 Lena Batoon
Lena Batoon Laurie K. McCauley
Laurie K. McCauley
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 03 November 2021
Sec. Bone Research
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.763846
This article is part of the Research Topic Cancer and Bone Metastasis, Volume II View all 9 articles
The skeleton is a common site for cancer metastases with the bone microenvironment providing the appropriate conditions for cancer cell colonization. Once in bone, cancer cells effectively manipulate their microenvironment to support their growth and survival. Despite previous efforts to improve treatment modalities, skeletal metastases remain with poor prognoses. This warrants an improved understanding of the mechanisms leading to bone metastasis that will aid development of effective treatments. Macrophages in the tumor microenvironment are termed tumor associated macrophages (TAMs) and their crosstalk with cancer cells is critical in regulating tumorigenicity in multiple cancers. In bone metastases, this crosstalk is also being increasingly implicated but the specific signaling pathways remain incompletely understood. Here, we summarize the reported functions, interactions, and signaling of macrophages with cancer cells during the metastatic cascade to bone. Specifically, we review and discuss how these specific interactions impact macrophages and their profiles to promote tumor development. We also discuss the potential of targeting this crosstalk to inhibit disease progression. Finally, we identify the remaining knowledge gaps that will need to be addressed in order to fully consider therapeutic targeting to improve clinical outcomes in cancer patients.
Bone metastases are common complications of solid tumors particularly in patients with cancers of the breast, prostate or lung (1, 2). Despite previous efforts to improve cancer diagnosis and treatment modalities, skeletal metastases remain with poor prognoses and high mortality and only about 10% one-year survival after bone metastasis diagnosis e.g. in lung cancer patients (3). Furthermore, such metastases result in considerable morbidity as they can cause limb dysfunction, impaired mobility, pathological fractures, spinal cord compression, and severe pain, significantly affecting patients’ quality of life (2, 4). Management of metastatic bone diseases imposes a huge burden on health care systems due to the substantially high costs associated with extensive use of medical resources (5, 6). Collectively, this warrants an improved understanding of the mechanisms leading to bone metastasis and progression that will aid development of effective treatments to prevent and alleviate the morbidity and mortality associated with bone metastases.
While cancer cell formation is a product of genetic/epigenetic aberrations in normal cells (7), development and progression requires corruption of the normal microenvironment by cancer cells to support their growth, survival and metastatic colonization. The metastatic process begins with intravasation of cancer cells from the primary site into the lymphatic or vascular system before extravasation of these disseminated tumor cells (DTCs) into compatible secondary sites that support their growth (8) (Figure 1). Some tumors, such as prostate (9), pancreatic (10), and oral squamous cell (11) cancers, have been shown to migrate along nerves (12) or the abluminal surface of the endothelium (13). The final stage of the metastatic cascade is colonization of the distant organ which is usually achieved through three key steps: colonization, dormancy and outgrowth (8). Despite bone being a common site of metastasis in addition to liver and lung, the exact mechanisms influencing metastatic progression to the skeleton remain unclear. In accordance with the ‘seed and soil’ hypothesis (14), it is widely accepted that the bone microenvironment provides an ideal niche allowing metastatic tumor cells to thrive.
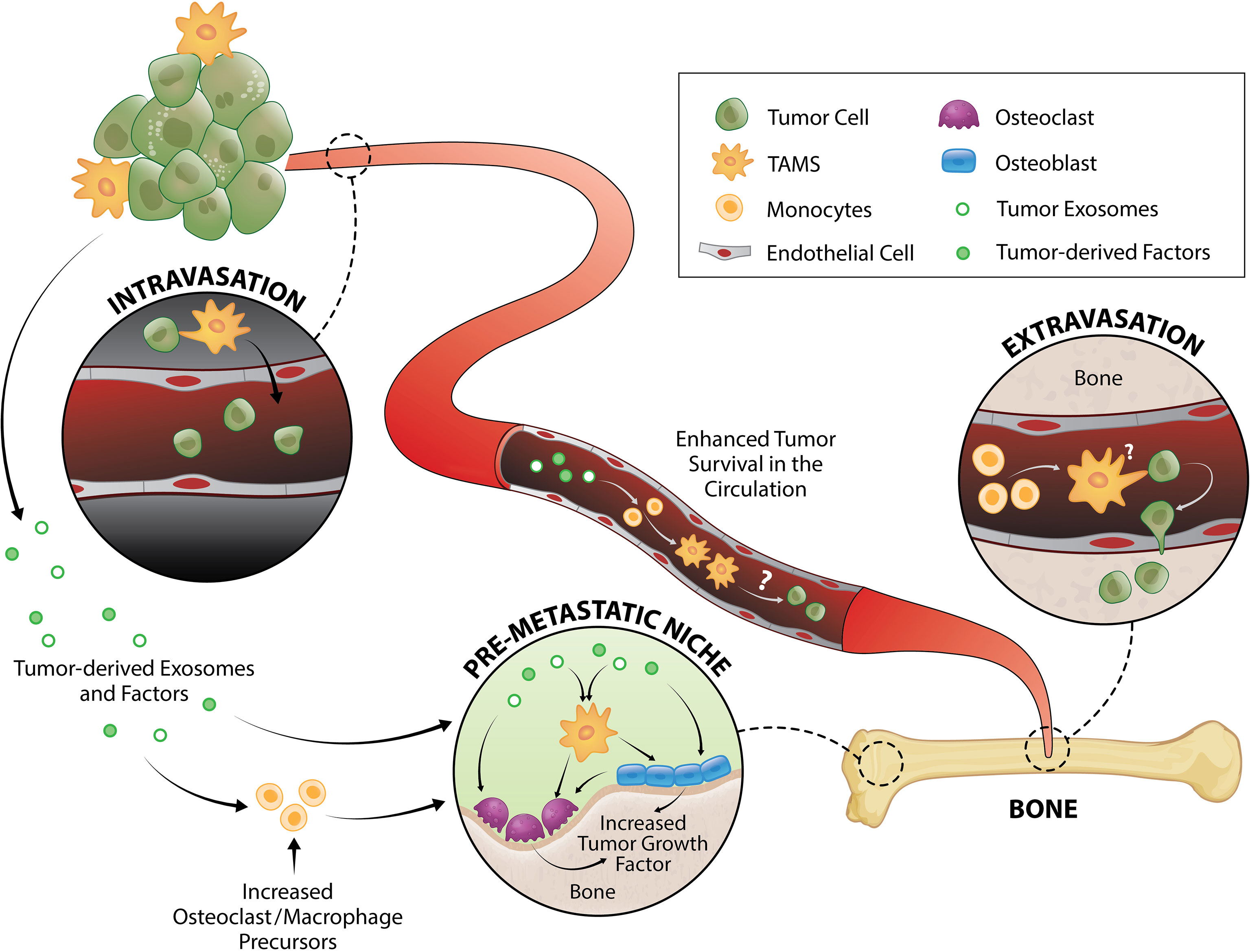
Figure 1 TAM and tumor crosstalk promotes the early metastatic cascade. TAMs contribute to tumor intravasation, extravasation and pre-metastatic niche formation. Direct interaction between TAMs and tumor cells induce migration from primary tumor site to the vessel wall and subsequently, tumors egress into the circulation where they generate fragments that recruit macrophages and monocytes required for successful early metastasis. Tumor-derived exosomes and factors also trigger pre-metastatic niche formation directly or by enhancing monocyte/macrophage recruitment that serves as osteoclast precursors or stimulate osteoblast function, both of which result to release of a myriad of tumor growth factors.
The bone microenvironment is a highly dynamic compartment consisting of diverse cell types as well as an extracellular matrix with a copious array of cytokines. Cells within this microenvironment include the bona fide bone cells (osteoblasts, osteocytes and osteoclasts), hematopoietic and immune cells (notably macrophages, natural killer (NK) cells, B cells and T cells), stromal cells, adipocytes, fibroblasts and endothelial cells (15). Interactions between these cells regulate multiple physiological processes including hematopoiesis, bone remodeling as well as bone and bone marrow homeostasis. Once in bone, cancer cells secrete cytokines and growth factors to interact with these cells, hijacking their normal functions to provide tumor growth requirements. This creates a pathological crosstalk causing lesions that are either osteoblastic (increased bone formation), osteolytic (bone destruction) or mixed (16, 17) depending on the primary mechanism of interference with normal bone remodeling. There is mounting evidence demonstrating that interaction between tumor cells and the supportive stroma plays a crucial role in development and progression of bone metastasis (18–22).
Macrophages are abundant immune cells in the bone microenvironment. They are heterogeneous myeloid cells (23) expressing diverse and adaptive transcriptomes (24, 25), and thus, are capable of rapidly responding to environmental changes (26). Macrophages are present within virtually all tissues (27) throughout life (termed “tissue resident macrophages”) contributing to tissue homeostasis, tissue specific physiology and innate immune surveillance. They can be replenished by the circulating monocyte pool (28) but some are established during embryonic development, persist into adulthood and have the ability to self-renew (29–31). Both monocyte-derived (32–36) and tissue-resident embryonic-derived (36, 37) macrophages have been shown to contribute to tumor development. Three subsets of tissue resident macrophages in the bone have been characterized; erythroblastic island macrophages (EIM), hematopoietic stem cell (HSC) niche macrophages and osteal macrophages (osteomacs). EIM and HSC niche macrophages are located in the bone marrow (38) while osteomacs are present on human and mouse osteal tissues (39).
According to the conventional binary classification, upon tissue insults, various signals may “activate” macrophages into either classically activated pro-inflammatory “M1” [induced by interferon- gamma (IFN-γ), lipopolysaccharide (LPS), tumor-necrosis factor-alpha (TNF-α)] or alternatively activated anti-inflammatory “M2” (40). Based on in vitro studies, M2 macrophages can be subdivided into four distinct subtypes depending on the nature of inducing agent and the expressed markers: M2a, M2b, M2c and M2d (41, 42). However, whether these subtypes occur in vivo remains unclear. It should also be noted that the actual activation state of macrophages is much more complex than the M1 and M2 classification – a concept which originated from the significant biases in macrophage polarization between C57Bl/6 and BALB/c mice (43, 44). Macrophages from various mouse strains differ markedly in their gene expression profiles (45, 46) however these differences were not known at the time the concept was proposed. Furthermore, the M1/M2 nomenclature is not well-supported by large-scale transcriptomic data which instead favors a broad spectrum of activation states (47, 48). Hence for the remainder of this review, we will use “M1-like” and “M2-like” to refer to these conventional activation states rather than M1 or M2 alone which are considered over-simplified.
Macrophages are a major component of the tumor microenvironment, representing 50% of the tumor mass (49). The original and early hypotheses proposed that macrophages are involved in antitumor immunity, however there is substantial clinical and experimental evidence that in majority of cases, macrophages in the tumor microenvironment, termed tumor-associated macrophages (TAMs), enhance progression to malignancy. Once cancers leave the primary site, monocyte-derived macrophages are recruited to support the metastatic cascade. These macrophages have been termed metastasis-associated macrophages (MAMs). Primary (50) and review (51–53) articles have acknowledged the existence of both macrophage types suggesting differences to be based on origin with TAMs arising from resident macrophages at the primary tumor site and MAMs differentiating from inflammatory monocytes/macrophages at the metastatic site. However, this idea is challenged by recent evidence demonstrating that TAMs can be sourced from both monocytes and resident macrophages (36, 37, 54, 55). Furthermore, while TAMs and MAMs have been proposed to be distinct, for example due to different fibroblast activation protein alpha (FAP) expression (56), molecular markers that fully differentiate TAMs and MAMs are still lacking. Therefore, further studies are required to better understand the similar/distinct phenotypes of TAMs and MAMs and to clarify literature inconsistencies, although it is likely that their molecular signatures vary across different cancer types.
It is generally believed that M1-like macrophages have anti-tumor activity while M2-like macrophages promote tumor progression. In many types of human cancers, the density of macrophages is strongly correlated with poor prognosis (57–61) particularly those with M2-like polarization (60, 62–64). Macrophage-derived exosomes have also been shown to facilitate tumor metastasis and development (65–67). Furthermore, macrophage proliferation, differentiation and survival are regulated by colony stimulating factor 1 (CSF1) receptor (CSF1R) which can be activated by two ligands: CSF1 and interleukin (IL)-34 both of which have been associated with poor patient outcomes. For example, overexpression of CSF1 in breast (68, 69), prostate (70), pancreatic (71), hepatocellular (72) and colorectal (73) cancers, and IL-34 expression in hepatocellular carcinoma (74), lung (75) and colorectal (76) cancers are associated with disease progression and unfavorable prognosis. Notably, serum levels of CSF1 are increased in prostate cancer patients with bone metastasis (70). Consequently, targeting macrophages and their tumor-promoting functions are major areas of research in the pursuit of successful therapy (77).
Intravasation, and therefore generation of circulating tumor cells (CTCs), is one the first key steps in the development of distant metastases. This process can be separated into two stages: 1) invasive migration of tumor cells through the extracellular matrix to the vessel wall, and 2) penetration through the vessel wall. Two TAM subsets have been identified that support these processes: migratory macrophages guide cancer cells toward blood vessels and perivascular macrophages assist their entry into the circulation (Figure 1). A recent study showed that C-C motif chemokine receptor type 2 (CCR2)-recruited monocytes differentiate into migratory TAMs that assist with cancer cell motility through the extracellular matrix before differentiating into perivascular macrophages via C-X-C motif chemokine receptor 4 (CXCR4)/CXCL12 signaling (78). CXCR4/CXCL12 has long been known as a key signaling pathway in breast and prostate cancer metastasis to bone (79, 80). In vitro (81, 82) and in vivo (82) imaging demonstrated that direct contact between macrophages and cancer cells facilitate cancer cell migration from the primary tumor site. Upon physical contact, macrophages induce RhoA signaling in tumor cells to form invadopodia (82) - actin-rich membrane protrusions that degrade the extracellular matrix. This interaction is further promoted by a positive feedback loop whereby cancer cells produce CSF1 and TAMs express epidermal growth factor (EGF) (81, 83) which is a positive regulator of RhoA signaling (84, 85). Once in the blood vessel, vascular endothelial growth factor A (VEGFA) signaling from TIE2+ perivascular macrophages (86, 87) causes vascular leakiness, allowing tumor cell egress.
CTCs travel as single cells or in clusters (88) in the circulation where they are exposed to various insults including immune destruction and mechanical forces due to fluid shear stress. Very little is known about the survival mechanisms that CTCs employ while in the circulation, but putative mechanisms have been proposed and reviewed elsewhere (89–91). Using an intravital two-photon lung imaging model in mice, it has been shown that CTCs generate fragments that serve as immune-interacting intermediates to recruit phagocytic myeloid cells that were predominantly macrophages and monocytes for successful early metastasis (92) (Figure 1). Although CTCs use various strategies to survive in the circulation, their metastatic potential ultimately depends on rapid extravasation into another tissue. Cancer cell extravasation appears to be one of the least studied steps in the metastatic cascade perhaps due to the difficulty in investigating this event that occurs well hidden within intact organs. As CTCs encounter capillary beds, where extravasation typically occurs (93, 94), they undergo physical arrest and are thought to form adhesive interactions with their surface receptors to the complementary sites on the endothelium (95, 96). CTCs then transmigrate via paracellular migration where they extend projections between adjacent endothelial cells into the extravascular space before moving their cell body, nucleus, and trailing edge across the endothelium (93, 97–99). In the bone marrow, the sinusoidal capillaries are lined with endothelial cells and discontinuous basal lamina (100) which may facilitate simpler extravasation and therefore contribute to high metastatic incidence in bone (1, 2).
MAMs have been implicated in breast cancer cell extravasation in the lung (50, 101). This population originated from circulating monocytes and is characterized as F4/80lowCD11bhighLy6Clow macrophage population (50, 102). While the specific mechanism underlying the macrophage contribution in CTC extravasation is not fully understood, it has been shown that direct tumor cell-macrophage interaction is required for this process (101) (Figure 1). Of note, elevated peripheral blood monocytes (macrophage precursors) have been associated with poorer disease prognosis in several cancer types (103–108), which could be due to monocytes serving as MAMs precursors. Furthermore, overexpression of the monocyte chemoattractant protein-1 (MCP-1, also known as CC chemokine ligand 2/CCL2 which is the ligand for CCR2) in human prostate cancer cells increases macrophage accumulation and enhances bone metastasis (109). In breast cancer bone metastasis, MAMs are largely derived from Ly6C+CCR2+ monocytes and they express high levels of CD204 and IL4R (35). While it was shown that these MAMs promote bone metastasis in an IL4R-dependent manner (35), it is unknown whether they are directly involved in extravasation to bone or other steps in the metastatic cascade.
Bone marrow-derived macrophages (BMDMs) have been implicated in tumor growth and metastasis such as in metastatic lung cancer (110, 111). Using a murine model, Cho et al. demonstrated that co-injection with mammary carcinoma cells with bone marrow-derived M2-like macrophages promoted tumor growth and lung metastasis (110). Given the bone and its marrow are abundant in resident macrophages, it is likely that they provide an attractive microenvironment for metastases because bone and bone marrow macrophages can largely contribute to formation of a pro-tumorigenic niche.
It is now being increasingly recognized that organs of future metastasis are not passive receivers of CTCs; instead, they have been primed and actively modified to support incoming cancer cell needs even prior to metastatic spread. These primed sites are termed “pre-metastatic niches” and they form as a result of tumor-secreted factors and tumor-shed extracellular vesicles (112) (Figure 1). Osteoclasts have been implicated in pre-metastatic niche formation for bone metastases. They are myeloid-derived cells commonly known as the bone-resorbing cells. While some still regard osteoclasts as a resident macrophage in bone, in vivo evidence support that osteomacs and osteoclasts are distinct mature myeloid cell types (113, 114). In metastatic breast cancer, tumors secrete lysyl oxidase (LOX) that triggers osteoclastic bone resorption resulting to formation of pre-metastatic lesions (115, 116) as well as release of bone-matrix stored growth factors including insulin-like growth factors (IGFs), transforming growth factor-β (TGFβ), fibroblast growth factor (FGFs), and bone morphogenetic proteins (BMPs) which all have pro-tumorigenic effects (117–119). Recently, Yuan et al. also demonstrated that breast cancer exosomes contribute to pre-metastatic niche formation in the bone by transferring exosomal miR-21 to osteoclasts (120) which triggers their activation and survival (121). Osteoclastogenesis in metastatic bone cancer has been suggested to be mediated by CD137L-CD137 signaling pathway where CD137 enhances monocyte/macrophage migration and differentiation to osteoclasts (122) (Figure 1).
After arriving in the bone microenvironment, cancer cell survival is determined by their ability to resist immunity and other bone tissue defenses and settle within the specialized local niches. Newly disseminated cancer cells are particularly vulnerable to immune surveillance by macrophages and T cells (Figure 2). In a colorectal cancer model, TAMs have been shown to be pivotal constructors of tumor collagenous extracellular matrix, upregulating synthesis and assembly of collagens (collagen types 1, VI, XIV), and instructing their deposition in areas of tumor development (32). Coordinated tumor and tumor-stromal cell interactions subsequently remodel these extracellular matrix components forming a physical barrier to evade immune surveillance (123).
The phagocytic function of macrophages plays an essential role in bone homeostasis (124) where they clear pathogens, debris as well as apoptotic cells (efferocytosis). Under normal physiology, efferocytosis serves as a waste disposal mechanism but also a pro-resolving phenotype in macrophages (125). In bone metastasis, efferocytosis of apoptotic cancer cells via milk fat globule-EGF facto 8 (MFG-E8) bridge protein induces an M2-like phenotype in TAMs (126) which has a well-documented crucial role in immunosuppression including influencing T cell recruitment and function (127). Efferocytic M2-like macrophages under normal circumstances (128, 129) and M2-like TAMs in cancer setting (130) have been shown to secrete TGF-β1 which can, 1) directly inhibit cytotoxic T cell (CTL) expression of cytolytic gene products (perforin, granzyme A, granzyme B, Fas ligand, IFN-γ) required for cytotoxicity (131) or, 2) act indirectly by stimulating differentiation of regulatory T cells (Tregs) (132) (Figure 2). In vitro, macrophage efferocytosis of prostate cancer cells induced expression of inflammatory cytokines including CCL5, CXCL1, CXCL5 and IL-6 (133) which can orchestrate growth of bone metastases (133–137). In particular, CXCL5 has been shown to accelerate growth of metastatic prostate cancer in bone (133).
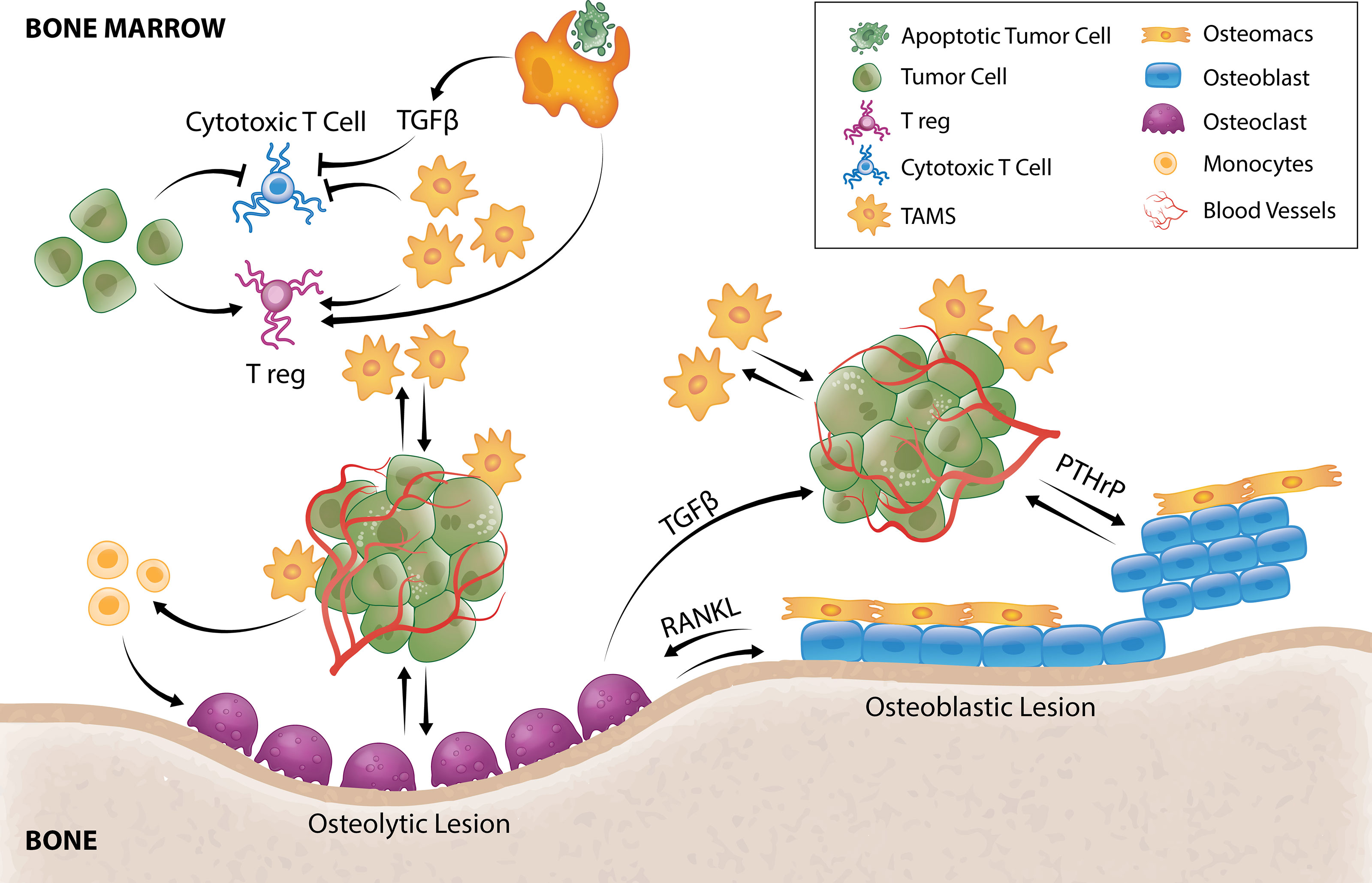
Figure 2 TAM and tumor crosstalk promotes tumor survival and outgrowth in bone. Efferocytosis of apoptotic tumor cells by TAMs results in TGFβ secretion that inhibits cytotoxic T cells and stimulates Tregs. TAMs and tumor cells can also directly hijack immune checkpoint pathways to negatively regulate T-cell anti-tumor function. TAMs secrete a multitude of pro-angiogenic factors that induce tumor angiogenesis allowing delivery of oxygen, nutrients and growth factors. Tumor cells can directly stimulate pro-tumorigenic osteoblastic or osteolytic lesion formation and/or recruit TAMs or osteoclast precursors to support these processes. In osteolytic lesions, tumors secrete factors that directly stimulate osteoclastic-mediated resorption to release bone-derived tumor growth factors or stimulate osteoclastogenesis by promoting osteoblast-derived RANKL. Osteoclastogenesis also releases TGFβ from the bone matrix that directly stimulates tumor cells to secrete PTHrP that can enhance osteoblasts function to form osteoblastic lesions or enhance osteoblast production of RANKL to form osteolytic lesions. Osteomacs which are abundant within osteoblastic lesions also support osteoblast-mediated bone anabolism, suggesting that they could be a key driver of tumor-induced bone formation.
Tumor cells are also well-known to hijack the programmed cell death-1 (PD-1)/PD-1 ligand (PD-L1) pathway to escape immunosurveillance (138). In fact, it is one of the best-studied and most promising immune checkpoint drug targets with benefits reported in different cancer types (139), although bone metastases appear to impair its efficacy (140). PD-1 is an immune checkpoint receptor expressed by activated T cells while tumor cells frequently express PD-L1. PD-1/PD-L1 engagement has been shown to negatively affect CD8+ T cell activity (141), allowing tumors to escape T cell-mediated cell death. TAMs also express high levels of PD-L1 in different types of cancers (142–145), indicating that they could also directly suppress T cell cytotoxic functions which is reflected by improved anti-tumor T cell activity following macrophage depletion (145–147). Recently, Gordon et al. showed that TAMs from human colorectal cancer samples and colon cancer cell line CT26 also express high levels of PD-1 (148). PD-1 expression in TAMs negatively correlated with phagocytic potency against tumor cells while blockade of PD-1/PD-L1 improved phagocytosis and reduced tumor growth (148). Together, these suggest that the PD-1/PD-L1 pathway has a significant role in TAM function and tumor survival.
Another immune checkpoint pathway that could negatively regulate T-cell anti-tumor function in the setting of cancer is cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) and macrophage Clever-1. CTLA-4 upregulation and binding with CD80/CD86 promotes T cell anergy (149), and cancer cells (150–152) and TAMs (153) have been shown to express CD80 and CD86. The macrophage Clever-1, a multifunctional adhesion and scavenger receptor expressed by immunosuppressive macrophages and TAMs, has also been shown to have inhibitory effect on CD8+ T cells (154). Viitala et al. showed that genetic deficiency of macrophage Clever-1 or its blockage resulted in activation of CD8+ T cells and impaired tumor growth (154). Interestingly, these immunostimulatory effects were comparable with PD-1 checkpoint inhibition (154). Overall, cancer cells can either suppress anti-tumor immune responses directly or indirectly by employing other cells such as TAMs (Figure 2).
The location of DTCs in the bone could be the key to their fate. Tumor cells delivered to the bone remodeling compartments (metaphyseal trabecular bone regions) will be exposed to a microenvironment rich in growth factors that promote growth and survival and thus, they may proliferate immediately (155). Conversely, those arriving in the quiescent region (endosteal surface) will encounter a microenvironment that promotes tumor cell dormancy (155). Given that the inactive endosteal surface predominates (156), it is conceivable that DTCs mainly undergo dormancy once arrested in bone. Indeed, DTCs are detected in the bone marrow of breast (157–159) or prostate (160) cancer patients that neither correlated with tumor stage nor size which could indicate tumor cells have entered dormancy. Similarly, DTCs have been detected in bone marrow aspirates of patients with ovarian and endometrial cancer that did not correlate with established clinicopathological factors (161). Moreover, the metaphyseal trabecular region of long bones is normoxic while the diaphyseal region is more hypoxic (162) and hypoxia regulates key tumor dormancy factors (163). Hypoxia also supports macrophage recruitment via hypoxic tumor-derived cytokines (164, 165) and modifies polarization of macrophages, selectively promoting M2-like phenotype in multiple cancers (166–169) through activation of ERK signaling (166).
Recently, an in vitro study showed that macrophage exosomes regulate dormancy of breast cancer cells in bone marrow stroma (170). Exosomes from M2-like macrophages were shown to sustain quiescence and reduce proliferation of cancer cells while M1-like macrophage-derived exosomes reversed dormancy by NFκB activation (170). In mice with subcutaneous tumors and patients with non-small cell lung cancer (NSCLC), TAMs have been shown to exacerbate tumor hypoxia via AMP-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) activation (171) which could perpetuate tumor dormancy.
Tumor dormancy can last for years and decades, but what is perhaps the most clinically relevant phase is the exit from dormancy, also termed the “reactivation” phase, that requires escape from the dormant state to proliferate actively and form micrometastases. This process is nursed by the surrounding supportive niches (172). The osteoblastic niche has been shown to play an important role in controlling tumor cell dormancy (173, 174) through the Wnt5a/ROR2/SIAH2 signaling axis (174) while remodeling of the endosteal niche by osteoclasts have been shown to induce reactivation (173). Data is lacking that directly demonstrates the role of TAMs in tumor reactivation in bone metastasis setting. However, this would not be surprising given it has been shown that: 1) the tumor dormancy-reactivation process is reversible (173), 2) TAMs provide or stimulate tumor dormancy signals, and 3) inhibition of dormancy signals reawakens dormant cancer cells (175). Of note, tumor-derived vascular cell adhesion molecule 1 (VCAM-1) has been shown to promote transition from indolence to overt metastasis by recruiting CD11b+ monocytes and increasing osteoclast activity (176). Recently, abscisic acid was reported to regulate dormancy of prostate cancer disseminated tumor cells in the bone marrow (177) and separately studies identified abscisic acid as an inducer of the M1 phenotype (178). Further studies that investigate the contribution of TAMs in dormancy and tumor reactivation are warranted.
Following reactivation, the final step of metastasis occurs when DTCs proliferate, becoming independent of the microenvironment and ultimately modifying it to support outgrowth. While a hypoxic environment induces dormancy, angiogenesis is required for tumor proliferation as it allows delivery of oxygen, nutrients and growth factors. For decades, macrophages have been implicated in neovascularization of tumors (179). In healthy tissues, blood vessels are in a quiescent state and angiogenesis is only transiently activated in response to certain stimuli. Conversely, in tumor progression, an “angiogenic switch” results with continuous sprouting of de novo vessels. The tumor vasculature differs from a normal vascular network as it is characterized by hyperpermeability, excessive and convoluted branching and erratic blood flow (180). TAMs are important for this angiogenic switch as they represent a potent source of a multitude of other pro-angiogenic factors including those from the EF-hand calcium-binding cytosolic (S100A) protein family, semaphorins family and chitinase-like proteins (181). They have also been shown to promote angiogenesis in human tumors (182, 183) and in animal models of breast (184) and prostate (185) cancers. Tumor cells, under hypoxic conditions, have been proposed to produce oncometabolites including lactate and succinate that induce a pro-angiogenetic phenotype in TAMs (186). M2-like TAMs secrete VEGF-A (183) which is considered a major mediator of tumor angiogenesis (187) and an indicator of metastatic potential to bone in malignant prostate cancer (188). Conversely, repolarization of TAMs towards an M1-like phenotype leads to tumor vessel normalization (189, 190). For new capillaries to sprout, degradation of the host vessel at specific sites needs to occur and this process is mediated by proteinases such as matrix metalloproteinases (MMPs). In some tumors, TAMs have appeared to be a major source of MMP9 which mediates extracellular matrix degradation and releases VEGF-A from the extracellular matrix reservoir (18, 191–193). The release of VEGF-A can then induce a positive feedback loop on angiogenesis by further recruiting pro-angiogenic and immunosuppressive macrophages through their VEGF receptor (VEGFR1) (194, 195).
Significant effort has been made into characterizing the mechanisms associated with overt tumor growth in the skeleton – a paradigm commonly referred to as the “vicious cycle” (196). Herein, tumor cells can stimulate excessive bone formation (osteoblastic) or resorption (osteolytic) leading to disruption of bone integrity and production of factors that fuel cancer proliferation which then results to further bone formation or destruction (118). Osteolytic bone lesions are usually characteristic of the majority of breast cancers and non-small cell lung cancer while osteoblastic lesions are common in prostate and small cell lung cancers (197). Of note, in breast (198) and prostate (199) cancer patients, both type of lesions can be present. Tumor cells can instigate bone formation by producing osteogenic factors (Figure 2) including BMPs, EGFs, endothelin-1 and platelet derived growth factor (PDGF) (200). In turn, activated osteoblasts can produce pro-tumorigenic factors (Figure 2) including IL-6, CCL2 and VEGF (201). Prostate cancer cell exosomes have been shown to stimulate bone formation by inhibiting osteoclast fusion and differentiation (202) causing remodeling imbalance that ultimately results in net bone formation. Prostate cancer cells also produce CCL2 to recruit macrophages/TAMs or osteoclasts (109) that assist with pro-tumorigenic lesion formation. In vitro, bone marrow macrophages and prostate tumor cell interaction upregulates cathepsin K expression in macrophages which promotes tumor progression in bone (203).
CD68+ TAMs are present within patients’ prostate cancer skeletal lesions where they are directly associated with woven bone (204). Similarly, in a mouse model of prostate cancer bone metastasis, F4/80+ macrophages were abundant within the skeletal lesions and osteomacs were directly associated with de novo pathological bone (204). Osteomacs, including their efferocytic function, support osteoblast-mediated bone anabolism (113), suggesting that they could be a key driver of tumor-induced bone formation. This idea is strongly supported by the significant reduction in pathological woven bone deposition when CD169+ osteomacs/TAMs were depleted (204). Nonetheless, further investigation into the specific molecular interaction between osteoblasts and osteomacs are required to understand how these pathways are modulated in the tumor setting.
In metastatic cancers with osteolytic lesions, tumors secrete factors such as IL-8, IL-11, CSF1 and TNFα which directly stimulate osteoclastic-mediated resorption (Figure 2) to release bone-derived tumor growth factors (117). Recently, tumor-derived monoamine oxidase A (MAOA) was also shown to stimulate osteoclastogenesis by promoting osteoblast-derived receptor activator nuclear kappa B ligand (RANKL) and IL6 expression (22). As a result of osteoclastic bone resorption, TGFβ is released from the bone matrix and directly stimulates tumor cells to secrete parathyroid hormone–related protein (PTHrP) (205) (Figure 2). PTHrP has dual effects on bone remodeling. It stimulates osteoblasts which can result in osteoblastic lesion formation (206, 207) but it is also a potent stimulator of osteoclastogenesis by enhancing osteoblast production of RANKL (Figure 2) and CCL2 (208, 209). There is currently very limited literature on the contribution of TAMs in the formation of osteolytic lesions, but CD68+ macrophages have been reported within osteolytic lesions of prostate cancer patients (210). Furthermore, Movila et al. demonstrated that macrophage migration inhibitory factor (MIF) mainly produced by macrophages within the bone lytic site acts as a chemoattractant to osteoclast precursors, leading to further recruitment of osteoclasts to sustain bone destruction (211).
Bone metastases remain incurable and current management are focused on minimizing pain, resolving or minimizing the risk of developing skeletal related events (SREs) and inhibiting tumor progression. Current treatment usually includes palliative radiotherapy (212) and systemic chemotherapy which directly targets malignant cells. Bone metastasis harbors resistance to chemotherapeutic drugs perhaps contributed to by bone being less perfused than other organs and thus, drugs which are administered intravenously do not reach the site in sufficient doses. In addition, pre-clinical work suggests that chemotherapies could condition the microenvironment via macrophage influx to be more receptive to metastasis (213). Efforts have been made in the utilization of bone-targeted nanoparticles loaded with anti-cancer drug which have demonstrated potential in inhibiting bone metastases (214–216). Extensive research aimed at understanding the “vicious cycle” that occurs in metastatic bone diseases has also led to approval of drugs that target the remodeling imbalance including bisphosphonates and the RANKL inhibitor denosumab, both of which target osteoclast function (217). However, while these drugs improve patients’ quality of life by reducing pain, fractures and inhibiting development of new lesions (218–220), metastatic bone cancer still progresses and thus, they have little or no benefit in overall survival (218–221). Therefore, management of bone metastases remains a significant clinical challenge and effective treatments are still an unmet clinical need. TAMs are involved in all steps of the metastatic cascade to the skeleton and have tumor-permissive and immunosuppressive characteristics. Therefore, targeting TAMs and their pro-tumorigenic functions has a promising potential in anti-cancer therapy (222) (Figure 3).
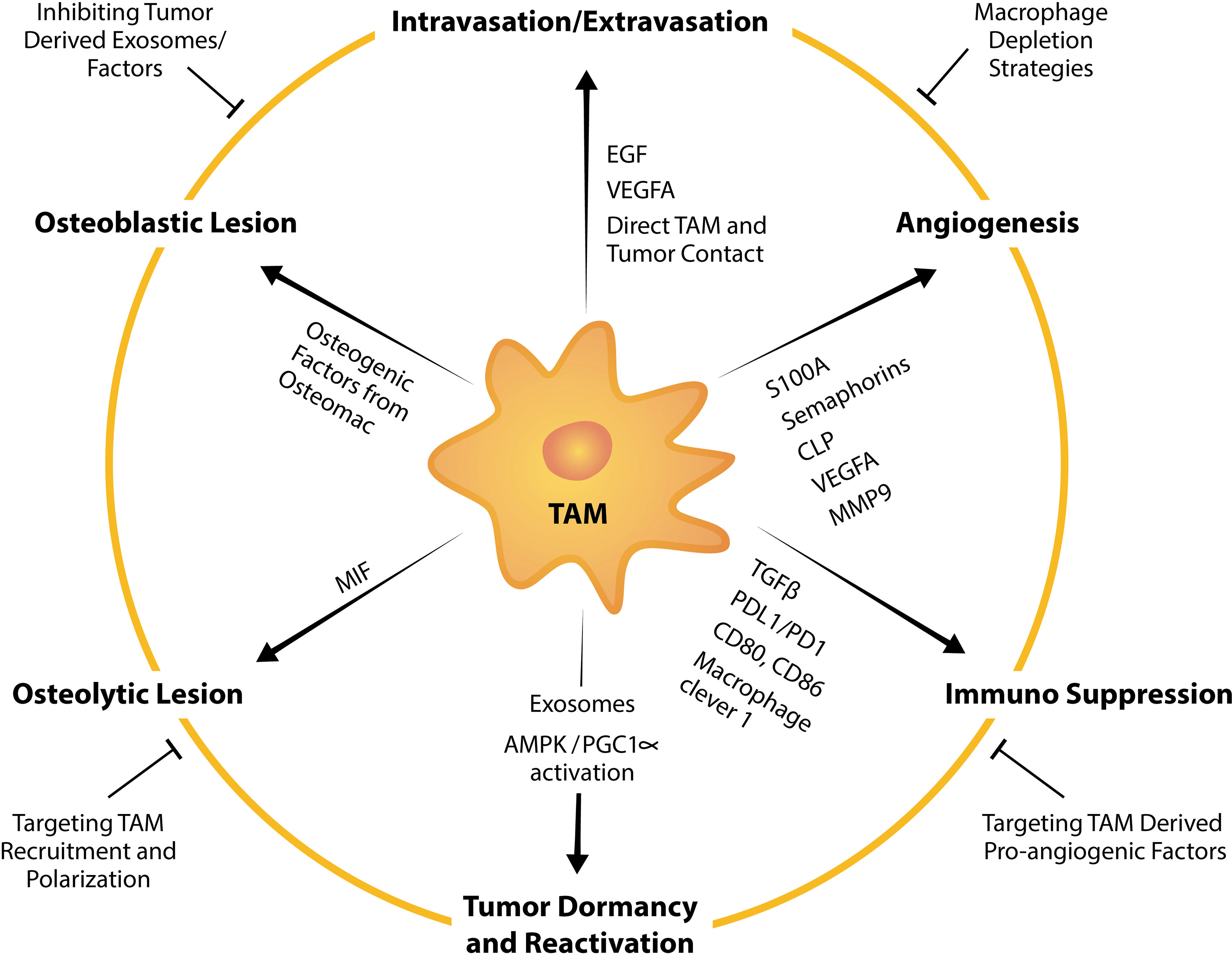
Figure 3 TAM factors and markers that facilitate tumor bone metastasis and strategies to suppress TAM-mediated promotion of metastasis. TAMs influence tumor intravasation, extravasation, angiogenesis, dormancy, reactivation, formation of osteolytic and osteoblastic lesions, and immunosuppression by expressing or releasing pro-tumorigenic factors. For example, TAMs are a potent source of a multitude of pro-angiogenic factors including VEGFA, MMP9 and those from the EF-hand calcium-binding cytosolic (S100A) protein family, semaphorins family and chitinase-like proteins. Strategies to target TAM pro-tumorigenic functions summarized in this schematic include broad macrophage depletion strategies, targeting pro-angiogenic factors from TAMs, inhibiting TAM recruitment, TAM repolarization and inhibiting tumor-derived exosomes or factors that instigate TAM and tumor pathological crosstalk.
Targeting the CSF1/CSF1R inhibitors has gained the most attention in this context with various approaches currently under clinical development for treatment of several cancer types including advanced castration-resistant prostate cancer (CRPC) with bone metastases (223). Most of the agents targeting the CSF1/CSF1R signaling axis are CSF1R inhibitors (224) with only two current clinical-stage programs targeting CSF1 and none targeting IL-34 thus far (223). Emerging data on the tolerability of CSF1/CSF1R-targeting agents indicate a relatively safe profile, with some studies reporting no dose-limiting toxicities (225–227). CSF1/CSF1R inhibitors have been shown to deplete TAMs (224, 226, 228) and reduce M2-like macrophage recruitment (229, 230). While these agents are not under clinical trial for metastatic bone disease other than advanced CRPC (NCT01499043), preclinical outcomes have been promising especially given CSF1R inhibitors have an additional benefit of targeting osteoclasts (231, 232). In a mouse model of metastatic lung cancer with osteolytic bone lesions, knockdown of CSF1 reduced the incidence of bone metastases (233). Similarly, in a rat model of mammary adenocarcinoma (234) and mouse models of breast cancer (232, 235, 236), treatment with CSF1R inhibitors prevented bone metastases and formation of osteolytic lesions. CSF1R targeting has also been demonstrated to successfully abrogate TAM infiltration and thus disrupt tumor promotion in animal models of prostate cancer (237, 238), however, whether these drugs have a role in inhibiting osteoblastic lesion formation remains underexplored. Overall, targeting the CSF1-CSF1R axis appear to be a promising strategy, however, it would be problematic to systemically deplete macrophages for a long period and thus, a more targeted approach would be ideal.
An example of a more directed approach are bisphosphonates which are bone-targeted drugs routinely used in metastatic disease with bone involvement to prevent or delay SREs and improve quality of life (219, 239). Given they effectively inhibit osteoclasts (240), bisphosphonates could have direct effect on preventing premetastatic niche formation and tumor proliferation by interfering with the “vicious cycle”. Interestingly, real-time intravital imaging has shown that bisphosphonates in extra-skeletal tumors are mainly taken up by TAMs (241). Moreover, bisphosphonates have been shown to inhibit macrophage proliferation and induce apoptosis in vitro (242, 243). Such effect on TAMs/macrophages could therefore contribute to the anti-tumor effects of these drugs.
The most investigated bisphosphonate to target TAMs in preclinical studies is the encapsulated clodronate (clodronate liposomes) which are preferentially taken up by macrophages owing to their phagocytic activities. In fact, clodronate (BONEFOS®) is approved for use in treatment of tumor-induced osteolysis in 67 counties though it remains commercially underdeveloped in the United States. This strategy has shown promise in inhibiting bone metastases in animal models of metastatic lung (243, 244), prostate (204) and breast cancers (245). In metastatic liver cancer model, macrophage depletion by clodronate liposomes significantly inhibited tumor progression, angiogenesis and lung metastasis (246). In mouse models of breast cancer, Zoledronic acid, another type of bisphosphonate, was shown to reduce TAM number and repolarized TAMs to M1-like anti-tumoral phenotype, reduce mammary carcinogenesis (247) and skeletal metastases (245). While these preclinical studies were promising, the clinical benefit of bisphosphonates is still limited to managing skeletal complications without improving patient’s overall survival. Current efforts are focused on utilizing bisphosphonates for a more targeted delivery of anti-cancer drugs to bone. While most studies on bisphosphonate-drug conjugates have been conducted in vitro, preliminary results are promising and reviewed elsewhere (248).
Targeting the CCL2-CCR2 axis has also been attractive given its role in TAM recruitment. CCL2 promotes bone metastasis in experimental models of prostate cancer (249, 250) while its inhibition hinders TAM recruitment (251, 252) and correlates with reduced tumor burden (253). In in vivo models of metastatic prostate cancer, treatment with neutralizing anti-CCL2 antibodies either reduced systemic tumor burden including bone lesions (254) or completely inhibit bone metastases (255). CCL2 is increased by treatment with the chemotherapeutic drug docetaxel and protects prostate cancer cells from docetaxel-induced toxicity (256) but when combined with CCL2 blockade, docetaxel had striking impact on tumor suppression (254, 255). This anti-tumor efficacy demonstrated in preclinical studies were preceded by establishment of clinical trials in solid and metastatic cancers (257), however, the outcomes have somewhat been disappointing. The anti-CCL2 monoclonal antibody carlumab failed to inhibit tumor growth in early-stage clinical trials in prostate cancer as it was unable to sustain CCL2 blockade due to induction of compensatory mechanisms (258). A humanized neutralizing anti-CCR2 monoclonal antibody also went through a clinical trial for treatment of bone metastasis from solid tumors (NCT01015560). While the treatment was well-tolerated, only 14 out of 43 patients had considerable reduction in urine n-telopeptide, a biomarker of bone turnover rate, and the anti-tumor outcomes have not been disclosed despite study completion. Therefore, there is not enough evidence to support efficacy of CCL2/CCR2 targeting in skeletal metastases. Of note, interruption of CCL2 inhibition in models of metastatic breast cancer accelerated metastases and death (259). This highlights our incomplete understanding of the CCL2-CCR2 signaling network and that caution should be taken when considering targeting this axis in metastatic diseases.
Macrophages are extremely plastic; they can have pro-tumoral characteristics or be tumoricidal. This indicate that strategies which indiscriminately target TAMs/macrophages might only be partially effective, could induce undesirable side effects and long-term toxicities. Reprogramming TAMs could therefore be a more efficacious approach, providing a strategy where pro-tumoral macrophages can be re-educated towards an anti-tumoral phenotype to create a microenvironment that reject rather than nurture tumor cells. There are several strategies currently used to reprogram TAMs to be anti-tumorigenic in preclinical and clinical investigation including toll-like receptor agonists and monoclonal antibodies that induce TAM anti-tumor effects (260), and Hu et al. have recently identified many more (261). Trabectedin is an anti-cancer agent that in addition to directly targeting certain types of cancer cells, it also induces apoptosis in monocytes and macrophages. In an experimental model of prostate cancer, it was found to reduce M2-like macrophages in the marrow and skeletal metastatic tumor growth in bone (262). There is a paucity of clinical trials that investigate macrophage targeting strategies in the context of metastatic bone disease, though macrophage reprogramming in the context of efferocytosis has shown promise in preclinical studies. Uptake of apoptotic prostate cancer cells by BMDMs induced an inflammatory response that promoted bone metastatic growth but treatment with IFN-γ reprogrammed macrophages towards an M1-like state that mitigated the pro-tumoral inflammatory response (263). Chemotherapy and radiotherapy induce apoptosis in cancer cells, resulting to increased efferocytosis and subsequent suppression of inflammatory responses. Therefore, combining conventional chemotherapy and radiotherapy with an efferocytosis-targeted treatment could be a promising therapeutic approach that should be the focus of future research. While re-educating TAMs hold promise in several cancer types (260), further studies are required to fully understand its efficacy in metastatic bone diseases especially given very recent report on transcriptomically-defined “M1” macrophages associated with an aggressive cancer biology (264). Interestingly, blockade of PD-1/PD-L1, the most commonly used immune checkpoint blockage therapy in the clinic that has achieved resolution of malignancies (265), polarizes macrophage towards an M1-like phenotype (266–268) and increases TAM phagocytic potency against tumor cells (148). In addition, combining anti-PD1 therapy with metformin-loaded macrophage-derived microparticles that potently polarized TAMs from M2-like to M1-like state, boosts anti-cancer efficacy (269). Recently, PD-1 blockage was also demonstrated to have an added benefit of inhibiting osteoclastogenesis resulting in reduced bone destruction and pain (193). Of note, there are also reports indicating that TAMs might limit anti-PD-1 treatments for example through preventing CD8+ T cells from reaching tumor cells (147) or by removing anti-PD-1 antibodies from T cells through Fc-Fcγ receptors binding (270), though the latter can be prevented by blocking Fc/Fcγ receptor interactions (270).
Other strategies are focused on inhibiting the tumor-promoting functions of TAMs including angiogenesis (Figure 3). Anti-angiogenic therapies which are largely VEGF inhibitors are used in the clinic for several cancers (271) with the first anti-angiogenic drug Bevacixumab (Avastin®) approved for use in 2004 by the Food and Drug Administration (FDA). While short-term relief from tumor growth is achieved in many patients, primary or acquired resistance is not uncommon and this is currently under intensive investigation (272). The existence of several novel angiogenesis regulators, such as those secreted by TAMs (181), that were not considered in the therapeutic approaches might explain the limited efficacy of current anti-angiogenic therapy. Recent studies implicate TAMs in decreased efficacy of anti-angiogenic therapy (273, 274) suggesting that targeting angiogenesis is more complex than originally thought. Findings from in vivo studies suggest that combinatory targeting of VEGF and other alternative effectors (275, 276) or combining anti-angiogenic drugs with TAM-targeted agents are more effective approaches (252, 277, 278), however, these are yet to be examined in metastatic bone diseases.
TAMs promote skeletal metastases via crosstalk with tumor cells which occurs at all stages of the metastatic cascade. Therefore, whilst cure of metastatic bone diseases remains elusive, research to date strongly support that TAMs are an attractive target. Knowledge is still lacking on how they can be appropriately targeted to effectively “cure” bone metastases. Research efforts should be focused on further understanding the underlying cellular and molecular mechanisms governing TAM and cancer interactions and how these can be directed to prevent metastatic progression. Once tumor cells are established in the bone, it is clear that a cure cannot be developed until the fundamental drivers of dormancy, resistant mechanisms, reactivation and outgrowth are discovered. Although there is increasing appreciation of TAM participation in these events, further investigation of the associated complex processes is crucial. Improved understanding of TAM contribution to these events could potentially identify effective strategies that can be used to rebalance the bone microenvironment to allow conventional cancer therapies to destroy metastatic tumor cells without compromising the skeleton, ultimately curing this terminal pathology.
Both authors identified the focus and overall direction of the review. LB composed an original version of the paper and LM provided additional writing and editing. Both authors reviewed the final version. Figures were composed by LB with the assistance of LM and a graphic artist (Ken Rieger). All authors contributed to the article and approved the submitted version.
This work was supported in part by the NIH: NCI and a University of Queensland Research Fellowship Scheme.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Brodowicz T, O'Byrne K, Manegold C. Bone Matters in Lung Cancer. Ann Oncol (2012) 23(9):2215–22. doi: 10.1093/annonc/mds009
2. Ibrahim T, Farolfi A, Mercatali L, Ricci M, Amadori D. Metastatic Bone Disease in the Era of Bone-Targeted Therapy: Clinical Impact. Tumori (2013) 99(1):1–9. doi: 10.1700/1248.13780
3. Svensson E, Christiansen CF, Ulrichsen SP, Rørth MR, Sørensen HT. Survival After Bone Metastasis by Primary Cancer Type: A Danish Population-Based Cohort Study. BMJ Open (2017) 7(9):e016022. doi: 10.1136/bmjopen-2017-016022
4. Hong S, Youk T, Lee SJ, Kim KM, Vajdic CM. Bone Metastasis and Skeletal-Related Events in Patients With Solid Cancer: A Korean Nationwide Health Insurance Database Study. PloS One (2020) 15(7):e0234927. doi: 10.1371/journal.pone.0234927
5. Li TT, Shore ND, Mehra M, Todd MB, Saadi R, Leblay G, et al. Impact of Subsequent Metastases on Costs and Medical Resource Use for Prostate Cancer Patients Initially Diagnosed With Localized Disease. Cancer (2017) 123(18):3591–601. doi: 10.1002/cncr.30784
6. Skov Dalgaard K, Gammelager H, Sværke C, Kurics T, Cetin K, Christiansen CF. Hospital Use Among Patients With Lung Cancer Complicated by Bone Metastases and Skeletal- Related Events: A Population-Based Cohort Study in Denmark. Clin Epidemiol (2015) 7:363–8. doi: 10.2147/clep.S78301
7. Timp W, Feinberg AP. Cancer as a Dysregulated Epigenome Allowing Cellular Growth Advantage at the Expense of the Host. Nat Rev Cancer (2013) 13(7):497–510. doi: 10.1038/nrc3486
8. Obenauf AC, Massagué J. Surviving at a Distance: Organ-Specific Metastasis. Trends Cancer (2015) 1(1):76–91. doi: 10.1016/j.trecan.2015.07.009
9. Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic Nerve Development Contributes to Prostate Cancer Progression. Science (2013) 341(6142):1236361. doi: 10.1126/science.1236361
10. Marchesi F, Piemonti L, Mantovani A, Allavena P. Molecular Mechanisms of Perineural Invasion, a Forgotten Pathway of Dissemination and Metastasis. Cytokine Growth Factor Rev (2010) 21(1):77–82. doi: 10.1016/j.cytogfr.2009.11.001
11. Schmitd LB, Beesley LJ, Russo N, Bellile EL, Inglehart RC, Liu M, et al. Redefining Perineural Invasion: Integration of Biology With Clinical Outcome. Neoplasia (2018) 20(7):657–67. doi: 10.1016/j.neo.2018.04.005
12. Roh J, Muelleman T, Tawfik O, Thomas SM. Perineural Growth in Head and Neck Squamous Cell Carcinoma: A Review. Oral Oncol (2015) 51(1):16–23. doi: 10.1016/j.oraloncology.2014.10.004
13. Lugassy C, Kleinman HK, Engbring JA, Welch DR, Harms JF, Rufner R, et al. Pericyte-Like Location of GFP-Tagged Melanoma Cells: Ex Vivo and In Vivo Studies of Extravascular Migratory Metastasis. Am J Pathol (2004) 164(4):1191–8. doi: 10.1016/s0002-9440(10)63207-5
14. Fidler IJ. The Pathogenesis of Cancer Metastasis: The 'Seed and Soil' Hypothesis Revisited. Nat Rev Cancer (2003) 3(6):453–8. doi: 10.1038/nrc1098
15. Buenrostro D, Mulcrone PL, Owens P, Sterling JA. The Bone Microenvironment: A Fertile Soil for Tumor Growth. Curr Osteoporos Rep (2016) 14(4):151–8. doi: 10.1007/s11914-016-0315-2
16. Gurkan G, Sarikaya I, Sarikaya A. Semiquantitative Assessment of Osteoblastic, Osteolytic, and Mixed Lytic-Sclerotic Bone Lesions on Fluorodeoxyglucose Positron Emission Tomography/Computed Tomography and Bone Scintigraphy. World J Nucl Med (2019) 18(2):132–6. doi: 10.4103/wjnm.WJNM_31_18
17. Suzuki A, Kashiwagi N, Doi H, Ishii K, Doi K, Kitano M, et al. Patterns of Bone Metastases From Head and Neck Squamous Cell Carcinoma. Auris Nasus Larynx (2020) 47(2):262–7. doi: 10.1016/j.anl.2019.08.001
18. Liu H, He J, Koh SP, Zhong Y, Liu Z, Wang Z, et al. Reprogrammed Marrow Adipocytes Contribute to Myeloma-Induced Bone Disease. Sci Transl Med (2019) 11(494):eaau9087. doi: 10.1126/scitranslmed.aau9087
19. Price TT, Burness ML, Sivan A, Warner MJ, Cheng R, Lee CH, et al. Dormant Breast Cancer Micrometastases Reside in Specific Bone Marrow Niches That Regulate Their Transit to and From Bone. Sci Transl Med (2016) 8(340):340ra73–ra73. doi: 10.1126/scitranslmed.aad4059
20. Sethi N, Dai X, Winter CG, Kang Y. Tumor-Derived Jagged1 Promotes Osteolytic Bone Metastasis of Breast Cancer by Engaging Notch Signaling in Bone Cells. Cancer Cell (2011) 19(2):192–205. doi: 10.1016/j.ccr.2010.12.022
21. Wang H, Yu C, Gao X, Welte T, Muscarella Aaron M, Tian L, et al. The Osteogenic Niche Promotes Early-Stage Bone Colonization of Disseminated Breast Cancer Cells. Cancer Cell (2015) 27(2):193–210. doi: 10.1016/j.ccell.2014.11.017
22. Wu JB, Yin L, Shi C, Li Q, Duan P, Huang J-M, et al. MAOA-Dependent Activation of Shh-IL6-RANKL Signaling Network Promotes Prostate Cancer Metastasis by Engaging Tumor-Stromal Cell Interactions. Cancer Cell (2017) 31(3):368–82. doi: 10.1016/j.ccell.2017.02.003
23. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity (2010) 32:593-604.
24. Randolph GJ, Mowat AM, Gordon S, Hume DA, Geissmann F. Unravelling Mononuclear Phagocyte Heterogeneity. Nat Reviews Immunol (2010) 10:453-60.
25. Wynn TA, Murray PJ. Protective and Pathogenic Functions of Macrophagesubsets. Nat Rev Immunol (2011) 11:723-37. doi: . doi: 10.1038/nri3073
26. Liddiard K, Taylor PR. Understanding Local Macrophage Phenotypes In Disease: Shape-Shifting Macrophages. Nat Med (2015) 21:119-20.
27. Grabert K, Sehgal A, Irvine KM, Wollscheid-Lengeling E, Ozdemir DD, Stables J, et al. A Transgenic Line That Reports CSF1R Protein Expression Provides a Definitive Marker for the Mouse Mononuclear Phagocyte System. J Immunol (2020) 205(11):3154–66. doi: 10.4049/jimmunol.2000835
28. Hume DA, Irvine KM, Pridans C. The Mononuclear Phagocyte System: The Relationship Between Monocytes and Macrophages. Trends Immunol (2019) 40(2):98–112. doi: 10.1016/j.it.2018.11.007
29. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-Resident Macrophages Originate From Yolk-Sac-Derived Erythro-Myeloid Progenitors. Nature (2015) 518(7540):547–51. doi: 10.1038/nature13989
30. Hoeffel G, Chen J, Lavin Y, Low D, Almeida Francisca F, See P, et al. C-Myb+ Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity (2015) 42(4):665–78. doi: 10.1016/j.immuni.2015.03.011
31. Schulz C, Perdiguero EG, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science (2012) 336(6077):86–90. doi: 10.1126/science.1219179
32. Afik R, Zigmond E, Vugman M, Klepfish M, Shimshoni E, Pasmanik-Chor M, et al. Tumor Macrophages Are Pivotal Constructors of Tumor Collagenous Matrix. J Exp Med (2016) 213(11):2315–31. doi: 10.1084/jem.20151193
33. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The Cellular and Molecular Origin of Tumor-Associated Macrophages. Science (2014) 344(6186):921–5. doi: 10.1126/science.1252510
34. Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature (2011) 475(7355):222–5. doi: 10.1038/nature10138
35. Ma RY, Zhang H, Li XF, Zhang CB, Selli C, Tagliavini G, et al. Monocyte-Derived Macrophages Promote Breast Cancer Bone Metastasis Outgrowth. J Exp Med (2020) 217(11):e20191820. doi: 10.1084/jem.20191820
36. Loyher PL, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z, et al. Macrophages of Distinct Origins Contribute to Tumor Development in the Lung. J Exp Med (2018) 215(10):2536–53. doi: 10.1084/jem.20180534
37. Zhu Y, Herndon JM, Sojka DK, Kim K-W, Knolhoff BL, Zuo C, et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate From Embryonic Hematopoiesis and Promote Tumor Progression. Immunity (2017) 47(2):323–38.e6. doi: 10.1016/j.immuni.2017.07.014
38. Kaur S, Raggatt LJ, Batoon L, Hume DA, Levesque J-P, Pettit AR. Role of Bone Marrow Macrophages in Controlling Homeostasis and Repair in Bone and Bone Marrow Niches. Semin Cell Dev Biol (2017) 61:12–21. doi: 10.1016/j.semcdb.2016.08.009
39. Chang MK, Raggatt LJ, Alexander KA, Kuliwaba JS, Fazzalari NL, Schroder K, et al. Osteal Tissue Macrophages are Intercalated Throughout Human and Mouse Bone Lining Tissues and Regulate Osteoblast Function In Vitro and In Vivo. J Immunol (2008) 181(2):1232–44. doi: 10.4049/jimmunol.181.2.1232
40. Sica A, Mantovani A. Macrophage Plasticity and Polarization: In Vivo Veritas. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/JCI59643
41. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage Phenotypes in Atherosclerosis. Immunol Rev (2014) 262(1):153–66. doi: 10.1111/imr.12218
42. Rőszer T. Understanding the Mysterious M2 Macrophage Through Activation Markers and Effector Mechanisms. Mediators Inflammation (2015) 2015:816460. doi: 10.1155/2015/816460
43. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J Immunol (2000) 164(12):6166–73. doi: 10.4049/jimmunol.164.12.6166
44. Murray Peter J, Allen Judith E, Biswas Subhra K, Fisher Edward A, Gilroy Derek W, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
45. Heinz S, Romanoski CE, Benner C, Allison KA, Kaikkonen MU, Orozco LD, et al. Effect of Natural Genetic Variation on Enhancer Selection and Function. Nature (2013) 503(7477):487–92. doi: 10.1038/nature12615
46. Raza S, Barnett MW, Barnett-Itzhaki Z, Amit I, Hume DA, Freeman TC. Analysis of the Transcriptional Networks Underpinning the Activation of Murine Macrophages by Inflammatory Mediators. J Leukoc Biol (2014) 96(2):167–83. doi: 10.1189/jlb.6HI0313-169R
47. Hume DA. The Many Alternative Faces of Macrophage Activation. Front Immunol (2015) 6:370. doi: 10.3389/fimmu.2015.00370
48. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017) 79(1):541–66. doi: 10.1146/annurev-physiol-022516-034339
49. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell (2018) 174(5):1293–308.e36. doi: 10.1016/j.cell.2018.05.060
50. Kitamura T, Doughty-Shenton D, Cassetta L, Fragkogianni S, Brownlie D, Kato Y, et al. Monocytes Differentiate to Immune Suppressive Precursors of Metastasis-Associated Macrophages in Mouse Models of Metastatic Breast Cancer. Front Immunol (2018) 8:2004. doi: 10.3389/fimmu.2017.02004
51. Mendoza-Reinoso V, McCauley LK, Fournier PGJ. Contribution of Macrophages and T Cells in Skeletal Metastasis. Cancers (2020) 12(4):1014. doi: 10.3390/cancers12041014
52. Argyle D, Kitamura T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front Immunol (2018) 9:2629. doi: 10.3389/fimmu.2018.02629
53. Prenen H, Mazzone M. Tumor-Associated Macrophages: A Short Compendium. Cell Mol Life Sci (2019) 76(8):1447–58. doi: 10.1007/s00018-018-2997-3
54. Müller A, Brandenburg S, Turkowski K, Müller S, Vajkoczy P. Resident Microglia, and Not Peripheral Macrophages, Are the Main Source of Brain Tumor Mononuclear Cells. Int J Cancer (2015) 137(2):278–88. doi: 10.1002/ijc.29379
55. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res (2017) 77(9):2266–78. doi: 10.1158/0008-5472.CAN-16-2310
56. Muliaditan T, Caron J, Okesola M, Opzoomer JW, Kosti P, Georgouli M, et al. Macrophages are Exploited From an Innate Wound Healing Response to Facilitate Cancer Metastasis. Nat Commun (2018) 9(1):2951. doi: 10.1038/s41467-018-05346-7
57. Campbell MJ, Tonlaar NY, Garwood ER, Huo D, Moore DH, Khramtsov AI, et al. Proliferating Macrophages Associated With High Grade, Hormone Receptor Negative Breast Cancer and Poor Clinical Outcome. Breast Cancer Res Treat (2011) 128(3):703–11. doi: 10.1007/s10549-010-1154-y
58. Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, et al. Dual Prognostic Significance of Tumour-Associated Macrophages in Human Pancreatic Adenocarcinoma Treated or Untreated With Chemotherapy. Pancreas (2016) 65(10):1710–20. doi: 10.1136/gutjnl-2015-309193
59. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-Associated Macrophages and Survival in Classic Hodgkin's Lymphoma. N Engl J Med (2010) 362(10):875–85. doi: 10.1056/NEJMoa0905680
60. Kumar AT, Knops A, Swendseid B, Martinez-Outschoom U, Harshyne L, Philp N, et al. Prognostic Significance of Tumor-Associated Macrophage Content in Head and Neck Squamous Cell Carcinoma: A Meta-Analysis. Front Oncol (2019) 9:656. doi: 10.3389/fonc.2019.00656
61. Zhang Y, Cheng S, Zhang M, Zhen L, Pang D, Zhang Q, et al. High-Infiltration of Tumor-Associated Macrophages Predicts Unfavorable Clinical Outcome for Node-Negative Breast Cancer. PloS One (2013) 8(9):e76147. doi: 10.1371/journal.pone.0076147
62. Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, et al. Significance of M2-Polarized Tumor-Associated Macrophage in Pancreatic Cancer. J Surg Res (2011) 167(2):e211–9. doi: 10.1016/j.jss.2009.05.026
63. Lan C, Huang X, Lin S, Huang H, Cai Q, Wan T, et al. Expression of M2-Polarized Macrophages is Associated With Poor Prognosis for Advanced Epithelial Ovarian Cancer. Technol Cancer Res Treat (2013) 12(3):259–67. doi: 10.7785/tcrt.2012.500312
64. Zhang B, Yao G, Zhang Y, Gao J, Yang B, Rao Z, et al. M2-Polarized Tumor-Associated Macrophages are Associated With Poor Prognoses Resulting From Accelerated Lymphangiogenesis in Lung Adenocarcinoma. Clinics (Sao Paulo) (2011) 66(11):1879–86. doi: 10.1590/s1807-59322011001100006
65. Yin Z, Ma T, Huang B, Lin L, Zhou Y, Yan J, et al. Macrophage-Derived Exosomal microRNA-501-3p Promotes Progression of Pancreatic Ductal Adenocarcinoma Through the TGFBR3-Mediated TGF-β Signaling Pathway. J Exp Clin Cancer Res (2019) 38(1):310. doi: 10.1186/s13046-019-1313-x
66. Lan J, Sun L, Xu F, Liu L, Hu F, Song D, et al. M2 Macrophage-Derived Exosomes Promote Cell Migration and Invasion in Colon Cancer. Cancer Res (2019) 79(1):146–58. doi: 10.1158/0008-5472.Can-18-0014
67. Walker ND, Elias M, Guiro K, Bhatia R, Greco SJ, Bryan M, et al. Exosomes From Differentially Activated Macrophages Influence Dormancy or Resurgence of Breast Cancer Cells Within Bone Marrow Stroma. Cell Death Dis (2019) 10(2):59. doi: 10.1038/s41419-019-1304-z
68. Ławicki S, Szmitkowski M, Wojtukiewicz M. The Pretreatment Plasma Level and Diagnostic Utility of M-CSF in Benign Breast Tumor and Breast Cancer Patients. Clin Chim Acta (2006) 371(1):112–6. doi: 10.1016/j.cca.2006.02.033
69. Aharinejad S, Salama M, Paulus P, Zins K, Berger A, Singer CF. Elevated CSF1 Serum Concentration Predicts Poor Overall Survival in Women With Early Breast Cancer. Endocr Relat Cancer (2013) 20(6):777–83. doi: 10.1530/erc-13-0198
70. Ide H, Hatake K, Terado Y, Tsukino H, Okegawa T, Nutahara K, et al. Serum Level of Macrophage Colony-Stimulating Factor is Increased in Prostate Cancer Patients With Bone Metastasis. Hum Cell (2008) 21(1):1–6. doi: 10.1111/j.1749-0774.2007.00042.x
71. Groblewska M, Mroczko B, Wereszczyńska-Siemiatkowska U, Myśliwiec P, Kedra B, Szmitkowski M. Serum Levels of Granulocyte Colony-Stimulating Factor (G-CSF) and Macrophage Colony-Stimulating Factor (M-CSF) in Pancreatic Cancer Patients. Clin Chem Lab Med (2007) 45(1):30–4. doi: 10.1515/cclm.2007.025
72. Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ, et al. High Expression of Macrophage Colony-Stimulating Factor in Peritumoral Liver Tissue Is Associated With Poor Survival After Curative Resection of Hepatocellular Carcinoma. J Clin Oncol (2008) 26(16):2707–16. doi: 10.1200/jco.2007.15.6521
73. Mroczko B, Groblewska M, Wereszczyńska-Siemiatkowska U, Okulczyk B, Kedra B, Łaszewicz W, et al. Serum Macrophage-Colony Stimulating Factor Levels in Colorectal Cancer Patients Correlate With Lymph Node Metastasis and Poor Prognosis. Clin Chim Acta (2007) 380(1-2):208–12. doi: 10.1016/j.cca.2007.02.037
74. Noda Y, Kawaguchi T, Korenaga M, Yoshio S, Komukai S, Nakano M, et al. High Serum Interleukin-34 Level Is a Predictor of Poor Prognosis in Patients With non-Viral Hepatocellular Carcinoma. Hepatol Res (2019) 49(9):1046–53. doi: 10.1111/hepr.13350
75. Baghdadi M, Endo H, Takano A, Ishikawa K, Kameda Y, Wada H, et al. High Co-Expression of IL-34 and M-CSF Correlates With Tumor Progression and Poor Survival in Lung Cancers. Sci Rep (2018) 8(1):418. doi: 10.1038/s41598-017-18796-8
76. Kobayashi T, Baghdadi M, Han N, Murata T, Hama N, Otsuka R, et al. Prognostic Value of IL-34 in Colorectal Cancer Patients. Immunol Med (2019) 42(4):169–75. doi: 10.1080/25785826.2019.1691429
77. DeNardo DG, Ruffell B. Macrophages as Regulators of Tumour Immunity and Immunotherapy. Nat Rev Immunol (2019) 19(6):369–82. doi: 10.1038/s41577-019-0127-6
78. Arwert EN, Harney AS, Entenberg D, Wang Y, Sahai E, Pollard JW, et al. A Unidirectional Transition From Migratory to Perivascular Macrophage is Required for Tumor Cell Intravasation. Cell Rep (2018) 23(5):1239–48. doi: 10.1016/j.celrep.2018.04.007
79. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of Chemokine Receptors in Breast Cancer Metastasis. Nature (2001) 410(6824):50–6. doi: 10.1038/35065016
80. Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the Stromal Cell-Derived Factor-1/CXCR4 Pathway in Prostate Cancer Metastasis to Bone. Cancer Res (2002) 62(6):1832–7.
81. Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, et al. Macrophages Promote the Invasion of Breast Carcinoma Cells via a Colony-Stimulating Factor-1/Epidermal Growth Factor Paracrine Loop. Cancer Res (2005) 65(12):5278–83. doi: 10.1158/0008-5472.Can-04-1853
82. Roh-Johnson M, Bravo-Cordero JJ, Patsialou A, Sharma VP, Guo P, Liu H, et al. Macrophage Contact Induces RhoA GTPase Signaling to Trigger Tumor Cell Intravasation. Oncogene (2014) 33(33):4203–12. doi: 10.1038/onc.2013.377
83. Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A Paracrine Loop Between Tumor Cells and Macrophages Is Required for Tumor Cell Migration in Mammary Tumors. Cancer Res (2004) 64(19):7022–9. doi: 10.1158/0008-5472.Can-04-1449
84. Kusama T, Mukai M, Iwasaki T, Tatsuta M, Matsumoto Y, Akedo H, et al. Inhibition of Epidermal Growth Factor-Induced RhoA Translocation and Invasion of Human Pancreatic Cancer Cells by 3-Hydroxy-3-Methylglutaryl-Coenzyme a Reductase Inhibitors. Cancer Res (2001) 61(12):4885–91.
85. Mateus AR, Seruca R, Machado JC, Keller G, Oliveira MJ, Suriano G, et al. EGFR Regulates RhoA-GTP Dependent Cell Motility in E-Cadherin Mutant Cells. Hum Mol Genet (2007) 16(13):1639–47. doi: 10.1093/hmg/ddm113
86. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 Identifies a Hematopoietic Lineage of Proangiogenic Monocytes Required for Tumor Vessel Formation and a Mesenchymal Population of Pericyte Progenitors. Cancer Cell (2005) 8(3):211–26. doi: 10.1016/j.ccr.2005.08.002
87. Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian B-Z, et al. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage–Derived VEGFA. Cancer Discovery (2015) 5(9):932–43. doi: 10.1158/2159-8290.CD-15-0012
88. Umer M, Vaidyanathan R, Nguyen NT, Shiddiky MJA. Circulating Tumor Microemboli: Progress in Molecular Understanding and Enrichment Technologies. Biotechnol Adv (2018) 36(4):1367–89. doi: 10.1016/j.biotechadv.2018.05.002
89. Amintas S, Bedel A, Moreau-Gaudry F, Boutin J, Buscail L, Merlio J-P, et al. Circulating Tumor Cell Clusters: United We Stand Divided We Fall. Int J Mol Sci (2020) 21(7):2653. doi: 10.3390/ijms21072653
90. Wang W-C, Zhang X-F, Peng J, Li X-F, Wang A-L, Bie Y-Q, et al. Survival Mechanisms and Influence Factors of Circulating Tumor Cells. BioMed Res Int (2018) 2018:6304701. doi: 10.1155/2018/6304701
91. Strilic B, Offermanns S. Intravascular Survival and Extravasation of Tumor Cells. Cancer Cell (2017) 32(3):282–93. doi: 10.1016/j.ccell.2017.07.001
92. Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, et al. Visualization of Immediate Immune Responses to Pioneer Metastatic Cells in the Lung. Nature (2016) 531(7595):513–7. doi: 10.1038/nature16985
93. Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, et al. Real-Time Imaging Reveals the Single Steps of Brain Metastasis Formation. Nat Med (2010) 16(1):116–22. doi: 10.1038/nm.2072
94. Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, et al. Visualizing Extravasation Dynamics of Metastatic Tumor Cells. J Cell Sci (2010) 123(Pt 13):2332–41. doi: 10.1242/jcs.069443
95. Ito S, Nakanishi H, Ikehara Y, Kato T, Kasai Y, Ito K, et al. Real-Time Observation of Micrometastasis Formation in the Living Mouse Liver Using a Green Fluorescent Protein Gene-Tagged Rat Tongue Carcinoma Cell Line. Int J Cancer (2001) 93(2):212–7. doi: 10.1002/ijc.1318
96. Schlüter K, Gassmann P, Enns A, Korb T, Hemping-Bovenkerk A, Hölzen J, et al. Organ-Specific Metastatic Tumor Cell Adhesion and Extravasation of Colon Carcinoma Cells With Different Metastatic Potential. Am J Pathol (2006) 169(3):1064–73. doi: 10.2353/ajpath.2006.050566
97. Morris VL, Koop S, MacDonald IC, Schmidt EE, Grattan M, Percy D, et al. Mammary Carcinoma Cell Lines of High and Low Metastatic Potential Differ Not in Extravasation But in Subsequent Migration and Growth. Clin Exp Metastasis (1994) 12(6):357–67. doi: 10.1007/bf01755879
98. Leong HS, Robertson AE, Stoletov K, Leith SJ, Chin CA, Chien AE, et al. Invadopodia are Required for Cancer Cell Extravasation and are a Therapeutic Target for Metastasis. Cell Rep (2014) 8(5):1558–70. doi: 10.1016/j.celrep.2014.07.050
99. Yamauchi K, Yang M, Jiang P, Xu M, Yamamoto N, Tsuchiya H, et al. Development of Real-Time Subcellular Dynamic Multicolor Imaging of Cancer-Cell Trafficking in Live Mice With a Variable-Magnification Whole-Mouse Imaging System. Cancer Res (2006) 66(8):4208–14. doi: 10.1158/0008-5472.Can-05-3927
100. Inoue S, Osmond DG. Basement Membrane of Mouse Bone Marrow Sinusoids Shows Distinctive Structure and Proteoglycan Composition: A High Resolution Ultrastructural Study. Anat Rec (2001) 264(3):294–304. doi: 10.1002/ar.1166
101. Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A Distinct Macrophage Population Mediates Metastatic Breast Cancer Cell Extravasation, Establishment and Growth. PloS One (2009) 4(8):e6562. doi: 10.1371/journal.pone.0006562
102. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-Induced Chemokine Cascade Promotes Breast Cancer Metastasis by Enhancing Retention of Metastasis-Associated Macrophages. J Exp Med (2015) 212(7):1043–59. doi: 10.1084/jem.20141836
103. Feng F, Zheng G, Wang Q, Liu S, Liu Z, Xu G, et al. Low Lymphocyte Count and High Monocyte Count Predicts Poor Prognosis of Gastric Cancer. BMC Gastroenterol (2018) 18(1):148. doi: 10.1186/s12876-018-0877-9
104. Hayashi T, Fujita K, Nojima S, Hayashi Y, Nakano K, Ishizuya Y, et al. Peripheral Blood Monocyte Count Reflecting Tumor-Infiltrating Macrophages Is a Predictive Factor of Adverse Pathology in Radical Prostatectomy Specimens. Prostate (2017) 77(14):1383–8. doi: 10.1002/pros.23398
105. Lee YY, Choi CH, Sung CO, Do IG, Huh S, Song T, et al. Prognostic Value of Pre-Treatment Circulating Monocyte Count in Patients With Cervical Cancer: Comparison With SCC-Ag Level. Gynecol Oncol (2012) 124(1):92–7. doi: 10.1016/j.ygyno.2011.09.034
106. Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin Cancer Res (2013) 19(13):3404–15. doi: 10.1158/1078-0432.Ccr-13-0525
107. Sasaki A, Iwashita Y, Shibata K, Matsumoto T, Ohta M, Kitano S. Prognostic Value of Preoperative Peripheral Blood Monocyte Count in Patients With Hepatocellular Carcinoma. Surgery (2006) 139(6):755–64. doi: 10.1016/j.surg.2005.10.009
108. Shigeta K, Kosaka T, Kitano S, Yasumizu Y, Miyazaki Y, Mizuno R, et al. High Absolute Monocyte Count Predicts Poor Clinical Outcome in Patients With Castration-Resistant Prostate Cancer Treated With Docetaxel Chemotherapy. Ann Surg Oncol (2016) 23(12):4115–22. doi: 10.1245/s10434-016-5354-5
109. Mizutani K, Sud S, McGregor NA, Martinovski G, Rice BT, Craig MJ, et al. The Chemokine CCL2 Increases Prostate Tumor Growth and Bone Metastasis Through Macrophage and Osteoclast Recruitment. Neoplasia (2009) 11(11):1235–42. doi: 10.1593/neo.09988
110. Cho HJ, Jung JI, Lim DY, Kwon GT, Her S, Park JH, et al. Bone Marrow-Derived, Alternatively Activated Macrophages Enhance Solid Tumor Growth and Lung Metastasis of Mammary Carcinoma Cells in a Balb/C Mouse Orthotopic Model. Breast Cancer Res (2012) 14(3):R81–R. doi: 10.1186/bcr3195
111. Kawai H, Tsujigiwa H, Siar CH, Nakano K, Takabatake K, Fujii M, et al. Characterization and Potential Roles of Bone Marrow-Derived Stromal Cells in Cancer Development and Metastasis. Int J Med Sci (2018) 15(12):1406–14. doi: 10.7150/ijms.24370
112. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-Metastatic Niches: Organ-Specific Homes for Metastases. Nat Rev Cancer (2017) 17(5):302–17. doi: 10.1038/nrc.2017.6
113. Batoon L, Millard SM, Raggatt LJ, Pettit AR. Osteomacs and Bone Regeneration. Curr Osteoporos Rep (2017) 15(4):385–95. doi: 10.1007/s11914-017-0384-x
114. Batoon L, Millard SM, Wullschleger ME, Preda C, Wu AC, Kaur S, et al. CD169(+) Macrophages Are Critical for Osteoblast Maintenance and Promote Intramembranous and Endochondral Ossification During Bone Repair. Biomaterials (2019) 196:51–66. doi: 10.1016/j.biomaterials.2017.10.033
115. Cox TR, Rumney RMH, Schoof EM, Perryman L, Høye AM, Agrawal A, et al. The Hypoxic Cancer Secretome Induces Pre-Metastatic Bone Lesions Through Lysyl Oxidase. Nature (2015) 522(7554):106–10. doi: 10.1038/nature14492
116. Monteiro AC, Leal AC, Gonçalves-Silva T, Mercadante AC, Kestelman F, Chaves SB, et al. T Cells Induce Pre-Metastatic Osteolytic Disease and Help Bone Metastases Establishment in a Mouse Model of Metastatic Breast Cancer. PloS One (2013) 8(7):e68171. doi: 10.1371/journal.pone.0068171
117. Weilbaecher KN, Guise TA, McCauley LK. Cancer to Bone: A Fatal Attraction. Nat Rev Cancer (2011) 11(6):411–25. doi: 10.1038/nrc3055
118. Esposito M, Guise T, Kang Y. The Biology of Bone Metastasis. Cold Spring Harb Perspect Med (2018) 8(6):a031252. doi: 10.1101/cshperspect.a031252
119. Juárez P, Guise TA. TGF-β in Cancer and Bone: Implications for Treatment of Bone Metastases. Bone (2011) 48(1):23–9. doi: 10.1016/j.bone.2010.08.004
120. Yuan X, Qian N, Ling S, Li Y, Sun W, Li J, et al. Breast Cancer Exosomes Contribute to Pre-Metastatic Niche Formation and Promote Bone Metastasis of Tumor Cells. Theranostics (2021) 11(3):1429–45. doi: 10.7150/thno.45351
121. Smieszek A, Marcinkowska K, Pielok A, Sikora M, Valihrach L, Marycz K. The Role of miR-21 in Osteoblasts-Osteoclasts Coupling In Vitro. Cells (2020) 9(2):479. doi: 10.3390/cells9020479
122. Jiang P, Gao W, Ma T, Wang R, Piao Y, Dong X, et al. CD137 Promotes Bone Metastasis of Breast Cancer by Enhancing the Migration and Osteoclast Differentiation of Monocytes/Macrophages. Theranostics (2019) 9(10):2950–66. doi: 10.7150/thno.29617
123. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat Commun (2020) 11(1):5120. doi: 10.1038/s41467-020-18794-x
124. Sinder BP, Pettit AR, McCauley LK. Macrophages: Their Emerging Roles in Bone. J Bone Miner Res (2015) 30(12):2140–9. doi: 10.1002/jbmr.2735
125. Kourtzelis I, Hajishengallis G, Chavakis T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front Immunol (2020) 11:553. doi: 10.3389/fimmu.2020.00553
126. Soki FN, Koh AJ, Jones JD, Kim YW, Dai J, Keller ET, et al. Polarization of Prostate Cancer-Associated Macrophages Is Induced by Milk Fat Globule-EGF Factor 8 (MFG-E8)-Mediated Efferocytosis. J Biol Chem (2014) 289(35):24560–72. doi: 10.1074/jbc.M114.571620
127. Quaranta V, Schmid MC. Macrophage-Mediated Subversion of Anti-Tumour Immunity. Cells (2019) 8(7):747. doi: 10.3390/cells8070747
128. Michalski MN, Koh AJ, Weidner S, Roca H, McCauley LK. Modulation of Osteoblastic Cell Efferocytosis by Bone Marrow Macrophages. J Cell Biochem (2016) 117(12):2697–706. doi: 10.1002/jcb.25567
129. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages That Have Ingested Apoptotic Cells In Vitro Inhibit Proinflammatory Cytokine Production Through Autocrine/Paracrine Mechanisms Involving TGF-Beta, PGE2, and PAF. J Clin Invest (1998) 101(4):890–8. doi: 10.1172/JCI1112
130. Liu Z, Kuang W, Zhou Q, Zhang Y. TGF-β1 Secreted by M2 Phenotype Macrophages Enhances the Stemness and Migration of Glioma Cells via the SMAD2/3 Signalling Pathway. Int J Mol Med (2018) 42(6):3395–403. doi: 10.3892/ijmm.2018.3923
131. Thomas DA, Massagué J. TGF-Beta Directly Targets Cytotoxic T Cell Functions During Tumor Evasion of Immune Surveillance. Cancer Cell (2005) 8(5):369–80. doi: 10.1016/j.ccr.2005.10.012
132. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of Peripheral CD4+CD25- Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-Beta Induction of Transcription Factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi: 10.1084/jem.20030152
133. Roca H, Jones JD, Purica MC, Weidner S, Koh AJ, Kuo R, et al. Apoptosis-Induced CXCL5 Accelerates Inflammation and Growth of Prostate Tumor Metastases in Bone. J Clin Invest (2018) 128(1):248–66. doi: 10.1172/JCI92466
134. Marcuzzi E, Angioni R, Molon B, Calì B. Chemokines and Chemokine Receptors: Orchestrating Tumor Metastasization. Int J Mol Sci (2018) 20(1):96. doi: 10.3390/ijms20010096
135. Harmer D, Falank C, Reagan MR. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss and Multiple Myeloma. Front Endocrino (2019) 9:788. doi: 10.3389/fendo.2018.00788
136. Hardaway AL, Herroon MK, Rajagurubandara E, Podgorski I. Marrow Adipocyte-Derived CXCL1 and CXCL2 Contribute to Osteolysis in Metastatic Prostate Cancer. Clin Exp Metastasis (2015) 32(4):353–68. doi: 10.1007/s10585-015-9714-5
137. Urata S, Izumi K, Hiratsuka K, Maolake A, Natsagdorj A, Shigehara K, et al. C-C Motif Ligand 5 Promotes Migration of Prostate Cancer Cells in the Prostate Cancer Bone Metastasis Microenvironment. Cancer Sci (2018) 109(3):724–31. doi: 10.1111/cas.13494
138. Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD-L1 Expression in the Tumor Microenvironment. J Hematol Oncol (2021) 14(1):10. doi: 10.1186/s13045-020-01027-5
139. Ribas A, Wolchok JD. Cancer Immunotherapy Using Checkpoint Blockade. Science (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
140. Landi L, D’Incà F, Gelibter A, Chiari R, Grossi F, Delmonte A, et al. Bone Metastases and Immunotherapy in Patients With Advanced Non-Small-Cell Lung Cancer. J Immunother Cancer (2019) 7(1):316. doi: 10.1186/s40425-019-0793-8
141. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell (2017) 170(6):1120–33.e17. doi: 10.1016/j.cell.2017.07.024
142. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated Monocytes in Peritumoral Stroma of Hepatocellular Carcinoma Foster Immune Privilege and Disease Progression Through PD-L1. J Exp Med (2009) 206(6):1327–37. doi: 10.1084/jem.20082173
143. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas Promote Immunosuppression Through Induction of B7-H1 Expression in Tumor-Associated Macrophages. Clin Cancer Res (2013) 19(12):3165–75. doi: 10.1158/1078-0432.Ccr-12-3314
144. Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer ARL, Bajor DL, et al. Induction of T-Cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res (2015) 3(4):399–411. doi: 10.1158/2326-6066.CIR-14-0215
145. Jing W, Guo X, Wang G, Bi Y, Han L, Zhu Q, et al. Breast Cancer Cells Promote CD169+ Macrophage-Associated Immunosuppression Through JAK2-Mediated PD-L1 Upregulation on Macrophages. Int Immunopharmacol (2020) 78:106012. doi: 10.1016/j.intimp.2019.106012
146. Duong L, Radley-Crabb HG, Gardner JK, Tomay F, Dye DE, Grounds MD, et al. Macrophage Depletion in Elderly Mice Improves Response to Tumor Immunotherapy, Increases Anti-Tumor T Cell Activity and Reduces Treatment-Induced Cachexia. Front Genet (2018) 9:526. doi: 10.3389/fgene.2018.00526
147. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages Impede CD8 T Cells From Reaching Tumor Cells and Limit the Efficacy of Anti–PD-1 Treatment. Proc Natl Acad Sci USA (2018) 115(17):E4041–E50. doi: 10.1073/pnas.1720948115
148. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature (2017) 545(7655):495–9. doi: 10.1038/nature22396
149. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol (2016) 39(1):98–106. doi: 10.1097/COC.0000000000000239
150. Chang C-S, Chang JH, Hsu NC, Lin H-Y, Chung C-Y. Expression of CD80 and CD86 Costimulatory Molecules are Potential Markers for Better Survival in Nasopharyngeal Carcinoma. BMC Cancer (2007) 7(1):88. doi: 10.1186/1471-2407-7-88
151. Scarpa M, Marchiori C, Scarpa M, Castagliuolo I. CD80 Expression is Upregulated by TP53 Activation in Human Cancer Epithelial Cells. OncoImmunology (2021) 10(1):1907912. doi: 10.1080/2162402X.2021.1907912
152. Tirapu I, Huarte E, Guiducci C, Arina A, Zaratiegui M, Murillo O, et al. Low Surface Expression of B7-1 (CD80) is an Immunoescape Mechanism of Colon Carcinoma. Cancer Res (2006) 66(4):2442–50. doi: 10.1158/0008-5472.CAN-05-1681
153. Sun D, Luo T, Dong P, Zhang N, Chen J, Zhang S, et al. CD86+/CD206+ Tumor-Associated Macrophages Predict Prognosis of Patients With Intrahepatic Cholangiocarcinoma. PeerJ (2020) 8:e8458–e. doi: 10.7717/peerj.8458
154. Viitala M, Virtakoivu R, Tadayon S, Rannikko J, Jalkanen S, Hollmén M. Immunotherapeutic Blockade of Macrophage Clever-1 Reactivates the CD8+ T-Cell Response Against Immunosuppressive Tumors. Clin Cancer Res (2019) 25(11):3289–303. doi: 10.1158/1078-0432.CCR-18-3016
155. Croucher PI, McDonald MM, Martin TJ. Bone Metastasis: The Importance of the Neighbourhood. Nat Rev Cancer (2016) 16(6):373–86. doi: 10.1038/nrc.2016.44
156. Andersen TL, Sondergaard TE, Skorzynska KE, Dagnaes-Hansen F, Plesner TL, Hauge EM, et al. A Physical Mechanism for Coupling Bone Resorption and Formation in Adult Human Bone. Am J Pathol (2009) 174(1):239–47. doi: 10.2353/ajpath.2009.080627
157. Hüsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic Spread is an Early Step in Breast Cancer. Cancer Cell (2008) 13(1):58–68. doi: 10.1016/j.ccr.2007.12.003
158. Tjensvoll K, Oltedal S, Heikkilä R, Kvaløy JT, Gilje B, Reuben JM, et al. Persistent Tumor Cells in Bone Marrow of non-Metastatic Breast Cancer Patients After Primary Surgery are Associated With Inferior Outcome. BMC Cancer (2012) 12:190. doi: 10.1186/1471-2407-12-190
159. Domschke C, Diel IJ, Englert S, Kalteisen S, Mayer L, Rom J, et al. Prognostic Value of Disseminated Tumor Cells in the Bone Marrow of Patients With Operable Primary Breast Cancer: A Long-Term Follow-Up Study. Ann Surg Oncol (2013) 20(6):1865–71. doi: 10.1245/s10434-012-2814-4
160. Köllermann J, Weikert S, Schostak M, Kempkensteffen C, Kleinschmidt K, Rau T, et al. Prognostic Significance of Disseminated Tumor Cells in the Bone Marrow of Prostate Cancer Patients Treated With Neoadjuvant Hormone Treatment. J Clin Oncol (2008) 26(30):4928–33. doi: 10.1200/jco.2007.15.0441
161. Banys M, Solomayer EF, Becker S, Krawczyk N, Gardanis K, Staebler A, et al. Disseminated Tumor Cells in Bone Marrow may Affect Prognosis of Patients With Gynecologic Malignancies. Int J Gynecol Cancer (2009) 19(5):948–52. doi: 10.1111/IGC.0b013e3181a23c4c
162. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of Angiogenesis and Osteogenesis by a Specific Vessel Subtype in Bone. Nature (2014) 507(7492):323–8. doi: 10.1038/nature13145
163. Todd VM, Johnson RW. Hypoxia in Bone Metastasis and Osteolysis. Cancer Lett (2020) 489:144–54. doi: 10.1016/j.canlet.2020.06.004
164. Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, et al. Macrophages are Recruited to Hypoxic Tumor Areas and Acquire a Pro-Angiogenic M2-Polarized Phenotype via Hypoxic Cancer Cell Derived Cytokines Oncostatin M and Eotaxin. Oncotarget (2014) 5(14):5350–68. doi: 10.18632/oncotarget.2110
165. Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding Macrophage Entry Into Hypoxic Tumor Areas by Sema3A/Nrp1 Signaling Blockade Inhibits Angiogenesis and Restores Antitumor Immunity. Cancer Cell (2013) 24(6):695–709. doi: 10.1016/j.ccr.2013.11.007
166. Zhang J, Cao J, Ma S, Dong R, Meng W, Ying M, et al. Tumor Hypoxia Enhances non-Small Cell Lung Cancer Metastasis by Selectively Promoting Macrophage M2 Polarization Through the Activation of ERK Signaling. Oncotarget (2014) 5(20):9664–77. doi: 10.18632/oncotarget.1856
167. Ke X, Chen C, Song Y, Cai Q, Li J, Tang Y, et al. Hypoxia Modifies the Polarization of Macrophages and Their Inflammatory Microenvironment, and Inhibits Malignant Behavior in Cancer Cells. Oncol Lett (2019) 18(6):5871–8. doi: 10.3892/ol.2019.10956
168. Leblond MM, Gérault AN, Corroyer-Dulmont A, MacKenzie ET, Petit E, Bernaudin M, et al. Hypoxia Induces Macrophage Polarization and Re-Education Toward an M2 Phenotype in U87 and U251 Glioblastoma Models. OncoImmunology (2016) 5(1):e1056442. doi: 10.1080/2162402X.2015.1056442
169. Qi L, Chen J, Yang Y, Hu W. Hypoxia Correlates With Poor Survival and M2 Macrophage Infiltration in Colorectal Cancer. Front Oncol (2020) 10:566430(2491). doi: 10.3389/fonc.2020.566430
170. Yu-Lee L-Y, Lee Y-C, Pan J, Lin S-C, Pan T, Yu G, et al. Bone Secreted Factors Induce Cellular Quiescence in Prostate Cancer Cells. Sci Rep (2019) 9(1):18635. doi: 10.1038/s41598-019-54566-4
171. Jeong H, Kim S, Hong B-J, Lee C-J, Kim Y-E, Bok S, et al. Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res (2019) 79(4):795–806. doi: 10.1158/0008-5472.CAN-18-2545
172. Giancotti Filippo G. Mechanisms Governing Metastatic Dormancy and Reactivation. Cell (2013) 155(4):750–64. doi: 10.1016/j.cell.2013.10.029
173. Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, et al. Osteoclasts Control Reactivation of Dormant Myeloma Cells by Remodelling the Endosteal Niche. Nat Commun (2015) 6:8983. doi: 10.1038/ncomms9983
174. Ren D, Dai Y, Yang Q, Zhang X, Guo W, Ye L, et al. Wnt5a Induces and Maintains Prostate Cancer Cells Dormancy in Bone. J Exp Med (2019) 216(2):428–49. doi: 10.1084/jem.20180661
175. Park S-Y, Nam J-S. The Force Awakens: Metastatic Dormant Cancer Cells. Exp Mol Med (2020) 52(4):569–81. doi: 10.1038/s12276-020-0423-z
176. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 Promotes Osteolytic Expansion of Indolent Bone Micrometastasis of Breast Cancer by Engaging α4β1-Positive Osteoclast Progenitors. Cancer Cell (2011) 20(6):701–14. doi: 10.1016/j.ccr.2011.11.002
177. Jung Y, Cackowski FC, Yumoto K, Decker AM, Wang Y, Hotchkin M, et al. Abscisic Acid Regulates Dormancy of Prostate Cancer Disseminated Tumor Cells in the Bone Marrow. Neoplasia (2021) 23(1):102–11. doi: 10.1016/j.neo.2020.11.009
178. Chaqour J, Lee S, Ravichandra A, Chaqour B. Abscisic Acid - An Anti-Angiogenic Phytohormone That Modulates the Phenotypical Plasticity of Endothelial Cells and Macrophages. J Cell Sci (2018) 131(3):210492. doi: 10.1242/jcs.210492
179. Polverini PJ, Leibovich SJ. Induction of Neovascularization In Vivo and Endothelial Proliferation In Vitro by Tumor-Associated Macrophages. Lab Invest (1984) 51(6):635–42.
180. Schaaf MB, Garg AD, Agostinis P. Defining the Role of the Tumor Vasculature in Antitumor Immunity and Immunotherapy. Cell Death Dis (2018) 9(2):115. doi: 10.1038/s41419-017-0061-0
181. Larionova I, Kazakova E, Gerashchenko T, Kzhyshkowska J. New Angiogenic Regulators Produced by TAMs: Perspective for Targeting Tumor Angiogenesis. Cancers (2021) 13(13):3253. doi: 10.3390/cancers13133253
182. Badawi MA, Abouelfadl DM, El-Sharkawy SL, El-Aal WEA, Abbas NF. Tumor-Associated Macrophage (TAM) and Angiogenesis in Human Colon Carcinoma. Open Access Maced J Med Sci (2015) 3(2):209–14. doi: 10.3889/oamjms.2015.044
183. Hwang I, Kim JW, Ylaya K, Chung EJ, Kitano H, Perry C, et al. Tumor-Associated Macrophage, Angiogenesis and Lymphangiogenesis Markers Predict Prognosis of Non-Small Cell Lung Cancer Patients. J Transl Med (2020) 18(1):443. doi: 10.1186/s12967-020-02618-z
184. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages Regulate the Angiogenic Switch in a Mouse Model of Breast Cancer. Cancer Res (2006) 66(23):11238–46. doi: 10.1158/0008-5472.Can-06-1278
185. Halin S, Rudolfsson SH, Van Rooijen N, Bergh A. Extratumoral Macrophages Promote Tumor and Vascular Growth in an Orthotopic Rat Prostate Tumor Model. Neoplasia (2009) 11(2):177–86. doi: 10.1593/neo.81338
186. Kes MMG, Van den Bossche J, Griffioen AW, Huijbers EJM. Oncometabolites Lactate and Succinate Drive Pro-Angiogenic Macrophage Response in Tumors. Biochim Biophys Acta Rev Cancer (2020) 1874(2):188427. doi: 10.1016/j.bbcan.2020.188427
187. Goel HL, Mercurio AM. VEGF Targets the Tumour Cell. Nat Rev Cancer (2013) 13(12):871–82. doi: 10.1038/nrc3627
188. Häggström S, Bergh A, Damber JE. Vascular Endothelial Growth Factor Content in Metastasizing and Nonmetastasizing Dunning Prostatic Adenocarcinoma. Prostate (2000) 45(1):42–50. doi: 10.1002/1097-0045(20000915)45:1<42::aid-pros5>3.0.co;2-e
189. Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG Inhibits Tumor Growth and Metastasis by Inducing Macrophage Polarization and Vessel Normalization Through Downregulation of PlGF. Cancer Cell (2011) 19(1):31–44. doi: 10.1016/j.ccr.2010.11.009
190. Jarosz-Biej M, Kamińska N, Matuszczak S, Cichoń T, Pamuła-Piłat J, Czapla J, et al. M1-Like Macrophages Change Tumor Blood Vessels and Microenvironment in Murine Melanoma. PloS One (2018) 13(1):e0191012–e. doi: 10.1371/journal.pone.0191012
191. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix Metalloproteinase-9 Triggers the Angiogenic Switch During Carcinogenesis. Nat Cell Biol (2000) 2(10):737–44. doi: 10.1038/35036374
192. Huang S, Van Arsdall M, Tedjarati S, McCarty M, Wu W, Langley R, et al. Contributions of Stromal Metalloproteinase-9 to Angiogenesis and Growth of Human Ovarian Carcinoma in Mice. J Natl Cancer Inst (2002) 94(15):1134–42. doi: 10.1093/jnci/94.15.1134
193. Wang B, Li Q, Wang J, Zhao S, Nashun B, Qin L, et al. Plasmodium Infection Inhibits Tumor Angiogenesis Through Effects on Tumor-Associated Macrophages in a Murine Implanted Hepatoma Model. Cell Commun Signaling (2020) 18(1):157–. doi: 10.1186/s12964-020-00570-5
194. Ceci C, Atzori MG, Lacal PM, Graziani G. Role of VEGFs/VEGFR-1 Signaling and its Inhibition in Modulating Tumor Invasion: Experimental Evidence in Different Metastatic Cancer Models. Int J Mol Sci (2020) 21(4):1388. doi: 10.3390/ijms21041388
195. Sekiguchi K, Ito Y, Hattori K, Inoue T, Hosono K, Honda M, et al. VEGF Receptor 1-Expressing Macrophages Recruited From Bone Marrow Enhances Angiogenesis in Endometrial Tissues. Sci Rep (2019) 9(1):7037. doi: 10.1038/s41598-019-43185-8
196. Guise TA. The Vicious Cycle of Bone Metastases. J Musculoskelet Neuronal Interact (2002) 2(6):570–2.
197. Macedo F, Ladeira K, Pinho F, Saraiva N, Bonito N, Pinto L, et al. Bone Metastases: An Overview. Oncol Rev (2017) 11(1):321. doi: 10.4081/oncol.2017.321
198. Quattrocchi CC, Piciucchi S, Sammarra M, Santini D, Vincenzi B, Tonini G, et al. Bone Metastases in Breast Cancer: Higher Prevalence of Osteosclerotic Lesions. Radiol Med (2007) 112(7):1049–59. doi: 10.1007/s11547-007-0205-x
199. Roudier MP, Morrissey C, True LD, Higano CS, Vessella RL, Ott SM. Histopathological Assessment of Prostate Cancer Bone Osteoblastic Metastases. J Urol (2008) 180(3):1154–60. doi: 10.1016/j.juro.2008.04.140
200. Ottewell PD. The Role of Osteoblasts in Bone Metastasis. J Bone Oncol (2016) 5(3):124–7. doi: 10.1016/j.jbo.2016.03.007
201. Bussard KM, Venzon DJ, Mastro AM. Osteoblasts are a Major Source of Inflammatory Cytokines in the Tumor Microenvironment of Bone Metastatic Breast Cancer. J Cell Biochem (2010) 111(5):1138–48. doi: 10.1002/jcb.22799
202. Karlsson T, Lundholm M, Widmark A, Persson E. Tumor Cell-Derived Exosomes From the Prostate Cancer Cell Line TRAMP-C1 Impair Osteoclast Formation and Differentiation. PloS One (2016) 11(11):e0166284. doi: 10.1371/journal.pone.0166284
203. Herroon MK, Rajagurubandara E, Rudy DL, Chalasani A, Hardaway AL, Podgorski I. Macrophage Cathepsin K Promotes Prostate Tumor Progression in Bone. Oncogene (2013) 32(12):1580–93. doi: 10.1038/onc.2012.166
204. Wu AC, He Y, Broomfield A, Paatan NJ, Harrington BS, Tseng H-W, et al. CD169+ Macrophages Mediate Pathological Formation of Woven Bone in Skeletal Lesions of Prostate Cancer. J Pathol (2016) 239(2):218–30. doi: 10.1002/path.4718
205. Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, et al. TGF-Beta Signaling Blockade Inhibits PTHrP Secretion by Breast Cancer Cells and Bone Metastases Development. J Clin Invest (1999) 103(2):197–206. doi: 10.1172/JCI3523
206. Miao D, He B, Jiang Y, Kobayashi T, Sorocéanu MA, Zhao J, et al. Osteoblast-Derived PTHrP Is a Potent Endogenous Bone Anabolic Agent That Modifies the Therapeutic Efficacy of Administered PTH 1-34. J Clin Invest (2005) 115(9):2402–11. doi: 10.1172/jci24918
207. Liao J, Li X, Koh AJ, Berry JE, Thudi N, Rosol TJ, et al. Tumor Expressed PTHrP Facilitates Prostate Cancer-Induced Osteoblastic Lesions. Int J Cancer (2008) 123(10):2267–78. doi: 10.1002/ijc.23602
208. Ma YL, Cain RL, Halladay DL, Yang X, Zeng Q, Miles RR, et al. Catabolic Effects of Continuous Human PTH (1–38) In Vivo is Associated With Sustained Stimulation of RANKL and Inhibition of Osteoprotegerin and Gene-Associated Bone Formation. Endocrinology (2001) 142(9):4047–54. doi: 10.1210/endo.142.9.8356
209. Li X, Qin L, Bergenstock M, Bevelock LM, Novack DV, Partridge NC. Parathyroid Hormone Stimulates Osteoblastic Expression of MCP-1 to Recruit and Increase the Fusion of Pre-Osteoclasts. J Biol Chem (2007) 282(45):33098–106. doi: 10.1074/jbc.M611781200
210. Ihle CL, Provera MD, Straign DM, Smith EE, Edgerton SM, Van Bokhoven A, et al. Distinct Tumor Microenvironments of Lytic and Blastic Bone Metastases in Prostate Cancer Patients. J Immunother Cancer (2019) 7(1):293. doi: 10.1186/s40425-019-0753-3
211. Movila A, Ishii T, Albassam A, Wisitrasameewong W, Howait M, Yamaguchi T, et al. Macrophage Migration Inhibitory Factor (MIF) Supports Homing of Osteoclast Precursors to Peripheral Osteolytic Lesions. J Bone Miner Res (2016) 31(9):1688–700. doi: 10.1002/jbmr.2854
212. De Felice F, Piccioli A, Musio D, Tombolini V. The Role of Radiation Therapy in Bone Metastases Management. Oncotarget (2017) 8(15):25691–9. doi: 10.18632/oncotarget.14823
213. Park SI, Liao J, Berry JE, Li X, Koh AJ, Michalski ME, et al. Cyclophosphamide Creates a Receptive Microenvironment for Prostate Cancer Skeletal Metastasis. Cancer Res (2012) 72(10):2522–32. doi: 10.1158/0008-5472.Can-11-2928
214. Ramanlal Chaudhari K, Kumar A, Megraj Khandelwal VK, Ukawala M, Manjappa AS, Mishra AK, et al. Bone Metastasis Targeting: A Novel Approach to Reach Bone Using Zoledronate Anchored PLGA Nanoparticle as Carrier System Loaded With Docetaxel. J Control Release (2012) 158(3):470–8. doi: 10.1016/j.jconrel.2011.11.020
215. Adjei IM, Sharma B, Peetla C, Labhasetwar V. Inhibition of Bone Loss With Surface-Modulated, Drug-Loaded Nanoparticles in an Intraosseous Model of Prostate Cancer. J Control Release (2016) 232:83–92. doi: 10.1016/j.jconrel.2016.04.019
216. Vijayaraghavalu S, Gao Y, Rahman MT, Rozic R, Sharifi N, Midura RJ, et al. Synergistic Combination Treatment to Break Cross Talk Between Cancer Cells and Bone Cells to Inhibit Progression of Bone Metastasis. Biomaterials (2020) 227:119558. doi: 10.1016/j.biomaterials.2019.119558
217. Esposito M, Kang Y. Targeting Tumor-Stromal Interactions in Bone Metastasis. Pharmacol Ther (2014) 141(2):222–33. doi: 10.1016/j.pharmthera.2013.10.006
218. Coleman R, Cameron D, Dodwell D, Bell R, Wilson C, Rathbone E, et al. Adjuvant Zoledronic Acid in Patients With Early Breast Cancer: Final Efficacy Analysis of the AZURE (BIG 01/04) Randomised Open-Label Phase 3 Trial. Lancet Oncol (2014) 15(9):997–1006. doi: 10.1016/s1470-2045(14)70302-x
219. D'Oronzo S, Wood S, Brown JE. The Use of Bisphosphonates to Treat Skeletal Complications in Solid Tumours. Bone (2021) 147:115907. doi: 10.1016/j.bone.2021.115907
220. Jakob T, Tesfamariam YM, Macherey S, Kuhr K, Adams A, Monsef I, et al. Bisphosphonates or RANK-Ligand-Inhibitors for Men With Prostate Cancer and Bone Metastases: A Network Meta-Analysis. Cochrane Database Syst Rev (2020) 12(12):Cd013020. doi: 10.1002/14651858.CD013020.pub2
221. Wirth M, Tammela T, Cicalese V, Gomez Veiga F, Delaere K, Miller K, et al. Prevention of Bone Metastases in Patients With High-Risk Nonmetastatic Prostate Cancer Treated With Zoledronic Acid: Efficacy and Safety Results of the Zometa European Study (ZEUS). Eur Urol (2015) 67(3):482–91. doi: 10.1016/j.eururo.2014.02.014
222. Anfray C, Ummarino A, Andón FT, Allavena P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells (2019) 9(1):46. doi: 10.3390/cells9010046
223. Cannarile MA, Weisser M, Jacob W, Jegg A-M, Ries CH, Rüttinger D. Colony-Stimulating Factor 1 Receptor (CSF1R) Inhibitors in Cancer Therapy. J Immunother Cancer (2017) 5(1):53. doi: 10.1186/s40425-017-0257-y
224. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting Tumor-Associated Macrophages With Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell (2014) 25(6):846–59. doi: 10.1016/j.ccr.2014.05.016
225. Cassier PA, Italiano A, Gomez-Roca CA, Le Tourneau C, Toulmonde M, Cannarile MA, et al. CSF1R Inhibition With Emactuzumab in Locally Advanced Diffuse-Type Tenosynovial Giant Cell Tumours of the Soft Tissue: A Dose-Escalation and Dose-Expansion Phase 1 Study. Lancet Oncol (2015) 16(8):949–56. doi: 10.1016/s1470-2045(15)00132-1
226. Gomez-Roca C, Cassier P, Italiano A, Cannarile M, Ries C, Brillouet A, et al. Phase I Study of RG7155, a Novel Anti-CSF1R Antibody, in Patients With Advanced/Metastatic Solid Tumors. J Clin Oncol (2015) 33:3005. doi: 10.1200/jco.2015.33.15_suppl.3005
227. von Tresckow B, Morschhauser F, Ribrag V, Topp MS, Chien C, Seetharam S, et al. An Open-Label, Multicenter, Phase I/II Study of JNJ-40346527, a CSF-1R Inhibitor, in Patients With Relapsed or Refractory Hodgkin Lymphoma. Clin Cancer Res (2015) 21(8):1843–50. doi: 10.1158/1078-0432.Ccr-14-1845
228. MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An Antibody Against the Colony-Stimulating Factor 1 Receptor Depletes the Resident Subset of Monocytes and Tissue- and Tumor-Associated Macrophages But Does Not Inhibit Inflammation. Blood (2010) 116(19):3955–63. doi: 10.1182/blood-2010-02-266296
229. Ngiow SF, Meeth KM, Stannard K, Barkauskas DS, Bollag G, Bosenberg M, et al. Co-Inhibition of Colony Stimulating Factor-1 Receptor and BRAF Oncogene in Mouse Models of BRAF(V600E) Melanoma. Oncoimmunology (2016) 5(3):e1089381. doi: 10.1080/2162402x.2015.1089381
230. Valero JG, Matas-Céspedes A, Arenas F, Rodriguez V, Carreras J, Serrat N, et al. The Receptor of the Colony-Stimulating Factor-1 (CSF-1R) is a Novel Prognostic Factor and Therapeutic Target in Follicular Lymphoma. Leukemia (2021) 35:2635–49. doi: 10.1038/s41375-021-01201-9
231. Mun SH, Park PSU, Park-Min KH. The M-CSF Receptor in Osteoclasts and Beyond. Exp Mol Med (2020) 52(8):1239–54. doi: 10.1038/s12276-020-0484-z
232. Fend L, Accart N, Kintz J, Cochin S, Reymann C, Le Pogam F, et al. Therapeutic Effects of Anti-CD115 Monoclonal Antibody in Mouse Cancer Models Through Dual Inhibition of Tumor-Associated Macrophages and Osteoclasts. PloS One (2013) 8(9):e73310. doi: 10.1371/journal.pone.0073310
233. Hung JY, Horn D, Woodruff K, Prihoda T, LeSaux C, Peters J, et al. Colony-Stimulating Factor 1 Potentiates Lung Cancer Bone Metastasis. Lab Invest (2014) 94(4):371–81. doi: 10.1038/labinvest.2014.1
234. Manthey CL, Johnson DL, Illig CR, Tuman RW, Zhou Z, Baker JF, et al. JNJ-28312141, a Novel Orally Active Colony-Stimulating Factor-1 Receptor/FMS-Related Receptor Tyrosine Kinase-3 Receptor Tyrosine Kinase Inhibitor With Potential Utility in Solid Tumors, Bone Metastases, and Acute Myeloid Leukemia. Mol Cancer Ther (2009) 8(11):3151–61. doi: 10.1158/1535-7163.Mct-09-0255
235. Murray LJ, Abrams TJ, Long KR, Ngai TJ, Olson LM, Hong W, et al. SU11248 Inhibits Tumor Growth and CSF-1R-Dependent Osteolysis in an Experimental Breast Cancer Bone Metastasis Model. Clin Exp Metastasis (2003) 20(8):757–66. doi: 10.1023/b:clin.0000006873.65590.68
236. Hiraga T, Nakamura H. Imatinib Mesylate Suppresses Bone Metastases of Breast Cancer by Inhibiting Osteoclasts Through the Blockade of C-Fms Signals. Int J Cancer (2009) 124(1):215–22. doi: 10.1002/ijc.23903
237. Escamilla J, Schokrpur S, Liu C, Priceman SJ, Moughon D, Jiang Z, et al. CSF1 Receptor Targeting in Prostate Cancer Reverses Macrophage-Mediated Resistance to Androgen Blockade Therapy. Cancer Res (2015) 75(6):950–62. doi: 10.1158/0008-5472.Can-14-0992
238. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R Signaling Blockade Stanches Tumor-Infiltrating Myeloid Cells and Improves the Efficacy of Radiotherapy in Prostate Cancer. Cancer Res (2013) 73(9):2782–94. doi: 10.1158/0008-5472.Can-12-3981
239. Stresing V, Daubiné F, Benzaid I, Mönkkönen H, Clézardin P. Bisphosphonates in Cancer Therapy. Cancer Lett (2007) 257(1):16–35. doi: 10.1016/j.canlet.2007.07.007
240. Rogers MJ, Mönkkönen J, Munoz MA. Molecular Mechanisms of Action of Bisphosphonates and New Insights Into Their Effects Outside the Skeleton. Bone (2020) 139:115493. doi: 10.1016/j.bone.2020.115493
241. Junankar S, Shay G, Jurczyluk J, Ali N, Down J, Pocock N, et al. Real-Time Intravital Imaging Establishes Tumor-Associated Macrophages as the Extraskeletal Target of Bisphosphonate Action in Cancer. Cancer Discovery (2015) 5(1):35–42. doi: 10.1158/2159-8290.Cd-14-0621
242. Moreau MF, Guillet C, Massin P, Chevalier S, Gascan H, Baslé MF, et al. Comparative Effects of Five Bisphosphonates on Apoptosis of Macrophage Cells In Vitro. Biochem Pharmacol (2007) 73(5):718–23. doi: 10.1016/j.bcp.2006.09.031
243. Rogers TL, Holen I. Tumour Macrophages as Potential Targets of Bisphosphonates. J Transl Med (2011) 9:177. doi: 10.1186/1479-5876-9-177
244. Hiraoka K, Zenmyo M, Watari K, Iguchi H, Fotovati A, Kimura YN, et al. Inhibition of Bone and Muscle Metastases of Lung Cancer Cells by a Decrease in the Number of Monocytes/Macrophages. Cancer Sci (2008) 99(8):1595–602. doi: 10.1111/j.1349-7006.2008.00880.x
245. Daubiné F, Le Gall C, Gasser J, Green J, Clézardin P. Antitumor Effects of Clinical Dosing Regimens of Bisphosphonates in Experimental Breast Cancer Bone Metastasis. J Natl Cancer Inst (2007) 99(4):322–30. doi: 10.1093/jnci/djk054
246. Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, et al. Depletion of Tumor-Associated Macrophages Enhances the Effect of Sorafenib in Metastatic Liver Cancer Models by Antimetastatic and Antiangiogenic Effects. Clin Cancer Res (2010) 16(13):3420–30. doi: 10.1158/1078-0432.Ccr-09-2904
247. Coscia M, Quaglino E, Iezzi M, Curcio C, Pantaleoni F, Riganti C, et al. Zoledronic Acid Repolarizes Tumour-Associated Macrophages and Inhibits Mammary Carcinogenesis by Targeting the Mevalonate Pathway. J Cell Mol Med (2010) 14(12):2803–15. doi: 10.1111/j.1582-4934.2009.00926.x
248. Xing L, Ebetino FH, Boeckman RK, Srinivasan V, Tao J, Sawyer TK, et al. Targeting Anti-Cancer Agents to Bone Using Bisphosphonates. Bone (2020) 138:115492. doi: 10.1016/j.bone.2020.115492
249. Lu Y, Chen Q, Corey E, Xie W, Fan J, Mizokami A, et al. Activation of MCP-1/CCR2 Axis Promotes Prostate Cancer Growth in Bone. Clin Exp Metastasis (2009) 26(2):161–9. doi: 10.1007/s10585-008-9226-7
250. Li X, Loberg R, Liao J, Ying C, Snyder LA, Pienta KJ, et al. A Destructive Cascade Mediated by CCL2 Facilitates Prostate Cancer Growth in Bone. Cancer Res (2009) 69(4):1685–92. doi: 10.1158/0008-5472.CAN-08-2164
251. Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 Axis Recruits Tumor Associated Macrophages to Induce Immune Evasion Through PD-1 Signaling in Esophageal Carcinogenesis. Mol Cancer (2020) 19(1):41. doi: 10.1186/s12943-020-01165-x
252. Cho HR, Kumari N, Thi Vu H, Kim H, Park C-K, Choi SH. Increased Antiangiogenic Effect by Blocking CCL2-Dependent Macrophages in a Rodent Glioblastoma Model: Correlation Study With Dynamic Susceptibility Contrast Perfusion MRI. Sci Rep (2019) 9(1):11085. doi: 10.1038/s41598-019-47438-4
253. Li M, Knight DA L, MJ S, Stewart TJ. A Role for CCL2 in Both Tumor Progression and Immunosurveillance. Oncoimmunology (2013) 2(7):e25474. doi: 10.4161/onci.25474
254. Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, et al. Targeting CCL2 With Systemic Delivery of Neutralizing Antibodies Induces Prostate Cancer Tumor Regression In Vivo. Cancer Res (2007) 67(19):9417–24. doi: 10.1158/0008-5472.CAN-07-1286
255. Kirk PS, Koreckij T, Nguyen HM, Brown LG, Snyder LA, Vessella RL, et al. Inhibition of CCL2 Signaling in Combination With Docetaxel Treatment has Profound Inhibitory Effects on Prostate Cancer Growth in Bone. Int J Mol Sci (2013) 14(5):10483–96. doi: 10.3390/ijms140510483
256. Qian DZ, Rademacher BL, Pittsenbarger J, Huang CY, Myrthue A, Higano CS, et al. CCL2 is Induced by Chemotherapy and Protects Prostate Cancer Cells From Docetaxel-Induced Cytotoxicity. Prostate (2010) 70(4):433–42. doi: 10.1002/pros.21077
257. Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 Signaling Axis in Cancer Metastasis. Oncotarget (2016) 7(19):28697–710. doi: 10.18632/oncotarget.7376
258. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 Study of Carlumab (CNTO 888), a Human Monoclonal Antibody Against CC-Chemokine Ligand 2 (CCL2), in Metastatic Castration-Resistant Prostate Cancer. Invest New Drugs (2013) 31(3):760–8. doi: 10.1007/s10637-012-9869-8
259. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 Inhibition Accelerates Breast Cancer Metastasis by Promoting Angiogenesis. Nature (2014) 515(7525):130–3. doi: 10.1038/nature13862
260. Cassetta L, Pollard JW. Targeting Macrophages: Therapeutic Approaches in Cancer. Nat Rev Drug Discovery (2018) 17(12):887–904. doi: 10.1038/nrd.2018.169
261. Hu G, Su Y, Kang BH, Fan Z, Dong T, Brown DR, et al. High-Throughput Phenotypic Screen and Transcriptional Analysis Identify New Compounds and Targets for Macrophage Reprogramming. Nat Commun (2021) 12(1):773. doi: 10.1038/s41467-021-21066-x
262. Jones JD, Sinder BP, Paige D, Soki FN, Koh AJ, Thiele S, et al. Trabectedin Reduces Skeletal Prostate Cancer Tumor Size in Association With Effects on M2 Macrophages and Efferocytosis. Neoplasia (2019) 21(2):172–84. doi: 10.1016/j.neo.2018.11.003
263. Mendoza-Reinoso V, Baek DY, Kurutz A, Rubin JR, Koh AJ, McCauley LK, et al. Unique Pro-Inflammatory Response of Macrophages During Apoptotic Cancer Cell Clearance. Cells (2020) 9(2):429. doi: 10.3390/cells9020429
264. Oshi M, Tokumaru Y, Asaoka M, Yan L, Satyananda V, Matsuyama R, et al. M1 Macrophage and M1/M2 Ratio Defined by Transcriptomic Signatures Resemble Only Part of Their Conventional Clinical Characteristics in Breast Cancer. Sci Rep (2020) 10(1):16554. doi: 10.1038/s41598-020-73624-w
265. Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 Inhibitors as a Form of Cancer Immunotherapy: A Comprehensive Review of Registration Trials and Future Considerations. J Immunother Cancer (2018) 6(1):8. doi: 10.1186/s40425-018-0316-z
266. Dhupkar P, Gordon N, Stewart J, Kleinerman ES. Anti-PD-1 Therapy Redirects Macrophages From an M2 to an M1 Phenotype Inducing Regression of OS Lung Metastases. Cancer Med (2018) 7(6):2654–64. doi: 10.1002/cam4.1518
267. Sun NY, Chen YL, Wu WY, Lin HW, Chiang YC, Chang CF, et al. Blockade of PD-L1 Enhances Cancer Immunotherapy by Regulating Dendritic Cell Maturation and Macrophage Polarization. Cancers (Basel) (2019) 11(9). doi: 10.3390/cancers11091400
268. Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling During Successful Immune-Checkpoint Cancer Therapy. Cell (2018) 175(4):1014–30.e19. doi: 10.1016/j.cell.2018.09.030
269. Wei Z, Zhang X, Yong T, Bie N, Zhan G, Li X, et al. Boosting Anti-PD-1 Therapy With Metformin-Loaded Macrophage-Derived Microparticles. Nat Commun (2021) 12(1):440. doi: 10.1038/s41467-020-20723-x
270. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In Vivo Imaging Reveals a Tumor-Associated Macrophage–Mediated Resistance Pathway in Anti–PD-1 Therapy. Sci Transl Med (2017) 9(389):eaal3604. doi: 10.1126/scitranslmed.aal3604
271. Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int J Mol Sci (2018) 19(4):1232. doi: 10.3390/ijms19041232
272. Lugano R, Ramachandran M, Dimberg A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell Mol Life Sci (2020) 77(9):1745–70. doi: 10.1007/s00018-019-03351-7
273. Dost Gunay FS, Kırmızı BA, Ensari A, İcli F, Akbulut H. Tumor-Associated Macrophages and Neuroendocrine Differentiation Decrease the Efficacy of Bevacizumab Plus Chemotherapy in Patients With Advanced Colorectal Cancer. Clin Colorectal Cancer (2019) 18(2):e244–e50. doi: 10.1016/j.clcc.2018.12.004
274. Dalton HJ, Pradeep S, McGuire M, Hailemichael Y, Ma S, Lyons Y, et al. Macrophages Facilitate Resistance to Anti-VEGF Therapy by Altered VEGFR Expression. Clin Cancer Res (2017) 23(22):7034–46. doi: 10.1158/1078-0432.CCR-17-0647
275. Kloepper J, Riedemann L, Amoozgar Z, Seano G, Susek K, Yu V, et al. Ang-2/VEGF Bispecific Antibody Reprograms Macrophages and Resident Microglia to Anti-Tumor Phenotype and Prolongs Glioblastoma Survival. Proc Natl Acad Sci U.S.A. (2016) 113(16):4476–81. doi: 10.1073/pnas.1525360113
276. Peterson TE, Kirkpatrick ND, Huang Y, Farrar CT, Marijt KA, Kloepper J, et al. Dual Inhibition of Ang-2 and VEGF Receptors Normalizes Tumor Vasculature and Prolongs Survival in Glioblastoma by Altering Macrophages. Proc Natl Acad Sci U.S.A. (2016) 113(16):4470–5. doi: 10.1073/pnas.1525349113
277. Achyut BR, Shankar A, Iskander AS, Ara R, Angara K, Zeng P, et al. Bone Marrow Derived Myeloid Cells Orchestrate Antiangiogenic Resistance in Glioblastoma Through Coordinated Molecular Networks. Cancer Lett (2015) 369(2):416–26. doi: 10.1016/j.canlet.2015.09.004
Keywords: macrophage, tumor microenvironment, skeletal metastasis, bone marrow, myeloid derived suppressor cells, efferocytosis, angiogenesis
Citation: Batoon L and McCauley LK (2021) Cross Talk Between Macrophages and Cancer Cells in the Bone Metastatic Environment. Front. Endocrinol. 12:763846. doi: 10.3389/fendo.2021.763846
Received: 24 August 2021; Accepted: 11 October 2021;
Published: 03 November 2021.
Edited by:
Julie A. Rhoades (Sterling), Vanderbilt University, United StatesReviewed by:
Hanna Taipaleenmäki, University Medical Center Hamburg-Eppendorf, GermanyCopyright © 2021 Batoon and McCauley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurie K. McCauley, bWNjYXVsZXlAdW1pY2guZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.