 Wensheng Che
Wensheng Che Ming Zhao
Ming Zhao Xiaoqing Li4
Xiaoqing Li4 William C. Cho
William C. Cho Shan Yu
Shan Yu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 29 November 2022
Sec. Gut Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1002916
This article is part of the Research Topic The NAFLD-MAFLD Conundrum View all 8 articles
There is a continuously rising incidence of non-alcoholic fatty liver disease (NAFLD) around the world, which parallels the increasing incidence of metabolic diseases. NAFLD is a range of liver conditions that contains simple non-alcoholic fatty liver and advanced non-alcoholic steatohepatitis. In serious cases, NAFLD may develop into cirrhosis or even liver cancer. NAFLD has an intense relationship with metabolic syndrome, type 2 diabetes mellitus. It is known that gut microbiota, and functional molecules such as adenosine monophosphate-activated protein kinase JNK, and peroxisome proliferator-activated receptors (PPARs) in progressing and treating NAFLD. Traditionally, the conventional and effective therapeutic strategy is lifestyle intervention. Nowadays, new medicines targeting specific molecules, such as farnesoid X receptor, PPARs, and GLP-1 receptor, have been discovered and shown beneficial effects on patients with NAFLD. In this article, we focus on the molecular mechanisms and therapeutic approaches to NAFLD.
Non-alcoholic fatty liver disease (NAFLD), defined by the National Institute of Diabetes and Digestive and Kidney Diseases, is a condition in which excess fat builds up in the liver, and it is not caused by heavy alcohol consumption. In patients with NAFLD, there is fat accumulation in the liver without inflammation or liver damage. Besides, it is characterized by hepatic steatosis and necroinflammation with different stages of fibrosis known as non-alcoholic steatohepatitis (NASH) (1). The incidence of NAFLD has risen rapidly in the last decade and has become a major health issue: NAFLD affects 25% of the adult population worldwide (2). In addition, NAFLD has seen a significant rise in liver-associated mortality and morbidity among liver-related diseases (3). Notably, NAFLD can occur in non-obese or even lean populations (10.8%) in Asia (4), and NAFLD is associated with extrahepatic diseases, such as cardiovascular disease (CVD) and type 2 diabetes mellitus (T2DM). NAFLD results in insulin resistance due to controlling lipid accumulation and mitochondrial function (5). In advanced stage of NAFLD, NASH is characterized by steatosis, inflammation, and liver damage, often accompanied by pericellular fibrosis (6). NASH elevates the incidence of cirrhosis, hepatic failure, and even hepatocellular carcinoma (HCC). Hyperglycemia and toxic lipids such as ceramides, diacylglycerol (DAG), free fatty acid (FFA), and cholesterol in hepatocytes may result in deleterious effects (glucolipotoxicity), which may change NAFLD from simple steatosis to NASH through mechanisms including cellular death, oxidative stress, and mitochondrial disorders (7).
A series of studies have reported that NAFLD is highly associated with metabolic syndrome (8). Metabolic syndrome (hyperglycemia, dyslipidemia, hypertension, and insulin resistance) is a crucial risk factor for NAFLD progression (9, 10). Metabolites of FFAs contribute to hepatocyte injuries such as endoplasmic reticulum (ER) stress, inflammation, apoptosis, and ballooning (11), which finally leads to NAFLD or even NASH (Figure 1). Obesity, with reference to body mass index (BMI), has been thought of as a key risk factor for NAFLD for decades. Interestingly, it has been reported that almost 40% of NAFLD patients do not have obesity (12), suggesting that BMI may not be considered a criterion for NAFLD diagnosis. A meta-analysis also showed a much weaker relationship between obesity and the incidence of severe liver diseases compared to other risk factors, such as insulin resistance and dyslipidemia (13). However, the incidence of metabolic syndromes is more prevalent in obese people (14), and NAFLD patients with obesity seem to have a poor prognosis (15). Obesity, or higher BMI, may contribute to NAFLD in a non-directive way, for example, through metabolic syndrome; this requires further in-depth study.
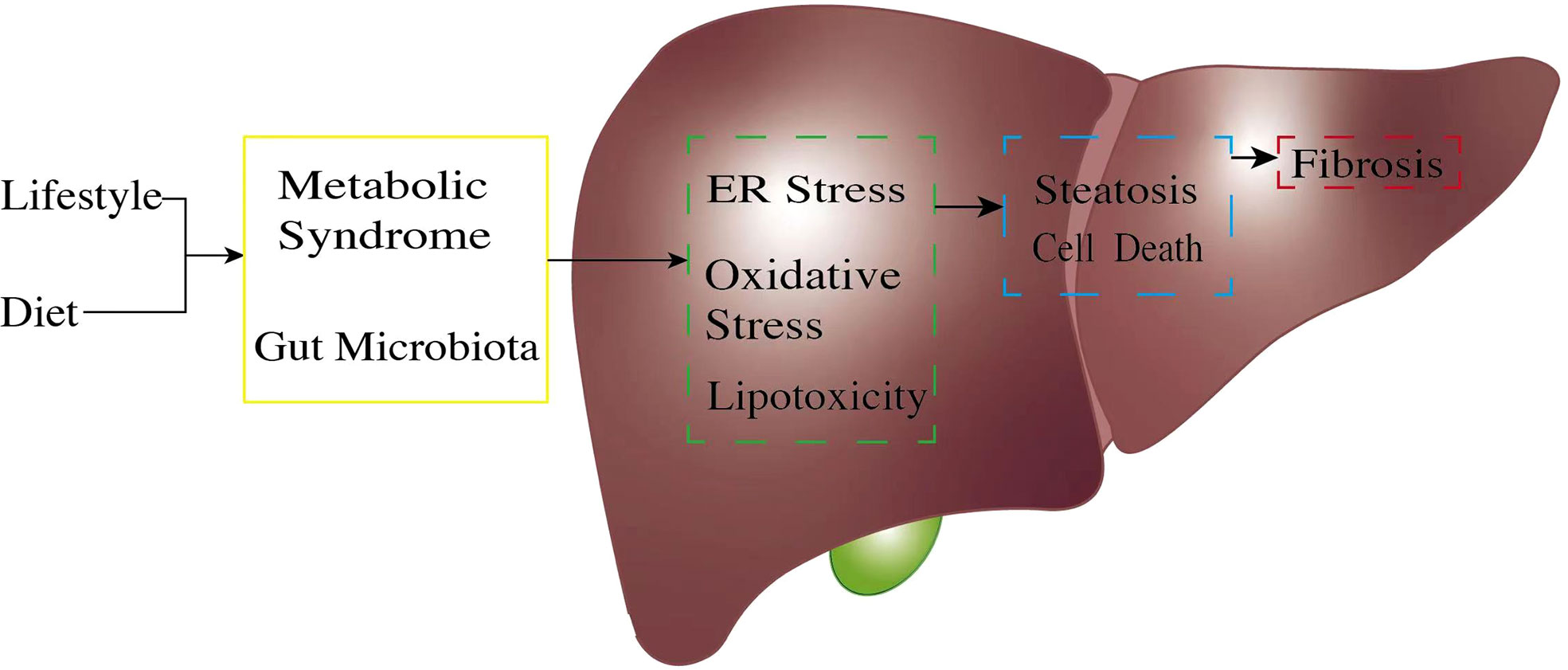
Figure 1 The pathogenic process of non-alcoholic fatty liver disease (NAFLD). The pathogenetic process of NAFLD always starts from unsatisfactory customs in daily life. An unhealthy lifestyle and poor diet, exceptionally high fructose, and a high-fat diet will trigger gut microbiota dysregulation. Unbalanced gut microbiota exposes liver cells to an endotoxic environment by producing short-chain fatty acids and secondary bile acids. At the same time, metabolic syndrome, through factors such as obesity and insulin resistance, causes excessive free fatty acid production because of unsatisfactory energy homeostasis. Once the body reaches its limit for lipid management, this will cause lipid accumulation in the liver cells; expose liver cells to high ER stress, oxidative stress, and lipotoxic condition (green box); and result in hepatocyte inflammation. These pro-inflammatory stimuli generate multiple pathways, leading to further hepatocyte injury (blue box) and continuous inflammation. Cell damage finally causes liver fibrosis (red box). ER, endoplasmic reticulum.
Because of the intense relationship between NAFLD and metabolic diseases, it has been suggested to re-classify fatty liver diseases by whether metabolic dysfunction coexists, naming it as “metabolic dysfunction-associated fatty liver disease (MAFLD)” (16). This classification is probably a better description of patients and improves clinical care and scientific research.
The gut microbiota is an “invisible organ” in the body and a key player in host metabolism and immune regulation (17). The gut microbiota benefits our bodies in several ways, not only contributing to absorbing nutrients and energy, but also producing some molecules, including short-chain fatty acids (SCFAs), secondary bile acids (BAs), and choline derivatives. These molecules can regulate host metabolism through multiple molecular mechanisms (18, 19).
The gut microbiota changes under conditions such as a high-fat diet (HFD), the use of antibiotics, or exposure to toxic substances (20). A recent report also confirmed a strong connection between gut microbiota and NAFLD in mice and humans (21).
NAFLD shows an alteration of the gut microbiota profile, and the occurrence of chronic liver disease is elevated by the overgrowth of extremely small intestinal bacteria (22). For example, the fecal microbiota transplantation (FMP) test demonstrated that the germ-free C57BL/6J mice that transplanted the gut microbiome of HFD mice displayed a higher possibility of steatosis than these transplanted bacteria from normal mice (23).
Industrialized nations are bearing witness to an obesity epidemic. An increasing number of patients with serious chronic diseases, such as NAFLD, lead to liver dysfunction and metabolic syndrome, which in turn cause T2DM and vascular complications (24).
In the past decades, the dietary composition in many countries has become focused on higher fat and lower carbohydrates from a traditional diet with a high percentage of carbohydrates and lower fat. The nutritional transition was highly associated with dramatically increased incidence of obesity, NAFLD, T2DM, and cardiovascular disease (25). A high-fat diet could promote NAFLD progression by causing gut microbiota dysregulation (26). A study tested whether a different percentage of fat intake changes the structure of the gut microbiota, decreases the proportion of Firmicutes, and increases Alistipes and Bacteroides. However, a low-fat diet showed contrasting results (27).
Oxidative stress is also strongly associated with NAFLD. Oxidative stress reflects the lack of balance between the reactive oxygen species (ROS) and the eliminating capacity of the antioxidant system (28).
Under physiological conditions, the amount of ROS is kept at a homeostatic status to promote physiological redox signaling. Normal physiological levels of ROS act as signaling molecules involved in cell metabolism, survival, immune defense, proliferation, and differentiation through the modulation of transcription factors and epigenetic pathways (29). However, once under oxidative stress, excessive ROS triggers pathological redox signaling, causing cellular injury in various diseases, significantly when cellular ROS production changes into further toxic ROS species such as HO• (30, 31). Even a little elevation of ROS will lead to cytotoxicity and oxidative stress in the cells. Overproduced ROS can lead to lipid peroxidation, reduced mitochondrial and peroxisomal oxidation of fatty acids, and cytokine release. Furthermore, increased ROS output and oxidative stress are considered underlying mechanisms of insulin resistance (32, 33). Reduction of nuclear factor E2-related factor 2 (Nrf2), a redox-sensitive transcription factor and the principal regulator of the redox balance, and upregulation of the NF-κB signaling pathway have been observed simultaneously with the presence of ROS (32). Also, it has been discovered that the downregulation of Nrf2 and upregulated NF-κB lead to insulin resistance (32). Oxidative stress is associated with insulin resistance, chronic inflammation, and hepatic fibrosis. In addition, it has been shown that the Nrf2 pathway is essential for mitochondrial and ER homeostasis due to its ability to mediate the expression of detoxifying enzymes, which may indicate the relationship between oxidative stress, ER stress, and mitochondrial dysfunction (34). In the condition of NAFLD, impaired redox status and ROS accumulation are the origins of hepatic fat accumulation, thereby leading to hepatic metabolic impairment and NASH progression. Thus, maintaining cellular redox homeostasis is considered one of the therapeutic strategies for NASH (35).
Inflammation is a process responding to injury or infection, which leads to the secretion of various inflammatory factors, such as cytokines, chemokines, and eicosanoids. Liver inflammatory response is an important factor in NAFLD occurrence and progression. The persistence of inflammatory activity over time results in chronic inflammatory changes that exacerbate tissue injury and result in an abnormal response, which in NAFLD develops into NASH and liver fibrosis (36).
Liver inflammation in NAFLD can be triggered by extrahepatic (e.g., adipose tissue) and/or intrahepatic (e.g., lipotoxicity, oxidative stress, and cell death) factors (36). Studies showed that when under a high-fat diet or upon FFA treatment, Kupffer cells are activated by steatotic hepatocyte released extracellular vesicle and direct effect of FFA, producing a high amount of pro-inflammatory cytokines such as TNF-α and IFN (37). Notably, other cells are also known as important mediators of inflammation and NAFLD progression. NASH is characterized by B-cell and T-cell infiltration of the liver. T cells could potentially stimulate hepatic macrophages regulated through releasing of cytokines, and alterations in regulatory T cell and hepatic dendritic cell homeostasis can trigger immune responses that drive the progression of NASH (38). This evidence supports inflammation being a key pathophysiological mechanism and a target for therapeutic intervention.
In a family cohort study, the risk of advanced fibrosis in the NAFLD group was 12 times more than in the control group (39). Some single-nucleotide polymorphisms (SNPs), including rs738409 c.444 C>G p.I148, rs58542926 c.499 G>A p.E167K, rs1260326 c.1337 C>T p.P446L, rs641738 g.54173068 C>T/c.50 G>A p.G17E, and rs62305723 c.778 C>T p.P260S, have been discovered as being robustly associated with the pathogenesis of NAFLD (40), and these genetic variants showed moderate-to-large effect sizes in glucose and lipid homeostasis pathways for the development of NAFLD (41). The rs738409 c.444 C>G p.I148 is considered the most common genetic predisposition in NASH. One of the major findings is that the PNPLA3 I148M variant increases susceptibility to the whole spectrum of liver damage related to NAFLD, from steatosis to NASH, fibrosis, and HCC (42). Carrying this variant increases the risk of liver-associated mortality (43). These findings represent that genetic factor is probably an important risk factor for the development of NAFLD. These genes also have an association with therapeutic approaches. For example, PNPLA3 I148M carriers showed higher effectiveness through lifestyle intervention and seemed to receive less benefit from omega-3 supplements than certain other SNPs (44–46).
Recently, targeting these genes to treat NAFLD has come under the spotlight. Targeting PNPLA3 (p.I148M) at RNA levels through small hairpin RNAs (shRNAs) or antisense oligonucleotides (ASOs) could provide long-lasting suppression of the risk variant expression in carriers. PNPLA3 silencing caused a significant reduction of liver steatosis, inflammation, and fibrosis in mice fed with a NASH-inducing diet (47). An ASO compound called ION839 (also known as AZD2693) is currently under investigation in phase I trials (NCT04142424 and NCT04483947).
Degradation of gene production also could be a viable therapeutic intervention. Momelotinib, previously identified to treat myeloproliferative neoplasm, showed downregulation of PNPLA3 mRNA, representing a new and effective therapeutic approach for NASH (48).
The identification of the relationship between genes and the development of NAFLD may explore new therapeutic options, and the future genome-wide studies may reveal additional mechanisms.
Adenosine monophosphate-activated protein kinase (AMPK) is a well-known energy-sensing kinase that manages the energy balance. Therefore, it is necessary for cell survival with marginal energy supplements (49–51). The AMPK activity is allosterically regulated by AMP/ATP ratio, with activation during nutrition shortage and inactivation under obesity conditions, hyperglycemia (52–54), and hyperinsulinemia (55, 56).
AMPK is activated through phosphorylation on the NH2-terminus (Thr172) (57), which is mediated by its upstream kinase liver kinase B1 (LKB1) or the AMP binding with γ-unit allosterically (58, 59), as well as exercise directly. The AMPK activation not only reduces the AMP/ATP ratio but also induces lipolysis and lipid oxidation to consume whole-body energy (60, 61). Therefore, AMPK activation could benefit NAFLD. AMPK facilitates a variety of biological processes, including oxidative phosphorylation, autophagy, uptake, and utilization of glucose and FFA while reducing anabolism, such as protein synthesis by inhibiting mTOR signaling, cholesterol by inhibiting HMA-CoA reductase, and fatty acids by phosphorylating acetyl CoA carboxylase (ACC) (49, 62–66). These processes are closely correlated with hepatic lipid homeostasis and the pathogenesis of NAFLD. AMPK is dysregulated in obese humans. Decreased AMPK activity in adipose tissue is connected with whole-body insulin resistance, suggesting that AMPK activity in adipose tissue may be necessary for NAFLD (67, 68). In a healthy population, de novo lipogenesis (DNL) contributes less than 5% to liver triglyceride (TG) content, while it contributes up to 26% of liver TG in humans with NAFLD (69, 70). Researchers have discovered that AMPK phosphorylates and inactivates ACC to downregulate the rate-limiting step of DNL (50, 71–73). A-769662, an AMPK agonist, showed an abolishment effect in NAFLD, which restored hepatic fatty acid oxidation and ameliorated liver lipid accumulation (74). These studies indicate that AMPK may be a key player in the pathogenesis of NAFLD (Figure 2).
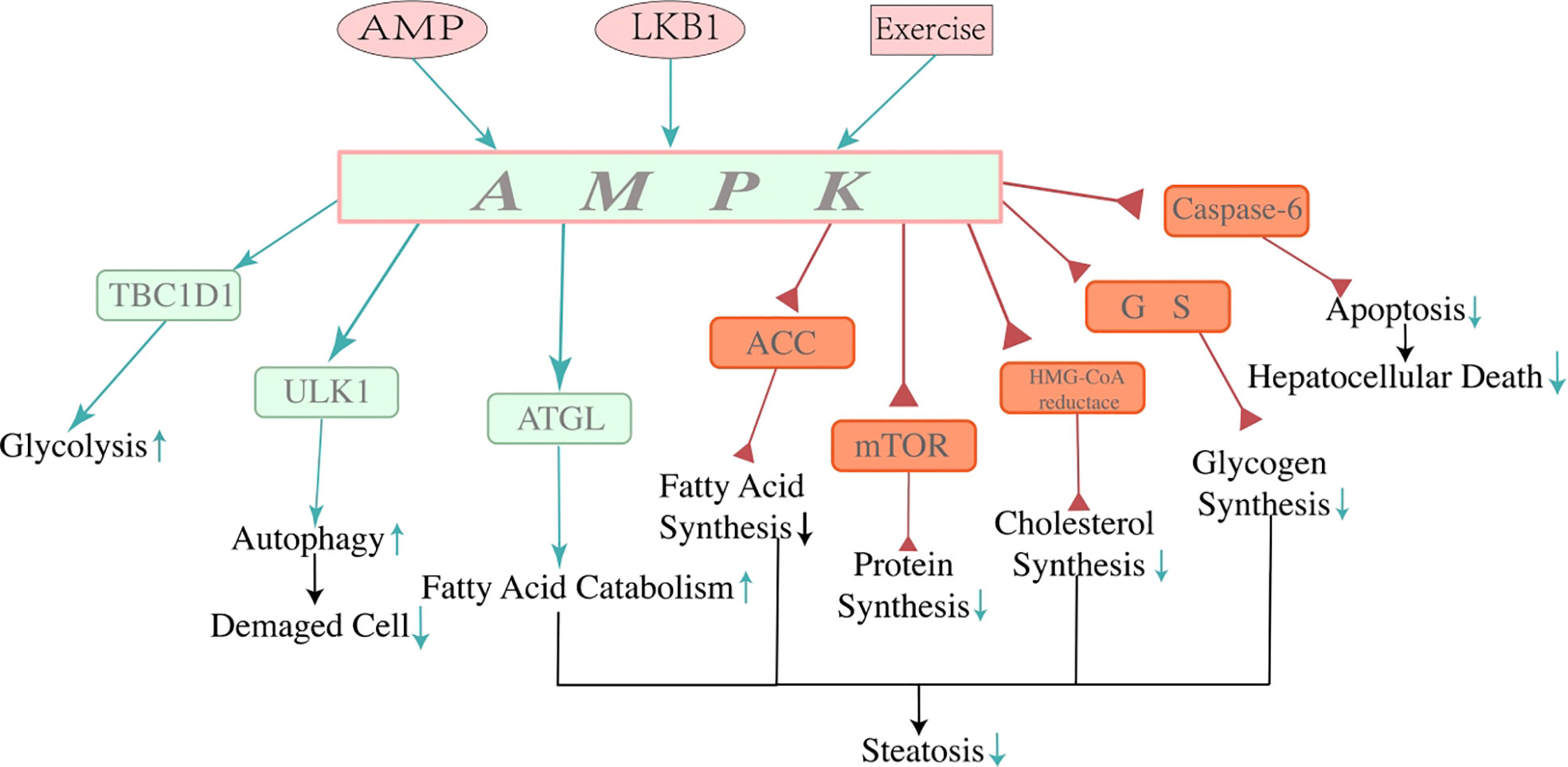
Figure 2 Mechanisms of adenosine 5′-monophosphate-activated protein kinase. AMPK is a crucial controller of energy metabolism. AMP, LKB1, and exercise can activate AMPK directly or indirectly. AMPK regulates protein, fat, and carbohydrate metabolism. Activation of AMPK restrains lipogenesis, promoting fatty acid catabolism in the liver through multiple mechanisms of energy regulation, thus decreasing hepatocyte steatosis and contributing to the consumption of the energy of the whole body. Due to its function in energy regulation, activation of AMPK reduces lipid accumulation by reducing lipid synthesis and stimulating lipid catabolism, which alleviated hepatocyte steatosis and showed a beneficial effect on NAFLD. In addition, the function of AMPK reducing hepatocyte apoptosis by inhibiting activation of caspase-6 has been found recently, which may indicate a new approach to improve NAFLD. Three pink icons at the top of the image are approaches to activating AMPK. The green arrows represent the positive effect, and the red triangles represent the negative effect. NAFLD, non-alcoholic fatty liver disease.
The activity of AMPK in hepatocytes is reduced within NAFLD (74). A recent study showed that the downregulation of AMPK in hepatocytes results in more hepatocellular death and fibrosis due to the upregulation of caspase-6 (75). AMPK is phosphorylated by AMPK-related protein kinase 5 (ARK5) and thus prevents hepatocytes from apoptosis by inhibiting the cleavage of procaspase-6 (76, 77). Inversely, when the activity of AMPK is reduced due to overnutrition, hyperglycemia, diabetes, and NAFLD, caspase-6 would gain functions to sustain the caspase cascade. The cascade activates caspase-3 and caspase-7, further leading hepatocytes to caspase-mediated apoptosis (78). Thus, AMPK not only is a crucial energy-sensing protein, but that it may also participate in cell apoptosis regulation in NAFLD/NASH.
c-Jun N-terminal kinase (JNK) is a member of the mitogen-activated protein kinase (MAPK) family and is capable of promoting apoptosis via Bcl-2 interacting mediator of cell death (Bim), p53 upregulated modulator of apoptosis (PUMA), and members of pro-apoptotic Bcl-2 family (79). JNK is a vital mediator of insulin resistance and FA-induced lipotoxicity (80), and its activation mediates apoptosis in hepatocytes (81). It has been reported that the ASK-1–JNK axis induced TNF-α related apoptosis in steatotic hepatocytes from a mouse model of NAFLD (56). The prolonged activation of JNK could be alleviated by NF-κB-mediated upregulation of anti-apoptotic genes, such as Bcl-xl and cFLIP (82), which may be a therapeutic strategy for NASH. In conclusion, JNK is essential for regulating apoptosis in NAFLD and NASH. Interestingly, JNK also contributes to protective responses such as hepatocyte proliferation and liver regeneration (83).
Peroxisome proliferator-activated receptors (PPARs) belong to the nuclear receptor superfamily, which is considered a fatty acid sensor, thus modulating carbohydrate metabolism and energy usage (Figure 3). PPARs are also reported as regulators of NAFLD development and treatment (84–86). PPARα, an isoform of PPARs, is negatively correlated with the severity of NASH (87), which may be linked with metabolism modulation, such as fatty acid metabolism, ketogenesis, and β-oxidation (88, 89). In addition, PPARα showed a countering effect on pro-inflammatory cytokines that were characterized by the downregulation of IL-6, TNF-α, and COX2 (90). In the PPARα knock-out mice, it showed an increment in triglyceride, oxidative stress, inflammation, and hepatocyte death (91). The PPARα activation was shown to improve NAFLD through AMPK-PPARα signaling through aerobic training (92). PPARγ is predominantly expressed in adipose tissue and macrophages and is capable of regulating adipocyte differentiation, lipid synthesis, and lipolysis (93). In NAFLD, disrupted PPARγ restricted macrophage polarization to the M2 phenotype, thus inducing hepatocyte steatosis, inflammation, and fibrosis. Thiazolidinediones (TZDs), a PPARγ agonist, were reported to affect NASH by increasing insulin sensitivity (94, 95).
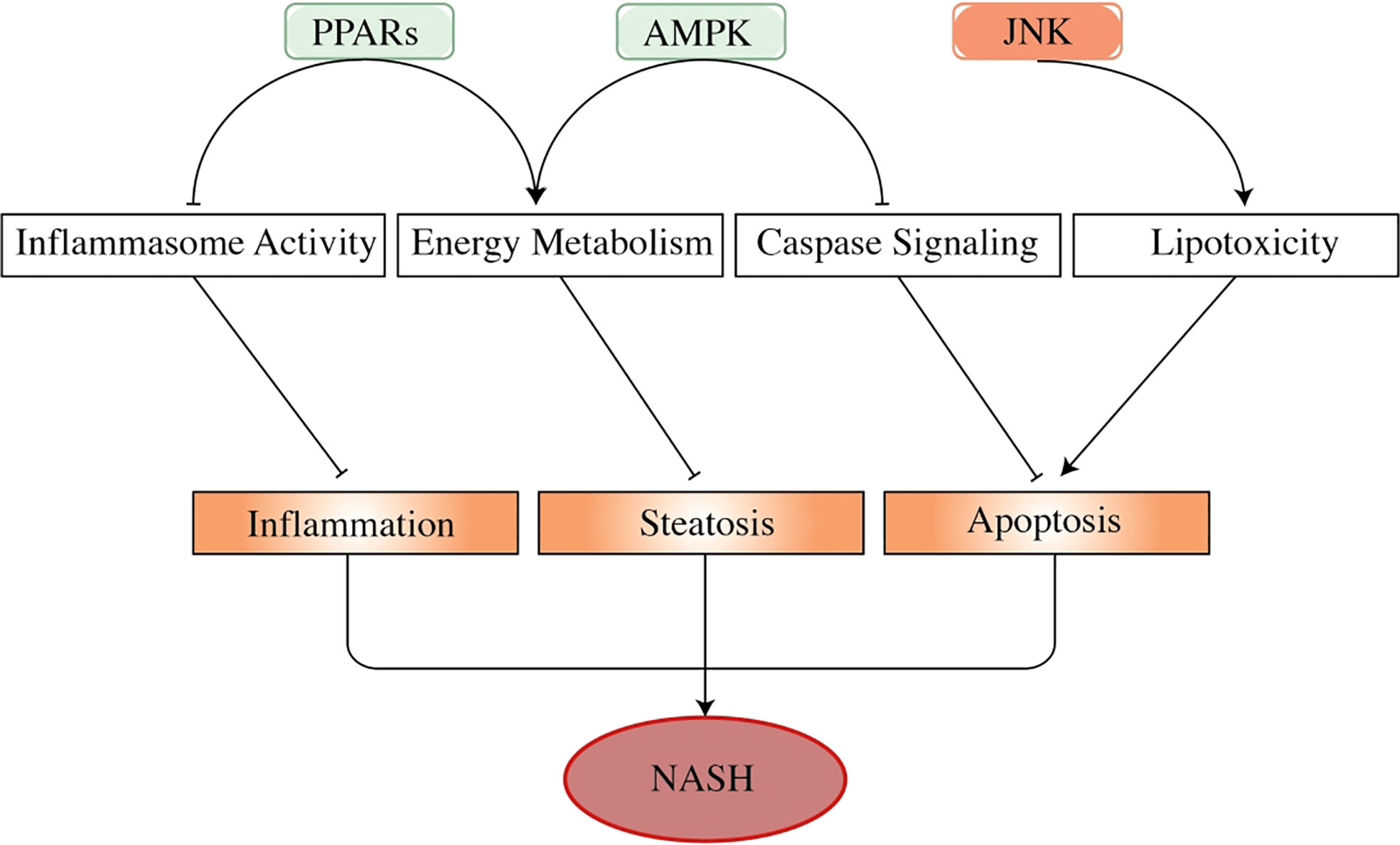
Figure 3 The effects of adenosine 5′-monophosphate-activated protein kinase, peroxisome proliferator-activated receptors, and c-Jun N-terminal kinase in the progress of non-alcoholic steatohepatitis. AMPK, PPARs, and JNK are important factors in NASH progression. As energy sensors, PPARs and AMPK regulate energy usage to prevent the liver from developing steatosis. PPARs also show an inhibitory effect on inflammasome activity (such as TNF-α and IL-6), thus regulating liver inflammation. Activated AMPK attenuates hepatocyte apoptosis by inhibiting the activity of the caspase pathway, especially caspase-6. By contrast, activated JNK promotes liver cell apoptosis by mediating fatty acid-related lipotoxicity and inducing TNF-α activation. These factors play a part in hepatocyte pathogenesis, regulating the progression of NAFLD and NASH. In this figure, arrows represent promoting effect, and short lines imply repressing effect. PPARs, peroxisome proliferator-activated receptors; NASH, non-alcoholic steatohepatitis; NAFLD, non-alcoholic fatty liver disease.
Although multiple clinical trials for NAFLD/NASH have been in process in recent years, there is currently no drug that has been approved by the United States Food and Drug Administration (U. S. FDA) (6). The current most effective way to alleviate NAFLD is still exercise and dietary intervention (96). Meanwhile, the drugs already approved for treating other diseases are repurposed for NAFLD/NASH treatment, saving time and cost. Notably, phase III clinical trials of promising drugs of these kinds are ongoing.
Lifestyle (exercise and dietary) intervention is still a good therapeutic choice for NAFLD. Several studies have demonstrated that caloric restriction and exercise could improve NAFLD through weight loss (97). Lifestyle intervention showed an impressive reduction in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) and amelioration in steatosis and cirrhosis (97). In addition, exercise stimulates AMPK directly, which restricts the synthesis of fatty acid, promotes fatty acid catabolism, and ameliorates hepatocyte apoptosis (75, 98). In addition, lifestyle intervention could reduce inflammation, ballooning, and fibrosis. However, achieving NASH improvement through weight loss has become challenging because of work-related stress and the difficulty of changing lifestyles in the modern era (99).
In addition to caloric restriction, the rational dietary structure is a hotspot for NAFLD/NASH improvement. The rampancy of NAFLD shows a parallel increase in the prevalence of a diet that contains high fat and low fiber. In contrast, a diet that contains whole grains with a high proportion of fiber showed a great effect on weight loss and blood lipid profile (100). It also modulates the composition of gut microbiota (49), which may further have an impact on NAFLD.
It has been reported that ω-3 poly-unsaturated fatty acids (PUFAs) could ameliorate liver fat and AST (101), and a lower content of ω-3 PUFAs was detected in patients with NAFLD (102). These findings highlighted made the diet with higher content of mono-unsaturated fatty acids (MUFAs) and ω-3 fatty acids a hotspot, such as nuts, olive oil, fish, and wine. Compared with other types of diet, these foods showed impressive results in weight loss, reduction of ALT, and improvement of insulin resistance in NAFLD patients (103, 104).
Obeticholic acid is a selective farnesoid X receptor agonist. Activation of the farnesoid X receptor can reduce fibrosis and inflammation in NASH by regulating bile acid metabolism (Figure 4) (105, 106). Usage of obeticholic acid for 18 months showed a significant amelioration in fibrosis and histological problems such as hepatocyte ballooning, lobular inflammation, and reduction of ALT and AST level in a phase III trial (NCT02548351) (107). Obeticholic acid showed mild adverse effects, but the most common one is pruritus. Another mechanism of obeticholic acid in NASH treatment has been discovered that inhibits NLRP3 inflammasome in macrophage activation and suppresses lipid accumulation without the participation of the farnesoid X receptor (108).
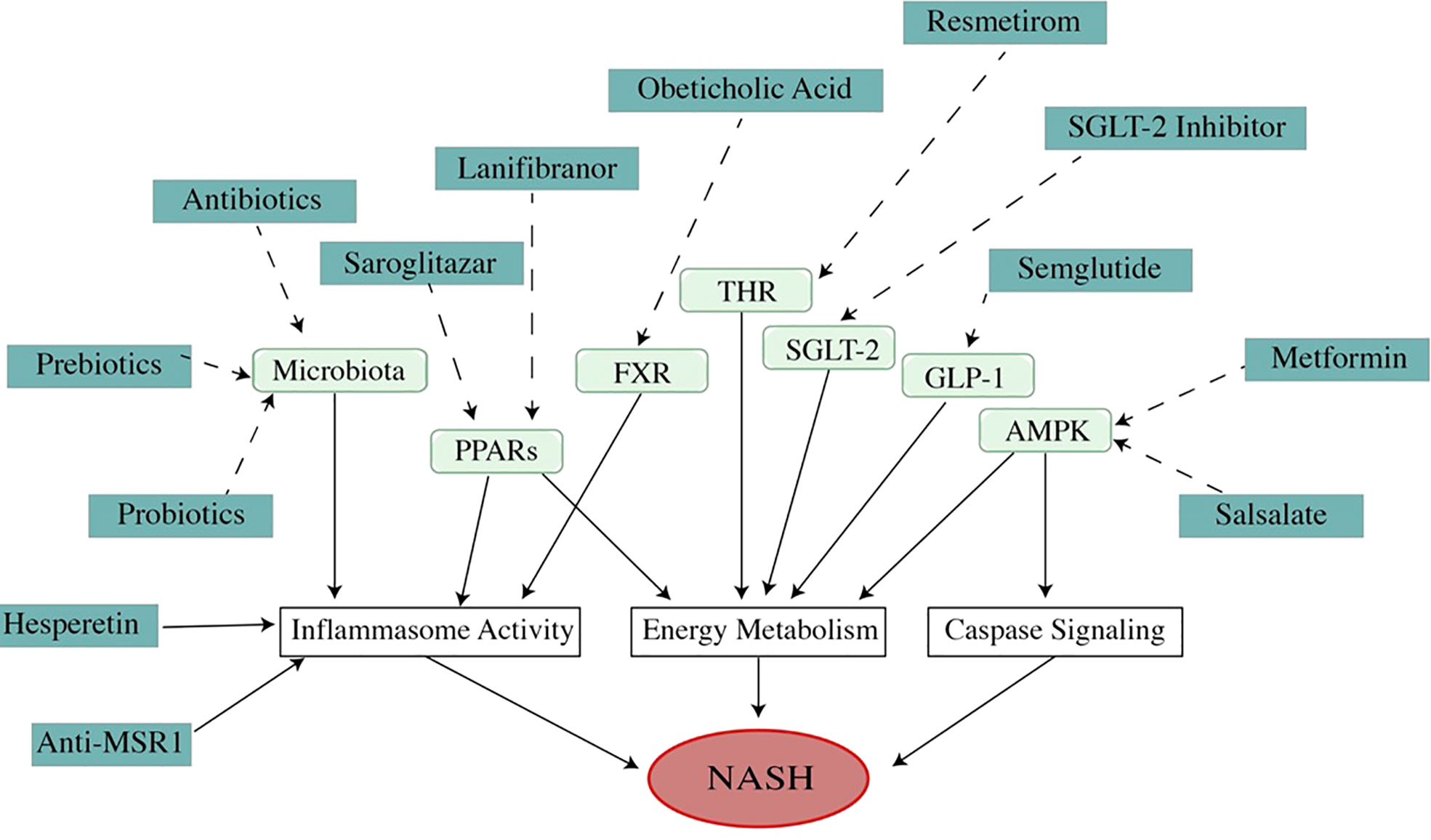
Figure 4 Mechanism of medications (dosage is listed in Table 1). As a multi-factor disease, the pathogenesis of NAFLD/NASH involves many pathways. Modulating these pathways with different medications can improve NAFLD/NASH. As a metabolic disease, the theme of NAFLD treatment is to regulate energy metabolism. A high percentage of ongoing drug experiments are related to energy management. However, the medicines that improve NAFLD in other mechanisms, such as regulating apoptosis or inflammation, are also attracting attention because of their impressive effects in treatment. Unfortunately, all of these drugs are undergoing trials, and no drug has been approved by U. S. FDA).
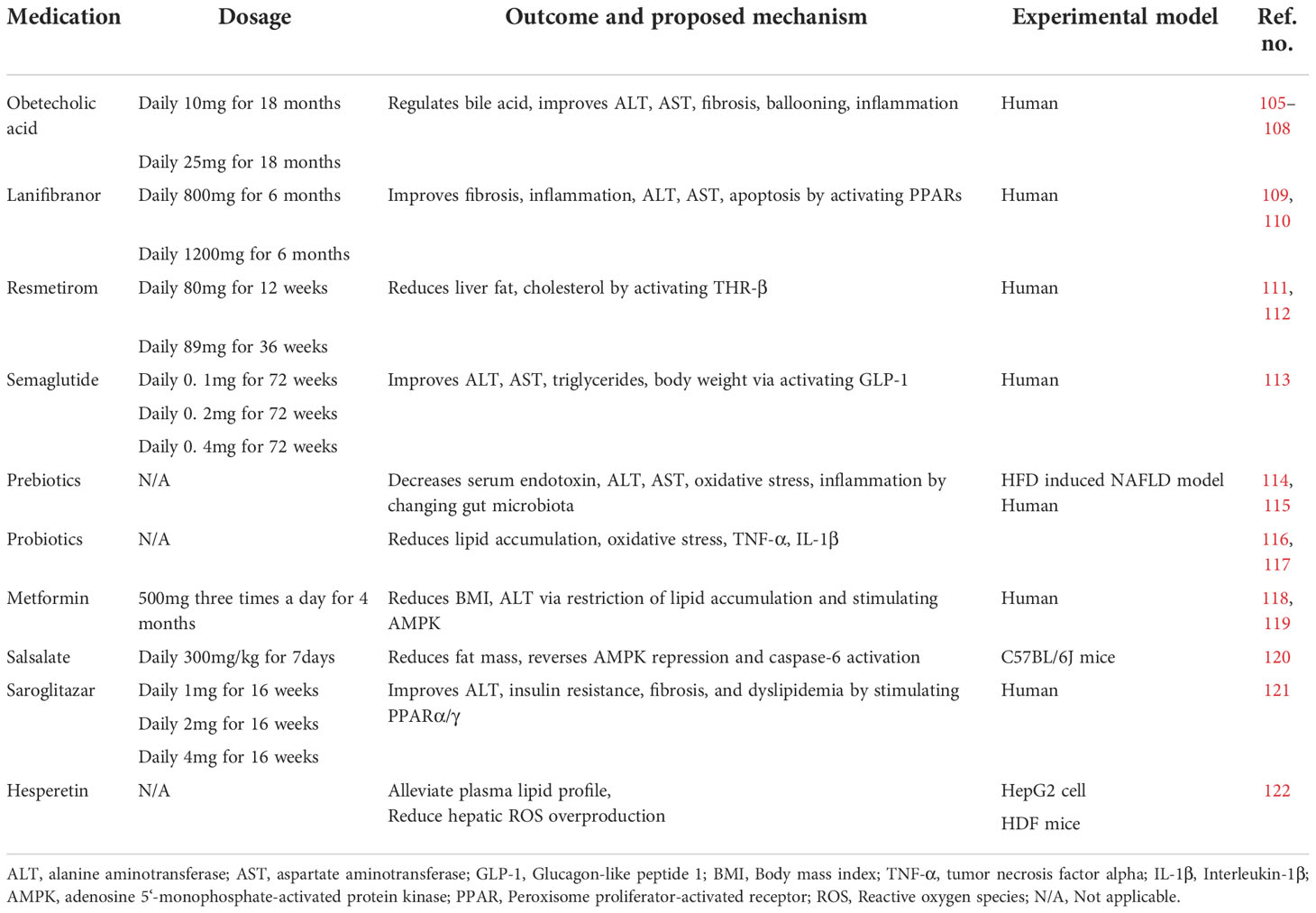
Table 1 Medications treatment.
Lanifibranor is an anti-fibrotic drug that activates PPAR-α, PPAR-β, and PPAR-δ, called a pan-PPAR agonist. Lanifibranor improved insulin resistance, fibrosis, and inflammation in preclinical trials (109, 110). Oral intake of 800 or 1,200 mg once daily of lanifibranor for 6 months showed impressive amelioration in steatosis, activity, and fibrosis (SAF) score, ALT, AST level, and markers of apoptosis and steatosis in a phase IIb trial (NCT03008070) (123). Some scholars suggest that there is no sufficient evidence to support the efficacy of lanifibranor because there is only one randomized controlled trial for the drug (124). A phase III trial of lanifibranor in the treatment of NASH is ongoing to investigate its safety and long-term efficacy.
Resmetirom is a thyroid hormone receptor β (THR-β) agonist. THR-β is the most important receptor of thyroxine in the liver, regulating cholesterol metabolism (111). Many studies demonstrated that activation of THR-β reduces triglyceride, cholesterol, apoptosis, and insulin resistance in an animal model (125–127). Usage of resmetirom for 12 or 36 weeks orally significantly alleviated liver fat content evaluated by MRI-PDFF in a phase II trial (NCT02912260) (112). At the same time, resmetirom showed relatively mild side effects (diarrhea and nausea). Resmetirom has also been demonstrated to reduce ALT and AST reduction. A phase III trial is ongoing to investigate its efficacy on NASH and stage F2–F3 fibrosis patients.
Semaglutide is a glucagon-like protein-1 (GLP-1) receptor agonist approved for T2DM treatment and chronic weight management (128). A number of trials have recently assessed the possible beneficial hepatic effects of injectable GLP-1 receptor agonists for NAFLD. An updated meta-analysis of randomized controlled trials showed that GLP-1 receptor agonists reduce liver fat content and serum liver enzyme level, thus improving NAFLD (129). Semaglutide has shown its therapeutic effect for NASH in phase II clinical trials in 2021 (113). Compared with the placebo, it showed the ability to ameliorate obesity, T2DM, ALT, AST, fibrosis, and liver histology. However, semaglutide also showed side effects, such as constipation, nausea, vomiting, decreased appetite, and abdominal pain. Moreover, neoplasm and fatal cardiovascular events were observed during the trial. The American Association for the Study of Liver Disease practice guidelines published in 2018 suggested that it is premature to consider GLP-1 receptor agonists to treat NAFLD or NASH (130). Nevertheless, at the same time, some researchers suggest that the effect of GLP-1 receptor agonists for NAFLD treatment is attractive, especially in patients with coexisting type 2 diabetes or obesity (124). The usage of semaglutide for NAFLD requires further observation.
Saroglitazar is a PPARα/γ agonist shown to improve NAFLD/NASH (131). Due to its double-sensitizing effect, saroglitazar not only increases β-oxidation and reduces triglyceride synthesis but also increases insulin sensitivity (121). In a randomized controlled, double-blind phase II trial, saroglitazar ameliorated ALT secretion, insulin resistance, and dyslipidemia significantly in NAFLD/NASH patients (132). Of note, even though limited cases of severe adverse effects were observed, a mild, dose-dependent weight gain was observed in the saroglitazar group.
Metformin is widely used in T2DM as a first-line therapy, which suppresses hepatic gluconeogenesis (133–135). Metformin showed an inhibitory effect in lipogenesis and lipid oxidation in hepatocytes (74), restricting lipid accumulation in NAFLD. Metformin showed a significant effect on weight loss in a human trial (136) and an amelioration effect on ALT levels (118, 119). In addition, metformin activates AMPK through the phosphorylation at Thr172 to inhibit gluconeogenesis in the liver (137). Meanwhile, metformin inhibited the cleavage and activation of pro-apoptotic factors, such as caspase-3 and caspase-7, thus ameliorating apoptosis in the NASH model (138). Meanwhile, some guidelines recommend against using metformin as a specific treatment for NASH because metformin was shown to not improve liver histology in adult NAFLD patients (124).
Salsalate, a member of the salicylates family, is a weak non-steroidal anti-inflammatory drug (NSAID) used for inflammatory and non-inflammatory disorders (139, 140). It has been reported that salsalate strongly phosphorylated AMPK and ACC and restored activation of AMPK and caspase-6 improved NAFLD in the HFD mouse model (120). Additionally, salsalate elevates the resting expenditure of energy (141, 142). These mechanisms indicate that salsalate may improve symptoms of NASH/NAFLD simultaneously. Furthermore, salsalate may also contribute to NASH improvement via COX-2, which may interact with TNF-α and IL-6 to promote hepatocellular apoptosis (143).
Several types of antibiotics are being tested for NAFLD counteraction due to the association between gut microbiota and fatty liver disease. Long-term usage of antibiotics reduces gut bacteria diversity and downregulates liver inflammation (144). Rifaximin, an antibiotic that is non-absorbable in the intestines, induced a significant reduction of AST, ALT, low-density lipoprotein (LDL), and BMI in NASH patients (145). However, antibiotic application in NAFLD is of concern because antibiotics not only kill the pernicious bacteria, but also destroy the probiotics. Moreover, the most concern issue is antibiotic resistance due to frequent abuse in daily life.
Prebiotics are incompletely digested food ingredients that guide gut microbiota in a beneficial manner (146). Prebiotic feeding is an effective therapeutic strategy for NAFLD that works by increasing the population of probiotics in the gut (114, 115). The underlying mechanism may include oxidative stress reduction, inflammation alleviation, glucose tolerance, and triglyceride accumulation by adjusting gut microbiota (147–149). In addition, prebiotics also stimulate SCFA production, which is beneficial for NAFLD.
Numerous animal studies, as well as clinical trials, have demonstrated the effect of probiotics on NAFLD improvement. Probiotics are a series of non-pathogenic microbes that positively impact the host (116). Probiotics improve NAFLD by reducing lipid accumulation, oxidative stress, and inflammatory cytokines such as TNF-α and IL-1β (116, 117). However, probiotics are raising concerns because of biosafety, and just a few bacteria have been proven to have therapeutic effects. Personalized use of probiotics based on gut microbiota tests may be an effective way of NAFLD treatment because of the different gut microbiota biological structures among individuals.
Hesperetin (3′,5,7-trihydroxy-4′-methoxyflavanone) is a citrus flavonoid belonging to the flavanone class and is abundant in oranges, lemons, and grape juice. A recent study discovered that hesperetin alleviated oleic acid-induced hepatotoxicity and oxidative stress in vitro and plasma lipid profile, including TG, total cholesterol (TC), and LDL-C, in HFD-induced NAFLD rat model (122). Hesperetin showed its effect in reducing fatty acid-induced hepatic ROS overproduction and oxidative damage. Also, hesperetin showed inhibition of fatty acid-induced NF-κB activation and subsequent inflammation by reducing ROS overproduction (122). Anti-macrophage scavenger receptor-1 (anti-MSR1) antibody (MSR1 inhibitor) may be an important therapeutic approach for the treatment of NAFLD requiring clinical investigations.
MSR1 is an important receptor for the uptake of lipids in macrophages, leading to an inflammatory response and metabolic changes throughout the body. An MSR1 antibody showed a reduction of hepatic inflammation and changes in hepatic lipid metabolism by reducing hepatic lipid-laden foamy macrophages in vivo and ex vivo (150). Targeting MSR1 using monoclonal antibody therapy in an obesity-associated NAFLD mouse model and human liver slices displayed prevention of foamy macrophage formation and inflammation by regulating the JNK signaling pathway (150). MSR1 plays a critical role in lipid-induced inflammation and the MSR1 inhibitor may be an interesting therapeutic approach for the treatment of NAFLD in the future.
Sodium-glucose cotransporter-2 (SGLT-2) inhibitors (such as dapagliflozin, empagliflozin, ipragliflozin, and canagliflozin) are oral glucose-lowering medicines approved for the treatment of T2DM. Recent studies showed beneficial effects in people with NAFLD (124). By reducing the renal capacity to reabsorb filtered glucose, SGLT-2 inhibitors lessen the ability to reabsorb filtered glucose in the kidney, thereby lowering serum glucose and accompanying some additional benefits such as weight loss and blood pressure control (151, 152). Recently, a meta-analysis of several placebo-controlled or randomized control trials that used various SGLT-2 inhibitors for the treatment of NAFLD has displayed that usage of SGLT-2 inhibitor for 24 weeks significantly decreased ALT, gamma-glutamyl transferase (GGT) level, and liver fat content, as well as body weight in NAFLD patients (152).
NAFLD has become one of the major public health issues throughout the world. The rampancy of NAFLD worldwide is highly associated with a changed lifestyle and rising incidence of metabolic problems such as obesity or T2DM. We should pay attention to dietary structure and exercise to prevent metabolic disease and NAFLD. Exercise and dietary intervention is the current therapeutic strategy Nevertheless, the pace of newly discovered molecules correlated with NAFLD, such as AMPK, PPARs, and JNK, may act as alternative approaches to NAFLD treatment. Researchers are focusing on the discovery of medications that regulate energy metabolism. In the future, combination therapy with dietary intervention, exercise, and medicine will probably be the mainstream and effective therapeutic strategy because NAFLD is a complex disease that involves many pathways in pathogenesis. Besides, liver biopsy is regarded as the most reliable diagnostic approach. However, a biopsy is a snapshot which involves invasive operation and cannot provide continuous monitoring of changes in disease. Therefore, a repeatable, non-invasive approach that is capable of precisely diagnosing and staging is needed. Some extracellular biomarkers, such as circulating nucleic acid fragments, have been identified that could help NAFLD diagnosis. Cell-free non-coding RNA showed its different expression profile among different stages of NAFLD patients (153). Recent studies have discovered that plasma exosome and some exosome markers such as CD9, CD36, and CD63 are significantly increased in NAFLD patients, especially those liver-derived exosome markers, compared with controls (154, 155). Exosomes are spotlighted as an effective target for NAFLD diagnosis due to their effect on lipid metabolism. These newly discovered biomarkers are showing their potential in recent studies but still need further validation. Also, low-cost pharmacotherapy is required because of the relation to lifestyle and the prevalence of NAFLD. We look for reliable and cost-effective diagnostic approaches because a considerable number of patients worldwide are not properly diagnosed. We summarized our views on potential research directions for NAFLD in Table 2.

Table 2 Potential future perspectives.
WSC drafted the manuscript and figures. MZ conducted the literature research and revised the entire manuscript language. XL revised the manuscript and figures. CL acquired the funding and conducted the concept design. SY and WCC revised the manuscript and supervised the study.
Funding was provided by the Postdoctoral Scientific Research Developmental Fund (No. LBH-Q20112) of Heilongjiang.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Armstrong MJ, Adams LA, Canbay A, Syn W. Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology (2014) 59:1174–97. doi: 10.1002/hep.26717
2. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease–meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology (2016) 64:73–84. doi: 10.1002/hep.28431
3. Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms, and pharmacological implications. Gut (2020) 69:gutjnl–2020-320622. doi: 10.1136/gutjnl-2020-320622
4. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United kingdom, and United States for the period 2016–2030. J Hepatol (2018) 69:896–904. doi: 10.1016/j.jhep.2018.05.036
5. Tanase DM, Gosav EM, Costea CF, Ciocoiu M, Lacatusu CM, Maranduca MA, et al. The intricate relationship between type 2 diabetes mellitus (T2DM), insulin resistance (IR), and nonalcoholic fatty liver disease (NAFLD). J Diabetes Res (2020) 2020:3920196. doi: 10.1155/2020/3920196
6. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
7. Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolis (2016) 65:1049–61. doi: 10.1016/j.metabol.2016.02.014
8. Marchesini G, Marzocchi R, Agostini F, Bugianesi E. Nonalcoholic fatty liver disease and the metabolic syndrome. Curr Opin Lipidol (2005) 16:421–7. doi: 10.1097/01.mol.0000174153.53683.f2
9. Lindenmeyer CC, McCullough AJ. The natural history of nonalcoholic fatty liver disease–an evolving view. Clin Liver Dis (2018) 22:11–21. doi: 10.1016/j.cld.2017.08.003
10. Zelber-Sagi S, Lotan R, Shlomai A, Webb M, Harrari G, Buch A, et al. Predictors for incidence and remission of NAFLD in the general population during a seven-year prospective follow-up. J Hepatol (2012) 56:1145–51. doi: 10.1016/j.jhep.2011.12.011
11. Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology (2010) 52:774–88. doi: 10.1002/hep.23719
12. Ye Q, Zou B, Yeo YH, Li J, Huang DQ, Wu Y, et al. Global prevalence, incidence, and outcomes of non-obese or lean non-alcoholic fatty liver disease: A systematic review and meta-analysis. Lancet Gastroenterol Hepatol (2020) 5:739–52. doi: 10.1016/s2468-1253(20)30077-7
13. Jarvis H, Craig D, Barker R, Spiers G, Stow D, Anstee QM, et al. Metabolic risk factors and incident advanced liver disease in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of population-based observational studies. PloS Med (2020) 17:e1003100. doi: 10.1371/journal.pmed.1003100
14. Sinha N, Mukhopadhyay S, Sau M. Metabolic syndrome is not uncommon among lean non-alcoholic fatty liver disease patients as compared with those with obesity. Indian J Gastroenterol (2020) 39:75–83. doi: 10.1007/s12664-020-01020-3
15. Lu F-B, Hu E-D, Xu L-M, Chen L, Wu J-L, Li H, et al. The relationship between obesity and the severity of non-alcoholic fatty liver disease: systematic review and meta-analysis. Expert Rev Gastroent (2018) 12:1–12. doi: 10.1080/17474124.2018.1460202
16. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol (2020) 73:202–9. doi: 10.1016/j.jhep.2020.03.039
17. Chen H-T, Huang H-L, Li Y-Q, Xu H-M, Zhou Y-J. Therapeutic advances in non-alcoholic fatty liver disease: A microbiota-centered view. World J Gastroentero (2020) 26:1901–11. doi: 10.3748/wjg.v26.i16.1901
18. Porras D, Nistal E, Martínez-Flórez S, González-Gallego J, García-Mediavilla MV, Sánchez-Campos S. Intestinal microbiota modulation in obesity-related non-alcoholic fatty liver disease. Front Physiol (2018) 9:1813. doi: 10.3389/fphys.2018.01813
19. Petrov PD, García-Mediavilla MV, Guzmán C, Porras D, Nistal E, Martínez-Flórez S, et al. A network involving gut microbiota, circulating bile acids, and hepatic metabolism genes that protects against non-alcoholic fatty liver disease. Mol Nutr Food Res (2019) 63:1900487. doi: 10.1002/mnfr.201900487
20. Pan X, Wen SW, Kaminga AC, Liu A. Gut metabolites and inflammation factors in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Sci Rep-Uk (2020) 10:8848. doi: 10.1038/s41598-020-65051-8
21. Park JS, Seo JH, Youn H-S. Gut microbiota and clinical disease: Obesity and nonalcoholic fatty liver disease. Pediatr Gastroenterol Hepatol Nutr (2013) 16:22–7. doi: 10.5223/pghn.2013.16.1.22
22. Lian C-Y, Zhai Z-Z, Li Z-F, Wang L. High fat diet-triggered non-alcoholic fatty liver disease: A review of proposed mechanisms. Chem-Biol Interact (2020) 330:109199. doi: 10.1016/j.cbi.2020.109199
23. Roy TL, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut (2013) 62:1787. doi: 10.1136/gutjnl-2012-303816
24. Wilson AS, Koller KR, Ramaboli MC, Nesengani LT, Ocvirk S, Chen C, et al. Diet and the human gut microbiome: An international review. Digest Dis Sci (2020) 65:723–40. doi: 10.1007/s10620-020-06112-w
25. Du SF, Wang HJ, Zhang B, Zhai FY, Popkin BM. China In the period of transition from scarcity and extensive undernutrition to emerging nutrition-related non-communicable diseases, 1949–1992. Obes Rev (2014) 15:8–15. doi: 10.1111/obr.12122
26. Kashtanova DA, Tkacheva ON, Doudinskaya EN, Strazhesko ID, Kotovskaya YV, Popenko AS, et al. Gut microbiota in patients with different metabolic statuses: Moscow study. Microorg (2018) 6:98. doi: 10.3390/microorganisms6040098
27. Wan Y, Wang F, Yuan J, Li J, Jiang D, Zhang J, et al. Effects of dietary fat on gut microbiota and faecal metabolites, and their relationship with cardiometabolic risk factors: A 6-month randomised controlled-feeding trial. Gut (2019) 68:1417. doi: 10.1136/gutjnl-2018-317609
28. Takaki A, Kawai D, Yamamoto K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int J Mol Sci (2013) 14:20704–28. doi: 10.3390/ijms141020704
29. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res (2018) 122:877–902. doi: 10.1161/circresaha.117.311401
30. Zhang L, Wang X, Cueto R, Effi C, Zhang Y, Tan H, et al. Biochemical basis and metabolic interplay of redox regulation. Redox Biol (2019) 26:101284. doi: 10.1016/j.redox.2019.101284
31. Campbell EL, Colgan SP. Control and dysregulation of redox signalling in the gastrointestinal tract. Nat Rev Gastroentero (2019) 16:106–20. doi: 10.1038/s41575-018-0079-5
32. Arroyave-Ospina JC, Wu Z, Geng Y, Moshage H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: Implications for prevention and therapy. Antioxidants (2021) 10:174. doi: 10.3390/antiox10020174
33. Zhao Q, Liu J, Deng H, Ma R, Liao J-Y, Liang H, et al. Targeting mitochondria-located circRNA SCAR alleviates NASH via reducing mROS output. Cell (2020) 183:76–93.e22. doi: 10.1016/j.cell.2020.08.009
34. Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci (2014) 39:199–218. doi: 10.1016/j.tibs.2014.02.002
35. Ouyang X, Han S-N, Zhang J-Y, Dioletis E, Nemeth BT, Pacher P, et al. Digoxin suppresses pyruvate kinase M2-promoted HIF-1α transactivation in steatohepatitis. Cell Metab (2018) 27:339–50.e3. doi: 10.1016/j.cmet.2018.01.007
36. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroentero (2018) 15:349–64. doi: 10.1038/s41575-018-0009-6
37. Bergheim I, Weber S, Vos M, Krämer S, Volynets V, Kaserouni S, et al. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J Hepatol (2008) 48:983–92. doi: 10.1016/j.jhep.2008.01.035
38. Sutti S, Albano E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat Rev Gastroentero (2020) 17:81–92. doi: 10.1038/s41575-019-0210-2
39. Caussy C, Soni M, Cui J, Bettencourt R, Schork N, Chen C-H, et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J Clin Invest (2017) 127:2697–704. doi: 10.1172/jci93465
40. Valenti LVC, Baselli GA. Genetics of nonalcoholic fatty liver disease: A 2018 update. Curr Pharm Design (2019) 24:4566–73. doi: 10.2174/1381612825666190119113836
41. Carlsson B, Lindén D, Brolén G, Liljeblad M, Bjursell M, Romeo S, et al. Review article: the emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis. Aliment Pharm Therap (2020) 51:1305–20. doi: 10.1111/apt.15738
42. Trépo E, Valenti L. Update on NAFLD genetics: From new variants to the clinic. J Hepatol (2020) 72:1196–209. doi: 10.1016/j.jhep.2020.02.020
43. Unalp-Arida A, Ruhl CE. Patatin-like phospholipase domain-containing protein 3 I148M and liver fat and fibrosis scores predict liver disease mortality in the US. Population. Hepatol (2020) 71:820–34. doi: 10.1002/hep.31032
44. Shen J, Wong GL, Chan HL, Chan RS, Chan H, Chu WC, et al. PNPLA3 gene polymorphism and response to lifestyle modification in patients with nonalcoholic fatty liver disease. J Gastroen Hepatol (2015) 30:139–46. doi: 10.1111/jgh.12656
45. Oscarsson J, Önnerhag K, Risérus U, Sundén M, Johansson L, Jansson P-A, et al. Effects of free omega-3 carboxylic acids and fenofibrate on liver fat content in patients with hypertriglyceridemia and non-alcoholic fatty liver disease: A double-blind, randomized, placebo-controlled study. J Clin Lipidol (2018) 12:1390–1403.e4. doi: 10.1016/j.jacl.2018.08.003
46. Scorletti E, West AL, Bhatia L, Hoile SP, McCormick KG, Burdge GC, et al. Treating liver fat and serum triglyceride levels in NAFLD, effects of PNPLA3 and TM6SF2 genotypes: Results from the WELCOME trial. J Hepatol (2015) 63:1476–83. doi: 10.1016/j.jhep.2015.07.036
47. Lindén D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andréasson A-C, et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol Metab (2019) 22:49–61. doi: 10.1016/j.molmet.2019.01.013
48. Schwartz BE, Rajagopal V, Smith C, Cohick E, Whissell G, Gamboa M, et al. Discovery and targeting of the signaling controls of PNPLA3 to effectively reduce transcription, expression, and function in pre-clinical NAFLD/NASH settings. Cells (2020) 9:2247. doi: 10.3390/cells9102247
49. Hardie DG. AMP-activated protein kinase–an energy sensor that regulates all aspects of cell function. Gene Dev (2011) 25:1895–908. doi: 10.1101/gad.17420111
50. Woods A, Munday MR, Scott J, Yang X, Carlson M, Carling D. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J Biol Chem (1994) 269:19509–15. doi: 10.1016/S0021-9258(17)32198-1
51. Carling D, Aguan K, Woods A, Verhoeven AJ, Beri RK, Brennan CH, et al. Mammalian AMP-activated protein kinase is homologous to yeast and plant protein kinases involved in the regulation of carbon metabolism. J Biol Chem (1994) 269:11442–8. doi: 10.1016/S0021-9258(19)78143-5
52. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Bio (2012) 13:251–62. doi: 10.1038/nrm3311
53. Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle*. J Biol Chem (2006) 281:18933–41. doi: 10.1074/jbc.m512831200
54. Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, et al. AMPK inhibition in health and disease. Crit Rev Biochem Mol (2010) 45:276–95. doi: 10.3109/10409238.2010.488215
55. Valentine RJ, Coughlan KA, Ruderman NB, Saha AK. Insulin inhibits AMPK activity and phosphorylates AMPK Ser485/491 through akt in hepatocytes, myotubes and incubated rat skeletal muscle. Arch Biochem Biophys (2014) 562:62–9. doi: 10.1016/j.abb.2014.08.013
56. Steinberg GR, Michell BJ, van Denderen BJW, Watt MJ, Carey AL, Fam BC, et al. Tumor necrosis factor α-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab (2006) 4:465–74. doi: 10.1016/j.cmet.2006.11.005
57. Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase*. J Biol Chem (1996) 271:27879–87. doi: 10.1074/jbc.271.44.27879
58. Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, et al. Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol (2003) 2:28–8. doi: 10.1186/1475-4924-2-28
59. Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun (2013) 4:3017. doi: 10.1038/ncomms4017
60. Boon H, Bosselaar M, Praet SFE, Blaak EE, Saris WHM, Wagenmakers AJM, et al. Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia (2008) 51:1893. doi: 10.1007/s00125-008-1108-7
61. Nielsen TS, Jessen N, Jørgensen JOL, Møller N, Lund S. Dissecting adipose tissue lipolysis: molecular regulation and implications for metabolic disease. J Mol Endocrinol (2014) 52:R199–222. doi: 10.1530/jme-13-0277
62. Yuan H-X, Xiong Y, Guan K-L. Nutrient sensing, metabolism, and cell growth control. Mol Cell (2013) 49:379–87. doi: 10.1016/j.molcel.2013.01.019
63. Garcia D, Shaw RJ. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell (2017) 66:789–800. doi: 10.1016/j.molcel.2017.05.032
64. Hardie DG. Keeping the home fires burning†: AMP-activated protein kinase. J Roy Soc Interface (2018) 15:20170774. doi: 10.1098/rsif.2017.0774
65. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev (2009) 89:1025–78. doi: 10.1152/physrev.00011.2008
66. Hardie DG, Schaffer BE, Brunet A. AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol (2016) 26:190–201. doi: 10.1016/j.tcb.2015.10.013
67. Gauthier M-S, O’Brien EL, Bigornia S, Mott M, Cacicedo JM, Xu XJ, et al. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem Bioph Res Co (2011) 404:382–7. doi: 10.1016/j.bbrc.2010.11.127
68. Xu XJ, Gauthier M-S, Hess DT, Apovian CM, Cacicedo JM, Gokce N, et al. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res (2012) 53:792–801. doi: 10.1194/jlr.p022905
69. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest (2005) 115:1343–51. doi: 10.1172/jci23621
70. Lambert JE, Ramos–Roman MA, Browning JD, Parks EJ. Increased De novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology (2014) 146:726–35. doi: 10.1053/j.gastro.2013.11.049
71. Carling D, Clarke PR, Zammit VA, HARDIE DG. Purification and characterization of the AMP-activated protein kinase. Eur J Biochem (1989) 186:129–36. doi: 10.1111/j.1432-1033.1989.tb15186.x
72. Hardie DG. Regulation of fatty acid and cholesterol metabolism by the AMP-activated protein kinase. Biochim Et Biophys Acta Bba - Lipids Lipid Metab (1992) 1123:231–8. doi: 10.1016/0005-2760(92)90001-c
73. Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen Z, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin–sensitizing effects of metformin. Nat Med (2013) 19:1649–54. doi: 10.1038/nm.3372
74. Boudaba N, Marion A, Huet C, Pierre R, Viollet B, Foretz M. AMPK re-activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. Ebiomedicine (2018) 28:194–209. doi: 10.1016/j.ebiom.2018.01.008
75. Zhao P, Sun X, Chaggan C, Liao Z, Wong K, He F, et al. An AMPK–caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science (2020) 367:652–60. doi: 10.1126/science.aay0542
76. Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature (2018) 559:637–41. doi: 10.1038/s41586-018-0350-5
77. Velázquez-Delgado EM, Hardy JA. Phosphorylation regulates assembly of the caspase-6 substrate-binding groove. Structure (2012) 20:742–51. doi: 10.1016/j.str.2012.02.003
79. Ibrahim SH, Akazawa Y, Cazanave SC, Bronk SF, Elmi NA, Werneburg NW, et al. Glycogen synthase kinase-3 (GSK-3) inhibition attenuates hepatocyte lipoapoptosis. J Hepatol (2011) 54:765–72. doi: 10.1016/j.jhep.2010.09.039
80. Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis*. J Biol Chem (2006) 281:12093–101. doi: 10.1074/jbc.m510660200
81. Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, Minicis SD, et al. C-jun n-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology (2009) 137:1467–77.e5. doi: 10.1053/j.gastro.2009.06.045
82. Cai N, Zhao X, Jing Y, Sun K, Jiao S, Chen X, et al. Autophagy protects against palmitate-induced apoptosis in hepatocytes. Cell Biosci (2014) 4:28. doi: 10.1186/2045-3701-4-28
83. Schwabe RF, Bradham CA, Uehara T, Hatano E, Bennett BL, Schoonhoven R, et al. C-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology (2003) 37:824–32. doi: 10.1053/jhep.2003.50135
84. Xu P, Zhai Y, Wang J. The role of PPAR and its cross-talk with CAR and LXR in obesity and atherosclerosis. Int J Mol Sci (2018) 19:1260. doi: 10.3390/ijms19041260
85. Cordoba-Chacon J. Loss of hepatocyte-specific PPARγ expression ameliorates early events of steatohepatitis in mice fed the methionine and choline-deficient diet. Ppar Res (2020) 2020:9735083. doi: 10.1155/2020/9735083
86. Strand E, Lysne V, Grinna ML, Bohov P, Svardal A, Nygård O, et al. Short-term activation of peroxisome proliferator-activated receptors α and γ induces tissue-specific effects on lipid metabolism and fatty acid composition in Male wistar rats. Ppar Res (2019) 2019:8047627. doi: 10.1155/2019/8047627
87. Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J Hepatol (2015) 63:164–73. doi: 10.1016/j.jhep.2015.02.019
88. Cave MC, Clair HB, Hardesty JE, Falkner KC, Feng W, Clark BJ, et al. Nuclear receptors and nonalcoholic fatty liver disease 1 1 this article is part of a special issue entitled: Xenobiotic nuclear receptors: New tricks for an old dog, edited by dr. wen xie. Biochim Et Biophys Acta Bba - Gene Regul Mech (2016) 1859:1083–99. doi: 10.1016/j.bbagrm.2016.03.002
89. Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr Rev (1999) 20:649–88. doi: 10.1210/edrv.20.5.0380
90. Ramanan S, Zhao W, Riddle DR, Robbins ME. Role of PPARs in radiation-induced brain injury. Ppar Res (2010) 2010:234975. doi: 10.1155/2010/234975
91. Abdelmegeed MA, Yoo S-H, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song B-J. PPARα expression protects Male mice from high fat–induced nonalcoholic fatty liver. J Nutr (2011) 141:603–10. doi: 10.3945/jn.110.135210
92. Diniz TA, Junior EA de L, Teixeira AA, Biondo LA, da Rocha LAF, Valadão IC, et al. Aerobic training improves NAFLD markers and insulin resistance through AMPK-PPAR-α signaling in obese mice. Life Sci (2021) 266:118868. doi: 10.1016/j.lfs.2020.118868
93. Li J, Guo C, Wu J. 15-Deoxy-Δ-12,14-Prostaglandin J2 (15d-PGJ2), an endogenous ligand of PPAR-γ: Function and mechanism. Ppar Res (2019) 2019:7242030. doi: 10.1155/2019/7242030
95. Wu L, Guo C, Wu J. Therapeutic potential of PPARγ natural agonists in liver diseases. J Cell Mol Med (2020) 24:2736–48. doi: 10.1111/jcmm.15028
96. Nascimbeni F, Pais R, Bellentani S, Day CP, Ratziu V, Loria P, et al. From NAFLD in clinical practice to answers from guidelines. J Hepatol (2013) 59:859–71. doi: 10.1016/j.jhep.2013.05.044
97. Koutoukidis DA, Astbury NM, Tudor KE, Morris E, Henry JA, Noreik M, et al. Association of weight loss interventions with changes in biomarkers of nonalcoholic fatty liver disease. JAMA Intern Med (2019) 179:1262–71. doi: 10.1001/jamainternmed.2019.2248
98. Kasper P, Breuer S, Hoffmann T, Vohlen C, Janoschek R, Schmitz L, et al. Maternal exercise mediates hepatic metabolic programming via activation of AMPK-PGC1α axis in the offspring of obese mothers. Cells (2021) 10:1247. doi: 10.3390/cells10051247
99. (EASL) EA for the S of the L, (EASD) EA for the S of D, (EASO) EA for the S of O, EASL–EASD–EASO. Clinical practice guidelines for the management of non-alcoholic fatty liver disease. Diabetologia (2016) 59:1121–40. doi: 10.1007/s00125-016-3902-y
100. Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science (2018) 359:1151–6. doi: 10.1126/science.aao5774
101. Parker HM, Johnson NA, Burdon CA, Cohn JS, O’Connor HT, George J. Omega-3 supplementation and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J Hepatol (2012) 56:944–51. doi: 10.1016/j.jhep.2011.08.018
102. Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, et al. Increase in long-chain polyunsaturated fatty acid n–6/n–3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (2004) 106:635–43. doi: 10.1042/cs20030326
103. Ryan MC, Itsiopoulos C, Thodis T, Ward G, Trost N, Hofferberth S, et al. The Mediterranean diet improves hepatic steatosis and insulin sensitivity in individuals with non-alcoholic fatty liver disease. J Hepatol (2013) 59:138–43. doi: 10.1016/j.jhep.2013.02.012
104. Angelico F, Ferro D, Baratta F. Is the Mediterranean diet the best approach to NAFLD treatment today? Nutrients (2021) 13:739. doi: 10.3390/nu13030739
105. Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology (2004) 127:1497–512. doi: 10.1053/j.gastro.2004.08.001
106. Gadaleta RM, van EKJ, Oldenburg B, Willemsen ECL, Renooij W, Murzilli S, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut (2011) 60:463. doi: 10.1136/gut.2010.212159
107. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet (2019) 394:2184–96. doi: 10.1016/s0140-6736(19)33041-7
108. Huang S, Wu Y, Zhao Z, Wu B, Sun K, Wang H, et al. A new mechanism of obeticholic acid on NASH treatment by inhibiting NLRP3 inflammasome activation in macrophage. Metabolis (2021) 120:154797. doi: 10.1016/j.metabol.2021.154797
109. Wettstein G, Luccarini J, Poekes L, Faye P, Kupkowski F, Adarbes V, et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol Commun (2017) 1:524–37. doi: 10.1002/hep4.1057
110. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages☆. J Hepatol (2020) 73:757–70. doi: 10.1016/j.jhep.2020.04.025
111. Taub R, Chiang E, Chabot-Blanchet M, Kelly MJ, Reeves RA, Guertin M-C, et al. Lipid lowering in healthy volunteers treated with multiple doses of MGL-3196, a liver-targeted thyroid hormone receptor-β agonist. Atherosclerosis (2013) 230:373–80. doi: 10.1016/j.atherosclerosis.2013.07.056
112. Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet (2019) 394:2012–24. doi: 10.1016/s0140-6736(19)32517-6
113. Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med (2020) 384:1113–24. doi: 10.1056/nejmoa2028395
114. Han R, Ma J, Li H. Mechanistic and therapeutic advances in non-alcoholic fatty liver disease by targeting the gut microbiota. Front Med-Prc (2018) 12:645–57. doi: 10.1007/s11684-018-0645-9
115. Cani P, Delzenne N. The role of the gut microbiota in energy metabolism and metabolic disease. Curr Pharm Design (2009) 15:1546–58. doi: 10.2174/138161209788168164
116. Xie C, Halegoua-DeMarzio D. Role of probiotics in non-alcoholic fatty liver disease: Does gut microbiota matter? Nutrients (2019) 11:2837. doi: 10.3390/nu11112837
117. Ritze Y, Bárdos G, Claus A, Ehrmann V, Bergheim I, Schwiertz A, et al. Lactobacillus rhamnosus GG protects against non-alcoholic fatty liver disease in mice. PloS One (2014) 9:e80169. doi: 10.1371/journal.pone.0080169
118. Marchesini G, Brizi M, Bianchi G, Tomassetti S, Zoli M, Melchionda N. Metformin in non-alcoholic steatohepatitis. Lancet (2001) 358:893–4. doi: 10.1016/s0140-6736(01)06042-1
119. Bugianesi E, Gentilcore E, Manini R, Natale S, Vanni E, Villanova N, et al. A randomized controlled trial of metformin versus vitamin e or prescriptive diet in nonalcoholic fatty liver disease. Am J Gastroenterol (2005) 100:1082–90. doi: 10.1111/j.1572-0241.2005.41583.x
120. Li J, Chen C, Zhang W, Bi J, Yang G, Li E. Salsalate reverses metabolic disorders in a mouse model of non-alcoholic fatty liver disease through AMPK activation and caspase-6 activity inhibition. Basic Clin Pharmacol (2021) 128:394–409. doi: 10.1111/bcpt.13535
121. Pai V, Paneerselvam A, Mukhopadhyay S, Bhansali A, Kamath D, Shankar V, et al. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of saroglitazar 2 and 4 mg compared to pioglitazone 45 mg in diabetic dyslipidemia (PRESS V). J Diabetes Sci Technol (2014) 8:132–41. doi: 10.1177/1932296813518680
122. Li J, Wang T, Liu P, Yang F, Wang X, Zheng W, et al. Hesperetin ameliorates hepatic oxidative stress and inflammation via the PI3K/AKT-Nrf2-ARE pathway in oleic acid-induced HepG2 cells and a rat model of high-fat diet-induced NAFLD. Food Funct (2021) 12:3898–918. doi: 10.1039/d0fo02736g
123. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. N Engl J Med (2021) 385:1547–58. doi: 10.1056/nejmoa2036205
124. Mantovani A, Byrne CD, Targher G. Efficacy of peroxisome proliferator-activated receptor agonists, glucagon-like peptide-1 receptor agonists, or sodium-glucose cotransporter-2 inhibitors for treatment of non-alcoholic fatty liver disease: a systematic review. Lancet Gastroenterol Hepatol (2022) 7:367–78. doi: 10.1016/s2468-1253(21)00261-2
125. Grover GJ, Mellström K, Ye L, Malm J, Li Y-L, Bladh L-G, et al. Selective thyroid hormone receptor-β activation: A strategy for reduction of weight, cholesterol, and lipoprotein (a) with reduced cardiovascular liability. Proc Natl Acad Sci (2003) 100:10067–72. doi: 10.1073/pnas.1633737100
126. Johansson L, Rudling M, Scanlan TS, Lundåsen T, Webb P, Baxter J, et al. Selective thyroid receptor modulation by GC-1 reduces serum lipids and stimulates steps of reverse cholesterol transport in euthyroid mice. Proc Natl Acad Sci (2005) 102:10297–302. doi: 10.1073/pnas.0504379102
127. Morkin E, Pennock GD, Spooner PH, Bahl JJ, Goldman S. Clinical and experimental studies on the use of 3,5-diiodothyropropionic acid, a thyroid hormone analogue, in heart failure. Thyroid (2002) 12:527–33. doi: 10.1089/105072502760143935
128. Mahapatra MK, Karuppasamy M, Sahoo BM, Bahl JJ. Semaglutide, a glucagon like peptide-1 receptor agonist with cardiovascular benefits for management of type 2 diabetes. Rev Endocr Metab Disord (2022) 23(3):521–39. doi: 10.1007/s11154-021-09699-1
129. Mantovani A, Petracca G, Beatrice G, Csermely A, Lonardo A, Targher G. Glucagon-like peptide-1 receptor agonists for treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: An updated meta-analysis of randomized controlled trials. Metabolites (2021) 11:73. doi: 10.3390/metabo11020073
130. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American association for the study of liver diseases. Hepatology (2018) 67:328–57. doi: 10.1002/hep.29367
131. Jani RH, Kansagra K, Jain MR, Patel H. Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects. Clin Drug Invest (2013) 33:809–16. doi: 10.1007/s40261-013-0128-3
132. Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K, et al. Saroglitazar, a PPAR-α/γ agonist, for treatment of NAFLD: A randomized controlled double-blind phase 2 trial. Hepatology (2021) 74:1809–24. doi: 10.1002/hep.31843
133. Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes (2000) 49:2063–9. doi: 10.2337/diabetes.49.12.2063
134. Inzucchi SE, Maggs DG, Spollett GR, Page SL, Rife FS, Walton V, et al. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med (1998) 338:867–73. doi: 10.1056/nejm199803263381303
135. Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: An update. Ann Intern Med (2002) 137:25. doi: 10.7326/0003-4819-137-1-200207020-00009
136. Feng W, Bi Y, Li P, Yin T, Gao C, Shen S, et al. Effects of liraglutide, metformin and gliclazide on body composition in patients with both type 2 diabetes and non-alcoholic fatty liver disease: A randomized trial. J Diabetes Invest (2019) 10:399–407. doi: 10.1111/jdi.12888
137. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest (2001) 108:1167–74. doi: 10.1172/jci13505
138. Geng Y, Villanueva AH, Oun A, Buist-Homan M, Blokzijl H, Faber KN, et al. Protective effect of metformin against palmitate-induced hepatic cell death. Biochim Et Biophys Acta Bba - Mol Basis Dis (2020) 1866:165621. doi: 10.1016/j.bbadis.2019.165621
139. Cryer B, Goldschmiedt M, Redfern JS, Feldman M. Comparison of salsalate and aspirin on mucosal injury and gastroduodenal mucosal prostaglandins. Gastroenterology (1990) 99:1616–21. doi: 10.1016/0016-5085(90)90465-d
140. Scheiman JM, Elta GH. Gastroduodenal mucosal damage with salsalate versus aspirin: Results of experimental models and endoscopic studies in humans. Semin Arthritis Rheu (1990) 20:121–7. doi: 10.1016/0049-0172(90)90025-b
141. Meex RCR, Phielix E, Moonen-Kornips E, Schrauwen P, Hesselink MKC. Stimulation of human whole-body energy expenditure by salsalate is fueled by higher lipid oxidation under fasting conditions and by higher oxidative glucose disposal under insulin-stimulated conditions. J Clin Endocrinol Metab (2011) 96:1415–23. doi: 10.1210/jc.2010-1816
142. Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, et al. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci (2008) 1:36–43. doi: 10.1111/j.1752-8062.2008.00026.x
143. Cheng Q, Li N, Chen M, Zheng J, Qian Z, Wang X, et al. Cyclooxygenase-2 promotes hepatocellular apoptosis by interacting with TNF-α and IL-6 in the pathogenesis of nonalcoholic steatohepatitis in rats. Digest Dis Sci (2013) 58:2895–902. doi: 10.1007/s10620-013-2823-6
144. Janssen AWF, Houben T, Katiraei S, Dijk W, Boutens L, van der BN, et al. Modulation of the gut microbiota impacts nonalcoholic fatty liver disease: a potential role for bile acids. J Lipid Res (2017) 58:1399–416. doi: 10.1194/jlr.m075713
145. Gangarapu V, Ince AT, Baysal B, Kayar Y, Kiliç U, Gök Ö, et al. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur J Gastroen Hepat (2015) 27:840–5. doi: 10.1097/meg.0000000000000348
146. Vallianou N, Stratigou T, Christodoulatos GS, Dalamaga M. Understanding the role of the gut microbiome and microbial metabolites in obesity and obesity-associated metabolic disorders: Current evidence and perspectives. Curr Obes Rep (2019) 8:317–32. doi: 10.1007/s13679-019-00352-2
147. Koopman N, Molinaro A, Nieuwdorp M, Holleboom AG. Review article: can bugs be drugs? the potential of probiotics and prebiotics as treatment for non-alcoholic fatty liver disease. Aliment Pharm Therap (2019) 50:628–39. doi: 10.1111/apt.15416
148. Yan F, Cao H, Cover TL, Whitehead R, Washington MK, Polk DB. Soluble proteins produced by probiotic bacteria regulate intestinal epithelial cell survival and growth. Gastroenterology (2007) 132:562–75. doi: 10.1053/j.gastro.2006.11.022
149. Neyrinck AM, Possemiers S, Verstraete W, Backer FD, Cani PD, Delzenne NM. Dietary modulation of clostridial cluster XIVa gut bacteria (Roseburia spp.) by chitin–glucan fiber improves host metabolic alterations induced by high-fat diet in mice. J Nutr Biochem (2012) 23:51–9. doi: 10.1016/j.jnutbio.2010.10.008
150. Govaere O, Petersen SK, Martinez-Lopez N, Wouters J, Haele MV, Mancina RM, et al. Macrophage scavenger receptor 1 mediates lipid-induced inflammation in non-alcoholic fatty liver disease. J Hepatol (2022) 76:1001–12. doi: 10.1016/j.jhep.2021.12.012
151. Scheen AJ. Sodium–glucose cotransporter type 2 inhibitors for the treatment of type 2 diabetes mellitus. Nat Rev Endocrinol (2020) 16:556–77. doi: 10.1038/s41574-020-0392-2
152. Mantovani A, Petracca G, Csermely A, Beatrice G, Targher G. Sodium-glucose cotransporter-2 inhibitors for treatment of nonalcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Metabolites (2020) 11:22. doi: 10.3390/metabo11010022
153. Mauro SD, Scamporrino A, Petta S, Urbano F, Filippello A, Ragusa M, et al. Serum coding and noncoding RNAs as biomarkers of NAFLD and fibrosis severity. Liver Int (2019) 39:1742–54. doi: 10.1111/liv.14167
154. Zhang J, Tan J, Wang M, Wang Y, Dong M, Ma X, et al. Lipid-induced DRAM recruits STOM to lysosomes and induces LMP to promote exosome release from hepatocytes in NAFLD. Sci Adv (2021) 7:eabh1541. doi: 10.1126/sciadv.abh1541
155. Yan C, Tian X, Li J, Liu D, Ye D, Xie Z, et al. A high-fat diet attenuates AMPK α1 in adipocytes to induce exosome shedding and nonalcoholic fatty liver development in vivo. Diabetes (2021) 70:577–88. doi: 10.2337/db20-0146

Keywords: non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), gut microbiota, metabolic diseases, adenosine monophosphate-activated protein kinase
Citation: Che W, Zhao M, Li X, Li C, Cho WC and Yu S (2022) Current insights in molecular characterization of non-alcoholic fatty liver disease and treatment. Front. Endocrinol. 13:1002916. doi: 10.3389/fendo.2022.1002916
Received: 25 July 2022; Accepted: 14 October 2022;
Published: 29 November 2022.
Edited by:
Andrea Dalbeni, Verona University Hospital, ItalyCopyright © 2022 Che, Zhao, Li, Li, Cho and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunlong Li, Y2h1bmxvbmc4MUAxNjMuY29t; William C. Cho, d2lsbGlhbWNzY2hvQGdtYWlsLmNvbQ==; Y2hvY3NAaGEub3JnLmhr; Shan Yu, eXVzaGFuQGhyYm11LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.