are negative for CgA (106); Type IV gNET are very rare, but are highly malignant and exhibit very high Ki67 staining. The tumor cells may lack CgA expression but stain positive for other NE markers such as synaptophysin (SYP) and NSE (106).
Pancreatic neuroendocrine tumors
These are rare neoplasms that represent 1-2% of all pancreatic tumors (107). They were originally thought to arise from the islets of Langerhans, however, evidence suggest an origin from the pluripotent stem cells in the pancreatic ductal/acinar system (108). pNETs produce a range of hormones, including insulin, glucagon, somatostatin, and vasoactive intestinal peptide (VIP) (107). Although most pNETs occur sporadically, about 10% are associated with underlying genetic syndromes including multiple endocrine neoplasia type I (MEN1), type IV (MEN4), von Hippel-Lindau disease (VHL), neurofibromatosis type I (NF1), or tuberous sclerosis complex (TSC) (109, 110). Like the gNETs, pNETs are also classified into functional and non-functional tumors. Functional tumors elicit systemic symptoms through excessive secretion of hormones.
Lung neuroendocrine tumors
These are a heterogenous family of neoplasms in the lung, that arises from the Kulchitzky cells of the bronchial mucosa (111). They are classified into four distinct histologic variants, namely, typical carcinoid (TC), atypical carcinoid (AC), large cell neuroendocrine carcinoma (LCNEC) and small cell lung carcinoma (SCLC) (112).
SCLC is the most aggressive form of lung cancer. SCLC was originally thought to arise de novo from resident neuroendocrine lung cells, but recent evidence from model systems suggests alternative cells of origin such as alveolar type 2 cells (113–115). Similar to NEPC, SCLC can also emerge following targeted therapy for lung adenocarcinoma. For example, resistance to epidermal growth factor receptor (EGFR) inhibitors can result through tumor cell transdifferentiation to SCLC phenotypes which no longer depend on EGFR signaling (116). SCLC is well known to produce a variety of paraneoplastic syndromes that result from the production of hormones such as adrenocorticotrophic hormone (ACTH) (117).
Notably, SCLC shares a strong similarity of chromatin structure and gene expression with NEPC (118). A detailed study of various small cell neuroendocrine cancers (SCNCs) across multiple tissues, shows that these cancers share a convergence of molecular signatures (119, 120). Further, as tumor cells progress towards a small cell neuroendocrine (SCN) phenotype through transdifferentiation, they become increasingly independent of tissue of origin and cluster with SCNCs derived from different tissue types (120). Progress in the clinical treatment of SCNCs has been very slow and improving the outcomes of these aggressive tumors by exploiting the mechanisms underlying their genesis has yet to be fully realized.
Merkel cell carcinoma
Merkel cells are highly specialized cells located in the epidermis that function as pressure receptors and may originate from neural crest cells or from epidermal progenitors (121). While originally considered to be the cell of origin of MCC, which is also classified as primary neuroendocrine carcinoma of the skin, primary small cell carcinoma of the skin and trabecular carcinoma of the skin, more recent studies indicate that these tumors arise from a Merkel cell precursor or from resident fibroblasts via transdifferentiation (121, 122). Drivers of MCC include the Merkel cell polyoma virus – accounting for ~80% of MCCs, and the combination of TP53 and RB1 loss, which occur in the remainder (123–126). MCCs share many features with NE carcinomas arising in other tissues including the expression of SYP and CGA and the neural transcription factor NEUROD1 (27).
In summary NETs are a diverse group of neoplasms, distinguished by site of origin, degree of aggressiveness and function. Although site-specific, most of the NETs express immunohistochemical markers like CGA and SYP. These tumors also produce a similar range of bioactive compounds or hormones (127) that may influence tumor cells at distant sites or produce pathological host effects that are collectively termed as paraneoplastic syndrome (PNS). A PNS may be endocrine – resulting from a specific hormone produced by the cancer, or immune mediated. Though PNS are rare when considering all human cancers, they occur more frequently in NETs. Several well-characterized PNS results from the secretion of excess hormones such as ACTH and others that produce neurologic alterations due to the production of autoantibodies (117). For most of these NETs, surgery and chemotherapy remain the primary curative option if the cancer is identified while localized to the primary site. This is, however, not feasible in NEPC as most patients do not present with localized, organ-confined disease.
The endocrine and paracrine characteristics of NEPC
Analogous to benign NE cells found in the normal prostate, NEPC cells are also capable of secreting a wide range of neuropeptides and other factors (11, 12, 51). To date, studies defining the role(s) of NEPC derived secreted products with respect to PC pathogenesis and response to treatment are limited, but the published reports indicate the importance of further work in this field. Collectively, more than 20 distinct NEPC-derived secreted factors have been identified that are capable of exerting effects on PC adenocarcinoma (Table 2). Recently, it was discovered that neuropeptide Y has a paracrine effect on PC cells by influencing apoptosis, motility, and resistance to radiation therapy (63). The cytokine Interleukin 8 (IL-8) is produced by NEPC cells and is capable of activating non-NE PC cells via the IL-8 receptor CXCR1 with downstream signaling that is capable of driving androgen-independent proliferation and tumor cell invasion (71) (Figure 2). Adrenomedullin, a multifunctioning peptide, is produced by PC cells after androgen depletion, and exerts autocrine signaling that induces a NE-like transdifferentiation phenotype switch (69). The neuropeptide Bombesin/Gastrin Releasing Peptide (GRP) which is expressed in NEPC, exerts mitogenic effects toward PC cells via bombesin receptor (BB2) signaling and may also contribute to androgen-independent growth (74) (Figure 2).
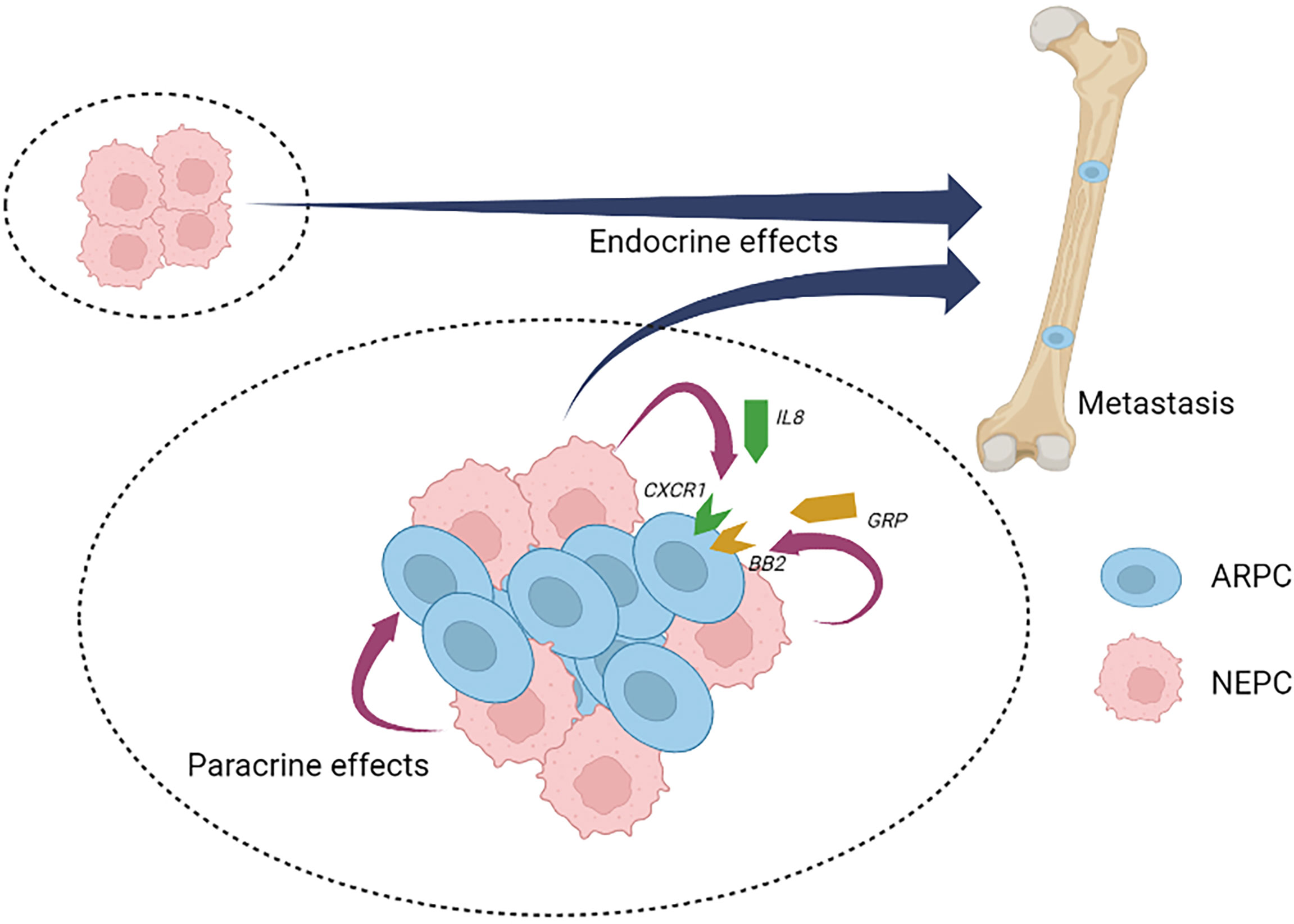
Figure 2 Neuroendocrine prostate cancer paracrine and endocrine signaling. Neuroendocrine prostate cancer (NEPC) cells produce and secrete a spectrum of peptides and proteins with paracrine effects that influence local cell types and endocrine effects that influence distant cell types, tissues and organs. NEPC-derived factors have the potential to promote the progression and therapy resistance of non-NEPC ARPC cells within heterogenous tumors (intra-tumor heterogeneity) or in situations where inter-tumor heterogeneity occurs. NEPC, neuroendocrine prostate cancer; ARPC, AR active prostate adenocarcinoma; IL8, interleukin 8; CXCR1, the IL8 receptor C-X-C motif chemokine receptor 1; GRP, gastrin releasing peptide; BB2, the GRP/Bombesin receptor 2.
The NEPC disease state is often associated with metastatic dissemination. Caveolin-1 is an oncogenic membrane protein associated with extracellular matrix organization, cell migration and signaling (128). In prostate cancer cells, caveolin-1was shown to exert paracrine effects that increase PC proliferative activity aiding in perineural invasion and reduced apoptosis (129). The presence of caveolin-1 in tumor derived exosomes also has a paracrine effect on PC cells, driving the induction of cancer stem cell phenotypes, epithelial- mesenchymal transition, and neuroendocrine differentiation (130). As discussed previously, NEPC cells have been shown to maintain ARPC adenocarcinoma tumor growth after castration, by releasing uncharacterized factors that act to increase AR expression and activity via paracrine and endocrine signaling (98). NEPC cells have also been shown to promote the development of adenocarcinoma pulmonary metastasis (99). Gelsolin is a multifunctional actin-binding protein (131), that shows an increased expression as a response to extracellular factors produced by NEPC cells. Gelsolin overexpression promotes epithelial cell invasion and an increase in cell migration (99).
As mentioned before, NEPC cells can also exert systemic effects through PNS. Though overall extremely rare, a range of PNSs have been shown to arise in the context of aggressive and metastatic PC, and notably NEPC [reviewed by Hong et al. (132)]. Though unusual, the following paraneoplastic syndromes have been attributed to PC: (1) The syndrome of inappropriate antidiuretic hormone secretion (SIADH) is a cause of hyponatremia (133). Patients with SIADH have an elevated antidiuretic hormone (ADH) level in the serum, which then acts on the distal tubules and collecting ducts of the nephron and in turn increase water resorption (132). Although SIADH is very rare in PC patients, there have been a few clinical cases reported (133), and PC tumor cells have been shown to express ADH (134). (2) Cushing’s syndrome is caused by an excess of circulating serum cortisol as a result of excess adrenocorticotrophic hormone (ACTH) (132). In PC, Cushing’s syndrome is primarily associated NE-differentiation to small cell carcinoma (135). (3) Humoral hypercalcemia, is caused by the inappropriate release of parathyroid hormone related peptide (PTHrP) by the tumor cells, which stimulates bone resorption throughout the skeletal system (132). Although very rare in typical PC, NEPC cells have been reported to synthesize and secrete PTHrP with both paracrine signaling effects toward ARPC and endocrine effects contributing to hypercalcemia (84, 136). Several other syndromes resulting from autoimmune responses have been reported in rare instances to be associated with PC and NEPC including Evan’s syndrome which involves immune-mediated hemolytic anemia and thrombocytopenia (137); exfoliative dermatitis (138); polymyalgia rheumatica (139); myasthenia gravis (140); dermatomyositis (141); paraneoplastic jaundice (142) and others (132).
Role of nerves in the development of prostate cancer
In addition to the potential for NE cells to exert effects on non-NE tumor cells via paracrine and/or endocrine effects, nerve cells have also been shown to influence tumor cell behaviors. This section will briefly summarize what is known regarding the role of neural signaling in prostate cancer progression (Figure 3). In addition to fibroblasts, endothelial cells and immune cells, neurons and nerve fibers are integral and functional components of tumor microenvironments (143). The processes of neurogenesis (increased numbers of neurons/nerves) and axonogenesis (tumor induced neural sprouting within or toward tumor microenvironments) can be driven by neurotrophic growth factors released by cancer cells and are emerging as hallmarks of aggressive cancer types (143, 144). The involvement of nerves in cancer has been studied in the context of perineural invasion (PNI), which is the process of neoplastic invasion of nerves contributing to metastatic spread (145, 146). However, until recently, nerves were generally considered passive components of cancers (144). Studies from multiple cancer models have now demonstrated the active involvement of the parasympathetic nervous system (PSNS) and sympathetic nervous system (SNS) in cancer progression and tumorigenesis (147) PSNS and SNS are components of the autonomic nervous system. SNS controls the “flight or fight” response and PSNS controls the “rest and digest” processes. Cholinergic fibers of the PSNS transmits impulses to other nerve cells or muscle fibers by transmitting acetylcholine. Adrenergic fibers of the SNS regulates the function of nearby and distant muscles and also components of the central nervous system by transmitting epinephrine or norepinephrine.
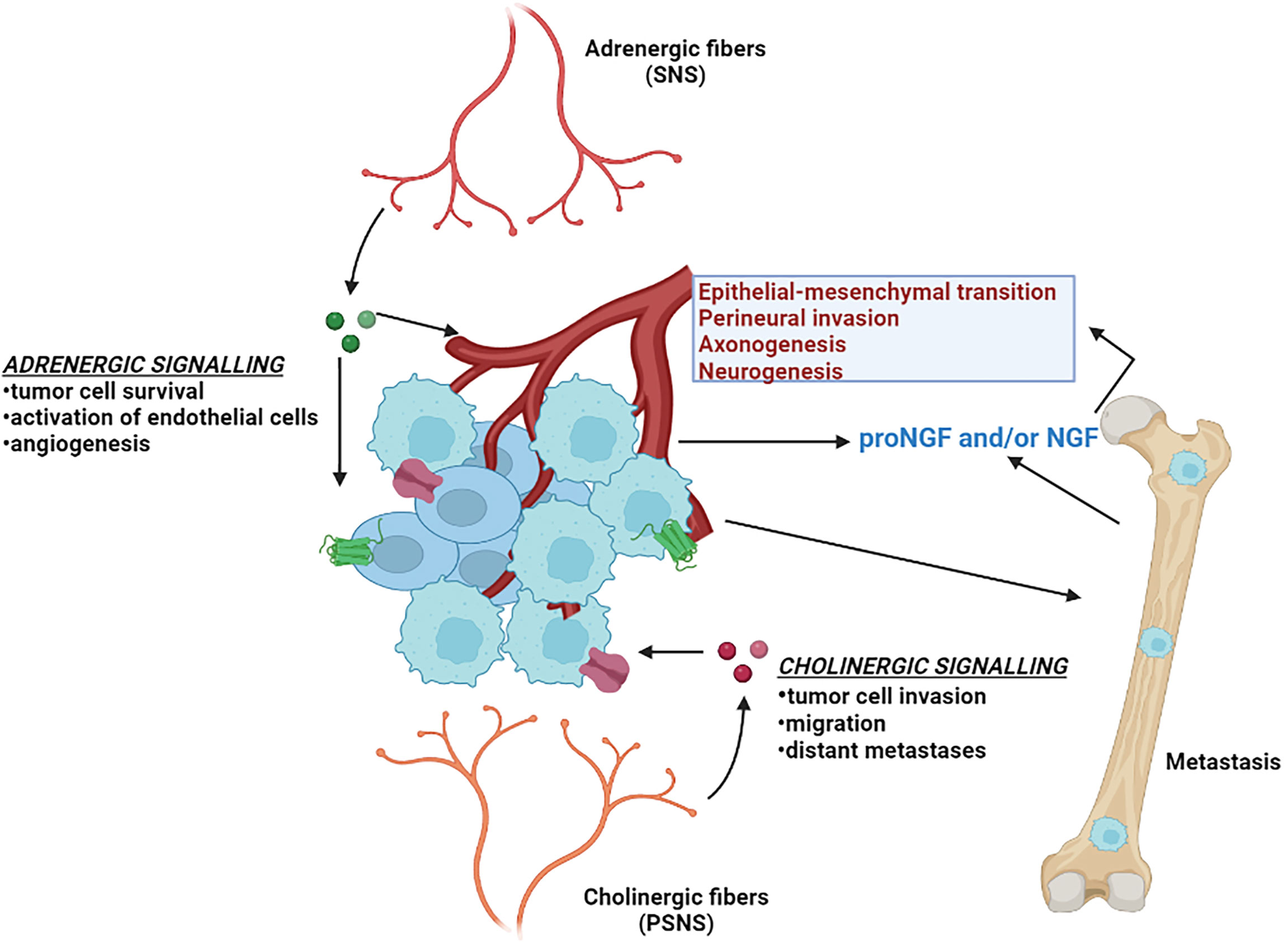
Figure 3 Neural signaling and prostate cancer. Different aspects of prostate cancer development and progression are supported by the autonomic nervous system. The adrenergic fibers of the sympathetic nervous system (SNS) release noradrenaline, that stimulate the beta-adrenergic receptors and supports angiogenesis and tumor cell survival. The cholinergic fibers of the parasympathetic nervous system (PSNS) secrete acetylcholine, that stimulates the cholinergic receptors and contributes to the pathogenesis of aggressive and malignant prostate cancer tumor variant. Invasive prostate cancer cells also secrete neurotrophic factors such as nerve growth factor (NGF) that further stimulates the growth of the autonomic nerve growth into the tumor microenvironment.
Several landmark studies regarding tumor innervation and its effect in PC have been reported in the last several years and have been reviewed in-depth (144, 148, 149). In-vitro co-culture experiments with dorsal root ganglia showed an increased proliferation of human PC cell lines (150), suggesting an interdependence of carcinoma cells and neurons in PNI contributing to PC progression. The importance of nerves in PC progression was further confirmed when surgical denervation showed inhibition of prostate tumor development in mouse models (151). The same group also parsed out the roles of the two distinct autonomic nerve types: (i) adrenergic fibers of the SNS in promoting tumor cell survival and establishing the initial stages of cancer development by acting through the β2 and β3-adrenergic receptors, and (ii) cholinergic fibers of PSNS in supporting tumor cell invasion, migration and distant metastases through stromal Chrm1 (cholinergic receptor muscarinic 1)-mediated signals (151). In alignment with these findings, clinical evidence has demonstrated that patients with spinal cord injuries resulting in functional denervation of the prostate have lower incidence rates of PC (152).
The mechanisms by which nerves influence the pathogenesis of solid tumors is beginning to be understood. Tumors rely on angiogenesis to expand beyond certain physiological constraints relating to oxygenation and the delivery and elimination of metabolites (153). Adrenergic nerve fibers release noradrenaline into the tumor microenvironment that stimulates β2-adrenergic receptor expression, resulting in the activation of endothelial cells and angiogenesis, which in turn supports PC growth (154). PCs of higher Gleason grade have been reported by exhibit greater innervations than PCs of lower grade or benign prostatic hyperplasia (144). Various neurotrophic growth factors produced by cancer cells can contribute to the increased axonogenesis in PC. Overexpression of the precursor of nerve growth factor (proNGF) has been reported in PC accompanied by increases in nerve density (155). Furthermore, it was shown that granulocyte colony-stimulating factor (G-CSF) supports the survival of sympathetic nerve fibers and promotes aberrant outgrowth of parasympathetic nerve fibers in PC models (156).
Several studies have evaluated the role of nerve growth factor (NGF) in the development of CRPC and NEPC disease states. Tropomyosin receptor kinase A (TrkA) receptors activated via nerve growth factor (NGF) signaling have been shown to mediate proliferation, invasiveness and epithelial-mesenchymal transition (EMT) in CRPC cells (157). In the context of ARPC treatment resistance, ADT has been shown to activate the transcription factor ZBTB46, which consequently regulates the activation of NGF. NGF in turn promotes NEPC differentiation by interacting with Chrm4 (cholinergic receptor muscarinic 4) (158). Another recent study reported that patients who subsequently developed metastatic CRPC had elevated adrenergic nerve fiber innervation in the primary prostate tumors. High levels of the neurotransmitter norepinephrine, which is produced by sympathetic nerves, was shown to induce NE-like alterations in PC cells, and these effects were effectively inhibited by β2-adrenenergic receptor blocker propranolol (159).
Conclusions and future directions
Neuroendocrine prostate cancer, whether developing de novo, or through transdifferentiation, carries a very poor prognosis with rapid disease progression and very limited survival. As the frequency of metastatic NEPC appears to be increasing in the setting of more potent AR pathway blockade, new treatment approaches are needed. A notable feature of NE tumors involves their ability to exert effects toward other tumor cell types and benign host cells through endocrine and paracrine mechanisms. These secreted proteins provide a communication network between cancer cells and their adjacent microenvironment that may serve to drive tumor progression and treatment resistance. Preclinical studies have identified the potential therapeutic benefit of inhibiting the activity of the signaling pathways activated by these NE-derived molecules. However, the full repertoire of NEPC-derived secreted factors – the secretome - remains to be identified and characterized. A recent comprehensive secretome study of different subtypes of SCLC underscores the benefit of understanding the aspects of tumor biology that have extracellular influence (160). A thorough understanding of the NEPC secretome: individual factors and combinations - has the potential to widen our understanding of peptides/proteins that can act in an endocrine/paracrine manner to create tumor macro- and microenvironments conducive to tumor survival and growth. Characterizing the interactions between NEPC and ARPC cells also has the potential to identify key drivers of cancer progression and therapy resistance that could serve as effective targets for future drug development.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author contributions
Writing and editing of the manuscript, TA and PN. All authors contributed to the article and approved the submitted version.
Funding
We gratefully acknowledge support from the Pacific Northwest Prostate Cancer SPORE CA097186; NCI R01 CA234715, 1R01 CA266452, PC170350P1, the Prostate Cancer Foundation and the Institute for Prostate Cancer Research.
Acknowledgments
We thank members of the Nelson laboratory, John Lee and Michael Haffner for helpful comments and suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Culp MB, Soerjomataram I, Efstathiou JA, Bray F, Jemal A. Recent global patterns in prostate cancer incidence and mortality rates. Eur Urol (2020) 77(1):38–52. doi: 10.1016/j.eururo.2019.08.005
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
3. Gleason DF. Histologic grading of prostate cancer: a perspective. Hum Pathol (1992) 23(3):273–9. doi: 10.1016/0046-8177(92)90108-F
4. Nelson PS. Molecular states underlying androgen receptor activation: a framework for therapeutics targeting androgen signaling in prostate cancer. J Clin Oncol (2012) 30(6):644–6. doi: 10.1200/JCO.2011.39.1300
5. Craft N, Sawyers CL. Mechanistic concepts in androgen-dependence of prostate cancer. Cancer Metastasis Rev (1998) 17(4):421–7. doi: 10.1023/A:1006141806801
6. Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol (2005) 23(32):8253–61. doi: 10.1200/JCO.2005.03.4777
7. Fine SW. Neuroendocrine tumors of the prostate. Mod Pathol (2018) 31(S1):S122–32. doi: 10.1038/modpathol.2017.164
8. di Sant'Agnese PA. Neuroendocrine differentiation in human prostatic carcinoma. Hum Pathol (1992) 23:287–96. doi: 10.1016/0046-8177(92)90110-O
9. Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma. Prostate (1999) 39(2):135–48. doi: 10.1002/(SICI)1097-0045(19990501)39:2<135::AID-PROS9>3.0.CO;2-S
10. Szczyrba J, Niesen A, Wagner M, Wandernoth PM, Aumuller G, Wennemuth G. Neuroendocrine cells of the prostate derive from the neural crest. J Biol Chem (2017) 292(5):2021–31. doi: 10.1074/jbc.M116.755082
11. Sun Y, Niu J, Huang J. Neuroendocrine differentiation in prostate cancer. Am J Transl Res (2009) 1(2):148–62.
12. Butler W, Huang J. Neuroendocrine cells of the prostate: Histology, biological functions, and molecular mechanisms. Precis Clin Med (2021) 4(1):25–34. doi: 10.1093/pcmedi/pbab003
13. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med (2016) 22(3):298–305. doi: 10.1038/nm.4045
14. Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest (2019) 129(10):4492. doi: 10.1172/JCI128212
15. Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science (2017) 355(6320):84–8. doi: 10.1126/science.aah4307
16. Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science (2017) 355(6320):78–83. doi: 10.1126/science.aah4199
17. Gingrich JR, Barrios RJ, Kattan MW, Nahm HS, Finegold MJ, Greenberg NM. Androgen-independent prostate cancer progression in the TRAMP model. Cancer Res (1997) 57(21):4687–91.
18. Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell (2017) 32(4):474–89 e6. doi: 10.1016/j.ccell.2017.09.003
19. Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov (2011) 1(6):487–95. doi: 10.1158/2159-8290.CD-11-0130
20. Beltran H, Tagawa ST, Park K, MacDonald T, Milowsky MI, Mosquera JM, et al. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol (2012) 30(36):e386–9. doi: 10.1200/JCO.2011.41.5166
21. Lapuk AV, Wu C, Wyatt AW, McPherson A, McConeghy BJ, Brahmbhatt S, et al. From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J Pathol (2012) 227(3):286–97. doi: 10.1002/path.4047
22. Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, Tagawa ST, et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia (2013) 15(1):1–10. doi: 10.1593/neo.121550
23. Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res Off J Am Assoc Cancer Res (2014) 20(11):2846–50. doi: 10.1158/1078-0432.CCR-13-3309
24. Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: A multi-institutional prospective study. J Clin Oncol (2018) 36(24):2492–503. doi: 10.1200/JCO.2017.77.6880
25. Beltran H, Hruszkewycz A, Scher HI, Hildesheim J, Isaacs J, Yu EY, et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin Cancer Res (2019) 25(23):6916–24. doi: 10.1158/1078-0432.CCR-19-1423
26. Brady L, Kriner M, Coleman I, Morrissey C, Roudier M, True LD, et al. Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat Commun (2021) 12(1):1426. doi: 10.1038/s41467-021-21615-4
27. Corella AN, Cabiliza Ordonio MVA, Coleman I, Lucas JM, Kaipainen A, Nguyen HM, et al. Identification of therapeutic vulnerabilities in small cell neuroendocrine prostate cancer. Clin Cancer Res (2019) 26(7):1667. doi: 10.1158/1078-0432.CCR-19-0775
28. Patel GK, Chugh N, Tripathi M. Neuroendocrine differentiation of prostate cancer-an intriguing example of tumor evolution at play. Cancers (Basel) (2019) 11(10):1405 doi: 10.3390/cancers11101405.
29. Nouruzi S, Ganguli D, Tabrizian N, Kobelev M, Sivak O, Namekawa T, et al. ASCL1 activates neuronal stem cell-like lineage programming through remodeling of the chromatin landscape in prostate cancer. Nat Commun (2022) 13(1):2282. doi: 10.1038/s41467-022-29963-5
30. Osborne JK, Larsen JE, Shields MD, Gonzales JX, Shames DS, Sato M, et al. NeuroD1 regulates survival and migration of neuroendocrine lung carcinomas via signaling molecules TrkB and NCAM. Proc Natl Acad Sci U S A (2013) 110(16):6524–9. doi: 10.1073/pnas.1303932110
31. Xin Z, Zhang Y, Jiang Z, Zhao L, Fan L, Wang Y, et al. Insulinoma-associated protein 1 is a novel sensitive and specific marker for small cell carcinoma of the prostate. Hum Pathol (2018) 79:151–9. doi: 10.1016/j.humpath.2018.05.014
32. Erickson LA, Lloyd RV. Practical markers used in the diagnosis of endocrine tumors. Adv Anat Pathol (2004) 11(4):175–89. doi: 10.1097/01.pap.0000131824.77317.a7
33. Sugimura Y, Cunha GR, Donjacour AA. Morphogenesis of ductal networks in the mouse prostate. Biol Reprod (1986) 34(5):961–71. doi: 10.1095/biolreprod34.5.961
34. Cunha GR, Donjacour AA, Cooke PS, Mee S, Bigsby RM, Higgins SJ, et al. The endocrinology and developmental biology of the prostate. Endocr Rev (1987) 8(3):338–62. doi: 10.1210/edrv-8-3-338
35. Henry GH, Malewska A, Joseph DB, Malladi VS, Lee J, Torrealba J, et al. A cellular anatomy of the normal adult human prostate and prostatic urethra. Cell Rep (2018) 25(12):3530–42 e5. doi: 10.1016/j.celrep.2018.11.086
36. Abdulfatah E, Fine SW, Lotan T, Mehra R. De novo neuroendocrine features in prostate cancer. Hum Pathol (2022) 127:112. doi: 10.1016/j.humpath.2022.07.002
37. Peinetti N, Scalerandi MV, Cuello Rubio MM, Leimgruber C, Nicola JP, Torres AI, et al. The response of prostate smooth muscle cells to testosterone is determined by the subcellular distribution of the androgen receptor. Endocrinology (2018) 159(2):945–56. doi: 10.1210/en.2017-00718
38. Bonollo F, Thalmann GN, Kruithof-de Julio M, Karkampouna S. The role of cancer-associated fibroblasts in prostate cancer tumorigenesis. Cancers (Basel) (2020) 12(7):1887. doi: 10.3390/cancers12071887
39. Trojan L, Thomas D, Friedrich D, Grobholz R, Knoll T, Alken P, et al. Expression of different vascular endothelial markers in prostate cancer and BPH tissue: an immunohistochemical and clinical evaluation. Anticancer Res (2004) 24(3a):1651–6. doi: 10.1016/S1569-9056(04)90362-4
40. Bates M, Kovalenko B, Wilson EL, Moscatelli D. Endothelial cells support the growth of prostate tissue in vivo. Prostate (2008) 68(8):893–901. doi: 10.1002/pros.20762
41. Alsaid B, Bessede T, Karam I, Abd-Alsamad I, Uhl JF, Benoit G, et al. Coexistence of adrenergic and cholinergic nerves in the inferior hypogastric plexus: anatomical and immunohistochemical study with 3D reconstruction in human male fetus. J Anat (2009) 214(5):645–54. doi: 10.1111/j.1469-7580.2009.01071.x
42. Cao J, Zhu X, Zhao X, Li XF, Xu R. Neutrophil-to-Lymphocyte ratio predicts PSA response and prognosis in prostate cancer: A systematic review and meta-analysis. PloS One (2016) 11(7):e0158770. doi: 10.1371/journal.pone.0158770
43. Dang T, Liou GY. Macrophage cytokines enhance cell proliferation of normal prostate epithelial cells through activation of ERK and akt. Sci Rep (2018) 8(1):7718. doi: 10.1038/s41598-018-26143-8
44. Kurita T, Medina RT, Mills AA, Cunha GR. Role of p63 and basal cells in the prostate. Development (2004) 131(20):4955–64. doi: 10.1242/dev.01384
45. Zhang D, Zhao S, Li X, Kirk JS, Tang DG. Prostate luminal progenitor cells in development and cancer. Trends Cancer (2018) 4(11):769–83. doi: 10.1016/j.trecan.2018.09.003
46. Joseph DB, Turco AE, Vezina CM, Strand DW. Progenitors in prostate development and disease. Dev Biol (2021) 473:50–8. doi: 10.1016/j.ydbio.2020.11.012
47. Joseph DB, Henry GH, Malewska A, Reese JC, Mauck RJ, Gahan JC, et al. Single-cell analysis of mouse and human prostate reveals novel fibroblasts with specialized distribution and microenvironment interactions. J Pathol (2021) 255(2):141–54. doi: 10.1002/path.5751
48. Falkmer S, Askensten U, Grimelius L, Abrahamsson PA. Cytochemical markers and DNA content of neuroendocrine cells in carcinoma of the prostate gland during tumour progression. Acta Histochem Suppl (1990) 38:127–32.
49. Bonkhoff H, Wernert N, Dhom G, Remberger K. Relation of endocrine-paracrine cells to cell proliferation in normal, hyperplastic, and neoplastic human prostate. Prostate (1991) 19(2):91–8. doi: 10.1002/pros.2990190202
50. Bonkhoff H, Stein U, Remberger K. Endocrine-paracrine cell types in the prostate and prostatic adenocarcinoma are postmitotic cells. Hum Pathol (1995) 26(2):167–70. doi: 10.1016/0046-8177(95)90033-0
51. Abrahamsson PA. Neuroendocrine differentiation and hormone-refractory prostate cancer. Prostate Suppl (1996) 6:3–8. doi: 10.1002/(SICI)1097-0045(1996)6+<3::AID-PROS2>3.0.CO;2-T
52. Grigore AD, Ben-Jacob E, Farach-Carson MC. Prostate cancer and neuroendocrine differentiation: more neuronal, less endocrine? Front Oncol (2015) 5:37. doi: 10.3389/fonc.2015.00037
53. Huang J, Wu C, di Sant'Agnese PA, Yao JL, Cheng L, Na Y. Function and molecular mechanisms of neuroendocrine cells in prostate cancer. Anal Quant Cytol Histol (2007) 29(3):128–38.
54. Giridhar, et al. Serum chromogranin-a-based prognosis in metastatic castration- resistant prostate cancer. Prostate Cancer Prostatic Dis (2018) 21(3):431. doi: 10.1038/s41391-018-0046-9
55. Guo, et al. Chromogranin a is a predictor of prognosis in patients with prostate cancer: a systematic review and meta-analysis. Cancer Manage Res (2019) 11:2747. doi: 10.2147/CMAR.S190678
56. Pruneri, et al. Chromogranin a and b and secretogranin II in prostatic adenocarcinomas: Neuroendocrine expression in patients untreated and treated with androgen deprivation therapy. Prostate (1998) 34(2):113. doi: 10.1002/(SICI)1097-0045(19980201)34:2<113::AID-PROS5>3.0.CO;2-L
57. Yuan, et al. Androgen deprivation induces human prostate epithelial neuroendocrine differentiation of androgen-sensitive LNCaP cells. Endocrine-Related Cancer (2006) 13(1):151. doi: 10.1677/erc.1.01043
58. Pal, et al. Synaptophysin expression on circulating tumor cells in patients with castration resistant prostate cancer undergoing treatment with abiraterone acetate or enzalutamide. Urologic Oncology: Semin Original Investigations (2018) 36(4):162.e1. doi: 10.1016/j.urolonc.2017.12.006
59. Isgro, et al. Neuron-specific enolase as a biomarker: Biochemical and clinical aspects. Adv Exp Med Biol (2015) 867:125. doi: 10.1007/978-94-017-7215-0_9
60. Kim, et al. FOXA1 inhibits prostate cancer neuroendocrine differentiation. Oncogene (2017) 36(28):4072. doi: 10.1038/onc.2017.50
61. Adolf, et al. Secretagogin is a new neuroendocrine marker in the human prostate. Prostate (2007) 67(5):472. doi: 10.1002/pros.20523
62. Flores-Morales, et al. Proteogenomic characterization of patient- derived xenografts highlights the role of REST in neuroendocrine differentiation of castration- resistant prostate cancer. Clin Cancer Res (2019) 25(2):595. doi: 10.1158/1078-0432.CCR-18-0729
63. Ding Y, Lee M, Gao Y, Bu P, Coarfa C, Miles B, et al. Neuropeptide y nerve paracrine regulation of prostate cancer oncogenesis and therapy resistance. Prostate (2021) 81(1):58–71. doi: 10.1002/pros.24081
64. Nagakawa, et al. Effect of prostatic neuropeptides on invasion and migration of PC-3 prostate cancer cells. Cancer Lett (1998) 133(1):27. doi: 10.1016/S0304-3835(98)00186-4
65. Zhu, et al. Neuronal calcitonin gene-related peptide promotes prostate tumor growth in the bone microenvironment. Peptides (2021) 135:170423. doi: 10.1016/j.peptides.2020.170423
66. Shah, et al. Calcitonin promotes in vivo metastasis of prostate cancer cells by altering cell signaling, adhesion, and inflammatory pathways. Endocrine-Related Cancer (2008) 15(4):953. doi: 10.1677/ERC-08-0136
67. Aldahish, et al. Calcitonin induces stem cell-like phenotype in prostate cancer cells. Endocrine-Related Cancer (2019) 26(11):815. doi: 10.1530/ERC-19-0333
68. Boules, et al. Diverse roles of neurotensin agonists in the central nervous system. Front Endocrinol (2013). doi: 10.3389/fendo.2013.00036
69. Berenguer C, Boudouresque F, Dussert C, Daniel L, Muracciole X, Grino M, et al. Adrenomedullin, an autocrine/paracrine factor induced by androgen withdrawal, stimulates 'neuroendocrine phenotype' in LNCaP prostate tumor cells. Oncogene (2008) 27(4):506–18. doi: 10.1038/sj.onc.1210656
70. Berenguer-Daize, et al. Adrenomedullin blockade suppresses growth of human hormone–independent prostate tumor xenograft in mice. Clin Cancer Res (2013) 19(22):6138. doi: 10.1158/1078-0432.CCR-13-0691
71. Huang J, Yao JL, Zhang L, Bourne PA, Quinn AM, di Sant'Agnese PA, et al. Differential expression of interleukin-8 and its receptors in the neuroendocrine and non-neuroendocrine compartments of prostate cancer. Am J Pathol (2005) 166(6):1807–15. doi: 10.1016/S0002-9440(10)62490-X
72. Guo, et al. IL-8 promotes proliferation and inhibition of apoptosis via STAT3/AKT/NF-;κB pathway in prostate cancer. Mol Med Rep (2017) 16(6):9035. doi: 10.3892/mmr.2017.7747
73. Merkens, et al. Aggressive variants of prostate cancer: underlying mechanisms of neuroendocrine transdifferentiation. J Exp Clin Cancer Res (2022) 41(1):46. doi: 10.1186/s13046-022-02255-y
74. Qiao J, Grabowska MM, Forestier-Roman IS, Mirosevich J, Case TC, Chung DH, et al. Activation of GRP/GRP-r signaling contributes to castration-resistant prostate cancer progression. Oncotarget (2016) 7(38):61955–69. doi: 10.18632/oncotarget.11326
75. Case, et al. Blocking GRP/GRP-r signaling decreases expression of androgen receptor splice variants and inhibits tumor growth in castration-resistant prostate cancer. Trans Oncol (2021) 14(11):101213. doi: 10.1016/j.tranon.2021.101213
76. Hope, et al. Somatostatin imaging of neuroendocrine-differentiated prostate cancer. Clin Nucl Med (2015) 40(6):540. doi: 10.1097/RLU.0000000000000776
77. Mori, et al. Imaging somatostatin receptor activity in neuroendocrine-differentiated prostate cancer. Internal Med (2018) 57(21):3123. doi: 10.2169/internalmedicine.0630-17
78. Ampofo, et al. Regulatory mechanisms of somatostatin expression. Int J Mol Sci (2020) 21(11):4170. doi: 10.3390/ijms21114170
79. Kitajima, et al. Pelvic MRI, FDG-PET/CT, and somatostatin receptor scintigraphy findings of treatment- related neuroendocrine-differentiated prostate cancer. Case Rep Oncol (2021) 14(1):397. doi: 10.1159/000511070
80. Collado, et al. Vasoactive intestinal peptide (VIP) induces c-fos expression in LNCaP prostate cancer cells through a mechanism that involves Ca2+ signalling. implications in angiogenesis and neuroendocrine differentiation. Biochim Biophys Acta (BBA) - Mol Cell Res (2005) 1744(2):224. doi: 10.1016/j.bbamcr.2005.04.009
81. Gutierrez-Canas, et al. Vasoactive intestinal peptide InducesNeuroendocrine DifferentiationintheLNCaPProstateCancerCellLine ThroughPKA,ERK, andPI3K. Prostate (2005) 63(1):44. doi: 10.1002/pros.20173
82. Harper, et al. Vascular endothelial growth factor (VEGF) expression in prostatic tumours and its relationship to neuroendocrine cells. Br J Cancer (1996) 74(6):910. doi: 10.1038/bjc.1996.456
83. Shariat, et al. Association of preoperative plasma levels of vascular endothelial growth factor and soluble vascular cell adhesion molecule-1 with lymph node status and biochemical progression after radical prostatectomy. J Clin Oncol (2004) 22(9):1655. doi: 10.1200/JCO.2004.09.142
84. Cui Y, Sun Y, Hu S, Luo J, Li L, Li X, et al. Neuroendocrine prostate cancer (NEPCa) increased the neighboring PCa chemoresistance via altering the PTHrP/p38/Hsp27/androgen receptor (AR)/p21 signals. Oncogene (2016) 35(47):6065–76. doi: 10.1038/onc.2016.135
85. DaSilva, et al. The neuroendocrine-derived peptide parathyroid hormone–related protein promotes prostate cancer cell growth by stabilizingthe androgen receptor. Cancer Res (2009) 69(18):7402. doi: 10.1158/0008-5472.CAN-08-4687
86. Park, et al. Parathyroid hormone–related protein drives a CD11bþGr1þ cell–mediated positive feedback loop to support prostate cancer growth. Cancer Res (2013) 73(22):6574. doi: 10.1158/0008-5472.CAN-12-4692
87. Ongkeko, et al. Parathyroid hormone related-protein promotes epithelial-to-Mesenchymal transition in prostate cancer. PloS One (2014) 9(1):e85803. doi: 10.1371/journal.pone.0085803
88. Sheaff, et al. phCG as a prognostic marker in adenocarcinoma of the prostate. J Clin Pathol (1996) 49(4):329. doi: 10.1136/jcp.49.4.329
89. Li, et al. Human chorionic gonadotropin b induces migration and invasion via activating ERK1/2 and MMP-2 in human prostate cancer DU145 cells. PloS One (2013) 8(2):e54592. doi: 10.1371/journal.pone.0054592
90. Dizeyi, et al. Serotonin activates MAP kinase and PI3K/Akt signaling pathways in prostate cancer cell lines. Urologic Oncology: Semin Original Investigations (2011) 29(4):436. doi: 10.1016/j.urolonc.2009.09.013
91. Shinka, et al. Serotonin synthesis and metabolism-related molecules in a human prostate cancer cell line. Oncol Lett (2011) 2(2):211. doi: 10.3892/ol.2011.244
92. Tang, et al. Adipocytes promote prostate cancer stem cell self-renewal through amplification of the cholecystokinin autocrine loop. Oncotarget (2016) 7(4):4939. doi: 10.18632/oncotarget.6643
93. Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. N Engl J Med (2003) 349(4):366–81. doi: 10.1056/NEJMra021562
94. Knudsen BS, Vasioukhin V. Mechanisms of prostate cancer initiation and progression. Adv Cancer Res (2010) 109:1–50. doi: 10.1016/B978-0-12-380890-5.00001-6
95. Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev (2000) 14(19):2410–34. doi: 10.1101/gad.819500
96. Kelly K, Balk SP. Reprogramming to resist. Science (2017) 355(6320):29–30. doi: 10.1126/science.aam5355
97. Sinha S, Nelson PS. The path of most resistance: Transdifferentiation underlies exceptional nonresponses to androgen receptor pathway inhibition in prostate cancer. Cancer Discov (2017) 7(7):673–4. doi: 10.1158/2159-8290.CD-17-0481
98. Jin RJ, Wang Y, Masumori N, Ishii K, Tsukamoto T, Shappell SB, et al. NE-10 neuroendocrine cancer promotes the LNCaP xenograft growth in castrated mice. Cancer Res (2004) 64(15):5489–95. doi: 10.1158/0008-5472.CAN-03-3117
99. Uchida K, Masumori N, Takahashi A, Itoh N, Kato K, Matusik RJ, et al. Murine androgen-independent neuroendocrine carcinoma promotes metastasis of human prostate cancer cell line LNCaP. Prostate (2006) 66(5):536–45. doi: 10.1002/pros.20369
100. Vinik AI, Woltering EA, Warner RR, Caplin M, O'Dorisio TM, Wiseman GA, et al. NANETS consensus guidelines for the diagnosis of neuroendocrine tumor. Pancreas (2010) 39(6):713–34. doi: 10.1097/MPA.0b013e3181ebaffd
101. Barakat MT, Meeran K, Bloom SR. Neuroendocrine tumours. Endocr Relat Cancer (2004) 11(1):1–18. doi: 10.1677/erc.0.0110001
102. Ponzoni M, Bachetti T, Corrias MV, Brignole C, Pastorino F, Calarco E, et al. Recent advances in the developmental origin of neuroblastoma: an overview. J Exp Clin Cancer Res (2022) 41(1):92. doi: 10.1186/s13046-022-02281-w
103. Dias AR, Azevedo BC, Alban LBV, Yagi OK, Ramos M, Jacob CE, et al. Gastric neuroendocrine tumor: Review and update. Arq Bras Cir Dig (2017) 30(2):150–4. doi: 10.1590/0102-6720201700020016
104. Carvao J, Dinis-Ribeiro M, Pimentel-Nunes P, Libanio D. Neuroendocrine tumors of the gastrointestinal tract: A focused review and practical approach for gastroenterologists. GE Port J Gastroenterol (2021) 28(5):336–48. doi: 10.1159/000512089
105. Oberg KE. Gastrointestinal Neuroendocrine Tumors. Annals of Oncol (2010) 21 Suppl 7:vii72–80. doi: 10.1093/annonc/mdq290
106. Li TT, Qiu F, Qian ZR, Wan J, Qi XK, Wu BY. Classification, clinicopathologic features and treatment of gastric neuroendocrine tumors. World J Gastroenterol (2014) 20(1):118–25. doi: 10.3748/wjg.v20.i1.118
107. McKenna LR, Edil BH. Update on pancreatic neuroendocrine tumors. Gland Surg (2014) 3(4):258–75. doi: 10.3978/j.issn.2227-684X.2014.06.03
108. Vortmeyer AO, Huang S, Lubensky I, Zhuang Z. Non-islet origin of pancreatic islet cell tumors. J Clin Endocrinol Metab (2004) 89(4):1934–8. doi: 10.1210/jc.2003-031575
109. Yachida S, Vakiani E, White CM, Zhong Y, Saunders T, Morgan R, et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol (2012) 36(2):173–84. doi: 10.1097/PAS.0b013e3182417d36
110. de Wilde RF, Edil BH, Hruban RH, Maitra A. Well-differentiated pancreatic neuroendocrine tumors: from genetics to therapy. Nat Rev Gastroenterol Hepatol (2012) 9(4):199–208. doi: 10.1038/nrgastro.2012.9
111. Chong S, Lee KS, Chung MJ, Han J, Kwon OJ, Kim TS. Neuroendocrine tumors of the lung: clinical, pathologic, and imaging findings. Radiographics (2006) 26(1):41–57. doi: 10.1148/rg.261055057
112. Pelosi G, Sonzogni A, Harari S, Albini A, Bresaola E, Marchio C, et al. Classification of pulmonary neuroendocrine tumors: new insights. Transl Lung Cancer Res (2017) 6(5):513–29. doi: 10.21037/tlcr.2017.09.04
113. Park KS, Liang MC, Raiser DM, Zamponi R, Roach RR, Curtis SJ, et al. Characterization of the cell of origin for small cell lung cancer. Cell Cycle (2011) 10(16):2806–15. doi: 10.4161/cc.10.16.17012
114. Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell (2011) 19(6):754–64. doi: 10.1016/j.ccr.2011.04.019
115. Ferone G, Song JY, Krijgsman O, van der Vliet J, Cozijnsen M, Semenova EA, et al. FGFR1 oncogenic activation reveals an alternative cell of origin of SCLC in Rb1/p53 mice. Cell Rep (2020) 30(11):3837–50 e3. doi: 10.1016/j.celrep.2020.02.052
116. Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol (2015) 16(4):e165–72. doi: 10.1016/S1470-2045(14)71180-5
117. Soomro Z, Youssef M, Yust-Katz S, Jalali A, Patel AJ, Mandel J. Paraneoplastic syndromes in small cell lung cancer. J Thorac Dis (2020) 12(10):6253–63. doi: 10.21037/jtd.2020.03.88
118. Cejas P, Xie Y, Font-Tello A, Lim K, Syamala S, Qiu X, et al. Subtype heterogeneity and epigenetic convergence in neuroendocrine prostate cancer. Nat Commun (2021) 12(1):5775. doi: 10.1038/s41467-021-26042-z
119. Park JW, Lee JK, Sheu KM, Wang L, Balanis NG, Nguyen K, et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science (2018) 362(6410):91–5. doi: 10.1126/science.aat5749
120. Balanis NG, Sheu KM, Esedebe FN, Patel SJ, Smith BA, Park JW, et al. Pan-cancer convergence to a small-cell neuroendocrine phenotype that shares susceptibilities with hematological malignancies. Cancer Cell (2019) 36(1):17–34 e7. doi: 10.1016/j.ccell.2019.06.005
121. Becker JC, Stang A, DeCaprio JA, Cerroni L, Lebbe C, Veness M, et al. Merkel cell carcinoma. Nat Rev Dis Primers (2017) 3:17077. doi: 10.1038/nrdp.2017.77
122. Dika E, Pellegrini C, Lambertini M, Patrizi A, Ventura A, Baraldi C, et al. Merkel cell carcinoma: an updated overview of clinico-pathological aspects, molecular genetics and therapy. Eur J Dermatol (2021) 31(6):691–701. doi: 10.1684/ejd.2021.4170
123. Hodgson NC. Merkel cell carcinoma: Changing incidence trends. J Surg Oncol (2005) 89(1):1–4. doi: 10.1002/jso.20167
124. Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human merkel cell carcinoma. Science (2008) 319(5866):1096–100. doi: 10.1126/science.1152586
125. Albores-Saavedra J, Batich K, Chable-Montero F, Sagy N, Schwartz AM, Henson DE. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutaneous Pathol (2010) 37(1):20–7. doi: 10.1111/j.1600-0560.2009.01370.x
126. Goh G, Walradt T, Markarov V, Blom A, Riaz N, Doumani R, et al. Mutational landscape of MCPyV-positive and MCPyV-negative merkel cell carcinomas with implications for immunotherapy. Oncotarget (2015) 7(3):3403. doi: 10.18632/oncotarget.6494
127. Oronsky B, Ma PC, Morgensztern D, Carter CA. Nothing but NET: A review of neuroendocrine tumors and carcinomas. Neoplasia (2017) 19(12):991–1002. doi: 10.1016/j.neo.2017.09.002
128. Nwosu ZC, Ebert MP, Dooley S, Meyer C. Caveolin-1 in the regulation of cell metabolism: a cancer perspective. Mol Cancer (2016) 15(1):71. doi: 10.1186/s12943-016-0558-7
129. Ayala GE, Dai H, Tahir SA, Li R, Timme T, Ittmann M, et al. Stromal antiapoptotic paracrine loop in perineural invasion of prostatic carcinoma. Cancer Res (2006) 66(10):5159–64. doi: 10.1158/0008-5472.CAN-05-1847
130. Lin CJ, Yun EJ, Lo UG, Tai YL, Deng S, Hernandez E, et al. The paracrine induction of prostate cancer progression by caveolin-1. Cell Death Dis (2019) 10(11):834. doi: 10.1038/s41419-019-2066-3
131. Bucki R, Levental I, Kulakowska A, Janmey PA. Plasma gelsolin: function, prognostic value, and potential therapeutic use. Curr Protein Pept Sci (2008) 9(6):541–51. doi: 10.2174/138920308786733912
132. Hong MK, Kong J, Namdarian B, Longano A, Grummet J, Hovens CM, et al. Paraneoplastic syndromes in prostate cancer. Nat Rev Urol (2010) 7(12):681–92. doi: 10.1038/nrurol.2010.186
133. Lee KS, Kang TW, Ma SK, Kim SW, Kim NH, Choi KC. SIADH associated with prostate cancer. Electrolyte Blood Press (2007) 5(1):47–9. doi: 10.5049/EBP.2007.5.1.47
134. Yamazaki T, Suzuki H, Tobe T, Sekita N, Kito H, Ichikawa T, et al. Prostate adenocarcinoma producing syndrome of inappropriate secretion of antidiuretic hormone. Int J Urol (2001) 8(9):513–6. doi: 10.1046/j.1442-2042.2001.00362.x
135. Nimalasena S, Freeman A, Harland S. Paraneoplastic cushing's syndrome in prostate cancer: a difficult management problem. BJU Int (2008) 101(4):424–7. doi: 10.1111/j.1464-410X.2007.07294.x
136. Wu CH, Lan YJ, Wang CH, Wu MS. Hypercalcemia in prostate cancer with positive neuron-specific enolase stain. Ren Fail (2004) 26(3):325–7. doi: 10.1081/JDI-200026727
137. Sidda A, Manu G, Abdallah M, Griswold D, Alsharedi M, Pacioles T. Paraneoplastic Evans syndrome in a patient with prostate cancer with small cell transformation. Cureus (2022) 14(4):e24505. doi: 10.7759/cureus.24505
138. Ghafouri SR, Escriba-Omar A, Wahdatyar I, Whited N, Hakim MN, Gaur S, et al. Exfoliative dermatitis as a para-neoplastic syndrome of prostate adenocarcinoma: A rare case report with literature review. Cancer Diagn Progn (2021) 1(4):289–95. doi: 10.21873/cdp.10038
139. Kawase M, Nakane K, Namiki S, Takeuchi Y, Ueda S, Kawase K, et al. A case of localized prostate cancer associated with polymyalgia rheumatica with marked symptomatic improvement after robot-assisted radical prostatectomy. Case Rep Urol (2021) 2021:8026883. doi: 10.1155/2021/8026883
140. Pereira MS, Escarigo MC, Correia Azevedo P, Delerue F. Myasthenia gravis and prostatic neoplasia: a rare association. BMJ Case Rep (2021) 14(5):e242416. doi: 10.1136/bcr-2021-242416
141. Papagoras C, Arelaki S, Botis I, Chrysafis I, Giannopoulos S, Skendros P. Co-Occurrence of dermatomyositis and polycythemia unveiling rare de Novo neuroendocrine prostate tumor. Front Oncol (2018) 8:534. doi: 10.3389/fonc.2018.00534
142. Vieira AC, Alvarenga MJ, Santos JC, Silva AM. Paraneoplastic jaundice and prostate cancer. BMJ Case Rep (2017) 2017:bcr2016218001. doi: 10.1136/bcr-2016-218001
143. Wang W, Li L, Chen N, Niu C, Li Z, Hu J, et al. Nerves in the tumor microenvironment: Origin and effects. Front Cell Dev Biol (2020) 8:601738. doi: 10.3389/fcell.2020.601738
144. Faulkner S, Jobling P, March B, Jiang CC, Hondermarck H. Tumor neurobiology and the war of nerves in cancer. Cancer Discov (2019) 9(6):702–10. doi: 10.1158/2159-8290.CD-18-1398
145. Liebig C, Ayala G, Wilks JA, Berger DH, Albo D. Perineural invasion in cancer: a review of the literature. Cancer (2009) 115(15):3379–91. doi: 10.1002/cncr.24396
146. Bautista M, Krishnan A. The autonomic regulation of tumor growth and the missing links. Front Oncol (2020) 10:744. doi: 10.3389/fonc.2020.00744
147. Reavis HD, Chen HI, Drapkin R. Tumor innervation: Cancer has some nerve. Trends Cancer (2020) 6(12):1059–67. doi: 10.1016/j.trecan.2020.07.005
148. March B, Faulkner S, Jobling P, Steigler A, Blatt A, Denham J, et al. Tumour innervation and neurosignalling in prostate cancer. Nat Rev Urol (2020) 17(2):119–30. doi: 10.1038/s41585-019-0274-3
149. Gysler SM, Drapkin R. Tumor innervation: peripheral nerves take control of the tumor microenvironment. J Clin Invest (2021) 131(11):e147276. doi: 10.1172/JCI147276
150. Ayala GE, Wheeler TM, Shine HD, Schmelz M, Frolov A, Chakraborty S, et al. In vitro dorsal root ganglia and human prostate cell line interaction: redefining perineural invasion in prostate cancer. Prostate (2001) 49(3):213–23. doi: 10.1002/pros.1137
151. Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science (2013) 341(6142):1236361. doi: 10.1126/science.1236361
152. Rutledge A, Jobling P, Walker MM, Denham JW, Hondermarck H. Spinal cord injuries and nerve dependence in prostate cancer. Trends Cancer (2017) 3(12):812–5. doi: 10.1016/j.trecan.2017.10.001
153. Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature (1989) 339(6219):58–61. doi: 10.1038/339058a0
154. Zahalka AH, Arnal-Estape A, Maryanovich M, Nakahara F, Cruz CD, Finley LWS, et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science (2017) 358(6361):321–6. doi: 10.1126/science.aah5072
155. Pundavela J, Demont Y, Jobling P, Lincz LF, Roselli S, Thorne RF, et al. ProNGF correlates with Gleason score and is a potential driver of nerve infiltration in prostate cancer. Am J Pathol (2014) 184(12):3156–62. doi: 10.1016/j.ajpath.2014.08.009
156. Dobrenis K, Gauthier LR, Barroca V, Magnon C. Granulocyte colony-stimulating factor off-target effect on nerve outgrowth promotes prostate cancer development. Int J Cancer (2015) 136(4):982–8. doi: 10.1002/ijc.29046
157. Di Donato M, Cernera G, Migliaccio A, Castoria G. Nerve growth factor induces proliferation and aggressiveness in prostate cancer cells. Cancers (Basel) (2019) 11(6):784. doi: 10.3390/cancers11060784
158. Chen WY, Wen YC, Lin SR, Yeh HL, Jiang KC, Chen WH, et al. Nerve growth factor interacts with CHRM4 and promotes neuroendocrine differentiation of prostate cancer and castration resistance. Commun Biol (2021) 4(1):22. doi: 10.1038/s42003-020-01549-1
159. Dwivedi S, Bautista M, Shrestha S, Elhasasna H, Chaphekar T, Vizeacoumar FS, et al. Sympathetic signaling facilitates progression of neuroendocrine prostate cancer. Cell Death Discovery (2021) 7(1):364. doi: 10.1038/s41420-021-00752-1
Keywords: prostate cancer, secretion, paracrine, endocrine, neuroendocrine
Citation: Arman T and Nelson PS (2022) Endocrine and paracrine characteristics of neuroendocrine prostate cancer. Front. Endocrinol. 13:1012005. doi: 10.3389/fendo.2022.1012005
Received: 04 August 2022; Accepted: 24 October 2022;
Published: 11 November 2022.
Edited by:
Amina Zoubeidi, University of British Columbia, CanadaReviewed by:
Paramita M Ghosh, University of California, Davis, United StatesAyesha Shafi, Center for Prostate Disease Research (CPDR), United States
Copyright © 2022 Arman and Nelson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter S. Nelson, cG5lbHNvbkBmcmVkaHV0Y2gub3Jn