 Jamila Zammouri1Camille Vatier1,2Emilie Capel1Martine Auclair1Caroline Storey-London3Elise Bismuth3Héléna Mosbah1,2Bruno Donadille1,2Sonja Janmaat1,2Bruno Fève1,2
Jamila Zammouri1Camille Vatier1,2Emilie Capel1Martine Auclair1Caroline Storey-London3Elise Bismuth3Héléna Mosbah1,2Bruno Donadille1,2Sonja Janmaat1,2Bruno Fève1,2 Isabelle Jéru1,2,4
Isabelle Jéru1,2,4 Corinne Vigouroux1,2,4*
Corinne Vigouroux1,2,4*- 1Sorbonne University, Inserm UMR_S 938, Saint–Antoine Research Centre, Cardiometabolism and Nutrition University Hospital Institute (ICAN), Paris, France
- 2Endocrinology Department, Assistance Publique–Hôpitaux de Paris, Saint–Antoine Hospital, National Reference Centre for Rare Diseases of Insulin Secretion and Insulin Sensitivity (PRISIS), Paris, France
- 3Assistance Publique–Hôpitaux de Paris, Robert Debré Hospital, Pediatric Endocrinology Department, National Competence Centre for Rare Diseases of Insulin Secretion and Insulin Sensitivity (PRISIS), Paris, France
- 4Genetics Department, Assistance Publique–Hôpitaux de Paris, La Pitié-Salpêtrière Hospital, Paris, France
Lipodystrophy syndromes are rare diseases originating from a generalized or partial loss of adipose tissue. Adipose tissue dysfunction results from heterogeneous genetic or acquired causes, but leads to similar metabolic complications with insulin resistance, diabetes, hypertriglyceridemia, nonalcoholic fatty liver disease, dysfunctions of the gonadotropic axis and endocrine defects of adipose tissue with leptin and adiponectin deficiency. Diagnosis, based on clinical and metabolic investigations, and on genetic analyses, is of major importance to adapt medical care and genetic counseling. Molecular and cellular bases of these syndromes involve, among others, altered adipocyte differentiation, structure and/or regulation of the adipocyte lipid droplet, and/or premature cellular senescence. Lipodystrophy syndromes frequently present as systemic diseases with multi-tissue involvement. After an update on the main molecular bases and clinical forms of lipodystrophy, we will focus on topics that have recently emerged in the field. We will discuss the links between lipodystrophy and premature ageing and/or immuno-inflammatory aggressions of adipose tissue, as well as the relationships between lipomatosis and lipodystrophy. Finally, the indications of substitutive therapy with metreleptin, an analog of leptin, which is approved in Europe and USA, will be discussed.
Introduction
Lipodystrophy syndromes are rare diseases characterized by generalized or segmental lack of adipose tissue, and by insulin resistance-related metabolic complications such as diabetes, hypertriglyceridemia, hepatic steatosis, and ovarian hyperandrogenism in women. Besides their different clinical presentation with generalized or partial lipoatrophy, accompanied or not by fat overgrowth in other body areas, lipodystrophy syndromes are highly heterogeneous diseases in several other aspects. Specific subtypes of lipodystrophy are associated with additional clinical signs and complications, with, among others, neurological or cardiovascular involvement, showing that lipodystrophy syndromes are frequently complex multisystem diseases (1–3). The onset of lipodystrophy may be precocious, in early infancy, or delayed in late childhood or adulthood. This review will mainly focus on genetic forms of lipodystrophies. Other forms of lipodystrophies, that will not be covered by this review, result from iatrogenic therapies and/or other environmental factors. This is the case for HIV-related lipodystrophies, due to multifactorial mechanisms resulting, among others, from HIV infection and antiretroviral agents (4). Glucocorticoid therapy leads to body fat redistribution and insulin resistance (5). The identification of causative pathogenic variants in more than 20 genes leading to monogenic forms of lipodystrophies has highlighted several determinants of adipose tissue pathophysiology. This field of research, still highly productive, indicates adipose tissue as a major actor to ensure proper whole-body insulin sensitivity (3, 6).
Main Molecular Causes of Lipodystrophy Syndromes and Their Impact on Adipose Tissue Functions and Insulin Response
Main Molecular Causes of Lipodystrophy Syndromes
Lipodystrophy syndromes include different congenital to adult-onset diseases, with either generalized or partial lipoatrophy. More than 20 genes are involved in monogenic lipodystrophy syndromes (6–8). Although lipodystrophy syndromes have been considered as ultra-rare diseases, with a prevalence of less than 5 cases per million (9), they are largely underdiagnosed, and systematic genetic screening suggests that 1/7000 individuals could be affected, with a majority of partial forms (10). Table 1 indicates the main monogenic lipodystrophy syndromes, their specific phenotypic features and the main functions of involved genes. The diversity of molecular causes of lipodystrophy reflects both clinical heterogeneity and close pathophysiological relationships of these diseases. Indeed, beyond the diversity of clinical forms, lipodystrophy syndromes share adipose tissue dysfunction as a key pathophysiological feature, with gene pathogenic variants mostly affecting adipocyte development, differentiation and/or functions (Figure 1).
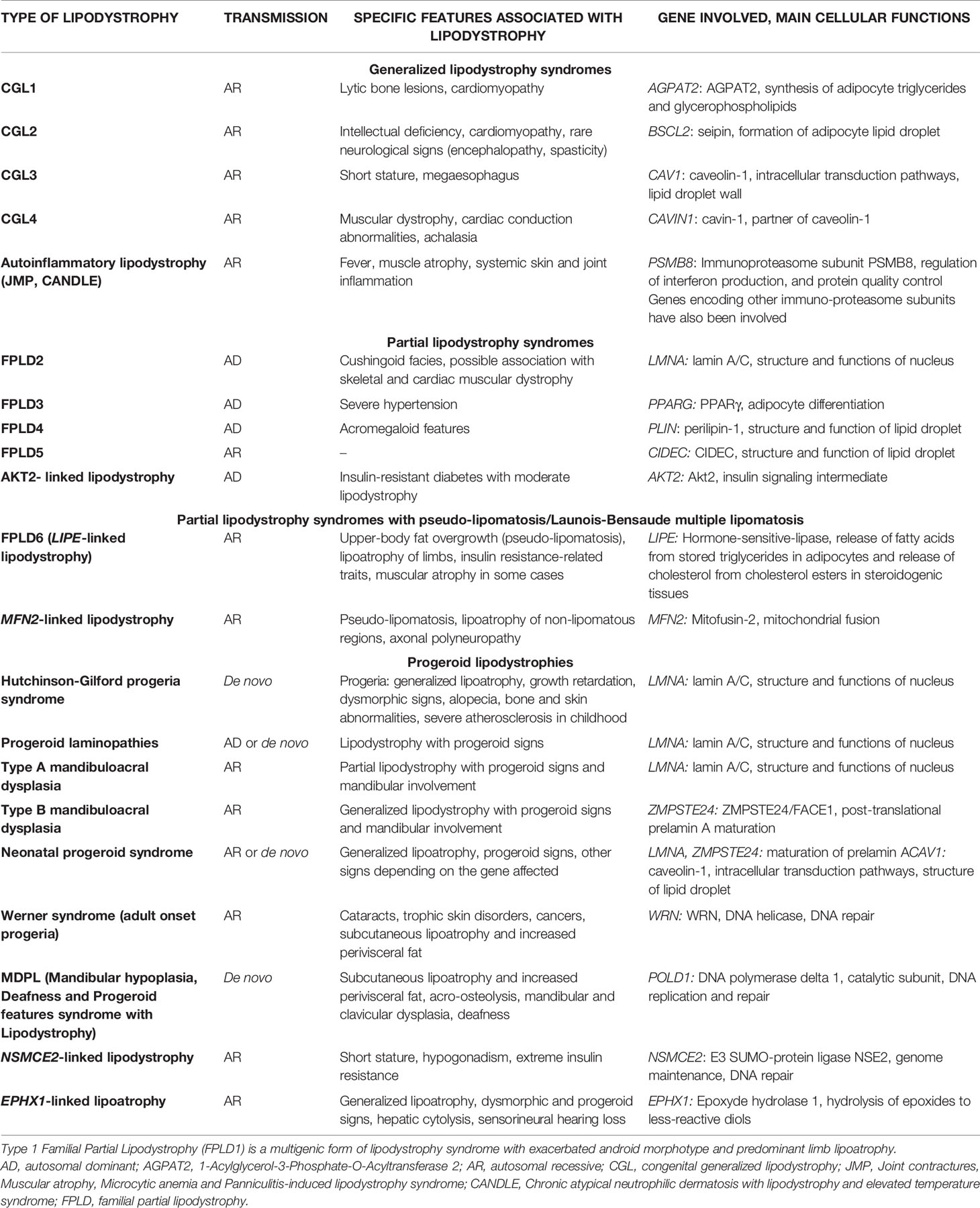
Table 1 Main forms of monogenic lipodystrophy syndromes.
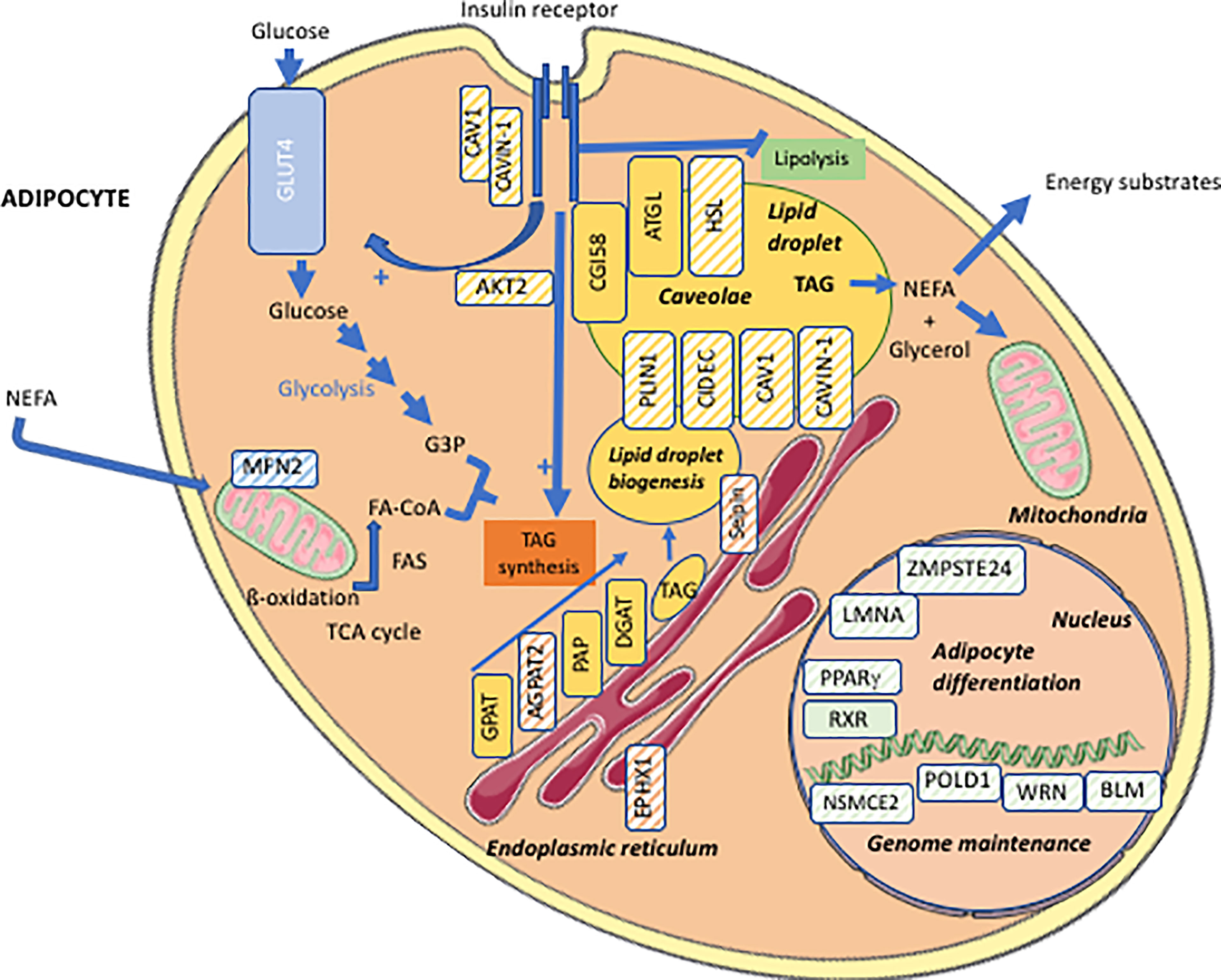
Figure 1 Cellular targets of the main molecular defects responsible for lipodystrophy syndromes. Adipocyte schematic representation with localization of the main proteins involved in the molecular pathophysiology of lipodystrophy syndromes (hatched symbols). AGPAT2, 1-acylglycerol-3-phosphate-O-acyltransferase 2; AKT2, serine/threonine-protein kinase 2; ATGL, adipose triglyceride lipase; BLM, Bloom syndrome protein; CAV1, caveolin-1; CAVIN1, cavin-1; CGI58, comparative gene identification-58, also known as α/β-hydrolase domain-containing 5 (ABHD5); DGAT, diacylglycerol acyltransferase; EPHX1, epoxide hydrolase 1; FA, fatty acid; FA-CoA, fatty acid-coenzyme A; FAS, fatty acid synthase; G3P, glycerol-3-phosphate; GLUT4, glucose transporter 4; GPAT, glycerol-3-phosphate acyltransferase; HSL, hormone-sensitive lipase; LMNA, lamin A/C; MFN2, mitofusin-2; NEFA, non-esterified fatty acids; NSMCE2, E3 SUMO-protein ligase NSE2; PAP, phosphatidic acid phosphatase; PLIN1, perilipin-1; POLD1, DNA polymerase delta 1, catalytic subunit; PPARγ, peroxisome proliferator-activated receptor gamma; RXR, retinoid X receptor; TAG, triacylglycerol; TCA cycle, tricarboxylic acid cycle; WRN, WRN RecQ like helicase.
Congenital generalized lipodystrophy syndromes (CGL or Berardinelli-Seip Congenital Lipodystrophy) are autosomal recessive diseases, mainly observed in patients from consanguineous families. They are mainly due to null variants in AGPAT2 encoding 1-acylglycerol-3-phosphate-O-acyltransferase 2, involved in triglyceride and phospholipid synthesis, or in BSCL2 encoding seipin, an endoplasmic reticulum membrane protein which contributes to lipid droplet biogenesis (11–16). CGL3 and CGL4 are due to genetic defects in caveolin-1 or cavin-1 respectively, involved in the formation of cell plasma membrane microdomains called caveolae, that initiate several signaling pathways. Caveolin-1 and cavin-1 are also localized at the adipocyte lipid droplet and contribute to intracellular fluxes of lipids (17, 18) (Figure 2). Most familial partial lipodystrophies (FPLD) are transmitted as autosomal dominant diseases due to loss-of-function or dominant negative mutations, with initial clinical manifestations occurring from late childhood onwards. Apart FPLD1, which is probably a multigenic form of lipodystrophy syndrome (19), FPLD2 due to LMNA pathogenic variants, is the most frequent form of genetically determined partial lipodystrophy (10). Generalized or partial lipoatrophy is a clinical feature of several diseases with accelerated ageing called progeroid syndromes (Table 1).
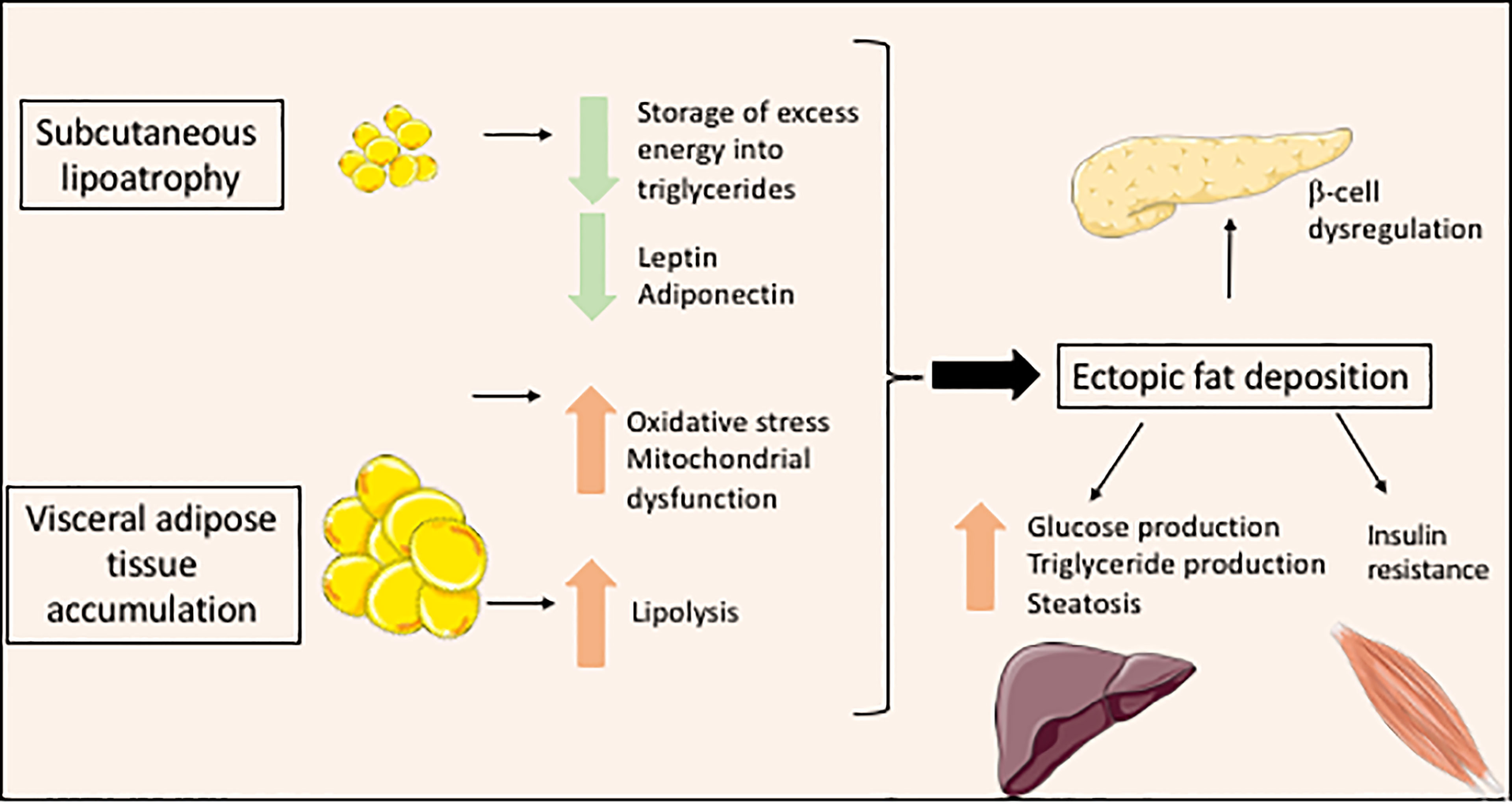
Figure 2 Metabolic consequences of lipodystrophy leading to cellular lipotoxicity.
Lipodystrophy and Lipotoxicity
Lipoatrophy is a main contributor of metabolic complications associated with lipodystrophy syndromes. Adipocytes represent 20 to 40% of the cell population of adipose tissue and 90% of adipose tissue volume (20, 21). They arise from mesenchymal stem cells, adipocyte differentiation being carried out under the control of several adipogenic transcription factors. PPARγ is a major adipogenic factor, which regulates the expression of several genes of lipid metabolism and modulates both insulin sensitivity and secretory functions of adipocytes (21, 22). White adipocytes are the most abundant adipocytes in humans. They are characterized by a single voluminous lipid vacuole surrounding a neutral lipid core mainly composed of triglycerides and cholesterol esters. The adipocyte lipid droplet is coated with a monolayer of phospholipids and with proteins belonging to the perilipin family, which play important structural and functional roles. Indeed, white adipocytes have a crucial role in the regulation of energy balance and systemic metabolic homeostasis. In response to nutritional and hormonal signals, the adipocyte lipid droplet is able to store excess energy as triglycerides in the postprandial state, and then to release fatty acids from triglycerides, providing energy substrates for other organs. Adequate storage of energy in adipocytes protects other organs from lipotoxicity due to lipid overload, which leads to oxidative stress, mitochondrial dysfunction, apoptosis and tissue dysfunction (23). In lipodystrophy syndromes, the very limited adipose tissue expandability, due to the major decrease in the capacity of adipose tissue to store lipids, exposes non-adipose organs to lipotoxicity even in well-balanced diet conditions. This results in muscle insulin resistance due to disruption of insulin signaling (24), in non-alcoholic fatty liver disease, as well as in myocardial, endothelial and pancreatic beta-cell dysfunction (25–27). Adipocytes are also important autocrine, paracrine, and endocrine cells that produce numerous adipokines. Among others, leptin regulates satiety by acting on hypothalamic neurons, and modulates carbohydrate and lipid metabolism by acting on muscle, liver adipose tissue and pancreatic beta-cells (28, 29). Integrated effects of leptin ensure an efficient protection of non-adipose tissues from lipid accumulation (30). Adiponectin produced by adipose tissue increases fatty acid oxidation and glucose transport in muscle, and decreases hepatic gluconeogenesis. In lipodystrophy syndromes, the lack of functional subcutaneous fat drives multiple metabolic alterations resulting from altered metabolic and secretory functions of adipocytes. Decreased adipose tissue expandability, and leptin deficiency induce ectopic accumulation of fat upon increased energy intake, even during the physiological postprandial state (Figure 2). Furthermore, decreased serum levels of adiponectin, although not to the same extent in the different forms of lipodystrophy (31), contribute to insulin resistance and hepatic steatosis associated with lipodystrophy (32). Altered production of other adipokines by lipodystrophic adipose tissue has also been described in several studies, mostly related to HIV-related forms of lipodystrophy (4). It could lead to adipose tissue inflammation and fibrosis, and to a state of systemic low-grade inflammation with insulin resistance (33). In lipodystrophy syndromes, insulin signaling pathways are strongly impacted by mechanisms linked to cellular lipotoxicity and metabolic inflexibility (23, 24). Increased lipid fluxes activate hepatic production of very-low-density lipoproteins, triglycerides and glucose, and impair muscular glucose uptake (6, 34, 35). Leptin deficiency also increases appetite, which worsens metabolic profile. In some forms of partial lipodystrophies, redistribution of body fat, with increased visceral fat and decreased subcutaneous fat, particularly of the lower limbs, also contributes to metabolic dysfunction. Indeed, subcutaneous adipose tissue is physiologically more sensitive to insulin. It has been shown that subcutaneous adipose tissue of the lower part of the body is protective against diabetes and cardiovascular diseases in the general population (36, 37). Conversely, visceral adipose tissue is more sensitive than subcutaneous adipose tissue to adipocyte lipolysis. Furthermore, visceral adipose tissue directly releases fatty acids into the portal vein during lipolysis, which are captured by the liver, leading to an increased risk of liver lipotoxicity, liver steatosis and insulin resistance (Figure 2). Visceral fat is more susceptible to chronic inflammation and fibrosis, and produces lower amounts of leptin than subcutaneous tissue (38). Excess visceral, but not subcutaneous fat, is involved in adiposity-related hypertension (39). Mitochondrial dysfunction and oxidative stress, which are frequently observed cellular consequences of lipodystrophy, also decrease cellular responses to insulin (40–42) (Figure 2).
Main Clinical Features of Generalized and Partial Lipodystrophies
The diagnosis of lipodystrophy syndromes is based first and foremost on clinical examination. Since objective clinical measures are still lacking to document an abnormal development of subcutaneous fat, the clinical diagnosis of lipodystrophy is highly dependent on the clinician’s self-experience. It can be particularly difficult in men affected with partial lipodystrophy syndromes, in whom the diagnosis could be missed or substantially delayed. Indeed, the nosological framework is poorly defined between an android distribution of fat, commonly observed in the general population, which is a major risk factor for insulin resistance-related diseases (43), and the lipodystrophic phenotype. This is particularly striking for Type 1 Familial Partial Lipodystrophy (FPLD1), characterized by a central repartition of fat with lipoatrophy of limbs and severe insulin resistance, which is probably of polygenic origin (19). However, several clinical features are common to lipodystrophy syndromes, so that a distinctive clinical picture may be identified.
Lipoatrophy
The identification of generalized or segmental lipoatrophy, in the absence of malnutrition, is a major step for diagnosis.
In generalized forms of lipodystrophy syndromes, lipoatrophy of the face is striking. The patient’ facies is emaciated due to the absence of Bichat fat pads. Acromegaloid features, with protruding eyebrow arches, cheekbones, and lower jaw, and thick facial traits, are also observed, especially in congenital forms of lipoatrophy, but also in some partial forms of the disease (8, 44). These clinical signs are due to an increased visibility of bone structures in the absence of adipose tissue, and to the stimulation of growth factor pathways by excess insulin. Hands and feet may also be thickened. The lack of subcutaneous adipose tissue also increases the visibility of muscles (athletic appearance) and of veins (pseudo-veinomegaly) in limbs, trunk and abdomen. In addition, it should be noted that, at least in some cases, the volume and mass of skeletal muscle are truly increased (45, 46) (Figure 3A). Ingrown toenails are possible consequences of severe lipoatrophy of feet. Hypomastia is common in women, secondary to loss of breast adipose tissue and/or to hyperandrogenism.
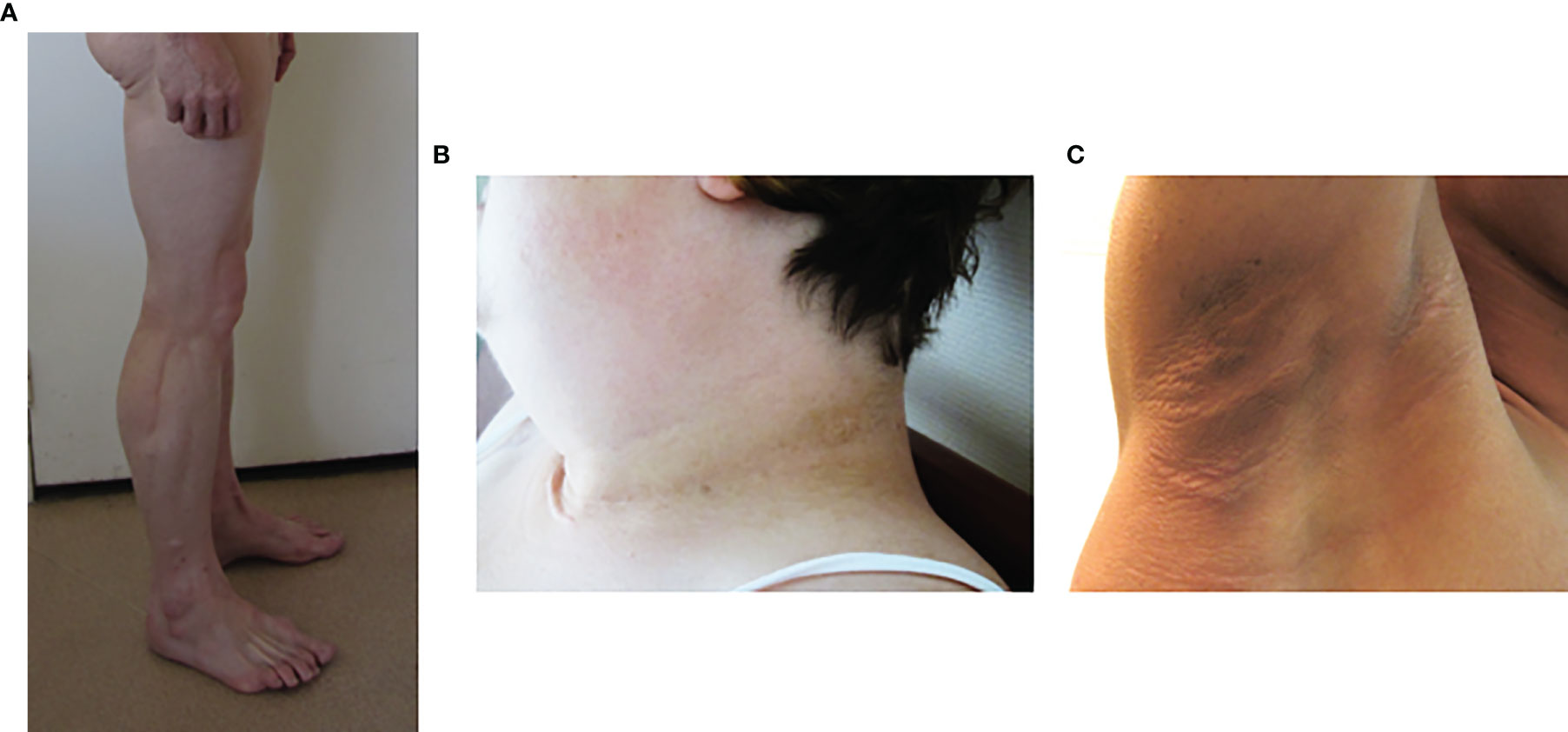
Figure 3 Phenotypic features of lipodystrophy syndromes. (A) Muscular hypertrophy and lipoatrophy of limbs in Type 2 Familial Partial Lipodystrophy (Dunnigan syndrome). (B, C) Cervical and axillary acanthosis nigricans in patients with lipodystrophy due to LMNA (B) or BSCL2 (C) pathogenic variants.
Fat Accumulation
In most forms of partial lipodystrophy syndromes, lipoatrophy mainly affects lower limbs and can coexist with areas of fat accumulation. Dunnigan syndrome (FPLD2), due to pathogenic variants in the LMNA gene encoding Type A lamins, is characterized by lipoatrophy of limbs and trunk with cushingoid features of face and neck, i.e. increased supraclavicular fat pads, double chin, buffalo hump, and with perineal accumulation of adipose tissue. The general morphotype is strikingly android, with a biacromial diameter greater than the bitrochanteric diameter (8, 47). Muscle hypertrophy, contrasting with the usual amyotrophy associated with hypercortisolism, may suggest the diagnosis of lipodystrophy syndrome (Figure 3A). Fat overgrowth may lead to massive pseudo-lipomatous regions in upper body and proximal limb areas, contrasting with lipoatrophy of non-lipomatous regions, in specific genetic forms of partial lipodystrophy (48–55). On the contrary, in Barraquer-Simons acquired partial lipodystrophy syndrome, a progressive lipoatrophy develops in upper parts of the body (face, trunk and upper limbs), while adipose tissue accumulates in lower limbs.
Insulin Resistance-Associated Clinical Signs
Acanthosis nigricans, a brownish hyperkeratotic thickening of the skin, and acrochorda (skin tags), are very common clinical signs of insulin resistance in lipodystrophy syndromes (Figures 3B, C). These skin lesions are usually located in cervical, axillary and inguinal folds, but may be very extensive in some patients. Insulin resistance frequently leads to ovarian hyperandrogenism in women, with hirsutism, irregular menses and polycystic ovaries (47, 56, 57). Hepatomegaly, resulting from dysmetabolic liver steatosis, is common. Hypertriglyceridemia can be complicated, or even revealed, by acute pancreatitis (7, 58).
Cardiovascular Signs
High blood pressure is frequent, and can be very severe, in particular in partial lipodystrophies associated with pathogenic variants of PPARG encoding the adipogenic factor PPARγ (peroxisome proliferator-activated receptor gamma) (59–61). The risk of atherosclerosis is strongly increased, which could result from insulin resistance, diabetes, and hypertension (62), but also from genetic variants that directly target the vascular wall, as observed in LMNA-related lipodystrophies (40, 63, 64). In addition to non-specific diabetic cardiomyopathy and atherosclerosis, several cardiovascular complications can be observed in the different forms of lipodystrophies. Patients with congenital generalized lipoatrophy may suffer from hypertrophic cardiomyopathy, with or without hypertension, associated with ectopic cardiac fat and/or lipotoxicity (65, 66). Pathogenic variants in LMNA are responsible for lipodystrophy syndromes with early atherosclerosis and/or with dilated cardiomyopathy, rhythm and/or conduction disorders and/or extensive calcifications of cardiac valves (67–72). A regular cardiovascular screening, with cardiac ultrasound and stress test, and, if needed, coronary CT angiogram, 24-hour ECG monitoring, and/or cardiac MRI is required in most patients with lipodystrophy syndromes (7, 8).
Other Clinical Signs
Depending on their molecular cause, lipodystrophy syndrome can be associated with several other clinical signs. Moderate intellectual disability can be observed in type 2 Congenital Generalized Lipoatrophy (CGL) due to BSCL2 pathogenic variants encoding seipin (11). Digestive signs are frequent in neonates or infants with CGL. In late infancy or adolescence, dysphagia can reveal megaesophagus, due to esophageal achalasia, in CGL due to pathogenic variants of CAVIN1 or CAV1, encoding proteins involved in the formation of caveolae at the cell plasma membrane (73–77). Growth disorders, dysmorphic features with micrognathia, beaked nose, dental crowding, prominent eyes, dystrophic bones, and/or signs suggesting accelerated aging such as precocious whitening and/or loss of hair, sclerodermatous skin appearance, joint limitations, and/or muscle atrophy are hallmarks of progeroid lipodystrophies (67, 78, 79). Other signs such as precocious cataracts, trophic skin disorders, hypogonadism, predisposition to cancer can be associated in progeroid lipodystrophy syndromes due to defects in DNA repair (80, 81). Muscle functional defects are observed in some patients with lipodystrophy due to pathogenic variants in LMNA, CAVIN1, or PSMB8, among other genes (67, 74, 82–85). Systemic inflammatory signs (fever, multiorgan inflammatory involvement affecting joints, skin, heart) are prominent features of lipodystrophies associated with rare autoinflammatory syndromes of genetic origin (86). The occurrence of some lipodystrophy syndromes in the context of panniculitis or autoimmune diseases, suggests that adipose tissue could be a target of immune tolerance disruption (87).
Biological Characteristics of Lipodystrophy Syndromes
Insulin resistance, resulting from adipose tissue dysfunction and subsequent ectopic lipid deposition, is one of the hallmarks of lipodystrophy syndromes. High serum insulin levels, with normal or increased plasma glucose, are detected in the fasting state and/or during oral glucose tolerance test. Patients with diabetes display preserved or even increased C-peptide levels for a long time, and/or frequently require very high doses of insulin to achieve glucose control. Hypertriglyceridemia is also very frequent, and can reach very high values, associated with a high risk of acute pancreatitis. Low HDL-cholesterol is almost always present. Increased liver enzymes are also common features, due to liver steatosis or fibrosis. Biological signs of adipose dysfunction include decreased serum adiponectin, and either barely detectable leptin levels in generalized lipodystrophies, or lower leptin levels than predicted by body mass index in partial lipodystrophies. Creatine phosphokinase may be elevated, especially in patients with lipodystrophies and muscular dystrophy. Ovarian hyperandrogenism results from insulin resistance, with high levels of total and free testosterone and of Δ4-androstenedione, decreased sex-hormone binding globulin, and increased luteinizing hormone to follicle-stimuling hormone ratio (7, 8).
Imaging Investigations
Although the diagnosis of lipodystrophy is mainly based on clinical examination, the objective measurement of fat mass by dual energy-ray absorptiometry (DEXA) is useful to determine the severity of lipoatrophy and, in partial forms of lipodystrophies, to document the altered segmental distribution of fat (Figure 4A). Abdominal ultrasound, computed tomodensitometry (CT), or magnetic resonance imagery (MRI) are required to search for liver steatosis and signs of cirrhosis (Figure 4B). In women, pelvic ultrasound may reveal ovaries of increased volume and/or with multiple follicles. Bone imaging can show precocious non-specific degenerative radiographic abnormalities in patients with familial partial lipodystrophies. Several bone lesions such as osteolysis, osteosclerosis or pseudo-osteopoikilosis are frequently present in generalized forms of lipodystrophy, and may lead to misdiagnosis (88, 89). Muscle imaging (CT or MRI) may show muscular hypertrophy and/or fatty degeneration, with lack of subcutaneous adipose tissue. Imaging investigations may be completed by an electromyogram to search for neuropathic and/or myopathic signs.
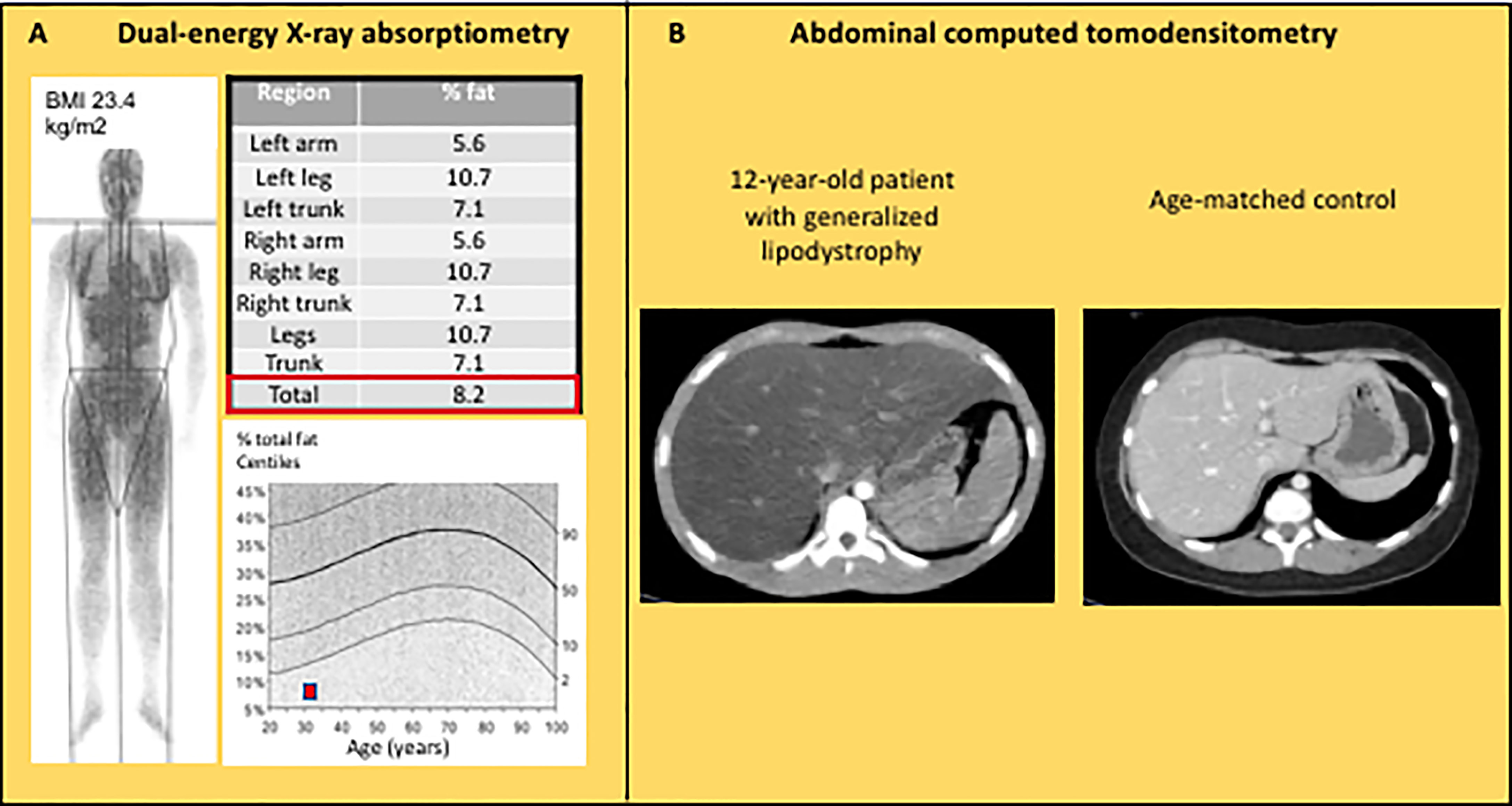
Figure 4 Imaging features in lipodystrophy syndromes. (A) Dual energy-ray absorptiometry (DEXA) in a 31 year-old patient with CGL1, showing a major decrease in total and segmental fat mass. (B) Abdominal computed tomodensitometry in a 12 year-old patient with acquired generalized lipodystrophy showing homogeneous hepatomegaly with low attenuation of the parenchyma (Hounsfield units: -13), and absence of subcutaneous adipose tissue.
Lipodystrophy and Ageing
Remodeling of body fat occurs during physiological ageing, with decrease in subcutaneous gluteofemoral adipose tissue, increase in visceral fat depots and ectopic deposition of lipids. Together with pro-inflammatory changes in adipose tissue, these lipodystrophy-like features contribute to age-related insulin resistance (90).
Accelerated ageing is probably one of the most important pathophysiological mechanisms of primary lipodystrophies. Arguments in favor of this hypothesis first came from studies of LMNA-associated diseases. LMNA encodes Type A lamins, nuclear proteins that interact with chromatin and regulate several nuclear functions including epigenetic cell developmental programs (91) (Figure 1). LMNA pathogenic variants cause several different laminopathies including muscular dystrophy, cardiomyopathy, neuropathy, lipodystrophy, and syndromes of accelerated aging (progeria and progeroid syndromes). Both typical FPLD2/Dunnigan syndrome, characterized by partial lipodystrophy and insulin resistance-related complications, and the extremely severe Hutchinson-Gilford accelerated ageing syndrome with generalized lipoatrophy are due to LMNA variants. In addition to lipodystrophy, several other clinical features, although of very different severity, are part of the clinical spectrum of both diseases. This is the case for early atherosclerosis, leading to frequent cardiovascular events before the age of 50 in patients with FPLD2, and to death at a mean age of 15 in Hutchinson-Gilford progeria (63, 92). In Hutchinson-Gilford progeria, but also in FPLD2, early atherosclerosis is due not only to metabolic risk factors, but also to direct pro-senescent effects of LMNA pathogenic variants on endothelial and vascular smooth muscle cells (40, 63, 93, 94). Clinical features of accelerated ageing are observed in patients with complex progeroid forms of LMNA-linked lipodystrophies, with a large continuum of severity between Dunnigan syndrome and Hutchinson-Gilford progeria (67, 71). At the cellular level, LMNA pathogenic variants impair the fate of several mesodermal lineages, such as endothelial vascular cells (64), myoblasts (95), cardiomyocytes (96), and adipocytes (97–101), and are involved in several signaling pathways which accelerate aging processes (102, 103).
The relationships between lipodystrophy and increased cellular senescence were further demonstrated by the identification of several pathogenic variants in genes involved in DNA repair as molecular causes of lipodystrophy syndromes (Table 1 and Figure 1). As discussed above, lipodystrophies due to genetic alterations in DNA repair are clinically expressed as progeroid syndromes, with lipodystrophy, insulin resistance and related metabolic alterations, and signs of premature ageing. However, additional clinical features, specific to each different genetic subtype, may be part of the phenotype. Among others, cataracts, which are not part of the phenotype of LMNA-associated progeroid syndromes, are a typical feature of Werner progeria syndrome (81) and MDPL (Mandibular hypoplasia, Deafness, Progeroid features, and Lipodystrophy) syndrome (80, 104, 105). These diseases are due, respectively, to biallelic pathogenic variants in WRN encoding the WRN DNA helicase and exonuclease, and to heterozygous loss-of-function variants in POLD1, encoding a catalytic subunit of DNA polymerase δ, both enzymes playing a major role in maintaining genome stability. Lipodystrophy is associated with a predisposition to cancer in Werner syndrome, as well as in Bloom syndrome, due to mutations in BLM encoding a DNA helicase, or in ataxia-telangiectasia with altered DNA synthesis and excision-repair pathways (106). Cultured fibroblasts from patients with Werner or MDPL syndromes present signs of premature senescence (81, 107). Importantly, premature senescence was shown to impair adipogenesis in human pluripotent stem cells lacking either WRN or BLM helicases (108). Several other DNA replication/repair-associated lipodystrophies have been described (7, 78, 109, 110), frequently associated with short stature, hypogonadism, and trophic skin disorders, among other progeroid signs.
Premature senescence and oxidative stress, directly resulting from defects in genes involved in cellular ageing and/or genome stability, or from consequences of other genetic defects impacting adipocytes (79), are thus probably important pathophysiological determinants of lipodystrophies.
Lipodystrophy and Immuno-Inflammatory Diseases
Lipodystrophy syndromes can develop during the course of systemic immune and/or inflammatory diseases, suggesting that adipose tissue dysfunction could result from auto-immune and/or inflammatory damages. This is the case in CANDLE (Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature) syndrome and related auto-inflammatory genetic diseases, due to genetic defects in immunoproteasome subunits (86, 111). Dysregulation of the interferon pathway is a key pathophysiological link between the molecular causes of these diseases and their clinical expression (112). Lipodystrophy with metabolic complications is also part of the phenotype of several autoimmune diseases. Generalized lipodystrophy with severe hyperinsulinemia and leptin deficiency has been described in a child with APECED (Autoimmune PolyEndocrinopathy-Candidiasis-Ectodermal Dystrophy) due to pathogenic variant in AIRE resulting in the disruption of immune tolerance (113). The term “acquired lipodystrophy”, which designates lipodystrophy syndromes without any known genetic cause, underlies several auto-immune diseases with adipose tissue involvement (114–116). Autoantibodies directed against perilipin-1, an important lipid droplet protein which regulates lipolysis, could be involved in adipocyte dysfunction (87). Barraquer-Simons partial lipodystrophy syndrome, which predominantly affects women, is associated in one third of the cases with decreased C3 and/or C4 complement factors, and with membranoproliferative glomerulopathy due to C3 nephritic autoantibodies (117, 118).
Recently, lipodystrophy syndromes have been reported during the course of targeted cancer therapy using immune checkpoint inhibitors (119–122). These agents, by releasing nonspecific immunosuppressive pathways, are known to lead to multiorgan auto-immune related adverse events. Inflammatory features and infiltration of adipose tissue with CD3+ and/or cytotoxic CD8+ lymphocytes have been identified in anti-PD1-related lipodystrophy syndromes, strongly suggesting that disrupted immune tolerance to adipocyte self-antigens could be the cause of lipodystrophy (119–122).
To note, HIV-related lipodystrophies which are reviewed elsewhere (4), also result from altered adipocyte differentiation and inflammatory dysregulation (123).
Lipodystrophy and Lipomatosis: Some Shared Pathophysiological Mechanisms?
In specific genetic forms of partial lipodystrophy, due to MFN2 or LIPE biallelic pathogenic variants, fat overgrowth may lead to massive pseudo-lipomatous regions in upper body and proximal limb areas, leading to the diagnosis of multiple symmetric lipomatosis. However, recent studies have shown that fat accumulates in non-encapsulated pseudo-lipomas, which differ from the organization of typical lipomas, and that patients also present with lipoatrophy of non-pseudo-lipomatous region and with lipodystrophy-related metabolic complications. Therefore, multiple symmetric lipomatosis could be, at least in MFN2 and LIPE-related forms, an exacerbated form of partial lipodystrophy (49–55).
MFN2-associated lipodystrophy is a mitochondrial disease due to biallelic pathogenic variants including a specific MFN2 p.Arg707Trp substitution. MFN2 encodes mitofusin-2, a transmembrane protein of the outer mitochondrial membrane whose oligomerization drives mitochondrial fusion (124) (Figure 1). Lipoatrophy, low serum leptin and adiponectin levels, as well as adipose tissue mitochondrial defects, oxidative stress and increased expression of some thermogenic markers, provide evidence of adipose tissue dysfunction in patients with MFN2-associated lipodystrophy (52, 54). Apart from lipodystrophy/pseudo-lipomatosis, patients may present with other clinical signs of mitochondrial involvement, including Charcot-Marie-Tooth peripheral neuropathy.
LIPE biallelic pathogenic variants may also lead to pseudo-lipomatous forms of partial lipodystrophy (49, 50, 53, 55). LIPE encodes the key lipolytic enzyme hormone-sensitive lipase (HSL) (Figure 1), and LIPE pathogenic variants leading to lipodystrophy act through loss-of-function mechanisms. Functional studies using adipose stem cells have shown that defective lipolysis and impaired adipocyte differentiation, but also mitochondrial dysfunction, could contribute to pathophysiological mechanisms in LIPE-related lipodystrophy syndrome (55). Importantly, although the metabolic phenotype and potential lipodystrophy signs were not systematically investigated, some patients carrying mtDNA mutations responsible for the myoclonic epilepsy and ragged red fibers (MERRF) syndrome were also reported with multiple symmetric lipomatosis (125). Mitochondrial defects have also been described in the most frequent form of multiple symmetric lipomatosis, associated with excessive alcohol consumption (126).
Therefore, mitochondrial alterations could lead to both pseudo-lipomatous and/or lipodystrophic diseases. Whether other mitochondriopathies induce lipodystrophic diseases in humans, as shown in mice, requires further investigations (127).
Metreleptin Treatment of Metabolic Complications Associated With Lipodystrophy Syndromes
Lipodystrophy syndromes are multi-tissue diseases, which require a multidisciplinary management. Regarding metabolic alterations, dietary measures and physical activity are very important therapeutic tools. Indeed, avoiding a positive energy balance leading to ectopic lipid infiltration is a major objective to prevent and/or treat metabolic alterations in the context of adipose tissue failure (7). To date, no medication has proven to be effective to cure lipoatrophy. Metformin is frequently used as a first-line pharmacological drug to decrease insulin resistance. Statins are frequently prescribed to reduce the cardiovascular risk, and fenofibrate can be added in case of insufficient response on triglycerides. Medium chain fatty acid supplementation is used to reduce hypertriglyceridemia in children. Very limited data are available regarding the effects of antidiabetic drugs in patients with lipodystrophic diabetes. Very high doses of insulin therapy are frequently used, due to severe insulin resistance.
The orphan drug metreleptin, a recombinant leptin analog, is the only specific treatment for the metabolic complications of lipodystrophy syndromes. Metreleptin, administered by subcutaneous injection once daily, is used as a hormone replacement therapy in patients with leptin deficiency. Metreleptin therapy obtained a marketing authorization to treat the complications of leptin deficiency in patients with lipodystrophy in Japan in 2013, in USA in 2014 (for generalized forms), and in Europe in 2018. Although metreleptin was not studied in placebo-controlled trials in the context of rare diseases, and although it does not lead to the reconstitution of lacking adipose tissue, it was shown effective, as an adjunct to diet, on metabolic and hepatic parameters in generalized lipodystrophy syndromes (Figure 5). In patients with lipodystrophy, metreleptin replacement therapy prevents ectopic storage of lipids, by decreasing food intake due to central effects, and by directly increasing peripheral insulin sensitivity (128–131). Metreleptin therapy has been shown to increase insulin sensitivity and insulin secretion, to reduce hypertriglyceridemia, hyperglycemia, HbA1c and fatty liver disease, and to improve quality of life (3, 6, 7, 130, 132–134) (Figure 5). Other recent studies have shown that metreleptin could also improve cardiac hypertrophy by reducing lipotoxicity and glucose toxicity (135), and decrease mortality risk in patients with lipodystrophy (134). In accordance with the different severity of leptin deficiency, metreleptin therapy seems more efficient in generalized than in partial forms of lipodystrophy (136). In a recent post-hoc statistical analysis of two clinical trials conducted at NIH, HbA1c improved by a mean 2.16 percentage point after 12 months of treatment with metreleptin in patients with generalized forms of lipodystrophy (n=59), but only by a mean 0.61 percentage point in patients with partial lipodystrophy (n=36) (137). Therefore, in patients with partial lipodystrophies, metreleptin is recommended in selected patients, with severe hypoleptinemia, HbA1c > 8% and/or serum triglyceride > 500 mg/dL, for whom standard treatments have failed to achieve adequate metabolic control (7). Further studies are needed to determine specific predictive factors for metreleptin response in patients with partial lipodystrophies.
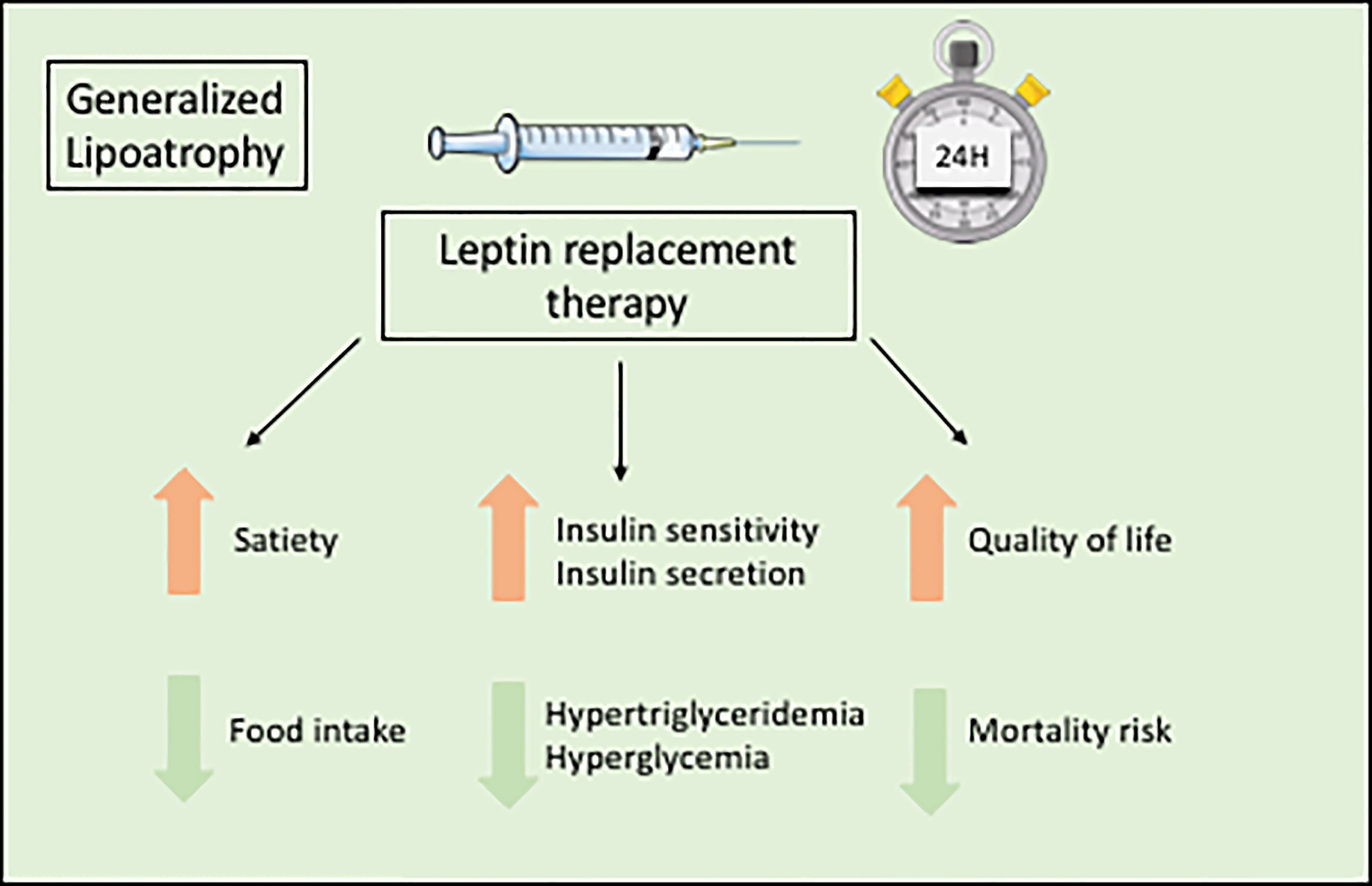
Figure 5 Benefits of metreleptin replacement therapy in generalized lipodystrophies.
Metreleptin therapy is well tolerated in the majority of patients. The dose of metreleptin is adapted to metabolic responses and to tolerance, with particular attention to the extent of weight loss, which is an expected effect of the treatment but should be controlled. Common adverse effects mainly comprise localized skin reaction, pain at injection sites, and hypoglycemia when the decrease of other antidiabetic treatments is not sufficiently anticipated. Very rare cases of lymphoma have been reported in patients with autoimmune forms of lipodystrophy treated with metreleptin, without any established causal relationship with the treatment (138). Although they display only exceptionally neutralizing effects, circulating anti-leptin autoantibodies frequently develop under treatment. Anti-leptin autoantibodies interfere with enzyme-linked immunosorbent assays for serum leptin, complicating the correct interpretation of leptinemia in treated patients. Further studies are needed to identify other consequences of anti-leptin autoantibodies (6).
Conclusion
Lipodystrophy syndromes are rare and heterogeneous diseases. Their diagnosis is difficult and can be significantly delayed, since adipose tissue is not systematically investigated during clinical exam, and several symptoms are nonspecific (7). Most clinical forms of lipodystrophy remain genetically unexplained. Next generation sequencing technologies with exome or genome analysis will probably allow for discovering new causative genetic variants in the near future, and lead to a better understanding of the pathophysiology in these rare diseases. However, the interpretation of genetic variants is increasingly challenging (44). Closely associated genetic, clinical and fundamental research, as well as broad collaborations are needed to explore new pathophysiological determinants of lipodystrophy syndromes, and improve the care of affected patients.
Author Contributions
JZ and CVi wrote the original draft. CVa, EC, MA, CS-L, EB, HM, BD, SJ, BF, and IJ reviewed and edited the manuscript and the figures. All authors contributed to the article and approved the submitted version.
Funding
Our group is supported by the French Ministry of Solidarity and Health, Assistance-Publique Hôpitaux de Paris, Sorbonne University, the Institut National de la Santé et de la Recherche Médicale (Inserm), and the Fondation pour la Recherche Médicale (EQU201903006878), France.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the members of the French National Reference Network for Rare Diseases of Insulin Secretion and Insulin Sensitivity (PRISIS), for their collaborative contribution to diagnosis and care of patients with lipodystrophy in France. We thank Dr Alexandra Ntorkou and Prof. Monique Elmaleh from Assistance Publique-Hôpitaux de Paris, Robert Debré Hospital, Pediatric Radiology Department, Paris, France, for help with radiological illustrations.
Glossary
References
1. Capeau J, Magré J, Caron-Debarle M, Lagathu C, Antoine B, Béréziat V, et al. Human Lipodystrophies: Genetic and Acquired Diseases of Adipose Tissue. Endocr Dev (2010) 19:1–20. doi: 10.1159/000316893
2. Garg A. Clinical Review: Lipodystrophies: Genetic and Acquired Body Fat Disorders. J Clin Endocrinol Metab (2011) 96:3313–25. doi: 10.1210/jc.2011-1159
3. Akinci B, Meral R, Oral EA. Phenotypic and Genetic Characteristics of Lipodystrophy: Pathophysiology, Metabolic Abnormalities, and Comorbidities. Curr Diabetes Rep (2018) 18:143. doi: 10.1007/s11892-018-1099-9
4. Koethe JR, Lagathu C, Lake JE, Domingo P, Calmy A, Falutz J, et al. HIV and Antiretroviral Therapy-Related Fat Alterations. Nat Rev Dis Primers (2020) 6:48. doi: 10.1038/s41572-020-0181-1
5. Fardet L, Antuna-Puente B, Vatier C, Cervera P, Touati A, Simon T, et al. Adipokine Profile in Glucocorticoid-Treated Patients: Baseline Plasma Leptin Level Predicts Occurrence of Lipodystrophy. Clin Endocrinol (Oxf) (2013) 78:43–51. doi: 10.1111/j.1365-2265.2012.04348.x
6. Lim K, Haider A, Adams C, Sleigh A, Savage DB. Lipodistrophy: A Paradigm for Understanding the Consequences of “Overloading” Adipose Tissue. Physiol Rev (2021) 101:907–93. doi: 10.1152/physrev.00032.2020
7. Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab (2016) 101:4500–11. doi: 10.1210/jc.2016-2466
8. Sollier C, Vatier C, Capel E, Lascols O, Auclair M, Janmaat S, et al. Lipodystrophic Syndromes: From Diagnosis to Treatment. Ann Endocrinol (Paris) (2020) 81:51–60. doi: 10.1016/j.ando.2019.10.003
9. Chiquette E, Oral EA, Garg A, Araujo-Vilar D, Dhankhar P. Estimating the Prevalence of Generalized and Partial Lipodystrophy: Findings and Challenges. Diabetes Metab Syndr Obes (2017) 10:375–83. doi: 10.2147/DMSO.S130810
10. Gonzaga-Jauregui C, Ge W, Staples J, Van Hout C, Yadav A, Colonie R, et al. Clinical and Molecular Prevalence of Lipodystrophy in an Unascertained Large Clinical Care Cohort. Diabetes (2020) 69:249–58. doi: 10.2337/db19-0447
11. Magré J, Delepine M, Khallouf E, Gedde-Dahl T Jr, Van Maldergem L, Sobel E, et al. Identification of the Gene Altered in Berardinelli-Seip Congenital Lipodystrophy on Chromosome 11q13. Nat Genet (2001) 28:365–70. doi: 10.1038/ng585
12. Agarwal AK, Arioglu E, De Almeida S, Akkoc N, Taylor SI, Bowcock AM, et al. AGPAT2 Is Mutated in Congenital Generalized Lipodystrophy Linked to Chromosome 9q34. Nat Genet (2002) 31:21–3. doi: 10.1038/ng880
13. Dollet L, Magré J, Cariou B, Prieur X. Function of Seipin: New Insights From Bscl2/seipin Knockout Mouse Models. Biochimie (2014) 96:166–72. doi: 10.1016/j.biochi.2013.06.022
14. Salo VT, Belevich I, Li S, Karhinen L, Vihinen H, Vigouroux C, et al. Seipin Regulates ER-Lipid Droplet Contacts and Cargo Delivery. EMBO J (2016) 35:2699–716. doi: 10.15252/embj.201695170
15. Salo VT, Li S, Vihinen H, Holtta-Vuori M, Szkalisity A, Horvath P, et al. Seipin Facilitates Triglyceride Flow to Lipid Droplet and Counteracts Droplet Ripening via Endoplasmic Reticulum Contact. Dev Cell (2019) 50:478–93.e479. doi: 10.1016/j.devcel.2019.05.016
16. Ceccarini G, Magno S, Pelosini C, Ferrari F, Sessa MR, Scabia G, et al. Congenital Generalized Lipoatrophy (Berardinelli-Seip Syndrome) Type 1: Description of Novel AGPAT2 Homozygous Variants Showing the Highly Heterogeneous Presentation of the Disease. Front Endocrinol (Lausanne) (2020) 11:39. doi: 10.3389/fendo.2020.00039
17. Le Lay S, Blouin CM, Hajduch E, Dugail I. Filling Up Adipocytes With Lipids. Lessons From Caveolin-1 Deficiency. Biochim Biophys Acta (2009) 1791:514–8. doi: 10.1016/j.bbalip.2008.10.008
18. Pol A, Morales-Paytuvi F, Bosch M, Parton RG. Non-Caveolar Caveolins - Duties Outside the Caves. J Cell Sci (2020) 133:jcs241562. doi: 10.1242/jcs.241562
19. Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, et al. Integrative Genomic Analysis Implicates Limited Peripheral Adipose Storage Capacity in the Pathogenesis of Human Insulin Resistance. Nat Genet (2017) 49:17–26. doi: 10.1038/ng.3714
20. Cinti S. The Adipose Organ. Prostaglandins Leukot Essent Fatty Acids (2005) 73:9–15. doi: 10.1016/j.plefa.2005.04.010
21. Rosen ED, Spiegelman BM. What We Talk About When We Talk About Fat. Cell (2014) 156:20–44. doi: 10.1016/j.cell.2013.12.012
22. Billon N, Dani C. Developmental Origins of the Adipocyte Lineage: New Insights From Genetics and Genomics Studies. Stem Cell Rev Rep (2012) 8:55–66. doi: 10.1007/s12015-011-9242-x
23. Carobbio S, Pellegrinelli V, Vidal-Puig A. Adipose Tissue Function and Expandability as Determinants of Lipotoxicity and the Metabolic Syndrome. Adv Exp Med Biol (2017) 960:161–96. doi: 10.1007/978-3-319-48382-5_7
24. Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, et al. Role of Diacylglycerol Activation of PKCtheta in Lipid-Induced Muscle Insulin Resistance in Humans. Proc Natl Acad Sci USA (2014) 111:9597–602. doi: 10.1073/pnas.1409229111
25. Leggat J, Bidault G, Vidal-Puig A. Lipotoxicity: A Driver of Heart Failure With Preserved Ejection Fraction? Clin Sci (2021) 135(9):2265–83. doi: 10.1042/CS20210127
26. Symons JD, Abel ED. Lipotoxicity Contributes to Endothelial Dysfunction: A Focus on the Contribution From Ceramide. Rev Endocr Metab Disord (2013) 14:59–68. doi: 10.1007/s11154-012-9235-3
27. Lytrivi M, Castell AL, Poitout V, Cnop M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J Mol Biol (2020) 432:1514–34. doi: 10.1016/j.jmb.2019.09.016
28. Friedman JM. Leptin and the Endocrine Control of Energy Balance. Nat Metab (2019) 1:754–64. doi: 10.1038/s42255-019-0095-y
29. Kahn BB. Adipose Tissue, Inter-Organ Communication, and the Path to Type 2 Diabetes: The 2016 Banting Medal for Scientific Achievement Lecture. Diabetes (2019) 68:3–14. doi: 10.2337/dbi18-0035
30. Unger RH, Roth MG. A New Biology of Diabetes Revealed by Leptin. Cell Metab (2015) 21:15–20. doi: 10.1016/j.cmet.2014.10.011
31. Antuna-Puente B, Boutet E, Vigouroux C, Lascols O, Slama L, Caron-Debarle M, et al. Higher Adiponectin Levels in Patients With Berardinelli-Seip Congenital Lipodystrophy Due to Seipin as Compared With 1-Acylglycerol-3-Phosphate-O-Acyltransferase-2 Deficiency. J Clin Endocrinol Metab (2010) 95:1463–8. doi: 10.1210/jc.2009-1824
32. Polyzos SA, Perakakis N, Mantzoros CS. Fatty Liver in Lipodystrophy: A Review With a Focus on Therapeutic Perspectives of Adiponectin and/or Leptin Replacement. Metabolism (2019) 96:66–82. doi: 10.1016/j.metabol.2019.05.001
33. Kahn CR, Wang G, Lee KY. Altered Adipose Tissue and Adipocyte Function in the Pathogenesis of Metabolic Syndrome. J Clin Invest (2019) 129:3990–4000. doi: 10.1172/JCI129187
34. Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of Insulin Resistance in A-ZIP/F-1 Fatless Mice. J Biol Chem (2000) 275:8456–60. doi: 10.1074/jbc.275.12.8456
35. Samuel VT, Petersen KF, Shulman GI. Lipid-Induced Insulin Resistance: Unravelling the Mechanism. Lancet (2010) 375:2267–77. doi: 10.1016/S0140-6736(10)60408-4
36. Vasan SK, Osmond C, Canoy D, Christodoulides C, Neville MJ, Di Gravio C, et al. Comparison of Regional Fat Measurements by Dual-Energy X-Ray Absorptiometry and Conventional Anthropometry and Their Association With Markers of Diabetes and Cardiovascular Disease Risk. Int J Obes (2018) 42:850–7. doi: 10.1038/ijo.2017.289
37. Stefan N. Causes, Consequences, and Treatment of Metabolically Unhealthy Fat Distribution. Lancet Diabetes Endocrinol (2020) 8:616–27. doi: 10.1016/S2213-8587(20)30110-8
38. Arner P. Regional Differences in Protein Production by Human Adipose Tissue. Biochem Soc Trans (2001) 29:72–5. doi: 10.1042/bst0290072
39. Malden D, Lacey B, Emberson J, Karpe F, Allen N, Bennett D, et al. Body Fat Distribution and Systolic Blood Pressure in 10,000 Adults With Whole-Body Imaging: UK Biobank and Oxford BioBank. Obesity (2019) 27:1200–6. doi: 10.1002/oby.22509
40. Afonso P, Auclair M, Boccara F, Vantyghem MC, Katlama C, Capeau J, et al. LMNA Mutations Resulting in Lipodystrophy and HIV Protease Inhibitors Trigger Vascular Smooth Muscle Cell Senescence and Calcification: Role of ZMPSTE24 Downregulation. Atherosclerosis (2016) 245:200–11. doi: 10.1016/j.atherosclerosis.2015.12.012
41. Sieprath T, Darwiche R, De Vos WH. Lamins as Mediators of Oxidative Stress. Biochem Biophys Res Commun (2012) 421:635–9. doi: 10.1016/j.bbrc.2012.04.058
42. Guénantin AC, Briand N, Bidault G, Afonso P, Béréziat V, Vatier C, et al. Nuclear Envelope-Related Lipodystrophies. Semin Cell Dev Biol (2014) 29:148–57. doi: 10.1016/j.semcdb.2013.12.015
43. Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the Metabolic Syndrome: A Joint Interim Statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation (2009) 120:1640–5. doi: 10.1161/CIRCULATIONAHA.109.192644
44. Jéru I, Vantyghem MC, Bismuth E, Cervera P, Barraud S. PLIN1-Study Group, Et al. Diagnostic Challenge in PLIN1-Associated Familial Partial Lipodystrophy. J Clin Endocrinol Metab (2019) 104:6025–32. doi: 10.1210/jc.2019-00849
45. Garg A, Stray-Gundersen J, Parsons D, Bertocci LA. Skeletal Muscle Morphology and Exercise Response in Congenital Generalized Lipodystrophy. Diabetes Care (2000) 23:1545–50. doi: 10.2337/diacare.23.10.1545
46. Ji H, Weatherall P, Adams-Huet B, Garg A. Increased Skeletal Muscle Volume in Women With Familial Partial Lipodystrophy, Dunnigan Variety. J Clin Endocrinol Metab (2013) 98:E1410–3. doi: 10.1210/jc.2013-1297
47. Vantyghem MC, Balavoine AS, Douillard C, Defrance F, Dieudonne L, Mouton F, et al. How to Diagnose a Lipodystrophy Syndrome. Ann Endocrinol (Paris) (2012) 73:170–89. doi: 10.1016/j.ando.2012.04.010
48. Mancuso M, Orsucci D, Angelini C, Bertini E, Carelli V, Comi GP, et al. Phenotypic Heterogeneity of the 8344A>G mtDNA "MERRF" Mutation. Neurology (2013) 80:2049–54. doi: 10.1212/WNL.0b013e318294b44c
49. Farhan SM, Robinson JF, McIntyre AD, Marrosu MG, Ticca AF, Loddo S, et al. A Novel LIPE Nonsense Mutation Found Using Exome Sequencing in Siblings With Late-Onset Familial Partial Lipodystrophy. Can J Cardiol (2014) 30:1649–54. doi: 10.1016/j.cjca.2014.09.007
50. Albert JS, Yerges-Armstrong LM, Horenstein RB, Pollin TI, Sreenivasan UT, Chai S, et al. Null Mutation in Hormone-Sensitive Lipase Gene and Risk of Type 2 Diabetes. N Engl J Med (2014) 370:2307–15. doi: 10.1056/NEJMoa1315496
51. Sawyer SL, Cheuk-Him Ng A, Innes AM, Wagner JD, Dyment DA, Tetreault M, et al. Homozygous Mutations in MFN2 Cause Multiple Symmetric Lipomatosis Associated With Neuropathy. Hum Mol Genet (2015) 24:5109–14. doi: 10.1093/hmg/ddv229
52. Rocha N, Bulger DA, Frontini A, Titheradge H, Gribsholt SB, Knox R, et al. Human Biallelic MFN2 Mutations Induce Mitochondrial Dysfunction, Upper Body Adipose Hyperplasia, and Suppression of Leptin Expression. Elife (2017) 6:e23813. doi: 10.7554/eLife.23813
53. Zolotov S, Xing C, Mahamid R, Shalata A, Sheikh-Ahmad M, Garg A. Homozygous LIPE Mutation in Siblings With Multiple Symmetric Lipomatosis, Partial Lipodystrophy, and Myopathy. Am J Med Genet A (2017) 173:190–4. doi: 10.1002/ajmg.a.37880
54. Capel E, Vatier C, Cervera P, Stojkovic T, Disse E, Cottereau AS, et al. MFN2-Associated Lipomatosis: Clinical Spectrum and Impact on Adipose Tissue. J Clin Lipidol (2018) 12:1420–35. doi: 10.1016/j.jacl.2018.07.009
55. Sollier C, Capel E, Aguilhon C, Smirnov V, Auclair M, Douillard C, et al. LIPE-Related Lipodystrophic Syndrome: Clinical Features and Disease Modeling Using Adipose Stem Cells. Eur J Endocrinol (2021) 184:155–68. doi: 10.1530/EJE-20-1013
56. Young J, Morbois-Trabut L, Couzinet B, Lascols O, Dion E, Béréziat V, et al. Type A Insulin Resistance Syndrome Revealing a Novel Lamin A Mutation. Diabetes (2005) 54:1873–8. doi: 10.2337/diabetes.54.6.1873
57. Le Dour C, Schneebeli S, Bakiri F, Darcel F, Jacquemont ML, Maubert MA, et al. A Homozygous Mutation of Prelamin-A Preventing Its Farnesylation and Maturation Leads to a Severe Lipodystrophic Phenotype: New Insights Into the Pathogenicity of Nonfarnesylated Prelamin-A. J Clin Endocrinol Metab (2011) 96:E856–62. doi: 10.1210/jc.2010-2234
58. Handelsman Y, Oral EA, Bloomgarden ZT, Brown RJ, Chan JL, Einhorn D, et al. The Clinical Approach to the Detection of Lipodystrophy - An AACE Consensus Statement. Endocr Pract (2013) 19:107–16. doi: 10.4158/endp.19.1.v767575m65p5mr06
59. Hegele RA, Leff T. Unbuckling Lipodystrophy From Insulin Resistance and Hypertension. J Clin Invest (2004) 114:163–5. doi: 10.1172/JCI22382
60. Auclair M, Vigouroux C, Boccara F, Capel E, Vigeral C, Guerci B, et al. Peroxisome Proliferator-Activated Receptor-Gamma Mutations Responsible for Lipodystrophy With Severe Hypertension Activate the Cellular Renin-Angiotensin System. Arterioscler Thromb Vasc Biol (2013) 33:829–38. doi: 10.1161/ATVBAHA.112.300962
61. Lambadiari V, Kountouri A, Maratou E, Liatis S, Dimitriadis GD, Karpe F. Case Report: Metreleptin Treatment in a Patient With a Novel Mutation for Familial Partial Lipodystrophy Type 3, Presenting With Uncontrolled Diabetes and Insulin Resistance. Front Endocrinol (Lausanne) (2021) 12:684182. doi: 10.3389/fendo.2021.684182
62. Hegele RA. Premature Atherosclerosis Associated With Monogenic Insulin Resistance. Circulation (2001) 103:2225–9. doi: 10.1161/01.cir.103.18.2225
63. Bidault G, Garcia M, Vantyghem MC, Ducluzeau PH, Morichon R, Thiyagarajah K, et al. Lipodystrophy-Linked LMNA P.R482W Mutation Induces Clinical Early Atherosclerosis and In Vitro Endothelial Dysfunction. Arterioscler Thromb Vasc Biol (2013) 33:2162–71. doi: 10.1161/ATVBAHA.113.301933
64. Briand N, Guenantin AC, Jeziorowska D, Shah A, Mantecon M, Capel E, et al. The Lipodystrophic Hotspot Lamin A P.R482W Mutation Deregulates the Mesodermal Inducer T/Brachyury and Early Vascular Differentiation Gene Networks. Hum Mol Genet (2018) 27:1447–59. doi: 10.1093/hmg/ddy055
65. Lupsa BC, Sachdev V, Lungu AO, Rosing DR, Gorden P. Cardiomyopathy in Congenital and Acquired Generalized Lipodystrophy: A Clinical Assessment. Med (Baltimore) (2010) 89:245–50. doi: 10.1097/MD.0b013e3181e9442f
66. Nelson MD, Victor RG, Szczepaniak EW, Simha V, Garg A, Szczepaniak LS. Cardiac Steatosis and Left Ventricular Hypertrophy in Patients With Generalized Lipodystrophy as Determined by Magnetic Resonance Spectroscopy and Imaging. Am J Cardiol (2013) 112:1019–24. doi: 10.1016/j.amjcard.2013.05.036
67. Garg A, Speckman RA, Bowcock AM. Multisystem Dystrophy Syndrome Due to Novel Missense Mutations in the Amino-Terminal Head and Alpha-Helical Rod Domains of the Lamin a/C Gene. Am J Med (2002) 112:549–55. doi: 10.1016/s0002-9343(02)01070-7
68. Caux F, Dubosclard E, Lascols O, Buendia B, Chazouillères O, Cohen A, et al. A New Clinical Condition Linked to a Novel Mutation in Lamins A and C With Generalized Lipoatrophy, Insulin-Resistant Diabetes, Disseminated Leukomelanodermic Papules, Liver Steatosis, and Cardiomyopathy. J Clin Endocrinol Metab (2003) 88:1006–13. doi: 10.1210/jc.2002-021506
69. Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM, Duboc D. Primary Prevention of Sudden Death in Patients With Lamin a/C Gene Mutations. N Engl J Med (2006) 354:209–10. doi: 10.1056/NEJMc052632
70. Kwapich M, Lacroix D, Espiard S, Ninni S, Brigadeau F, Kouakam C, et al. Cardiometabolic Assessment of Lamin a/C Gene Mutation Carriers: A Phenotype-Genotype Correlation. Diabetes Metab (2019) 45:382–9. doi: 10.1016/j.diabet.2018.09.006
71. Mosbah H, Vatier C, Boccara F, Jéru I, Lascols O, Vantyghem MC, et al. Looking at New Unexpected Disease Targets in LMNA-Linked Lipodystrophies in the Light of Complex Cardiovascular Phenotypes: Implications for Clinical Practice. Cells (2020) 9:765. doi: 10.3390/cells9030765
72. Treiber G, Flaus Furmaniuk A, Guilleux A, Medjane S, Bonfanti O, Schneebeli S, et al. A Recurrent Familial Partial Lipodystrophy Due to a Monoallelic or Biallelic LMNA Founder Variant Highlights the Multifaceted Cardiac Manifestations of Metabolic Laminopathies. Eur J Endocrinol (2021) 185:453–62. doi: 10.1530/EJE-21-0282
73. Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, et al. Association of a Homozygous Nonsense Caveolin-1 Mutation With Berardinelli-Seip Congenital Lipodystrophy. J Clin Endocrinol Metab (2008) 93:1129–34. doi: 10.1210/jc.2007-1328
74. Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, et al. Human PTRF Mutations Cause Secondary Deficiency of Caveolins Resulting in Muscular Dystrophy With Generalized Lipodystrophy. J Clin Invest (2009) 119:2623–33. doi: 10.1172/JCI38660
75. Rajab A, Straub V, McCann LJ, Seelow D, Varon R, Barresi R, et al. Fatal Cardiac Arrhythmia and Long-QT Syndrome in a New Form of Congenital Generalized Lipodystrophy With Muscle Rippling (CGL4) Due to PTRF-CAVIN Mutations. PLoS Genet (2010) 6:e1000874. doi: 10.1371/journal.pgen.1000874
76. van der Pol RJ, Benninga MA, Magre J, Van Maldergem L, Rotteveel J, van der Knaap MS, et al. Berardinelli-Seip Syndrome and Achalasia: A Shared Pathomechanism? Eur J Pediatr (2015) 174:975–80. doi: 10.1007/s00431-015-2556-y
77. Karhan AN, Zammouri J, Auclair M, Capel E, Apaydin FD, Ates F, et al. Biallelic CAV1 Null Variants Induce Congenital Generalized Lipodystrophy With Achalasia. Eur J Endocrinol (2021) 185:841–54. doi: 10.1530/EJE-21-0915
78. Hennekam RCM. Pathophysiology of Premature Aging Characteristics in Mendelian Progeroid Disorders. Eur J Med Genet (2020) 63:104028. doi: 10.1016/j.ejmg.2020.104028
79. Gautheron J, Morisseau C, Chung WK, Zammouri J, Auclair M, Baujat G, et al. EPHX1 Mutations Cause a Lipoatrophic Diabetes Syndrome Due to Impaired Epoxide Hydrolysis and Increased Cellular Senescence. Elife (2021) 10:e68445. doi: 10.7554/eLife.68445
80. Weedon MN, Ellard S, Prindle MJ, Caswell R, Lango Allen H, Oram R, et al. An in-Frame Deletion at the Polymerase Active Site of POLD1 Causes a Multisystem Disorder With Lipodystrophy. Nat Genet (2013) 45:947–50. doi: 10.1038/ng.2670
81. Donadille B, D'Anella P, Auclair M, Uhrhammer N, Sorel M, Grigorescu R, et al. Partial Lipodystrophy With Severe Insulin Resistance and Adult Progeria Werner Syndrome. Orphanet J Rare Dis (2013) 8:106. doi: 10.1186/1750-1172-8-106
82. Vantyghem MC, Pigny P, Maurage CA, Rouaix-Emery N, Stojkovic T, Cuisset JM, et al. Patients With Familial Partial Lipodystrophy of the Dunnigan Type Due to a LMNA R482W Mutation Show Muscular and Cardiac Abnormalities. J Clin Endocrinol Metab (2004) 89:5337–46. doi: 10.1210/jc.2003-031658
83. Decaudain A, Vantyghem MC, Guerci B, Hecart AC, Auclair M, Reznik Y, et al. New Metabolic Phenotypes in Laminopathies: LMNA Mutations in Patients With Severe Metabolic Syndrome. J Clin Endocrinol Metab (2007) 92:4835–44. doi: 10.1210/jc.2007-0654
84. Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, et al. PSMB8 Encoding the Beta5i Proteasome Subunit Is Mutated in Joint Contractures, Muscle Atrophy, Microcytic Anemia, and Panniculitis-Induced Lipodystrophy Syndrome. Am J Hum Genet (2010) 87:866–72. doi: 10.1016/j.ajhg.2010.10.031
85. Shastry S, Delgado MR, Dirik E, Turkmen M, Agarwal AK, Garg A. Congenital Generalized Lipodystrophy, Type 4 (CGL4) Associated With Myopathy Due to Novel PTRF Mutations. Am J Med Genet A (2010) 152A:2245–53. doi: 10.1002/ajmg.a.33578
86. Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, et al. Additive Loss-of-Function Proteasome Subunit Mutations in CANDLE/PRAAS Patients Promote Type I IFN Production. J Clin Invest (2015) 125:4196–211. doi: 10.1172/JCI81260
87. Corvillo F, Aparicio V, Lopez-Lera A, Garrido S, Araujo-Vilar D, de Miguel MP, et al. Autoantibodies Against Perilipin 1 as a Cause of Acquired Generalized Lipodystrophy. Front Immunol (2018) 9:2142. doi: 10.3389/fimmu.2018.02142
88. Teboul-Coré S, Rey-Jouvin C, Miquel A, Vatier C, Capeau J, Robert JJ, et al. Bone Imaging Findings in Genetic and Acquired Lipodystrophic Syndromes: An Imaging Study of 24 Cases. Skeletal Radiol (2016) 45:1495–506. doi: 10.1007/s00256-016-2457-9
89. Freire EBL, Madeira MP, Lima G, Fernandes VO, Aguiar LB, Fontenele JPU, et al. Misdiagnosis of Paget's Disease of Bone in a Congenital Generalized Lipodystrophy Patient: Case Report. Front Endocrinol (Lausanne) (2021) 12:683697. doi: 10.3389/fendo.2021.683697
90. Zamboni M, Nori N, Brunelli A, Zoico E. How Does Adipose Tissue Contribute to Inflammageing? Exp Gerontol (2021) 143:111162. doi: 10.1016/j.exger.2020.111162
91. Briand N, Collas P. Lamina-Associated Domains: Peripheral Matters and Internal Affairs. Genome Biol (2020) 21:85. doi: 10.1186/s13059-020-02003-5
92. Gordon LB, Kleinman ME, Massaro J, D'Agostino RB Sr, Shappell H, Gerhard-Herman M, et al. Clinical Trial of the Protein Farnesylation Inhibitors Lonafarnib, Pravastatin, and Zoledronic Acid in Children With Hutchinson-Gilford Progeria Syndrome. Circulation (2016) 134:114–25. doi: 10.1161/CIRCULATIONAHA.116.022188
93. Hamczyk MR, Villa-Bellosta R, Quesada V, Gonzalo P, Vidak S, Nevado RM, et al. Progerin Accelerates Atherosclerosis by Inducing Endoplasmic Reticulum Stress in Vascular Smooth Muscle Cells. EMBO Mol Med (2019) 11:e9736. doi: 10.15252/emmm.201809736
94. Bidault G, Garcia M, Capeau J, Morichon R, Vigouroux C, Béréziat V. Progerin Expression Induces Inflammation, Oxidative Stress and Senescence in Human Coronary Endothelial Cells. Cells (2020) 9:1201. doi: 10.3390/cells9051201
95. Perovanovic J, Dell'Orso S, Gnochi VF, Jaiswal JK, Sartorelli V, Vigouroux C, et al. Laminopathies Disrupt Epigenomic Developmental Programs and Cell Fate. Sci Transl Med (2016) 8:335ra358. doi: 10.1126/scitranslmed.aad4991
96. Shah PP, Lv W, Rhoades JH, Poleshko A, Abbey D, Caporizzo MA, et al. Pathogenic LMNA Variants Disrupt Cardiac Lamina-Chromatin Interactions and De-Repress Alternative Fate Genes. Cell Stem Cell (2021) 28:938–54.e939. doi: 10.1016/j.stem.2020.12.016
97. Boguslavsky RL, Stewart CL, Worman HJ. Nuclear Lamin A Inhibits Adipocyte Differentiation: Implications for Dunnigan-Type Familial Partial Lipodystrophy. Hum Mol Genet (2006) 15:653–63. doi: 10.1093/hmg/ddi480
98. Oldenburg A, Briand N, Sorensen AL, Cahyani I, Shah A, Moskaug JO, et al. A Lipodystrophy-Causing Lamin A Mutant Alters Conformation and Epigenetic Regulation of the Anti-Adipogenic MIR335 Locus. J Cell Biol (2017) 216:2731–43. doi: 10.1083/jcb.201701043
99. Friesen M, Cowan CA. FPLD2 LMNA Mutation R482W Dysregulates iPSC-Derived Adipocyte Function and Lipid Metabolism. Biochem Biophys Res Commun (2018) 495:254–60. doi: 10.1016/j.bbrc.2017.11.008
100. Pellegrini C, Columbaro M, Schena E, Prencipe S, Andrenacci D, Iozzo P, et al. Altered Adipocyte Differentiation and Unbalanced Autophagy in Type 2 Familial Partial Lipodystrophy: An In Vitro and In Vivo Study of Adipose Tissue Browning. Exp Mol Med (2019) 51:1–17. doi: 10.1038/s12276-019-0289-0
101. Najdi F, Kruger P, Djabali K. Impact of Progerin Expression on Adipogenesis in Hutchinson-Gilford Progeria Skin-Derived Precursor Cells. Cells (2021) 10:1598. doi: 10.3390/cells10071598
102. Sanchez P, Infante A, de Eguino GR, Fuentes-Maestre JA, Garcia-Verdugo JM, Rodriguez CI. Age-Related Lipid Metabolic Signature in Human LMNA-Lipodystrophic Stem Cell-Derived Adipocytes. J Clin Endocrinol Metab (2015) 100:E964–3. doi: 10.1210/jc.2014-4528
103. Cenni V, Capanni C, Mattioli E, Schena E, Squarzoni S, Bacalini MG, et al. Lamin A Involvement in Ageing Processes. Ageing Res Rev (2020) 62:101073. doi: 10.1016/j.arr.2020.101073
104. Shastry S, Simha V, Godbole K, Sbraccia P, Melancon S, Yajnik CS, et al. A Novel Syndrome of Mandibular Hypoplasia, Deafness, and Progeroid Features Associated With Lipodystrophy, Undescended Testes, and Male Hypogonadism. J Clin Endocrinol Metab (2010) 95:E192–7. doi: 10.1210/jc.2010-0419
105. Nicolas E, Golemis EA, Arora S. POLD1: Central Mediator of DNA Replication and Repair, and Implication in Cancer and Other Pathologies. Gene (2016) 590:128–41. doi: 10.1016/j.gene.2016.06.031
106. Karakasilioti I, Kamileri I, Chatzinikolaou G, Kosteas T, Vergadi E, Robinson AR, et al. DNA Damage Triggers a Chronic Autoinflammatory Response, Leading to Fat Depletion in NER Progeria. Cell Metab (2013) 18:403–15. doi: 10.1016/j.cmet.2013.08.011
107. Murdocca M, Spitalieri P, De Masi C, Udroiu I, Marinaccio J, Sanchez M, et al. Functional Analysis of POLD1 P.Ser605del Variant: The Aging Phenotype of MDPL Syndrome Is Associated With an Impaired DNA Repair Capacity. Aging (Albany NY) (2021) 13:4926–45. doi: 10.18632/aging.202680
108. Goh KJ, Chen JH, Rocha N, Semple RK. Human Pluripotent Stem Cell-Based Models Suggest Preadipocyte Senescence as a Possible Cause of Metabolic Complications of Werner and Bloom Syndromes. Sci Rep (2020) 10:7490. doi: 10.1038/s41598-020-64136-8
109. Payne F, Colnaghi R, Rocha N, Seth A, Harris J, Carpenter G, et al. Hypomorphism in Human NSMCE2 Linked to Primordial Dwarfism and Insulin Resistance. J Clin Invest (2014) 124:4028–38. doi: 10.1172/JCI73264
110. Knapp KM, Jenkins DE, Sullivan R, Harms FL, von Elsner L, Ockeloen CW, et al. MCM Complex Members MCM3 and MCM7 Are Associated With a Phenotypic Spectrum From Meier-Gorlin Syndrome to Lipodystrophy and Adrenal Insufficiency. Eur J Hum Genet (2021) 29:1110–20. doi: 10.1038/s41431-021-00839-4
111. Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M, et al. A Mutation in the Immunoproteasome Subunit PSMB8 Causes Autoinflammation and Lipodystrophy in Humans. J Clin Invest (2011) 121:4150–60. doi: 10.1172/JCI58414
112. D'Angelo DM, Di Filippo P, Breda L, Chiarelli F. Type I Interferonopathies in Children: An Overview. Front Pediatr (2021) 9:631329. doi: 10.3389/fped.2021.631329
113. Sorkina E, Frolova E, Rusinova D, Polyakova S, Roslavtseva E, Vasilyev E, et al. Progressive Generalized Lipodystrophy as a Manifestation of Autoimmune Polyglandular Syndrome Type 1. J Clin Endocrinol Metab (2016) 101:1344–7. doi: 10.1210/jc.2015-3722
114. Misra A, Garg A. Clinical Features and Metabolic Derangements in Acquired Generalized Lipodystrophy: Case Reports and Review of the Literature. Med (Baltimore) (2003) 82:129–46. doi: 10.1097/00005792-200303000-00007
115. Savage DB, Semple RK, Clatworthy MR, Lyons PA, Morgan BP, Cochran EK, et al. Complement Abnormalities in Acquired Lipodystrophy Revisited. J?Clin Endocrinol Metab (2009) 94:10–6. doi: 10.1210/jc.2008-1703
116. Ceccarini G, Magno S, Gilio D, Pelosini C, Santini F. Autoimmunity in Lipodystrophy Syndromes. Presse Med (2021) 50:104073. doi: 10.1016/j.lpm.2021.104073
117. Corvillo F, Akinci B. An Overview of Lipodystrophy and the Role of the Complement System. Mol Immunol (2019) 112:223–32. doi: 10.1016/j.molimm.2019.05.011
118. Corvillo F, Ceccarini G, Nozal P, Magno S, Pelosini C, Garrido S, et al. Immunological Features of Patients Affected by Barraquer-Simons Syndrome. Orphanet J Rare Dis (2020) 15:9. doi: 10.1186/s13023-019-1292-1
119. Jehl A, Cugnet-Anceau C, Vigouroux C, Legeay AL, Dalle S, Harou O, et al. Acquired Generalized Lipodystrophy: A New Cause of Anti-PD-1 Immune-Related Diabetes. Diabetes Care (2019) 42:2008–10. doi: 10.2337/dc18-2535
120. Falcao CK, Cabral MCS, Mota JM, Arbache ST, Costa-Riquetto AD, Muniz DQB, et al. Acquired Lipodystrophy Associated With Nivolumab in a Patient With Advanced Renal Cell Carcinoma. J Clin Endocrinol Metab (2019) 104:3245–8. doi: 10.1210/jc.2018-02221
121. Haddad N, Vidal-Trecan T, Baroudjian B, Zagdanski AM, Arangalage D, Battistella M, et al. Acquired Generalized Lipodystrophy Under Immune Checkpoint Inhibition. Br J Dermatol (2020) 182:477–80. doi: 10.1111/bjd.18124
122. Eigentler T, Lomberg D, Machann J, Stefan N. Lipodystrophic Nonalcoholic Fatty Liver Disease Induced by Immune Checkpoint Blockade. Ann Intern Med (2020) 172:836–7. doi: 10.7326/L19-0635
123. Srinivasa S, Garcia-Martin R, Torriani M, Fitch KV, Carlson AR, Kahn CR, et al. Altered Pattern of Circulating miRNAs in HIV Lipodystrophy Perturbs Key Adipose Differentiation and Inflammation Pathways. JCI Insight (2021) 6:e150399. doi: 10.1172/jci.insight.150399
124. Mattie S, Riemer J, Wideman JG, McBride HM. A New Mitofusin Topology Places the Redox-Regulated C Terminus in the Mitochondrial Intermembrane Space. J Cell Biol (2018) 217:507–15. doi: 10.1083/jcb.201611194
125. Altmann J, Buchner B, Nadaj-Pakleza A, Schafer J, Jackson S, Lehmann D, et al. Expanded Phenotypic Spectrum of the M.8344A>G “MERRF” Mutation: Data From the German mitoNET Registry. J Neurol (2016) 263:961–72. doi: 10.1007/s00415-016-8086-3
126. Enzi G, Busetto L, Ceschin E, Coin A, Digito M, Pigozzo S. Multiple Symmetric Lipomatosis: Clinical Aspects and Outcome in a Long-Term Longitudinal Study. Int J Obes Relat Metab Disord (2002) 26:253–61. doi: 10.1038/sj.ijo.0801867
127. Kobayashi M, Nezu Y, Tagawa R, Higami Y. Mitochondrial Unfolded Protein Responses in White Adipose Tissue: Lipoatrophy, Whole-Body Metabolism and Lifespan. Int J Mol Sci (2021) 22:2854. doi: 10.3390/ijms22062854
128. Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, et al. Leptin-Replacement Therapy for Lipodystrophy. N Engl J Med (2002) 346:570–8. doi: 10.1056/NEJMoa012437
129. Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, et al. Leptin Reverses Insulin Resistance and Hepatic Steatosis in Patients With Severe Lipodystrophy. J Clin Invest (2002) 109:1345–50. doi: 10.1172/JCI15001
130. Brown RJ, Valencia A, Startzell M, Cochran E, Walter PJ, Garraffo HM, et al. Metreleptin-Mediated Improvements in Insulin Sensitivity Are Independent of Food Intake in Humans With Lipodystrophy. J Clin Invest (2018) 128:3504–16. doi: 10.1172/JCI95476
131. Baykal AP, Parks EJ, Shamburek R, Syed-Abdul MM, Chacko S, Cochran E, et al. Leptin Decreases De Novo Lipogenesis in Patients With Lipodystrophy. JCI Insight (2020) 5:e137180. doi: 10.1172/jci.insight.137180
132. Vatier C, Fetita S, Boudou P, Tchankou C, Deville L, Riveline J, et al. One-Year Metreleptin Improves Insulin Secretion in Patients With Diabetes Linked to Genetic Lipodystrophic Syndromes. Diabetes Obes Metab (2016) 18:693–7. doi: 10.1111/dom.12606
133. Vatier C, Kalbasi D, Vantyghem MC, Lascols O, Jéru I, Daguenel A, et al. Adherence With Metreleptin Therapy and Health Self-Perception in Patients With Lipodystrophic Syndromes. Orphanet J Rare Dis (2019) 14:177. doi: 10.1186/s13023-019-1141-2
134. Cook K, Adamski K, Gomes A, Tuttle E, Kalden H, Cochran E, et al. Effects of Metreleptin on Patient Outcomes and Quality of Life in Generalized and Partial Lipodystrophy. J Endocr Soc (2021) 5:bvab019. doi: 10.1210/jendso/bvab019
135. Nguyen ML, Sachdev V, Burklow TR, Li W, Startzell M, Auh S, et al. Leptin Attenuates Cardiac Hypertrophy in Patients With Generalized Lipodystrophy. J Clin Endocrinol Metab (2021) 106:e4327–39. doi: 10.1210/clinem/dgab499
136. Diker-Cohen T, Cochran E, Gorden P, Brown RJ. Partial and Generalized Lipodystrophy: Comparison of Baseline Characteristics and Response to Metreleptin. J Clin Endocrinol Metab (2015) 100:1802–10. doi: 10.1210/jc.2014-4491
137. Adamski K, Cook K, Gupta D, Morris E, Tuttle E, Carr E, et al. Effects of Metreleptin in Patients With Lipodystrophy With and Without Baseline Concomitant Medication Use. Curr Med Res Opin (2021) 37:1881–9. doi: 10.1080/03007995.2021.1976125
Keywords: lipodystrophy, insulin resistance, diabetes, adipose tissue, genetics, senescence, lipomatosis, immunity
Citation: Zammouri J, Vatier C, Capel E, Auclair M, Storey-London C, Bismuth E, Mosbah H, Donadille B, Janmaat S, Fève B, Jéru I and Vigouroux C (2022) Molecular and Cellular Bases of Lipodystrophy Syndromes. Front. Endocrinol. 12:803189. doi: 10.3389/fendo.2021.803189
Received: 27 October 2021; Accepted: 09 December 2021;
Published: 03 January 2022.
Edited by:
Gaetano Santulli, Columbia University, United StatesReviewed by:
Giamila Fantuzzi, University of Illinois at Chicago, United StatesJoan Villarroya, University of Barcelona, Spain
Copyright © 2022 Zammouri, Vatier, Capel, Auclair, Storey-London, Bismuth, Mosbah, Donadille, Janmaat, Fève, Jéru and Vigouroux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Corinne Vigouroux, Y29yaW5uZS52aWdvdXJvdXhAaW5zZXJtLmZy