 Sharon Baumel-Alterzon
Sharon Baumel-Alterzon Donald K. Scott
Donald K. Scott
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 15 September 2022
Sec. Diabetes: Molecular Mechanisms
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1011187
This article is part of the Research TopicProgression to Diabetes: Molecular and Cellular MechanismsView all 10 articles
The beta-cell identity gene, pancreatic duodenal homeobox 1 (Pdx1), plays critical roles in many aspects of the life of beta-cells including differentiation, maturation, function, survival and proliferation. High levels of reactive oxygen species (ROS) are extremely toxic to cells and especially to beta-cells due to their relatively low expression of antioxidant enzymes. One of the major mechanisms for beta-cell dysfunction in type-2 diabetes results from oxidative stress-dependent inhibition of PDX1 levels and function. ROS inhibits Pdx1 by reducing Pdx1 mRNA and protein levels, inhibiting PDX1 nuclear localization, and suppressing PDX1 coactivator complexes. The nuclear factor erythroid 2-related factor (Nrf2) antioxidant pathway controls the redox balance and allows the maintenance of high Pdx1 levels. Therefore, pharmacological activation of the Nrf2 pathway may alleviate diabetes by preserving Pdx1 levels.
Since its first introduction into the scientific community in 1945, reactive oxygen species (ROS) have been the focus of numerous studies (1). ROS are defined as oxygen-derived atoms or molecules that possess one or more unpaired electrons, making them highly reactive (2). While the cytoplasm, cell membrane, endoplasmic reticulum (ER) and peroxisome are capable of producing ROS, up to 90% of cellular ROS (composed mostly of H2O2 and) are generated in the mitochondria due to incomplete reduction of oxygen to water in the electron transport chain (ETC) (3). High levels of ROS are extremely toxic to the cell, leading to DNA breaks, lipid peroxidation, protein aggregation, protein denaturation and protein fragmentation (4). Therefore, most cells are equipped with a battery of antioxidant genes that have the power to neutralize this threat, maintaining a fine equilibrium between ROS production and an antioxidant defense. In cases where ROS levels exceed the cell’s ability to detoxify it and the equilibrium is breached, the cell enters into a pathophysiological condition called “oxidative stress” (5). Oxidative stress takes part in the etiology of many diseases, contributing to the initiation and progression of cancer, vascular-related diseases, respiratory diseases, neurodegenerative disorders, digestive diseases, kidney diseases, chronic inflammatory disorders, aging and diabetes (5–7). Diabetes is linked to various cellular stresses that generate ROS, such as hyperglycemia, hyperlipidemia, hypoxia, inflammation, and ER stress (8, 9) (Figure 1). Indeed, pancreatic islets of type-2 diabetic patients often present increased levels of oxidative stress markers (such as oxidative DNA damage marker, 8-hydroxy-2′-deoxyguanosine) and this, at least in part, is due to the fact that beta-cells express low levels of several antioxidant genes, making them highly vulnerable to increased ROS levels (7, 10). In line with that, high ROS levels reduce functional beta-cell mass by increasing beta-cell apoptosis, reducing beta-cell proliferation, and damaging beta-cell function (7). Therefore, and perhaps as a compensatory mechanism, beta-cells protect themselves against oxidative stress by activating the nuclear factor erythroid 2-related factor (Nrf2) antioxidant signaling pathway. This activation is performed by ROS which oxidize critical cysteine in the NRF2 inhibitor, KEAP1, thus inhibiting NRF2 degradation (7, 11). Remarkably, apart from providing protection against oxidative stress, NRF2 regulates beta-cell mitochondrial biogenesis and activity, provides anti-inflammatory effects, promotes beta-cell function and stimulates beta-cell proliferation (7, 11).
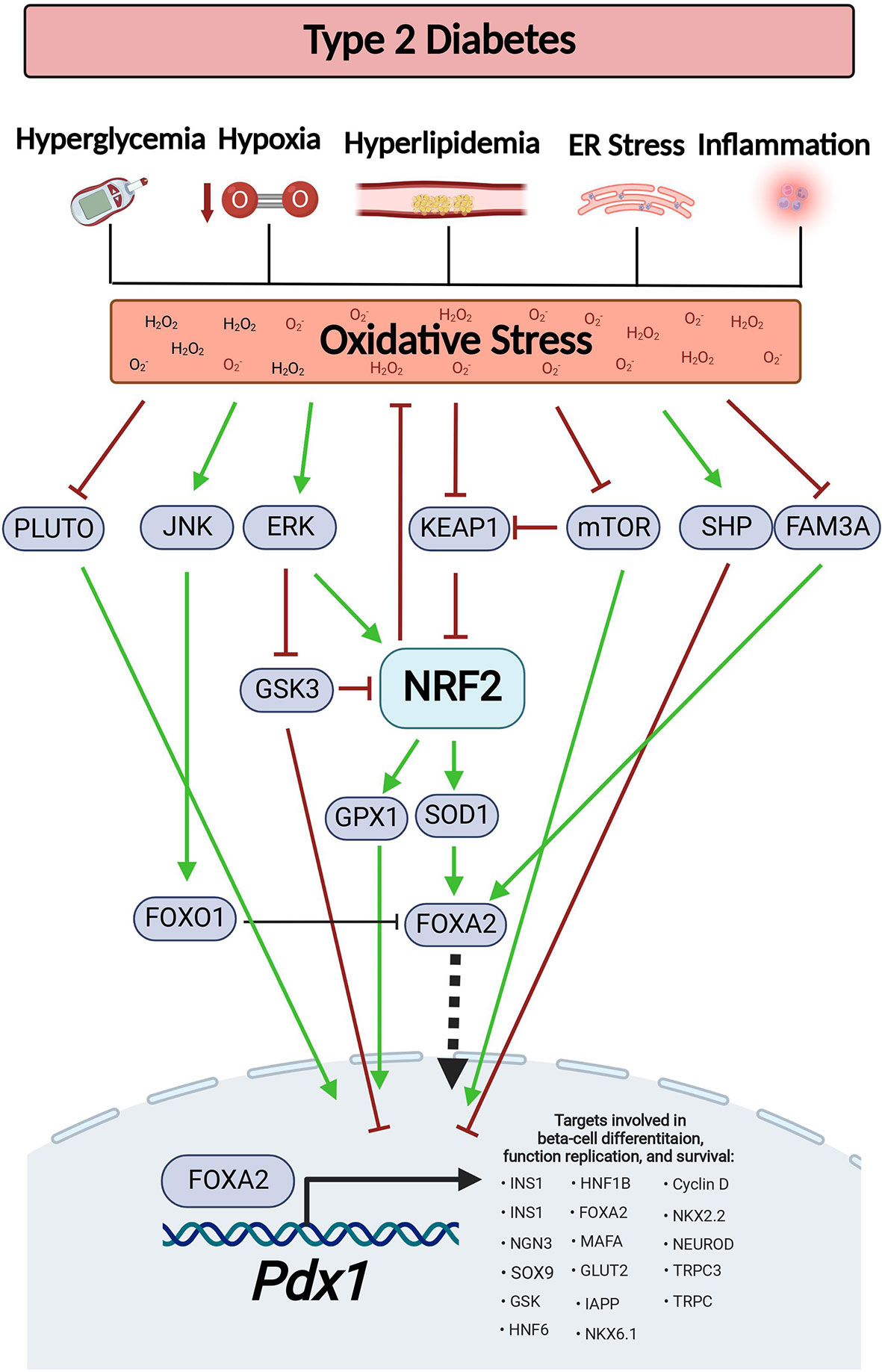
Figure 1 Oxidative stress-dependent pathways affect Pdx1 levels during diabetes. Type-2 diabetes is associated with various pathological conditions that generate ROS, such as hyperglycemia, hyperlipidemia, hypoxia, inflammation, and ER stress. This results in oxidative stress. Oxidative stress reduces the expression level of PLUTO lncRNA, stimulates JNK-dependent FOXO1 activation, inhibits mTOCR signaling, increases SHP expression and reduces FAM3A levels, all of which results in decreased PDX1 levels. On the other hand, in order to maintain ROS at appropriate levels, oxidative stress also serves as a signal for Nrf2 activation by inhibition of Keap1 and by activation of the ERK signaling. This results in increased expression of the Nrf2 target genes, Sod1 and Gpx, which restore PDX1 levels. This figure was generated using BioRender.com.
Beta-cells express a unique set of key marker genes that define their identity and function by contributing to the expression and secretion of insulin (12). One of these genes is the duodenal homeobox 1 (Pdx1), a protein that plays many distinct roles in the life of a beta-cell, including involvement in beta-cell differentiation, beta-cell function, beta-cell survival and beta-cell proliferation (13–18). To do so, PDX1 controls the expression of other genes that play important roles in the beta-cell fate. These genes include insulin (Ins1, Ins2), neurogenin 3 (Ngn3), SRY-box transcription factor 9 (Sox9), hepatocyte nuclear factor 6 and 1b (Hnf6, Hnf1b), forkhead box protein a2 (Foxa2), V-Maf musculoaponeurotic fibrosarcoma oncogene homolog A (MafA), NK2 homeobox 2 (Nkx2.2), neurogenic differentiation (NeuroD), solute carrier family 2 member 2 (Glut2, Slc2a2), glucokinase (Gck), islet amyloid polypeptide (Iapp), NK6 homeobox 1 (Nkx6.1), cyclin D1 (Ccnd1), cyclin D2 (Ccnd2), and the transient receptor potential cation channel family 3 and 6 (Trpc3,6) (13, 16, 17, 19). In agreement with these findings, homozygous Pdx1 knockout (Pdx1-/-) mice exhibit pancreatic agenesis while heterozygous Pdx1 knockout (Pdx1+/-) mice present with beta-cell ER stress, beta-cell apoptosis, impaired insulin secretion, decreased beta-cell proliferation and reduced beta-cell mass (20–25). Additionally, tamoxifen-inducible beta-cell specific Pdx1 deletion in adult mice results in hyperglycemia and transdifferentiation of beta-cells into alpha-like cells (26). In humans, 5% of diagnosed type-2 diabetes patients present with genetic defects in PDX1 and mutations in PDX1 define a subset of maturity-onset diabetes of the young 4 (MODY 4) (27, 28).
Defective expression of Pdx1 has been documented in diabetes and this is attributed to oxidative stress (9, 29–31). Beta-cell specific deletion of Nrf2 in mice under diabetogenic situation results in increased oxidative stress and reduced Pdx1 expression. Conversely, activation of NRF2 using genetic or pharmacological approaches increases Pdx1 expression (11). These findings suggest that the NRF2 pathway is essential for preserving PDX1 levels, thus contributing to the maintenance of functional beta-cell mass. In this review, we aim to bring together all the current knowledge about the effect of physiological changes in redox balance on beta-cell fate via regulation on PDX1 levels.
During embryogenesis, Pdx1 is expressed in two waves. The first wave begins at early stages of pancreatic development (E8.5 in mouse, gestational week 4 in human), with the appearance of dorsal and ventral foregut endoderm buds that later transform into a fully developed pancreas. At that stage, PDX1 is detected in the pancreas epithelium (13, 32–34). Mutations and deletions of Pdx1 during the first wave results in pancreas agenesis in both mice and humans. Accordingly, Pdx1 deletion does not affect the development of the endoderm buds. Rather, deletion of Pdx1 inhibits the morphogenesis of the buds creating defects in the development of pancreatic epithelium (13, 20, 35, 36). Interestingly, the appearance of early premature insulin and glucagon positive cells in cells that lack PDX1 suggests that unlike in the second wave of Pdx1 expression, the first wave of Pdx1 expression is not involved in the development of endocrine cells (34, 36).
At the second wave of Pdx1 expression, which occurs at late gestation (beginning at E13.5 in mouse, gestational week 12 in human), PDX1 stimulates the expression of another developmental factor, Ngn3. NGN3 levels, which significantly increase in mouse at E13.5 and reach maximal levels at E15.5, are responsible for the specification of endocrine progenitors into endocrine cells that will later form the pancreatic islets (13, 37, 38). Inducible Pdx1 deletion at these ages blocks the formation of acinar cells and islets in mouse embryos (35). Additionally, beta-cell specific Pdx1 deletion increases glucagon and somatostatin positive cells at the expense of insulin positive cells resulting in diabetes (34). Following the second wave of Pdx1 expression, PDX1 levels remain constant towards and during adulthood (in healthy non-diabetic settings). In adults, PDX1 is restricted to beta-cells where it works to maintain beta-cell maturity and function (13, 33).
Some levels of ROS are necessary for normal cellular function. For example, moderate levels of ROS (up to ~100 nM H2O2) are needed for stem cells to maintain their ability to go through cell differentiation and to keep their “stemness”. The underlying mechanism involves the oxidation of tyrosine and cysteine residues which activates protein kinases, protein phosphatases, and signaling factors that take part in cell differentiation (39). This might explain the formation of moderated ROS levels (starting at E12.5 and peaking at E15.5) by NADPH oxidase 4 (Nox4) activity during the development of embryonic mouse pancreas (40). Nevertheless, accumulation of higher ROS levels (above ~100 μM H2O2) may lead to cell senescence or death (39). Furthermore, prompt increase of oxidative stress may pose a risk on normal pancreatic development since PDX1 levels are negatively regulated by ROS (for further details on the mechanisms see Figure 1 and Table 1 below) (41–45). This highlights the need to maintain ROS at appropriate levels to support pancreatic development and differentiation without damaging the cells. Surprisingly two NRF2 antioxidant target genes, catalase (Cat) and superoxide dismutase 1 (Sod1), are upregulated during pancreatic development (40). This suggests that NRF2 helps maintaining the required redox balance in the embryonic pancreas, a theory that has not been tested yet. In support of that, SOD1 stimulates Pdx1 expression by increasing the expression of Forkhead Box Protein A2 (Foxa2), one of the main Pdx1 transcriptional regulators (46). SOD1 can also support Pdx1 transcription by increasing H3 acetylation and H3K4 methylation on the Pdx1 promoter, thus providing an open active chromatin (46). Intriguingly, during pancreatic development, ROS activates the ERK signaling pathway which can further activate the NRF2 pathway and increase Pdx1 expression, resulting in differentiation of endocrine progenitors into beta-cells (47–49). Overall, these findings suggest that by maintaining normal redox balance, the NRF2 pathway supports appropriate PDX1 levels that promote normal pancreatic development.
After birth, both mouse and human neonates exhibit a sharp burst of beta-cell proliferation. This peak of proliferation, which is temporary, substantially declines with age (50). Weaning is believed to be the main trigger for the loss of beta-cell proliferative capabilities, and it coincides with the gradual maturation of beta-cells (51, 52). Mechanistically, the transition from fat-rich maternal milk to carbohydrate-rich diet inhibits the proliferative factor, mammalian target of rapamycin complex1 (mTORC1) and activates 5’-adenosine monophosphate–activated protein kinase (AMPK), which promotes beta-cell maturity. On the other hand, continuous supplementation of milk-fat rich diet to mice during weaning into adulthood maintains high mTORC1 levels and beta-cell proliferation (53). Consistent with these findings, beta-cell specific deletion of mtorc1 in mice leads to impaired beta-cell proliferation, reduced beta-cell survival and decreased beta-cell mass (54), while activation of mTORC1 stimulates mouse beta-cell proliferation by increasing cyclin D2 expression (55). Interestingly, branched chain amino acids can activate the mTORC1-Rab1A axis to maintain PDX1 protein stability and increase its nuclear localization (56, 57). Moreover, beta-cell-specific overexpression of kinase-dead mTOR mutants results in decreased Pdx1 mRNA and protein levels (58). These findings link mTOR-stimulated beta-cell proliferation during postnatal ages with PDX1 activity. Oxidative stress and conditions that generate ROS such as ER stress and hypoxia, can inhibit mTOR signaling (59). In postnatal Akita mice, ER stress inhibits mTORC1 leading to reduced beta-cell proliferation, as well to decreased PDX1 protein levels and downregulation of PDX1 target genes. However, restoration of mTORC1 activity in these mice did not affect PDX1 protein levels, suggesting that ER stress affects PDX1 levels by mTORC1-independent mechanisms (60). Although the exact mechanism by which mTORC1 regulates Pdx1 is unknown, mTORC1 can control NRF2 levels by p62-dependent degradation of its inhibitor, KEAP1 (7, 61), suggesting that NRF2 might be involved.
PDX1 binding sites are found in several genes that are associated with cell replication, such as Nasp, Bard1, Mnx1, and Mcm7 (17). This suggest that PDX1 on its own can stimulate beta-cell proliferation. Accordingly, overexpression of Pdx1 in primary rat islets increases beta-cell proliferation by upregulation of cyclin D1 and D2 expression (16), which are essential for beta-cell proliferation during postnatal ages (62, 63), over-nutrition (64, 65), and pregnancy (66). Interestingly, oxidative stress reduces cyclins D1 and D2 expression while activation of the NRF2 antioxidant pathway increases cyclin D1 (9, 11). On the other hand, mice expressing mutated PDX1 develop diabetes at weaning concomitantly with reduced beta-cell proliferation and beta-cell area (17). Thus, regulation of redox-balance is important for PDX1-stimulated beta-cell proliferation.
During adulthood, PDX1 switches roles and takes part in beta-cell identity and maturation. For example, PDX1 controls insulin gene transcription by forming a transcriptional activation complex with neuronal differentiation 1 (Neurod1) and by upregulation of MafA and Ngn3 expression, which are needed for insulin transcription (13). MAFA can further enhance glucose-stimulated insulin secretion (GSIS) to maintain glucose homeostasis (67). PDX1 can also upregulate the expression of other factors that are necessary for GSIS, such as Glut2 and glucokinase (28). Indeed, two missense mutations in the Pdx1 transactivation domain inhibit GSIS (68). Lastly, as previously mentioned, PDX1 positively regulates the expression of various beta-cell identity genes (13, 16, 17, 19). This might explain why beta-cell specific Pdx1 deletion results in an altered transcriptional profile that resembles alpha-like cells. This includes downregulation of Ins1 and Glut2 genes while upregulation of Gcg (glucagon) and MafB (26). These findings suggest that physiological situations that increase oxidative stress may inhibit PDX1 from maintaining beta-cell identity and function, and eventually lead to diabetes (as described in part 4).
One of the major mechanisms for beta-cell dysfunction in type-2 diabetes involves the inhibition of Pdx1 by oxidative stress (41–44). For example, chronic exposure of syrian hamster islet cell line HIT-T15 to high glucose concentrations reduces Pdx1 mRNA and protein levels (44, 69). Additionaly, type-2 diabetic rodent models, such as db/db mice as well as mice and rats fed on high fat diet (HFD), display reduced Pdx1 mRNA and/or protein expression as well as decreased PDX1 nuclear localization (53, 70–72). Similarly, beta-cell specific deletion of Nrf2 in mice fed on HFD results in increased oxidative stress, reduced Pdx1 mRNA levels and inhibition of PDX1 translocation into the nucleus (11). Conversely, treatment of obese diabetic C57BL/KsJ-db/db mice or Zucker diabetic rats with antioxidant agents restores PDX1 nuclear localization and PDX1 transcriptional activity (73, 74). Likewise, overexpression of glutathione peroxidase 1 (Gpx1), a Nrf2 antioxidant target gene, increases Pdx1 mRNA and protein levels in mouse beta-cells (75).
The mechanisms behind ROS-dependent reduction of PDX1 levels vary (Figure 1 and Table 1). For example, oxidative stress can lead to activation of forkhead box protein O1 (FOXO1) by c-Jun N-terminal kinase (JNK) or by acetylation of FOXO1. As a result, FOXO1 goes through phosphorylation, nuclear translocation and subsequent activation (76, 88). FOXO1 then mediates inhibition of FOXA2-transcriptional activation of Pdx1 and inhibits PDX1 nuclear translocation (76, 89–91). Moreover, islets from mice fed on HFD exhibit nuclear exclusion and reduced activity of FOXA2 (92) and db/db mouse islets show reduced levels of family with sequence similarity 3 member A (FAM3A), a factor that upregulates PDX1 levels via activation of CaM-FOXA2 pathway (85). Oxidative stress can reduce PDX1 levels by additional mechanisms. For example, during conditions that generate ROS, such as high glucose concentrations, ER stress and treatment with streptozotocin (STZ) (7), orphan nuclear receptor small heterodimer partner (SHP) downregulates Pdx1 at the mRNA levels (78, 82, 84). In addition, mutations in Klf11 transcription factor which are associated with maturity-onset diabetes of the young 7 (MODY7), lead to promoter repression of the antioxidant gene catalase 1 (Cat1), and to a reduction in Pdx1 transcription in beta-cells (93, 94). Saturated fatty-acids, such as palmitic-acid, can stimulate the production of H2O2 in beta-cells, leading to beta-cell death (95). Interestingly, recent findings show that incubation of beta-cells with palmitic acid leads to sequestration of PDX1 into stress-granules. This sequestration prevents PDX1 from translocating to the nucleus and transcribing its target genes. Moreover, inhibition of the stress-granules formation in HFD fed mice results in increased PDX1 nuclear localization and improved glucose tolerance as well as GSIS (81).
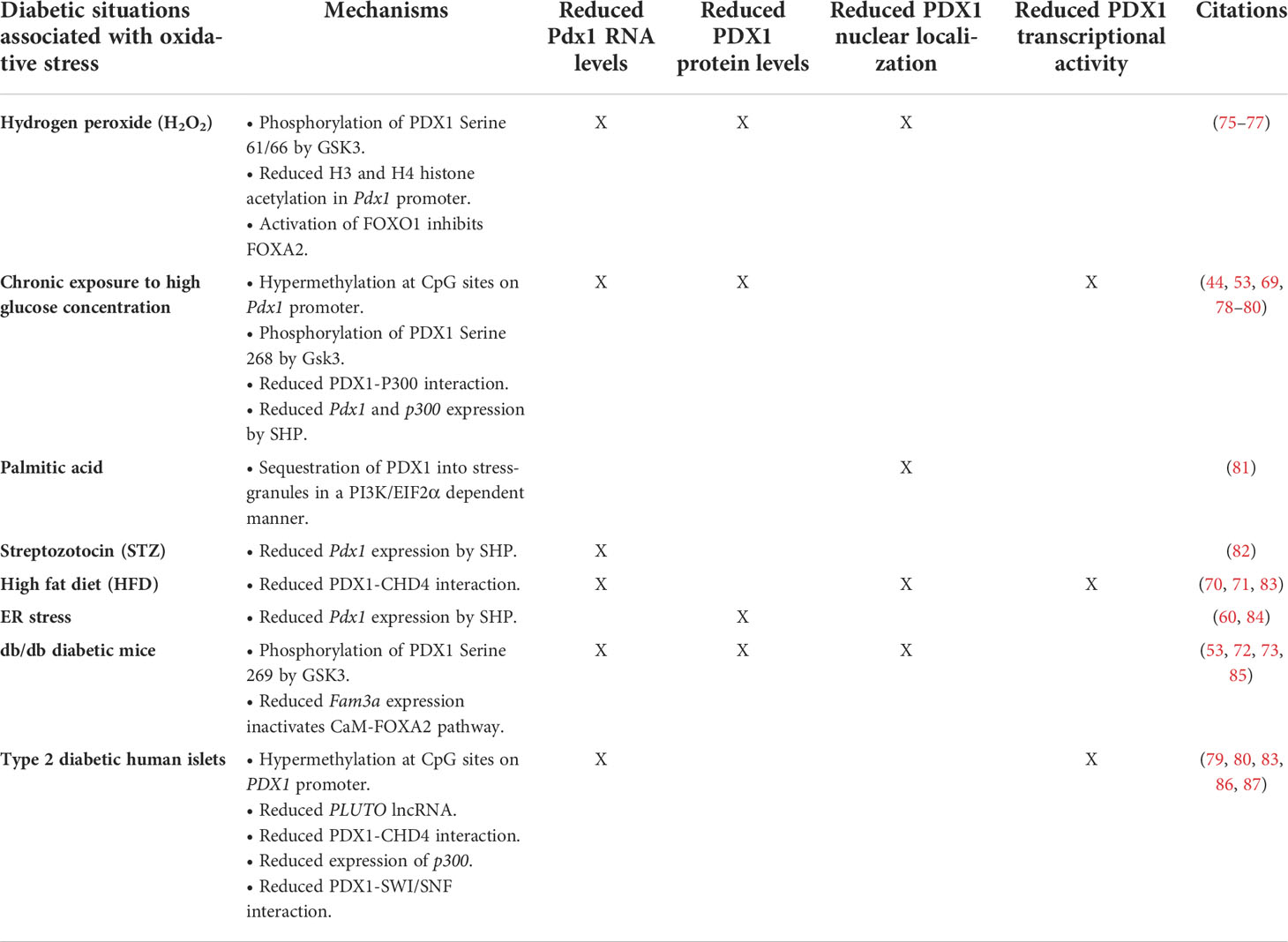
Table 1 Ros-dependent mechanisms that reduce Pdx1 levels and activity during diabetes.
At the post-transcriptional level, oxidative stress increases activity of glycogen synthase kinase 3 (GSK3), a known NRF2 inhibitor, which then phosphorylates PDX1 serine 61 and/or serine 66 resulting in PDX1 protein degradation (7, 77). Additionally, INS1e rat beta-cell-like insulinoma cells and human islets chronically exposed to high glucose display GSK3-mediated phosphorylation of PDX1 serine 268, resulting in PDX1 degradation and reduced expression of PDX1 target genes (53). Oxidative stress also reduces H3 and H4 histone acetylation in the Pdx1 promoter, leading to transcriptional silencing due to tightly packed chromatin (75). Islets from type-2 diabetic donors show higher methylation status at ten CpG sites on the PDX1 promoter. This is associated with reduced PDX1 mRNA levels. The same phenomenon is observed in rat insulinoma beta-like cells (INS 832/13) incubated chronically at high glucose concentrations (19, 79). Additionally, treatment of human stem cells with the DNA methylation inhibitor, 5-aza-2′-deoxycytidine, increases PDX1 nuclear levels (19, 96). Human and mouse pancreatic islets express hundreds of long non-coding RNAs (lncRNAs), some of which are playing important roles in beta-cell differentiation and function. One of the most characterized ones is a beta-cell specific lncRNA called PLUTO, which positively regulates PDX1 transcription. PLUTO, exhibits a marked reduction of expression in islets from type-2 diabetic donors, positioning it as another mechanism for reducing PDX1 levels under ROS-associated pathological conditions (86).
Apart from affecting PDX1 abundance, conditions associated with increased ROS can affect PDX1 transcriptional activity by targeting coactivator complexes associated with PDX1 (97). For example, decreased interaction between PDX1 and the chromodomain helicase DNA-binding 4 (CHD4) ATPase subunit of the NuRD complex is observed in both islets of type-2 donors and in mice fed on HFD (83). Similarly, in human type-2 diabetic beta-cells, there is a significant reduction in PDX1 binding to the ATP-dependent SWI/SNF chromatin remodeling complex, a complex that is needed for pancreas development and beta-cell identity (87, 98). PDX1 also interacts with histone acetyltransferases p300 and CBP (p300/CBP) to stimulate expression of PDX1 target genes, including insulin (99). INS-1E cells incubated in high glucose concentrations and islets from type-2 donors display reduced levels of p300 due to protein degradation (80), a situation that may hamper PDX1 transcriptional activity.
Based on their ability to reduce hyperglycemia and body weight, the U.S. food and drug administration (FDA) approved the use of Glucagon-like peptide-1 receptor (GLP-1R) agonists as a treatment for type-2 patients (100). Interestingly, treatments of islets from a rat model of intrauterine growth retardation (IUGR) or from a “catch up growth” Wistar rat model with the GLP-1R agonists, Exendin 4 and Liraglutide, increase Pdx1 transcription by increasing H3 histone acetylation, increasing H3K4me3 levels and by reducing H3K9me2 levels in Pdx1 promoter (101, 102). Additionally, GLP-1 itself stimulates PDX1 nuclear translocation via cAMP-dependent PKA pathway and activates NRF2 through the PKA-dependent ERK activation pathway, suggesting that NRF2 might be involved in this regulation (7, 103).
To conclude, published data indicate that alteration in redox-balance leads to dysregulated PDX1 levels and activity, which can result in diabetes. Expression of several NRF2 target genes, as well as treatment with CDDO-Me, an NRF2 pharmacological activator, maintain PDX1 abundance by reducing oxidative stress (11, 40, 75, 104). These findings suggest that activation of the NRF2 pathway may alleviate diabetes by preserving PDX1 levels. Additional studies are needed to further explore the role of NRF2 in maintaining PDX1 levels during embryonic, postnatal and adult life. Interestingly, CDDO derivatives were also shown to contribute to the maintenance of functional beta-cell mass by promoting beta-cell proliferation, reducing beta-cell oxidative damages, increasing islet cell viability, improving insulin content, stimulating insulin secretion and reducing secretion of proinflammatory cytokines (7, 11). Furthermore, treatment of db/db and streptozotocin-induced diabetic mouse models with CDDO derivatives improves diabetes outcome (105–107) and these compounds have been tested under several clinical trials to improve chronic kidney disease in diabetic patients (7).This places NRF2 as a potential therapeutic target for type-2 diabetes.
SB-A designed content, wrote text, and produced the figure. DKS reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Institutes ofHeath/The National INstitute of Diabetes Digestive and Kidney Diseases R01DK114338 andR01DK130300 (to DKS) and K01128387 (to SB-A).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Alfadda AA, Sallam RM. Reactive oxygen species in health and disease. J BioMed Biotechnol (2012) 2012:936486. doi: 10.1155/2012/936486
2. Zhang C, Wang X, Du J, Gu Z, Zhao Y. Reactive oxygen species-regulating strategies based on nanomaterials for disease treatment. Adv Sci (Weinh) (2021) 8(3):2002797. doi: 10.1002/advs.202002797
3. Tirichen H, Yaigoub H, Xu W, Wu C, Li R, Li Y. Mitochondrial reactive oxygen species and their contribution in chronic kidney disease progression through oxidative stress. Front Physiol (2021) 12:627837. doi: 10.3389/fphys.2021.627837
4. Len JS, Koh WSD, Tan SX. The roles of reactive oxygen species and antioxidants in cryopreservation. Biosci Rep (2019) 39(8):BSR20191601. doi: 10.1042/BSR20191601
5. Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V, et al. Oxidative stress: Harms and benefits for human health. Oxid Med Cell Longev (2017) 2017:8416763. doi: 10.1155/2017/8416763
6. Liu Z, Ren Z, Zhang J, Chuang CC, Kandaswamy E, Zhou T, et al. Role of ros and nutritional antioxidants in human diseases. Front Physiol (2018) 9:477. doi: 10.3389/fphys.2018.00477
7. Baumel-Alterzon S, Katz LS, Brill G, Garcia-Ocana A, Scott DK. Nrf2: The master and captain of beta cell fate. Trends Endocrinol Metab (2020) 32(1):7–19. doi: 10.1016/j.tem.2020.11.002
8. Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, et al. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci (2020) 21(17):6275. doi: 10.3390/ijms21176275
9. Wang J, Wang H. Oxidative stress in pancreatic beta cell regeneration. Oxid Med Cell Longev (2017) 2017:1930261. doi: 10.1155/2017/1930261
10. Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes (2005) 54(3):727–35. doi: 10.2337/diabetes.54.3.727
11. Baumel-Alterzon S, Katz LS, Brill G, Jean-Pierre C, Li Y, Tse I, et al. Nrf2 regulates beta-cell mass by suppressing beta-cell death and promoting beta-cell proliferation. Diabetes (2022) 71(5):989–1011. doi: 10.2337/db21-0581
12. Mostafa D, Yanagiya A, Georgiadou E, Wu Y, Stylianides T, Rutter GA, et al. Loss of beta-cell identity and diabetic phenotype in mice caused by disruption of Cnot3-dependent mrna deadenylation. Commun Biol (2020) 3(1):476. doi: 10.1038/s42003-020-01201-y
13. Zhu Y, Liu Q, Zhou Z, Ikeda Y. Pdx1, neurogenin-3, and mafa: Critical transcription regulators for beta cell development and regeneration. Stem Cell Res Ther (2017) 8(1):240. doi: 10.1186/s13287-017-0694-z
14. Yao X, Li K, Liang C, Zhou Z, Wang J, Wang S, et al. Tectorigenin enhances Pdx1 expression and protects pancreatic beta-cells by activating erk and reducing er stress. J Biol Chem (2020) 295(37):12975–92. doi: 10.1074/jbc.RA120.012849
15. Fujimoto K, Polonsky KS. Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes Obes Metab (2009) 11 Suppl 4:30–7. doi: 10.1111/j.1463-1326.2009.01121.x
16. Hayes HL, Moss LG, Schisler JC, Haldeman JM, Zhang Z, Rosenberg PB, et al. Pdx-1 activates islet alpha- and beta-cell proliferation Via a mechanism regulated by transient receptor potential cation channels 3 and 6 and extracellular signal-regulated kinases 1 and 2. Mol Cell Biol (2013) 33(20):4017–29. doi: 10.1128/MCB.00469-13
17. Spaeth JM, Gupte M, Perelis M, Yang YP, Cyphert H, Guo S, et al. Defining a novel role for the Pdx1 transcription factor in islet beta-cell maturation and proliferation during weaning. Diabetes (2017) 66(11):2830–9. doi: 10.2337/db16-1516
18. Zhu X, Oguh A, Gingerich MA, Soleimanpour SA, Stoffers DA, Gannon M. Cell cycle regulation of the Pdx1 transcription factor in developing pancreas and insulin-producing beta-cells. Diabetes (2021) 70(4):903–16. doi: 10.2337/db20-0599
19. Liu J, Lang G, Shi J. Epigenetic regulation of pdx-1 in type 2 diabetes mellitus. Diabetes Metab Syndr Obes (2021) 14:431–42. doi: 10.2147/DMSO.S291932
20. Vinogradova TV, Sverdlov ED. Pdx1: A unique pancreatic master regulator constantly changes its functions during embryonic development and progression of pancreatic cancer. Biochem (Mosc) (2017) 82(8):887–93. doi: 10.1134/S000629791708003X
21. Sachdeva MM, Claiborn KC, Khoo C, Yang J, Groff DN, Mirmira RG, et al. Pdx1 (Mody4) regulates pancreatic beta cell susceptibility to er stress. Proc Natl Acad Sci U.S.A. (2009) 106(45):19090–5. doi: 10.1073/pnas.0904849106
22. Brissova M, Blaha M, Spear C, Nicholson W, Radhika A, Shiota M, et al. Reduced pdx-1 expression impairs islet response to insulin resistance and worsens glucose homeostasis. Am J Physiol Endocrinol Metab (2005) 288(4):E707–14. doi: 10.1152/ajpendo.00252.2004
23. Brissova M, Shiota M, Nicholson WE, Gannon M, Knobel SM, Piston DW, et al. Reduction in pancreatic transcription factor pdx-1 impairs glucose-stimulated insulin secretion. J Biol Chem (2002) 277(13):11225–32. doi: 10.1074/jbc.M111272200
24. Sun J, Mao L, Yang H, Ren D. Critical role for the Tsc1-Mtorc1 pathway in beta-cell mass in Pdx1-deficient mice. J Endocrinol (2018) 238(2):151–63. doi: 10.1530/JOE-18-0015
25. Jara MA, Werneck-De-Castro JP, Lubaczeuski C, Johnson JD, Bernal-Mizrachi E. Pancreatic and duodenal homeobox-1 (Pdx1) contributes to beta-cell mass expansion and proliferation induced by Akt/Pkb pathway. Islets (2020) 12(2):32–40. doi: 10.1080/19382014.2020.1762471
26. Gao T, McKenna B, Li C, Reichert M, Nguyen J, Singh T, et al. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab (2014) 19(2):259–71. doi: 10.1016/j.cmet.2013.12.002
27. Urakami T. Maturity-onset diabetes of the young (Mody): Current perspectives on diagnosis and treatment. Diabetes Metab Syndr Obes (2019) 12:1047–56. doi: 10.2147/DMSO.S179793
28. Kushner JA, Ye J, Schubert M, Burks DJ, Dow MA, Flint CL, et al. Pdx1 restores beta cell function in Irs2 knockout mice. J Clin Invest (2002) 109(9):1193–201. doi: 10.1172/JCI14439
29. Moin ASM, Butler AE. Alterations in beta cell identity in type 1 and type 2 diabetes. Curr Diabetes Rep (2019) 19(9):83. doi: 10.1007/s11892-019-1194-6
30. Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H. Glucose toxicity in beta-cells: Type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes (2003) 52(3):581–7. doi: 10.2337/diabetes.52.3.581
31. Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, et al. Inactivation of specific beta cell transcription factors in type 2 diabetes. J Clin Invest (2013) 123(8):3305–16. doi: 10.1172/JCI65390
32. Astro V, Adamo A. Epigenetic control of endocrine pancreas differentiation in vitro: Current knowledge and future perspectives. Front Cell Dev Biol (2018) 6:141. doi: 10.3389/fcell.2018.00141
33. Jorgensen MC, Ahnfelt-Ronne J, Hald J, Madsen OD, Serup P, Hecksher-Sorensen J. An illustrated review of early pancreas development in the mouse. Endocr Rev (2007) 28(6):685–705. doi: 10.1210/er.2007-0016
34. Gannon M, Ables ET, Crawford L, Lowe D, Offield MF, Magnuson MA, et al. Pdx-1 function is specifically required in embryonic beta cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Dev Biol (2008) 314(2):406–17. doi: 10.1016/j.ydbio.2007.10.038
35. Holland AM, Hale MA, Kagami H, Hammer RE, MacDonald RJ. Experimental control of pancreatic development and maintenance. Proc Natl Acad Sci U.S.A. (2002) 99(19):12236–41. doi: 10.1073/pnas.192255099
36. Ahlgren U, Jonsson J, Edlund H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in Ipf1/Pdx1-deficient mice. Development (1996) 122(5):1409–16. doi: 10.1242/dev.122.5.1409
37. Oliver-Krasinski JM, Kasner MT, Yang J, Crutchlow MF, Rustgi AK, Kaestner KH, et al. The diabetes gene Pdx1 regulates the transcriptional network of pancreatic endocrine progenitor cells in mice. J Clin Invest (2009) 119(7):1888–98. doi: 10.1172/JCI37028
38. White P, May CL, Lamounier RN, Brestelli JE, Kaestner KH. Defining pancreatic endocrine precursors and their descendants. Diabetes (2008) 57(3):654–68. doi: 10.2337/db07-1362
39. Sies H, Jones DP. Reactive oxygen species (Ros) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol (2020) 21(7):363–83. doi: 10.1038/s41580-020-0230-3
40. Liang J, Wu SY, Zhang D, Wang L, Leung KK, Leung PS. Nadph oxidase-dependent reactive oxygen species stimulate beta-cell regeneration through differentiation of endocrine progenitors in murine pancreas. Antioxid Redox Signal (2016) 24(8):419–33. doi: 10.1089/ars.2014.6135
41. Yaribeygi H, Sathyapalan T, Atkin SL, Sahebkar A. Molecular mechanisms linking oxidative stress and diabetes mellitus. Oxid Med Cell Longev (2020) 2020:8609213. doi: 10.1155/2020/8609213
42. Kaneto H, Matsuoka TA. Involvement of oxidative stress in suppression of insulin biosynthesis under diabetic conditions. Int J Mol Sci (2012) 13(10):13680–90. doi: 10.3390/ijms131013680
43. Newsholme P, Keane KN, Carlessi R, Cruzat V. Oxidative stress pathways in pancreatic beta-cells and insulin-sensitive cells and tissues: Importance to cell metabolism, function, and dysfunction. Am J Physiol Cell Physiol (2019) 317(3):C420–C33. doi: 10.1152/ajpcell.00141.2019
44. Robertson R, Zhou H, Zhang T, Harmon JS. Chronic oxidative stress as a mechanism for glucose toxicity of the beta cell in type 2 diabetes. Cell Biochem Biophys (2007) 48(2-3):139–46. doi: 10.1007/s12013-007-0026-5
45. Kawamori D, Kajimoto Y, Kaneto H, Umayahara Y, Fujitani Y, Miyatsuka T, et al. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor pdx-1 through activation of c-jun Nh(2)-terminal kinase. Diabetes (2003) 52(12):2896–904. doi: 10.2337/diabetes.52.12.2896
46. Wang X, Vatamaniuk MZ, Roneker CA, Pepper MP, Hu LG, Simmons RA, et al. Knockouts of Sod1 and Gpx1 exert different impacts on murine islet function and pancreatic integrity. Antioxid Redox Signal (2011) 14(3):391–401. doi: 10.1089/ars.2010.3302
47. Hoarau E, Chandra V, Rustin P, Scharfmann R, Duvillie B. Pro-Oxidant/Antioxidant balance controls pancreatic beta-cell differentiation through the Erk1/2 pathway. Cell Death Dis (2014) 5:e1487. doi: 10.1038/cddis.2014.441
48. Liang C, Hao F, Yao X, Qiu Y, Liu L, Wang S, et al. Hypericin maintians Pdx1 expression Via the erk pathway and protects islet beta-cells against glucotoxicity and lipotoxicity. Int J Biol Sci (2019) 15(7):1472–87. doi: 10.7150/ijbs.33817
49. Papaiahgari S, Kleeberger SR, Cho HY, Kalvakolanu DV, Reddy SP. Nadph oxidase and erk signaling regulates hyperoxia-induced Nrf2-are transcriptional response in pulmonary epithelial cells. J Biol Chem (2004) 279(40):42302–12. doi: 10.1074/jbc.M408275200
50. Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, et al. Formation of a human beta-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab (2012) 97(9):3197–206. doi: 10.1210/jc.2012-1206
51. Jacovetti C, Matkovich SJ, Rodriguez-Trejo A, Guay C, Regazzi R. Postnatal beta-cell maturation is associated with islet-specific microrna changes induced by nutrient shifts at weaning. Nat Commun (2015) 6:8084. doi: 10.1038/ncomms9084
52. Jaafar R, Tran S, Shah AN, Sun G, Valdearcos M, Marchetti P, et al. Mtorc1 to ampk switching underlies beta-cell metabolic plasticity during maturation and diabetes. J Clin Invest (2019) 129(10):4124–37. doi: 10.1172/JCI127021
53. Sacco F, Seelig A, Humphrey SJ, Krahmer N, Volta F, Reggio A, et al. Phosphoproteomics reveals the Gsk3-Pdx1 axis as a key pathogenic signaling node in diabetic islets. Cell Metab (2019) 29(6):1422–32.e3. doi: 10.1016/j.cmet.2019.02.012
54. Blandino-Rosano M, Barbaresso R, Jimenez-Palomares M, Bozadjieva N, Werneck-de-Castro JP, Hatanaka M, et al. Loss of Mtorc1 signalling impairs beta-cell homeostasis and insulin processing. Nat Commun (2017) 8:16014. doi: 10.1038/ncomms16014
55. Balcazar N, Sathyamurthy A, Elghazi L, Gould A, Weiss A, Shiojima I, et al. Mtorc1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J Biol Chem (2009) 284(12):7832–42. doi: 10.1074/jbc.M807458200
56. Zhang X, Wang X, Yuan Z, Radford SJ, Liu C, Libutti SK, et al. Amino acids-Rab1a-Mtorc1 signaling controls whole-body glucose homeostasis. Cell Rep (2021) 34(11):108830. doi: 10.1016/j.celrep.2021.108830
57. Fan J, Yuan Z, Burley SK, Libutti SK, Zheng XFS. Amino acids control blood glucose levels through mtor signaling. Eur J Cell Biol (2022) 101(3):151240. doi: 10.1016/j.ejcb.2022.151240
58. Alejandro EU, Bozadjieva N, Blandino-Rosano M, Wasan MA, Elghazi L, Vadrevu S, et al. Overexpression of kinase-dead mtor impairs glucose homeostasis by regulating insulin secretion and not beta-cell mass. Diabetes (2017) 66(8):2150–62. doi: 10.2337/db16-1349
59. Wang J, Yang X, Zhang J. Bridges between mitochondrial oxidative stress, er stress and mtor signaling in pancreatic beta cells. Cell Signal (2016) 28(8):1099–104. doi: 10.1016/j.cellsig.2016.05.007
60. Riahi Y, Israeli T, Yeroslaviz R, Chimenez S, Avrahami D, Stolovich-Rain M, et al. Inhibition of Mtorc1 by er stress impairs neonatal beta-cell expansion and predisposes to diabetes in the akita mouse. Elife (2018) 7:e38472. doi: 10.7554/eLife.38472
61. Kageyama S, Saito T, Obata M, Koide RH, Ichimura Y, Komatsu M. Negative regulation of the Keap1-Nrf2 pathway by a P62/Sqstm1 splicing variant. Mol Cell Biol (2018) 38(7):e00642-17. doi: 10.1128/MCB.00642-17
62. Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, et al. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol (2005) 25(9):3752–62. doi: 10.1128/MCB.25.9.3752-3762.2005
63. Tschen SI, Zeng C, Field L, Dhawan S, Bhushan A, Georgia S. Cyclin D2 is sufficient to drive beta cell self-renewal and regeneration. Cell Cycle (2017) 16(22):2183–91. doi: 10.1080/15384101.2017.1319999
64. Lakshmipathi J, Alvarez-Perez JC, Rosselot C, Casinelli GP, Stamateris RE, Rausell-Palamos F, et al. Pkczeta is essential for pancreatic beta-cell replication during insulin resistance by regulating mtor and cyclin-D2. Diabetes (2016) 65(5):1283–96. doi: 10.2337/db15-1398
65. Stamateris RE, Sharma RB, Hollern DA, Alonso LC. Adaptive beta-cell proliferation increases early in high-fat feeding in mice, concurrent with metabolic changes, with induction of islet cyclin D2 expression. Am J Physiol Endocrinol Metab (2013) 305(1):E149–59. doi: 10.1152/ajpendo.00040.2013
66. Salazar-Petres ER, Sferruzzi-Perri AN. Pregnancy-induced changes in beta-cell function: What are the key players? J Physiol (2022) 600(5):1089–117. doi: 10.1113/JP281082
67. Aguayo-Mazzucato C, Koh A, El Khattabi I, Li WC, Toschi E, Jermendy A, et al. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia (2011) 54(3):583–93. doi: 10.1007/s00125-010-2026-z
68. Wang X, Sterr M, Ansarullah, Burtscher I, Bottcher A, Beckenbauer J, et al. Point mutations in the Pdx1 transactivation domain impair human beta-cell development and function. Mol Metab (2019) 24:80–97. doi: 10.1016/j.molmet.2019.03.006
69. Olson LK, Redmon JB, Towle HC, Robertson RP. Chronic exposure of hit cells to high glucose concentrations paradoxically decreases insulin gene transcription and alters binding of insulin gene regulatory protein. J Clin Invest (1993) 92(1):514–9. doi: 10.1172/JCI116596
70. Reimer MK, Ahren B. Altered beta-cell distribution of pdx-1 and glut-2 after a short-term challenge with a high-fat diet in C57bl/6j mice. Diabetes (2002) 51 Suppl 1:S138–43. doi: 10.2337/diabetes.51.2007.s138
71. Glavas MM, Hui Q, Tuduri E, Erener S, Kasteel NL, Johnson JD, et al. Early overnutrition reduces Pdx1 expression and induces beta cell failure in Swiss Webster mice. Sci Rep (2019) 9(1):3619. doi: 10.1038/s41598-019-39177-3
72. Bosma KJ, Andrei SR, Katz LS, Smith AA, Dunn JC, Ricciardi VF, et al. Pharmacological blockade of the Ep3 prostaglandin E2 receptor in the setting of type 2 diabetes enhances beta-cell proliferation and identity and relieves oxidative damage. Mol Metab (2021) 54:101347. doi: 10.1016/j.molmet.2021.101347
73. Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka T, Fujitani Y, Umayahara Y, et al. Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic beta-cells against glucose toxicity. Diabetes (1999) 48(12):2398–406. doi: 10.2337/diabetes.48.12.2398
74. Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in hit-T15 cells and zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U.S.A. (1999) 96(19):10857–62. doi: 10.1073/pnas.96.19.10857
75. Wang XD, Vatamaniuk MZ, Wang SK, Roneker CA, Simmons RA, Lei XG. Molecular mechanisms for hyperinsulinaemia induced by overproduction of selenium-dependent glutathione peroxidase-1 in mice. Diabetologia (2008) 51(8):1515–24. doi: 10.1007/s00125-008-1055-3
76. Kawamori D, Kaneto H, Nakatani Y, Matsuoka TA, Matsuhisa M, Hori M, et al. The forkhead transcription factor Foxo1 bridges the jnk pathway and the transcription factor pdx-1 through its intracellular translocation. J Biol Chem (2006) 281(2):1091–8. doi: 10.1074/jbc.M508510200
77. Boucher MJ, Selander L, Carlsson L, Edlund H. Phosphorylation marks Ipf1/Pdx1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J Biol Chem (2006) 281(10):6395–403. doi: 10.1074/jbc.M511597200
78. Park KG, Lee KM, Seo HY, Suh JH, Kim HS, Wang L, et al. Glucotoxicity in the ins-1 rat insulinoma cell line is mediated by the orphan nuclear receptor small heterodimer partner. Diabetes (2007) 56(2):431–7. doi: 10.2337/db06-0753
79. Yang BT, Dayeh TA, Volkov PA, Kirkpatrick CL, Malmgren S, Jing X, et al. Increased DNA methylation and decreased expression of pdx-1 in pancreatic islets from patients with type 2 diabetes. Mol Endocrinol (2012) 26(7):1203–12. doi: 10.1210/me.2012-1004
80. Ruiz L, Gurlo T, Ravier MA, Wojtusciszyn A, Mathieu J, Brown MR, et al. Proteasomal degradation of the histone acetyl transferase P300 contributes to beta-cell injury in a diabetes environment. Cell Death Dis (2018) 9(6):600. doi: 10.1038/s41419-018-0603-0
81. Zhang M, Yang C, Zhu M, Qian L, Luo Y, Cheng H, et al. Saturated fatty acids entrap Pdx1 in stress granules and impede islet beta cell function. Diabetologia (2021) 64(5):1144–57. doi: 10.1007/s00125-021-05389-4
82. Noh JR, Hwang JH, Kim YH, Kim KS, Gang GT, Kim SW, et al. The orphan nuclear receptor small heterodimer partner negatively regulates pancreatic beta cell survival and hyperglycemia in multiple low-dose streptozotocin-induced type 1 diabetic mice. Int J Biochem Cell Biol (2013) 45(8):1538–45. doi: 10.1016/j.biocel.2013.05.004
83. Davidson RK, Weaver SA, Casey N, Kanojia S, Hogarth E, Schneider Aguirre R, et al. The Chd4 subunit of the nurd complex regulates Pdx1-controlled genes involved in beta-cell function. J Mol Endocrinol (2022) 69(2):329–41. doi: 10.1530/JME-22-0011
84. Seo HY, Kim YD, Lee KM, Min AK, Kim MK, Kim HS, et al. Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases insulin gene expression Via up-regulation of orphan nuclear receptor small heterodimer partner. Endocrinology (2008) 149(8):3832–41. doi: 10.1210/en.2008-0015
85. Yang W, Chi Y, Meng Y, Chen Z, Xiang R, Yan H, et al. Fam3a plays crucial roles in controlling Pdx1 and insulin expressions in pancreatic beta cells. FASEB J (2020) 34(3):3915–31. doi: 10.1096/fj.201902368RR
86. Akerman I, Tu Z, Beucher A, Rolando DMY, Sauty-Colace C, Benazra M, et al. Human pancreatic beta cell lncrnas control cell-specific regulatory networks. Cell Metab (2017) 25(2):400–11. doi: 10.1016/j.cmet.2016.11.016
87. McKenna B, Guo M, Reynolds A, Hara M, Stein R. Dynamic recruitment of functionally distinct Swi/Snf chromatin remodeling complexes modulates Pdx1 activity in islet beta cells. Cell Rep (2015) 10(12):2032–42. doi: 10.1016/j.celrep.2015.02.054
88. Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, et al. Foxo1 protects against pancreatic beta cell failure through neurod and mafa induction. Cell Metab (2005) 2(3):153–63. doi: 10.1016/j.cmet.2005.08.004
89. Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH 3rd, Wright CV, et al. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest (2002) 110(12):1839–47. doi: 10.1172/JCI16857
90. Glauser DA, Schlegel W. The emerging role of foxo transcription factors in pancreatic beta cells. J Endocrinol (2007) 193(2):195–207. doi: 10.1677/JOE-06-0191
91. Kikuchi O, Kobayashi M, Amano K, Sasaki T, Kitazumi T, Kim HJ, et al. Foxo1 gain of function in the pancreas causes glucose intolerance, polycystic pancreas, and islet hypervascularization. PloS One (2012) 7(2):e32249. doi: 10.1371/journal.pone.0032249
92. Ohtsubo K, Chen MZ, Olefsky JM, Marth JD. Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat Med (2011) 17(9):1067–75. doi: 10.1038/nm.2414
93. Fernandez-Zapico ME, van Velkinburgh JC, Gutierrez-Aguilar R, Neve B, Froguel P, Urrutia R, et al. Mody7 gene, Klf11, is a novel P300-dependent regulator of pdx-1 (Mody4) transcription in pancreatic islet beta cells. J Biol Chem (2009) 284(52):36482–90. doi: 10.1074/jbc.M109.028852
94. Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, Dina C, Hamid YH, Joly E, et al. Role of transcription factor Klf11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci U.S.A. (2005) 102(13):4807–12. doi: 10.1073/pnas.0409177102
95. Elsner M, Gehrmann W, Lenzen S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes (2011) 60(1):200–8. doi: 10.2337/db09-1401
96. Elsharkawi I, Parambath D, Saber-Ayad M, Khan AA, El-Serafi AT. Exploring the effect of epigenetic modifiers on developing insulin-secreting cells. Hum Cell (2020) 33(1):1–9. doi: 10.1007/s13577-019-00292-y
97. Davidson RK, Kanojia S, Spaeth JM. The contribution of transcriptional coregulators in the maintenance of beta-cell function and identity. Endocrinology (2021) 162(2):bqaa213. doi: 10.1210/endocr/bqaa213
98. Spaeth JM, Liu JH, Peters D, Guo M, Osipovich AB, Mohammadi F, et al. The Pdx1-bound Swi/Snf chromatin remodeling complex regulates pancreatic progenitor cell proliferation and mature islet beta-cell function. Diabetes (2019) 68(9):1806–18. doi: 10.2337/db19-0349
99. Mosley AL, Corbett JA, Ozcan S. Glucose regulation of insulin gene expression requires the recruitment of P300 by the beta-Cell-Specific transcription factor pdx-1. Mol Endocrinol (2004) 18(9):2279–90. doi: 10.1210/me.2003-0463
100. Latif W, Lambrinos KJ, Rodriguez R. Compare and contrast the glucagon-like peptide-1 receptor agonists (Glp1ras). Treasure Island (FL: Statpearls (2022).
101. Gao M, Deng XL, Liu ZH, Song HJ, Zheng J, Cui ZH, et al. Liraglutide protects beta-cell function by reversing histone modification of pdx-1 proximal promoter in catch-up growth Male rats. J Diabetes Complications (2018) 32(11):985–94. doi: 10.1016/j.jdiacomp.2018.08.002
102. Pinney SE, Jaeckle Santos LJ, Han Y, Stoffers DA, Simmons RA. Exendin-4 increases histone acetylase activity and reverses epigenetic modifications that silence Pdx1 in the intrauterine growth retarded rat. Diabetologia (2011) 54(10):2606–14. doi: 10.1007/s00125-011-2250-1
103. Wang X, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 causes pancreatic duodenal homeobox-1 protein translocation from the cytoplasm to the nucleus of pancreatic beta-cells by a cyclic adenosine Monophosphate/Protein kinase a-dependent mechanism. Endocrinology (2001) 142(5):1820–7. doi: 10.1210/endo.142.5.8128
104. Muscogiuri G, Salmon AB, Aguayo-Mazzucato C, Li M, Balas B, Guardado-Mendoza R, et al. Genetic disruption of Sod1 gene causes glucose intolerance and impairs beta-cell function. Diabetes (2013) 62(12):4201–7. doi: 10.2337/db13-0314
105. Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T, et al. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol Cell Biol (2013) 33(15):2996–3010. doi: 10.1128/MCB.00225-13
106. Masuda Y, Vaziri ND, Li S, Le A, Hajighasemi-Ossareh M, Robles L, et al. The effect of Nrf2 pathway activation on human pancreatic islet cells. PloS One (2015) 10(6):e0131012. doi: 10.1371/journal.pone.0131012
Keywords: Pdx1, ROS, Nrf2, oxidative-stress, beta-cells, diabetes
Citation: Baumel-Alterzon S and Scott DK (2022) Regulation of Pdx1 by oxidative stress and Nrf2 in pancreatic beta-cells. Front. Endocrinol. 13:1011187. doi: 10.3389/fendo.2022.1011187
Received: 03 August 2022; Accepted: 26 August 2022;
Published: 15 September 2022.
Edited by:
Carlos Guillén, Department of Biochemistry and Molecular Biology (UCM), SpainReviewed by:
Teresa Pereira, Uppsala University, SwedenCopyright © 2022 Baumel-Alterzon and Scott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sharon Baumel-Alterzon, c2hhcm9uLmFsdGVyem9uQG1zc20uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.