
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 01 December 2021
Sec. Cancer Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.764512
This article is part of the Research Topic The Role of Genetic Alterations in Thyroid Cancer View all 6 articles
 Anna Reimer Hansen1
Anna Reimer Hansen1 Line Borgwardt1
Line Borgwardt1 Åse Krogh Rasmussen2
Åse Krogh Rasmussen2 Christian Godballe3,4
Christian Godballe3,4 Morten Møller Poulsen5Filipe G. Vieira1Jes Sloth Mathiesen3,4
Morten Møller Poulsen5Filipe G. Vieira1Jes Sloth Mathiesen3,4 Maria Rossing1,6*
Maria Rossing1,6*Activating variants in the receptor tyrosine kinase REarranged during Transfection (RET) cause multiple endocrine neoplasia type 2 (MEN 2), an autosomal dominantly inherited cancer-susceptibility syndrome. The variant c.166C>A, p.Leu56Met in RET was recently reported in two patients with medullary thyroid cancer (MTC). The presence of a pheochromocytoma in one of the patients, suggested a possible pathogenic role of the variant in MEN 2A. Here, we present clinical follow up of a Danish RET Leu56Met cohort. Patients were evaluated for signs of MEN 2 according to a set of predefined criteria. None of the seven patients in our cohort exhibited evidence of MEN 2. Furthermore, we found the Leu56Met variant in our in-house diagnostic cohort with an allele frequency of 0.59%, suggesting that it is a common variant in the population. Additionally, none of the patients who harbored the allele were listed in the Danish MTC and MEN 2 registries. In conclusion, our findings do not support a pathogenic role of the Leu56Met variant in MEN 2.
REarranged during Transfection (RET) is a receptor tyrosine kinase that plays essential roles in several intracellular pathways as well as processes such as embryonic development of the enteric nervous system. The RET protein, which is encoded by the RET gene on chromosome 10, is composed of three functional domains. These include an intracellular tyrosine kinase domain, a transmembrane domain and an extracellular ligand binding domain, containing cadherin-like domains and a cysteine-rich domain important for receptor dimerization (1). Germline loss-of-function variants in RET cause Hirschsprung’s disease (HSCR), a congenital intestinal malformation (2), while activating variants cause multiple endocrine neoplasia type 2 (MEN 2), an autosomal dominantly inherited cancer-susceptibility syndrome (3, 4). The MEN 2A subtype is characterized by medullary thyroid cancer (MTC) with pheochromocytoma, primary hyperparathyroidism (PHPT) and in some cases, cutaneous lichen amyloidosis and HSCR, while MEN2B is characterized by MTC, pheochromocytoma, gastrointestinal and/or mucosal neuromas and a marfanoid habitus (1, 5).
Several recurrent hotspot variants in RET have been identified in MEN 2 patients, and there appears to be some genotype-phenotype correlation, allowing for MTC risk stratification and possible early surgical intervention in affected individuals (6). However, for variants identified more sporadically, the causality may not be as clear, resulting in possible misclassification of the variants and subsequent inaccurate patient management (7–10).
The variant c.166C>A, p.Leu56Met in exon 2 of RET (NM_020975.5) was initially reported in a patient with HSCR (11) but was recently identified in two patients with MTC (12). The coinciding presence of a pheochromocytoma in one of the patients led the authors to suggest a possible pathogenic role of the Leu56Met variant in MEN 2A. In order to determine if the variant is associated with MEN 2, we present clinical follow up of a Danish RET Leu56Met cohort and the allele frequency of the variant from our in-house diagnostic cohort.
Our cohort consisted of patients who underwent genetic testing for MEN 2, MTC, PHPT or pheochromocytoma at the Center for Genomic Medicine, Copenhagen University Hospital, between 2014 and 2019. Carriers of RET Leu56Met were selected from the cohort and evaluated for symptoms of MEN 2 by reviewing patient files and by subsequent biochemical follow up at the Department of Endocrinology, Copenhagen University Hospital. An additional patient from Department of Endocrinology, Aarhus University Hospital was included and biochemical follow up was conducted. None of the Leu56Met carriers harbored additional variants in RET.
Evidence of MEN 2 in RET Leu56Met carriers was evaluated by the following criteria, as previously described (10) with additional criteria to include MEN2B: (i) the patient demonstrates more than one MEN 2 manifestation, including histologically verified MTC, histologically verified pheochromocytoma, histologically verified gastrointestinal or mucosal neuromas, histologically verified HSCR, biochemically verified PHPT, and clinically or histologically verified cutaneous lichen amyloidosis, or (ii) the patient has one MEN 2 manifestation and a relative with MTC and the RET Leu56Met variant.
Data was compiled from patients for whom RET sequencing was performed at our department until 2020, as part of a larger gene panel for hereditary endocrinological diseases, renal cancers and malignant melanoma. These patients constituted our in-house diagnostic cohort. Data from this cohort was bioinformatically processed to identify any carriers of the Leu56Met variant.
Genetic testing was conducted by next generation sequencing as previously described (13). This study was approved by the local Ethics Committee in the capital region of Denmark (H-4-2010-050) as well as the Danish Health Authority (3‐3013‐395/3) and the Danish Data Protection Agency (18/17801).
We identified seven unrelated RET Leu56Met carriers in our cohort (Table 1). None of the carriers exhibited more than one MEN 2A or MEN 2B manifestation or had any family history of MTC. Moreover, none of the patients were diagnosed with HSCR or cutaneous lichen amyloidosis. Consequently, the Leu56Met carriers from our cohort did not fulfill the predefined criteria and were not considered to show evidence of MEN 2.
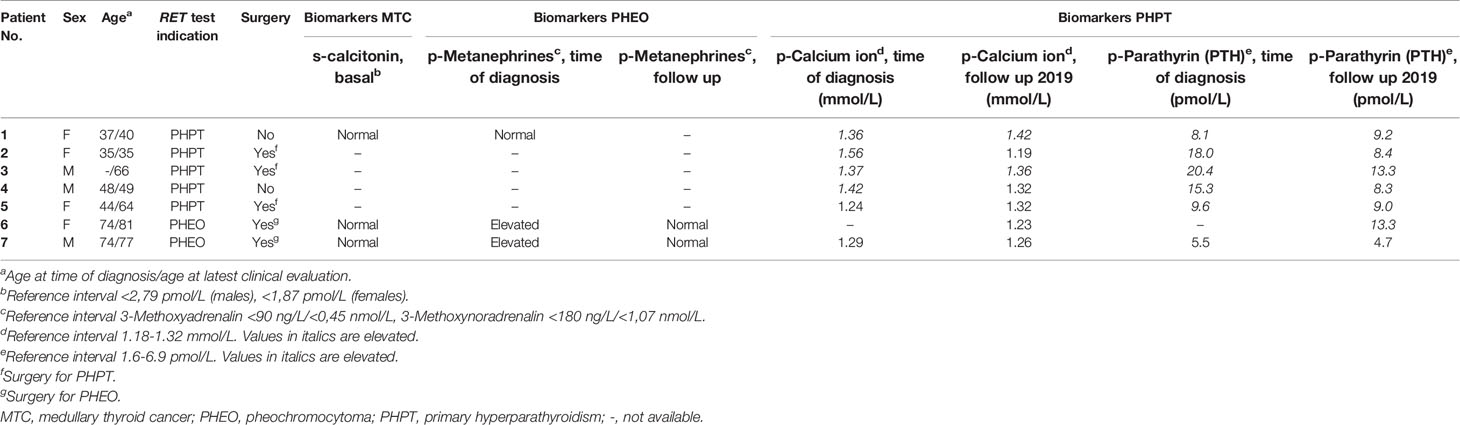
Table 1 RET Leu56Met carriers identified on suspicion of hereditary PHPT, MTC or PHEO.
We further screened our in-house cohort of diagnostic samples for the Leu56Met variant. In this cohort the variant was found with an allele frequency of 0.59% (36 of 6066 alleles). None of the patients who harbored the allele were found in the Danish MTC cohort (1960–2014) (14) or the Danish MEN 2 cohorts (1901–2021) (15, 16, Unpublished data).
In this study, we present a cohort of Danish carriers of the RET Leu56Met variant. None of the carriers display evidence of MEN 2 or MTC according to our predefined criteria. As some of the patients who only presented with PHPT were relatively young, it is possible that they could develop MTC later in life. However, a large study recently reported PHPT as a first manifestation of MEN 2A in less than 1% of index cases (17), rendering this possibility unlikely. Our analysis of the cohort is based on biomarkers to assess the presence of MTC. While non-secretory MTCs have been reported, these are exceedingly rare (18), and have never been identified as part of hereditary MTC in our MEN 2 cohort. Additionally, we find the Leu56Met variant with an allele frequency of 0.59% in our in-house diagnostic cohort. None of the patients who harbored the allele were listed in the Danish MTC and MEN 2 registries. Relatives of the Leu56Met carriers in our cohort were not examined for carrier status, however no family history of MEN 2 was reported. Considering these factors, the Leu56Met variant is most likely a benign variant. Thus, our findings dispute the previously suggested association between the Leu56Met variant and MEN 2A (12).
The RET Leu56Met variant was first described in a patient with short segment HSCR, but no MEN 2 manifestations (11). Subsequently, the Leu56Met variant has also been described in a fetus with bilateral renal agenesis (19), a patient with congenital abnormalities of the kidneys and urinary tract also carrying a frameshift variant in TBC1D1 (20), and in a family with periodontal disease also carrying a variant in IRF8 (21). Furthermore, the RET Leu56Met variant has previously been detected in control subjects. Johnston et al. found the Leu56Met variant in 6 of 571 individuals (1.1%) not selected for a personal or family history of cancer (22). Another large study of a healthy ancestrally diverse cohort identified Leu56Met with an allele frequency of 0.76% in the European subpopulation (23). The frequent findings of the variant in these studies indicate that Leu56Met is a common variant in the general population. Indeed, it has been reported in gnomAD with an allele frequency of 0.48% in the non-Finnish European population, including one individual homozygous for the variant1. In comparison, two of the known pathogenic variants causative of MEN 2, p.Cys634Tyr and p.Met918Thr (6) are both reported in gnomAD with allele frequencies below 0.001%. Even the possibly low penetrant pathogenic variant p.Val804Met (24) is reported with an allele frequency of 0.014% in gnomAD. The relatively high allele frequency of the Leu56Met variant in our in-house diagnostic cohort suggests that it is a common variant without clinical significance in the Danish population.
Leu56 is located in exon 2 of RET, while the pathogenic variants that give rise to MEN 2 are primarily reported in exons 5, 8, 10, 11, 13, 14, 15, and 16 (5). RET is comprised of three functional domains, and Leu56 is positioned within the extracellular cadherin-like domain, where loss-of-function variants were originally identified in HSCR patients (25). Mutation of this residue could potentially result in loss of function by misfolding of the protein and subsequent degradation in the endoplasmic reticulum, leading to a lack of secretion to the cell surface. However, RET protein containing the Leu56Met variant was shown to have a secretion efficiency similar to that of wildtype RET (26). Additionally, RET Leu56Met showed no significant difference in RET phosphorylation, or phosphorylation of downstream targets ERK and STAT3, compared to wildtype protein. In fact, the Leu56Met variant was applied as a negative or nonpathogenic control in these functional studies (27, 28). Furthermore, in silico analyses overall do not support a damaging effect of the amino acid substitution, although there is some discrepancy between the various tools (REVEL: 0.5, range from 0-1 where higher values predict pathogenicity; CADD: 8.7, range from 1-99 where higher values predict pathogenicity; BLOSUM62 score: 2, positive score indicates that the specific amino acid alteration is more likely to occur; Grantham distance: 15, range from 5-215 where higher values predict pathogenicity) (29–32).
A diagnosis of MEN 2 entails major individual implications with regards to treatment and follow-up, including the psychological aspects of a lifelong risk of developing MEN 2 related manifestations (5, 33, 34). Thus, it is important to correctly distinguish between pathogenic variants in RET and benign polymorphisms found in the population. Objectively, it is possible that Leu56Met represents a frequent pathogenic variant with very low penetrance, thus conveying a low lifetime risk of developing MTC, as recently reported for the pathogenic RET variant p.Val804Met (24). However, most reported pathogenic RET variants are highly penetrant and confer a moderate to high lifetime risk of MTC (6). The Leu56Met variant could also, although not inherently pathogenic, potentially act as a genetic modifier and increase the risk of other cancers. The male patient 1 described by Paragliola et al. was diagnosed with both MTC and pheochromocytoma, and thus MEN 2A cannot be ruled out. However, the diagnosis of pheochromocytoma in a patient without abnormal levels of metanephrines and catecholamines is highly unusual. Taken together, the results from our study and larger population studies (23, 35), suggests that the identification of the RET Leu56Met variant in the patient reported in Paragliola et al. most likely reflects the high allele frequency in the general population, rather than a pathogenic finding causative of MEN 2.
Based on results from our cohorts, we found no evidence to support a causative role of the RET Leu56Met variant in MEN 2. Consequently, we suggest that Leu56Met is most likely a benign variant.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Ethics Committee in the capital region of Denmark (H-4-2010-050), Danish Health Authority (3‐3013‐395/3), Danish Data Protection Agency (18/17801). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
AH drafted the manuscript, coordinated the study and carried out the data analysis. MR, LB and JM conceived the study, facilitated the collaboration and participated in the drafting of the manuscript. FV carried out the bioinfomatic processing and participated in drafting the manuscript. ÅR and MP participated in data collection and drafting of the manuscript. CG participated in review of patient cohorts and drafting of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mathiesen JS, Effraimidis G, Rossing M, Rasmussen ÅK, Hoejberg L, Bastholt L, et al. Multiple Endocrine Neoplasia Type 2: A Reveiw. Semin Cancer Biol (2021), S1044–579X(21)00085-7. doi: 10.1016/j.semcancer.2021.03.035
2. Pasini B, Borrello MG, Greco A, Bongarzone I, Luo Y, Mondellini P, et al. Loss of Function Effect of RET Mutations Causing Hirschsprung Disease. Nat Genet (1995) 10(1):35–40. doi: 10.1038/ng0595-35
3. Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, et al. Germ-Line Mutations of the RET Proto-Oncogene in Multiple Endocrine Neoplasia Type 2A. Nature (1993) 363(6428):458–60. doi: 10.1038/363458a0
4. Santoro M, Carlomagno F, Romano A, Bottaro DP, Dathan NA, Grieco M, et al. Activation of RET as a Dominant Transforming Gene by Germline Mutations of MEN2A and MEN2B. Science (1995) 267(5196):381–3. doi: 10.1126/science.7824936
5. Elisei R, Alevizaki M, Conte-Devolx B, Frank-Raue K, Leite V, Williams GR. European Thyroid Association Guidelines for Genetic Testing and Its Clinical Consequences in Medullary Thyroid Cancer. Eur Thyroid J (2013) 1(4):216–31. doi: 10.1159/000346174
6. Wells SA Jr, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association Guidelines for the Management of Medullary Thyroid Carcinoma. Thyroid (2015) 25(6):567–610. doi: 10.1089/thy.2014.0335
7. Fitze G, Schierz M, Bredow J, Saeger HD, Roesner D, Schackert HK. Various Penetrance of Familial Medullary Thyroid Carcinoma in Patients With RET Protooncogene Codon 790/791 Germline Mutations. Ann Surg (2002) 236(5):570–5. doi: 10.1097/00000658-200211000-00006
8. Machens A, Spitschak A, Lorenz K, Pützer BM, Dralle H. Germline RET Sequence Variation I852M and Occult Medullary Thyroid Cancer: Harmless Polymorphism or Causative Mutation? Clin Endocrinol (Oxf) (2011) 75(6):801–5. doi: 10.1111/j.1365-2265.2011.04158.x
9. Mathiesen JS, van Overeem Hansen T, Rasmussen ÅK, Hjortshøj TD, Kiss K, Larsen SR, et al. Novel Somatic RET Mutation Questioning the Causality of the RET I852M Germline Sequence Variant in Multiple Endocrine Neoplasia 2a. Thyroid (2017) 27(8):1103–4. doi: 10.1089/thy.2017.0131
10. Høxbroe Michaelsen S, Ornstrup MJ, Poulsen MM, Bennedbaek FN, Gaustadnes M, Rossing M, et al. Long-Term Follow-Up of RET Y791F Carriers in Denmark 1994-2017: A National Cohort Study. J Surg Oncol (2019) 119(6):687–93. doi: 10.1002/jso.25371
11. Hofstra RM, Wu Y, Stulp RP, Elfferich P, Osinga J, Maas SM, et al. RET and GDNF Gene Scanning in Hirschsprung Patients Using Two Dual Denaturing Gel Systems. Hum Mutat (2000) 15(5):418–29. doi: 10.1002/(SICI)1098-1004(200005)15:5<418::AID-HUMU3>3.0.CO;2-2
12. Paragliola RM, Lovicu RM, Papi G, Capoluongo E, Minucci A, Canu G, et al. Medullary Thyroid Carcinoma With Exon 2 P.L56M RET Variant: Clinical Particular Features in Two Patients. Front Endocrinol (Lausanne) (2018) 9:398. doi: 10.3389/fendo.2018.00398
13. Bennedbæk M, Rossing M, Rasmussen ÅK, Gerdes AM, Skytte AB, Jensen UB, et al. Identification of Eight Novel SDHB, SDHC, SDHD Germline Variants in Danish Pheochromocytoma/Paraganglioma Patients. Hered Cancer Clin Pract (2016) 14:13. doi: 10.1186/s13053-016-0053-6
14. Mathiesen JS, Kroustrup JP, Vestergaard P, Stochholm K, Poulsen PL, Rasmussen ÅK, et al. Incidence and Prevalence of Sporadic and Hereditary MTC in Denmark 1960-2014: A Nationwide Study. Endocr Connect (2018) 7(6):829–39. doi: 10.1530/EC-18-0157
15. Mathiesen JS, Kroustrup JP, Vestergaard P, Stochholm K, Poulsen PL, Rasmussen ÅK, et al. Incidence and Prevalence of Multiple Endocrine Neoplasia 2A in Denmark 1901-2014: A Nationwide Study. Clin Epidemiol (2018) 10:1479–87. doi: 10.2147/CLEP.S174606
16. Mathiesen JS, Kroustrup JP, Vestergaard P, Madsen M, Stochholm K, Poulsen PL, et al. Incidence and Prevalence of Multiple Endocrine Neoplasia 2B in Denmark: A Nationwide Study. Endocr Relat Cancer (2017) 24(7):L39–42. doi: 10.1530/ERC-17-0122
17. Larsen LV, Mirebeau-Prunier D, Imai T, Alvarez-Escola C, Hasse-Lazar K, Censi S, et al. Primary Hyperparathyroidism as First Manifestation in Multiple Endocrine Neoplasia Type 2A: An International Multicenter Study. Endocr Connect (2020) 9(6):489–97. doi: 10.1530/EC-20-0163
18. Frank-Raue K, Machens A, Leidig-Bruckner G, Rondot S, Haag C, Schulze E, et al. Prevalence and Clinical Spectrum of Nonsecretory Medullary Thyroid Carcinoma in a Series of 839 Patients With Sporadic Medullary Thyroid Carcinoma. Thyroid (2013) 23(3):294–300. doi: 10.1089/thy.2012.0236
19. Jeanpierre C, Macé G, Parisot M, Morinière V, Pawtowsky A, Benabou M, et al. RET and GDNF Mutations Are Rare in Fetuses With Renal Agenesis or Other Severe Kidney Development Defects. J Med Genet (2011) 48(7):497–504. doi: 10.1136/jmg.2010.088526
20. Kosfeld A, Kreuzer M, Daniel C, Brand F, Schäfer AK, Chadt A, et al. Whole-Exome Sequencing Identifies Mutations of TBC1D1 Encoding a Rab-GTPase-Activating Protein in Patients With Congenital Anomalies of the Kidneys and Urinary Tract (CAKUT). Hum Genet (2016) 135(1):69–87. doi: 10.1007/s00439-015-1610-1
21. Thumbigere-Math V, Foster BL, Bachu M, Yoshii H, Brooks SR, Coulter A, et al. Inactivating Mutation in IRF8 Promotes Osteoclast Transcriptional Programs and Increases Susceptibility to Tooth Root Resorption. J Bone Miner Res (2019) 34(6):1155–68. doi: 10.1002/jbmr.3690
22. Johnston JJ, Rubinstein WS, Facio FM, Ng D, Singh LN, Teer JK, et al. Secondary Variants in Individuals Undergoing Exome Sequencing: Screening of 572 Individuals Identifies High-Penetrance Mutations in Cancer-Susceptibility Genes. Am J Hum Genet (2012) 91(1):97–108. doi: 10.1016/j.ajhg.2012.05.021
23. Bodian DL, McCutcheon JN, Kothiyal P, Huddleston KC, Iyer RK, Vockley JG, et al. Germline Variation in Cancer-Susceptibility Genes in a Healthy, Ancestrally Diverse Cohort: Implications for Individual Genome Sequencing. PloS One (2014) 9(4):e94554. doi: 10.1371/journal.pone.0094554
24. Loveday C, Josephs K, Chubb D, Gunning A, Izatt L, Tischkowitz M, et al. P.Val804Met, the Most Frequent Pathogenic Mutation in RET, Confers a Very Low Lifetime Risk of Medullary Thyroid Cancer. J Clin Endocrinol Metab (2018) 103(11):4275–82. doi: 10.1210/jc.2017-02529
25. Edery P, Lyonnet S, Mulligan LM, Pelet A, Dow E, Abel L, et al. Mutations of the RET Proto-Oncogene in Hirschsprung's Disease. Nature (1994) 367(6461):378–80. doi: 10.1038/367378a0
26. Kjaer S, Hanrahan S, Totty N, McDonald NQ. Mammal-Restricted Elements Predispose Human RET to Folding Impairment by HSCR Mutations. Nat Struct Mol Biol (2010) 17(6):726–31. doi: 10.1038/nsmb.1808
27. Widowati T, Melhem S, Patria SY, de Graaf BM, Sinke RJ, Viel M, et al. RET and EDNRB Mutation Screening in Patients With Hirschsprung Disease: Functional Studies and Its Implications for Genetic Counseling. Eur J Hum Genet (2016) 24(6):823–9. doi: 10.1038/ejhg.2015.214
28. Wang H, Li Q, Zhang Z, Xiao P, Li L, Jiang Q. Functional Studies on Novel RET Mutations and Their Implications for Genetic Counseling for Hirschsprung Disease. Front Genet (2019) 10:924. doi: 10.3389/fgene.2019.00924
29. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet (2016) 99(4):877–85. doi: 10.1016/j.ajhg.2016.08.016
30. Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat Genet (2014) 46(3):310–5. doi: 10.1038/ng.2892
31. Henikoff S, Henikoff JG. Amino Acid Substitution Matrices From Protein Blocks. Proc Natl Acad Sci USA (1992) 89(22):10915–9. doi: 10.1073/pnas.89.22.10915
32. Grantham R. Amino Acid Difference Formula to Help Explain Protein Evolution. Science (New York NY) (1974) 185(4154):862–4. doi: 10.1126/science.185.4154.862
33. Mongelli MN, Peipert BJ, Goswami S, Helenowski I, Yount SE, Sturgeon C. Quality of Life in Multiple Endocrine Neoplasia Type 2A Compared With Normative and Disease Populations. Surgery (2018) 164(3):546–52. doi: 10.1016/j.surg.2018.04.036
34. Rodrigues KC, Toledo RA, Coutinho FL, Nunes AB, Maciel RMB, Hoff AO, et al. Assessment of Depression, Anxiety, Quality of Life, and Coping in Long-Standing Multiple Endocrine Neoplasia Type 2 Patients. Thyroid (2017) 27(5):693–706. doi: 10.1089/thy.2016.0148
Keywords: multiple endocrine neoplasia type 2, medullary thyroid cancer, RET, Leu56Met, Genetics
Citation: Hansen AR, Borgwardt L, Rasmussen ÅK, Godballe C, Poulsen MM, Vieira FG, Mathiesen JS and Rossing M (2021) Germline RET Leu56Met Variant Is Likely Not Causative of Multiple Endocrine Neoplasia Type 2. Front. Endocrinol. 12:764512. doi: 10.3389/fendo.2021.764512
Received: 25 August 2021; Accepted: 02 November 2021;
Published: 01 December 2021.
Edited by:
Joachim Feldkamp, University Hospital OWL of Bielefeld University, GermanyReviewed by:
Friedhelm Raue, Independent Researcher, Heidelberg, GermanyCopyright © 2021 Hansen, Borgwardt, Rasmussen, Godballe, Poulsen, Vieira, Mathiesen and Rossing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Rossing, Y2Fyb2xpbmUubWFyaWEucm9zc2luZ0ByZWdpb25oLmRr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.