
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 05 November 2021
Sec. Reproduction
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.763863
This article is part of the Research Topic Disruptors on Male Reproduction - Emerging Risk Factors View all 19 articles
 Eva Zatecka1†
Eva Zatecka1† Romana Bohuslavova2†
Romana Bohuslavova2† Eliska Valaskova1Hasmik Margaryan1
Eliska Valaskova1Hasmik Margaryan1 Fatima Elzeinova1Alena Kubatova1Simona Hylmarova2,3
Fatima Elzeinova1Alena Kubatova1Simona Hylmarova2,3 Jana Peknicova1‡
Jana Peknicova1‡ Gabriela Pavlinkova2*‡
Gabriela Pavlinkova2*‡Diabetes is a chronic metabolic disorder characterized by hyperglycemia and associated with many health complications due to the long-term damage and dysfunction of various organs. A consequential complication of diabetes in men is reproductive dysfunction, reduced fertility, and poor reproductive outcomes. However, the molecular mechanisms responsible for diabetic environment-induced sperm damage and overall decreased reproductive outcomes are not fully established. We evaluated the effects of type 2 diabetes exposure on the reproductive system and the reproductive outcomes of males and their male offspring, using a mouse model. We demonstrate that paternal exposure to type 2 diabetes mediates intergenerational and transgenerational effects on the reproductive health of the offspring, especially on sperm quality, and on metabolic characteristics. Given the transgenerational impairment of reproductive and metabolic parameters through two generations, these changes likely take the form of inherited epigenetic marks through the germline. Our results emphasize the importance of improving metabolic health not only in women of reproductive age, but also in potential fathers, in order to reduce the negative impacts of diabetes on subsequent generations.
Type 2 diabetes mellitus (T2D) combines insulin resistance and insulin secretion deficiency with the main characteristics of carbohydrate, protein, and lipid metabolism disorders (1, 2). The progression of the disease is clinically manifested by increasing weight gain, impaired glucose tolerance, and high plasma insulin, triglycerides, and fasting glucose. The worsening glycemic control causes the development of long-term complications, including diabetic retinopathy, nephropathy, peripheral neuropathy, autonomic neuropathy, and increases risk of cardiovascular disease (2). T2D is also an established risk factor for sexual dysfunction in both men and women (3). In men, diabetes is associated with erectile dysfunction, androgen deficiency, and disruption of the hypothalamic-pituitary-gonadal axis (4–6). Additionally, clinical data from IVF clinics show that type 1 and type 2 diabetic male patients have lower in vitro fertilization success rates, suggesting that diabetes-exposed sperm are damaged, including increased sperm nuclear DNA fragmentation, altered sperm morphology, and reduced sperm motility (7–10). With the increasing prevalence and incidence of T2D, more men in their reproductive years will be affected, contributing to the increasing prevalence of subfertility. However, the molecular mechanisms responsible for diabetic environment-induced sperm damage and overall decreased reproductive outcomes are not fully established.
In addition, parental exposure to the diabetic environment represents a higher risk of adverse effects for the offspring, influencing effects on their cardiovascular and metabolic health, and increased susceptibility to metabolic and cardiovascular disorders later in life (11–18). The epidemiological evidence is supported by experimental animal studies that demonstrate the transmission of diabetic phenotypes to the offspring (19–24). The effects of maternal exposure to diabetes combine the direct effects of in utero exposure, genetic effects (nuclear and mitochondrial DNA), and epigenetic modifications of germ cells. Metabolic phenotypes can also be transmitted via the paternal lineage. Paternal effects that result from environmental exposures represent predominantly epigenetic modifications in sperm, although particular environmental factors can affect the composition of seminal fluid, which can produce placental and developmental effects (25). For example, high fat diet exposure of male rats reprograms ß cells in offspring (26), high-fat diet-induced paternal obesity modulates the sperm microRNA content and DNA methylation status (27), and paternal T2D alters DNA methylation patterns in sperm, involving changes in methylation of insulin signaling genes (28).
Although the molecular mechanisms associated with the transmission of the diabetic phenotype to the offspring have been investigated, the impact of paternal T2D exposure on the reproductive outcomes of subsequent generations remains unclear. Previously, we showed that paternal type I diabetes induced transgenerational changes in the testes, and increased sperm damage in the offspring across two generations (29). Here, we describe T2D exposure effects on the reproductive system and the reproductive outcome of fathers and their offspring, using a mouse model. This is the first complex analysis of the metabolic and reproductive system phenotypes of TD2 fathers and their descendants, demonstrating how paternal exposure to TD2 mediates intergenerational and transgenerational effects on the reproductive health of the offspring.
This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). The experimental protocol was approved by the Animal Care and Use Committee of the Institute of Molecular Genetics, CAS and carried out in accordance with the relevant guidelines and regulations. We used a T2D mouse model generated by a combination of high-fat diet and a low dose of streptozotocin (28, 30). Male inbred C57BL/6J mice (purchased from Charles River, Germany), aged 6 weeks, were divided into two groups and were fed either a high-fat diet (HFD: 60% of the metabolizable energy coming from fat, 20% from carbohydrates, and 20% from protein; ssniff® EF acc. D12492 from Ssniff Spezialdiäten GmbH, Germany) or a control standard chow diet (11% of the metabolizable energy coming from fat, 65% from carbohydrates, and 20% from protein, #1320 diet, Altromin, Germany). After 9 weeks of HFD, intraperitoneal injection of 100 mg/kg body weight of streptozotocin (STZ; Sigma) was applied. Body weight, and blood samples were taken after 6-h fasting at indicated time points during the experiment (Experimental schematics in Figure 1). Blood glucose levels were measured in animals by a glucometer (COUNTOUR TS, Bayer, Switzerland); blood glucose levels maintained above 13.9 mmol/L are classified as diabetic. Mice were kept under standard experimental conditions with constant temperature (23–24°C). Glucose tolerance tests (GTT) were performed in 6h-fasted males. Mice were injected intraperitoneally with glucose (2 g/kg). Blood glucose was measured at 0, 15, 30, 60, and 120 min after glucose administration. After 12 weeks in the experiment, T2D males were glucose intolerant with blood glucose levels maintained above 13.9 mmol/L.
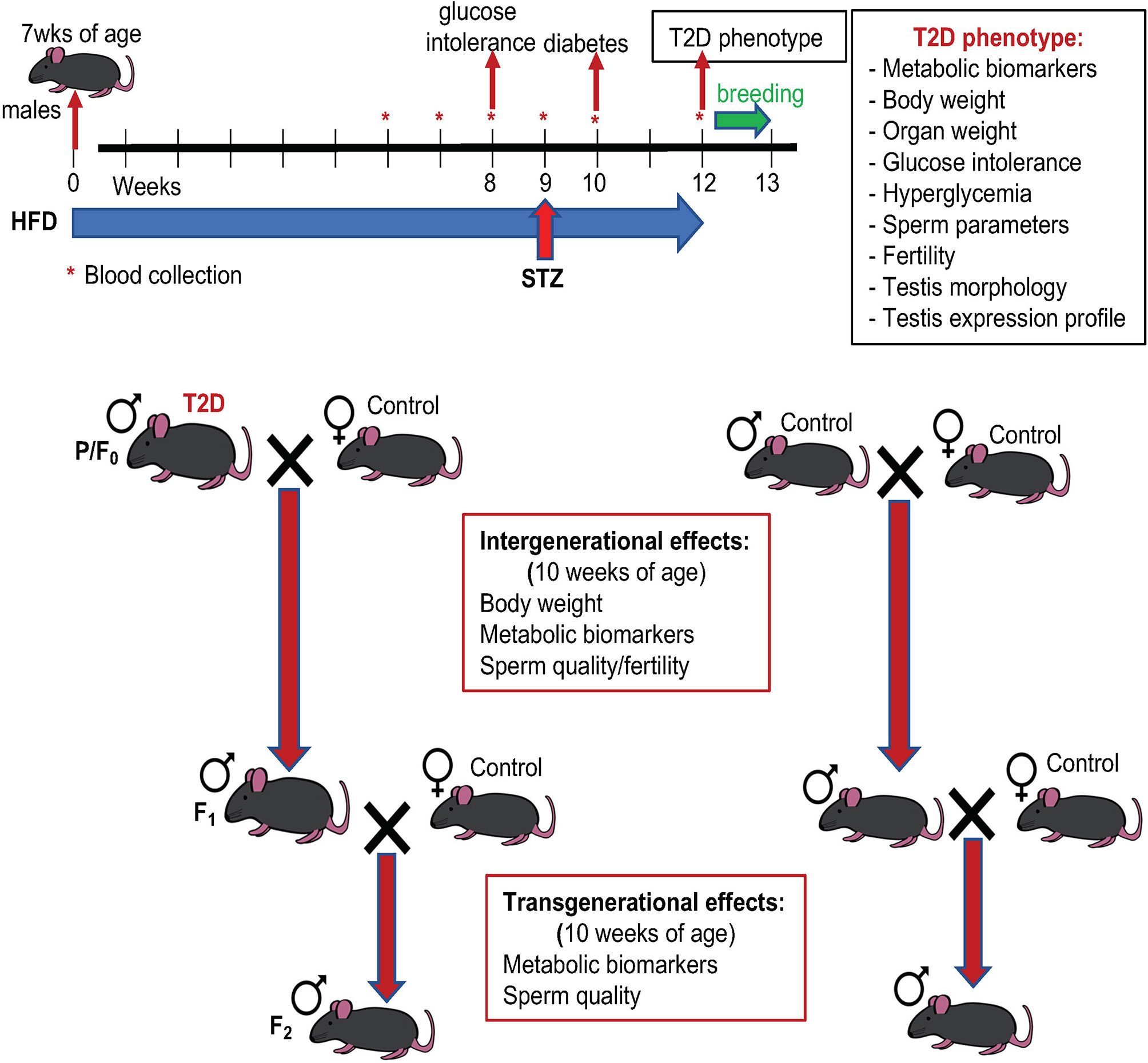
Figure 1 Experimental schematics. Type 2 diabetes (T2D) was induced by a combination of high fat diet (HFD) and a low dose of streptozotocin (STZ): sexually matured males (7 weeks old) were kept on HFD for 12 weeks before mating and injected with STZ at week 9 of the HFD experiment. T2D males and standard diet (control) fed males (parental generation, P/F0) were mated with standard diet fed females. After one week mating pairs were separated, males were killed, and tissues were collected for further analyses. The F1 male offspring were mated at 9 weeks of age, after one week mating pairs were separated. F1 males were killed, and analyses were performed at 10 weeks of age. Effects of diabetic paternal environment was also evaluated in the second (F2) male offspring generation at 10 weeks of age. Both F1 and F2 males were maintained on standard diet.
At 18 weeks of age, male founders (P generation) were mated with females fed with the standard chow diet. Individual males were placed in a cage with 1 healthy adult female. The animals were kept together for a week with minimalized disruptions and therefore, a vaginal plug was not checked in female mice. During this week all mice were fed with a standard chow diet. Immediately after separation, the males were killed, tissues and blood were collected for analyses. The females were housed individually during the gestation period and the litter size was recorded. Only male pups, the F1 offspring, were left in the litters to be used for following up transgenerational studies. At 9 weeks of age, randomly selected F1 males were mated with females to produce the F2 offspring. The offspring were fed only by the regular control diet. To minimize any additional environmental influence, molecular and cellular analyses were performed in both F1 and F2 male offspring at 10 weeks of age.
Blood serum was collected from non-diabetic and T2D males following a 6-h fast and was analyzed using a fully automated chemistry analyzer, Beckman Coulter AU480 (Beckman) according to the manufacturer’s protocol in the Core Facility for Phenogenomics, Biocev. System Beckman Coulter AU480 reagents used for the quantification: glucose (OSR6621), AST (OSR6009), ALT (OSR6007), TG (OSR60118), insulin (33410), PAI-1 (plasminogen kit 20009000), and LDL (ODC0024).
Mouse sperm cells were obtained from the left and right cauda epididymis. Spermatozoa from both caudae were left to release into warm (37 °C) PBS in a CO2 incubator for 15 min. The resulting mixture was poured through a 30 μm filter (Partec) to obtain only the sperm fraction and PBS was added to 1 mL of final volume (31). Sperm were counted in a Bürker chamber under the light microscope, and sperm concentration, sperm morphology, and sperm head separation were determined as described previously (32). Sperm morphology was evaluated in 200 cells per each animal. Sperm viability was analyzed with SYBR14 from Live/Dead sperm viability kit (Molecular Probes, USA) and by flow cytometry (LSRII, blue laser 488 nm, Becton Dickinson, USA), minimum 15,000 events were evaluated. An annexin V–FITC apoptosis kit (Sigma) and chromomycin A3 staining (CMA3, Sigma) were used to assess the sperm damage (29). The molecular and functional quality of sperm was further evaluated by the expression of two molecular biomarkers, sperm acrosomal transitional endoplasmic reticulum ATPase (TER ATPase, TERA) and glyceraldehyde-3-phosphate dehydrogenase-S (GAPDS), using monoclonal antibodies Hs-14 (33) and Hs-8 (34), respectively (EXBIO Ltd., Czechia). These antibodies are used for pathological human sperm and reproductive diagnostics, and evaluation of dysfunctional spermatogenesis (33, 35–37). A minimum of 200 spermatozoa per each animal was examined with a Nikon Eclipse E400 fluorescent microscope equipped with a Nikon Plan Apo VC 60/1.40 oil objective (Nikon Corporation Instruments Company) and photographed with a ProgRes MF CCD camera (Jenoptik, Germany) with the aid of NIS-ELEMENTS imaging software (Laboratory Imaging, Ltd.). Protamine 1 and 2 ratio was obtained from 4 x 10⁶ sperm cells per each sample, as described previously (29).
Dissected testes were fixed with 4% paraformaldehyde in PBS (pH 7.4) at 4°C overnight, and embedded in paraffin. Paraffin-embedded tissues were cut into 8 µm sections, de-paraffinized and rehydrated sections were stained with hematoxylin & eosin or used for immunohistochemistry. Citrate buffer (pH 6.0) was used as a heat-induced antigen retriever. Evaluation of meiotic cells and the 12 stages of production of spermatozoa in the mouse seminiferous epithelium was done using immunolabeling of the axial element of the synaptonemal complex (SYCP3, mouse anti-SCP3 #ab97672, 1:1000 dilution, Abcam). Rabbit anti-CX43 (#C6219, 1:2000 dilution, Sigma) was used to evaluate gap junctions. The secondary antibodies Alexa Fluor® 488 AffiniPure Goat Anti-Mouse IgG (Jackson ImmunoResearch Laboratories 115-545-146), and Alexa Fluor® 594 AffiniPure Goat Anti-Rabbit IgG (Jackson ImmunoResearch Laboratories 111-585-144) were used in a 1:400 dilution. Samples were incubated with primary antibodies at 4°C for 24 hours. After several PBS washes, secondary antibodies were added and incubated at 4°C for 24 hours. The sections were counterstained with Hoechst 33342 (#14533 Sigma) and imaged with a confocal microscope (ZEISS LSM 880 NLO) or with a fluorescence microscope (Nikon Eclipse E400). A relative quantification of Cx43 staining in Leydig cells was done using ImageJ software. Morphometric evaluations of seminiferous tubule diameter and thickness of the seminiferous epithelium were performed. The diameter of a seminiferous tubule was defined as the shortest distance between two parallel tangent lines of the outer edge of the tubule. Paraffin sections (8 μm) were stained with Periodic acid–Schiff to visualize advanced glycation end products (Periodic acid–Schiff (PAS) kit; 395B-1KT, Sigma).
RT-qPCR was performed as previously described (29). Briefly, cDNA was prepared using 2 μg of total RNA isolated from the testes. We used RevertAid Reverse Transcriptase (Thermo Fisher Scientific), 1 μL Oligo(dT) and random hexamer primers (Thermo Fisher Scientific) in reverse transcription (RT). Quantitative real-time PCR (qPCR) was performed with a final concentration of cDNA 10 ng/μL using a CFX 384 – qPCR cycler (BioRad). The relative expression of a target gene was calculated, based on the quantification cycle (Cq) difference (Δ) of an experimental sample versus control. The control was set at 100% and experimental samples were compared to the control (29). The β-actin (Actb) and Peptidylprolyl isomerase A (Ppia) genes were used as the reference genes. Primer sequences are listed in Supplementary Table S1.
The comparisons between diabetic and control mice were done using STATISTICA 7.0. (Statsoft, Czech Republic) and GraphPad Prism 7.0 (GraphPad Software, Inc., USA). Differences in organ weights were tested by ANCOVA with body weight as covariate. Differences in sperm parameters, testis morphology, and gene expression between control and T2D groups were assessed by t-test. One-way ANOVA, follow by Tukey’s post hoc tests for multiple comparisons was used to assess differences among offspring generations. Repeated measure-based parameters (such as weight gain over time, GTT, or blood glucose levels) were analyzed using two-way ANOVA for repeated measures. P value < 0.05 was considered to be statistically significant. Sample sizes and individual statistical results for all analyses are provided in the figure legends and tables.
We used a non-genetic type 2 diabetes (T2D) mouse model that manifests the metabolic abnormalities associated with human prediabetes and type 2 diabetes (28, 30). This model involves a combination of a high fat diet (HFD) to induce insulin resistance and hyperinsulinemia, and a low dose STZ, which reduces β-cell mass but does not cause diabetes in control-fed mice, to bring about glucose intolerance and hyperglycemia (28, 38) (Figure 1 Experimental schematics). Together these two stressors have been designed to mimic the pathology and the multi-genetic/environmental background of T2D in humans (39–41). We continuously monitored the metabolic changes in our T2D model. As expected, after 8 weeks of HFD, all male C57BL/6J male mice in the experiment had significantly higher fasting plasma insulin levels and were glucose intolerant, as shown by glucose tolerance tests, indicating compromised insulin response (Figure 2A). Body weight of the T2D mice increased progressively over time compared to control mice maintained on a normal standard chow diet (Figure 2B). After STZ treatment, mice became hyperglycemic with blood glucose levels maintained above 13.9 mmol/l, classified as diabetic (Figure 2C). Fasting plasma levels of glucose and insulin were significantly higher in the T2D group (Figure 3A). Correspondingly to the diabetic phenotype, after STZ treatment, plasma glucose levels were increased. Plasma insulin levels, which were initially increased in response to the fat-enriched diet in T2D mice, decreased after STZ-induced β-cell reduction but remained at higher levels than insulin levels in control mice. After 12 weeks, compared to control males, T2D mice displayed significant changes in serum metabolic biomarkers, including increased serum glucose, triglycerides, low-density lipoprotein (LDL), plasminogen activator inhibitor 1 (PAI-1, a marker of insulin resistance), metabolic liver enzymes, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (Figure 3B). Metabolic differences were also manifested by increased body weight, and liver weight of T2D males (Figure 3C). These physiological and metabolic changes confirmed decreased insulin sensitivity and impaired glucose tolerance associated with the development of type 2 diabetes mellitus. To analyze the effects of the T2D environment on the reproductive system and reproductive outcomes, we mated these T2D males or control males to female mice maintained on a control chow diet through the course of the experiment (Figure 1). After 1 week of mating, males were removed, limiting their influence on their progeny to the mating itself.
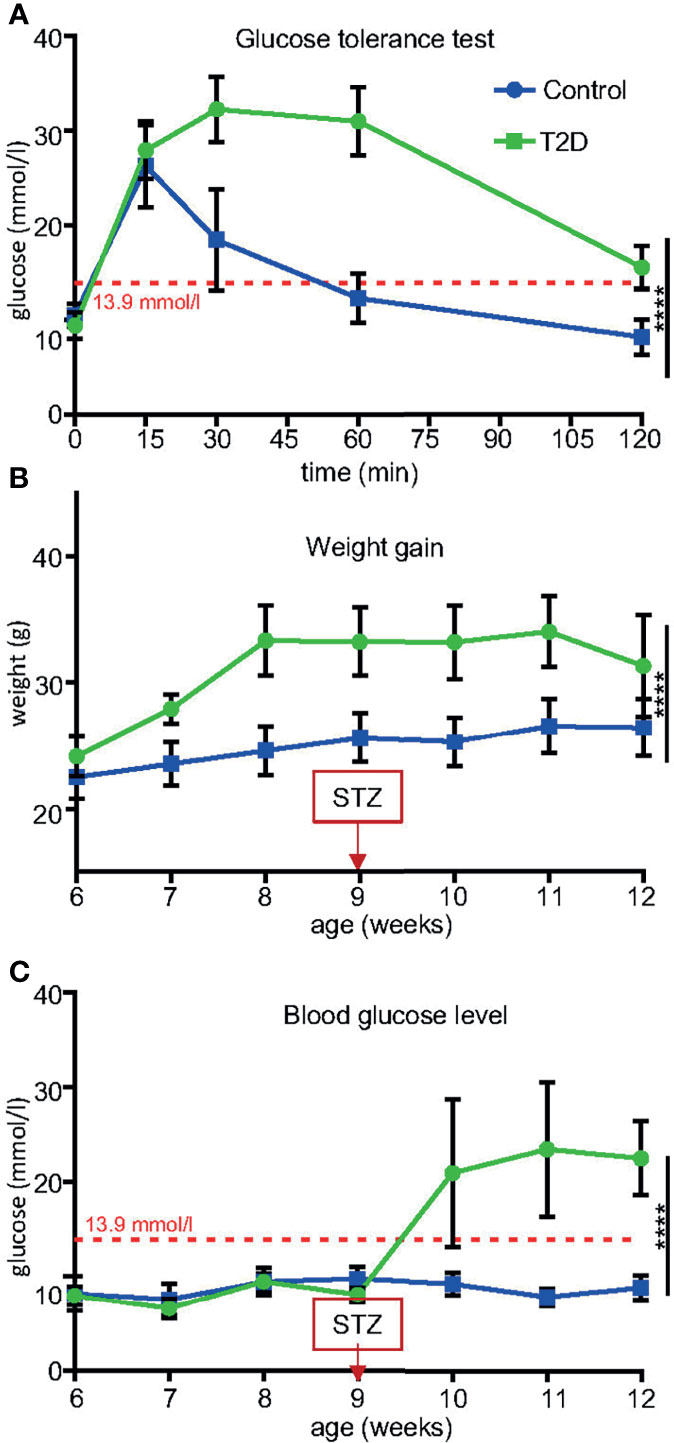
Figure 2 Temporal changes induced in T2D males before mating. (A) After 8 weeks of HFD, all males were glucose intolerant compared to control males fed by standard diet. (B) Body weight trajectories in T2D males and control males. Note decreased weight gain starting week 11 due to worsening diabetes (hyperglycemia). (C) Blood glucose levels significantly increased after a low dose of STZ treatment in combination with HFD, as measured using a glucometer in blood collected from the tail vein after 6-hr fast. Blood glucose levels maintained above 13.9 mmol/l are classified as diabetic (red broken line). Data are presented as the mean ± SD (n = 10 Control and 8 T2D mice). Differences between groups were tested by Two-Way ANOVA for repeated measures, showing significant interactions between time x treatment as repeated measures. ****P < 0.0001.
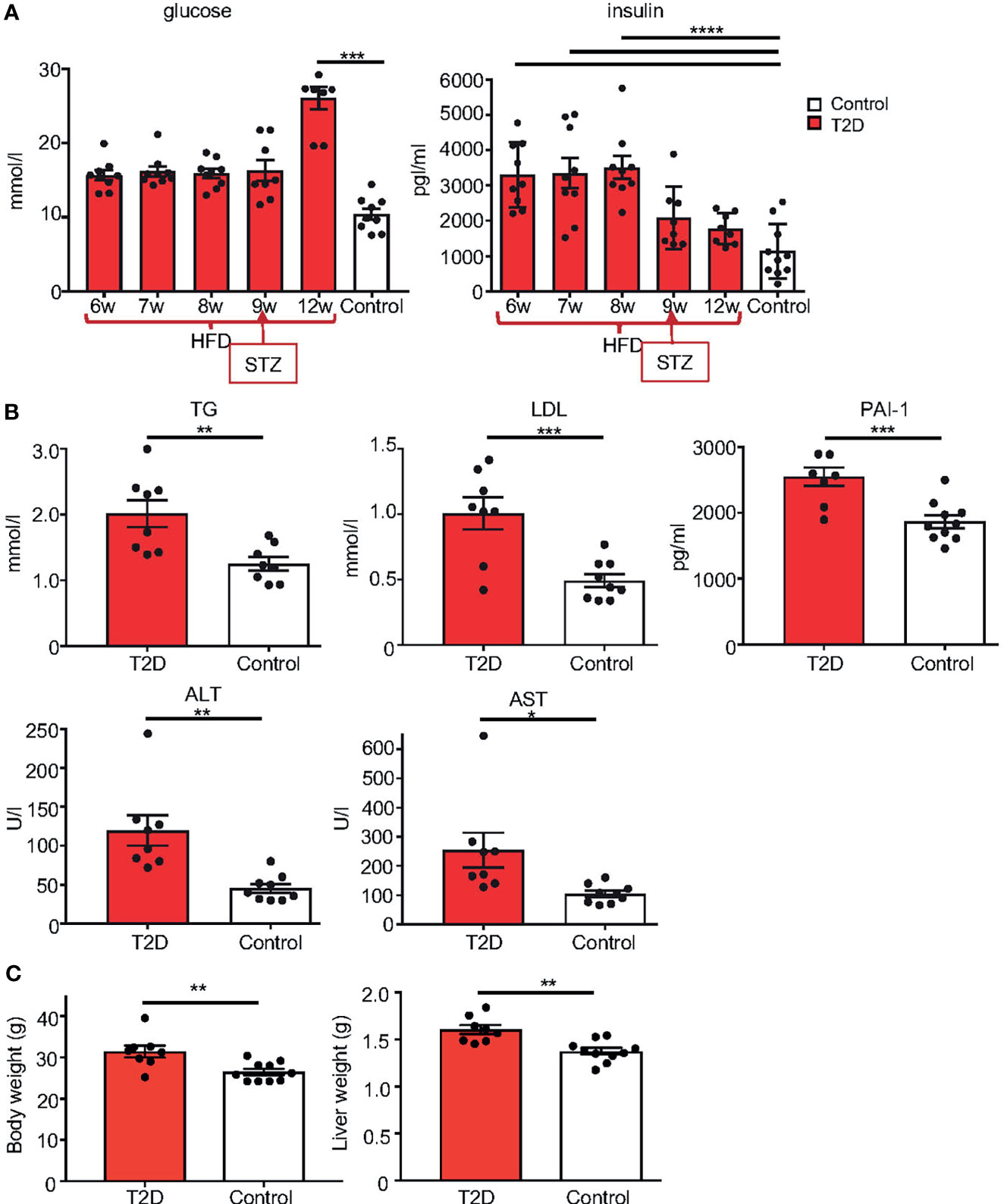
Figure 3 Metabolic phenotype of T2D mouse model. (A) Temporal changes in fasting plasma levels of glucose and insulin in T2D males compare to the average level of male controls. Differences were tested by one-way ANOVA with Tukey’s multiple comparisons test. (B) Fasting plasma levels of TG, triglycerides; LDL, low-density lipoprotein; marker of insulin resistance, PAI-1, plasminogen activator inhibitor 1; ALT, alanine aminotransferase; and AST, aspartate aminotransferase, in T2D males compared to controls at 12 weeks of age. (C) Body and liver weight. Data are presented as the mean ± SEM. Differences between groups were tested by t-test (GraphPad Prism 7.0). Differences in liver weight between groups were tested by ANCOVA with body weight as covariate (Ancova – STATISTICA 7.0). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
All diabetic males, with the exception of one mouse, were able to mate in the period of one week. The rate of pregnancy was in the limits of the rate of pregnancy in our C57BL/6J colony (typically 70–80%, unpublished observation), indicating that overall reproductive performance of T2D males was comparable to controls. However, the litter size of T2D males was more variable (5.63 ± 1.35, n = 6 litters) compared to controls (7 ± 0.53, n = 7 litters, Table 1). The weights of the reproductive organs, epididymis and seminal vesicles, and the anogenital distance (AGD), as an androgen-responsive outcome, were unaffected in T2D mice, except for the prostate (Supplementary Table S2).
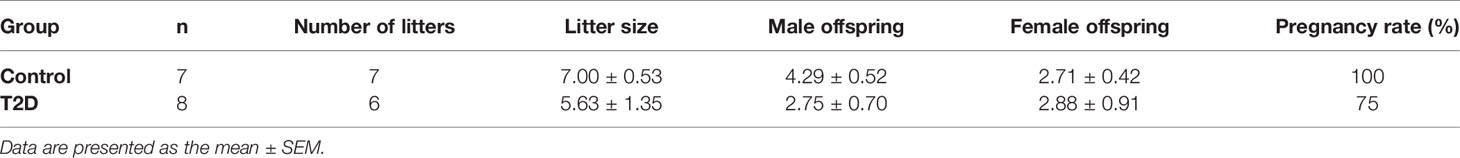
Table 1 Reproductive effects of T2D in males (parental generation, P).
We performed a sperm quality assessment of collected caudal epididymal sperm. No significant effects were found in sperm concentration, viability, apoptosis, or in the packaging quality of the chromatin (as assessed by chromomycin A3 staining) between T2D and control males (Supplementary Table S3). The abnormalities in sperm head morphology were increased in T2D males compared to controls (Figure 4A). To further assess sperm quality, we analyzed protamine 1 and protamine 2 ratios, which affect DNA stability, and play a role in the establishment of epigenetic marks (42, 43). The protamine 1 and protamine 2 ratios were altered in the paternal T2D generation (Figure 4A). Additionally, the expression of biomarkers of sperm dysfunction, transitional endoplasmic reticulum ATPase (TERA) and glyceraldehyde-3-phosphate dehydrogenase-S (GAPDS) was evaluated. TERA is expressed in the acrosomal part of the sperm head (33), whereas GAPDS is located mainly in the principal piece of the sperm flagellum and in lesser amount in the acrosome (34, 44). The expression of both TERA and GAPDS was reduced in sperm from T2D males compared to sperm from control males (Figures 4B, C).
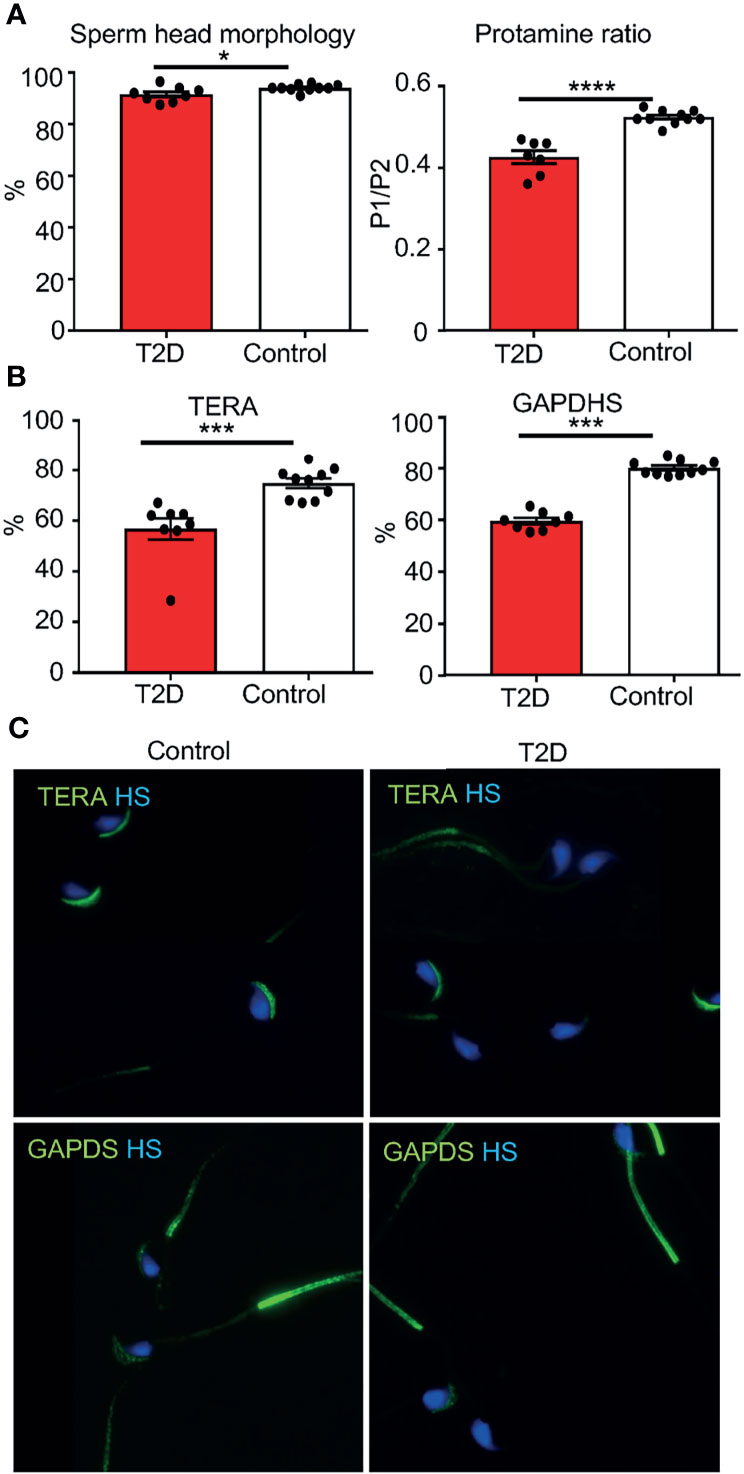
Figure 4 Changes in sperm parameters of the paternal T2D generation. (A) Evaluations of caudal epididymal sperm from T2D and control males: abnormalities in the sperm head morphology and protamine 1 and protamine 2 ratios. (B) Evaluation of the expression of TERA and GAPDS. (C) Representative images of immunolabeling of TERA in the sperm acrosome and GAPDS mainly localized in the principal piece of the flagellum of caudal epidymal sperm from T2D and control males (600x magnification). Data are presented as the mean ± SEM. Differences between groups were tested by t-test (GraphPad Prism 7.0). *P < 0.05, ***P < 0.001, ****P < 0.0001. TERA, transitional endoplasmatic reticulum ATPase; GAPDS, glyceraldehyde 3-phosphate dehydrogenase-S.
In mature testes, sperm are produced in the seminiferous epithelium during the cycle of I to XII stages of spermatogenesis, which are defined by the differentiation steps of germ cells, and occur continuously (45). In order to evaluate the morphology of the seminiferous epithelium at different stages of the differentiation of spermatogenic cells, we focused on four broad categories of the differentiation - spermatogonia, spermatocytes, spermatids, and spermatozoa (Schematic drawing in Figure 5A). We did not find any pronounced morphological abnormalities, such as testicular atrophy, deficiency, or loss of the seminiferous epithelium between the control and T2D testes. Immunolabeling of a meiotic marker, synaptonemal complex protein 3 (SYCP3) (46), revealed that T2D testes contained all stages of meiotic cells similar to controls (Figures 5B–D). Although all the distinct developmental stages of spermatogenesis were identified in similar percentages in both control and T2D testes, a significantly thinner germinal epithelium was found in all cycle stages in the testes of diabetic mice and the diameter of seminiferous tubules was reduced in diabetic testes compared to controls (Tables 2, 3 and Supplementary Table S4). To evaluate early markers of diabetes-induced tissue damage, the expression of gap junction protein connexin 43 (Cx43) and production of advanced glycation end products (AGE) in the testes was compared between control and T2D mice. Cx43 levels were increased specifically in the interstitial tissue containing Leydig cells, which are adjacent to the seminiferous tubules in the testes, suggesting altered intercellular communications between neighboring Leydig cells in the T2D testes (Figures 5E–G). Leydig cells are the primary source of testosterone or androgens in males, under the regulation of the hypothalamic-pituitary axis, and are vital for spermatogenesis (47). We also found an increased production of AGE in the testicular tissue from T2D males (Figures 5H–I”). As AGE are implicated in diabetes related complications, consequently, the formation of AGE may contribute to testicular dysfunction.
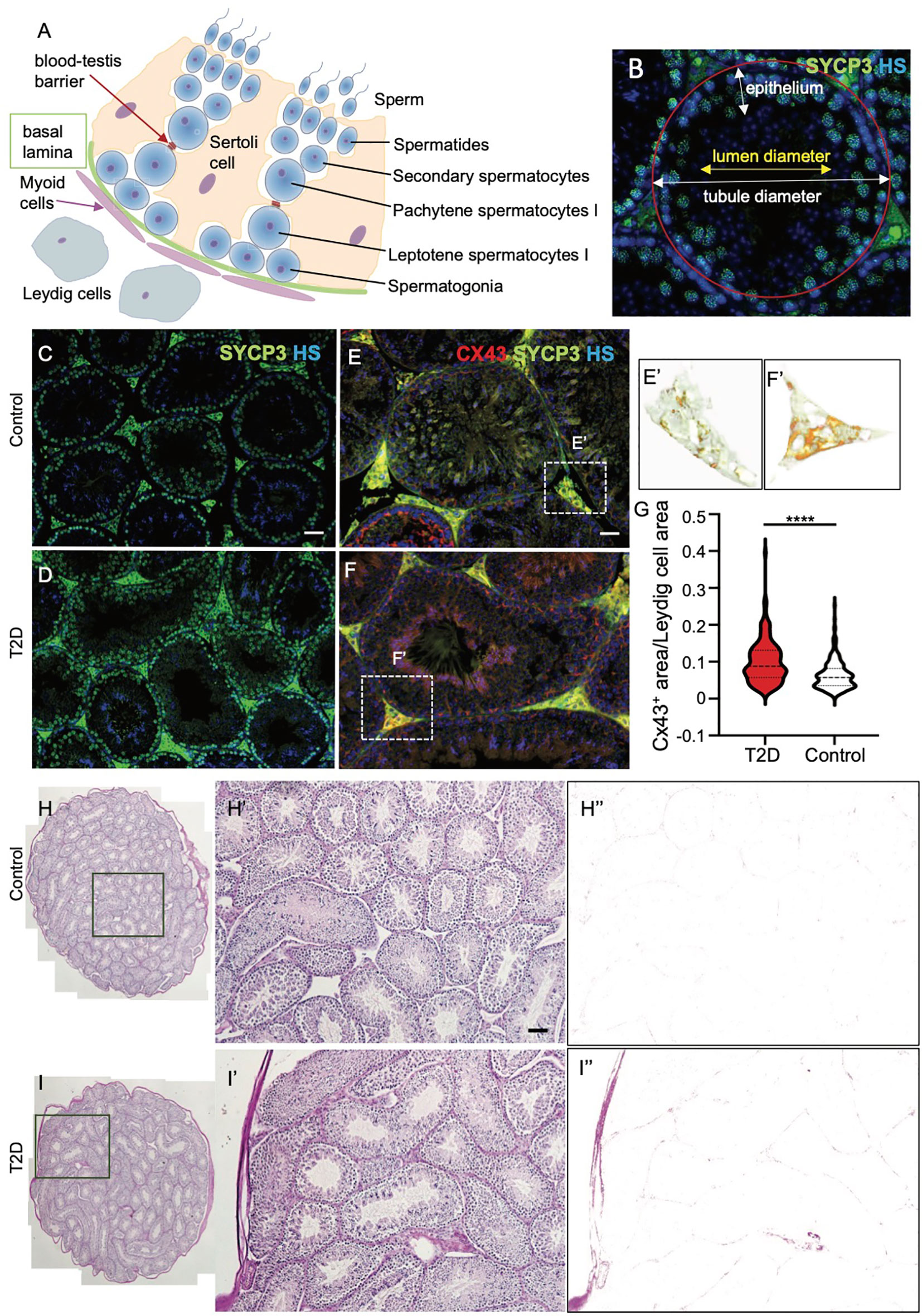
Figure 5 Morphological and expressional changes in the testes of T2D males. (A) Schematic cross-section through a testicular tubule, showing germ cells at different stages of spermatogenesis embedded within the somatic Sertoli cells. Leydig cells are present in the interstitium. The blood-testis barrier facilitates the migration of spermatocytes in addition to the maintenance of the microenvironment. (B) Morphological evaluation of seminiferous tubules included the measurements of tubule diameter and epithelium thickness for different spermatogenesis stages. (C, D) Representative images of cross sections of control and T2D testes after immunolabeling of meiotic marker SYCP3, an axial element of the synaptonemal complex, show the germinal epithelium lining seminiferous tubules at different stages of spermatogenesis. Nuclei are counterstained with Hoechst 33342 (HS, blue). (E–G) Immunolabeling of tissue sections shows increased Cx43 expression in the interstitial tissue of the testes of T2D males compared to controls. (E’, F’) Delineated Cx43+ area in the testis sections by ImageJ program. A relative quantification of staining determined as a ratio of Cx43+ areas per the total Leydig cell areas by ImageJ. Stacked violin plot shows the differential expression of Cx43 in control and T2D. Data are presented as the mean ± SEM (n = 6 controls/271 Leydig cell areas, and 6 T2D/198 Leydig cell areas). Differences between groups were tested by t-test (GraphPad Prism 7.0). ****P < 0.0001. Nuclei are counterstained with Hoechst 33342 (blue). (H–I’) Representative images of PAS staining of the control and T2D testes show increased advanced glycation end-products in the interstitial tissue of the testes from T2D males. (H”, I”) Delineated PAS+ area in the testes by ImageJ. Scale bars, 50 µm.

Table 2 Tubule diameter (μm) for different stages of sperm production in the testes.

Table 3 The seminiferous epithelium thickness (μm) for different stages of sperm production.
Having recognized the effects of T2D on testis morphology, we next wanted to assess the effects of the diabetic environment on mRNA expression in testicular tissue. Out of a selection of germ cell marker genes, we found two with altered expression: the expression of Sycp1 in spermatocytes was reduced and Prm1 levels were increased correspondingly with changes in the protamine ratio found in sperm from T2D males (Figure 6A). We also measured expression of a selection of blood testis barrier markers amongst which we found a significant increase in the expression of Cldn11, and Cdh2 in the testes from T2D males compared to control testes (Figures 6B, C).
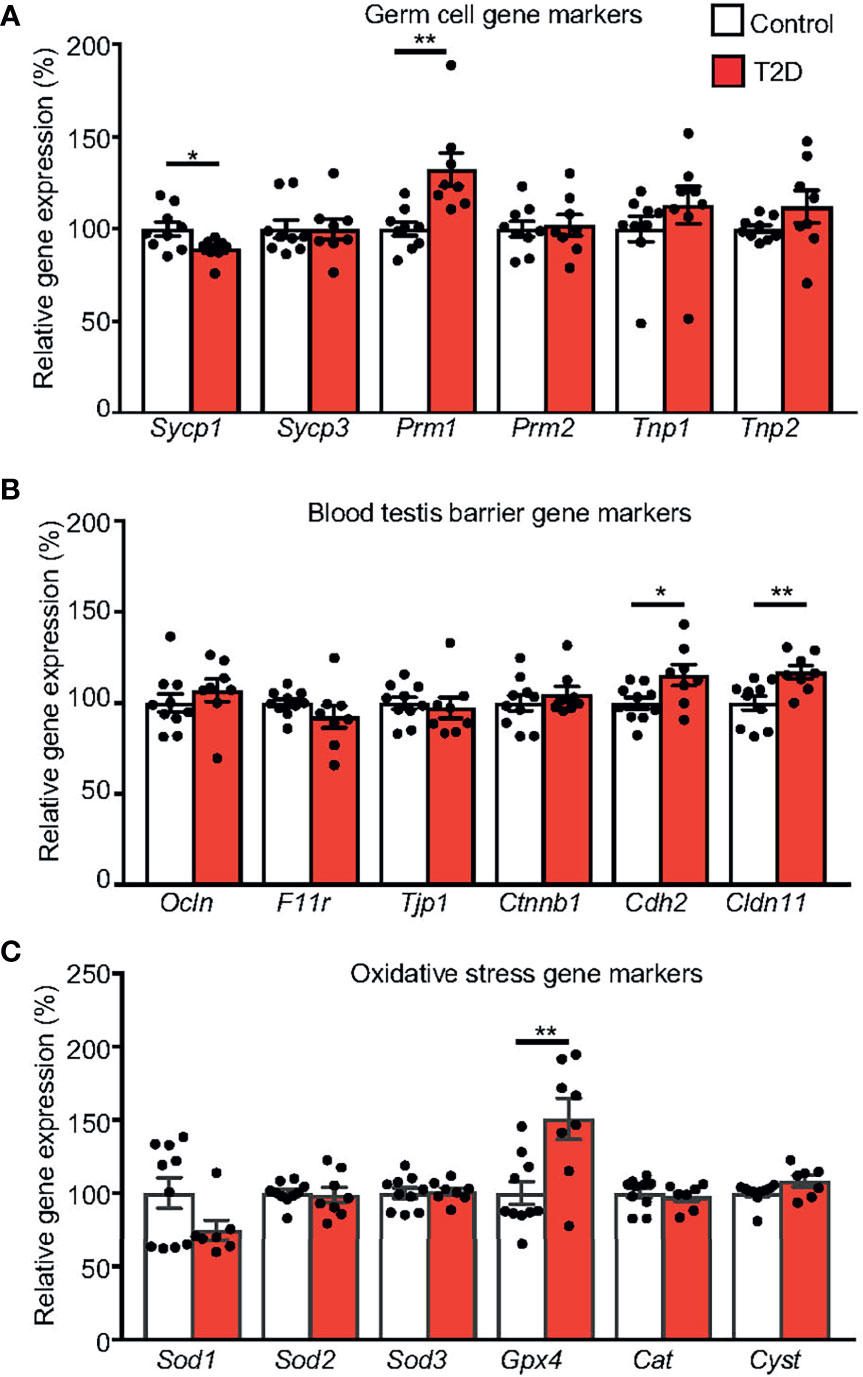
Figure 6 Expression changes in the testes of the parental generation. (A) qPCR analysis of markers for spermatogenesis and spermiogenesis: Sycp1, synaptonemal complex protein 1; Sycp3, synaptonemal complex protein 3; Prm1, protamine 1; Prm1, protamine 2; Tnp1, transition protein 1 and Tnp2, transition protein 2; (B) Blood testis barrier markers: Ocln, occludin; F11r, F11 receptor; Tjp1, tight junction protein 1; Ctnnb1, catenin beta 1; Cdh2, cadherin 2; Cldn11, claudin 11 and (C) oxidative stress markers: Sod1, Sod2, and Sod3, superoxide dismutase 1, 2, and 3; Gpx4, glutathione peroxidase 4; Cat, catalase; and Cyct, cytochrome c. The control group represents 100% of relative gene expression. The values are means ± SEM, tested by t-test (GraphPad Prism 7.0). *P < 0.05, **P < 0.01.
To assess whether the paternal response to a diabetic environment affects the reproductive system of male offspring, males on either diet were mated to females. Fathers were removed after mating, limiting influence on their offspring only to the mating itself. The females were housed individually during the gestation period and the litter size was recorded. All females and the offspring generations were fed by the standard, control diet. Only male pups, the F1 offspring, were left in the litters to be used for following up transgenerational studies. The F2 offspring were produced from the mating of randomly selected F1 males with control females (n = 10 males/group). To evaluate paternal effects and minimize any additional environmental influence, all molecular and cellular analyses of both F1 and F2 offspring were performed at 10 weeks of age, when adult male mice are considered sexually mature (48, 49).
First, to determine the potential effect of paternal T2D on the F1 and F2 generations, we examined physiological and metabolic changes in the offspring. At 10 weeks of age, the body and liver weights were significantly increased in the F1 offspring of T2D males (Figures 7A, B). Most analyzed serum biomarkers were unaffected in the F1 and F2 offspring with the exception of glucose and ALT (Figures 7C, D). Plasma glucose levels were increased in the F1 males at 10 weeks of age, however, these F1 males had no impairment of glucose tolerance, as shown by a glucose tolerance test. Interestingly, ALT was increased in F2 males, indicating a transgenerational effect of T2D exposure.
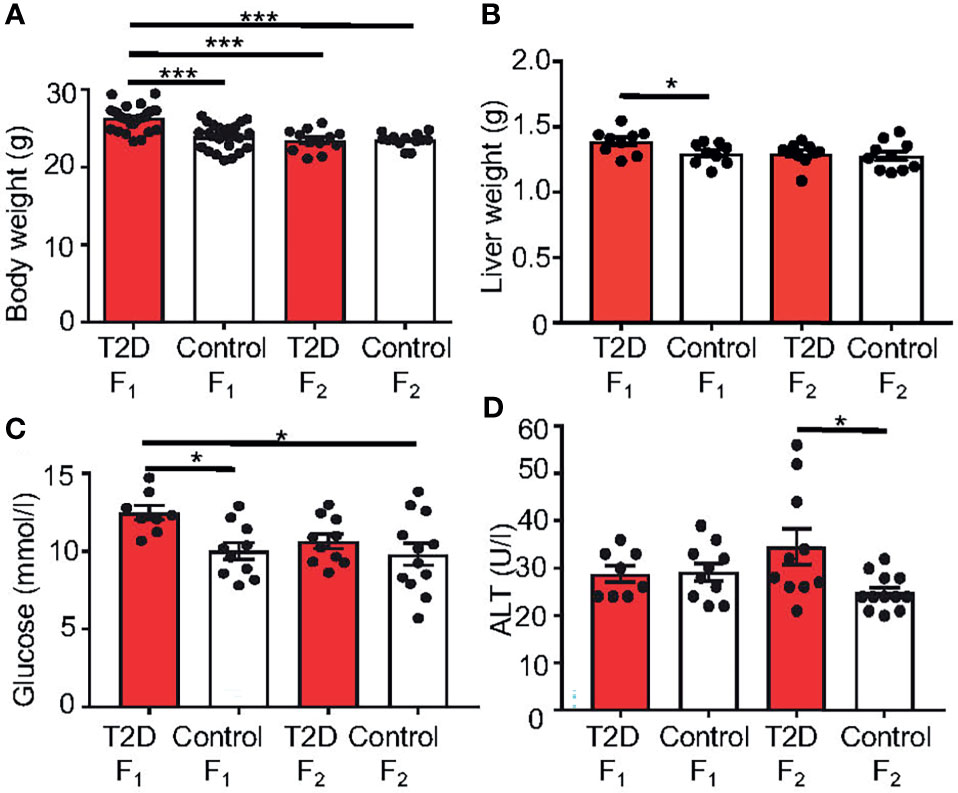
Figure 7 Changes in metabolic characteristics of the offspring generations. (A) Body weight and (B) liver weight at 10 weeks of age. Differences in liver weight between groups were tested by ANCOVA with body weight as covariate (Ancova – STATISTICA 7.0). (C) Fasting plasma glucose and (D) alanine aminotransferase (ALT) levels at 10 weeks of age. Data are presented as the mean ± SEM. Differences were tested by one-way ANOVA, follow by Tukey’s post hoc tests for multiple comparisons (GraphPad Prism 7.0). *P < 0.05, ***P < 0.001.
Next, we evaluated the effects of paternal T2D on the reproductive system and reproductive parameters of the F1 and F2 male offspring. The testicular morphology of the F1 offspring from control and T2D fathers was comparable, including the thickness of the germinal epithelium and diameter of the seminiferous tubules. Accordingly, the mRNA expression of selected genes in the testes was not different between the F1 offspring groups (Supplementary Figure S1). However, the weight of testes of the F1 offspring from T2D fathers was significantly smaller compared to the control F1 offspring (Supplementary Table S5). Additionally, significantly reduced fertility was found in the F1 males from the T2D parental generation with a pregnancy rate of 63% compared to a 90% pregnancy rate in the F1 control generation (Table 4). Of eight F1 males in the experiment, only five males were able to mate in the period of one week and produce progeny with the average litter size of 4.63 ± 1.41 (n = 5 litters) compared to the F1 control cohort with the litter size of 6.44 ± 1.14 (n = 9 litters). The sex ratio of born pups was not significantly different (Table 4).
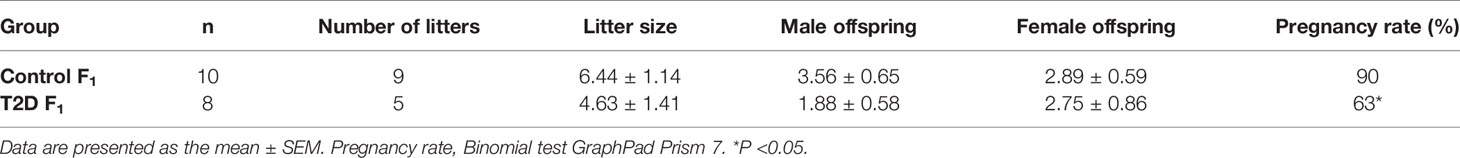
Table 4 Reproductive effects of T2D in parental generation on the F1 offspring.
Caudal epididymal sperm were collected from the F1 and F2 offspring for a sperm quality assessment. Similar to the paternal T2D males, we found no changes in sperm concentration, viability, and cell apoptosis in the F1 and F2 offspring generations (Supplementary Table S6). The packaging quality of the sperm chromatin in the F1 and F2 offspring was not affected by the T2D parental exposure, as evaluated by chromomycin A3 staining of sperm, and by protamine 1 and protamine 2 ratios (Supplementary Table S6). However, abnormalities in sperm head morphology were increased in both the F1 and F2 generations from T2D fathers compared to the control offspring generations (Figure 8A). Additionally, the levels of TERA and GAPDS were reduced in the offspring of diabetic males in both F1 and F2 generations (Figures 8B, C), indicating not only intergenerational but also transgenerational transmission of negative effects of the diabetic environment.
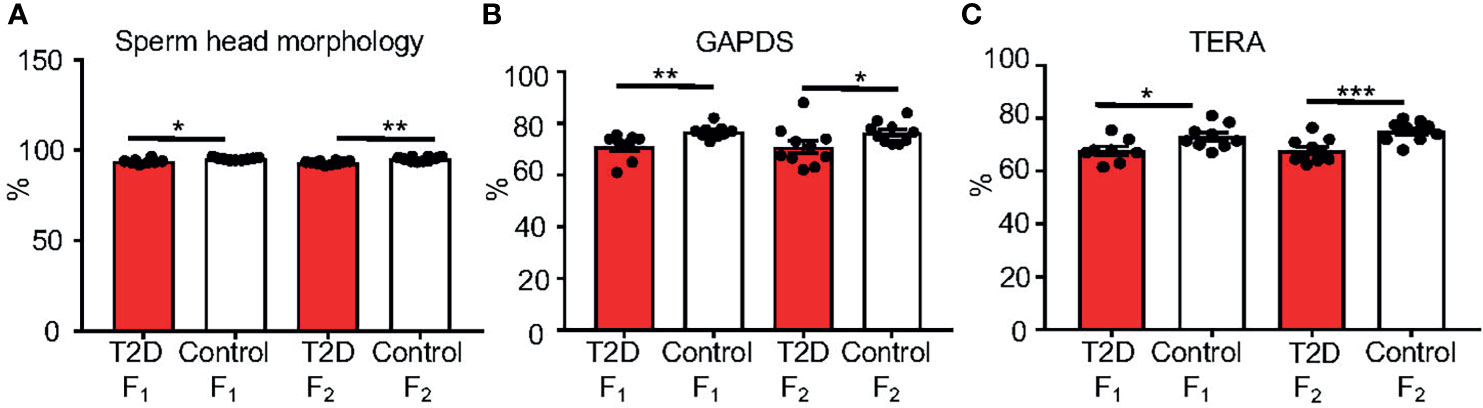
Figure 8 Changes in sperm characteristics of the F1 and F2 offspring. (A) Evaluations of sperm head abnormalities of caudal epididymal sperm from the offspring F1 and F2 generations from T2D and control fathers. (B) The protein levels of glyceraldehyde 3-phosphate dehydrogenase-S (GAPDS) and (C) transitional endoplasmatic reticulum ATPase (TERA), as detected by immunolabeling of caudal epidymal sperm from the F1 and F2 offspring. All analyses were done at 10 weeks of age. Data are presented as the mean ± SEM. (n = 10 for all groups except n = 9 for T2D F1 males/200 cells per each animal). Differences between groups were tested by t-test (GraphPad Prism 7.0). *P < 0.05, **P < 0.01, ***P < 0.001.
In this study, we show that T2D affects not only metabolic health but also the reproductive system through the male lineage to the F2 offspring. Paternal T2D exposure causes increased body weight gain and fasting plasma glucose, and significantly impaired reproductive functions of the F1 males and sperm abnormalities in the F1 male offspring. Furthermore, F2 males had altered sperm parameters. Thus, the observed transgenerational non-genetic transmission of sperm damage caused by T2D in fathers indicate programmed sperm perturbations likely in the form of inherited epigenetic marks.
Both fertility and paternally transmitted effects are associated with changes in the sperm epigenome, including non-coding RNAs, DNA methylation, histones, and protamines (28, 50–53). Epigenetic paternal inheritance involving non-coding RNAs has been established in paternal metabolic disorder models. The intergenerational transmission of a paternal HFD-induced metabolic disorder was linked to a subset of sperm transfer RNA-derived small RNAs (tsRNAs) (53). Another study found changes in sperm miRNA profiles and germ cell methylation status in two offspring generations following diet-induced paternal obesity (27). Differential DNA methylation profiles have been identified in multiple regions of the sperm DNA because of environment-induced epigenomic reprogramming (28, 54–56). For example, numerous genes were differently methylated in sperm of prediabetic/T2D fathers, with a significant proportion of differentially methylated genes overlapping with hypermethylated genes in pancreatic islets of the offspring and corresponding to the prediabetes-associated physiological and metabolic phenotype (28). In a model of caloric restriction during in utero development, the germline DNA methylation was altered in the adult male offspring (57). Histone variants and histone modifications represent additional modes of paternal transmission of environmental information to the offspring. In contrast to somatic cells, male germ cells have more histone variants and their incorporation into the nucleosome may influence the activation of germline-specific genes and repression of the somatic gene expression program [reviewed in (50, 58)]. Some of the histone variants are exclusively detected on germ cells in testes and seem to also be important in the chromatin condensation process (59). The altered histone modifications may also contribute to transgenerational phenotypes through the male germline, as shown in a transgenic mouse model with histone demethylase KDM1A overexpression, resulting in increased H3K4me3 histone methylation in sperm, a transgenerational phenotype of impaired fertility, and severe birth defects in offspring (51). Similarly, deletion of the H3K4me2 demethylase leads to a transgenerational progressive decline in spermatogenesis (60). Specific histone modifications promote chromatin remodeling and histone-to-protamine replacement, which reshapes the nucleus and compacts chromatin [reviewed in (50, 58)]. The histone-to-protamine transition is a tightly controlled process, as premature expression of protamine 1 or disruption of protamine genes results in male infertility in mice (52, 61). Like histones, protamines have post-translational modifications, which are essential for the histone-to-protamine transitions, and abnormalities in protamine modifications may result in impaired spermiogenesis (62, 63). Epigenetic changes during histone-to-protamine transition may not only lead to reduced fertility but are also transmitted to the offspring (52, 62). Protamine 2-deficient mice demonstrate reduced integrity of sperm DNA, altered compaction of the chromatin, and developmental arrest of embryos (64). Furthermore, alterations in the protamine 1/protamine 2 ratio are related to infertility in humans (43, 65), correlate with worsened assisted reproduction outcomes (66), and aberrant protamine replacement may affect the sperm epigenome [reviewed in (65)]. The role of a ‘protamine code’ in addition to the ‘histone code’ in transgenerational epigenetic inheritance remains to be further investigated. Although adverse metabolic phenotypes correlate with reduced sperm concentration, motility, and increase sperm DNA damage in humans (67–69), a causal relationship by which paternal exposures affect the phenotype of offspring has not been established. Nevertheless, epigenetic changes in sperm are implicated as potential mediators of paternal effects and it is essential to understand how epigenetic patterning is produced and how it is influenced by environmental factors resulting in a greater susceptibility to disease in offspring.
Our study used a well characterized model of type 2 diabetes (28, 30, 70). A combination of high-fat diet and low dose STZ resulted in increased body weight gain, glucose intolerance, and altered metabolic parameters, associated with insulin resistance and hyperglycemia, replicating metabolic changes in T2D patients [reviewed in (71)]. The major advantage of this T2D model is that it provides a more accurate model of the human multi-genetic/environmental T2D phenotype compared to monogenic mouse models, such as the mutant with deficiency in leptin (39, 40) or with deficiency in the leptin receptor (41).
We demonstrated that paternal T2D exposure induces a metabolic shift in glucose and fat metabolism in F1 male offspring, as demonstrated by increased plasma glucose together with increased liver weight and body weight. Although the T2D metabolic perturbation phenotype is gradually reversed in the subsequent male generations, increased plasma levels of liver enzyme ALT in the F2 males indicate a higher risk for susceptibility to metabolic syndrome and T2D (72, 73).
For the first time, we provide a complex molecular and morphological analysis of the effects of TD2 on the male reproductive system and the effects of TD2 paternal exposure on the reproductive health of two subsequent male offspring generations. Although the testis morphology of T2D mice appeared grossly normal without any atrophy or destruction of the seminiferous epithelium, immunohistological analysis showed a significant reduction of seminiferous tubule diameter and thickness of the seminiferous epithelium for all cycle stages of sperm production in the testes of T2D mice (Tables 2, 3). In line with a lower severity of testicular damage in T2D males, we found only a few expression changes, particularly, altered expression of cell junction proteins, e.g. gap junction protein Cx43 in Leydig cells, and blood-testis-barrier components, tight junction protein, claudin 11 (74), and the adhesion junction component, cadherin 2 (75). Alternations in cell junctions, indicating abnormalities in cell-cell communications, are an early sign of diabetes-induced tissue damage (76). While the F1 offspring of T2D males had no detectable molecular and morphological changes in the testes, the weight of testes of the F1 offspring of T2D fathers was significantly smaller compared to the control F1 males.
A significant finding of this study is the transgenerational transmission of sperm perturbations from T2D fathers through the male lineage to the F2 offspring. The direct impact of metabolic diseases, such as obesity and diabetes, on sperm quality in men as well as in animal models is well documented. These negative changes include decreased sperm viability and motility, and increased DNA damage, and sperm morphology abnormalities (67, 77–81). Despite these observations, at present, there is only scant evidence for the impact of paternal exposure on sperm quality and the reproductive viability of subsequent generations. Previously, we showed that type 1 diabetes in male mice induces DNA damage, apoptosis, and alters nuclear protamine ratios in sperm in two subsequent male offspring generations (29). Another study, using a diet-induced obesity mouse model, demonstrated that increased sperm DNA damage and reduced motility was transmitted through the paternal line to the first male offspring generation (82). Similarly, Crisostomo et al. (83) reported irreversible changes in sperm quality in correlation with altered testicular metabolism induced by HFD feeding. Here we focused on the effects of T2D on reproductive health of the young adult offspring. We showed that T2D substantially decreased sperm quality in two subsequent F1 and F2 offspring male generations. A sperm impairment phenotype consisting of increased sperm head abnormalities and decreased expression of the sperm dysfunctional biomarkers, TERA and GAPDS, resulted in decreased reproductive function of the F1 males. Since similar sperm defects were found in the F2 offspring, we can speculate that the reproductive function of F2 males may also be compromised. TERA is a member of the AAA ATPase family of proteins, and is found in the sperm acrosome (33). Consistent with the role of TERA in capacitation and the acrosome reaction (84), decreased expression of TERA correlates with reduced fertility in humans (33, 35). GAPDS is one of the sperm specific glycolytic enzymes localized primarily in the principal piece of the sperm flagellum and secondary in the acrosomal part of the sperm head (34, 85). It is essential for sperm metabolism, motility and sperm-oocyte binding, and reduced GAPDS is associated with infertility (34, 35, 44, 86).
In conclusion, our data demonstrate that the impaired reproductive system and sperm quality of T2D fathers negatively effects the reproductive health of F1 male offspring, which persists into the subsequent F2 male generation. Given the transgenerational impairment of reproductive and metabolic parameters through two generations, these changes likely take the form of inherited epigenetic marks through the germline. One possible way could be via alterations in protamine 1 and protamine 2 ratios, as we observed changes in this ratio in parental generation. Nevertheless, further investigation of the epigenetic alterations in sperm that result from paternal exposure to T2D are warranted. Consequently, new strategies to improve metabolic health not only in women of reproductive age but also in potential fathers are necessary in order to reduce the negative impacts on subsequent generations.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by the Animal Care and Use Committee of the Institute of Molecular Genetics, CAS.
All authors have read and approved the manuscript. JP and GP conceived the study, designed experiments, and GP wrote the manuscript. FE, HM, and RB conducted animal study, morphological evaluations, and statistical analyses. EZ and EV designed and performed qPCR. SH and AK performed sperm analyses and Western blotting. HM, EZ, and RB performed immunohistological evaluations. All authors contributed to the article and approved the submitted version.
This work was supported by the institutional support of the Czech Academy of Sciences RVO: 86652036 and the Czech Health Research Council #NU20J-02-00035.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Biochemical parameter analyses were done by the Czech Centre for Phenogenomics supported by LM2015040. We thank Imaging Methods Core Facility at BIOCEV supported by the MEYS CR (Large RI Project LM2018129 Czech-BioImaging) and ERDF (project No. CZ.02.1.01/0.0/0.0/18_046/0016045) for its support with obtaining imaging data presented in this paper. We thank A. Pavlinek (King’s College London) for editing the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.763863/full#supplementary-material
1. Fonseca VA. Defining and Characterizing the Progression of Type 2 Diabetes. Diabetes Care (2009) 32 Suppl 2:S151–6. doi: 10.2337/dc09-S301
2. A. American Diabetes. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care (2014) 37 Suppl 1:S81–90. doi: 10.2337/dc14-S081
3. Maiorino MI, Bellastella G, Esposito K. Diabetes and Sexual Dysfunction: Current Perspectives. Diabetes Metab Syndr Obes (2014) 7:95–105. doi: 10.2147/DMSO.S36455
4. Hylmarova S, Stechova K, Pavlinkova G, Peknicova J, Macek M, Kvapil M. The Impact of Type 1 Diabetes Mellitus on Male Sexual Functions and Sex Hormone Levels. Endocr J (2020) 67:59–71. doi: 10.1507/endocrj.EJ19-0280
5. Andersson DP, Ekstrom U, Lehtihet M. Rigiscan Evaluation of Men With Diabetes Mellitus and Erectile Dysfunction and Correlation With Diabetes Duration, Age, BMI, Lipids and HbA1c. PloS One (2015) 10:e0133121. doi: 10.1371/journal.pone.0133121
6. Dhindsa S, Furlanetto R, Vora M, Ghanim H, Chaudhuri A, Dandona P. Low Estradiol Concentrations in Men With Subnormal Testosterone Concentrations and Type 2 Diabetes. Diabetes Care (2011) 34:1854–9. doi: 10.2337/dc11-0208
7. Mulholland J, Mallidis C, Agbaje I, McClure N. Male Diabetes Mellitus and Assisted Reproduction Treatment Outcome. Reprod BioMed Online (2011) 22:215–9. doi: 10.1016/j.rbmo.2010.10.005
8. La Vignera S, Condorelli R, Vicari E, D'Agata R, Calogero AE. Diabetes Mellitus and Sperm Parameters. J Androl (2012) 33:145–53. doi: 10.2164/jandrol.111.013193
9. Agbaje IM, Rogers DA, McVicar CM, McClure N, Atkinson AB, Mallidis C, et al. Insulin Dependant Diabetes Mellitus: Implications for Male Reproductive Function. Hum Reprod (2007) 22:1871–7. doi: 10.1093/humrep/dem077
10. Bhattacharya SM, Ghosh M, Nandi N. Diabetes Mellitus and Abnormalities in Semen Analysis. J Obstet Gynaecol Res (2014) 40:167–71. doi: 10.1111/jog.12149
11. Oyen N, Diaz LJ, Leirgul E, Boyd HA, Priest J, Mathiesen ER, et al. Prepregnancy Diabetes and Offspring Risk of Congenital Heart Disease: A Nationwide Cohort Study. Circulation (2016) 133:2243–53. doi: 10.1161/CIRCULATIONAHA.115.017465
12. Ornoy A, Reece EA, Pavlinkova G, Kappen C, Miller RK. Effect of Maternal Diabetes on the Embryo, Fetus, and Children: Congenital Anomalies, Genetic and Epigenetic Changes and Developmental Outcomes. Birth Defects Res C Embryo Today (2015) 105:53–72. doi: 10.1002/bdrc.21090
13. West NA, Crume TL, Maligie MA, Dabelea D. Cardiovascular Risk Factors in Children Exposed to Maternal Diabetes In Utero. Diabetologia (2011) 54:504–7. doi: 10.1007/s00125-010-2008-1
14. Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir MM, et al. Intrauterine Exposure to Diabetes Conveys Risks for Type 2 Diabetes and Obesity: A Study of Discordant Sibships. Diabetes (2000) 49:2208–11. doi: 10.2337/diabetes.49.12.2208
15. Martin AO, Simpson JL, Ober C, Freinkel N. Frequency of Diabetes Mellitus in Mothers of Probands With Gestational Diabetes: Possible Maternal Influence on the Predisposition to Gestational Diabetes. Am J Obstet Gynecol (1985) 151:471–5. doi: 10.1016/0002-9378(85)90272-8
16. Sobngwi E, Boudou P, Mauvais-Jarvis F, Leblanc H, Velho G, Vexiau P, et al. Effect of a Diabetic Environment In Utero on Predisposition to Type 2 Diabetes. Lancet (2003) 361:1861–5. doi: 10.1016/S0140-6736(03)13505-2
17. Manderson JG, Mullan B, Patterson CC, Hadden DR, Traub AI, McCance DR. Cardiovascular and Metabolic Abnormalities in the Offspring of Diabetic Pregnancy. Diabetologia (2002) 45:991–6. doi: 10.1007/s00125-002-0865-y
18. Loffredo CA, Wilson PD, Ferencz C. Maternal Diabetes: An Independent Risk Factor for Major Cardiovascular Malformations With Increased Mortality of Affected Infants. Teratology (2001) 64:98–106. doi: 10.1002/tera.1051
19. Cerychova R, Bohuslavova R, Papousek F, Sedmera D, Abaffy P, Benes V, et al. Adverse Effects of Hif1a Mutation and Maternal Diabetes on the Offspring Heart. Cardiovasc Diabetol (2018) 17:68. doi: 10.1186/s12933-018-0713-0
20. Dunn GA, Bale TL. Maternal High-Fat Diet Effects on Third-Generation Female Body Size via the Paternal Lineage. Endocrinology (2011) 152:2228–36. doi: 10.1210/en.2010-1461
21. Gniuli D, Calcagno A, Caristo ME, Mancuso A, Macchi V, Mingrone G, et al. Effects of High-Fat Diet Exposure During Fetal Life on Type 2 Diabetes Development in the Progeny. J Lipid Res (2008) 49:1936–45. doi: 10.1194/jlr.M800033-JLR200
22. Sasson IE, Vitins AP, Mainigi MA, Moley KH, Simmons RA. Pre-Gestational vs Gestational Exposure to Maternal Obesity Differentially Programs the Offspring in Mice. Diabetologia (2015) 58:615–24. doi: 10.1007/s00125-014-3466-7
23. Aerts L, Van Assche FA. Animal Evidence for the Transgenerational Development of Diabetes Mellitus. Int J Biochem Cell Biol (2006) 38:894–903. doi: 10.1016/j.biocel.2005.07.006
24. Huypens P, Sass S, Wu M, Dyckhoff D, Tschop M, Theis F, et al. Epigenetic Germline Inheritance of Diet-Induced Obesity and Insulin Resistance. Nat Genet (2016) 48:497–9. doi: 10.1038/ng.3527
25. Patti ME. Intergenerational Programming of Metabolic Disease: Evidence From Human Populations and Experimental Animal Models. Cell Mol Life Sci (2013) 70:1597–608. doi: 10.1007/s00018-013-1298-0
26. Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic High-Fat Diet in Fathers Programs Beta-Cell Dysfunction in Female Rat Offspring. Nature (2010) 467:963–6. doi: 10.1038/nature09491
27. Fullston T, Ohlsson Teague EM, Palmer NO, DeBlasio MJ, Mitchell M, Corbett M, et al. Paternal Obesity Initiates Metabolic Disturbances in Two Generations of Mice With Incomplete Penetrance to the F2 Generation and Alters the Transcriptional Profile of Testis and Sperm microRNA Content. FASEB J (2013) 27:4226–43. doi: 10.1096/fj.12-224048
28. Wei Y, Yang CR, Wei YP, Zhao ZA, Hou Y, Schatten H, et al. Paternally Induced Transgenerational Inheritance of Susceptibility to Diabetes in Mammals. Proc Natl Acad Sci USA (2014) 111:1873–8. doi: 10.1073/pnas.1321195111
29. Pavlinkova G, Margaryan H, Zatecka E, Valaskova E, Elzeinova F, Kubatova A, et al. Transgenerational Inheritance of Susceptibility to Diabetes-Induced Male Subfertility. Sci Rep (2017) 7:4940. doi: 10.1038/s41598-017-05286-0
30. Winzell MS, Ahren B. The High-Fat Diet-Fed Mouse: A Model for Studying Mechanisms and Treatment of Impaired Glucose Tolerance and Type 2 Diabetes. Diabetes (2004) 53 Suppl 3:S215–9. doi: 10.2337/diabetes.53.suppl_3.S215
31. Dostalova P, Zatecka E, Ded L, Elzeinova F, Valaskova E, Kubatova A, et al. Gestational and Pubertal Exposure to Low Dose of Di-(2-Ethylhexyl) Phthalate Impairs Sperm Quality in Adult Mice. Reprod Toxicol (2020) 96:175–84. doi: 10.1016/j.reprotox.2020.06.014
32. Elzeinova F, Novakova V, Buckiova D, Kubatova A, Peknicova J. Effect of Low Dose of Vinclozolin on Reproductive Tract Development and Sperm Parameters in CD1 Outbred Mice. Reprod Toxicol (2008) 26:231–8. doi: 10.1016/j.reprotox.2008.09.007
33. Capkova J, Margaryan H, Kubatova A, Novak P, Peknicova J. Target Antigens for Hs-14 Monoclonal Antibody and Their Various Expression in Normozoospermic and Asthenozoospermic Men. Basic Clin Androl (2015) 25:11. doi: 10.1186/s12610-015-0025-0
34. Margaryan H, Dorosh A, Capkova J, Manaskova-Postlerova P, Philimonenko A, Hozak P, et al. Characterization and Possible Function of Glyceraldehyde-3-Phosphate Dehydrogenase-Spermatogenic Protein GAPDHS in Mammalian Sperm. Reprod Biol Endocrinol (2015) 13:15. doi: 10.1186/s12958-015-0008-1
35. Tepla O, Peknicova J, Koci K, Mika J, Mrazek M, Elzeinova F. Evaluation of Reproductive Potential After Intracytoplasmic Sperm Injection of Varied Human Semen Tested by Antiacrosomal Antibodies. Fertil Steril (2006) 86:113–20. doi: 10.1016/j.fertnstert.2005.12.019
36. Capkova J, Kubatova A, Ded L, Tepla O, Peknicova J. Evaluation of the Expression of Sperm Proteins in Normozoospermic and Asthenozoospermic Men Using Monoclonal Antibodies. Asian J Androl (2016) 18:108–13. doi: 10.4103/1008-682X.151400
37. Peknicova J, Chladek D, Hozak P. Monoclonal Antibodies Against Sperm Intra-Acrosomal Antigens as Markers for Male Infertility Diagnostics and Estimation of Spermatogenesis. Am J Reprod Immunol (2005) 53:42–9. doi: 10.1111/j.1600-0897.2004.00245.x
38. Luo J, Quan J, Tsai J, Hobensack CK, Sullivan C, Hector R, et al. Nongenetic Mouse Models of Non-Insulin-Dependent Diabetes Mellitus. Metabolism (1998) 47:663–8. doi: 10.1016/S0026-0495(98)90027-0
39. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional Cloning of the Mouse Obese Gene and Its Human Homologue. Nature (1994) 372:425–32. doi: 10.1038/372425a0
40. D'Souza A M, Johnson JD, Clee SM, Kieffer TJ. Suppressing Hyperinsulinemia Prevents Obesity But Causes Rapid Onset of Diabetes in Leptin-Deficient Lep(ob/ob) Mice. Mol Metab (2016) 5:1103–12. doi: 10.1016/j.molmet.2016.09.007
41. Leifheit-Nestler M, Wagner NM, Gogiraju R, Didie M, Konstantinides S, Hasenfuss G, et al. Importance of Leptin Signaling and Signal Transducer and Activator of Transcription-3 Activation in Mediating the Cardiac Hypertrophy Associated With Obesity. J Transl Med (2013) 11:170. doi: 10.1186/1479-5876-11-170
42. Castillo J, Amaral A, Oliva R. Sperm Nuclear Proteome and Its Epigenetic Potential. Andrology (2014) 2:326–38. doi: 10.1111/j.2047-2927.2013.00170.x
43. Oliva R. Protamines and Male Infertility. Hum Reprod Update (2006) 12:417–35. doi: 10.1093/humupd/dml009
44. Miki K, Qu W, Goulding EH, Willis WD, Bunch DO, Strader LF, et al. Glyceraldehyde 3-Phosphate Dehydrogenase-S, a Sperm-Specific Glycolytic Enzyme, Is Required for Sperm Motility and Male Fertility. Proc Natl Acad Sci USA (2004) 101:16501–6. doi: 10.1073/pnas.0407708101
45. Eddy EM, O'Brien DA, Welch JE. Mammalian Sperm Development In Vivo and In Vitro. Boston: CRC Press (1991).
46. Yuan L, Liu JG, Zhao J, Brundell E, Daneholt B, Hoog C. The Murine SCP3 Gene Is Required for Synaptonemal Complex Assembly, Chromosome Synapsis, and Male Fertility. Mol Cell (2000) 5:73–83. doi: 10.1016/S1097-2765(00)80404-9
47. Zhou R, Wu J, Liu B, Jiang Y, Chen W, Li J, et al. The Roles and Mechanisms of Leydig Cells and Myoid Cells in Regulating Spermatogenesis. Cell Mol Life Sci (2019) 76:2681–95. doi: 10.1007/s00018-019-03101-9
48. Dutta S, Sengupta P. Men and Mice: Relating Their Ages. Life Sci (2016) 152:244–8. doi: 10.1016/j.lfs.2015.10.025
49. Flurkey K, Currer JM, Harrison DE. Mouse Models in Aging Research. Cambridge, Massachusetts: Academic Press (2007).
50. Kimmins S, Sassone-Corsi P. Chromatin Remodelling and Epigenetic Features of Germ Cells. Nature (2005) 434:583–9. doi: 10.1038/nature03368
51. Lismer A, Siklenka K, Lafleur C, Dumeaux V, Kimmins S. Sperm Histone H3 Lysine 4 Trimethylation Is Altered in a Genetic Mouse Model of Transgenerational Epigenetic Inheritance. Nucleic Acids Res (2020) 48:11380–93. doi: 10.1093/nar/gkaa712
52. Cho C, Willis WD, Goulding EH, Jung-Ha H, Choi YC, Hecht NB, et al. Haploinsufficiency of Protamine-1 or -2 Causes Infertility in Mice. Nat Genet (2001) 28:82–6. doi: 10.1038/ng0501-82
53. Chen Q, Yan M, Cao Z, Li X, Zhang Y, Shi J, et al. Sperm tsRNAs Contribute to Intergenerational Inheritance of an Acquired Metabolic Disorder. Science (2016) 351:397–400. doi: 10.1126/science.aad7977
54. Lambrot R, Xu C, Saint-Phar S, Chountalos G, Cohen T, Paquet M, et al. Low Paternal Dietary Folate Alters the Mouse Sperm Epigenome and Is Associated With Negative Pregnancy Outcomes. Nat Commun (2013) 4:2889. doi: 10.1038/ncomms3889
55. Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, et al. Paternally Induced Transgenerational Environmental Reprogramming of Metabolic Gene Expression in Mammals. Cell (2010) 143:1084–96. doi: 10.1016/j.cell.2010.12.008
56. Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic Transgenerational Actions of Endocrine Disruptors and Male Fertility. Science (2005) 308:1466–9. doi: 10.1126/science.1108190
57. Radford EJ, Ito M, Shi H, Corish JA, Yamazawa K, Isganaitis E, et al. In Utero Effects. In Utero Undernourishment Perturbs the Adult Sperm Methylome and Intergenerational Metabolism. Science (2014) 345:1255903. doi: 10.1126/science.1255903
58. Bao J, Bedford MT. Epigenetic Regulation of the Histone-to-Protamine Transition During Spermiogenesis. Reproduction (2016) 151:R55–70. doi: 10.1530/REP-15-0562
59. Martianov I, Brancorsini S, Catena R, Gansmuller A, Kotaja N, Parvinen M, et al. Polar Nuclear Localization of H1T2, a Histone H1 Variant, Required for Spermatid Elongation and DNA Condensation During Spermiogenesis. Proc Natl Acad Sci USA (2005) 102:2808–13. doi: 10.1073/pnas.0406060102
60. Greer EL, Beese-Sims SE, Brookes E, Spadafora R, Zhu Y, Rothbart SB, et al. A Histone Methylation Network Regulates Transgenerational Epigenetic Memory in C. Elegans. Cell Rep (2014) 7:113–26. doi: 10.1016/j.celrep.2014.02.044
61. Lee K, Haugen HS, Clegg CH, Braun RE. Premature Translation of Protamine 1 mRNA Causes Precocious Nuclear Condensation and Arrests Spermatid Differentiation in Mice. Proc Natl Acad Sci USA (1995) 92:12451–5. doi: 10.1073/pnas.92.26.12451
62. Wu JY, Ribar TJ, Cummings DE, Burton KA, McKnight GS, Means AR. Spermiogenesis and Exchange of Basic Nuclear Proteins Are Impaired in Male Germ Cells Lacking Camk4. Nat Genet (2000) 25:448–52. doi: 10.1038/78153
63. Brunner AM, Nanni P, Mansuy IM. Epigenetic Marking of Sperm by Post-Translational Modification of Histones and Protamines. Epigenet Chromatin (2014) 7:2. doi: 10.1186/1756-8935-7-2
64. Cho C, Jung-Ha H, Willis WD, Goulding EH, Stein P, Xu Z, et al. Protamine 2 Deficiency Leads to Sperm DNA Damage and Embryo Death in Mice. Biol Reprod (2003) 69:211–7. doi: 10.1095/biolreprod.102.015115
65. Carrell DT, Emery BR, Hammoud S. The Aetiology of Sperm Protamine Abnormalities and Their Potential Impact on the Sperm Epigenome. Int J Androl (2008) 31:537–45. doi: 10.1111/j.1365-2605.2008.00872.x
66. de Mateo S, Gazquez C, Guimera M, Balasch J, Meistrich ML, Ballesca JL, et al. Protamine 2 Precursors (Pre-P2), Protamine 1 to Protamine 2 Ratio (P1/P2), and Assisted Reproduction Outcome. Fertil Steril (2009) 91:715–22. doi: 10.1016/j.fertnstert.2007.12.047
67. Kort HI, Massey JB, Elsner CW, Mitchell-Leef D, Shapiro DB, Witt MA, et al. Impact of Body Mass Index Values on Sperm Quantity and Quality. J Androl (2006) 27:450–2. doi: 10.2164/jandrol.05124
68. Jensen TK, Andersson AM, Jorgensen N, Andersen AG, Carlsen E, Petersen JH, et al. Body Mass Index in Relation to Semen Quality and Reproductive Hormones Among 1,558 Danish Men. Fertil Steril (2004) 82:863–70. doi: 10.1016/j.fertnstert.2004.03.056
69. Magnusdottir EV, Thorsteinsson T, Thorsteinsdottir S, Heimisdottir M, Olafsdottir K. Persistent Organochlorines, Sedentary Occupation, Obesity and Human Male Subfertility. Hum Reprod (2005) 20:208–15. doi: 10.1093/humrep/deh569
70. Mansor LS, Gonzalez ER, Cole MA, Tyler DJ, Beeson JH, Clarke K, et al. Cardiac Metabolism in a New Rat Model of Type 2 Diabetes Using High-Fat Diet With Low Dose Streptozotocin. Cardiovasc Diabetol (2013) 12:136. doi: 10.1186/1475-2840-12-136
71. Fang JY, Lin CH, Huang TH, Chuang SY. In Vivo Rodent Models of Type 2 Diabetes and Their Usefulness for Evaluating Flavonoid Bioactivity. Nutrients (2019) 11:530. doi: 10.3390/nu11030530
72. Pinhas-Hamiel O, Zeitler P. Acute and Chronic Complications of Type 2 Diabetes Mellitus in Children and Adolescents. Lancet (2007) 369:1823–31. doi: 10.1016/S0140-6736(07)60821-6
73. Hanley AJ, Williams K, Festa A, Wagenknecht LE, D'Agostino RB Jr., Kempf J, et al. Elevations in Markers of Liver Injury and Risk of Type 2 Diabetes: The Insulin Resistance Atherosclerosis Study. Diabetes (2004) 53:2623–32. doi: 10.2337/diabetes.53.10.2623
74. Gow A, Southwood CM, Li JS, Pariali M, Riordan GP, Brodie SE, et al. CNS Myelin and Sertoli Cell Tight Junction Strands Are Absent in Osp/claudin-11 Null Mice. Cell (1999) 99:649–59. doi: 10.1016/S0092-8674(00)81553-6
75. Jiang X, Ma T, Zhang Y, Zhang H, Yin S, Zheng W, et al. Specific Deletion of Cdh2 in Sertoli Cells Leads to Altered Meiotic Progression and Subfertility of Mice. Biol Reprod (2015) 92:79. doi: 10.1095/biolreprod.114.126334
76. Eftekhari A, Vahed SZ, Kavetskyy T, Rameshrad M, Jafari S, Chodari L, et al. Cell Junction Proteins: Crossing the Glomerular Filtration Barrier in Diabetic Nephropathy. Int J Biol Macromol (2020) 148:475–82. doi: 10.1016/j.ijbiomac.2020.01.168
77. Chavarro JE, Toth TL, Wright DL, Meeker JD, Hauser R. Body Mass Index in Relation to Semen Quality, Sperm DNA Integrity, and Serum Reproductive Hormone Levels Among Men Attending an Infertility Clinic. Fertil Steril (2010) 93:2222–31. doi: 10.1016/j.fertnstert.2009.01.100
78. Palmer NO, Fullston T, Mitchell M, Setchell BP, Lane M. SIRT6 in Mouse Spermatogenesis Is Modulated by Diet-Induced Obesity. Reprod Fertil Dev (2011) 23:929–39. doi: 10.1071/RD10326
79. Bakos HW, Mitchell M, Setchell BP, Lane M. The Effect of Paternal Diet-Induced Obesity on Sperm Function and Fertilization in a Mouse Model. Int J Androl (2011) 34:402–10. doi: 10.1111/j.1365-2605.2010.01092.x
80. Navarro-Casado L, Juncos-Tobarra MA, Chafer-Rudilla M, de Onzono LI, Blazquez-Cabrera JA, Miralles-Garcia JM. Effect of Experimental Diabetes and STZ on Male Fertility Capacity. Study in rats. J Androl (2010) 31:584–92. doi: 10.2164/jandrol.108.007260
81. Crisostomo L, Rato L, Jarak I, Silva BM, Raposo JF, Batterham RL, et al. A Switch From High-Fat to Normal Diet Does Not Restore Sperm Quality But Prevents Metabolic Syndrome. Reproduction (2019) 158:377–87. doi: 10.1530/REP-19-0259
82. Fullston T, Palmer NO, Owens JA, Mitchell M, Bakos HW, Lane M. Diet-Induced Paternal Obesity in the Absence of Diabetes Diminishes the Reproductive Health of Two Subsequent Generations of Mice. Hum Reprod (2012) 27:1391–400. doi: 10.1093/humrep/des030
83. Crisostomo L, Jarak I, Rato LP, Raposo JF, Batterham RL, Oliveira PF, et al. Inheritable Testicular Metabolic Memory of High-Fat Diet Causes Transgenerational Sperm Defects in Mice. Sci Rep (2021) 11:9444. doi: 10.1038/s41598-021-88981-3
84. Ficarro S, Chertihin O, Westbrook VA, White F, Jayes F, Kalab P, et al. Phosphoproteome Analysis of Capacitated Human Sperm. Evidence of Tyrosine Phosphorylation of a Kinase-Anchoring Protein 3 and Valosin-Containing Protein/P97 During Capacitation. J Biol Chem (2003) 278:11579–89. doi: 10.1074/jbc.M202325200
85. Bunch DO, Welch JE, Magyar PL, Eddy EM, O'Brien DA. Glyceraldehyde 3-Phosphate Dehydrogenase-S Protein Distribution During Mouse Spermatogenesis. Biol Reprod (1998) 58:834–41. doi: 10.1095/biolreprod58.3.834
86. Zatecka E, Castillo J, Elzeinova F, Kubatova A, Ded L, Peknicova J, et al. The Effect of Tetrabromobisphenol A on Protamine Content and DNA Integrity in Mouse Spermatozoa. Andrology (2014) 2:910–7. doi: 10.1111/j.2047-2927.2014.00257.x
Keywords: sperm, diabetes, testes, fertility, offspring, molecular biomarkers, TERA, GAPDS
Citation: Zatecka E, Bohuslavova R, Valaskova E, Margaryan H, Elzeinova F, Kubatova A, Hylmarova S, Peknicova J and Pavlinkova G (2021) The Transgenerational Transmission of the Paternal Type 2 Diabetes-Induced Subfertility Phenotype. Front. Endocrinol. 12:763863. doi: 10.3389/fendo.2021.763863
Received: 24 August 2021; Accepted: 20 October 2021;
Published: 05 November 2021.
Edited by:
Honggang Li, Huazhong University of Science and Technology, ChinaReviewed by:
Francesca Mancuso, University of Perugia, ItalyCopyright © 2021 Zatecka, Bohuslavova, Valaskova, Margaryan, Elzeinova, Kubatova, Hylmarova, Peknicova and Pavlinkova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela Pavlinkova, Z3BhdmxpbmtvdmFAaWJ0LmNhcy5jeg==
†These authors share first authorship
‡These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.