 Jun Li1,2,3†
Jun Li1,2,3† Bing Wei1,2,3†
Bing Wei1,2,3† Junnan Feng1,2,3Xinxin Wu1,2,3Yuxi Chang1,2,3Yi Wang4Xiuli Yang5Haiyan Zhang6
Junnan Feng1,2,3Xinxin Wu1,2,3Yuxi Chang1,2,3Yi Wang4Xiuli Yang5Haiyan Zhang6 Sile Han1
Sile Han1 Cuiyun Zhang1,2,3Jiawen Zheng1,2,3
Cuiyun Zhang1,2,3Jiawen Zheng1,2,3 Harry J. M. Groen7
Harry J. M. Groen7 Anke van den Berg8
Anke van den Berg8 Jie Ma1,2,3Hongle Li1,2,3*
Jie Ma1,2,3Hongle Li1,2,3* Yongjun Guo1,2,3*
Yongjun Guo1,2,3*- 1Department of Molecular Pathology, Clinical Pathology Center, Affiliated Cancer Hospital of Zhengzhou University and Henan Cancer Hospital, Zhengzhou, China
- 2Henan Key Laboratory of Molecular Pathology, Zhengzhou, China
- 3Henan International Joint Laboratory of Cancer Molecular Genetics, Zhengzhou, China
- 4Department of Pathology, Clinical Pathology Center, Affiliated Cancer Hospital of Zhengzhou University and Henan Cancer Hospital, Zhengzhou, China
- 5Department of Oncology, First Affiliated Hospital of Nanyang Medical College, Nanyang, China
- 6Department of Pathology, First Affiliated Hospital of Nanyang Medical College, Nanyang, China
- 7Department of Pulmonary Diseases, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
- 8Department of Pathology and Medical Biology, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
Introduction: Transformation from lung adenocarcinoma (LUAD) to small cell lung cancer (SCLC) is one of the mechanisms responsible for acquired EGFR-TKIs resistance. Although it rarely happens this event determines a rapid disease deterioration and needs specific treatment.
Patient and method: We report a case of 75-year-old LUAD female with a p.L858R mutation in Epidermal Growth Factor Receptor (EGFR) who presented with SCLC transformation after responding to first line osimertinib treatment for only 6 months. To understand the underlying molecular mechanism, we retrospectively sequenced the first (LUAD) and the second (SCLC) biopsy using a 56 multi-gene panel. Immunohistochemistry (IHC) staining and Fluorescence In Situ Hybridization (FISH) was applied to confirm the genetic aberrations identified.
Results: EGFR p.E709A and p.L858R, Tumor Protein p53 (TP53) p.A159D and Retinoblastoma 1 (RB1) c.365-1G>A were detected in both the diagnostic LUAD and transformed SCLC samples. A high copy number gain for Proto-Oncogene C-Myc (MYC) and a Phosphoinositide 3-Kinase Alpha (PIK3CA) p.E545K mutation were found in the transformed sample specifically. Strong TP53 staining and negative RB1 staining were observed in both LUAD and SCLC samples, but FISH only identified MYC amplification in SCLC tissue.
Conclusion: We consider the combined presence of MYC amplification with mutations in TP53 and RB1 as drivers of SCLC transformation. Our results highlight the need to systematically evaluate TP53 and RB1 status in LUAD patients to offer a different therapeutic strategy.
Introduction
Transformation from EGFR mutant LUAD to SCLC is one of the mechanisms underlying the acquired resistance to EGFR tyrosine kinase inhibitors (TKIs) (1). The reported frequency of SCLC transformation ranges from 5% to 14% (2, 3), but the disease usually becomes highly aggressive when it occurs (4). Although the precursors of transformed SCLC cells are still inconclusive, it would be of great value to find markers that can predict the transformation risk efficiently. This is because the treatment strategies for SCLC are substantially different from LUAD (5). For limited stage disease, concurrent chemoradiation is usually used for SCLC treatment but surgery is rarely recommended; for LUAD, radical surgery is still the mainstay of treatment, although other adjuvant treatments, i.e., radiotherapy, chemotherapy and immunotherapy either given alone or in combination, have shown promising clinical outcomes. For advanced disease, chemotherapy is the mainstay of treatment for both LUAD and SCLC but with different regimens. Here, we report an LUAD case suffered from SCLC transformation with the treatment of osimertinib for only 6 months. Mutation screening was performed on the FFPE tissues from both primary LUAD and transformed SCLC malignancies to understand its evolution.
Case presentation
A 75-year-old non-smoking female presented with chronic cough and fatigue as her main complaint and was admitted for medical tests and treatment to the hospital in April 2020. Physical examination was good with a performance score of one. Computed tomography (CT) scanning of the chest revealed a 52×43mm mass with several small nodules in the right lower lobe, and positron emission tomography (PET)-CT confirmed the hypermetabolic status of the nodules. Metastatic lesions were observed at the 9th thoracic vertebrae and right 9th rib bone. Tumor tissue of the main tumor mass was collected by fine needle aspiration (FNA) and processed into a formalin-fixed, paraffin-embedded (FFPE) block for histopathology diagnosis and molecular testing. Hematoxylin-eosin (HE) staining revealed a non-small cell lung cancer, with tumor cells staining positive for cytokeratin AE1/AE3 (CK AE1/AE3), thyroid transcription factor 1 (TTF-1) and Napsin A, and negative for cytokeratin 5/6 (CK5/6), synaptophysin (Syn) and P40, supporting a diagnosis of lung adenocarcinoma. Ki-67 and programmed death-ligand 1 (PD-L1) expression were found in about 50% and 5% of the tumor cells respectively. Programmed cell death protein 1 (PD1) was negative. An amplification-refractory mutation system (ARMS) test identified the common mutation of NM_005228.5(EGFR): c.2573T>G (p.Leu858Arg) (EGFR p.L858R) with an allele frequency of 72.4% in the tumor (ACCB Biotech Ltd, China; detection limit, 0.1%). The patient received oral osimertinib at a once-daily dose of 80mg as the first-line treatment. Follow-up CT imaging revealed a partial tumor response that continued for 6 months. Thereafter the patient developed disease progression. Hematoxylin and Eosin (HE) staining of the re-biopsy tumor tissue showed a small round morphology of the cells with high nuclear/cytoplasmic ratio and positive staining of Syn and TTF-1, and no staining for CD56, Chromogranin A (CgA), Napsin A and P40. The percentage of Ki-67 expression positive cells increased to about 90% (Figures 1A, B). Targeted capture sequencing of 56 lung cancer related genes (Burning Rock Dx, Guangzhou China) revealed following mutations: EGFR p.L858R (56.7%) and NM_005228.5(EGFR):c.2126A>C (p.Glu709Ala) (EGFR p.E709A, 56.7%), NM_006218.4(PIK3CA):c.1633G>A (p.Glu545Lys) (PIK3CA p.E545K, 53.1%), NM_000321.2 (RB1): c.365-1G>A (RB1 c.365-1G>A, 92.1%) and NM_000546.6(TP53):c.476C>A (p.Ala159Asp) (TP53 p.A159D, 93.98%), as well as copy number changes in MYC (CN=55). With this knowledge we also performed targeted capture sequencing analysis on the first tumor biopsy tissue. This revealed a similar mutational profile in the diagnostic tissue sample with the presence of RB1 c.365-1G>A and the TP53 p.A159D in addition to the EGFR p.L858R and p.E709A.Amplification in EGFR (CN=4.2), instead of MYC, was observed in the diagnostic LUAD sample (Figures 1C, D). Subsequent fluorescence in situ hybridization (FISH) confirmed the MYC amplification in the SCLC sample, while the diagnostic sample did not have the MYC amplification (Figure 1E). Additional immunohistochemistry revealed a positive staining for TP53MUT (MAB-0674, Maixin Biotech., Fuzhou China) and negative staining for RB1 (MAB-0186, Maixin Biotech., Fuzhou China) on both the LUAD and transformed SCLC samples, consistent with the loss of function mutations caused by the TP53 p.A159D and the RB1 c.365-1G>A which leads to loss of a splice site (Figure 1F; Supplementary Figure 1).
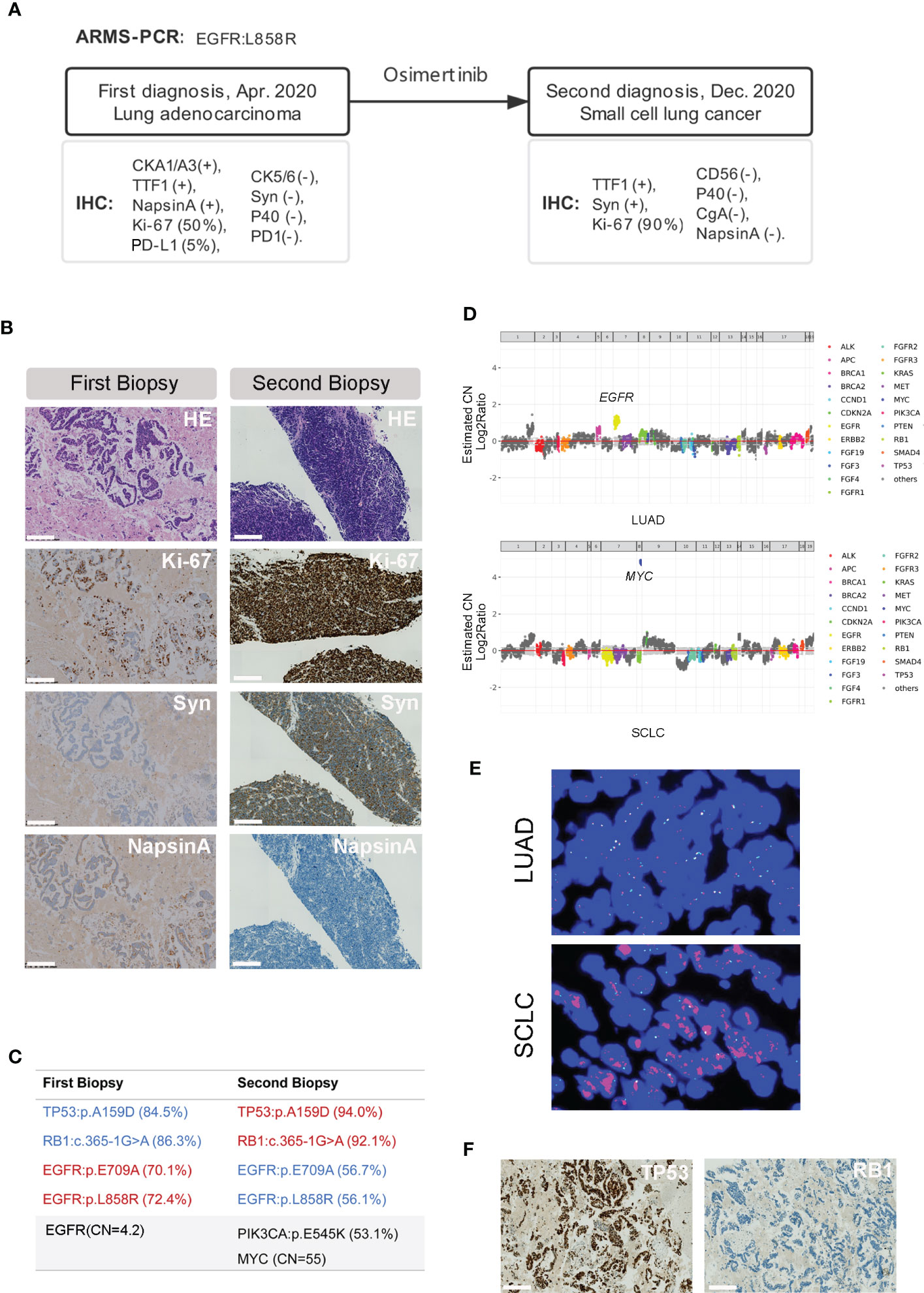
Figure 1 Pathology features and genetic aberrations present before and after SCLC transformation. (A) A brief history of the disease diagnosis and treatment in this patient. (B) HE and IHC staining of the tumor tissue before (first biopsy) and after (second biopsy) the SCLC transformation (10×). Scale bar indicates 200μm. (C) Genetic aberrations detected in the first and second tumor biopsy tissue using the targeted sequencing covering 56 lung cancer related genes. (D) MYC copy number (CN=55, upper) and EGFR copy number (CN=4.2) determined by modeling copy number variation and aneuploidy across the genome, the Y axis is indicated as log2 scale. (E) Example of MYC amplification detected by FISH in the primary LUAD (upper) and transformed SCLC (lower) tissue (40×). (F) IHC staining pattern of TP53 and RB-1 in the LUAD biopsy before transformation (10×). Scale bar indicates 200μm.
Then the patient was treated with cisplatin plus irinotecan hydrochloride, but unfortunately passed away 4 months later. Collectively, this case showed a transformation from EGFR mutant adenocarcinoma to an aggressive neuroendocrine small-cell lung cancer (SCLC) after first-line osimertinib treatment in 6 months.
Discussion
Functional loss of RB1 and TP53 are recurrent aberrations in transformed SCLC tumors, suggesting these aberrations might be highly predictive markers (1). However, TP53 and RB1 staining are not included in routine clinical care. Based on the updated CSCO and NCCN guidelines for non-small cell lung cancer, targeted sequencing panels usually only cover a subset of oncogenic driver genes, i.e., EGFR, Kirsten rat sarcoma virus (KRAS), Anaplastic lymphoma kinase (ALK), ROS Proto-Oncogene 1, Receptor Tyrosine Kinase (ROS1), B-Raf Proto-Oncogene, Serine/Threonine Kinase (BRAF), MET Proto-Oncogene, Receptor Tyrosine Kinase (MET) and Neurotrophin receptor tyrosine kinase (NTRK), and do not provide information on TP53 and RB1.
By targeted sequencing of the primary tumor tissue, we found both the RB1 and TP53 mutations were already present before start of osimertinib treatment. The EGFR p.L858R and p.E709A mutations were detected at comparable variant allele frequencies (VAFs) in both tumor samples, indicating that the mutations most likely occurred in cis. Although this specific combination of EGFR mutations is rare, mutations at E709 in EGFR usually present concurrently with other oncogenic EGFR mutations (6). Proliferation assay and colony formation assays revealed an oncogenic role of E790X in Ba/F3 and NIH-3T3 cells and dose-response experiments showed that E709K mutant cells were more sensitive to second-generation EGFR-TKIs than first- and third-generation drugs (7). Clinical data showed lower response rates of about 50% in patients with combined EGFR mutations including a p.E709X to first-generation EGFR-TKIs (8).
As compared to the primary LUAD tissue, the VAFs of EGFR p.L858R and p.E709A were decreased in the transformed SCLC tissue, and EGFR amplification was disappeared as well, indicating Osimertinib indeed inhibited the proliferation of the EGFR-mutant clone. In contrast, the VAFs of both TP53 p.A159D and RB1 c.365-1G>A increased, suggesting the transformed cells arise from a different clone. This observation is consistent with a series of cases have been reported that the original EGFR mutation could always be detected in the transformed SCLC tissues without exception (9–11). Unfortunately, the TP53 and RB1 status was unknown in these studies.
In recent years, several important studies pinpointed the essential role of TP53 and RB1 loss in the transformation from LUAD to SCLC. Offin et al. identified 43 out of 4112 lung cancer patients with concurrent EGFR/TP53/RB1 mutations in their primary tumors, of which 7 were suffered from SCLC transformation after EGFR-TKIs treatment, 4 patients had small cell histology at the initial diagnosis, and the other 32 never had SCLC transformation till the last review (12). Of the patients with concurrent EGFR/RB1 mutations (n=54), 11 patients didn’t have TP53 mutation and none of them had SCLC transformation. In a sub-group of 142 cancer patients with EGFR mutation but not TP53 and RB1, no transformation was observed. These results clearly showed EGFR/RB1/TP53-mutant patients had a significantly increased risk of SCLC transformation than the patients with mutations in one or two of these genes. In addition, EGFR/RB1/TP53-mutant patients showed worse clinical outcomes in both time to treatment discontinuation (TTD) and overall survival, as compared to the patients with only EGFR/TP53 or EGFR/RB1 mutations, indicating concurrent loss of RB1 and TP53 substantially changed the disease behavior. Actually, neither EGFR mutation, nor EGFR-TKIs treatment, is indispensable, since SCLC transformation were also observed in EGFR wild-type patients after immunotherapy, in anaplastic lymphoma kinase (ALK)-mutant patients after ALK-inhibitor treatment or immunotherapy (4).
Both TP53 and RB1 are well established tumor suppressor genes and mutated in a wide spectrum of cancers. A recent study investigating concurrent RB1/TP53 mutations in pan-cancer revealed that RB1 and TP53 co-mutations were significantly enriched in small cell carcinomas and neuroendocrine carcinomas (13). Studies in animal models have shed light on understanding the biological context of SCLC transformation after TP53 and RB1 loss. Different from TP53, RB1 appears to play a more flexible role in regulation lineage-specific cell differentiation, evidenced by an increased number of lung neuroendocrine cells in Rb1 conditional knockout mice during development (14). In addition, neuroendocrine markers are expressed in 39 out of 42 RB1-mutant SCLC cell lines, but are absence in 5/6 RB1 wild-type SCLC cell lines. In prostate cancer mouse models, Ku et al. showed Rb1 loss facilitated the lineage plasticity of prostate cancer then additional Tp53 inactivation enabled the cells insensitive to antiandrogen therapy (15). However, it’s noteworthy that some of the TP53/RB1-mutant LUAD patients never transformed into SCLC, suggesting other factors are also involved (12).
In this case, we identified a PIK3CA p.E545K and a MYC amplification (CN=55) to be specific for the transformed SCLC tissue, indicating these aberrations may contribute to disease progression. The E545K is one of the hotspot mutations in PIK3CA, presenting in about 2% of NSCLC patients. This mutation affects the helical binding domain of PIK3CA. Different from the classic oncogenic mutations, i.e., EGFR and KRAS, which are often mutually exclusive, PIK3CA mutations usually co-occur with other genetic aberrations in EGFR, KRAS or ALK etc. (16). Of note, the PIK3CA p.E545K is commonly observed in EGFR p.L858R mutant Chinese NSCLC patients (17). In a functional study, this mutation was shown to be sufficient to abrogate EGFR-TKI induced cellular apoptosis (18). Therefore, the occurrence of the PIK3CA p.E545K mutation might have attenuated the efficacy of osimertinib in this patient. Similar observations have been reported in other studies (19, 20). Although its role in the carcinogenesis of lung cancer is still controversial, it has been shown to promote the disease development in gallbladder and cervical cancer (21, 22).
MYC regulates a wide spectrum of biological processes involved in the initiation and progression of many cancers, and amplification of MYC has been observed in both lung adenocarcinoma and SCLC patients (23). MYC amplification has been associated with unfavorable clinical outcome and treatment resistance (24, 25). In SCLC, MYC amplification promotes cellular proliferation and inhibits cellular differentiation (26). We identified MYC amplification (CN=55) in the transformed SCLC tissue and not in the primary tumor. Besides MYC amplification, the SCLC sample also showed increased percentage of Ki-67 positive cells consistent with its role in proliferation. Of note. MYC was shown to promote rapid disease deterioration in Tp53/Rb1 null SCLC mice and induced dynamic evolution in NEUROD1-expressing neuroendocrine-low SCLC subtype (24, 27). Taken together, these studies suggest that SCLC transformation and rapid disease deterioration as observed in this case might be contributed by the TP53/RB1-mutant profile in combination with the amplification of MYC. Although previous studies suggest that SCLC transformation is determined by multiple factors beyond genetic changes, i.e., the origin and plasticity of the tumor cells (24, 27), TP53 and RB1 loss might be the key drivers in this case.
Considering the rapid disease progression and poor prognosis of patients with transformed SCLC, a lesson we learned from this case is the necessity to evaluate TP53 and RB1 status in EGFR mutated lung adenocarcinoma patients before start of EGFR-TKI treatment. This can be achieved by a high-throughput NGS test, or by IHC staining, which is more cost-effective. A critical question remains whether with the information currently available, we should treat these patients with chemotherapy in the first line or directly start with a combination of chemotherapy and osimertinib.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the ethics committee of Henan Cancer Hospital. The patients/participants provided their written informed consent to participate in this study. Written, informed consent was obtained from the participant for the publication of this case report (including all data and images).
Author contributions
YG and HL conceived and designed the study, JL and BW analysed the data and drafted the manuscript with the support from HG and AB, JF, CZ and JZ performed the sequencing and bioinformatic analysis, SH, YW, YC and JM performed the IHC staining, FISH test and analysed the result, XY and HZ collected the clinical data. All authors contributed to the article and approved the submitted version.
Funding
JL was supported by Henan provincial young researcher program (2021). The study was financially supported by Henan Provincial Health commission and the Department of science and technology (SBGJ202002020, SBGJ20211008, 212102310675, 222300420574, RKX202002007 and 201300310400).
Acknowledgments
The authors would like to thank all the other colleagues in the Department of Molecular Pathology at the Affiliated Cancer Hospital of Zhengzhou University for their helpful discussion.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.1006480/full#supplementary-material
References
1. Lee JK, Lee J, Kim S, Kim S, Youk J, Park S, et al. Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J Clin Oncol (2017) 35:3065–74. doi: 10.1200/JCO.2016.71.9096
2. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res (2013) 19:2240–7. doi: 10.1158/1078-0432.CCR-12-2246
3. Lai L, Meng W, Wei J, Zhang X, Tan Z, Lu Y, et al. Transformation of NSCLC to SCLC after 1st- and 3rd-generation EGFR-TKI resistance and response to EP regimen and erlotinib: 2 CARE-compliant case reports. Med (Baltimore) (2021) 100:e25046. doi: 10.1097/MD.0000000000025046
4. Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol (2015) 16:e165–72. doi: 10.1016/S1470-2045(14)71180-5
5. Heigener DF, Reck M. Lung cancer in 2017: Giant steps and stumbling blocks. Nat Rev Clin Oncol (2018) 15:71–2. doi: 10.1038/nrclinonc.2017.178
6. Kobayashi S, Canepa HM, Bailey AS, Nakayama S, Yamaguchi N, Goldstein MA, et al. Compound EGFR mutations and response to EGFR tyrosine kinase inhibitors. J Thorac Oncol (2013) 8:45–51. doi: 10.1097/JTO.0b013e3182781e35
7. Kobayashi Y, Togashi Y, Yatabe Y, Mizuuchi H, Jangchul P, Kondo C, et al. EGFR exon 18 mutations in lung cancer: Molecular predictors of augmented sensitivity to afatinib or neratinib as compared with first- or third-generation TKIs. Clin Cancer Res (2015) 21:5305–13. doi: 10.1158/1078-0432.CCR-15-1046
8. Harrison PT, Vyse S, Huang PH. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin Cancer Biol (2020) 61:167–79. doi: 10.1016/j.semcancer.2019.09.015
9. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3 (2011) 3(75):75ra26. doi: 10.1126/scitranslmed.3002003
10. Watanabe S, Sone T, Matsui T, Yamamura K, Tani M, Okazaki A, et al. Transformation to small-cell lung cancer following treatment with EGFR tyrosine kinase inhibitors in a patient with lung adenocarcinoma. Lung Cancer (2013) 82:370–2. doi: 10.1016/j.lungcan.2013.06.003
11. Popat S, Wotherspoon A, Nutting CM, Gonzalez D, Nicholson AG, O'Brien M. Transformation to "high grade" neuroendocrine carcinoma as an acquired drug resistance mechanism in EGFR-mutant lung adenocarcinoma. Lung Cancer (2013) 80:1–4. doi: 10.1016/j.lungcan.2012.12.019
12. Offin M, Chan JM, Tenet M, Rizvi HA, Shen R, Riely GJ, et al. Concurrent RB1 and TP53 alterations define a subset of EGFR-mutant lung cancers at risk for histologic transformation and inferior clinical outcomes. J Thorac Oncol (2019) 14:1784–93. doi: 10.1016/j.jtho.2019.06.002
13. Cai L, DeBerardinis RJ, Xiao G, Minna JD, Xie Y. A pan-cancer assessment of RB1/TP53 Co-mutations. Cancers (Basel) 14 (2022) 14(17):4199. doi: 10.3390/cancers14174199
14. Wikenheiser-Brokamp KA. Rb Family proteins differentially regulate distinct cell lineages during epithelial development. Development (2004) 131:4299–310. doi: 10.1242/dev.01232
15. Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science (2017) 355:78–83. doi: 10.1126/science.aah4199
16. Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, Pietanza MC, et al. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther (2012) 11:485–91. doi: 10.1158/1535-7163.MCT-11-0692
17. Li S, Li L, Zhu Y, Huang C, Qin Y, Liu H, et al. Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: a comprehensive mutation profiling from 5125 Chinese cohorts. Br J Cancer (2014) 110:2812–20. doi: 10.1038/bjc.2014.210
18. Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest (2006) 116:2695–706. doi: 10.1172/JCI28656
19. Eng J, Woo KM, Sima CS, Plodkowski A, Hellmann MD, Chaft JE, et al. Impact of concurrent PIK3CA mutations on response to EGFR tyrosine kinase inhibition in EGFR-mutant lung cancers and on prognosis in oncogene-driven lung adenocarcinomas. J Thorac Oncol (2015) 10:1713–9. doi: 10.1097/JTO.0000000000000671
20. Wu SG, Chang YL, Yu CJ, Yang PC, Shih JY. The role of PIK3CA mutations among lung adenocarcinoma patients with primary and acquired resistance to EGFR tyrosine kinase inhibition. Sci Rep (2016) 6:35249. doi: 10.1038/srep35249
21. Zhao S, Cao Y, Liu SB, Wang XA, Bao RF, Shu YJ, et al. The E545K mutation of PIK3CA promotes gallbladder carcinoma progression through enhanced binding to EGFR. J Exp Clin Cancer Res (2016) 35:97. doi: 10.1186/s13046-016-0370-7
22. Arjumand W, Merry CD, Wang C, Saba E, McIntyre JB, Fang S, et al. Phosphatidyl inositol-3 kinase (PIK3CA) E545K mutation confers cisplatin resistance and a migratory phenotype in cervical cancer cells. Oncotarget (2016) 7:82424–39. doi: 10.18632/oncotarget.10955
23. Masso-Valles D, Beaulieu ME, Soucek L. MYC, MYCL, and MYCN as therapeutic targets in lung cancer. Expert Opin Ther Targets (2020) 24:101–14. doi: 10.1080/14728222.2020.1723548
24. Mollaoglu G, Guthrie MR, Bohm S, Bragelmann J, Can I, Ballieu PM, et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell (2017) 31:270–85. doi: 10.1016/j.ccell.2016.12.005
25. Liu SY, Bao H, Wang Q, Mao WM, Chen Y, Tong X, et al. Genomic signatures define three subtypes of EGFR-mutant stage II-III non-small-cell lung cancer with distinct adjuvant therapy outcomes. Nat Commun (2021) 12:6450. doi: 10.1038/s41467-021-26806-7
26. Bragelmann J, Bohm S, Guthrie MR, Mollaoglu G, Oliver TG, Sos ML. Family matters: How MYC family oncogenes impact small cell lung cancer. Cell Cycle (2017) 16:1489–98. doi: 10.1080/15384101.2017.1339849
Keywords: small cell lung cancer transformation, lung adenocarcinoma, osimertinib, TP53 and RB1 loss, MYC amplification
Citation: Li J, Wei B, Feng J, Wu X, Chang Y, Wang Y, Yang X, Zhang H, Han S, Zhang C, Zheng J, Groen HJM, van den Berg A, Ma J, Li H and Guo Y (2022) Case report: TP53 and RB1 loss may facilitate the transformation from lung adenocarcinoma to small cell lung cancer by expressing neuroendocrine markers. Front. Endocrinol. 13:1006480. doi: 10.3389/fendo.2022.1006480
Received: 29 July 2022; Accepted: 29 November 2022;
Published: 13 December 2022.
Edited by:
Ben Nephew, Worcester Polytechnic Institute, United StatesReviewed by:
Khawla S. Al-Kuraya, King Faisal Specialist Hospital & Research Centre, Saudi ArabiaJie Wu, First Affiliated Hospital of Jinzhou Medical University, China
Copyright © 2022 Li, Wei, Feng, Wu, Chang, Wang, Yang, Zhang, Han, Zhang, Zheng, Groen, van den Berg, Ma, Li and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongjun Guo, Z3VveW9uZ2p1bkB6enUuZWR1LmNu; Hongle Li, bGxobDczQDE2My5jb20=
†These authors have contributed equally to this work