 Yongxiang Li
Yongxiang Li Qingyan Jiang
Qingyan Jiang Lina Wang
Lina Wang- 1Guangdong Provincial Key Laboratory of Animal Nutrition Control, South China Agricultural University, Guangzhou, China
- 2National Engineering Research Center for Breeding Swine Industry, College of Animal Science, South China Agricultural University, Guangzhou, China
- 3Children’s Nutrition Research Center, Department of Pediatrics, Baylor College of Medicine, Houston, TX, United States
Appetite is the basis for obtaining food and maintaining normal metabolism. Toll-like receptor 4 (TLR4) is an important receptor expressed in the brain that induces inflammatory signaling after activation. Inflammation is considered to affect the homeostatic and non-homeostatic systems of appetite, which are dominated by hypothalamic and mesolimbic dopamine signaling. Although the pathological features of many types of inflammation are known, their physiological functions in appetite are largely unknown. This review mainly addresses several key issues, including the structures of the homeostatic and non-homeostatic systems. In addition, the mechanism by which TLR4-induced inflammatory signaling contributes to these two systems to regulate appetite is also discussed. This review will provide potential opportunities to develop new therapeutic interventions that control appetite under inflammatory conditions.
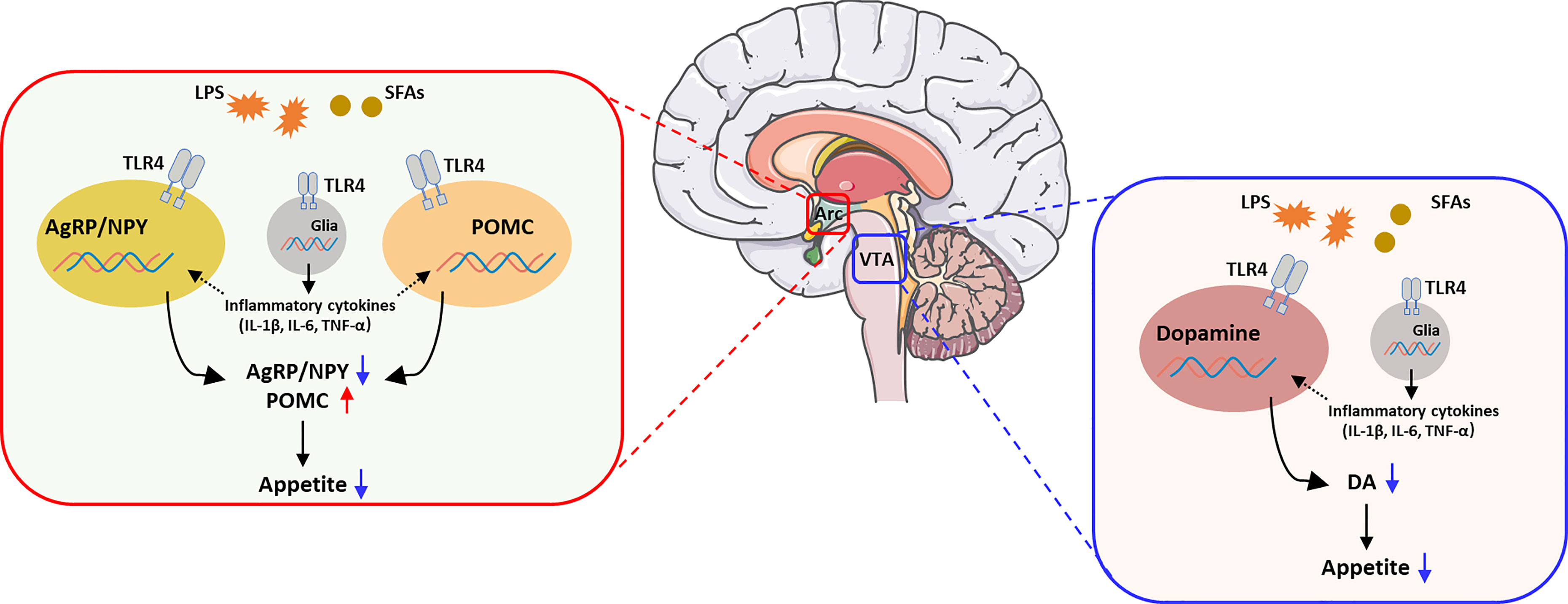
Graphical Abstract The activation of TLR4 in the regulation of appetite. Brain image was provided by smart.servier.com.
Introduction
Appetite plays an essential role in the regulation of energy balance. Metabolic requirements must be met by obtaining and consuming food. In this process, the brain plays a vital role in regulating energy metabolism. At the most basic level, the regulation of appetite by the central nervous system (CNS) is divided into two types: homeostasis and non-homeostasis. The homeostatic system controls feeding to resolve general energy deficiencies or meet other types of metabolic needs. However, the non-homeostatic system, also called the reward system, is typically driven by palatable foods with high fat or sugar contents. Food intake results from various physiological and behavioral processes that control hunger, satiety, and reward systems. Recently, the regulation of feeding by inflammatory signals such as TLR4 has been reported frequently. TLR4 is a transmembrane pattern recognition receptor that recognizes molecular patterns related to injury and pathogens (1, 2). TLR4 is expressed on cells of the innate immune system in the CNS, such as endothelial cells, microglia, and some astrocytes, as well as adult neurons (3, 4). Pattern recognition receptors have been extensively developed to recognize a wide variety of pathogen-related molecular patterns (PAMPs) associated with microbial pathogens or cell signals of danger or stress. Ligands bind to TLR4 and its accessory molecules, such as myeloid differentiation protein 2 (MD2) and cluster of differentiation 14 (CD14), to activate downstream intracellular signaling pathways, thus producing and releasing proinflammatory and neuroexcitatory mediators via MyD88-dependent or MyD88-independent intracellular pathways (5). Therefore, TLR4-induced inflammatory signaling provides a mechanistic link between the hypothalamus, mesolimbic dopamine (DA) system and appetite (6).
Here, we will focus on current insights into the regulation of appetite by TLR4-induced inflammatory signaling in the hypothalamus and mesolimbic DA system. In addition, we also explored recent publications to investigate the activation of TLR4-induced inflammatory signaling and its effect on appetite.
TLR4-Induced Inflammatory Signaling in the Hypothalamus Affects the Appetite Process
Energy Homeostasis and Inflammatory Signaling
Appropriate energy homeostasis results from a delicate balance between energy intake and expenditure. Substantial evidence indicates that arcuate nucleus (Arc) has emerged as a key regulator of energy homeostasis (7). The key to this process is two sets of interconnected Arc neurons (Figure 1). As described below, orexigenic neuropeptide Y (NPY)/agouti gene-related protein (AgRP) neurons and anorexigenic proopiomelanocortin (POMC) neurons play opposite but coordinated roles in controlling food intake. In addition, neurons that are distributed in the paraventricular hypothalamic nucleus (PVH), lateral hypothalamus (LH) and ventromedial hypothalamus (VMH) are also involved in regulating energy homeostasis (8–10). In most cases, our bodies are constantly adapting to changes in the external environment that are essential for survival. Normally, the existence of the immune system always maintains the normal function of organs. However, when the immune system is activated by pathogens or other stimuli, typical symptoms such as fever, anorexia and pain occur. Two types of proinflammatory responses to immune system activation have been identified: (1) acute inflammation, such as a bacterial infection that lasts for a few minutes to several hours, and (2) chronic inflammation, such as obesity induced by a high-fat diet (HFD), cancer, and chronic obstructive pulmonary disease (COPD), which ranges from days to years. Although peripheral inflammation plays a key role in regulating energy metabolism and appetite (11), central production of inflammatory mediators is more important than peripheral production in regulating appetite and body weight. Recent discoveries have indicated that the brain contains lymphatic vessels and thus forms a lymphatic drainage system (12). The discovery that lymphocytes are useful to examine diseases related to the CNS and the immune response in the break further overcomes the misconception of the brain as an immune-isolated organ (13).
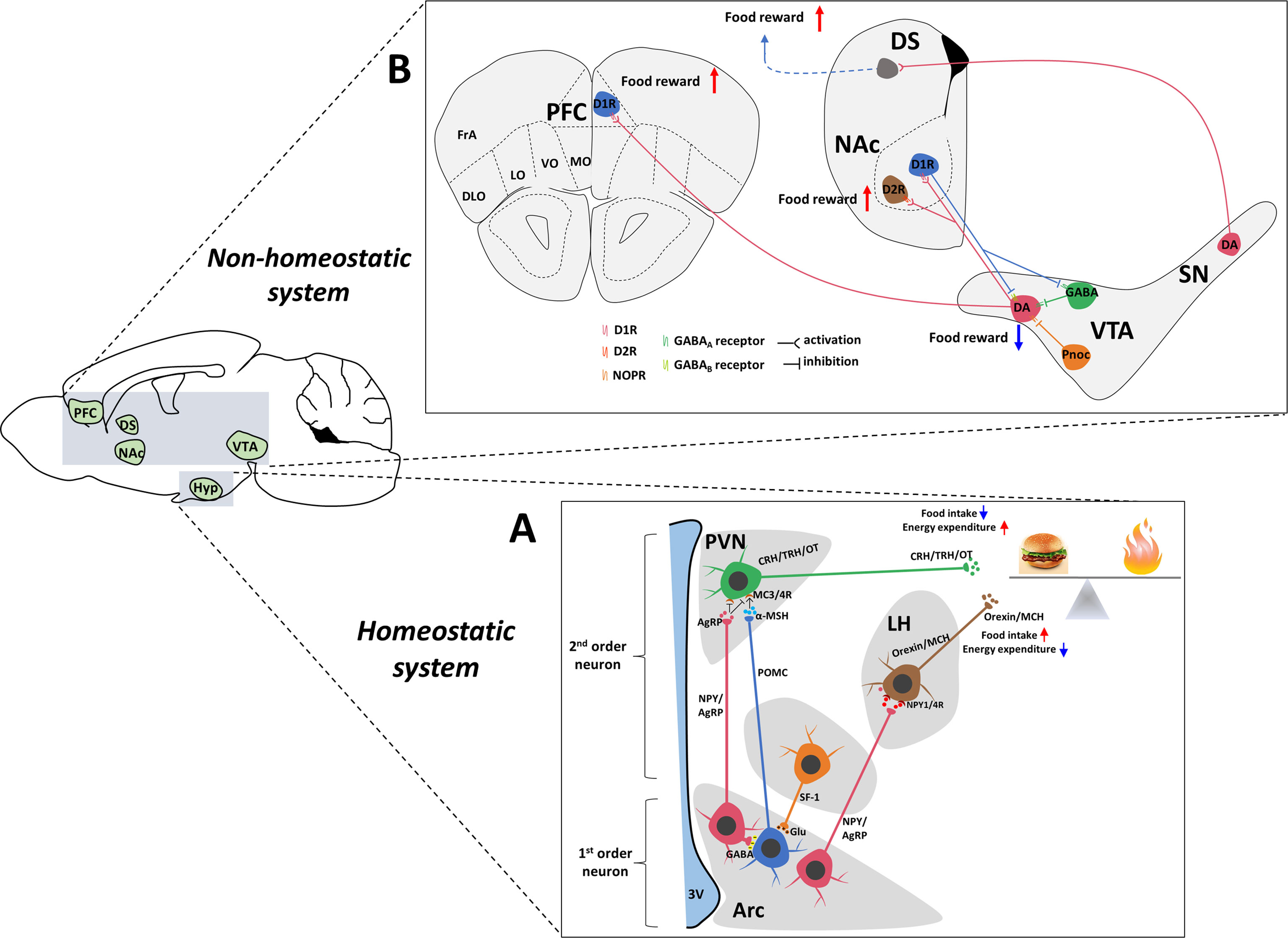
Figure 1 Central neuronal circuits involved in homeostatic and non-homeostatic systems. (A) Hypothalamic regulation of energy homeostasis. The Arc of the hypothalamus contains two groups of neurons: NPY/AgRP and POMC neurons. These neurons secrete orexigenic AgRP/NPY or anorexigenic POMC to the second-order neurons in the PVH and LH. In the PVN, neurons produce OT, TRH and CRH to decrease food intake. Neurons in the LH produce ORX and MCH to increase food intake. AgRP is an antagonist of anorectic MC4R expressed on neurons within the PVN. In the Arc, a local circuit exists in which POMC neurons receive GABAergic (inhibitory) input from NPY/AgRP neurons. The SF-1 nerve located in the VMH innervates POMC/CART neurons to enhance the anorexia function of POMC/CART neurons and is critically involved in the regulation of energy homeostasis. (B) The dopamine system, which contains connections between the midbrain and forebrain. The reward pathway includes dopaminergic neurons in the ventral tegmental area that project to the NAc and PFC. Dopamine, which is released from dopaminergic neurons in the VTA, binds to D1R or D2R in the NAc and D1R in the PFC, which contribute to food reward. In turn, D1R also inhibits DA and GABAergic neurons in the VTA through different GABA receptors to form negative feedback regulation. The VTA also contains GABAergic cells and Pnoc neurons that project to DA neurons to inhibit DA activity. In addition, DA neurons located in the SN also project to the DS, which potentially increases food reward behavior. Hyp, hypothalamus; Arc, arcuate nucleus; LHA, lateral hypothalamus area; PVN, paraventricular nucleus of the hypothalamus; VMH, ventromedial hypothalamus; 3V, 3rd ventricle; VTA, ventral tegmental area; NAc, nucleus accumbens; PFC, prefrontal cortex; DS, dorsal striatum; SN, substantia nigra; AgRP, agouti-related peptide; NPY, neuropeptide Y; POMC, proopiomelanocortin; Glu, glutamate; GABA, gamma-aminobutyric acid; MCH, melanin-concentrating hormone; α-MSH, α-melanocyte-stimulating hormone; CRH, corticotropin-releasing hormone; TRH, thyrotropin-releasing hormone; OT, oxytocin; DA, dopamine; D1R, dopamine 1 receptor; NOPR, nociceptin opioid peptide receptor.
Although we understand how to achieve the balance between pathology and physiology, the effect of inflammatory processes on homeostasis remains inadequate. Energy homeostasis and the immune system have many coordinated responses. Disease syndrome is a coordinated response by the brain to help the immune system combat infection, which is accompanied by typical symptoms such as fever, anorexia and pain. For example, fever is an increase in the core body temperature that enhances immune cell function to eliminate pathogens. In addition, sleepiness and achiness decrease energy expenditure by decreasing locomotor activity. Last, the reduction in food intake decreases glucose supply available to the infectious agent (14). Over the past few decades, research has highlighted the key role of proinflammatory cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) in controlling energy homeostasis in anorexic-cachexia syndrome. Most studies support the hypothesis that the hypothalamus plays a key role in reducing food intake and the development of illness with inflammation.
How Does the Inflammatory Signaling Decrease Appetite to Maintain the Energy Balance?
Chronic diseases, such as cancer, disrupt these very primitive and coordinated responses. In cancer-induced anorexia, central sensors of energy homeostasis are rapidly associated with increased energy expenditure and decreased food intake. The aim of this process is to reduce the glucose supply to cancer cells. However, rapidly proliferating cancer cells have lost their oxidative glycolysis capabilities, and thus they can develop a chronic malnutrition state (15). Notably, most of these causes are usually associated with “high-grade” inflammation. Even if the pathological states are benign and short term, they are also highly inflammatory and associated with decreased appetite or “anorexia”. Anorexia is a result of the classic defense of an organism against infections, also known as “sickness behavior”. (16, 17). The production of proinflammatory cytokines is induced by infectious factors, and higher TNF-α, IL-1β and IL-6 levels affect the surrounding organs to induce disease behavior and inflammation-related anorexia, which participate in the induction of disease behavior (18). Hypothalamus cannot regulate energy balance under increasing energy expenditure and chronic inflammation (19). Neuroinflammation and hypothalamic signal transduction were affected by chronic inflammation induced by cancer (19), HIV (20), COPD (21), and heart failure (22). This inflammatory response in the hypothalamus is elicited by elevated plasma cytokine levels entering the brain, as several cytokines are able to cross the blood–brain barrier (BBB), including TNF-α (23), IL-6 (24), IL-1α (25) and IL-1β (26). In addition, many receptors for these proinflammatory cytokines are expressed in the hypothalamus (27). Hypothalamic neurons in the Arc sense peripheral circulating factors, including cytokines, from the adjacent median eminence (ME), without protection of the blood–brain barrier.
Indeed, a common phenomenon appears to be that hypothalamic microglia, astrocytes and macrophage-like cells are activated in the hypothalamus in response to chronic inflammatory diseases (28, 29). Chronic administration of cytokines reproduces the characteristics of anorexia syndrome (30–32), while its development is inhibited by blocking the signaling of one of these cytokines, such as TNF-α, using neutralizing antibodies (33, 34). In animal models of inflammatory anorexia, interfering with inflammatory mediators reduces hypothalamic inflammation and prevents weight loss. For example, inhibiting the action of adenosine monophosphate protein kinase (AMPK) in the hypothalamus reduces hypothalamic inflammation in patients with cancer-related anorexia, which leads to increased food intake to improve overall survival (35). Brain-derived neurotrophic factor (BDNF) reduces hypothalamic inflammation and increases sympathetic activation, thereby inhibiting cancer growth (36, 37). In addition, administration of an IL-1β receptor antagonist blocks the effects of a peripheral injection of cytokines (38, 39) and prevents anorexia in animal models of cancer (40).
Many studies have shown that intraventricular (ICV) IL-1β injections cause profound behavioral changes in rodents (19). The injection of central or peripheral cytokines indicates that proinflammatory mediators exert their effect primarily at the central level rather than the peripheral level (41, 42). Therefore, the brain is a more complex and advanced organ, and the action of cytokine signaling overrides the effect of peripheral signaling to maintain normal function. Some studies have shown that endogenous IL-1β expressed in the brain mediates lipopolysaccharide (LPS)-induced anorexia by modulating the expression of cytokines in the hypothalamus (43). Multiple circulating cytokines and inflammatory mediators trigger CNS immune signaling after LPS injection in a short period instead of sustained hypothalamic inflammation or sickness behavior. More importantly, these animals only experienced a very short period of mild anorexia after LPS administration, and food intake and body weight recovered to normal levels after 6 hours (44, 45). LPS-induced anorexia depends on its central inflammatory mechanisms, and the central role of peripheral LPS may be mediated by certain cytokines and/or receptors expressed in the brain (45, 46). Therefore, these studies support the hypothesis that the hypothalamus senses metabolic and inflammatory signals to fully regulate energy homeostasis and systemic inflammation.
Inflammatory Signaling Affects Appetite-Related Peptides in the Homeostasis System
In LPS- or TNF-α-induced anorexia experimental models of acute inflammation, NPY expression in the hypothalamus is reduced, consistent with the observed decrease in food intake (47–49). Furthermore, the administration of NPY or blocking the induced inflammatory response prevents the development of anorexia (47, 50). Thus, NPY release is necessary to change food intake in a manner dependent on energy expenditure. However, NPY mRNA expression in the hypothalamus increases in animal models of chronic inflammatory diseases characterized by cachexia, such as cancer cachexia and arthritis (51–55). However, this increase in NPY mRNA levels did not correlate with a decrease in food intake (52, 54, 56, 57). It was associated with weight loss. In addition, NPY levels and NPY release are either unchanged (52) or decreased in these animal models (58–60). In these cases, NPY signaling is likely regulated at the posttranscriptional level. Therefore, we propose that NPY mRNA expression might represent a sensor for weight loss and the appropriate translation and release of NPY are subsequently required to determine changes in food intake and energy expenditure. The AgRP expression pattern is similar to NPY, as its mRNA expression increases (61) but secretion decreases in both acute and chronic inflammatory anorexia models. In addition, the changes in AgRP levels are associated with weight loss. In addition, genetic models of anorexia suggest that the disruption of NPY/AgRP signaling is partially due to axonal transport dysfunction (62). Altogether, these data indicate that inflammatory mediators affect NPY/AgRP mRNA expression in a posttranscriptional manner, including altered NPY translation, synthesis, packaging, or release, ultimately changing food intake.
Acute inflammation activates POMC neurons, which increase melanocortin-4 receptor (MC4R) expression (63) and POMC expression (64, 65), as shown in the LPS- and IL-1β-induced anorexia model. In addition, IL-1β binds to IL-1β receptor-expressing POMC neurons in the Arc of the hypothalamus to promote the effect of inflammation downstream (62). In fact, researchers have shown that the NF-κB pathway plays an important role in disease-induced anorexia and weight loss using different methods. Both the administration of AgRP and inhibition of the NF-κB pathway, especially in POMC neurons, significantly attenuates the effects of LPS on food intake and body weight. Therefore, POMC is a potential mediator of illness-induced anorexia and a downstream target of NF-κB. Interestingly, some researchers suggest that leptin-induced anorexia may also partially depend on the NF-κB pathway (64).
However, there are no obvious evidence that indicated the contribution of TLR4 in neurons or non-neuronal in the hypothalamus. On one hand, the activation of TLR4 signal in AgRP/NPY and POMC neurons decreased the expression of AgRP/NPY and increased the expression of POMC, which induced the reduction of appetite and suggested to be involved in obesity. On the other hand, the activation of TLR4 in non-neuronal cells, like microglia or astrocyte will increased the mRNA levels of specific inflammatory genes, which repression of TNF-α expression in the hypothalamic neurons (66). This evidence shown TLR4 in non-neuronal cells may affect the function of neurons in hypothalamus and potentially make a contribution of appetite and obesity.
Appetite in a Non-Homeostatic State is Regulated by Inflammatory Signaling
Increasing Inflammation Decreases Appetite
Consuming palatable foods is known to activate the reward system in the brain, which plays a critical role in appetite (67). The midbrain DA system has been suggested to play an important role in the regulation of reward-related behaviors (68, 69). DA neurons are located in the ventral tegmental area (VTA) and mainly project onto the nucleus accumbens (NAc) in the ventral striatum (70–73) and the prefrontal cortex (PFC) (73–76), which are associated with appetite. Although many factors influence appetite, inflammation is one of factors that should be considered (77, 78). Inflammation-induced sickness and behavioral changes are examples of inflammation altering motivational states (79). The host responds to the infection detected by innate immune cells and exhibits drastic behavioral changes, which facilitate the development of fever and fight against pathogens to maintain energy balance. Meanwhile, the results of many laboratory studies consistently show that innate immune system activation and release of inflammatory cytokines preferentially affect the reward circuit and basal ganglia DA level, leading to decreased appetite (80–82). Given the importance of DA in the reward system, a few studies have examined the relationship between inflammatory markers and symptoms/signs, which have shown an association between increased inflammation and decreased appetite (83–85).
These findings indicated that motivational symptoms like anergia and fatigue. Clinical data support the hypothesis that the effect of inflammation on appetite is driven by the action of cytokines on the DA system, as described below. In humans, much of the evidence is derived from the acute administration of inflammatory stimuli (such as endotoxin or typhoid vaccination) to healthy volunteers and the chronic administration of inflammatory cytokines [such as interferon alpha (IFN-α)] to patients as a treatment for certain cancers and infectious diseases (86, 87). These drugs induce the release of the inflammatory cytokines IL-6, IL-1β and TNF-α (88–90). IFN-α-treated patients have a reduced motivation to obtain food (86, 87). Neuroimaging studies have shown that the administration of inflammatory cytokines or cytokine attractants alters the activation of brain regions associated with appetite, including a decreased response to pleasurable food (82, 87, 91).
Initial neurochemical and behavioral studies reported that inflammation affects brain DA levels following acute or chronic injection of IFN-α in rodents. Based on these results, chronic administration of IFN-α decreases DA release in the striatum, which correlates with reduced appetite. In addition, long-term use of IFN-α reduces the reward associated with food but does not change sucrose consumption in monkeys. In vivo microdialysis was conducted to assess the effects of cytokines on the synaptic availability and release of striatal DA and to reveal the concrete effect of inflammation on appetite (92). Similar to the effect of IFN-α, peripheral administration of IL-1β to rodents also reduced appetite (85). Interestingly, endotoxin administration to rats reduces appetite but increases the motivation to obtain rest in a running wheel. Thus, the body will reduce energy consumption in order to survive in a pathological state.
A single intraperitoneal injection of LPS was administered to induce peripheral inflammation. Twenty-four hours after LPS administration, mice showed reduced appetite. Meanwhile, acute low-dose LPS (100 g/kg) systemic administration reduced the DA content in the NAc and increased extracellular DA metabolite levels (93, 94). As a result, LPS reduces incentive motivation for food rewards (95). The acute and chronic effects of LPS on brain DA levels are blocked by inhibiting or deleting genes encoding inflammatory cytokines such as TNF-α (96, 97). Inflammation-related medical disease models, such as experimental tumors, are associated with reduced brain DA levels (98, 99). Together, these results from animal studies suggest that various inflammatory stimuli consistently affect DA levels in the brain, leading to related changes in appetite. The DA precursor levodopa (L-DOPA) completely reverses IFN-α-induced reductions in striatal DA release observed using in vivo microanalysis, potentially indicating that cytokines may reduce DA synthesis (100). The effects of inflammation on DA and neural activation and metabolism in the reward circuit were examined, and IFN-α-treated subjects were administered [18F] fluorodopa (FDO-PA) (87). L-DOPA is absorbed by DA neurons and converted to DA by DA decarboxylase, which is then stored in vesicles and released. Interestingly, patients exhibited increased absorption and reduced turnover of L-DOPA in the ventral striatum after IFN-α treatment.
Our previous study has reported that TLR4 in dopamine neurons affect the food reward (food motivation) and food preference (HFD consumption) at the same time. In this process, deletion of TLR4 in dopamine neurons decreased the dopamine level in the brain, which contributes to the reduction of HFD preference. However, global TLR4 KO mice decreased expression of the key taste molecules cluster of differentiation 36 (CD36), phospholipase Cβ2 (PLCβ2) and transient receptor potential cation channel, superfamily M, member 5 (TRPM5) in tongue epithelium, which induced the change of food preferences. Therefore, TLR4 signaling in epithelium still can affect the food preference by decreasing the nutrient sensing (101).
The Mechanism by Which Inflammation Regulates Appetite
In summary, evidence from humans and animals suggests that inflammation is associated with the reduced DA availability and release, which exert functional effects on reward circuits. These changes are associated with fundamental changes in motivation and motor function. The mechanisms underlying the effects of cytokines on DA synthesis, release, reuptake, or receptor signaling should be considered to determine the changes in the DA system in the inflammatory process that correlate with reward and motivation. Inflammation and cytokines affect DA-related functions through multiple pathways. DA synthesis, packaging and release, reuptake and DA receptors may interact to reduce the DA signal in the basal ganglia. Therefore, the next sections mainly discuss the potential mechanisms by which inflammation affects DA neurotransmission (Figure 2).
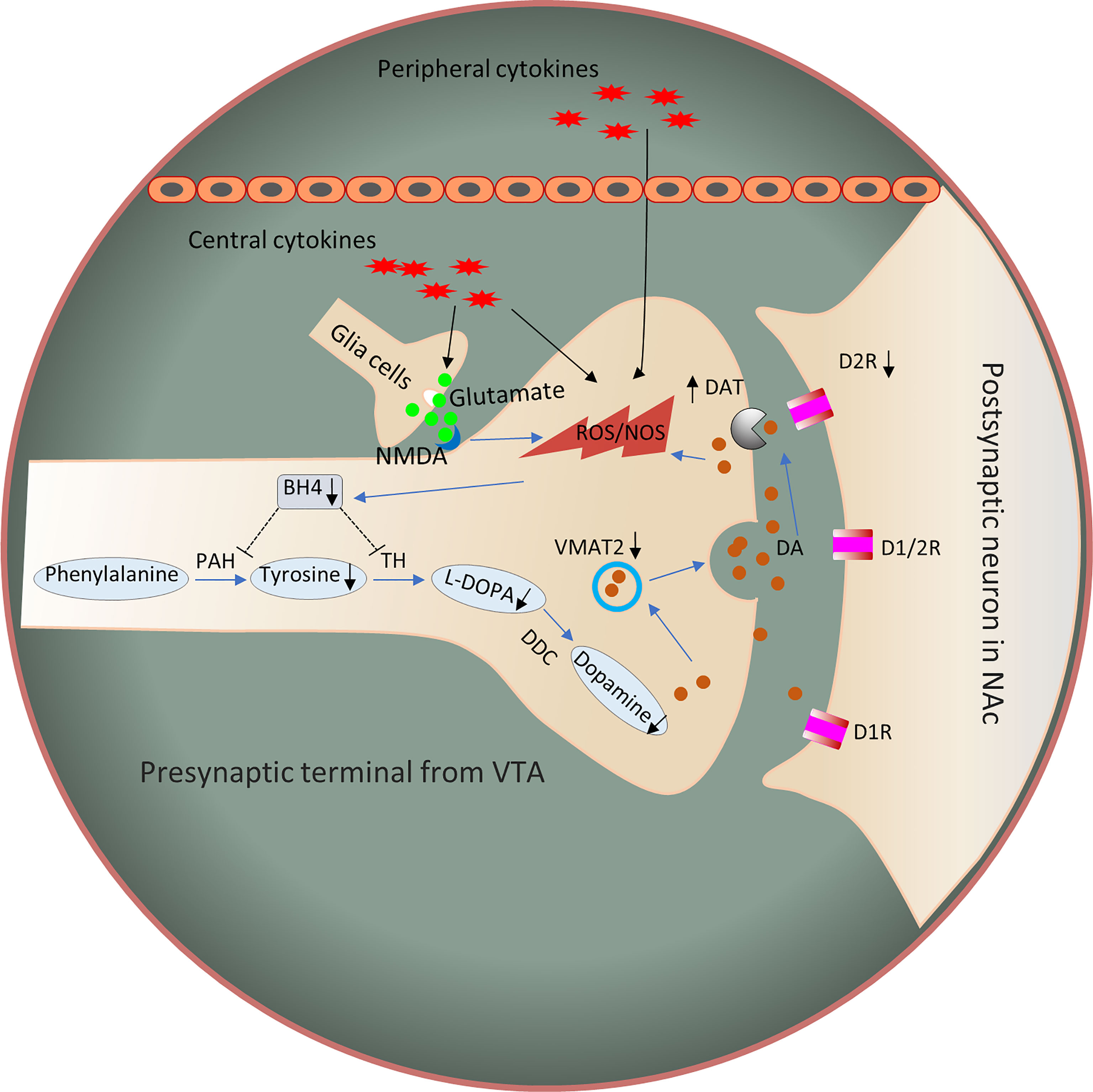
Figure 2 Potential mechanisms by which inflammation affects DA signaling through synthesis, release, and receptor function. Inflammation and cytokines released from the periphery or produced locally by activated microglia or infiltrating macrophages contribute to oxidative stress and the production of reactive oxygen species (ROS). Increased ROS and inflammation-induced nitric oxide levels contribute to the oxidation of BH4, an essential cofactor required for the conversion of phenylalanine to tyrosine and tyrosine to L-PODA, which are necessary for the synthesis of DA. In addition, the increased glutamate release and reduced reuptake by inflammation-induced glial cells, coupled with the activation of NMDARs, may cause excitotoxicity of glutamate. In turn, these changes lead to oxidative stress and decreased DA availability. Furthermore, some evidence has shown that inflammatory cytokines reduce the expression or function of VMAT2 and/or increase the expression or function of DAT and reduce DA signaling by reducing the levels of DA D2 receptors. Dysregulation of DAT and VMAT2 increases cytosolic DA levels, leading to auto-oxidation and ROS generation. D1R, dopamine 1 receptor; D2R, dopamine 2 receptor; DDC, dopamine decarboxylase; NMDAR, N-methyl-D-aspartic acid receptor; NOS, nitric oxide synthase; ROS, reactive oxygen species; PAH, phenylalanine hydroxylase; TH, tyrosine hydroxylase; BH4, 5,6,7,8-tetrahydrobiopterin; VMAT2, vesicular monoamine transporter 2; LPODC, L-3,4-dihydroxyphenylalanine.
DA Synthesis and Availability
DA synthesis relies on tyrosine hydroxylase (TH), which is the rate-limiting enzyme required for DA synthesis to convert tyrosine to L-DOPA. Phenylalanine hydroxylase (PAH) is an enzyme that converts phenylalanine to tyrosine. 5,6,7,8-Tetrahydrobiopterin (BH4) is a cofactor of aromatic amino acid monooxygenases. Both TH and PAH require the enzyme cofactor BH4 (102). Inflammation may decrease BH4 availability (103). Indeed, intramuscular administration of IFN-α in rats has been shown to reduce the concentration of BH4 in the CNS by stimulating nitric oxide (NO), while inhibition of NO reverses the inhibitory effect of IFN-α on brain concentrations of BH4 and DA (84). In addition, the administration of IFN-α also increases cerebrospinal fluid (CSF) IL-6 levels, which were also correlated with decreased BH4 levels in CSF (92). Evidence from IFN-α-treated patients has also revealed a reduction in BH4 levels (104). In addition, the peripheral blood phenylalanine/tyrosine ratio increases after IFN-α administration, which correlates with decreased DA synthesis and CSF levels of DA and its major metabolite HVA (92). However, no change was observed in the DOPAC/DA ratio after L-DOPA administration, which increases when DA is not packaged and secreted in synaptic vesicles and subsequently metabolized by monoamine oxidase (105). These findings are consistent with decreased levels of DA metabolites in the CSF of IFN-α-treated patients and monkeys (88, 106). Meanwhile, L-DOPA reverses IFN-α-induced reduction in DA release (100). Notably, IL-6 treatment has also been shown to reduce the BH4 content in sympathetic neurons (107).
Another mechanism may be that cytokines affect glutamate neurotransmission to change the function of the basal ganglia and DA. In addition, inflammatory cytokines increase extracellular glutamate levels by reducing the level of the excitatory amino acid transporter GLT-1 (increasing glutamate reuptake) and increasing glutamate release from astrocytes and activated microglia (108, 109). Then, glutamate binds to the N-methyl-D-aspartic acid receptor (NMDA) and potentially leads to excitotoxicity in the brain (110). As a result, oxidative stress increases and potentially contributes to the effects on BH4 and DA synthesis (106, 111).
DA Packaging, Release and Reuptake
In synapses, vesicular monoamine transporter 2 (VMAT2) packages cytoplasmic DA into vesicles for further release. The inflammatory cytokines IL-1β and TNF-α have been shown to decrease the expression of VMAT2 in rat enterochromaffin-like cells, whereas the administration of transforming growth factor-β (TGF-β), which is an immunomodulatory and anti-inflammatory growth factor, rescues VMAT2 expression (112). Therefore, inflammatory cytokines and inflammation may negatively affect VMAT2 expression and function. In addition, the administration of pituitary adenylate cyclase-activating polypeptide 38 (PACAP-38), an anti-inflammatory compound, in vivo reduces neuroinflammation and increases VMAT2 expression, which protects against DA neurotoxicity following chronic methamphetamine exposure (113). IFN-α and other cytokines activate p38 mitogen-activated protein kinase (MAPK) signaling, which plays an important role in the expression and function of the serotonin transporter in serotonin reuptake (114, 115). Recently, researchers have found that MAPK pathways also influence DAT (DA transporter). DAT is expressed in DA neurons, and the role of DAT is to induce DA reuptake by DA neurons after its release into synapses. DAT-expressing neurons transfected with activated MAPK kinase (MEK) exhibit increased DA reuptake. Administration of MEK inhibitors to rat striatal synaptosomes decreases DA reuptake in a dose- and time-dependent manner (116). Therefore, exposure to inflammatory cytokines reduces synaptic DA levels through a mechanism that may be associated with increasing DAT expression or function.
DA Receptor Expression and Function
Type D2 dopamine receptor (D2R) is a G-protein-coupled receptor located in postsynaptic dopaminergic neurons that is mainly involved in reward mediation and reward deficiency pathways (117). A recent study elegantly showed that D2R within the ventrolateral striatum plays an important role in motivated behavior (118). Conditional knockout of D2R reduces the progressive rate task breakpoint, while optogenetic inhibition of these neurons that express D2R causes a transient reduction in the breakpoint. Inflammation and cytokines may affect DA signaling by reducing the expression or function of DA receptors. Chronic administration of IFN-α in the striatum of monkeys decreases the binding of DA to D2 receptors (83).
Indirect Mechanism of TLR4 Expression in the Perphery
Although TLR4 expression in CNS plays an important role in eating disorder, the TLR4 in periphery still affects the process of appetite. In activity-based anorexia (ABA) model mice, the expression of TLR4 in colonic mucosa is higher. Meanwhile, the mucosal cytokines expression also increased during ABA mice. Interestingly, TLR4, MyD88, TLR adaptor molecule 1 (TRIF) and TRIF-related adaptor molecule (TRAM) remained unchanged in the hypothalamus, but increased the expression of IL-1β, IL-1β receptor 1 (IL-1R1) and Interleukin-1 receptor-associated kinase (IRAK-4) in hypothalamic (119). The indirect mechanisms may be the peripheral cytokines cross the blood-brain barrier and activate neuro-inflammation in the brain. In addition, another paper reported that global TLR4 KO mice decreased the expression of TLR4 in tongue gustatory epithelium, which may affect expression of taste molecules CD36, PLC2β and TRPM5 and change food intake (101).
The Role of TLR4 Activation in the Regulation of Appetite
As an innate immune receptor, TLR4 is well known for its response to LPS. However, it is also activated by nutritional signals, such as saturated fatty acids (SFAs), particularly lauric acid, palmitic acid, and stearic acid (120). TLR4-dependent priming senses the lcSFA and regulates gene expression and cellular metabolism (121). Chronic overconsumption of a HFD can increase the plasma levels of free fatty acids (122). Increased fatty acid concentrations in plasma are closely associated with metabolic syndrome, and SFAs activate innate immune responses and result in inflammation (123–125).
SFAs have been shown to cause hypothalamic inflammation (126), and chronic hypothalamic inflammation disrupts the function of brain circuits that control appetite, leading to an increase in food intake and weight gain (127). The dietary lipid composition will affect the degree of inflammation in the hypothalamus. For example, a diet high in saturated fat is associated with a higher risk of hypothalamic inflammation after 8 weeks than a diet high in unsaturated fat (128). In addition, fat from butter produces greater neuroinflammation than saturated fat from coconut oil, indicating that fat from different sources produces different hypothalamic inflammatory responses. SFAs exert major effects on neuro-immunity, enteroendocrine signaling, feeding homeostasis and appetite regulation. In addition, some studies attribute hypothalamic inflammation to SFAs. In this process, inflammatory signaling is activated by the SFA-TLR4 pathway. These free fatty acids bind to receptors on immune cells, activate inflammatory signaling pathways, and impair normal cellular signaling in the liver, pancreas, skeletal muscle, and white adipose tissue (129), resulting in the release of proinflammatory cytokines and chemokines.
Based on these findings, SFAs can activate TLR4 signal pathway, which plays an important role in physiological regulation of brain function (130, 131). SFA activates immune cells, including microglia, through TLR4 signal pathway (11). In the brain, palmitic acid activates microglia to release cytokines via the TLR4-activated signal transduction pathway, which leads to a decrease in hypothalamic cell activity, thus leading to the interruption of the neural circuit controlling appetite (132). Lauric acid, palmitate and stearic acid increase the release of TNF-α and IL-6 from astrocytes. Apparently, the increase in TNF-α and IL-6 levels in the hypothalamus mediates insulin and leptin resistance by increasing the level of the suppressor of cytokine signaling 3 (SOCS3) protein and phosphorylation of insulin resistance substrate and suppressing the Janus kinase/signal transducer and activator of transcription (JAK-STAT) pathway, which induces leptin receptor activation. In addition, the effect of palmitic acid on cytokine release requires TLR4 rather than CD36 or toll like receptor 2 (TLR2) (which are also palmitic acid receptors) and is independent of palmitate metabolism to palmitoyl-CoA (133). Accordingly, short-term ICV administration of stearic acid promotes TLR4 activation and the expression of endoplasmic reticulum (ER) stress-related proteins, while unsaturated fatty acids attenuate the inflammatory response. Stearic acid elicits AgRP expression and secretion via TLR4-dependent signaling pathways in hypothalamic N38 cells (134). However, the result has not been verified in animals. Thus, LCSFAs play a main role in hypothalamic inflammation and appetite (135). Additionally, chronic hypothalamic inflammation can also lead to leptin and insulin resistance, which will further weaken the homeostatic signaling of the hypothalamic circuit. However, evidence for the mechanism by which SFAs affect motivation in humans and animals is lacking. Previous studies reported that a single intra-VTA injection of palmitic acid does not affect food intake (136), but lauric acid increases appetite in mice (137). The proinflammatory state of glial cells is activated by SFAs via TLR4/NF-kB signaling in a microglial cell line. Therefore, SFAs activate TLR4 signaling, which is an important process for understanding how SFA-induced inflammation regulates appetite.
Conclusion
In this review, we showed that inflammatory signals in the hypothalamus and mesolimbic DA system play different roles in regulating appetite. In summary, cytokine release induced by inflammatory signals decreases NPY and AgRP expression and increases POMC expression to decrease food intake. In addition, inflammatory cytokines may affect multiple aspects of DA neurotransmission, resulting in reduced synthesis and impaired DA receptor signaling and/or packaging or release, all of which interact to reduce DA function, which contributes to appetite (Figure 3). As ligands, SFAs also trigger inflammation in the brain though TLR4 to affect appetite.
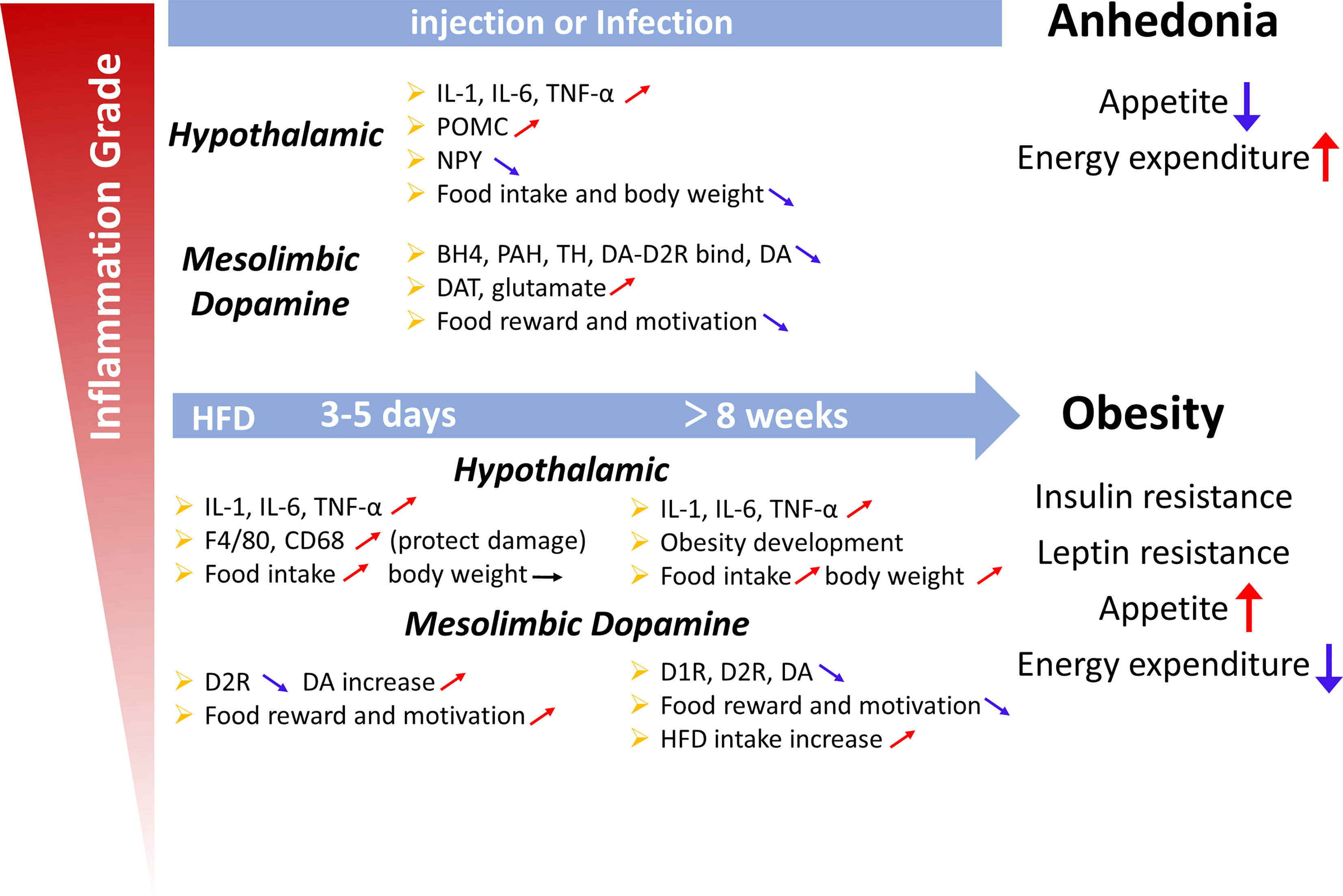
Figure 3 Summary diagram illustrating the links between inflammation in the hypothalamus and midbrain dopamine system with the deregulation of appetite. Upper panel: Lipopolysaccharide or cytokines injections, illnesses such as cancer, or infections induce high-grade inflammation. An acute increase in the local production of cytokines and chemokines is observed in the hypothalamic and midbrain dopamine systems. Cytokines, including IL-1, IL-6 and TNF-α, change the levels of neuropeptides (POMC, NPY and dopamine) involved in the hypothalamic and dopamine systems, which are associated with appetite and weight loss (the sickness behavior associated with a high-intensity infection). Lower panel: Short (3 days) or chronic (>8 weeks) HFD consumption is associated with low-grade inflammation. The increase in cytokine levels induced by a HFD also changes the levels of neuropeptides involved in the hypothalamic and dopamine systems, which appears to be associated with overeating and body weight gain and the development of obesity, leptin resistance and insulin resistance, potentially increasing appetite, and weight. However, in the first 3 days, the weight was not changed, which may protect organs from HFD-induced damage.
Perspectives and Future Directions
Appetite is controlled by several complex regulatory mechanisms involving both homeostatic and non-homeostatic processes. Additionally, inflammation is seen as a way for tissues to try to return to normal in response to infection or disruption. Such studies have provided evidence in understanding the full impact of TLR4-induced inflammatory signaling on appetite and how it may increase the risk of developing obesity and related health problems. Importantly, the interfering of appetite can be achieved by regulating inflammatory signaling in hypothalamus and dopamine system, which could guide development of novel therapies to treat diseases about appetite. More importantly, the link between SFAs and CNS immune system also provides an exciting new direction for the study of eating behaviors and the pathophysiology of obesity.
Author Contributions
YL wrote the manuscript. QJ provided constructive comments for the figure drawing. LW revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (32072779).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kaisho T, Akira S. Toll-Like Receptors and Their Signaling Mechanism in Innate Immunity. Acta Odontol Scand (2001) 59(3):124–30. doi: 10.1080/000163501750266701
2. Takeda K, Akira S. Toll Receptors and Pathogen Resistance. Cell Microbiol (2003) 5(3):143–53. doi: 10.1046/j.1462-5822.2003.00264.x
3. Buchanan MM, Hutchinson M, Watkins LR, Yin H. Toll-Like Receptor 4 in CNS Pathologies. J Neurochem (2010) 114(1):13–27. doi: 10.1111/j.1471-4159.2010.06736.x
4. Wadachi R, Hargreaves KM. Trigeminal Nociceptors Express TLR-4 and CD14: A Mechanism for Pain Due to Infection. J Dent Res (2006) 85(1):49–53. doi: 10.1177/154405910608500108
5. Yirmiya R, Goshen I. Immune Modulation of Learning, Memory, Neural Plasticity and Neurogenesis. Brain Behav Immun (2011) 25(2):181–213. doi: 10.1016/j.bbi.2010.10.015
6. Hotamisligil GS. Inflammation, Metaflammation and Immunometabolic Disorders. Nature (2017) 542(7640):177–85. doi: 10.1038/nature21363
7. Morton GJ, Meek TH, Schwartz MW. Neurobiology of Food Intake in Health and Disease. Nat Rev Neurosci (2014) 15(6):367–78. doi: 10.1038/nrn3745
8. Fekete C, Legradi G, Mihaly E, Tatro JB, Rand WM, Lechan RM. Alpha-Melanocyte Stimulating Hormone Prevents Fasting-Induced Suppression of Corticotropin-Releasing Hormone Gene Expression in the Rat Hypothalamic Paraventricular Nucleus. Neurosci Lett (2000) 289(2):152–6. doi: 10.1016/s0304-3940(00)01256-8
9. Giraud N, Blackledge M, Goldman M, Bockmann A, Lesage A, Penin F, et al. Quantitative Analysis of Backbone Dynamics in a Crystalline Protein From Nitrogen-15 Spin-Lattice Relaxation. J Am Chem Soc (2005) 127(51):18190–201. doi: 10.1021/ja055182h
10. Choi YH, Fujikawa T, Lee J, Reuter A, Kim KW. Revisiting the Ventral Medial Nucleus of the Hypothalamus: The Roles of SF-1 Neurons in Energy Homeostasis. Front Neurosci (2013) 7:71. doi: 10.3389/fnins.2013.00071
11. Maldonado-Ruiz R, Fuentes-Mera L, Camacho A. Central Modulation of Neuroinflammation by Neuropeptides and Energy-Sensing Hormones During Obesity. BioMed Res Int (2017) 2017:7949582. doi: 10.1155/2017/7949582
12. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and Functional Features of Central Nervous System Lymphatic Vessels. Nature (2015) 523(7560):337–41. doi: 10.1038/nature14432
13. Ransohoff RM, Brown MA. Innate Immunity in the Central Nervous System. J Clin Invest (2012) 122(4):1164–71. doi: 10.1172/JCI58644
14. Vetrivelan R, Fuller PM, Yokota S, Lu J, Saper CB. Metabolic Effects of Chronic Sleep Restriction in Rats. Sleep (2012) 35(11):1511–20. doi: 10.5665/sleep.2200
15. O’Neill LA, Hardie DG. Metabolism of Inflammation Limited by AMPK and Pseudo-Starvation. Nature (2013) 493(7432):346–55. doi: 10.1038/nature11862
16. Dantzer R. Cytokine, Sickness Behavior, and Depression. Immunol Allergy Clin N Am (2009) 29(2):247–64. doi: 10.1016/j.iac.2009.02.002
17. van Niekerk G, Isaacs AW, Nell T, Engelbrecht AM. Sickness-Associated Anorexia: Mother Nature’s Idea of Immunonutrition? Mediators Inflamm (2016) 2016:8071539. doi: 10.1155/2016/8071539
18. Alexander C, Rietschel ET. Bacterial Lipopolysaccharides and Innate Immunity. J Endotoxin Res (2001) 7(3):167–202.
19. Laviano A, Inui A, Meguid MM, Molfino A, Conte C, Rossi Fanelli F. NPY and Brain Monoamines in the Pathogenesis of Cancer Anorexia. Nutrition (2008) 24(9):802–5. doi: 10.1016/j.nut.2008.06.005
20. Perrella O, Carrieri PB, Guarnaccia D, Soscia M. Cerebrospinal Fluid Cytokines in AIDS Dementia Complex. J Neurol (1992) 239(7):387–8. doi: 10.1007/bf00812156
21. Cai C, Zhang HY, Le JJ, Dong JC, Cui Y, Xu CQ, et al. Inflammatory Airway Features and Hypothalamic-Pituitary-Adrenal Axis Function in Asthmatic Rats Combined With Chronic Obstructive Pulmonary Disease. Chin (Engl) (2010) 123(13):1720–6. doi: 10.3760/cma.j.issn.0366-6999.2010.13.020
22. Kang YM, Zhang ZH, Johnson RF, Yu Y, Beltz T, Johnson AK, et al. Novel Effect of Mineralocorticoid Receptor Antagonism to Reduce Proinflammatory Cytokines and Hypothalamic Activation in Rats With Ischemia-Induced Heart Failure. Circ Res (2006) 99(7):758–66. doi: 10.1161/01.RES.0000244092.95152.86
23. Gutierrez EG, Banks WA, Kastin AJ. Murine Tumor Necrosis Factor Alpha Is Transported From Blood to Brain in the Mouse. J Neuroimmunol (1993) 47(2):169–76. doi: 10.1016/0165-5728(93)90027-v
24. Banks WA, Kastin AJ, Gutierrez EG. Penetration of Interleukin-6 Across the Murine Blood-Brain Barrier. Neurosci Lett (1994) 179(1-2):53–6. doi: 10.1016/0304-3940(94)90933-4
25. Plotkin SR, Banks WA, Kastin AJ. Comparison of Saturable Transport and Extracellular Pathways in the Passage of Interleukin-1 Alpha Across the Blood-Brain Barrier. J Neuroimmunol (1996) 67(1):41–7. doi: 10.1016/0165-5728(96)00036-7
26. Banks WA, Ortiz L, Plotkin SR, Kastin AJ. Human Interleukin (IL) 1 Alpha, Murine IL-1 Alpha and Murine IL-1 Beta Are Transported From Blood to Brain in the Mouse by a Shared Saturable Mechanism. J Pharmacol Exp Ther (1991) 259(3):988–96.
27. Hopkins SJ, Rothwell NJ. Cytokines and the Nervous System. I: Expression and Recognition. Trends Neurosci (1995) 18(2):83–8. doi: 10.1016/0166-2236(95)80029-2
28. Rana I, Stebbing M, Kompa A, Kelly DJ, Krum H, Badoer E. Microglia Activation in the Hypothalamic PVN Following Myocardial Infarction. Brain Res (2010) 1326:96–104. doi: 10.1016/j.brainres.2010.02.028
29. Ronaldson PT, Bendayan R. HIV-1 Viral Envelope Glycoprotein Gp120 Triggers an Inflammatory Response in Cultured Rat Astrocytes and Regulates the Functional Expression of P-Glycoprotein. Mol Pharmacol (2006) 70(3):1087–98. doi: 10.1124/mol.106.025973
30. Mantovani G, Maccio A, Lai P, Massa E, Ghiani M, Santona MC. Cytokine Involvement in Cancer Anorexia/Cachexia: Role of Megestrol Acetate and Medroxyprogesterone Acetate on Cytokine Downregulation and Improvement of Clinical Symptoms. Crit Rev Oncog (1998) 9(2):99–106. doi: 10.1615/critrevoncog.v9.i2.10
31. Gelin J, Moldawer LL, Lonnroth C, Sherry B, Chizzonite R, Lundholm K. Role of Endogenous Tumor Necrosis Factor Alpha and Interleukin 1 for Experimental Tumor Growth and the Development of Cancer Cachexia. Cancer Res (1991) 51(1):415–21.
32. Matthys P, Billiau A. Cytokines and Cachexia. Nutrition (1997) 13(9):763–70. doi: 10.1016/s0899-9007(97)00185-8
33. Noguchi Y, Yoshikawa T, Matsumoto A, Svaninger G, Gelin J. Are Cytokines Possible Mediators of Cancer Cachexia? Surg Today (1996) 26(7):467–75. doi: 10.1007/bf00311551
34. Sherry BA, Gelin J, Fong Y, Marano M, Wei H, Cerami A, et al. Anticachectin/tumor Necrosis Factor-Alpha Antibodies Attenuate Development of Cachexia in Tumor Models. FASEB J (1989) 3(8):1956–62. doi: 10.1096/fasebj.3.8.2721856
35. Ropelle ER, Pauli JR, Zecchin KG, Ueno M, de Souza CT, Morari J, et al. A Central Role for Neuronal Adenosine 5’-Monophosphate-Activated Protein Kinase in Cancer-Induced Anorexia. Endocrinology (2007) 148(11):5220–9. doi: 10.1210/en.2007-0381
36. Liu X, McMurphy T, Xiao R, Slater A, Huang W, Cao L. Hypothalamic Gene Transfer of BDNF Inhibits Breast Cancer Progression and Metastasis in Middle Age Obese Mice. Mol Ther (2014) 22(7):1275–84. doi: 10.1038/mt.2014.45
37. Cao L, Liu X, Lin EJ, Wang C, Choi EY, Riban V, et al. Environmental and Genetic Activation of a Brain-Adipocyte BDNF/leptin Axis Causes Cancer Remission and Inhibition. Cell (2010) 142(1):52–64. doi: 10.1016/j.cell.2010.05.029
38. Kent S, Kelley KW, Dantzer R. Effects of Lipopolysaccharide on Food-Motivated Behavior in the Rat Are Not Blocked by an Interleukin-1 Receptor Antagonist. Neurosci Lett (1992) 145(1):83–6. doi: 10.1016/0304-3940(92)90209-p
39. Kent S, Bluthe RM, Dantzer R, Hardwick AJ, Kelley KW, Rothwell NJ, et al. Different Receptor Mechanisms Mediate the Pyrogenic and Behavioral Effects of Interleukin 1. Proc Natl Acad Sci USA (1992) 89(19):9117–20. doi: 10.1073/pnas.89.19.9117
40. Laviano A, Gleason JR, Meguid MM, Yang ZJ, Cangiano C, Rossi Fanelli F. Effects of Intra-VMN Mianserin and IL-1ra on Meal Number in Anorectic Tumor-Bearing Rats. J Investig Med (2000) 48(1):40–8.
41. Banks WA, Farr SA, La Scola ME, Morley JE. Intravenous Human Interleukin-1alpha Impairs Memory Processing in Mice: Dependence on Blood-Brain Barrier Transport Into Posterior Division of the Septum. J Pharmacol Exp Ther (2001) 299(2):536–41.
42. Kent S, Bluthe RM, Kelley KW, Dantzer R. Sickness Behavior as a New Target for Drug Development. Trends Pharmacol Sci (1992) 13(1):24–8. doi: 10.1016/0165-6147(92)90012-u
43. Laye S, Gheusi G, Cremona S, Combe C, Kelley K, Dantzer R, et al. Endogenous Brain IL-1 Mediates LPS-Induced Anorexia and Hypothalamic Cytokine Expression. Am J Physiol Regul Integr Comp Physiol (2000) 279(1):R93–8. doi: 10.1152/ajpregu.2000.279.1.R93
44. Chakravarty S, Herkenham M. Toll-Like Receptor 4 on Nonhematopoietic Cells Sustains CNS Inflammation During Endotoxemia, Independent of Systemic Cytokines. J Neurosci (2005) 25(7):1788–96. doi: 10.1523/JNEUROSCI.4268-04.2005
45. Wisse BE, Ogimoto K, Tang J, Harris MK Jr., Raines EW, Schwartz MW. Evidence That Lipopolysaccharide-Induced Anorexia Depends Upon Central, Rather Than Peripheral, Inflammatory Signals. Endocrinology (2007) 148(11):5230–7. doi: 10.1210/en.2007-0394
46. Ueta Y, Hashimoto H, Onuma E, Takuwa Y, Ogata E. Hypothalamic Neuropeptides and Appetite Response in Anorexia-Cachexia Animal. Endocr J (2007) 54(6):831–8. doi: 10.1507/endocrj.kr-111
47. Arruda AP, Milanski M, Romanatto T, Solon C, Coope A, Alberici LC, et al. Hypothalamic Actions of Tumor Necrosis Factor Alpha Provide the Thermogenic Core for the Wastage Syndrome in Cachexia. Endocrinology (2010) 151(2):683–94. doi: 10.1210/en.2009-0865
48. Iwasa T, Matsuzaki T, Kinouchi R, Fujisawa S, Murakami M, Kiyokawa M, et al. Neonatal LPS Injection Alters the Body Weight Regulation Systems of Rats Under Non-Stress and Immune Stress Conditions. Int J Dev Neurosci (2010) 28(1):119–24. doi: 10.1016/j.ijdevneu.2009.08.015
49. Sergeyev V, Broberger C, Hokfelt T. Effect of LPS Administration on the Expression of POMC, NPY, Galanin, CART and MCH mRNAs in the Rat Hypothalamus. Brain Res Mol Brain Res (2001) 90(2):93–100. doi: 10.1016/s0169-328x(01)00088-2
50. Kim YW, Kim KH, Ahn DK, Kim HS, Kim JY, Lee DC, et al. Time-Course Changes of Hormones and Cytokines by Lipopolysaccharide and Its Relation With Anorexia. J Physiol Sci (2007) 57(3):159–65. doi: 10.2170/physiolsci.RP003407
51. Nara-ashizawa N, Tsukada T, Maruyama K, Akiyama Y, Kajimura N, Nagasaki K, et al. Hypothalamic Appetite-Regulating Neuropeptide mRNA Levels in Cachectic Nude Mice Bearing Human Tumor Cells. Metabolism (2001) 50(10):1213–9. doi: 10.1053/meta.2001.26706
52. Chance WT, Xiao C, Dayal R, Sheriff S. Alteration of NPY and Y1 Receptor in Dorsomedial and Ventromedial Areas of Hypothalamus in Anorectic Tumor-Bearing Rats. Peptides (2007) 28(2):295–301. doi: 10.1016/j.peptides.2006.10.018
53. Plata-Salaman CR, Ilyin SE, Gayle D. Brain Cytokine mRNAs in Anorectic Rats Bearing Prostate Adenocarcinoma Tumor Cells. Am J Physiol (1998) 275(2):R566–73. doi: 10.1152/ajpregu.1998.275.2.R566
54. Nara-ashizawa N, Tsukada T, Maruyama K, Akiyama Y, Kajimura N, Yamaguchi K. Response of Hypothalamic NPY mRNAs to a Negative Energy Balance Is Less Sensitive in Cachectic Mice Bearing Human Tumor Cells. Nutr Cancer (2001) 41(1-2):111–8. doi: 10.1080/01635581.2001.9680621
55. Gomez-SanMiguel AB, Martin AI, Nieto-Bona MP, Fernandez-Galaz C, Lopez-Menduina M, Villanua MA, et al. Systemic Alpha-Melanocyte-Stimulating Hormone Administration Decreases Arthritis-Induced Anorexia and Muscle Wasting. Am J Physiol Regul Integr Comp Physiol (2013) 304(10):R877–86. doi: 10.1152/ajpregu.00447.2012
56. Stofkova A, Zelezna B, Romzova M, Ulicna O, Kiss A, Skurlova M, et al. Effect of Feeding Status on Adjuvant Arthritis Severity, Cachexia, and Insulin Sensitivity in Male Lewis Rats. Mediators Inflamm (2010) 2010:398026. doi: 10.1155/2010/398026
57. Dwarkasing JT, Boekschoten MV, Argiles JM, van Dijk M, Busquets S, Penna F, et al. Differences in Food Intake of Tumour-Bearing Cachectic Mice Are Associated With Hypothalamic Serotonin Signalling. J Cachexia Sarcopenia Muscle (2015) 6(1):84–94. doi: 10.1002/jcsm.12008
58. McCarthy HD, McKibbin PE, Perkins AV, Linton EA, Williams G. Alterations in Hypothalamic NPY and CRF in Anorexic Tumor-Bearing Rats. Am J Physiol (1993) 264(4 Pt 1):E638–43. doi: 10.1152/ajpendo.1993.264.4.E638
59. Meguid MM, Ramos EJ, Laviano A, Varma M, Sato T, Chen C, et al. Tumor Anorexia: Effects on Neuropeptide Y and Monoamines in Paraventricular Nucleus. Peptides (2004) 25(2):261–6. doi: 10.1016/j.peptides.2004.01.012
60. Makarenko IG, Meguid MM, Gatto L, Chen C, Ugrumov MV. Decreased NPY Innervation of the Hypothalamic Nuclei in Rats With Cancer Anorexia. Brain Res (2003) 961(1):100–8. doi: 10.1016/s0006-8993(02)03850-7
61. Scarlett JM, Zhu X, Enriori PJ, Bowe DD, Batra AK, Levasseur PR, et al. Regulation of Agouti-Related Protein Messenger Ribonucleic Acid Transcription and Peptide Secretion by Acute and Chronic Inflammation. Endocrinology (2008) 149(10):4837–45. doi: 10.1210/en.2007-1680
62. Scarlett JM, Jobst EE, Enriori PJ, Bowe DD, Batra AK, Grant WF, et al. Regulation of Central Melanocortin Signaling by Interleukin-1 Beta. Endocrinology (2007) 148(9):4217–25. doi: 10.1210/en.2007-0017
63. Borges BC, Rorato R, Avraham Y, da Silva LE, Castro M, Vorobiav L, et al. Leptin Resistance and Desensitization of Hypophagia During Prolonged Inflammatory Challenge. Am J Physiol Endocrinol Metab (2011) 300(5):E858–69. doi: 10.1152/ajpendo.00558.2010
64. Jang PG, Namkoong C, Kang GM, Hur MW, Kim SW, Kim GH, et al. NF-kappaB Activation in Hypothalamic Pro-Opiomelanocortin Neurons Is Essential in Illness- and Leptin-Induced Anorexia. J Biol Chem (2010) 285(13):9706–15. doi: 10.1074/jbc.M109.070706
65. Endo M, Masaki T, Seike M, Yoshimatsu H. Involvement of Stomach Ghrelin and Hypothalamic Neuropeptides in Tumor Necrosis Factor-Alpha-Induced Hypophagia in Mice. Regul Pept (2007) 140(1-2):94–100. doi: 10.1016/j.regpep.2006.11.022
66. Kim SM, McIlwraith EK, Chalmers JA, Belsham DD. Palmitate Induces an Anti-Inflammatory Response in Immortalized Microglial BV-2 and IMG Cell Lines That Decreases TNFalpha Levels in Mhypoe-46 Hypothalamic Neurons in Co-Culture. Neuroendocrinology (2018) 107(4):387–99. doi: 10.1159/000494759
67. Berridge KC. Food Reward: Brain Substrates of Wanting and Liking. Neurosci Biobehav Rev (1996) 20(1):1–25. doi: 10.1016/0149-7634(95)00033-b
68. Liu JJ, Mukherjee D, Haritan D, Ignatowska-Jankowska B, Liu J, Citri A, et al. High on Food: The Interaction Between the Neural Circuits for Feeding and for Reward. Front Biol (Beijing) (2015) 10(2):165–76. doi: 10.1007/s11515-015-1348-0
69. Volkow ND, Wang GJ, Baler RD. Reward, Dopamine and the Control of Food Intake: Implications for Obesity. Trends Cognit Sci (2011) 15(1):37–46. doi: 10.1016/j.tics.2010.11.001
70. Edwards NJ, Tejeda HA, Pignatelli M, Zhang S, McDevitt RA, Wu J, et al. Circuit Specificity in the Inhibitory Architecture of the VTA Regulates Cocaine-Induced Behavior. Nat Neurosci (2017) 20(3):438–48. doi: 10.1038/nn.4482
71. Haber SN. The Place of Dopamine in the Cortico-Basal Ganglia Circuit. Neuroscience (2014) 282:248–57. doi: 10.1016/j.neuroscience.2014.10.008
72. Leemburg S, Canonica T, Luft A. Motor Skill Learning and Reward Consumption Differentially Affect VTA Activation. Sci Rep (2018) 8(1):687. doi: 10.1038/s41598-017-18716-w
73. Morales M, Margolis EB. Ventral Tegmental Area: Cellular Heterogeneity, Connectivity and Behaviour. Nat Rev Neurosci (2017) 18(2):73–85. doi: 10.1038/nrn.2016.165
74. Oades RD, Rivet JM, Taghzouti K, Kharouby M, Simon H, Le Moal M. Catecholamines and Conditioned Blocking: Effects of Ventral Tegmental, Septal and Frontal 6-Hydroxydopamine Lesions in Rats. Brain Res (1987) 406(1-2):136–46. doi: 10.1016/0006-8993(87)90778-5
75. Yang SS, Li YC, Coley AA, Chamberlin LA, Yu P, Gao WJ. Cell-Type Specific Development of the Hyperpolarization-Activated Current, Ih, in Prefrontal Cortical Neurons. Front Synaptic Neurosci (2018) 10:7. doi: 10.3389/fnsyn.2018.00007
76. Ikemoto S. Dopamine Reward Circuitry: Two Projection Systems From the Ventral Midbrain to the Nucleus Accumbens-Olfactory Tubercle Complex. Brain Res Rev (2007) 56(1):27–78. doi: 10.1016/j.brainresrev.2007.05.004
77. Felger JC, Treadway MT. Inflammation Effects on Motivation and Motor Activity: Role of Dopamine. Neuropsychopharmacology (2017) 42(1):216–41. doi: 10.1038/npp.2016.143
78. Karshikoff B, Sundelin T, Lasselin J. Role of Inflammation in Human Fatigue: Relevance of Multidimensional Assessments and Potential Neuronal Mechanisms. Front Immunol (2017) 8:21. doi: 10.3389/fimmu.2017.00021
79. Dantzer R, Kelley KW. Twenty Years of Research on Cytokine-Induced Sickness Behavior. Brain Behav Immun (2007) 21(2):153–60. doi: 10.1016/j.bbi.2006.09.006
80. Brydon L, Harrison NA, Walker C, Steptoe A, Critchley HD. Peripheral Inflammation Is Associated With Altered Substantia Nigra Activity and Psychomotor Slowing in Humans. Biol Psychiatry (2008) 63(11):1022–9. doi: 10.1016/j.biopsych.2007.12.007
81. Capuron L, Pagnoni G, Demetrashvili MF, Lawson DH, Fornwalt FB, Woolwine B, et al. Basal Ganglia Hypermetabolism and Symptoms of Fatigue During Interferon-Alpha Therapy. Neuropsychopharmacology (2007) 32(11):2384–92. doi: 10.1038/sj.npp.1301362
82. Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR. Inflammation-Induced Anhedonia: Endotoxin Reduces Ventral Striatum Responses to Reward. Biol Psychiatry (2010) 68(8):748–54. doi: 10.1016/j.biopsych.2010.06.010
83. Felger JC, Mun J, Kimmel HL, Nye JA, Drake DF, Hernandez CR, et al. Chronic Interferon-Alpha Decreases Dopamine 2 Receptor Binding and Striatal Dopamine Release in Association With Anhedonia-Like Behavior in Nonhuman Primates. Neuropsychopharmacology (2013) 38(11):2179–87. doi: 10.1038/npp.2013.115
84. Kitagami T, Yamada K, Miura H, Hashimoto R, Nabeshima T, Ohta T. Mechanism of Systemically Injected Interferon-Alpha Impeding Monoamine Biosynthesis in Rats: Role of Nitric Oxide as a Signal Crossing the Blood-Brain Barrier. Brain Res (2003) 978(1-2):104–14. doi: 10.1016/s0006-8993(03)02776-8
85. Nunes EJ, Randall PA, Estrada A, Epling B, Hart EE, Lee CA, et al. Effort-Related Motivational Effects of the Pro-Inflammatory Cytokine Interleukin 1-Beta: Studies With the Concurrent Fixed Ratio 5/Chow Feeding Choice Task. Psychopharmacol (Berl) (2014) 231(4):727–36. doi: 10.1007/s00213-013-3285-4
86. Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, et al. Neurobehavioral Effects of Interferon-Alpha in Cancer Patients: Phenomenology and Paroxetine Responsiveness of Symptom Dimensions. Neuropsychopharmacology (2002) 26(5):643–52. doi: 10.1016/S0893-133X(01)00407-9
87. Capuron L, Pagnoni G, Drake DF, Woolwine BJ, Spivey JR, Crowe RJ, et al. Dopaminergic Mechanisms of Reduced Basal Ganglia Responses to Hedonic Reward During Interferon Alfa Administration. Arch Gen Psychiatry (2012) 69(10):1044–53. doi: 10.1001/archgenpsychiatry.2011.2094
88. Capuron L, Neurauter G, Musselman DL, Lawson DH, Nemeroff CB, Fuchs D, et al. Interferon-Alpha-Induced Changes in Tryptophan Metabolism. Relationship to Depression and Paroxetine Treatment. Biol Psychiatry (2003) 54(9):906–14. doi: 10.1016/s0006-3223(03)00173-2
89. Felger JC, Alagbe O, Hu F, Mook D, Freeman AA, Sanchez MM, et al. Effects of Interferon-Alpha on Rhesus Monkeys: A Nonhuman Primate Model of Cytokine-Induced Depression. Biol Psychiatry (2007) 62(11):1324–33. doi: 10.1016/j.biopsych.2007.05.026
90. Miller AH, Maletic V, Raison CL. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol Psychiatry (2009) 65(9):732–41. doi: 10.1016/j.biopsych.2008.11.029
91. Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD. Inflammation Causes Mood Changes Through Alterations in Subgenual Cingulate Activity and Mesolimbic Connectivity. Biol Psychiatry (2009) 66(5):407–14. doi: 10.1016/j.biopsych.2009.03.015
92. Felger JC, Li L, Marvar PJ, Woolwine BJ, Harrison DG, Raison CL, et al. Tyrosine Metabolism During Interferon-Alpha Administration: Association With Fatigue and CSF Dopamine Concentrations. Brain Behav Immun (2013) 31:153–60. doi: 10.1016/j.bbi.2012.10.010
93. van Heesch F, Prins J, Konsman JP, Korte-Bouws GA, Westphal KG, Rybka J, et al. Lipopolysaccharide Increases Degradation of Central Monoamines: An In Vivo Microdialysis Study in the Nucleus Accumbens and Medial Prefrontal Cortex of Mice. Eur J Pharmacol (2014) 725:55–63. doi: 10.1016/j.ejphar.2014.01.014
94. Yen CH, Yeh YW, Liang CS, Ho PS, Kuo SC, Huang CC, et al. Reduced Dopamine Transporter Availability and Neurocognitive Deficits in Male Patients With Alcohol Dependence. PLoS One (2015) 10(6):e0131017. doi: 10.1371/journal.pone.0131017
95. Vichaya EG, Hunt SC, Dantzer R. Lipopolysaccharide Reduces Incentive Motivation While Boosting Preference for High Reward in Mice. Neuropsychopharmacology (2014) 39(12):2884–90. doi: 10.1038/npp.2014.141
96. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS Causes Chronic Neuroinflammation and Progressive Neurodegeneration. Glia (2007) 55(5):453–62. doi: 10.1002/glia.20467
97. Tian YY, An LJ, Jiang L, Duan YL, Chen J, Jiang B. Catalpol Protects Dopaminergic Neurons From LPS-Induced Neurotoxicity in Mesencephalic Neuron-Glia Cultures. Life Sci (2006) 80(3):193–9. doi: 10.1016/j.lfs.2006.09.010
98. Lebena A, Vegas O, Gomez-Lazaro E, Arregi A, Garmendia L, Beitia G, et al. Melanoma Tumors Alter Proinflammatory Cytokine Production and Monoamine Brain Function, and Induce Depressive-Like Behavior in Male Mice. Behav Brain Res (2014) 272:83–92. doi: 10.1016/j.bbr.2014.06.045
99. Uomoto M, Nishibori M, Nakaya N, Takeuchi Y, Iwagaki H, Tanaka N, et al. Changes in Monoamine Turnover in the Brain of Cachectic Mice Bearing Colon-26 Tumor Cells. J Neurochem (1998) 70(1):260–7. doi: 10.1046/j.1471-4159.1998.70010260.x
100. Felger JC, Hernandez CR, Miller AH. Levodopa Reverses Cytokine-Induced Reductions in Striatal Dopamine Release. Int J Neuropsychopharmacol (2015) 18(4). doi: 10.1093/ijnp/pyu084
101. Camandola S, Mattson MP. Toll-Like Receptor 4 Mediates Fat, Sugar, and Umami Taste Preference and Food Intake and Body Weight Regulation. Obes (Silver Spring) (2017) 25(7):1237–45. doi: 10.1002/oby.21871
102. Werner-Felmayer G, Golderer G, Werner ER. Tetrahydrobiopterin Biosynthesis, Utilization and Pharmacological Effects. Curr Drug Metab (2002) 3(2):159–73. doi: 10.2174/1389200024605073
103. Neurauter G, Schrocksnadel K, Scholl-Burgi S, Sperner-Unterweger B, Schubert C, Ledochowski M, et al. Chronic Immune Stimulation Correlates With Reduced Phenylalanine Turnover. Curr Drug Metab (2008) 9(7):622–7. doi: 10.2174/138920008785821738
104. Zoller H, Schloegl A, Schroecksnadel S, Vogel W, Fuchs D. Interferon-Alpha Therapy in Patients With Hepatitis C Virus Infection Increases Plasma Phenylalanine and the Phenylalanine to Tyrosine Ratio. J Interferon Cytokine Res (2012) 32(5):216–20. doi: 10.1089/jir.2011.0093
105. Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL, et al. Reduced Vesicular Storage of Dopamine Causes Progressive Nigrostriatal Neurodegeneration. J Neurosci (2007) 27(30):8138–48. doi: 10.1523/JNEUROSCI.0319-07.2007
106. Felger JC, Miller AH. Cytokine Effects on the Basal Ganglia and Dopamine Function: The Subcortical Source of Inflammatory Malaise. Front Neuroendocrinol (2012) 33(3):315–27. doi: 10.1016/j.yfrne.2012.09.003
107. Li W, Knowlton D, Woodward WR, Habecker BA. Regulation of Noradrenergic Function by Inflammatory Cytokines and Depolarization. J Neurochem (2003) 86(3):774–83. doi: 10.1046/j.1471-4159.2003.01890.x
108. Dantzer R, Walker AK. Is There a Role for Glutamate-Mediated Excitotoxicity in Inflammation-Induced Depression? J Neural Transm (Vienna) (2014) 121(8):925–32. doi: 10.1007/s00702-014-1187-1
109. Takaki J, Fujimori K, Miura M, Suzuki T, Sekino Y, Sato K. L-Glutamate Released From Activated Microglia Downregulates Astrocytic L-Glutamate Transporter Expression in Neuroinflammation: The ‘Collusion’ Hypothesis for Increased Extracellular L-Glutamate Concentration in Neuroinflammation. J Neuroinflamm (2012) 9:275. doi: 10.1186/1742-2094-9-275
110. Erhardt S, Lim CK, Linderholm KR, Janelidze S, Lindqvist D, Samuelsson M, et al. Connecting Inflammation With Glutamate Agonism in Suicidality. Neuropsychopharmacology (2013) 38(5):743–52. doi: 10.1038/npp.2012.248
111. Najjar S, Pearlman DM, Devinsky O, Najjar A, Zagzag D. Neurovascular Unit Dysfunction With Blood-Brain Barrier Hyperpermeability Contributes to Major Depressive Disorder: A Review of Clinical and Experimental Evidence. J Neuroinflamm (2013) 10:142. doi: 10.1186/1742-2094-10-142
112. Kazumori H, Ishihara S, Rumi MA, Ortega-Cava CF, Kadowaki Y, Kinoshita Y. Transforming Growth Factor-Alpha Directly Augments Histidine Decarboxylase and Vesicular Monoamine Transporter 2 Production in Rat Enterochromaffin-Like Cells. Am J Physiol Gastrointest Liver Physiol (2004) 286(3):G508–14. doi: 10.1152/ajpgi.00269.2003
113. Guillot TS, Richardson JR, Wang MZ, Li YJ, Taylor TN, Ciliax BJ, et al. PACAP38 Increases Vesicular Monoamine Transporter 2 (VMAT2) Expression and Attenuates Methamphetamine Toxicity. Neuropeptides (2008) 42(4):423–34. doi: 10.1016/j.npep.2008.04.003
114. Stoecklin G, Lu M, Rattenbacher B, Moroni C. A Constitutive Decay Element Promotes Tumor Necrosis Factor Alpha mRNA Degradation via an AU-Rich Element-Independent Pathway. Mol Cell Biol (2003) 23(10):3506–15. doi: 10.1128/mcb.23.10.3506-3515.2003
115. Zhu CB, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA. Interleukin-1 Receptor Activation by Systemic Lipopolysaccharide Induces Behavioral Despair Linked to MAPK Regulation of CNS Serotonin Transporters. Neuropsychopharmacology (2010) 35(13):2510–20. doi: 10.1038/npp.2010.116
116. Moron JA, Zakharova I, Ferrer JV, Merrill GA, Hope B, Lafer EM, et al. Mitogen-Activated Protein Kinase Regulates Dopamine Transporter Surface Expression and Dopamine Transport Capacity. J Neurosci (2003) 23(24):8480–8. doi: 10.1523/JNEUROSCI.23-24-08480.2003
117. Blum K, Sheridan PJ, Wood RC, Braverman ER, Chen TJ, Cull JG, et al. The D2 Dopamine Receptor Gene as a Determinant of Reward Deficiency Syndrome. J R Soc Med (1996) 89(7):396–400. doi: 10.1177/014107689608900711
118. Tsutsui-Kimura I, Takiue H, Yoshida K, Xu M, Yano R, Ohta H, et al. Dysfunction of Ventrolateral Striatal Dopamine Receptor Type 2-Expressing Medium Spiny Neurons Impairs Instrumental Motivation. Nat Commun (2017) 8:14304. doi: 10.1038/ncomms14304
119. Belmonte L, Achamrah N, Nobis S, Guerin C, Riou G, Bole-Feysot C, et al. A Role for Intestinal TLR4-Driven Inflammatory Response During Activity-Based Anorexia. Sci Rep (2016) 6:35813. doi: 10.1038/srep35813
120. Hwang DH, Kim JA, Lee JY. Mechanisms for the Activation of Toll-Like Receptor 2/4 by Saturated Fatty Acids and Inhibition by Docosahexaenoic Acid. Eur J Pharmacol (2016) 785:24–35. doi: 10.1016/j.ejphar.2016.04.024
121. Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, et al. Evidence That TLR4 Is Not a Receptor for Saturated Fatty Acids But Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab (2018) 27(5):1096–1110 e5. doi: 10.1016/j.cmet.2018.03.014
122. Boden G. Obesity, Insulin Resistance and Free Fatty Acids. Curr Opin Endocrinol Diabetes Obes (2011) 18(2):139–43. doi: 10.1097/MED.0b013e3283444b09
123. Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of Plasma Glucose, Free Fatty Acid, Lactate, and Insulin for 24 H in Patients With NIDDM. Diabetes (1988) 37(8):1020–4. doi: 10.2337/diab.37.8.1020
124. Averill MM, Bornfeldt KE. Lipids Versus Glucose in Inflammation and the Pathogenesis of Macrovascular Disease in Diabetes. Curr Diabetes Rep (2009) 9(1):18–25. doi: 10.1007/s11892-009-0005-x
125. Kennedy A, Martinez K, Chuang CC, LaPoint K, McIntosh M. Saturated Fatty Acid-Mediated Inflammation and Insulin Resistance in Adipose Tissue: Mechanisms of Action and Implications. J Nutr (2009) 139(1):1–4. doi: 10.3945/jn.108.098269
126. Kalin S, Heppner FL, Bechmann I, Prinz M, Tschop MH, Yi CX. Hypothalamic Innate Immune Reaction in Obesity. Nat Rev Endocrinol (2015) 11(6):339–51. doi: 10.1038/nrendo.2015.48
127. Jais A, Bruning JC. Hypothalamic Inflammation in Obesity and Metabolic Disease. J Clin Invest (2017) 127(1):24–32. doi: 10.1172/JCI88878
128. Maric T, Woodside B, Luheshi GN. The Effects of Dietary Saturated Fat on Basal Hypothalamic Neuroinflammation in Rats. Brain Behav Immun (2014) 36:35–45. doi: 10.1016/j.bbi.2013.09.011
129. Lumeng CN, Saltiel AR. Inflammatory Links Between Obesity and Metabolic Disease. J Clin Invest (2011) 121(6):2111–7. doi: 10.1172/JCI57132
130. Mishra BB, Gundra UM, Teale JM. Toll-Like Receptors in CNS Parasitic Infections. Curr Top Microbiol Immunol (2009) 336:83–104. doi: 10.1007/978-3-642-00549-7_5
131. Lester SN, Li K. Toll-Like Receptors in Antiviral Innate Immunity. J Mol Biol (2014) 426(6):1246–64. doi: 10.1016/j.jmb.2013.11.024
132. Delint-Ramirez I, Willoughby D, Hammond GR, Ayling LJ, Cooper DM. Palmitoylation Targets AKAP79 Protein to Lipid Rafts and Promotes Its Regulation of Calcium-Sensitive Adenylyl Cyclase Type 8. J Biol Chem (2015) 290(21):13041. doi: 10.1074/jbc.A111.243899
133. Gupta S, Knight AG, Gupta S, Keller JN, Bruce-Keller AJ. Saturated Long-Chain Fatty Acids Activate Inflammatory Signaling in Astrocytes. J Neurochem (2012) 120(6):1060–71. doi: 10.1111/j.1471-4159.2012.07660.x
134. Wang S, Xiang N, Yang L, Zhu C, Zhu X, Wang L, et al. Linoleic Acid and Stearic Acid Elicit Opposite Effects on AgRP Expression and Secretion via TLR4-Dependent Signaling Pathways in Immortalized Hypothalamic N38 Cells. Biochem Biophys Res Commun (2016) 471(4):566–71. doi: 10.1016/j.bbrc.2016.02.031
135. Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, et al. Saturated Fatty Acids Produce an Inflammatory Response Predominantly Through the Activation of TLR4 Signaling in Hypothalamus: Implications for the Pathogenesis of Obesity. J Neurosci (2009) 29(2):359–70. doi: 10.1523/JNEUROSCI.2760-08.2009
136. Hryhorczuk C, Sheng Z, Decarie-Spain L, Giguere N, Ducrot C, Trudeau LE, et al. Oleic Acid in the Ventral Tegmental Area Inhibits Feeding, Food Reward, and Dopamine Tone. Neuropsychopharmacology (2018) 43(3):607–16. doi: 10.1038/npp.2017.203
Keywords: TLR4, inflammation, hypothalamus, dopamine system, SFAs
Citation: Li Y, Jiang Q and Wang L (2021) Appetite Regulation of TLR4-Induced Inflammatory Signaling. Front. Endocrinol. 12:777997. doi: 10.3389/fendo.2021.777997
Received: 16 September 2021; Accepted: 04 November 2021;
Published: 24 November 2021.
Edited by:
Claire Joanne Stocker, Aston University, United KingdomReviewed by:
Martin Valdearcos, University of California, San Francisco, United StatesPrasad Dalvi, Gannon University, United States
Copyright © 2021 Li, Jiang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lina Wang, d2FuZ2xpbmFAc2NhdS5lZHUuY24=; Qingyan Jiang, cXlqaWFuZ0BzY2F1LmVkdS5jbg==