 Kyu Won Lee
Kyu Won Lee Yoochan Shin
Yoochan Shin Sungahn Lee
Sungahn Lee Sihoon Lee
Sihoon Lee
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 18 January 2022
Sec. Thyroid Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.803024
Consistent activation and functioning of thyroid hormones are essential to the human body as a whole, especially in controlling the metabolic rate of all organs and systems. Impaired sensitivity to thyroid hormones describes any process that interferes with the effectiveness of thyroid hormones. The genetic origin of inherited thyroid hormone defects and the investigation of genetic defects upon the processing of thyroid hormones are of utmost importance. Impaired sensitivity to thyroid hormone can be categorized into three conditions: thyroid hormone cell membrane transport defect (THCMTD), thyroid hormone metabolism defect (THMD), and thyroid hormone action defect (THAD). THMD is caused by defects in the synthesis and processing of deiodinases that convert the prohormone thyroxine (T4) to the active hormone triiodothyronine (T3). Deiodinase, a selenoprotein, requires unique translation machinery that is collectively composed of the selenocysteine (Sec) insertion sequence (SECIS) elements, Sec-insertion sequence-binding protein 2 (SECISBP2), Sec-specific eukaryotic elongation factor (EEFSEC), and Sec-specific tRNA (TRU-TCA1-1), which leads to the recognition of the UGA codon as a Sec codon for translation into the growing polypeptide. In addition, THMD could be expanded to the defects of enzymes that are involved in thyroid hormone conjugation, such as glucuronidation and sulphation. Paucity of inherited disorders in this category leaves them beyond the scope of this review. This review attempts to specifically explore the genomic causes and effects that result in a significant deficiency of T3 hormones due to inadequate function of deiodinases. Moreover, along with SECISBP2, TRU-TCA1-1, and deiodinase type-1 (DIO1) mutations, this review describes the variants in DIO2 single nucleotide polymorphism (SNP) and thyroid stimulating hormone receptor (TSHR) that result in the reduced activity of DIO2 and subsequent abnormal conversion of T3 from T4. Finally, this review provides additional insight into the general functionality of selenium supplementation and T3/T4 combination treatment in patients with hypothyroidism, suggesting the steps that need to be taken in the future.
Selenium as a basic element was first discovered by the Swedish chemist Jons Jacob Berzelius in 1817 (1, 2). Selenium is a trace element, and as such a “micronutrient” in humans and animals is generally obtained from the diet through food or other forms of supplementation (3, 4). We obtain dietary selenium in the form of selenomethionine (SeMet), selenocysteine (Sec), selenate, and selenite. Selenium metabolic systems play significant physiological roles in thyroid hormone metabolism, immunity, and antioxidant defense (4, 5). Selenium deficiency is associated with the occurrence, virulence, and progression of viral infectious diseases (6). In contrast, selenium supplementation resulted in immunostimulation, such as enhanced proliferation of activated T cells, activation of natural killer cells, and tumor regression mediated by cytotoxic lymphocytes (7, 8).
Selenium inadequacy is related to various types of diseases, such as cardiovascular disease (9–13), cancer (14–16), hepatopathy (17, 18), and arthropathy (19). Keshan disease is an endemic cardiomyopathy that occurs in selenium deficient areas in China and is prevented by sodium selenite supplementaion (9, 10, 12). Selenium deficiency is correlated with a significant increase in cancer incidence and mortality (14–16) and epidemiological evidence has accumulated on the cancer-preventing effects of selenium (20–22). Low selenium status is also characterized by liver injury (18) presumably resulting from elevated levels of oxidative stress (23). Oxidative stress caused by selenium deficiency plays a detrimental role in the development of joints (19). Selenium deficiency is the main cause of endemic Kashin–Beck disease (KBD), which is mainly presented as an arthropathy and reported in low-selenium areas of Far Eastern Asia. Furthermore, the pathogenesis of osteoarthritis (OA) may also be associated with oxidative stress caused by selenium deficiency (24–28).
Selenium is required for the production of thyroid hormone-metabolizing enzymes that are deiodinases, and selenium supplementation is thought to improve the function of thyrocytes (29). Impaired sensitivity to thyroid hormones, including genetic defects in thyroid hormone transport, metabolism, and action, describes disorders that interfere with the biological actions of thyroid hormone (30–32). Herein, we review the pathophysiology of impaired sensitivity to thyroid hormone. Of these, inherited disorders caused by thyroid hormone metabolism defects, mainly due to dysfunction of deiodinase, one of the selenoproteins, including the selenoprotein physiology, will be highlighted.
Selenium is a trace element. Selenium metabolism systematically proceeds in the order of absorption, transportation, transformation, and excretion of selenium (Figure 1). Selenium is taken from the diet in organic forms, Sec and SeMet, and inorganic forms Selenate and Selenite. Selenium is absorbed by small intestine and taken up by the liver, which synthesizes and exports the selenoprotein P (SELENOP), ultimately circulating in the bloodstream. SELENOP, with a number of Sec residues, carries selenium to other tissues and organs, and the transported selenium is converted to selenophosphate via intracellular selenium metabolic pathway (33, 34). Selenium is excreted through exhalation and urine. Selenosugars are key urinary metabolites for selenium excretion within the required to low-toxic range (35–37).
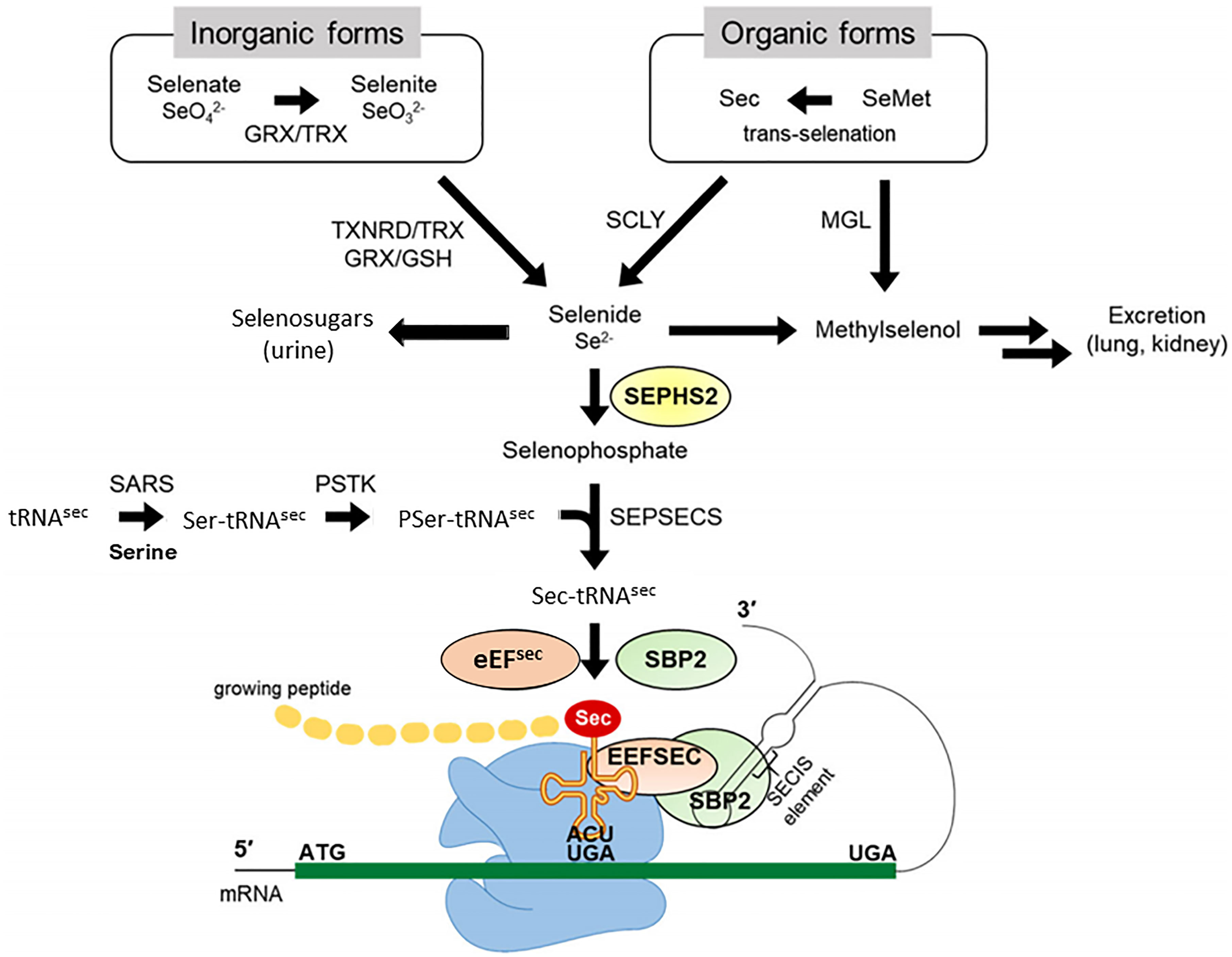
Figure 1 Selenium metabolic process in mammals. Selenium in organic forms, Sec and SeMet, and inorganic forms Selenate and Selenite taken from the diet, undergoes several conversion steps, and is incorporated into polypeptide chains, completing synthesis of selenoprotein. Selenide is synthesized from inorganic forms reduced by TXNRD/TRX or GRX/GSH systems and organic forms cleaved by SCLY. SEPHS2 synthesizes selenophosphate from selenide, and the subsequent reaction with PSer-TRU-TCA1-1 mediated by SEPSECS yields Sec-TRU-TCA1-1. SECISBP2 binds to SECIS located in the 3′UTR of a selenoprotein mRNA and mediates the transfer of Sec-TRU-TCA1-1 to the A-site of ribosome. Finally, Sec-TRU-TCA1-1 recognizes the UGA codon as the Sec integration codon. SeMet, selenomethionine; Sec, selenocysteine; GRX, glutathione reductase; TRX, thioredoxin; TXNRD, thioredoxin reductase; GSH, glutathione; MGL, methionine gamma-lyase; SCLY, selenocysteine lyase; SEPHS2, selenophosphate synthetase 2; SARS, seryl-tRNA synthetase; PSTK, phosphoseryl(Sep)-tRNA kinase; SEPSECS, Sep-tRNA: Sec-tRNA synthase; EEFSEC, Sec-specific eukaryotic elongation factor; SECISBP2, SECIS binding protein 2. Modified from Exp Mol Med. 2020 August; 52: 1198–1208.
Selenium plays a biological role mainly in the form of selenoproteins synthesized by the selenium metabolic system. Glutathione (GSH) and thioledoxin (TXN) systems first reduce ingested inorganic selenium to hydrogen selenide (H2Se). Sec amino acids converted from selenide are incorporated into specific sites of selenoproteins, such as the catalytic sites of the selenoenzyme. Mechanistically, selenophosphate is produced by the catalytic action of selenophosphate synthetase 2 (SEPHS2) through the reduction of hydrogen selenide. The subsequent reaction with phosphoseryl-tRNA (PSer-TRU-TCA1-1) yields Sec-TRU-TCA1-1. The intracellular machinery utilizing the UGA codon incorporates Sec amino acids into polypeptide chains. Selenocysteine insertion sequence binding protein 2 (SECISBP2) binds to the selenocysteine insertion sequence (SECIS) element located in the 3’-untranslated region (UTR) of selenoprotein mRNA, mediating the transfer of Sec-TRU-TCA1-1 to the A-site of the ribosome, which recognizes the UGA codon for integration of Sec. The selenoprotein translation machinery is collectively composed of SECIS elements, SECISBP2, Sec-specific eukaryotic elongation factor (EEFSEC), and aminoacylated Sec-TRU-TCA1-1, which can make the UGA codon recognized as a Sec codon and can be translated into the growing polypeptide (19).
Selenocystein (Sec) is the 21st proteinogenic amino acid, which was discovered by biochemist Thressa Stadtman at the National Institutes of Health (2, 38). Sec is a cysteine analogue with a selenium-containing selenol group in place of the sulfur-containing thiol group. After a long standing investigatory period, Sec has been confirmed as a new proteinogenic amino acid only after TRU-TCA1-1 was identified and characterized first in procaryote and later in mammalians (39–42).
Selenoprotein is a protein containing a Sec amino acid residue. The biological function of selenium is mostly demonstrated through the selenoprotein domain, which contains Sec residues. Twenty-four selenoproteins have been identified and characterized in mice. Targeted deletion of these selenoproteins has revealed their essential roles in developmental processes and in the pathogenesis of diseases (43). In the human genome, 25 selenoprotein genes have been identified so far (44). Selenoproteins can be classified into subfamilies based on their cellular functions, such as anti-oxidation [Glutathione peroxidase (GPX)-1, GPX2, GPX3, and GPX4], calcium metabolism (SELENOK, SELENOT), myogenesis (SELENON), protein AMPylation (SELENOO), protein folding (SELENOF, SELENOI, SELENOS), redox regulation [thioredoxin reductase (TXNRD)-1, TXNRD2, TXNRD3, methionine sulfoxide reductase (MSRB)-1, SELENOH, SELENOM, SELENOW], selenium transport and storage (SELENOP), selenophosphate synthesis (SEPHS2), and thyroid hormone metabolism [deiodinase (DIO)-1, DIO2, DIO3] (45, 46). The cellular functions of other selenoproteins, such as GPX6 and SELENOV, remain to be elucidated. GPXs, such as GPX1 (cytosolic GPX), GPX2 (gastrointestinal GPX), and GPX4 (phospholipid hydroperoxide GPX), promote the decomposition of a wide variety of peroxides, protecting the cells from oxidative damage (47, 48). TXNRDs use NADPH as an electron donor to return oxidized TXN to a reduced dithiol, where oxidation states have a decisive effect on regulating various cell behaviors, including proliferation and apoptosis (49). The physiological importance of TXNRD is further supported by the embryonic lethality of either Txnrd1 or Txnrd2 knockout mice (50, 51). DIOs regulate thyroid hormone metabolism by catalyzing the conversion of thyroid hormones from precursor thyroxine (T4) to biologically active triiodothyronine (T3) or inactive reverse T3 (rT3) (52). The expression levels of some selenoproteins are affected by the degree of selenium intake. Selenium deficient animals and human cell lines, for example, have decreased the transcription of selenoproteins, such as GPX1, DIOs, SELENOI, and SELENOW (53–55). Some selenoproteins, such as GPX1 and SELENOW, are more sensitive to selenium supplementation or deficiency. The hierarchy of selenoprotein expression is more obvious at a limited intracellular selenium level (3). Selenium deficiency in cells in culture has also shown to reduce selenoprotein transcript levels by nonsense-mediated decay (56).
Homeostatic regulation of the thyroid hormone economy is tightly maintained by a feedback control mechanism involving the hypothalamus, pituitary, and thyroid gland (H-P-T) axis, as shown in Figure 2. As thyroid hormones are inhibitory for H-P-T axis, the decreased supply of thyroid hormones decreases the inhibitory effect on this axis leading to its increased activity. Conversely, excess supply of thyroid hormones shuts down the system through the same H-P-T axis pathway, resulting in a restored steady state. Fine tuning of the local thyroid hormone requirement is controlled through three additional steps. First, thyroid hormone entry across the cell membrane through transmembrane transporters such as MCT8 and MCT10 through facilitated diffusion (57–59). Second, the formation of active T3 (triiodothyronine) by removal of one of the outer ring iodine atoms (5’-deiodination) from prohormone T4 (thyroxine), or inactive rT3 and T2 by inner ring (5-deiodination) from T4 and T3, respectively, provide additional levels of control (52). Finally, the integrity of thyroid hormone receptors (THRs), through which thyroid hormone action is mediated, determines the type and extent of thyroid hormone response. Thyroid hormone action occurs not only in the nucleus of the target cell, but also in the cytoplasm (60, 61). The former, known as a genomic effect, has been studied extensively (62, 63). There are two THRs (THR-alpha and THR-beta) encoded by separate genes located on chromosomes 17 and 3, respectively. Different isoforms are formed by alternative transcription and splicing. The receptors have structural and sequence similarities with DNA binding and T3 binding domains. Other regions of the molecules are involved in dimeric formation with another THR or another type of nuclear receptor, and in binding coactivators and corepressor cofactors (64, 65). In the nucleus, THRs act as transcription factors that regulate the expression of certain genes, which are recognized through the thyroid hormone response element. Binding of unliganded dimeric receptors (without T3) to the thyroid hormone response element and recruitment of corepressor proteins results in inhibition of the expression of genes that are positively regulated by T3. When T3 binds to a receptor, the THR molecule undergoes a steric change, resulting in the release of corepressor proteins, dissociation of dimers, and formation of heterodimers of THR and retinoid X receptors that then bind coactivator proteins. This change promotes the expression of target genes and ultimately increases the synthesis of certain proteins. Although THRs reside primarily in the nucleus, they shuttle rapidly between the nucleus and cytoplasm. Recently, cryptic cytoplasmic functions have been described to other THR subtypes, expanding the diverse cellular responses to thyroid hormone. (66). Thyroid hormones can also act through binding sites at plasma membrane such as integrin αvβ3 (67).
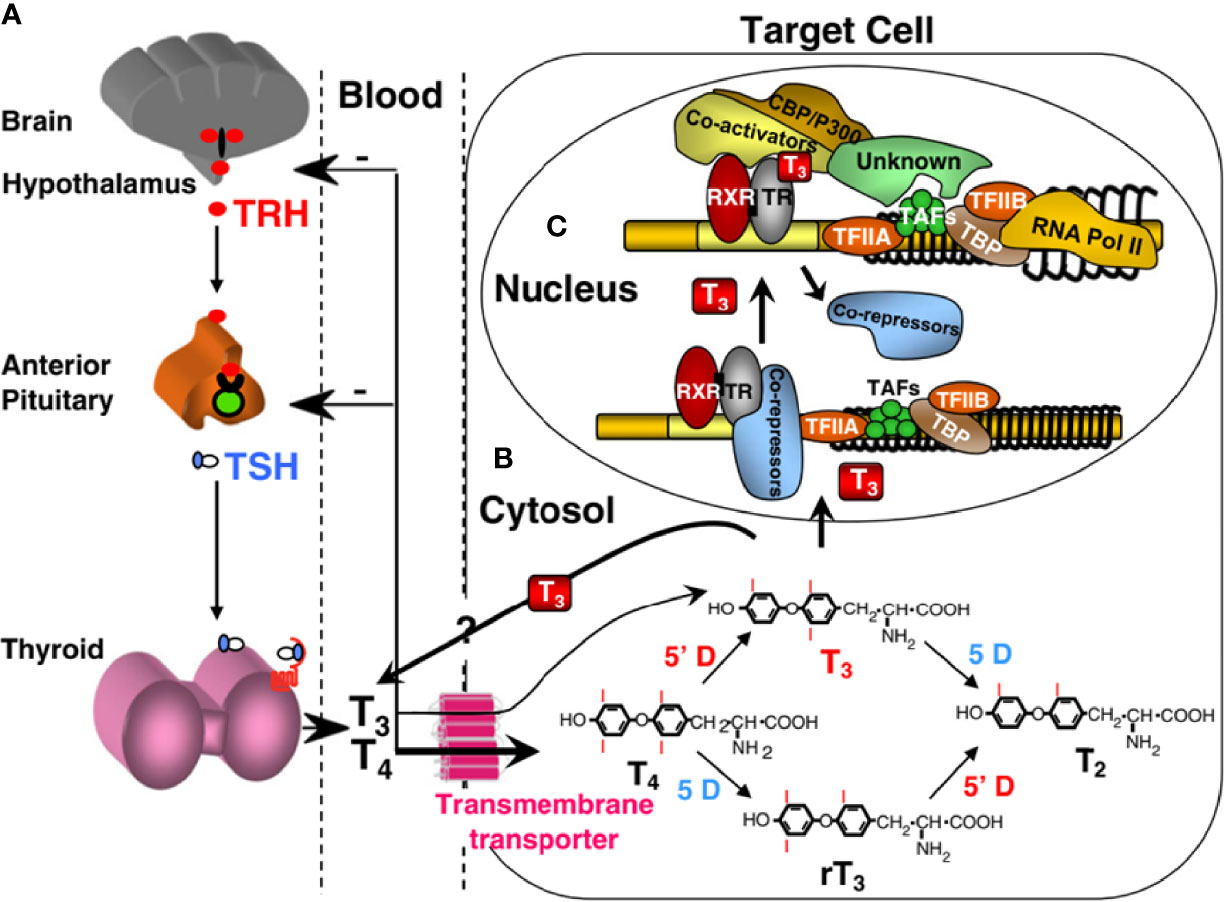
Figure 2 Regulation of TH supply, metabolism and genomic action. (A) Central feedback control that regulates the amount of TH in blood. (B) Intracellular metabolism of TH, regulating TH bioactivity. (C) Genomic action of TH. CBP/P300, cAMP-binding protein/general transcription adaptor; TFIIA and TFIIB, transcription intermediary factor II, A and B; TBP, TATA-binding protein; TAF, TBP-associated factor. Modified from Refetoff S, Dumitrescu AM. Syndromes of reduced sensitivity to thyroid hormone: genetic defects in hormone receptors, cell transporters and deiodination. Best Pract Res Clin Endocrinol Metab. 2007 Jun;21(2):277–305.
Impaired sensitivity to thyroid hormone refers to any process that interferes with the effects of thyroid hormone, including defects in the transport, metabolism, or action of thyroid hormone (30, 68–70). Each defect is named representing the step affected as detailed in the following sections: (32, 71, 72).
Defects in one of the cell transport proteins that allow thyroid hormones to enter cells can cause decreased intracellular levels of thyroid hormones. Defective cell transport proteins may not locate their normal sites on the cell membrane or transport hormones. This causes a disorder depending on the hormone transporter that is affected. For example, a defect in monocarboxylate transporter 8 (MCT8) results in elevated serum concentrations of T3 and low levels of T4 and rT3 (68). This transporter is also involved in thyroid hormone secretion from the thyroid gland (73).
T4, a major form secreted by the thyroid gland, is a prohormone that must be converted to active T3 in the cytoplasm. Any defect in the factors involved in this enzymatic deiodination reaction can reduce T3 production, impairing the sensitivity to thyroid hormones. Abnormalities in the synthesis or degradation of various deiodinases may be included in these defects. Patients in this category had low serum T3 and high T4 and rT3 concentrations. More details in this category of defects will be described in section 5.
Theoretically, for the thyroid hormone to enter the target cell to exhibit genomic effects, it must be transported into the nucleus and form a complex with the thyroid hormone receptor (THR) along with a series of cofactors to regulate the transcription of target genes. Therefore, it can interfere with the action of thyroid hormones due to disorders arising from defects in nuclear migration and various cofactors, but there have been no reports of patients showing such disorders in actual clinical practice.
Genomic thyroid hormone action is mediated through THRs, which act as transcription factors that activate or repress the transcription of certain target genes. Most RTHs are caused by THR defects (30). Mutant THR proteins have a reduced ability to bind cognate ligands or protein cofactors or bind to target genes. Mutations in the thyroid hormone receptor beta gene (THRB), which encodes the thyroid hormone receptor beta (THR-beta), are the most common cause of RTH and are defined as RTH-beta (74). In contrast, RTH-alpha, caused by mutations in the thyroid hormone receptor alpha gene (THRA), encoding thyroid hormone receptor alpha (THR-alpha), has rarely been reported (75).
Since SECISBP2 is epistatic to selenoprotein, SECISBP2 defects lead to low expression levels of selenoproteins due to poor Sec insertion and UGA decoding. To date, 12 families with SECISBP2 mutations have been identified. Three of them had homozygous alleles, while nine had compound heterozygous alleles. Altogether, 20 unique SECISBP2 mutations have been reported (70, 76–82)
Nearly all families from published cases exhibit common TFT abnormalities: elevated free T4 (FT4), elevated rT3, low free T3 (FT3), and normal or slightly elevated TSH levels. Such abnormalities are a universal indication of dysfunctional T4 to T3 conversion due to deficient DIOs from epistatic SECISBP2 mutations (83).
Other biochemical signatures include low serum selenium levels, reflecting deficiencies of SELENOP and GPX3. Similarly, all but one patient (Family 5) exhibited short stature and delayed skeletal development (78). These are the most common clinical features of SECISBP2 mutations. Growth retardation in Dio2 and Dio3 null mice suggests that the human phenotype is also mediated by abnormal thyroid hormone metabolism (84, 85). For more severe cases, failure to thrive was observed, while Family 4, arguably the most extreme case, exhibited multiple skeletal abnormalities such as craniofacial dysmorphism, bilateral clinodactyly, kyphoscoliosis, and leg asymmetry (77).
Symptoms unrelated to the skeleton and growth vary greatly depending on the severity of the mutations. Among the published cases, six suffered from delayed motor milestones and muscle weakness, especially in the legs (77–81). Families 5, 6, and 7 all exhibit characteristic phenotypes of SEPN1 myopathies due to defects in SELENON, affected axial muscles, adductors, and sartorius (78, 79). Additionally, families 7 and 10 have connective or fatty tissue infiltrating the adductor muscles (79, 81). In some cases, myopathy is severe enough to exhibit Gowers’ signs or a waddling gait. All families suffering from myopathy, except for family 6, also suffer from neurological symptoms (78). Intellectual disability is the most common, while families 5 and 10 each have milder neurological symptoms: delayed motor milestones and attention deficit disorder, respectively (78, 81). Neurological phenotypes are difficult to associate with a deficit of any specific selenoprotein.
In severe cases, bilateral hearing loss (either sensorineural or conductive), secretory otitis media, and rotatory vertigo have been observed (77–79). One hypothesis associates this phenotype with a DIO2 deficit (84). However, a more recent alternate hypothesis suggests that the buildup of reactive oxygen species (ROS) due to antioxidant GPX1 deficiency leads to cochlear damage and hearing loss (78).
Common metabolic phenotypes such as elevated fat mass index, obesity, and paradoxically increased systemic sensitivity to insulin have also been observed in adults and children (78). Gpx1 null mice exhibiting increased ROS levels become insulin sensitive (86). In contrast, mice overexpressing Gpx1 develop insulin resistance (87). Thus, it is likely that these phenotypes are the result of increased cellular ROS due to GPX1 deficiency. Overall, a link between selenoproteins and systemic insulin sensitivity has been suggested. Family 10 was noted as the patient exhibited impaired glucose tolerance despite GPX1 deficiency (81). These conditions are assumed to be independent of SECISBP2 mutations.
The only adult in the studies, proband of family 5, exhibits unique phenotypes such as azoospermia and photosensitivity (78). Azoospermia is likely a result of the underexpression of GPX4, thioredoxin/glutathione reductase (TGR), and selenium-containing protein V (SELENOV), the first two of which are integral in sperm development. Cutaneous photosensitivity is attributed to the underexpression of the antioxidant selenoprotein, which increases susceptibility to ROS generation from UV rays. The other characteristics and clinical features are summarized in Table 1.
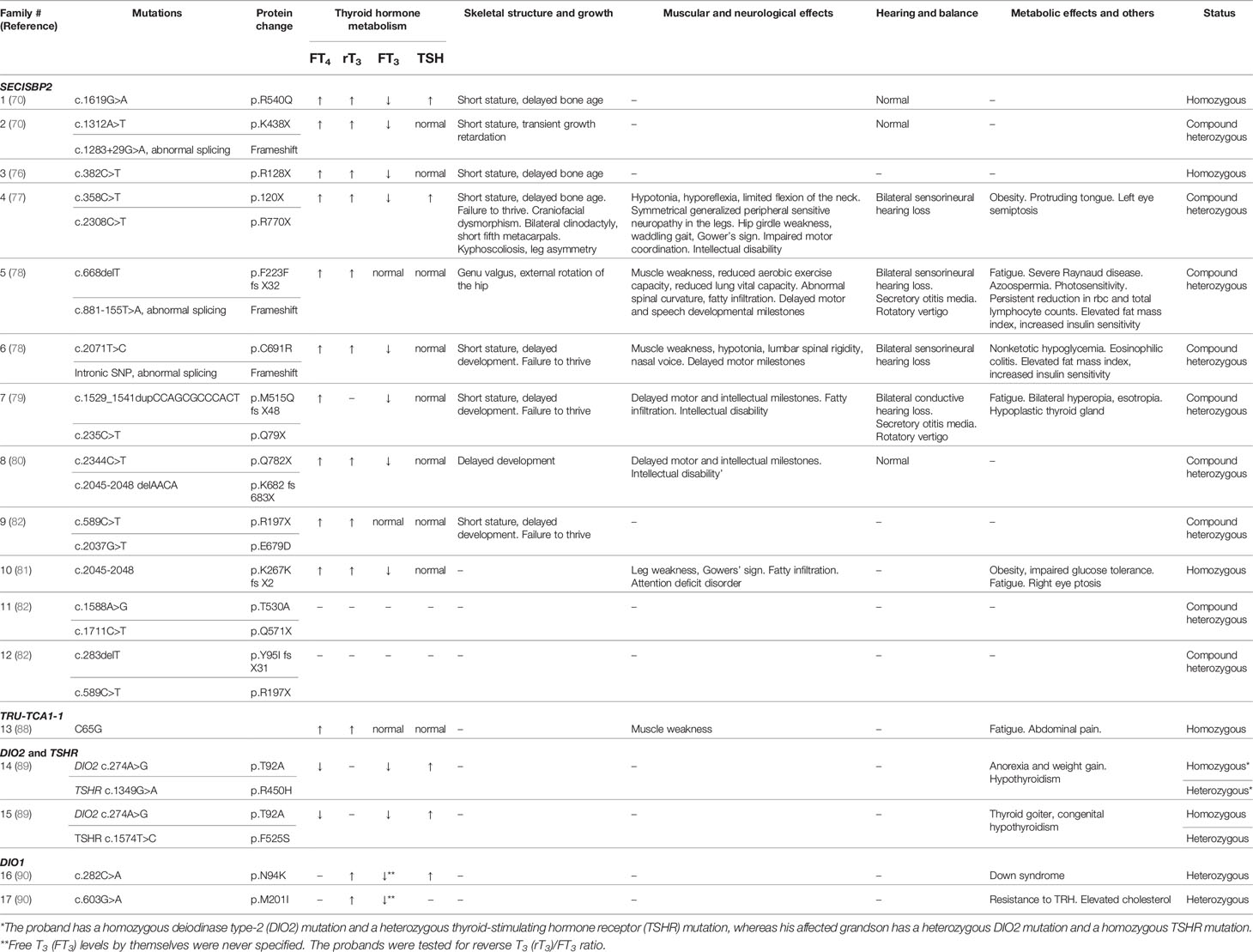
Table 1 Selenoprotein mutations affecting thyroid hormone metabolism defects (THMDs) and their clinical features.
Homozygosity mapping with known consanguinity in the proband (an 8-year-old boy who exhibited thyroid dysfunction [raised T4, normal T3, raised reverse T3]) identified a single interval in the proband, encompassing the chromosomal locus of only one gene (TRU-TCA1-1) in the Sec-incorporation pathway. Sequencing of this gene in the proband indicated homozygosity for a single nucleotide change, C65G in TRU-TCA1-1 (88). The early stages of tRNASec maturation involve sequential base modifications, yielding two major tRNASec isoforms containing either 5-methoxycarbonyl-methyluridine (mcm5U) and 5-methoxycarbonyl methyl-2’-O-methyluridine (mcm5Um) at position 34, situated in the anticodon loop. Isoform containing mcm5U is known for its role in synthesizing housekeeping selenoproteins, while another isoform containing mcm5Um is known to be responsible for the synthesis of stress-related selenoproteins (91, 92). Primary cells from the proband showed a marked reduction in the mcm5Um isoform of tRNASec, whereas the mcm5U isoform of tRNASec was relatively preserved. Radiolabeled tRNASec injection into Xenopus oocytes followed by chromatography analysis indicated significantly attenuated base modification in C65G TRU-TCA1-1 compared with the wild-type, indicating weakened subsequent methylation of mcm5U at position U34. This confirms that the maturation of mutant tRNASec is impaired. These findings indicate that a TRU-TCA1-1 mutation can selectively impair the synthesis of stress-related selenoproteins (88).
In an extensive case-finding study conducted in Korea, which targets patients who present inappropriately high TSH levels despite a high dose of levothyroxine (L-T4) and a drop in TSH levels only after addition of liothyronine (L-T3), two patients were noted (89). The first is a 68-year-old Asian man with symptoms of anorexia and weight gain for three months. Initial serum TSH was 85.50 mIU/L (0.27–4.20) and FT4 was 0.16 ng/dL (0.93–1.70). After diagnosis of hypothyroidism, he began to take L-T4 at 100 μg per day, but his serum TSH level was not suppressed. When L-T3 30 μg was administered three times a day instead of L-T4 100μg, the serum TSH level dramatically decreased. The second patient was a 75-year-old Asian woman who had suffered from a huge thyroid goiter (up to 10 cm in diameter) since her early twenties. Her thyroid function tests revealed extremely high TSH levels, inappropriately high FT4 and relatively low T3 levels; TSH 82 mIU/L, FT4>7.70 ng/dL, T3 64 ng/dL without thyrotoxic symptoms. Her goiter decreased in size dramatically when administered L-T3 30 μg twice a day, followed by a decrease in TSH level to 8.21 mIU/L and an increase in T3 level to 94 ng/dL without differences in FT4 levels. The TRH stimulation test revealed that it was less likely due to either RTH-or TSH-secreting pituitary adenoma for inappropriately increased TSH. Nucleotide sequencing of the first patient revealed a homozygous SNP in the DIO2 gene (c.274A>G, DIO2 T92A), which had previously been identified in a certain portion of the population as a significant SNP (93) and a heterozygous mutation in the TSHR gene (c.1349G>A, R450H) (94). Interestingly, the second patient also had a homozygous DIO2 T92A mutation, together with a heterozygous TSHR F525S mutation. Both TSHR R450H and F525S are loss-of-function mutations. Since it was first reported that DIO2 is regulated by TSH (95, 96), there have been reports supporting the same concept in various cell types (97, 98). Large cohort studies with normal thyroid function also support the notion that TSH regulates DIO2 both in pediatric and adult populations (99, 100). In the same context, one can hypothesize that decreased signal from TSH due to defects in TSHR as well as DIO2 dysfunctional SNP results in reduced activity of DIO2 and abnormal conversion of T3 from T4 (Figure 3). To prove this hypothesis, DIO2 activity was measured using fibroblasts obtained and primarily cultured from patients harboring the DIO2 T92A SNP together with the TSHR R450H mutation. Both the relative activity and mRNA expression of DIO2 were blunt in fibroblasts from the patient, confirming the hypothesis (89).
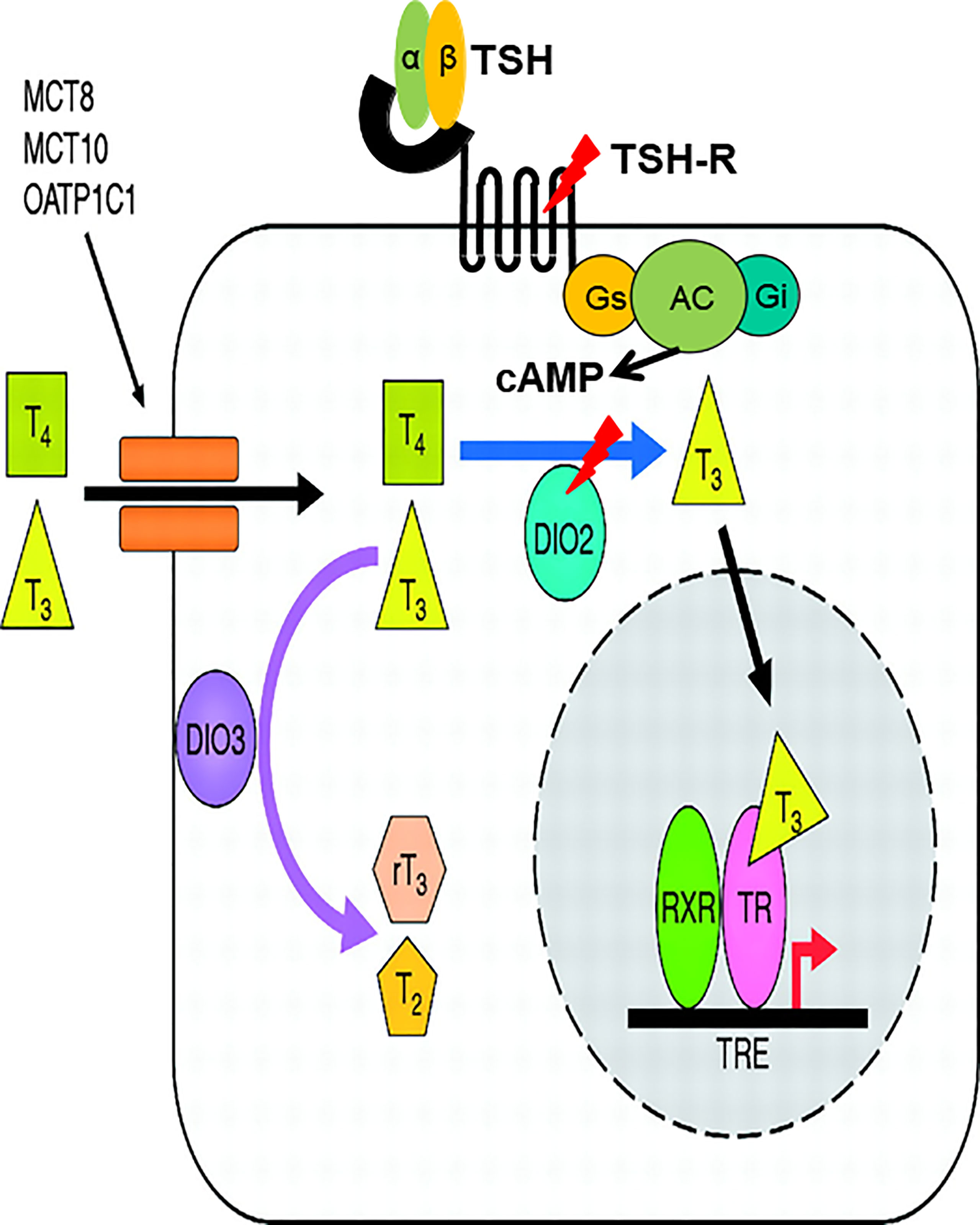
Figure 3 TSHR loss-of function mutations and homozygous DIO2 T92A SNP (“double hit”) cause decreased activity of DIO2, resulting in a novel form of abnormal thyroid hormone metabolism. cAMP produced by interaction of TSH and TSHR affects the cAMP response element located upstream of promoter region of DIO2. Lack of cAMP production caused by loss-of-function mutation of TSHR and DIO2 T92A SNP cooperatively causes decreased DIO2 enzymatic activity.
Next generation sequencing analyses of two patients from two unrelated families presenting with abnormal TH metabolism and elevated serum rT3 levels and rT3/T3 ratios revealed two missense DIO1 mutations (p.Asn94Lys and p.Met201Ile) (90). In silico prediction of these DIO1 mutants suggested pathogenic variants, and in vitro assessment of the functional activity of DIO1 mutants showed decreased enzymatic activities, confirming that this mutation is disease-causing. Heterozygous Dio1 knock-out mice back crossed >10 times into the WT 57BL/J5 strain showed elevated serum rT3 levels and elevated rT3/T3 ratios, consistent with the data observed in the affected patients. Advances and widespread sequencing technologies will allow us to identify more mutations in patients showing a bizarre thyroid function test.
The effect of selenium deficiency and selenium supplementation on general health, especially on thyroid function, remains to be elucidated (29). Several trials have been conducted in regions with different baseline selenium status in UK, New Zealand, USA, and rural Tibet (101–105). No definite effects or consistent adverse effects on thyroid function have been observed (102). In critical illnesses, such as severe sepsis or trauma injuries, selenium supplementation showed null or only moderate effects on thyroid function (106, 107). These findings suggest that selenium supplementation does not play an important role in determining the serum thyroid hormone status. Meanwhile, selenosis, i.e., an excess of Se intake, should be avoided, as Se in high dosages is toxic, potentially due to elevated selenoprotein P levels as observed in clinical trials with therapeutic dosages of selenite.
Identification of the clinical phenotypes of affected patients and the metabolic pathways responsible for them provides insight into targeted treatment options. Selenium supplementation could first be considered an ideal treatment modality for disorders caused by selenium and selenoprotein dysregulation. Administration of up to 400 µg of selenomethionine-rich yeast, but not sodium selenite, normalized the serum selenium concentration but did not recover normal thyroid hormone metabolism in patients with SECISBP2 mutations (108). The difference in Selenium bioavailability between selenomethionine and selenite results from the efficiency of Selenium incorporation. Selenomethionine seems to be more efficient because it can be non-specifically incorporated into all circulating serum proteins (109), whereas selenite is metabolized and inserted as Sec into the growing peptide chain of selenoproteins (110).
Hashimoto’s thyroiditis and Graves’ disease are the most common autoimmune thyroid diseases (111). Reduced serum selenium concentrations have been reported in patients with autoimmune thyroid disease (112, 113). Consequently, several studies have tested the efficacy of selenium supplementation in patients with Hashimoto’s thyroiditis. Three trials reported successful reductions in TPO autoantibodies titers and/or improvement of mood and well-being (114–117). As no consistent adverse events were observed, routine selenium supplementation in patients with Hashimoto’s thyroiditis was considered as a promising adjuvant treatment option (118). It is worth noting that experiences with subjects deficient in both iodine and selenium, and displaying increased disease symptoms when Se supplementation was initiated without at the same time raising iodine supply (so called myxedematous cretinism).
Selenium supplementation trials in patients with Graves’ disease are limited. One of the major and severe complications of Graves’ disease, ophthalmopathy (also known as orbitopathy) is an inflammatory process in nature and presents as a protrusion of one or both eyes. Increased intraorbital pressure results in proptosis and compressive neuropathy, and inflammation of the extraocular muscles causes diplopia (119). One randomized clinical trial of selenium supplementation in patients with mild Graves’ orbitopathy was performed, and the quality of life and eye disease parameters improved significantly after 6 months of treatment (120, 121). Recently, it was reported that selenium suppressed hyaluronan production, inflammatory cytokines, and intracellular ROS generation in cultured orbital fibroblasts of patients with Graves’ orbitopathy (GO), suggesting a basis for the use of selenium in the treatment of GO (122).
Approximately 5–10% or more of biochemically well-controlled hypothyroidism patients with levothyroxine (L-T4) treatment have persistent complaints, such as depression and impaired psychological wellbeing (123). There are various explanations for the discrepancies between thyroid function tests and clinical symptoms, but there is no definite answer yet. Instead, expectations for T3/T4 combination therapy have been raised as a realistic solution. Several trials using combined T3/T4 therapy have been conducted for comparison with L-T4 monotherapy in the past years. Some studies have shown a beneficial effect, such as patient preference or an improved metabolic profile (124–127), however, in general, patients on T3/T4 combination therapy do not have improved outcomes compared with those on L-T4 monotherapy (128, 129). Possible explanations for unsatisfactory results may include inadequate L-T4 and L-T3 doses or frequency of administration (130).
Moreover, individuals with genetic variations in thyroid hormone metabolism should be considered (130). A subgroup that could be targeted is individuals with common genetic variations in DIO2, such as T92A SNP, which encodes the deiodinase 2 enzyme that converts T4 to T3 locally in several tissues (131). In a study conducted in the UK, the T92A SNP in DIO2 was associated with lower baseline psychological well-being in patients on LT4 replacement therapy and with better response to T3/T4 combination therapy, compared with patients without the SNP on L-T4 replacement therapy (132). In contrast, results from a large population-based cohort study revealed no effect of the T92A SNP on quality of life or cognitive function measures (133).
Generally, L-T3 is much more effective in treating developmental symptoms in patients with SECISBP2 mutations. For example, delayed linear growth can be improved by L-T3 supplementation (76). Similarly, L-T3 improved growth, speech, and development, while normalizing TFT (78). Of particular interest is the treatment of Japanese patients (79). The proband was initially treated with GH alone, since 52 months of age, in the hope of normalizing the TFT and improving his growth. While six years of GH treatment increased his height SDS from -3.4 to -1.7, his TFT abnormalities persisted. Addition of L-T3 to the treatment for six months normalized TFT and advanced bone age. Later treatment of the proband with α-tocopherol (vitamin E) was shown to have decreased lipid peroxidation products and increased circulating white blood cells and neutrophils, strengthening the immune system.
The effect of L-T3 administration was also tested in two patients, as it was demonstrated to equally suppress serum TSH concentration, which was not sufficiently suppressed by L-T4 in T92A SNP in DIO2 and TSHR mutations (89).
While the American Thyroid Association guidelines generally disadvocate the routine use of T3/T4 combination therapy in patients with hypothyroidism (134), the European Thyroid Association guidelines state that a 3-month trial of T3/T4 combination might be considered experimentally in adherent, biochemically well-controlled patients who have persistent symptoms despite L-T4 treatment (135). Sufficiently powered prospective randomized controlled trials are therefore prerequisite before concrete conclusions can be drawn, especially considering the genetic variations responsible for thyroid hormone metabolism discussed in this review. Collectively, these findings could provide clinical relevance in a select population of hypothyroidism patients who might benefit from T3/T4 combination therapy.
The main reason for the increased interest in rare genetic diseases and their molecular genetic mechanisms is to facilitate the effective treatment of more common diseases related to it. In the treatment of hypothyroidism, there has always been much controversy about T4 treatment and T3/T4 combination therapy. It is the Se status and Se intake that may be critical for success or failure of the T3/T4 combination therapy, in view of Se deficiency potentially impairing sufficient DIO expression in target tissues for efficient T3 production (without this defect being reflected in circulating thyroid hormone concentrations). It is expected that a fundamental approach to this will become possible as knowledge accumulates through future studies. As our understanding of selenoprotein metabolism and action deepens, the scope for its clinical application expands. In the future, it is expected that a new era of the most appropriate thyroid hormone replacement therapy will further expand our understanding of the molecular mechanisms of action of selenium and selenoprotein, especially the entire process of thyroid hormone metabolism through DIOs and its related pathological conditions.
KL and SiL contributed to conception and design of the study. KL, YS, and SuL collected references. KL wrote the first draft of the manuscript. KL, YS, and SuL wrote sections of the manuscript. SiL wrote the final version of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This study was supported by the Basic Science Research Program, National Research Foundation of Korea (grant number NRF-2019R1F1A1063459 to SiL) and Korea Research-driven Hospitals Grant fostered for Gachon University Gil Medical Center (grant number 2018-5284 to SiL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Profs. Byeong Jae Lee, Jin-Hong Kim, and Dong-Yeob Shin for their insightful advices.
1. Berzelius JJ. Undersökning Af En Ny Mineral-Kropp, Funnen I De Orenare Sorterna Af Det I Falun Tillverkade Svafl Et. Afhandlingar I Fysik Kemi Och Mineral (1818) 6:42.
2. Flohe L. The Labour Pains of Biochemical Selenology: The History of Selenoprotein Biosynthesis. Biochim Biophys Acta (2009) 1790:1389–403. doi: 10.1016/j.bbagen.2009.03.031
3. Hatfield DL, Gladyshev VN. How Selenium has Altered Our Understanding of the Genetic Code. Mol Cell Biol (2002) 22:3565–76. doi: 10.1128/MCB.22.11.3565-3576.2002
4. Rayman MP. Selenium and Human Health. Lancet (2012) 379:1256–68. doi: 10.1016/S0140-6736(11)61452-9
5. Arthur JR, Mckenzie RC, Beckett GJ. Selenium in the Immune System. J Nutr (2003) 133:1457S–9S. doi: 10.1093/jn/133.5.1457S
6. Broome CS, Mcardle F, Kyle JA, Andrews F, Lowe NM, Hart CA, et al. An Increase in Selenium Intake Improves Immune Function and Poliovirus Handling in Adults With Marginal Selenium Status. Am J Clin Nutr (2004) 80:154–62. doi: 10.1093/ajcn/80.1.154
7. Wood SM, Beckham C, Yosioka A, Darban H, Watson RR. Beta-Carotene and Selenium Supplementation Enhances Immune Response in Aged Humans. Integr Med (2000) 2:85–92. doi: 10.1016/S1096-2190(00)00009-3
8. Hawkes WC, Kelley DS, Taylor PC. The Effects of Dietary Selenium on the Immune System in Healthy Men. Biol Trace Elem Res (2001) 81:189–213. doi: 10.1385/BTER:81:3:189
9. Chen X, Yang G, Chen J, Chen X, Wen Z, Ge K. Studies on the Relations of Selenium and Keshan Disease. Biol Trace Elem Res (1980) 2:91–107. doi: 10.1007/BF02798589
10. Neve J. Selenium as a Risk Factor for Cardiovascular Diseases. J Cardiovasc Risk (1996) 3:42–7. doi: 10.1177/174182679600300106
11. Alissa EM, Bahijri SM, Ferns GA. The Controversy Surrounding Selenium and Cardiovascular Disease: A Review of the Evidence. Med Sci Monit (2003) 9:RA9–18.
12. Loscalzo J. Keshan Disease, Selenium Deficiency, and the Selenoproteome. N Engl J Med (2014) 370:1756–60. doi: 10.1056/NEJMcibr1402199
13. Zhang X, Liu C, Guo J, Song Y. Selenium Status and Cardiovascular Diseases: Meta-Analysis of Prospective Observational Studies and Randomized Controlled Trials. Eur J Clin Nutr (2016) 70:162–9. doi: 10.1038/ejcn.2015.78
14. Willett WC, Polk BF, Morris JS, Stampfer MJ, Pressel S, Rosner B, et al. Prediagnostic Serum Selenium and Risk of Cancer. Lancet (1983) 2:130–4. doi: 10.1016/S0140-6736(83)90116-2
15. Patrick L. Selenium Biochemistry and Cancer: A Review of the Literature. Altern Med Rev (2004) 9:239–58.
16. Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN. Selenium and Selenocysteine: Roles in Cancer, Health, and Development. Trends Biochem Sci (2014) 39:112–20. doi: 10.1016/j.tibs.2013.12.007
17. Navarro-Alarcon M, Lopez-Ga de la Serrana H, Perez-Valero V, Lopez-Martinez MC. Selenium Concentrations in Serum of Individuals With Liver Diseases (Cirrhosis or Hepatitis): Relationship With Some Nutritional and Biochemical Markers. Sci Total Environ (2002) 291:135–41. doi: 10.1016/S0048-9697(01)01088-9
18. Guo CH, Chen PC, Ko WS. Status of Essential Trace Minerals and Oxidative Stress in Viral Hepatitis C Patients With Nonalcoholic Fatty Liver Disease. Int J Med Sci (2013) 10:730–7. doi: 10.7150/ijms.6104
19. Kang D, Lee J, Wu C, Guo X, Lee BJ, Chun JS, et al. The Role of Selenium Metabolism and Selenoproteins in Cartilage Homeostasis and Arthropathies. Exp Mol Med (2020) 52:1198–208. doi: 10.1038/s12276-020-0408-y
20. Clark LC, Combs GF Jr, Turnbull BW, Slate EH, Chalker DK, Chow J, et al. Effects of Selenium Supplementation for Cancer Prevention in Patients With Carcinoma of the Skin. A Randomized Controlled Trial. Nutritional Prevention of Cancer Study Group. JAMA (1996) 276:1957–63. doi: 10.1001/jama.1996.03540240035027
21. Zhuo H, Smith AH, Steinmaus C. Selenium and Lung Cancer: A Quantitative Analysis of Heterogeneity in the Current Epidemiological Literature. Cancer Epidemiol Biomarkers Prev (2004) 13:771–8.
22. Davis CD, Tsuji PA, Milner JA. Selenoproteins and Cancer Prevention. Annu Rev Nutr (2012) 32:73–95. doi: 10.1146/annurev-nutr-071811-150740
23. Lawrence RA, Burk RF. Glutathione Peroxidase Activity in Selenium-Deficient Rat Liver. Biochem Biophys Res Commun (1976) 71:952–8. doi: 10.1016/0006-291X(76)90747-6
24. Sasaki S, Iwata H, Ishiguro N, Habuchi O, Miura T. Low-Selenium Diet, Bone, and Articular Cartilage in Rats. Nutrition (1994) 10:538–43.
25. Li S, Cao J, Caterson B, Hughes CE. Proteoglycan Metabolism, Cell Death and Kashin-Beck Disease. Glycoconj J (2012) 29:241–8. doi: 10.1007/s10719-012-9421-2
26. Jordan JM. An Ongoing Assessment of Osteoarthritis in African Americans and Caucasians in North Carolina: The Johnston County Osteoarthritis Project. Trans Am Clin Climatol Assoc (2015) 126:77–86.
27. Yang L, Zhao GH, Yu FF, Zhang RQ, Guo X. Selenium and Iodine Levels in Subjects With Kashin-Beck Disease: A Meta-Analysis. Biol Trace Elem Res (2016) 170:43–54. doi: 10.1007/s12011-015-0463-8
28. Wang L, Yin J, Yang B, Qu C, Lei J, Han J, et al. Serious Selenium Deficiency in the Serum of Patients With Kashin-Beck Disease and the Effect of Nano-Selenium on Their Chondrocytes. Biol Trace Elem Res (2020) 194:96–104. doi: 10.1007/s12011-019-01759-7
29. Schomburg L. Selenium, Selenoproteins and the Thyroid Gland: Interactions in Health and Disease. Nat Rev Endocrinol (2011) 8:160–71. doi: 10.1038/nrendo.2011.174
30. Refetoff S, Weiss RE, Usala SJ. The Syndromes of Resistance to Thyroid Hormone. Endocr Rev (1993) 14:348–99. doi: 10.1210/er.14.3.348
31. Dumitrescu AM, Refetoff S. Impaired Sensitivity to Thyroid Hormone: Defects of Transport, Metabolism and Action,. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, De Herder WW, Dungan K, et al, editors. Endotext. South Dartmouth, MA: MDText.com, Inc. (2000).
32. Refetoff S, Bassett JH, Beck-Peccoz P, Bernal J, Brent G, Chatterjee K, et al. Classification and Proposed Nomenclature for Inherited Defects of Thyroid Hormone Action, Cell Transport, and Metabolism. J Clin Endocrinol Metab (2014) 99:768–70. doi: 10.1210/jc.2013-3393
33. Hill KE, Lloyd RS, Yang JG, Read R, Burk RF. The cDNA for Rat Selenoprotein P Contains 10 TGA Codons in the Open Reading Frame. J Biol Chem (1991) 266:10050–3. doi: 10.1016/S0021-9258(18)99185-4
34. Hill KE, Zhou J, Mcmahan WJ, Motley AK, Atkins JF, Gesteland RF, et al. Deletion of Selenoprotein P Alters Distribution of Selenium in the Mouse. J Biol Chem (2003) 278:13640–6. doi: 10.1074/jbc.M300755200
35. Mc CK, Portman OW. Excretion of Dimethyl Selenide by the Rat. J Biol Chem (1952) 195:277–82. doi: 10.1016/S0021-9258(19)50898-5
36. Byard JL. Trimethyl Selenide. A Urinary Metabolite of Selenite. Arch Biochem Biophys (1969) 130:556–60. doi: 10.1016/0003-9861(69)90070-8
37. Kobayashi Y, Ogra Y, Ishiwata K, Takayama H, Aimi N, Suzuki KT. Selenosugars Are Key and Urinary Metabolites for Selenium Excretion Within the Required to Low-Toxic Range. Proc Natl Acad Sci USA (2002) 99:15932–6. doi: 10.1073/pnas.252610699
38. Cone JE, Del Rio RM, Davis JN, Stadtman TC. Chemical Characterization of the Selenoprotein Component of Clostridial Glycine Reductase: Identification of Selenocysteine as the Organoselenium Moiety. Proc Natl Acad Sci USA (1976) 73:2659–63. doi: 10.1073/pnas.73.8.2659
39. Leinfelder W, Zehelein E, Mandrand-Berthelot MA, Bock A. Gene for a Novel tRNA Species That Accepts L-Serine and Cotranslationally Inserts Selenocysteine. Nature (1988) 331:723–5. doi: 10.1038/331723a0
40. Mizutani T, Hitaka T. The Conversion of Phosphoserine Residues to Selenocysteine Residues on an Opal Suppressor tRNA and Casein. FEBS Lett (1988) 232:243–8. doi: 10.1016/0014-5793(88)80425-3
41. Lee BJ, Worland PJ, Davis JN, Stadtman TC, Hatfield DL. Identification of a selenocysteyl-tRNA(Ser) in Mammalian Cells That Recognizes the Nonsense Codon, UGA. J Biol Chem (1989) 264:9724–7. doi: 10.1016/S0021-9258(18)81714-8
42. Mizutani T. Some Evidence of the Enzymatic Conversion of Bovine Suppressor phosphoseryl-tRNA to selenocysteyl-tRNA. FEBS Lett (1989) 250:142–6. doi: 10.1016/0014-5793(89)80707-0
43. Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, et al. Characterization of Mammalian Selenoproteomes. Science (2003) 300:1439–43. doi: 10.1126/science.1083516
45. Pitts MW, Hoffmann PR. Endoplasmic Reticulum-Resident Selenoproteins as Regulators of Calcium Signaling and Homeostasis. Cell Calcium (2018) 70:76–86. doi: 10.1016/j.ceca.2017.05.001
46. Sreelatha A, Yee SS, Lopez VA, Park BC, Kinch LN, Pilch S, et al. Protein AMPylation by an Evolutionarily Conserved Pseudokinase. Cell (2018) 175:809–21.e819. doi: 10.1016/j.cell.2018.08.046
47. Brigelius-Flohe R. Glutathione Peroxidases and Redox-Regulated Transcription Factors. Biol Chem (2006) 387:1329–35. doi: 10.1515/BC.2006.166
48. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell (2018) 172:409–22.e421. doi: 10.1016/j.cell.2017.11.048
49. Arner ES, Holmgren A. Physiological Functions of Thioredoxin and Thioredoxin Reductase. Eur J Biochem (2000) 267:6102–9. doi: 10.1046/j.1432-1327.2000.01701.x
50. Conrad M, Jakupoglu C, Moreno SG, Lippl S, Banjac A, Schneider M, et al. Essential Role for Mitochondrial Thioredoxin Reductase in Hematopoiesis, Heart Development, and Heart Function. Mol Cell Biol (2004) 24:9414–23. doi: 10.1128/MCB.24.21.9414-9423.2004
51. Jakupoglu C, Przemeck GK, Schneider M, Moreno SG, Mayr N, Hatzopoulos AK, et al. Cytoplasmic Thioredoxin Reductase Is Essential for Embryogenesis But Dispensable for Cardiac Development. Mol Cell Biol (2005) 25:1980–8. doi: 10.1128/MCB.25.5.1980-1988.2005
52. Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, et al. Cellular and Molecular Basis of Deiodinase-Regulated Thyroid Hormone Signaling. Endocr Rev (2008) 29:898–938. doi: 10.1210/er.2008-0019
53. Christensen MJ, Burgener KW. Dietary Selenium Stabilizes Glutathione Peroxidase mRNA in Rat Liver. J Nutr (1992) 122:1620–6. doi: 10.1093/jn/122.8.1620
54. Avissar N, Kerl EA, Baker SS, Cohen HJ. Extracellular Glutathione Peroxidase mRNA and Protein in Human Cell Lines. Arch Biochem Biophys (1994) 309:239–46. doi: 10.1006/abbi.1994.1108
55. Liang Y, Lin SL, Wang CW, Yao HD, Zhang ZW, Xu SW. Effect of Selenium on Selenoprotein Expression in the Adipose Tissue of Chickens. Biol Trace Elem Res (2014) 160:41–8. doi: 10.1007/s12011-014-0024-6
56. Seyedali A, Berry MJ. Nonsense-Mediated Decay Factors Are Involved in the Regulation of Selenoprotein mRNA Levels During Selenium Deficiency. RNA (2014) 20:1248–56. doi: 10.1261/rna.043463.113
57. Friesema EC, Jansen J, Jachtenberg JW, Visser WE, Kester MH, Visser TJ. Effective Cellular Uptake and Efflux of Thyroid Hormone by Human Monocarboxylate Transporter 10. Mol Endocrinol (2008) 22:1357–69. doi: 10.1210/me.2007-0112
58. Groeneweg S, Van Geest FS, Peeters RP, Heuer H, Visser WE. Thyroid Hormone Transporters. Endocr Rev (2020) 41:146–201. doi: 10.1210/endrev/bnz008
59. Van Geest FS, Gunhanlar N, Groeneweg S, Visser WE. Monocarboxylate Transporter 8 Deficiency: From Pathophysiological Understanding to Therapy Development. Front Endocrinol (Lausanne) (2021) 12:723750. doi: 10.3389/fendo.2021.723750
60. Bassett JH, Harvey CB, Williams GR. Mechanisms of Thyroid Hormone Receptor-Specific Nuclear and Extra Nuclear Actions. Mol Cell Endocrinol (2003) 213:1–11. doi: 10.1016/j.mce.2003.10.033
61. Flamant F, Cheng SY, Hollenberg AN, Moeller LC, Samarut J, Wondisford FE, et al. Thyroid Hormone Signaling Pathways: Time for a More Precise Nomenclature. Endocrinology (2017) 158:2052–7. doi: 10.1210/en.2017-00250
62. Zhang J, Lazar MA. The Mechanism of Action of Thyroid Hormones. Annu Rev Physiol (2000) 62:439–66. doi: 10.1146/annurev.physiol.62.1.439
63. Yen PM, Ando S, Feng X, Liu Y, Maruvada P, Xia X. Thyroid Hormone Action at the Cellular, Genomic and Target Gene Levels. Mol Cell Endocrinol (2006) 246:121–7. doi: 10.1016/j.mce.2005.11.030
64. Koenig RJ. Thyroid Hormone Receptor Coactivators and Corepressors. Thyroid (1998) 8:703–13. doi: 10.1089/thy.1998.8.703
65. Vella KR, Ramadoss P, Costa ESRH, Astapova I, Ye FD, Holtz KA, et al. Thyroid Hormone Signaling In Vivo Requires a Balance Between Coactivators and Corepressors. Mol Cell Biol (2014) 34:1564–75. doi: 10.1128/MCB.00129-14
66. Anyetei-Anum CS, Roggero VR, Allison LA. Thyroid Hormone Receptor Localization in Target Tissues. J Endocrinol (2018) 237:R19–34. doi: 10.1530/JOE-17-0708
67. Tedeschi L, Vassalle C, Iervasi G, Sabatino L. Main Factors Involved in Thyroid Hormone Action. Molecules (2021) 26:7337. doi: 10.3390/molecules26237337
68. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A Novel Syndrome Combining Thyroid and Neurological Abnormalities Is Associated With Mutations in a Monocarboxylate Transporter Gene. Am J Hum Genet (2004) 74:168–75. doi: 10.1086/380999
69. Friesema EC, Grueters A, Biebermann H, Krude H, Von Moers A, Reeser M, et al. Association Between Mutations in a Thyroid Hormone Transporter and Severe X-Linked Psychomotor Retardation. Lancet (2004) 364:1435–7. doi: 10.1016/S0140-6736(04)17226-7
70. Dumitrescu AM, Liao XH, Abdullah MS, Lado-Abeal J, Majed FA, Moeller LC, et al. Mutations in SECISBP2 Result in Abnormal Thyroid Hormone Metabolism. Nat Genet (2005) 37:1247–52. doi: 10.1038/ng1654
71. Refetoff S, Bassett JH, Beck-Peccoz P, Bernal J, Brent G, Chatterjee K, et al. Classification and Proposed Nomenclature for Inherited Defects of Thyroid Hormone Action, Cell Transport, and Metabolism. Thyroid (2014) 24:407–9. doi: 10.1089/thy.2013.3393.nomen
72. Refetoff S, Bassett JH, Beck-Peccoz P, Bernal J, Brent G, Chatterjee K, et al. Classification and Proposed Nomenclature for Inherited Defects of Thyroid Hormone Action, Cell Transport, and Metabolism. Eur Thyroid J (2014) 3:7–9. doi: 10.1159/000358180
73. Di Cosmo C, Liao XH, Dumitrescu AM, Philp NJ, Weiss RE, Refetoff S. Mice Deficient in MCT8 Reveal a Mechanism Regulating Thyroid Hormone Secretion. J Clin Invest (2010) 120:3377–88. doi: 10.1172/JCI42113
74. Beck-Peccoz P, Chatterjee VK. The Variable Clinical Phenotype in Thyroid Hormone Resistance Syndrome. Thyroid (1994) 4:225–32. doi: 10.1089/thy.1994.4.225
75. Bochukova E, Schoenmakers N, Agostini M, Schoenmakers E, Rajanayagam O, Keogh JM, et al. A Mutation in the Thyroid Hormone Receptor Alpha Gene. N Engl J Med (2012) 366:243–9. doi: 10.1056/NEJMoa1110296
76. Di Cosmo C, Mclellan N, Liao XH, Khanna KK, Weiss RE, Papp L, et al. Clinical and Molecular Characterization of a Novel Selenocysteine Insertion Sequence-Binding Protein 2 (SBP2) Gene Mutation (R128X). J Clin Endocrinol Metab (2009) 94:4003–9. doi: 10.1210/jc.2009-0686
77. Azevedo MF, Barra GB, Naves LA, Ribeiro Velasco LF, Godoy Garcia Castro P, De Castro LC, et al. Selenoprotein-Related Disease in a Young Girl Caused by Nonsense Mutations in the SBP2 Gene. J Clin Endocrinol Metab (2010) 95:4066–71. doi: 10.1210/jc.2009-2611
78. Schoenmakers E, Agostini M, Mitchell C, Schoenmakers N, Papp L, Rajanayagam O, et al. Mutations in the Selenocysteine Insertion Sequence-Binding Protein 2 Gene Lead to a Multisystem Selenoprotein Deficiency Disorder in Humans. J Clin Invest (2010) 120:4220–35. doi: 10.1172/JCI43653
79. Hamajima T, Mushimoto Y, Kobayashi H, Saito Y, Onigata K. Novel Compound Heterozygous Mutations in the SBP2 Gene: Characteristic Clinical Manifestations and the Implications of GH and Triiodothyronine in Longitudinal Bone Growth and Maturation. Eur J Endocrinol (2012) 166:757–64. doi: 10.1530/EJE-11-0812
80. Dumitrescu AM, Refetoff S. The Syndromes of Reduced Sensitivity to Thyroid Hormone. Biochim Biophys Acta (2013) 1830:3987–4003. doi: 10.1016/j.bbagen.2012.08.005
81. Catli G, Fujisawa H, Kirbiyik O, Mimoto MS, Gencpinar P, Ozdemir TR, et al. A Novel Homozygous Selenocysteine Insertion Sequence Binding Protein 2 (SECISBP2, SBP2) Gene Mutation in a Turkish Boy. Thyroid (2018) 28:1221–3. doi: 10.1089/thy.2018.0015
82. Dumitrescu AM, Korwutthikulrangsri M, Refetoff S. Impaired Sensitivity to Thyroid Hormone: Defects of Transport, Metabolism, and Action. In: Braverman LE, Cooper DS, Kopp P, editors. Werner & Ingbar’s The Thyroid: A Fundamental and Clinical Text, 11th ed. Lippincott Williams & Wilkins (2020).
83. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, Cellular and Molecular Biology, and Physiological Roles of the Iodothyronine Selenodeiodinases. Endocr Rev (2002) 23:38–89. doi: 10.1210/edrv.23.1.0455
84. Ng L, Goodyear RJ, Woods CA, Schneider MJ, Diamond E, Richardson GP, et al. Hearing Loss and Retarded Cochlear Development in Mice Lacking Type 2 Iodothyronine Deiodinase. Proc Natl Acad Sci USA (2004) 101:3474. doi: 10.1073/pnas.0307402101
85. Hernandez A, Martinez ME, Fiering S, Galton VA, St Germain D. Type 3 Deiodinase Is Critical for the Maturation and Function of the Thyroid Axis. J Clin Invest (2006) 116:476–84. doi: 10.1172/JCI26240
86. Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, et al. Reactive Oxygen Species Enhance Insulin Sensitivity. Cell Metab (2009) 10:260–72. doi: 10.1016/j.cmet.2009.08.009
87. Wang XD, Vatamaniuk MZ, Wang SK, Roneker CA, Simmons RA, Lei XG. Molecular Mechanisms for Hyperinsulinaemia Induced by Overproduction of Selenium-Dependent Glutathione Peroxidase-1 in Mice. Diabetologia (2008) 51:1515–24. doi: 10.1007/s00125-008-1055-3
88. Schoenmakers E, Carlson B, Agostini M, Moran C, Rajanayagam O, Bochukova E, et al. Mutation in Human Selenocysteine Transfer RNA Selectively Disrupts Selenoprotein Synthesis. J Clin Invest (2016) 126:992–6. doi: 10.1172/JCI84747
89. Park E, Jung J, Araki O, Tsunekawa K, Park SY, Kim J, et al. Concurrent TSHR Mutations and DIO2 T92A Polymorphism Result in Abnormal Thyroid Hormone Metabolism. Sci Rep (2018) 8:10090. doi: 10.1038/s41598-018-28480-0
90. Franca MM, German A, Fernandes GW, Liao XH, Bianco AC, Refetoff S, et al. Human Type 1 Iodothyronine Deiodinase (DIO1) Mutations Cause Abnormal Thyroid Hormone Metabolism. Thyroid (2021) 31:202–7. doi: 10.1089/thy.2020.0253
91. Carlson BA, Yoo MH, Tsuji PA, Gladyshev VN, Hatfield DL. Mouse Models Targeting Selenocysteine tRNA Expression for Elucidating the Role of Selenoproteins in Health and Development. Molecules (2009) 14:3509–27. doi: 10.3390/molecules14093509
92. Shetty SP, Copeland PR. Selenocysteine Incorporation: A Trump Card in the Game of mRNA Decay. Biochimie (2015) 114:97–101. doi: 10.1016/j.biochi.2015.01.007
93. Canani LH, Capp C, Dora JM, Meyer EL, Wagner MS, Harney JW, et al. The Type 2 Deiodinase a/G (Thr92Ala) Polymorphism Is Associated With Decreased Enzyme Velocity and Increased Insulin Resistance in Patients With Type 2 Diabetes Mellitus. J Clin Endocrinol Metab (2005) 90:3472–8. doi: 10.1210/jc.2004-1977
94. Narumi S, Muroya K, Abe Y, Yasui M, Asakura Y, Adachi M, et al. TSHR Mutations as a Cause of Congenital Hypothyroidism in Japan: A Population-Based Genetic Epidemiology Study. J Clin Endocrinol Metab (2009) 94:1317–23. doi: 10.1210/jc.2008-1767
95. Ishii H, Inada M, Tanaka K, Mashio Y, Naito K, Nishikawa M, et al. Induction of Outer and Inner Ring Monodeiodinases in Human Thyroid Gland by Thyrotropin. J Clin Endocrinol Metab (1983) 57:500–5. doi: 10.1210/jcem-57-3-500
96. Wu SY. Thyrotropin-Mediated Induction of Thyroidal Iodothyronine Monodeiodinases in the Dog. Endocrinology (1983) 112:417–24. doi: 10.1210/endo-112-2-417
97. Murakami M, Kamiya Y, Morimura T, Araki O, Imamura M, Ogiwara T, et al. Thyrotropin Receptors in Brown Adipose Tissue: Thyrotropin Stimulates Type II Iodothyronine Deiodinase and Uncoupling Protein-1 in Brown Adipocytes. Endocrinology (2001) 142:1195–201. doi: 10.1210/endo.142.3.8012
98. Morimura T, Tsunekawa K, Kasahara T, Seki K, Ogiwara T, Mori M, et al. Expression of Type 2 Iodothyronine Deiodinase in Human Osteoblast Is Stimulated by Thyrotropin. Endocrinology (2005) 146:2077–84. doi: 10.1210/en.2004-1432
99. Karavani G, Strich D, Edri S, Gillis D. Increases in Thyrotropin Within the Near-Normal Range Are Associated With Increased Triiodothyronine But Not Increased Thyroxine in the Pediatric Age Group. J Clin Endocrinol Metab (2014) 99:E1471–1475. doi: 10.1210/jc.2014-1441
100. Park SY, Park SE, Jung SW, Jin HS, Park IB, Ahn SV, et al. Free Triiodothyronine/Free Thyroxine Ratio Rather Than Thyrotropin Is More Associated With Metabolic Parameters in Healthy Euthyroid Adult Subjects. Clin Endocrinol (Oxf) (2017) 87:87–96. doi: 10.1111/cen.13345
101. Moreno-Reyes R, Mathieu F, Boelaert M, Begaux F, Suetens C, Rivera MT, et al. Selenium and Iodine Supplementation of Rural Tibetan Children Affected by Kashin-Beck Osteoarthropathy. Am J Clin Nutr (2003) 78:137–44. doi: 10.1093/ajcn/78.1.137
102. Hawkes WC, Keim NL, Diane Richter B, Gustafson MB, Gale B, Mackey BE, et al. High-Selenium Yeast Supplementation in Free-Living North American Men: No Effect on Thyroid Hormone Metabolism or Body Composition. J Trace Elem Med Biol (2008) 22:131–42. doi: 10.1016/j.jtemb.2007.11.005
103. Rayman MP, Thompson AJ, Bekaert B, Catterick J, Galassini R, Hall E, et al. Randomized Controlled Trial of the Effect of Selenium Supplementation on Thyroid Function in the Elderly in the United Kingdom. Am J Clin Nutr (2008) 87:370–8. doi: 10.1093/ajcn/87.2.370
104. Combs GF Jr., Midthune DN, Patterson KY, Canfield WK, Hill AD, Levander OA, et al. Effects of Selenomethionine Supplementation on Selenium Status and Thyroid Hormone Concentrations in Healthy Adults. Am J Clin Nutr (2009) 89:1808–14. doi: 10.3945/ajcn.2008.27356
105. Thomson CD, Campbell JM, Miller J, Skeaff SA, Livingstone V. Selenium and Iodine Supplementation: Effect on Thyroid Function of Older New Zealanders. Am J Clin Nutr (2009) 90:1038–46. doi: 10.3945/ajcn.2009.28190
106. Berger MM, Reymond MJ, Shenkin A, Rey F, Wardle C, Cayeux C, et al. Influence of Selenium Supplements on the Post-Traumatic Alterations of the Thyroid Axis: A Placebo-Controlled Trial. Intensive Care Med (2001) 27:91–100. doi: 10.1007/s001340000757
107. Angstwurm MW, Schopohl J, Gaertner R. Selenium Substitution has No Direct Effect on Thyroid Hormone Metabolism in Critically Ill Patients. Eur J Endocrinol (2004) 151:47–54. doi: 10.1530/eje.0.1510047
108. Schomburg L, Dumitrescu AM, Liao XH, Bin-Abbas B, Hoeflich J, Kohrle J, et al. Selenium Supplementation Fails to Correct the Selenoprotein Synthesis Defect in Subjects With SBP2 Gene Mutations. Thyroid (2009) 19:277–81. doi: 10.1089/thy.2008.0397
109. Burk RF, Norsworthy BK, Hill KE, Motley AK, Byrne DW. Effects of Chemical Form of Selenium on Plasma Biomarkers in a High-Dose Human Supplementation Trial. Cancer Epidemiol Biomarkers Prev (2006) 15:804–10. doi: 10.1158/1055-9965.EPI-05-0950
110. Xu XM, Carlson BA, Mix H, Zhang Y, Saira K, Glass RS, et al. Biosynthesis of Selenocysteine on Its tRNA in Eukaryotes. PloS Biol (2007) 5:e4. doi: 10.1371/journal.pbio.0050004
111. Weetman AP. Autoimmune Thyroid Disease. Autoimmunity (2004) 37:337–40. doi: 10.1080/08916930410001705394
112. Beckett GJ, Peterson FE, Choudhury K, Rae PW, Nicol F, Wu PS, et al. Inter-Relationships Between Selenium and Thyroid Hormone Metabolism in the Rat and Man. J Trace Elem Electrolytes Health Dis (1991) 5:265–7.
113. Kucharzewski M, Braziewicz J, Majewska U, Gozdz S. Concentration of Selenium in the Whole Blood and the Thyroid Tissue of Patients With Various Thyroid Diseases. Biol Trace Elem Res (2002) 88:25–30. doi: 10.1385/BTER:88:1:25
114. Gartner R, Gasnier BC, Dietrich JW, Krebs B. Selenium Supplementation in Patients With Autoimmune Thyroiditis Decreases Thyroid Peroxidase Antibodies Concentrations. J Clin Endocrinol Metab (2002) 87:1687–91. doi: 10.1210/jcem.87.4.8421
115. Duntas LH, Mantzou E, Koutras DA. Effects of a Six Month Treatment With Selenomethionine in Patients With Autoimmune Thyroiditis. Eur J Endocrinol (2003) 148:389–93. doi: 10.1530/eje.0.1480389
116. Turker O, Kumanlioglu K, Karapolat I, Dogan I. Selenium Treatment in Autoimmune Thyroiditis: 9-Month Follow-Up With Variable Doses. J Endocrinol (2006) 190:151–6. doi: 10.1677/joe.1.06661
117. Karanikas G, Schuetz M, Kontur S, Duan H, Kommata S, Schoen R, et al. No Immunological Benefit of Selenium in Consecutive Patients With Autoimmune Thyroiditis. Thyroid (2008) 18:7–12. doi: 10.1089/thy.2007.0127
118. Toulis KA, Anastasilakis AD, Tzellos TG, Goulis DG, Kouvelas D. Selenium Supplementation in the Treatment of Hashimoto’s Thyroiditis: A Systematic Review and a Meta-Analysis. Thyroid (2010) 20:1163–73. doi: 10.1089/thy.2009.0351
120. Bartalena L, Baldeschi L, Dickinson AJ, Eckstein A, Kendall-Taylor P, Marcocci C, et al. Consensus Statement of the European Group on Graves’ Orbitopathy (EUGOGO) on Management of Graves’ Orbitopathy. Thyroid (2008) 18:333–46. doi: 10.1089/thy.2007.0315
121. Marcocci C, Kahaly GJ, Krassas GE, Bartalena L, Prummel M, Stahl M, et al. Selenium and the Course of Mild Graves’ Orbitopathy. N Engl J Med (2011) 364:1920–31. doi: 10.1056/NEJMoa1012985
122. Kim BY, Jang SY, Choi DH, Jung CH, Mok JO, Kim CH. Anti-Inflammatory and Antioxidant Effects of Selenium on Orbital Fibroblasts of Patients With Graves Ophthalmopathy. Ophthalmic Plast Reconstr Surg (2021) 37:476–81. doi: 10.1097/IOP.0000000000001931
123. Saravanan P, Chau WF, Roberts N, Vedhara K, Greenwood R, Dayan CM. Psychological Well-Being in Patients on ‘Adequate’ Doses of L-Thyroxine: Results of a Large, Controlled Community-Based Questionnaire Study. Clin Endocrinol (Oxf) (2002) 57:577–85. doi: 10.1046/j.1365-2265.2002.01654.x
124. Bunevicius R, Kazanavicius G, Zalinkevicius R, Prange AJ Jr. Effects of Thyroxine as Compared With Thyroxine Plus Triiodothyronine in Patients With Hypothyroidism. N Engl J Med (1999) 340:424–9. doi: 10.1056/NEJM199902113400603
125. Appelhof BC, Fliers E, Wekking EM, Schene AH, Huyser J, Tijssen JG, et al. Combined Therapy With Levothyroxine and Liothyronine in Two Ratios, Compared With Levothyroxine Monotherapy in Primary Hypothyroidism: A Double-Blind, Randomized, Controlled Clinical Trial. J Clin Endocrinol Metab (2005) 90:2666–74. doi: 10.1210/jc.2004-2111
126. Escobar-Morreale HF, Botella-Carretero JI, Gomez-Bueno M, Galan JM, Barrios V, Sancho J. Thyroid Hormone Replacement Therapy in Primary Hypothyroidism: A Randomized Trial Comparing L-Thyroxine Plus Liothyronine With L-Thyroxine Alone. Ann Intern Med (2005) 142:412–24. doi: 10.7326/0003-4819-142-6-200503150-00007
127. Nygaard B, Jensen EW, Kvetny J, Jarlov A, Faber J. Effect of Combination Therapy With Thyroxine (T4) and 3,5,3’-Triiodothyronine Versus T4 Monotherapy in Patients With Hypothyroidism, A Double-Blind, Randomised Cross-Over Study. Eur J Endocrinol (2009) 161:895–902. doi: 10.1530/EJE-09-0542
128. Walsh JP, Shiels L, Lim EM, Bhagat CI, Ward LC, Stuckey BG, et al. Combined Thyroxine/Liothyronine Treatment Does Not Improve Well-Being, Quality of Life, or Cognitive Function Compared to Thyroxine Alone: A Randomized Controlled Trial in Patients With Primary Hypothyroidism. J Clin Endocrinol Metab (2003) 88:4543–50. doi: 10.1210/jc.2003-030249
129. Grozinsky-Glasberg S, Fraser A, Nahshoni E, Weizman A, Leibovici L. Thyroxine-Triiodothyronine Combination Therapy Versus Thyroxine Monotherapy for Clinical Hypothyroidism: Meta-Analysis of Randomized Controlled Trials. J Clin Endocrinol Metab (2006) 91:2592–9. doi: 10.1210/jc.2006-0448
130. Wiersinga WM. Paradigm Shifts in Thyroid Hormone Replacement Therapies for Hypothyroidism. Nat Rev Endocrinol (2014) 10:164–74. doi: 10.1038/nrendo.2013.258
131. Mcaninch EA, Bianco AC. New Insights Into the Variable Effectiveness of Levothyroxine Monotherapy for Hypothyroidism. Lancet Diabetes Endocrinol (2015) 3:756–8. doi: 10.1016/S2213-8587(15)00325-3
132. Panicker V, Saravanan P, Vaidya B, Evans J, Hattersley AT, Frayling TM, et al. Common Variation in the DIO2 Gene Predicts Baseline Psychological Well-Being and Response to Combination Thyroxine Plus Triiodothyronine Therapy in Hypothyroid Patients. J Clin Endocrinol Metab (2009) 94:1623–9. doi: 10.1210/jc.2008-1301
133. Wouters HJ, Van Loon HC, van der Klauw MM, Elderson MF, Slagter SN, Kobold AM, et al. No Effect of the Thr92Ala Polymorphism of Deiodinase-2 on Thyroid Hormone Parameters, Health-Related Quality of Life, and Cognitive Functioning in a Large Population-Based Cohort Study. Thyroid (2017) 27:147–55. doi: 10.1089/thy.2016.0199
134. Jonklaas J, Bianco AC, Bauer AJ, Burman KD, Cappola AR, Celi FS, et al. Guidelines for the Treatment of Hypothyroidism: Prepared by the American Thyroid Association Task Force on Thyroid Hormone Replacement. Thyroid (2014) 24:1670–751. doi: 10.1089/thy.2014.0028
Keywords: thyroid, thyroid hormone, selenium, selenoprotein, deiodinase, genetics
Citation: Lee KW, Shin Y, Lee S and Lee S (2022) Inherited Disorders of Thyroid Hormone Metabolism Defect Caused by the Dysregulation of Selenoprotein Expression. Front. Endocrinol. 12:803024. doi: 10.3389/fendo.2021.803024
Received: 27 October 2021; Accepted: 20 December 2021;
Published: 18 January 2022.
Edited by:
Noriyuki Koibuchi, Gunma University, JapanReviewed by:
Lutz Schomburg, Charité University Medicine Berlin, GermanyCopyright © 2022 Lee, Shin, Lee and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sihoon Lee, c2hsZWVtZEBnYWNob24uYWMua3I=, orcid.org/0000-0002-9444-5849
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.