
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 29 September 2022
Sec. Bone Research
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1005189
This article is part of the Research TopicRare Musculoskeletal Disorders: Disease Mechanisms and TherapiesView all 6 articles
 Miwa Yamazaki
Miwa Yamazaki Toshimi Michigami*
Toshimi Michigami*Since phosphorus is a component of hydroxyapatite, its prolonged deprivation affects bone mineralization. Fibroblast growth factor 23 (FGF23) is essential for maintaining phosphate homeostasis and is mainly produced by osteocytes. FGF23 increases the excretion of inorganic phosphate (Pi) and decreases the production of 1,25-dihydroxyvitamin D in the kidneys. Osteocytes are cells of osteoblastic lineage that have undergone terminal differentiation and become embedded in mineralized bone matrix. Osteocytes express FGF23 and other multiple genes responsible for hereditary hypophosphatemic rickets, which include phosphate-regulating gene homologous to endopeptidase on X chromosome (PHEX), dentin matrix protein 1 (DMP1), and family with sequence similarity 20, member C (FAM20C). Since inactivating mutations in PHEX, DMP1, and FAM20C boost the production of FGF23, these molecules might be considered as local negative regulators of FGF23. Mouse studies have suggested that enhanced FGF receptor (FGFR) signaling is involved in the overproduction of FGF23 in PHEX-deficient X-linked hypophosphatemic rickets (XLH) and DMP1-deficient autosomal recessive hypophosphatemic rickets type 1. Since FGFR is involved in the transduction of signals evoked by extracellular Pi, Pi sensing in osteocytes may be abnormal in these diseases. Serum levels of sclerostin, an inhibitor Wnt/β-catenin signaling secreted by osteocytes, are increased in XLH patients, and mouse studies have suggested the potential of inhibiting sclerostin as a new therapeutic option for the disease. The elucidation of complex abnormalities in the osteocytes of FGF23-related hypophosphatemic diseases will provide a more detailed understanding of their pathogenesis and more effective treatments.
In human adults, approximately 90% of total phosphorus in the body is stored in the skeleton as calcium/phosphate crystals, such as hydroxyapatite. Most of the remaining phosphorus exists in soft tissues, and less than 1% is distributed in extracellular fluid (ECF). Phosphorus is present as inorganic phosphate (Pi) in serum (1). Since Pi is indispensable for the synthesis of hydroxyapatite, its prolonged deficiency or wasting impairs skeletal mineralization (2).
Fibroblast growth factor 23 (FGF23) is produced in bone and increases renal Pi excretion by down-regulating the expression of type IIa and IIc sodium/Pi (Na+/Pi) co-transporters (NaPi-IIa and NaPi-IIc, respectively) (3). FGF23 also down-regulates the expression of 25-hydroxyvitamin D-1α hydroxylase and up-regulates that of 25-hydroxyvitamin D-24 hydroxylase, reducing the production of 1,25-dihydroxyvitamin D (1,25(OH)2D) (3). The excessive effects of FGF23 result in hypophosphatemic rickets/osteomalacia, which is characterized by renal Pi wasting, hypophosphatemia, inappropriately normal or low levels of serum 1,25(OH)2D (4). FGF23-related hypophosphatemic diseases include various genetic conditions, such as X-linked hypophosphatemic rickets (XLH, MIM #307800), autosomal dominant hypophosphatemic rickets (ADHR, MIM #193100), autosomal recessive hypophosphatemic rickets 1 (ARHR1, MIM #241520), ARHR2 (MIM #613312), and Raine syndrome (RNS, MIM #259775) (4).
Osteocytes are dendritic-shaped cells that terminally differentiate from a subpopulation of matrix-producing osteoblasts (5). They reside in lacunae, small cavities surrounded by a mineralized bone matrix, and communicate with other osteocytes and cells on bone surface including osteoblasts, by their long processes in canaliculi. Although research on their function has been hindered because of their location and inaccessibility, scientific advances in the past few decades have revealed that osteocytes play critical roles in the bone homeostasis and the regulation of Pi metabolism (5). The identification and characterization of the human osteocytic genes responsible for rare skeletal diseases have contributed to a more detailed understanding of osteocyte function. SOST was identified as the causative gene for sclerosteosis 1 (MIM #269500) and van Buchem disease (MIM #239100), which are characterized by a high bone mass, indicating that osteocytes regulate bone mass by producing sclerostin (6, 7). In addition to FGF23, several causative genes for hereditary hypophosphatemic rickets are highly expressed in osteocytes. Among them are the phosphate-regulating gene homologous to endopeptidase on X chromosome (PHEX), dentin matrix protein 1 (DMP1), and family with sequence similarity 20, member C (FAM20C). Inactivating mutations in these genes cause hypophosphatemia and impaired skeletal mineralization by increasing the production of FGF23 in osteocytes (8).
This article aims to summarize and discuss the current knowledge on the roles of osteocytes in the pathogenesis of hypophosphatemic rickets.
Pi homeostasis is mainly maintained by the balance between its absorption in the intestines, excretion from the kidneys, and deposition in and mobilization from bone and involves endocrine factors, such as 1,25(OH)2D, parathyroid hormone (PTH), and FGF23 (1). 1,25(OH)2D increases intestinal Pi absorption by up-regulating the expression of type IIb Na+/Pi co-transporter NaPi-IIb (1, 9). PTH increases Pi excretion by reducing the abundance of NaPi-IIa and NaPi-IIc in the brush border membrane of proximal tubules (1, 9). FGF23, whose main source are the osteocytes in bone, down-regulates the renal expression of NaPi-IIa and NaPi-IIc to increase Pi excretion and lower serum Pi levels. FGF23 also decreases the production of 1,25(OH)2D, which reduces intestinal Pi absorption and further lowers serum Pi levels (3).
Bioactive intact FGF23 is cleaved to be inactivated between Arg179 and Ser180 by a subtilisin-like proprotein convertase (10). The O-glycosylation of Thr178 in FGF23 prevents the cleavage-mediated inactivation of FGF23 (11). Intact FGF23 assays utilize two antibodies against the N- and C-terminal segments of the cleavage site and capture only bioactive intact proteins (12). On the other hand, C-terminal assays use antibodies against the C-terminal segments of the cleavage site, and detect both the intact protein and cleaved C-terminal fragments of FGF23 (13). FGF23 at physiological concentrations requires αKlotho as a cofactor to exert signals through FGF receptors (FGFR) in its distant target organs (14, 15).
In the adult skeleton, osteocytes represent more than 90% of all bone cells (5). They control bone mass by producing sclerostin encoded by the SOST gene. Sclerostin inhibits Wnt/β-catenin signaling by binding to low-density lipoprotein receptor-related protein 5 (LRP5) and LRP6 (16). Wnt/β-catenin signaling plays important roles in bone homeostasis: the activation of this pathway facilitates the commitment of mesenchymal progenitor cells to the osteoblast lineage and promotes the proliferation and differentiation of committed osteoblasts (17, 18). In addition, this pathway suppresses the differentiation and activation of osteoclasts by reducing the expression ratio of receptor activator of nuclear factor κ B ligand (RANKL) to osteoprotegerin in osteoblast lineage cells (17). A sclerostin deficiency due to inactivating variants and a large deletion in the regulatory sequence of the SOST gene cause sclerosteosis 1 and van Buchem disease, respectively, which are characterized by osteosclerosis and a high bone mass (6, 7). Mechanical force and PTH suppress the expression of sclerostin/SOST in osteocytes, leading to enhanced Wnt/β-catenin signaling and an increased bone mass (19–22).
Inhibition of sclerostin provides a bone anabolic treatment for bone diseases with a low bone mass. The anti-sclerostin antibody romosozumab is widely used to treat patients with osteoporosis at a high risk of fracture (23). A randomized phase 2a trial on adult patients with moderate osteogenesis imperfecta demonstrated that treatment with the anti-sclerostin antibody BPS804 increased the areal bone mineral density of the lumber spine by stimulating bone formation and reducing bone resorption (24).
RANKL, which plays an essential role in osteoclastogenesis and the activation of osteoclasts, is expressed in osteocytes and osteoblasts. The conditional deletion of RANKL from osteocytes in mice resulted in a progressive osteopetrotic phenotype after birth (25, 26), and studies using several Cre drivers suggested that osteocyte-derived RANKL, rather than osteoblast-derived RANKL, plays a major role in bone remodeling (27). Osteocyte-derived RANKL is up-regulated by aging and may be involved in the aging-related loss of cortical bone (28). Therefore, osteocytes are essential in the regulation of both bone formation and resorption.
Disorders associated with the excessive effects of FGF23 manifest hypophosphatemic rickets/osteomalacia characterized by renal Pi wasting and inappropriately normal or low levels of serum 1,25(OH)2D, and are collectively called FGF23-related hypophosphatemic rickets/osteomalacia (4). This includes various conditions such as tumor-induced osteomalacia caused by the overproduction of FGF23 due to phosphaturic mesenchymal tumors, hypophosphatemia associated with the intravenous administration of iron preparations, and genetic disorders, such as ADHR, XLH, ARHR1, ARHR2, and RNS (4).
ADHR is caused by missense mutations at the Arg176 or Arg179 residues within the RXXR/S motif of FGF23, which is recognized by subtilisin-like proprotein convertase (29, 30). Although these mutations are expected to confer resistance to the cleavage-mediated inactivation of FGF23, ADHR shows incomplete penetrance (31). The delayed onset of ADHR generally manifests in females after puberty, and an iron deficiency has been suggested to drive increases in serum FGF23 levels and manifestation of the disease (32). The iron status modified the plasma levels of intact and C-terminal fragments of FGF23 differently in ADHR patients and healthy subjects (32). In animal studies, a low-iron diet caused hypophosphatemia in ADHR model mice carrying FGF23[p.R179Q] mutation, along with increased serum levels of both intact FGF23 and C-terminal fragments. On the other hand, wild-type mice on a low-iron diet showed normal intact FGF23 levels and normophosphatemia despite elevated C-terminal FGF23 levels (33). In vitro experiments using an iron chelator have suggested that an iron deficiency up-regulates the expression of Fgf23 in osteoblastic cells via the activation of hypoxia inducible factor 1α (33). These findings suggest that, in an iron deficiency, the wild-type FGF23 protein is excessively inactivated by proteolytic cleavage to maintain normal serum Pi levels, whereas the impaired cleavage of FGF23 with an ADHR mutation increases bioactive FGF23 levels and promotes hypophosphatemia. Therefore, sensing of the iron status by osteocytes appears to be a critical step for the onset of ADHR.
XLH is the most common form of hereditary hypophosphatemic rickets and is caused by inactivating mutations in the PHEX gene (34). XLH patients have elevated serum levels of intact FGF23, resulting in renal Pi wasting, hypophosphatemia, and low to inappropriately normal levels of serum 1,25(OH)2D (35). Pediatric patients may manifest rickets, bone deformities, and a short stature, while adult patients may show osteomalacia, fractures, pseudo-fractures, enthesopathies (pathological changes in the insertion of tendons, ligaments, and joint capsules), osteoarthritis, hearing loss, and hyperparathyroidism (35). To date, more than 1000 PHEX mutations have been identified to be responsible for XLH (https://www.rarediseasegenes.com/).
Similar to FGF23, PHEX is expressed in osteoblast lineage cells with higher expression in osteocytes (8). Hypomineralized periosteocytic lesions are unique features in the bones of XLH patients, indicating abnormal osteocyte functions (36). Hypophosphatemic Hyp mice, which carry a large deletion in the Phex gene, reproduce elevated FGF23 levels and hypophosphatemia and are widely used as a murine model for XLH (37, 38). The conditional ablation of the Phex gene from osteoblasts and osteocytes using Cre recombinase driven by the Osteocalcin promoter resulted in an almost identical phenotype to that of Hyp mice, suggesting that impaired PHEX functions in osteoblasts and/or osteocytes alone are sufficient to cause the disease (39).
The PHEX protein is suggested to function as a cell surface-bound, zinc-dependent protease based on its structure (40): however, its precise physiological roles and the mechanisms by which its deficiency increases FGF23 levels remain elusive. Although there is no clear relationship between the PHEX genotype and clinical phenotype (41), a recent study using 3-dimensional modeling has suggested that serum FGF23 levels are higher in patients whose PHEX mutations affect the zinc-binding site or the cavity of the protein (42). Thus, these regions may be important for PHEX function to regulate FGF23.
Matrix extracellular phosphoglycoprotein (MEPE) belongs to the SIBLING (small integrin-binding ligand, N-linked glycoproteins) family. PHEX binds to MEPE via the acidic serine-aspartate rich MEPE-associated motif (ASARM) located at the C-terminal region of MEPE, immediately downstream of cathepsin B recognition site (43). ASARM peptides produced by proteolytic cleavage of MEPE inhibit mineralization (44). Serum levels of ASARM peptides are increased in Hyp mice, implicating that they are degraded by PHEX and that their accumulation might be involved in the defective skeletal mineralization in PHEX deficiency (45).
The ASARM motif is shared in other members of SIBLINGs family, such as osteopontin (OPN) and DMP1 (46). Synthesized OPN-ASARM peptides inhibited in vitro mineralization of osteoblasts by binding to hydroxyapatite, and addition of recombinant PHEX protein rescued this inhibition of mineralization (46). OPN is highly expressed in osteoblasts and osteocytes and can be a candidate of protein substrate for PHEX (47). A recent study demonstrated that the genetic ablation of OPN in Hyp mice partially rescued impaired skeletal mineralization without correcting hypophosphatemia (48), suggesting the involvement of OPN in the pathogenesis of XLH.
FGFR signaling is enhanced in Hyp bone (49). Comparisons of gene expression in osteocytes between Hyp mice and wild-type mice revealed marked increases in the osteocytic expression of canonical FGF ligands (Fgf1 and Fgf2), FGF receptors (Fgfr1-3), and early growth response 1, a downstream target for FGFR activation, in Hyp mice (8). Furthermore, the osteocyte-specific deletion of Fgfr1 partially restored the overproduction of FGF23 and ameliorated hypophosphatemia and rickets in Hyp mice (50). These findings suggest a pathogenic role for enhanced FGFR signaling in the overproduction of FGF23 in Phex-deficient osteocytes. This is of interest because previous studies suggested the involvement of FGFR in Pi sensing (1). In various cell types including osteoblasts, a treatment with high Pi activates FGFR to regulate gene expression (51–53). In the osteoblastic cell line MC3T3-E1, a treatment with an FGFR inhibitor abolished the up-regulation of Dmp1 by increasing extracellular Pi levels (53). In mice, the Pi-induced activation of FGFR1 up-regulated the Galnt3 expression in bone, resulting in an elevation of serum FGF23 levels (54). Enhanced FGFR signaling in Hyp osteocytes suggests that dysregulated Pi sensing might underlie the pathogenesis of XLH. In Hyp osteocytes, the expression of Dmp1 and Fam20c was also markedly up-regulated, indicating complex abnormalities in Phex-deficient osteocytes (8). Figure 1 shows complex abnormalities in Hyp osteocytes.
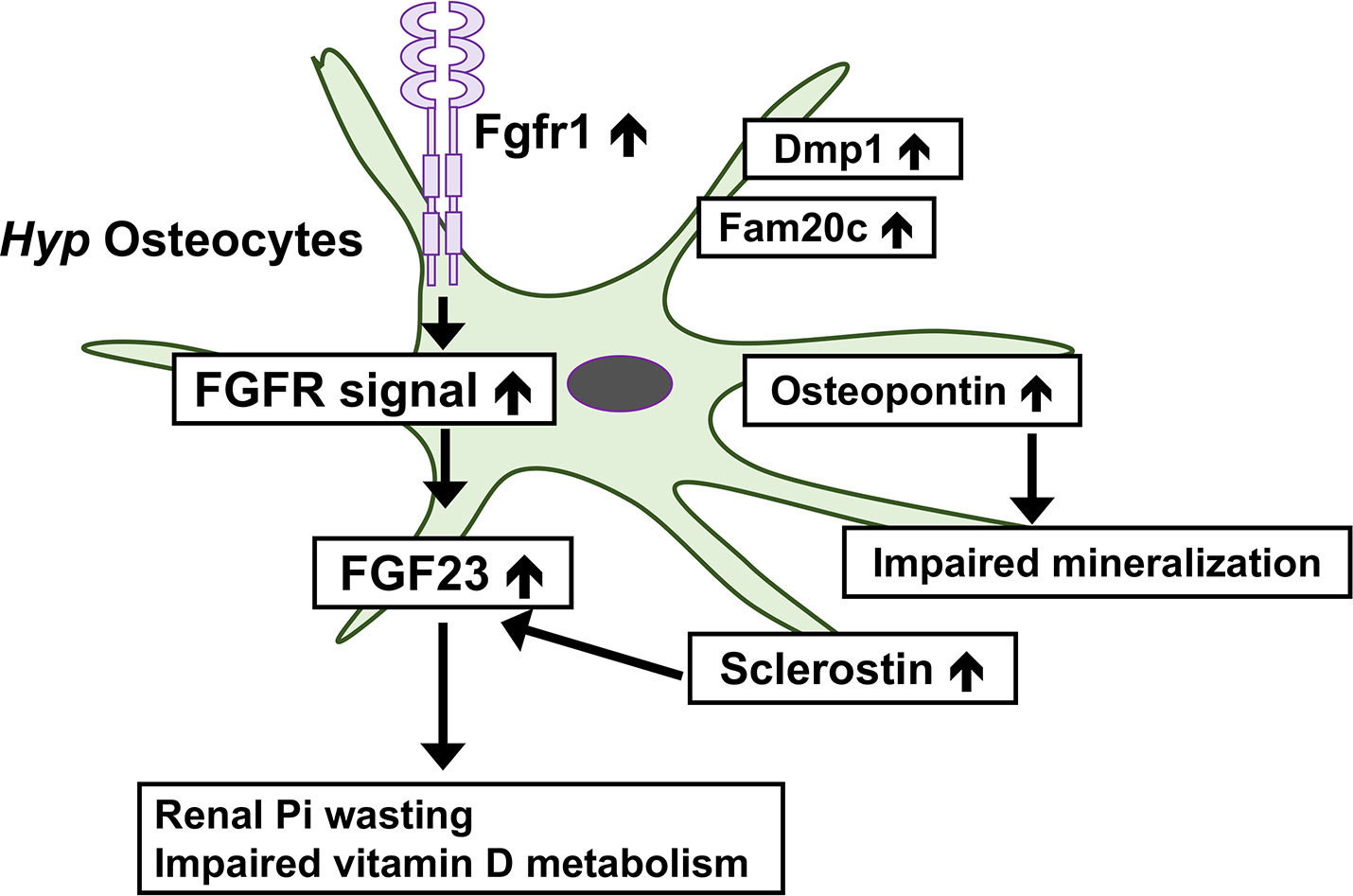
Figure 1 Complex abnormalities in osteocytes of Phex-deficient Hyp mice. In Hyp osteocytes, the expression of Fgfr1 is increased and enhanced FGFR signaling may underlie the overproduction of FGF23. The overproduction of FGF23 leads to the renal wasting of inorganic phosphate (Pi) and impaired vitamin D metabolism. Hyp osteocytes produce an increased amount of sclerostin, which may stimulate the production of FGF23. The production of osteopontin is up-regulated in Hyp osteocytes, which impairs skeletal mineralization. The expression of Dmp1 and Fam20c is also increased.
ARHR1 is caused by inactivating mutations in the DMP1 gene, which encodes an extracellular matrix protein belonging to the SIBLINGs family (55, 56). DMP1 is predominantly expressed in osteocytes and odontoblasts. Dmp1-null mice show phenotypes reproducing the clinical manifestation in ARHR1 and exhibit defective osteocyte maturation, increased FGF23 expression in osteocytes, and impaired skeletal mineralization (56). Similar to Phex-deficient Hyp mice, the activation of the FGFR pathway was suggested to be involved in the increased production of FGF23 in Dmp1-null mice (49). The overproduction of FGF23 may be attributed to enhanced FGFR signaling in osteocytes in both XLH and ARHR1.
It was reported that CRISPR/Cas9-mediated ablation of DMP1 in rabbit caused the phenotype similar to human ARHR1, including the elevated serum FGF23 and hypophosphatemic rickets. DMP1-knockout rabbits also exhibited severe defects in bone microarchitecture (57).
ARHR2 is caused by inactivating mutations in the ectonucleotide pyrophosphatase phosphodiesterase-1 (ENPP1) gene, which encodes an ectoenzyme that catalyzes the hydrolysis of ATP to AMP and inorganic pyrophosphate (PPi) (58–60). PPi inhibits mineralization, and inactivating mutations in ENPP1 cause disorders characterized by ectopic calcification, such as generalized arterial calcification of infancy (60, 61). ENPP1 is expressed in many tissues, and is highly expressed in chondrocytes, osteoblasts, and vascular smooth muscle cells (62). The mechanisms by which an ENPP1 deficiency leads to the overproduction of FGF23 currently remain elusive. PPi may regulate the FGF23 production: however, patients with hypophosphatasia, the disease caused by inactivating mutations in tissue non-specific alkaline phosphatase, have normal FGF23 levels along with elevated PPi levels (63). A recent mouse study demonstrated the increased expression of Fgf23 in the bones of Enpp1-deficient mice, and a negative correlation between Enpp1 and Fgf23 transcription was revealed by dosing Enpp1-deficient mice with ENPP1-Fc recombinant protein (64). These findings suggest that the lack of osteocyte-derived Enpp1 plays a role in the pathogenesis of ARHR2.
RNS is a disease of autosomal recessive inheritance, and the patients show craniofacial malformation, osteosclerotic bone dysplasia, and a poor prognosis. RNS patients who survive infancy may manifest hypophosphatemia related to FGF23 excess and dental abnormalities (65, 66). The gene responsible for RNS is FAM20C (also called DMP4), which encodes a kinase that phosphorylates various secreted proteins including FGF23 and members of the SIBLING family, such as DMP1 (10, 67, 68). The expression of Fam20c was higher in osteocytes than in osteoblasts, which was similar to the expression pattern of Fgf23, Dmp1 and Phex (8). Wang, et al. generated Fam20c conditional knockout mice, in which the gene was globally deleted or specifically inactivated in the mineralized tissues (69). Both mouse lines exhibited hypophosphatemic rickets along with elevated FGF23 levels in serum and bone. They also demonstrated the down-regulation of a number of marker genes in osteoblast/osteocyte lineage cells, which included Dmp1 and osteocalcin (69). Furthermore, knockout of Fam20c in immortalized murine osteoblasts resulted in the up-regulation of Fgf23 and down-regulation of Dmp1 (70). Thus, Fam20c-deficiency may lead to the overproduction of FGF23 in bone through thedown-regulation of the Dmp1 expression and impaired phosphorylation of Dmp1 protein. However, a transgenic overexpression of Dmp1 failed to rescue elevated FGF23 levels and hypophosphatemia of Fam20c knockout mice (71), suggesting the involvement of other mechanisms. Since it was reported that FAM20C phosphorylated FGF23 on Ser180, which prevented the O-glycosylation of FGF23 on Thr178 and accelerated cleavage-mediated inactivation (10),the impaired cleavage of FGF23 due to the reduced phosphorylation of FGF23 on Ser180 may be one of the pathogenesis of elevated intact FGF23 in RNS.
As conventional medical therapy for FGF23-related hypophosphatemic rickets/osteomalacia, oral Pi salts and active vitamin D analogues have been administered to correct hypophosphatemia and reduced levels of 1,25(OH)2D (35). A previous study reported that the initiation of conventional therapy in early infancy improved the growth and biochemical and radiological outcomes of pediatric patients with XLH; however, it failed to completely normalize skeletal development (72). Furthermore, conventional therapy did not prevent or attenuate enthesopathies or hearing loss in adult patients (35). Moreover, the administration of oral Pi salts and active vitamin D may cause adverse effects, such as hypercalciuria, nephrocalcinosis, and secondary/tertiary hyperparathyroidism (35).
Burosumab, a humanized anti-FGF23 monoclonal antibody, has been approved since 2018 for the treatment of XLH in several countries based on the promising findings of the clinical trials (73–76). The approved indication for burosumab differs among regions/countries; it is approved for patients with TIO in several countries. In Japan, it is approved for all patients with FGF23-related hypophosphatemic rickets/osteomalacia (4).
Since FGF23 and sclerostin are both expressed in osteocytes, it is reasonable to hypothesize that they may be related. Serum sclerostin levels were reported to be higher in XLH patients than in healthy controls (77, 78). An in vitro study suggested that sclerostin directly stimulated the production of FGF23 (79). The treatment of Hyp mice with an anti-sclerostin antibody suppressed serum intact FGF23 levels and increased serum Pi levels and bone mass (80, 81), suggesting the potential of inhibiting sclerostin as a treatment option for XLH.
In addition to their profound roles in bone homeostasis, osteocytes embedded in the bone matrix play a central role in Pi metabolism as the main source of FGF23. Osteocytes also express PHEX, DMP1, ENPP1, and FAM20C, and inactivating mutations cause FGF23-related hypophosphatemic rickets/osteomalacia. Although the mechanisms by which inactivating mutations in these genes cause the overproduction of FGF23 have not yet been clarified, enhanced FGFR signaling and abnormal Pi sensing might be involved in the pathogenesis of XLH and ARHR1. Serum sclerostin levels are elevated in XLH patients, indicating complex abnormalities in osteocytes. Further studies are needed to elucidate the precise roles of osteocytes in the pathogenesis of hereditary hypophosphatemic diseases, which will contribute to the development of better treatments for these conditions.
MY and TM developed the concept and prepared the manuscript. All authors contributed to the article and approved the submitted version.
Preparation of the manuscript was supported by a grant from Japan Society for the Promotion of Science (JSPS KAKENHI Grant Number 21K07835) to TM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Michigami T, Kawai M, Yamazaki M, Ozono K. Phosphate as a signaling molecule and its sensing mechanism. Physiol Rev (2018) 98(4):2317–48. doi: 10.1152/physrev.00022.2017
2. Michigami T, Ozono K. Roles of phosphate in skeleton. Front Endocrinol (Lausanne). (2019) 10:180. doi: 10.3389/fendo.2019.00180
3. Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin d metabolism. J Clin Invest. (2004) 113(4):561–8. doi: 10.1172/JCI200419081
4. Fukumoto S. FGF23-related hypophosphatemic rickets/osteomalacia: diagnosis and new treatment. J Mol Endocrinol (2021) 66(2):R57–65. doi: 10.1530/JME-20-0089
5. Delgado-Calle J, Bellido T. The osteocyte as a signaling cell. Physiol Rev (2022) 102(1):379–410. doi: 10.1152/physrev.00043.2020
6. Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet (2001) 68(3):577–89. doi: 10.1086/318811
7. Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van buchem disease. J Med Genet (2002) 39(2):91–7. doi: 10.1136/jmg.39.2.91
8. Miyagawa K, Yamazaki M, Kawai M, Nishino J, Koshimizu T, Ohata Y, et al. Dysregulated gene expression in the primary osteoblasts and osteocytes isolated from hypophosphatemic hyp mice. PloS One (2014) 9(4):e93840. doi: 10.1371/journal.pone.0093840
9. Wagner CA, Hernando N, Forster IC, Biber J. The SLC34 family of sodium-dependent phosphate transporters. Pflugers Arch (2014) 466(1):139–53. doi: 10.1007/s00424-013-1418-6
10. Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A. (2014) 111(15):5520–5. doi: 10.1073/pnas.1402218111
11. Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B, Bennett EP, et al. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem (2006) 281(27):18370–7. doi: 10.1074/jbc.M602469200
12. Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab (2002) 87(11):4957–60. doi: 10.1210/jc.2002-021105
13. Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med (2003) 348(17):1656–63. doi: 10.1056/NEJMoa020881
14. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature (2006) 444(7120):770–4. doi: 10.1038/nature05315
15. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem (2006) 281(10):6120–3. doi: 10.1074/jbc.C500457200
16. Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, et al. Sclerostin binds to LRP5/6 and antagonizes canonical wnt signaling. J Biol Chem (2005) 280(20):19883–7. doi: 10.1074/jbc.M413274200
17. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med (2013) 19(2):179–92. doi: 10.1038/nm.3074
18. Choi RB, Robling AG. The wnt pathway: An important control mechanism in bone’s response to mechanical loading. Bone (2021) 153:116087. doi: 10.1016/j.bone.2021.116087
19. Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of sost/sclerostin. J Biol Chem (2008) 283(9):5866–75. doi: 10.1074/jbc.M705092200
20. Sasaki F, Hayashi M, Mouri Y, Nakamura S, Adachi T, Nakashima T. Mechanotransduction via the Piezo1-akt pathway underlies sost suppression in osteocytes. Biochem Biophys Res Commun (2020) 521(3):806–13. doi: 10.1016/j.bbrc.2019.10.174
21. Leupin O, Kramer I, Collette NM, Loots GG, Natt F, Kneissel M, et al. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res (2007) 22(12):1957–67. doi: 10.1359/jbmr.070804
22. Wein MN, Liang Y, Goransson O, Sundberg TB, Wang J, Williams EA, et al. SIKs control osteocyte responses to parathyroid hormone. Nat Commun (2016) 7:13176. doi: 10.1038/ncomms13176
23. Singh S, Dutta S, Khasbage S, Kumar T, Sachin J, Sharma J, et al. A systematic review and meta-analysis of efficacy and safety of romosozumab in postmenopausal osteoporosis. Osteoporos Int (2022) 33(1):1–12. doi: 10.1007/s00198-021-06095-y
24. Glorieux FH, Devogelaer JP, Durigova M, Goemaere S, Hemsley S, Jakob F, et al. BPS804 anti-sclerostin antibody in adults with moderate osteogenesis imperfecta: Results of a randomized phase 2a trial. J Bone Miner Res (2017) 32(7):1496–504. doi: 10.1002/jbmr.3143
25. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med (2011) 17(10):1231–4. doi: 10.1038/nm.2452
26. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med (2011) 17(10):1235–41. doi: 10.1038/nm.2448
27. Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, et al. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PloS One (2015) 10(9):e0138189. doi: 10.1371/journal.pone.0138189
28. Kim HN, Xiong J, MacLeod RS, Iyer S, Fujiwara Y, Cawley KM, et al. Osteocyte RANKL is required for cortical bone loss with age and is induced by senescence. JCI Insight (2020) 5(19):e138815. doi: 10.1172/jci.insight.138815
29. ADHR-CONSORTIUM. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet (2000) 26(3):345–8. doi: 10.1038/81664
30. White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int (2001) 60(6):2079–86. doi: 10.1046/j.1523-1755.2001.00064.x
31. Imel EA, Hui SL, Econs MJ. FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. J Bone Miner Res (2007) 22(4):520–6. doi: 10.1359/jbmr.070107
32. Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab (2011) 96(11):3541–9. doi: 10.1210/jc.2011-1239
33. Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci U S A. (2011) 108(46):E1146–55. doi: 10.1073/pnas.1110905108
34. Francis F, Henning S, Korn B, Reinhardt R, de Jong P, Poustka A, et al. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. HYP Consortium. Nat Genet (1995) 11(2):130–6. doi: 10.1038/ng1095-130
35. Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. (2019) 15(7):435–55. doi: 10.1038/s41581-019-0152-5
36. Marie PJ, Glorieux FH. Relation between hypomineralized periosteocytic lesions and bone mineralization in vitamin d-resistant rickets. Calcif Tissue Int (1983) 35(4-5):443–8. doi: 10.1007/BF02405074
37. Beck L, Soumounou Y, Martel J, Krishnamurthy G, Gauthier C, Goodyer CG, et al. Pex/PEX tissue distribution and evidence for a deletion in the 3’ region of the pex gene in X-linked hypophosphatemic mice. J Clin Invest. (1997) 99(6):1200–9. doi: 10.1172/JCI119276
38. Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in hyp mice. Am J Physiol Endocrinol Metab (2006) 291(1):E38–49. doi: 10.1152/ajpendo.00008.2006
39. Yuan B, Takaiwa M, Clemens TL, Feng JQ, Kumar R, Rowe PS, et al. Aberrant phex function in osteoblasts and osteocytes alone underlies murine X-linked hypophosphatemia. J Clin Invest. (2008) 118(2):722–34. doi: 10.1172/JCI32702
40. Drezner MK. PHEX gene and hypophosphatemia. Kidney Int (2000) 57(1):9–18. doi: 10.1046/j.1523-1755.2000.00807.x
41. Park PG, Lim SH, Lee H, Ahn YH, Cheong HI, Kang HG. Genotype and phenotype analysis in X-linked hypophosphatemia. Front Pediatr (2021) 9:699767. doi: 10.3389/fped.2021.699767
42. Ishihara Y, Ohata Y, Takeyari S, Kitaoka T, Fujiwara M, Nakano Y, et al. Genotype-phenotype analysis, and assessment of the importance of the zinc-binding site in PHEX in Japanese patients with X-linked hypophosphatemic rickets using 3D structure modeling. Bone (2021) 153:116135. doi: 10.1016/j.bone.2021.116135
43. Rowe PS, Garrett IR, Schwarz PM, Carnes DL, Lafer EM, Mundy GR, et al. Surface plasmon resonance (SPR) confirms that MEPE binds to PHEX via the MEPE-ASARM motif: a model for impaired mineralization in X-linked rickets (HYP). Bone (2005) 36(1):33–46. doi: 10.1016/j.bone.2004.09.015
44. Addison WN, Nakano Y, Loisel T, Crine P, McKee MD. MEPE-ASARM peptides control extracellular matrix mineralization by binding to hydroxyapatite: an inhibition regulated by PHEX cleavage of ASARM. J Bone Miner Res (2008) 23(10):1638–49. doi: 10.1359/jbmr.080601
45. Bresler D, Bruder J, Mohnike K, Fraser WD, Rowe PS. Serum MEPE-ASARM-peptides are elevated in X-linked rickets (HYP): implications for phosphaturia and rickets. J Endocrinol (2004) 183(3):R1–9. doi: 10.1677/joe.1.05989
46. Addison WN, Masica DL, Gray JJ, McKee MD. Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J Bone Miner Res (2010) 25(4):695–705. doi: 10.1359/jbmr.090832
47. Barros NM, Hoac B, Neves RL, Addison WN, Assis DM, Murshed M, et al. Proteolytic processing of osteopontin by PHEX and accumulation of osteopontin fragments in hyp mouse bone, the murine model of X-linked hypophosphatemia. J Bone Miner Res (2013) 28(3):688–99. doi: 10.1002/jbmr.1766
48. Hoac B, Ostergaard M, Wittig NK, Boukpessi T, Buss DJ, Chaussain C, et al. Genetic ablation of osteopontin in osteomalacic hyp mice partially rescues the deficient mineralization without correcting hypophosphatemia. J Bone Miner Res (2020) 35(10):2032–48. doi: 10.1002/jbmr.4101
49. Martin A, Liu S, David V, Li H, Karydis A, Feng JQ, et al. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J (2011) 25(8):2551–62. doi: 10.1096/fj.10-177816
50. Xiao Z, Huang J, Cao L, Liang Y, Han X, Quarles LD. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PloS One (2014) 9(8):e104154. doi: 10.1371/journal.pone.0104154
51. Yamazaki M, Ozono K, Okada T, Tachikawa K, Kondou H, Ohata Y, et al. Both FGF23 and extracellular phosphate activate Raf/MEK/ERK pathway via FGF receptors in HEK293 cells. J Cell Biochem (2010) 111(5):1210–21. doi: 10.1002/jcb.22842
52. Kimata M, Michigami T, Tachikawa K, Okada T, Koshimizu T, Yamazaki M, et al. Signaling of extracellular inorganic phosphate up-regulates cyclin D1 expression in proliferating chondrocytes via the Na+/Pi cotransporter pit-1 and Raf/MEK/ERK pathway. Bone (2010) 47(5):938–47. doi: 10.1016/j.bone.2010.08.006
53. Nishino J, Yamazaki M, Kawai M, Tachikawa K, Yamamoto K, Miyagawa K, et al. Extracellular phosphate induces the expression of dentin matrix protein 1 through the FGF receptor in osteoblasts. J Cell Biochem (2017) 118(5):1151–63. doi: 10.1002/jcb.25742
54. Takashi Y, Kosako H, Sawatsubashi S, Kinoshita Y, Ito N, Tsoumpra MK, et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc Natl Acad Sci U S A. (2019) 116(23):11418–27. doi: 10.1073/pnas.1815166116
55. Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet (2006) 38(11):1248–50. doi: 10.1038/ng1868
56. Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet (2006) 38(11):1310–5. doi: 10.1038/ng1905
57. Liu T, Wang J, Xie X, Wang K, Sui T, Liu D, et al. DMP1 ablation in the rabbit results in mineralization defects and abnormalities in haversian Canal/Osteon microarchitecture. J Bone Miner Res (2019) 34(6):1115–28. doi: 10.1002/jbmr.3683
58. Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet (2010) 86(2):267–72. doi: 10.1016/j.ajhg.2010.01.006
59. Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet (2010) 86(2):273–8. doi: 10.1016/j.ajhg.2010.01.010
60. Ralph D, Levine MA, Richard G, Morrow MM, Flynn EK, Uitto J, et al. Mutation update: Variants of the ENPP1 gene in pathologic calcification, hypophosphatemic rickets, and cutaneous hypopigmentation with punctate keratoderma. Hum Mutat (2022) 43(9):1183–200. doi: 10.1002/humu.24391
61. Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet (2003) 34(4):379–81. doi: 10.1038/ng1221
62. Mackenzie NC, Huesa C, Rutsch F, MacRae VE. New insights into NPP1 function: lessons from clinical and animal studies. Bone (2012) 51(5):961–8. doi: 10.1016/j.bone.2012.07.014
63. Linglart A, Biosse-Duplan M. Hypophosphatasia. Curr Osteoporos Rep (2016) 14(3):95–105. doi: 10.1007/s11914-016-0309-0
64. Maulding ND, Kavanagh D, Zimmerman K, Coppola G, Carpenter TO, Jue NK, et al. Genetic pathways disrupted by ENPP1 deficiency provide insight into mechanisms of osteoporosis, osteomalacia, and paradoxical mineralization. Bone (2021) 142:115656. doi: 10.1016/j.bone.2020.115656
65. Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, et al. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res (2013) 28(6):1378–85. doi: 10.1002/jbmr.1850
66. Takeyari S, Yamamoto T, Kinoshita Y, Fukumoto S, Glorieux FH, Michigami T, et al. Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of raine syndrome. Bone (2014) 67:56–62. doi: 10.1016/j.bone.2014.06.026
67. Tagliabracci VS, Engel JL, Wen J, Wiley SE, Worby CA, Kinch LN, et al. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science (2012) 336(6085):1150–3. doi: 10.1126/science.1217817
68. Palma-Lara I, Perez-Ramirez M, Garcia Alonso-Themann P, Espinosa-Garcia AM, Godinez-Aguilar R, Bonilla-Delgado J, et al. FAM20C overview: Classic and novel targets, pathogenic variants and raine syndrome phenotypes. Int J Mol Sci (2021) 22(15):8039. doi: 10.3390/ijms22158039
69. Wang X, Wang S, Li C, Gao T, Liu Y, Rangiani A, et al. Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PloS Genet (2012) 8(5):e1002708. doi: 10.1371/journal.pgen.1002708
70. Liu C, Zhang H, Jani P, Wang X, Lu Y, Li N, et al. FAM20C regulates osteoblast behaviors and intracellular signaling pathways in a cell-autonomous manner. J Cell Physiol (2018) 233(4):3476–86. doi: 10.1002/jcp.26200
71. Wang X, Wang J, Yuan B, Lu Y, Feng JQ, Qin C. Overexpression of Dmp1 fails to rescue the bone and dentin defects in Fam20C knockout mice. Connect Tissue Res (2014) 55(4):299–303. doi: 10.3109/03008207.2014.923414
72. Makitie O, Doria A, Kooh SW, Cole WG, Daneman A, Sochett E. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab (2003) 88(8):3591–7. doi: 10.1210/jc.2003-030036
73. Carpenter TO, Whyte MP, Imel EA, Boot AM, Hogler W, Linglart A, et al. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med (2018) 378(21):1987–98. doi: 10.1056/NEJMoa1714641
74. Whyte MP, Carpenter TO, Gottesman GS, Mao M, Skrinar A, San Martin J, et al. Efficacy and safety of burosumab in children aged 1-4 years with X-linked hypophosphataemia: a multicentre, open-label, phase 2 trial. Lancet Diabetes Endocrinol (2019) 7(3):189–99. doi: 10.1016/S2213-8587(18)30338-3
75. Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet (2019) 393(10189):2416–27. doi: 10.1016/S0140-6736(19)30654-3
76. Insogna KL, Briot K, Imel EA, Kamenicky P, Ruppe MD, Portale AA, et al. A randomized, double-blind, placebo-controlled, phase 3 trial evaluating the efficacy of burosumab, an anti-FGF23 antibody, in adults with X-linked hypophosphatemia: Week 24 primary analysis. J Bone Miner Res (2018) 33(8):1383–93. doi: 10.1002/jbmr.3475
77. Palomo T, Glorieux FH, Rauch F. Circulating sclerostin in children and young adults with heritable bone disorders. J Clin Endocrinol Metab (2014) 99(5):E920–5. doi: 10.1210/jc.2013-3852
78. Hansen S, Shanbhogue VV, Jorgensen NR, Beck-Nielsen SS. Elevated bone remodeling markers of CTX and P1NP in addition to sclerostin in patients with X-linked hypophosphatemia: A cross-sectional controlled study. Calcif Tissue Int (2019) 104(6):591–8. doi: 10.1007/s00223-019-00526-z
79. Ito N, Prideaux M, Wijenayaka AR, Yang D, Ormsby RT, Bonewald LF, et al. Sclerostin directly stimulates osteocyte synthesis of fibroblast growth factor-23. Calcif Tissue Int (2021) 109(1):66–76. doi: 10.1007/s00223-021-00823-6
80. Carpenter KA, Ross RD. Sclerostin antibody treatment increases bone mass and normalizes circulating phosphate levels in growing hyp mice. J Bone Miner Res (2020) 35(3):596–607. doi: 10.1002/jbmr.3923
Keywords: phosphate, osteocyte, fibroblast growth factor 23, rickets, mutation
Citation: Yamazaki M and Michigami T (2022) Osteocytes and the pathogenesis of hypophosphatemic rickets. Front. Endocrinol. 13:1005189. doi: 10.3389/fendo.2022.1005189
Received: 28 July 2022; Accepted: 15 September 2022;
Published: 29 September 2022.
Edited by:
Lothar Seefried, Julius Maximilian University of Würzburg, GermanyReviewed by:
Xiaofang Wang, Texas A&M University Baylor College of Dentistry, United StatesCopyright © 2022 Yamazaki and Michigami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshimi Michigami, bWljaGlnYW1pQHdjaC5vcGhvLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.