
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 01 December 2021
Sec. Cancer Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.762095
This article is part of the Research Topic The Progress of Rare Lesions of the Sellar Region View all 23 articles
 Christopher S. Hong1
Christopher S. Hong1 Aladine A. Elsamadicy1
Aladine A. Elsamadicy1 Adeniyi Fisayo2Silvio E. Inzucchi3
Adeniyi Fisayo2Silvio E. Inzucchi3 Pallavi P. Gopal4Eugenia M. Vining5
Pallavi P. Gopal4Eugenia M. Vining5 E. Zeynep Erson-Omay1*
E. Zeynep Erson-Omay1* Sacit Bulent Omay1*
Sacit Bulent Omay1*Granular cell tumors of the pituitary belong to a rare family of neoplasms, arising from the posterior pituitary gland. Although considered benign, they may cause significant morbidity and residual disease after resection can lead to poor clinical outcomes. Currently, there is no known medical therapy for any posterior pituitary gland tumor, in part due to sparse molecular characterization of these lesions. We report data from whole exome sequencing of a case of granular cell tumor of the pituitary, performed under an institutional review board approved protocol. A 77 year-old female underwent resection of an incidentally diagnosed pituitary mass that was causing radiographic compression of the optic nerves with a subclinical temporal field defect and central hypothyroidism. The pathology of the resected specimen demonstrated a granular cell tumor of the posterior pituitary gland. Whole-exome sequencing revealed mutations predicted to be deleterious in key oncogenes, SETD2 and PAX8, both of which have been described in other cancers and could potentially be amenable to targeted therapies with existing approved drugs, including immune checkpoint inhibitors and histone deacetylase inhibitors, respectively. To our knowledge, this is the first comprehensive genomic characterization of granular cell tumor of the posterior pituitary gland. We report mutations in oncogenes predicted to be deleterious and reported in other cancers with potential for therapeutic targeting with existing pharmacologic agents. These data provide new insights into the molecular pathogenesis of GCT of the pituitary and may warrant further investigation.
Granular cell tumors (GCT) of the pituitary are rare, benign WHO I tumors. They, alongside pituicytomas, spindle cell oncocytomas, and sellar ependymomas comprise a family of tumors of the posterior pituitary gland (1). According to the latest edition of the WHO Classification of CNS Tumors, these pathologies may represent morphological variants of the same tumor with shared nuclear thyroid transcription factor-1 (TTF-1) expression and a lack of expression of pituitary hormones (2). Clinically, these tumors may cause symptoms related to local mass effect, including optic chiasm compression leading to visual field deficits, stalk compression with resultant hyperprolactinemia, and partial or even panhypopituitarism. Like pituitary adenomas, surgical resection of GCTs and other posterior pituitary tumors remains the mainstay of treatment (3). While considered benign, GCTs can be quite vascular (4–6), making complete surgical resection more difficult, and may lead to higher rates of tumor recurrence after subtotal resection and poorer survival outcomes (7, 8). The success of radiation therapy for residual tumors remains unclear, and likewise, there remain no known medical therapies for surgically refractory cases (7, 9, 10). As such, a better molecular understanding of these tumors is needed in order to develop potential novel targeted therapies.
To date, the molecular characterization of posterior pituitary tumors remain sparse and genomic sequencing have been comprised of only limited targeted sequencing panels (2, 11). To date, no genetic studies of GCTs of the pituitary have been performed. In this study, we report results from whole exome sequencing of a case of a GCT and discuss the relevant findings.
This study was conducted under an institutional review board approved protocol at Yale University. The patient’s blood and tumor tissue were collected after obtaining written informed consent. Histopathology, including immunohistochemical studies, were evaluated by a board-certified neuropathologist.
Whole exome sequencing and analysis was performed in accordance with our previously described methods at the Yale Cancer for Genome Analysis (YCGA) (12). Briefly, genomic DNA from the tumor and blood were isolated and coding regions were captured with IDT xGen Exome Research Panel v1 (Integrated DNA Technologies, Coralville, IA) with the additional spike-in of ~2,500 regions totaling ~620 kb of RefGene coding regions together with custom spikes designed specifically for cancer by Genomic Oncology Academic Laboratory (GOAL) Consortium. The captured regions were then sequenced on the Illumina NovaSeq6000 whole-exome sequencing platform with 2x100 base pair reads. Downstream analysis of raw reads, including alignment, duplicate marking, realignment, and base quality recalibration was performed according to “GATK Best Practice” recommendations (GATK v4.1.9). Somatic single nucleotide variant (SNV), insertion/deletions (INDEL) and, and copy number variations (CNV) were identified as previously described (12). Mean coverage of 130.1x was achieved for blood and 265.8x for tumor tissue.
A 77 year-old female with a recent diagnosis of ductal carcinoma of the breast in-situ with subsequent radiation and no radiographic evidence of recurrence was incidentally found to have a pituitary mass, measuring 1.6x1.5x2.6 cm on magnetic resonance imaging (MRI) of the brain, performed for a work-up of worsening hearing loss (Figures 1A–C). Review of systems was unremarkable and she was neurologically intact without any other complaints. Endocrine evaluation was notable for a mildly elevated prolactin of 51.1 ng/ml (normal range: 4.8-23.3 ng/mL), felt to be due to stalk effect, and decreased free T4 of 0.63 ng/dL (normal range: 0.80-1.70 ng/dL), the latter suggesting central hypothyroidism. She also demonstrated inappropriately low levels of LH and FSH gonadotropins for her menopausal status but of no clinical significance. The extent of pre-operative endocrine testing is outlined in Table 1. Neuro-ophthalmology testing revealed a temporal defect in the right eye, while left eye testing was limited given congenital vision loss on this side due to a macular scar. Given her visual deficit and radiographic compression of the optic nerves on MRI, surgical resection was recommended. The tumor was removed via an endoscopic endonasal approach and was noted intra-operatively to be well-encapsulated and originating from the posterior pituitary. The patient’s post-operative course was only remarkable for continued mildly reduced free T4 of 0.47 ng/dL and for a new low-normal morning cortisol level of 6.3 ug/dL (normal range: 6.0-18.4 ug/dL) for which she remained asymptomatic. She was started on levothyroxine and a prednisone taper, respectively. She was monitored for diabetes insipidus with serial sodium levels and urine output measurements but did not develop the condition. She also reported subjectively improved vision.
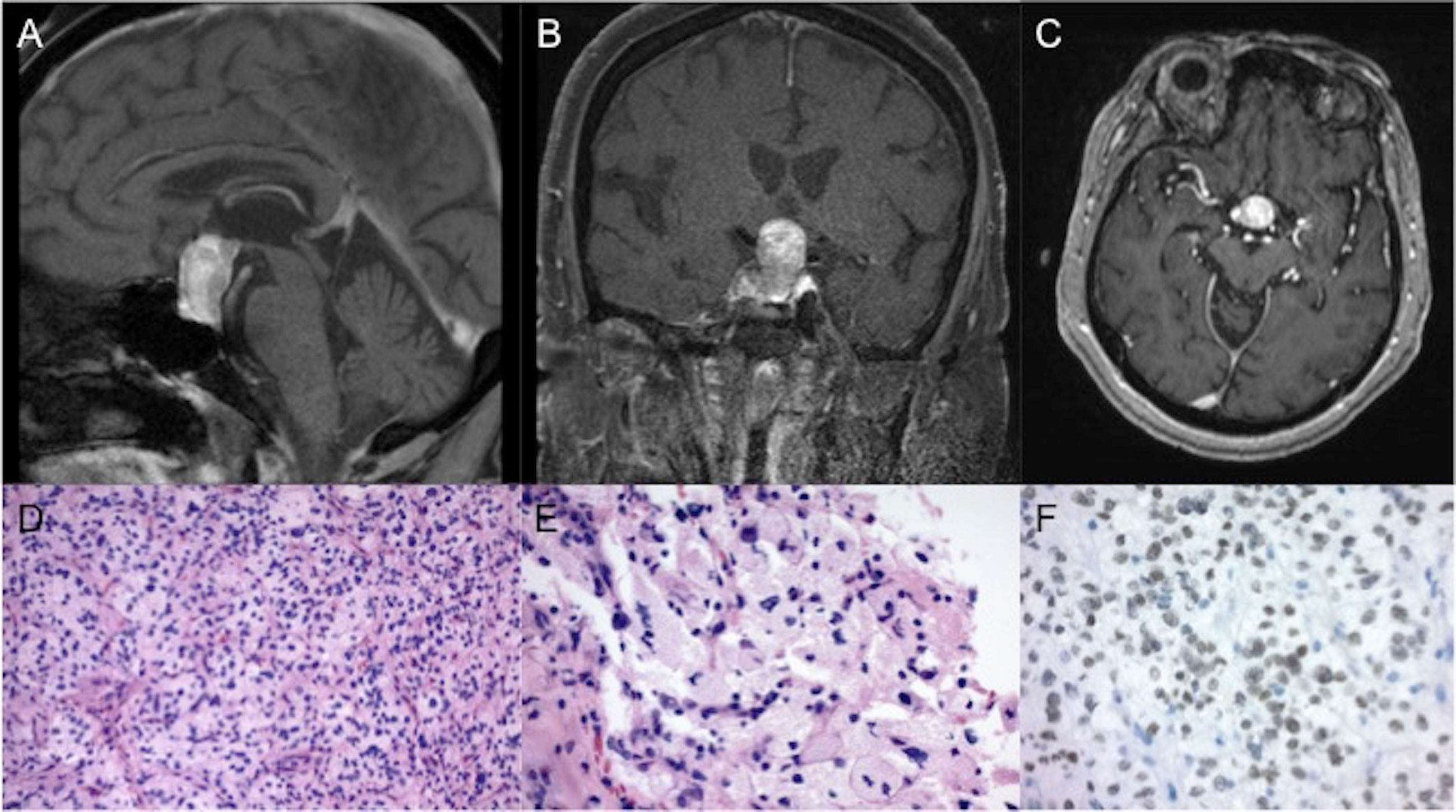
Figure 1 Imaging and pathology of the tumor. Representative (A) sagittal (B) coronal and (C) axial slices of a T1-weighted post-contrast MRI, obtained prior to surgery, showed a pituitary mass, measuring 1.6x1.5x2.6 cm. Final pathology of the resected tumor revealed a low grade neoplasm of polygonal cells with mild nuclear pleomorphism and granular cytoplasm on routine hematoxylin and eosin staining at (D) 200x and (E) 400x magnification. (F) Immunohistochemical stains revealed strong positivity for TTF-1 (magnification 400x).
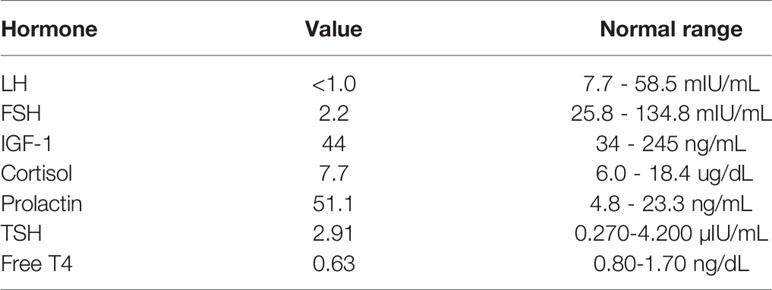
Table 1 Pre-operative endocrine testing of index patient.
Final pathology revealed a low grade neoplasm composed of polygonal cells with discrete cell borders, mild nuclear pleomorphism, and ample granular to foamy cytoplasm (Figures 1D, E). PAS and PAS with diastase stains showed scattered tumor cells with diastase resistant granules. Immunohistochemical stains revealed strong positivity for S100 and TTF-1 (Figure 1F) with focal positivity for CD68. Tumor cells were negative for GFAP, synaptophysin, keratins (Cam5.2, pancytokeratin), inhibin, carbonic anhydrase, PAX8, and D2-40. The Ki-67 index was low at 1-2%. Taken together, given positive TTF-1 staining in the absence of cytokeratin expression, the immunoprofile of the surgical specimen was consistent with a diagnosis of GCT of the pituitary.
Whole-exome sequencing (WES) was performed on the resected surgical specimen and matched blood in accordance with an institutional review board-approved protocol and utilizing previously described methods (13). No germline variants of potential pathological significance were identified. The analysis of somatic SNV/INDEL data revealed two somatic missense mutations in SETD2 (c.T2081C:p.V694A) and PAX8 (c.T527A:p.L176Q) with variant allele frequency (VAF) of 22.1% and 25.5%, respectively. Both variants are considered pathogenic by FATHMM algorithm (14). Detailed data are provided in Table 2. Chromosomal analysis revealed deletions in chromosomes 9, 10, and 13. Additionally, there was focal amplification on chromosome 12, involving PIK3C2G, a member of the class II PI3K kinase family, whose dysregulation may be involved in human metabolic diseases but its role in cancer remains unclear (15).

Table 2 Somatic genetic findings of index patient.
Outside of the posterior pituitary gland, GCTs have been described in many anatomic locations, most commonly in skin/subcutaneous soft tissues and the gastrointestinal tract and are thought to arise from Schwann cell origin (16). All GCTs stain positive for S100 and histologically show cells with granular eosinophilic cytoplasm, as demonstrated in our case. WES of GCTs remain limited, but recent hallmark studies have demonstrated loss-of-function mutations in vacuolar ATPase subunits, leading to aberrant lysosomal pH regulation and accumulation of intracytoplasmic granules, as observed histologically in GCTs (17–19). While these ATPase mutations may occur in up to 72% of GCTs (18), it is not known if they represent true driver mutations in tumorigenesis. We did not observe genomic alterations in ATPases or other genes integral to endosomal/lysosomal networks in our patient (17). It remains unclear whether GCTs of the posterior pituitary are true GCTs of similar Schwann cell origin. Alternatively, they have been proposed to belong to a morphologic spectrum of posterior pituitary tumors, all derived from pituicyte origin (20). Further genomic characterization of GCTs of the posterior pituitary may help elucidate this distinction.
Genomic characterization of posterior pituitary tumors have been limited to a handful of cases, given the rarity of these pathologies and limited sample sizes taken at time of surgery (11). For spindle cell oncocytomas and pituicytomas, individual mutations affecting the MAPK signaling pathway (SND1, FAT1, HRAS), as well as in other oncogenes (i.e. MEN1, TSC1, CBL, FZD7, PIK3GC, SBK1, CDKN2A/B, SKT11, SMARCA4, CIC, SMARCB1, NF1, NF2) have been reported in case reports or limited case series (11, 21, 22). Interestingly, BRAF mutations have been identified in two cases of spindle cell oncocytomas, which responded to targeted therapy with BRAF inhibitors (23, 24). However, to date, a comprehensive genetic study of GCTs of the pituitary has not been reported in the literature.
In this study, we found somatic mutations in SETD2 and PAX8, which are known tumor-related genes but not reported in tumors of the posterior pituitary. SETD2 is a histone methyltransferase whose normal function is critical for genomic stability and DNA damage repair (25). Clinically, SETD2 mutations are most prevalent in clear cell renal carcinomas but are also less commonly seen in hematopoietic cancers, high-grade glioma, melanoma, and adenocarcinomas of the lung and gastrointestinal system (25). SETD2 mutations have been correlated with shorter progression-free and overall survivals in metastatic renal cell carcinoma and breast cancers (26). Interestingly, SETD2 depletion in vitro led to microsatellite instability and increased mutational burden, characteristic of the mismatch repair deficient phenotype that may be amenable to immunotherapies (27). Indeed, in an analysis of The Cancer Genome Atlas (TCGA) pan-cancer cohort found that patients with tumors harboring SETD2 mutations responded favorably to treatment with immune checkpoint inhibitors, raising the possibility for targeted therapies in SETD2-mutated cancers (28).
PAX8 is a member of the paired box family of transcription factors, which encode proteins important in embryogenesis, particularly for thyroid and urogenital system development. In cancer, mutations in PAX8 have multiple downstream consequences affecting cell proliferation, adhesion, and angiogenesis (29). Clinically, positive immunohistochemical staining for PAX8 is widely used to diagnose primary renal cell tumors and mutations are also frequently seen in thyroid, ovarian, bladder, prostate, endometrial, cervical, and uterine cancers (29). In our case, the tumor did not stain for PAX8, as well as carbonic anhydrase and keratins, effectively ruling out a diagnosis of renal cell carcinoma. Although cases of pituitary metastasis have been reported from PAX8-mutated renal cell carcinoma (30), PAX8 mutations have not been reported in primary pituitary pathologies. In addition, PAX8 is a promising therapeutic target as its expression is restricted in normal tissues, compared to its role in cancer (31). Several preclinical studies have demonstrated suppression of cancer growth with genetic knockdown or pharmacologic inhibition of PAX8 (32, 33), including histone deacetylases (HDAC) inhibitors, which may epigenetically downregulate PAX8 transcripts and are a class of small molecule drugs now approved for cancer therapy (34).
This study reports comprehensive genomic characterization of a case of GCT of the pituitary. We found mutations predicted to be deleterious in known oncogenes, SETD2 and PAX8, previously not described in this pathology but have potential for therapeutic targeting with existing pharmacologic agents. The significance of these findings is limited by being a single case report, but genomic characterization of this rare tumor entity remains severely lacking. Further gene sequencing studies and mechanistic preclinical data are needed to better understand the pathophysiology and potential for molecularly based targeted therapies for GCTs of the pituitary.
The data used to support the findings of this study are available from the corresponding author upon request.
The studies involving human participants were reviewed and approved by Yale University Institutional Review Board. The patients/participants provided their written informed consent to participate in this study.
CH, ZE-O, and SB designed the study. All authors provided medical care to the patient. CH and ZE-O performed the genetic analysis. CH, ZE-O, and SB wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding for performing genomic sequencing and analysis of the data was provided by the Yale School of Medicine, Department of Neurosurgery Clinical Sequencing Funds.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mete O, Lopes MB. Overview of the 2017 WHO Classification of Pituitary Tumors. Endocr Pathol (2017) 28(3):228–43. doi: 10.1007/s12022-017-9498-z
2. Mete O, Lopes MB, Asa SL. Spindle Cell Oncocytomas and Granular Cell Tumors of the Pituitary Are Variants of Pituicytoma. Am J Surg Pathol (2013) 37(11):1694–9. doi: 10.1097/PAS.0b013e31829723e7
3. Ahmed AK, Dawood HY, Cote DJ, Bale TA, De Girolami U, Laws ER Jr., et al. Surgical Resection of Granular Cell Tumor of the Sellar Region: Three Indications. Pituitary (2019) 22(6):633–9. doi: 10.1007/s11102-019-00999-z
4. Aquilina K, Kamel M, Kalimuthu SG, Marks JC, Keohane C. Granular Cell Tumour of the Neurohypophysis: A Rare Sellar Tumour With Specific Radiological and Operative Features. Br J Neurosurg (2006) 20(1):51–4. doi: 10.1080/02688690600600996
5. Covington MF, Chin SS, Osborn AG. Pituicytoma, Spindle Cell Oncocytoma, and Granular Cell Tumor: Clarification and Meta-Analysis of the World Literature Since 1893. AJNR Am J Neuroradiol (2011) 32(11):2067–72. doi: 10.3174/ajnr.A2717
6. Gagliardi F, Spina A, Barzaghi LR, Bailo M, Losa M, Terreni MR, et al. Suprasellar Granular Cell Tumor of the Neurohypophysis: Surgical Outcome of a Very Rare Tumor. Pituitary (2016) 19(3):277–85. doi: 10.1007/s11102-016-0704-7
7. Ahmed AK, Dawood HY, Penn DL, Smith TR. Extent of Surgical Resection and Tumor Size Predicts Prognosis in Granular Cell Tumor of the Sellar Region. Acta Neurochir (Wien) (2017) 159(11):2209–16. doi: 10.1007/s00701-017-3337-3
8. Kleinschmidt-DeMasters BK, Lopes MB. Update on Hypophysitis and TTF-1 Expressing Sellar Region Masses. Brain Pathol (2013) 23(5):495–514. doi: 10.1111/bpa.12068
9. Guerrero-Perez F, Marengo AP, Vidal N, Iglesias P, Villabona C. Primary Tumors of the Posterior Pituitary: A Systematic Review. Rev Endocr Metab Disord (2019) 20(2):219–38. doi: 10.1007/s11154-019-09484-1
10. Osman M, Wild A. Spindle Cell Oncocytoma of the Anterior Pituitary Presenting With an Acute Clinical Course Due To Intraventricular Hemorrhage. A Case Report and Review of Literature. Am J Case Rep (2017) 18:894–901. doi: 10.12659/AJCR.903702
11. Barresi V, Simbolo M, Gessi M, Rossi S, Caffo M, Eccher A, et al. Clinical-Pathological, Immunohistochemical, and Genetic Characterization of a Series of Posterior Pituitary Tumors. J Neuropathol Exp Neurol (2021) 80(1):45–51. doi: 10.1093/jnen/nlaa139
12. Fomchenko EI, Erson-Omay EZ, Zhao A, Bindra RS, Huttner A, Fulbright RK, et al. DNMT3A Co-Mutation in an IDH1-Mutant Glioblastoma. Cold Spring Harb Mol Case Stud (2019) 5(4). doi: 10.1101/mcs.a004119
13. Hong CS, Kuzmik GA, Kundishora AJ, Elsamadicy AA, Koo AB, McGuone D, et al. Hypermutated Phenotype in Gliosarcoma of the Spinal Cord. NPJ Precis Oncol (2021) 5(1):8. doi: 10.1038/s41698-021-00143-w
14. Rogers MF, Shihab HA, Mort M, Cooper DN, Gaunt TR, Campbell C. FATHMM-XF: Accurate Prediction of Pathogenic Point Mutations via Extended Features. Bioinformatics (2018) 34(3):511–3. doi: 10.1093/bioinformatics/btx536
15. Gulluni F, De Santis MC, Margaria JP, Martini M, Hirsch E. Class II PI3K Functions in Cell Biology and Disease. Trends Cell Biol (2019) 29(4):339–59. doi: 10.1016/j.tcb.2019.01.001
16. Mobarki M, Dumollard JM, Dal Col P, Camy F, Peoc'h M, Karpathiou G. Granular Cell Tumor a Study of 42 Cases and Systemic Review of the Literature. Pathol Res Pract (2020) 216(4):152865. doi: 10.1016/j.prp.2020.152865
17. Franca JA, Gayden T, Bareke E, Santos JN, de Sousa SF, Bastos-Rodrigues L, et al. Whole-Exome Sequencing Reveals Novel Vacuolar ATPase Genes' Variants and Variants in Genes Involved in Lysosomal Biology and Autophagosomal Formation in Oral Granular Cell Tumors. J Oral Pathol Med (2021) 50(4):410–7. doi: 10.1111/jop.13148
18. Pareja F, Brandes AH, Basili T, Selenica P, Geyer FC, Fan D, et al. Loss-Of-Function Mutations in ATP6AP1 and ATP6AP2 in Granular Cell Tumors. Nat Commun (2018) 9(1):3533. doi: 10.1038/s41467-018-05886-y
19. Sekimizu M, Yoshida A, Mitani S, Asano N, Hirata M, Kubo T, et al. Frequent Mutations of Genes Encoding Vacuolar H(+) -ATPase Components in Granular Cell Tumors. Genes Chromosomes Cancer (2019) 58(6):373–80. doi: 10.1002/gcc.22727
20. Whipple SG, Savardekar AR, Rao S, Mahadevan A, Guthikonda B, Kosty JA. Primary Tumors of the Posterior Pituitary Gland: A Systematic Review of the Literature in Light of the New 2017 World Health Organization Classification of Pituitary Tumors. World Neurosurg (2021) 145:148–58. doi: 10.1016/j.wneu.2020.09.023
21. Miller MB, Bi WL, Ramkissoon LA, Kang YJ, Abedalthagafi M, Knoff DS, et al. MAPK Activation and HRAS Mutation Identified in Pituitary Spindle Cell Oncocytoma. Oncotarget (2016) 7(24):37054–63. doi: 10.18632/oncotarget.9244
22. Viaene AN, Lee EB, Rosenbaum JN, Nasrallah IM, Nasrallah MP. Histologic, Immunohistochemical, and Molecular Features of Pituicytomas and Atypical Pituicytomas. Acta Neuropathol Commun (2019) 7(1):69. doi: 10.1186/s40478-019-0722-6
23. Dawoud FM, Naylor RM, Giannini C, Swanson AA, Meyer FB, Uhm JH. TTF-1 Positive Posterior Pituitary Tumor: Limitations of Current Treatment and Potential New Hope in BRAF V600E Mutation Variants. Clin Neurol Neurosurg (2020) 196:106059. doi: 10.1016/j.clineuro.2020.106059
24. Sollfrank L, Lettmaier S, Erdmann M, Uslu U. Panniculitis Under Successful Targeted Inhibition of the MAPK/ERK Signaling Pathway in a Patient With BRAF V600E-Mutated Spindle Cell Oncocytoma of the Pituitary Gland. Anticancer Res (2019) 39(7):3955–9. doi: 10.21873/anticanres.13549
25. Fahey CC, Davis IJ. SETting the Stage for Cancer Development: SETD2 and the Consequences of Lost Methylation. Cold Spring Harb Perspect Med (2017) 7(5). doi: 10.1101/cshperspect.a026468
26. Newbold RF, Mokbel K. Evidence for a Tumour Suppressor Function of SETD2 in Human Breast Cancer: A New Hypothesis. Anticancer Res (2010) 30(9):3309–11.
27. Li F, Mao G, Tong D, Huang J, Gu L, Yang W, et al. The Histone Mark H3K36me3 Regulates Human DNA Mismatch Repair Through Its Interaction With MutSalpha. Cell (2013) 153(3):590–600. doi: 10.1016/j.cell.2013.03.025
28. Lu M, Zhao B, Liu M, Wu L, Li Y, Zhai Y, et al. Pan-Cancer Analysis of SETD2 Mutation and Its Association With the Efficacy of Immunotherapy. NPJ Precis Oncol (2021) 5(1):51. doi: 10.1038/s41698-021-00193-0
29. Fernandez LP, Lopez-Marquez A, Santisteban P. Thyroid Transcription Factors in Development, Differentiation and Disease. Nat Rev Endocrinol (2015) 11(1):29–42. doi: 10.1038/nrendo.2014.186
30. Gandhi GY, Fung R, Natter PE, Makary R, Balaji KC. Symptomatic Pituitary Metastasis as Initial Manifestation of Renal Cell Carcinoma: Case Report and Review of Literature. Case Rep Endocrinol (2020) 2020:8883864. doi: 10.1155/2020/8883864
31. Chaves-Moreira D, Morin PJ, Drapkin R. Unraveling the Mysteries of PAX8 in Reproductive Tract Cancers. Cancer Res (2021) 81(4):806–10. doi: 10.1158/0008-5472.CAN-20-3173
32. Hardy LR, Pergande MR, Esparza K, Heath KN, Onyuksel H, Cologna SM, et al. Proteomic Analysis Reveals a Role for PAX8 in Peritoneal Colonization of High Grade Serous Ovarian Cancer That can be Targeted With Micelle Encapsulated Thiostrepton. Oncogene (2019) 38(32):6003–16. doi: 10.1038/s41388-019-0842-2
33. Shi K, Yin X, Cai MC, Yan Y, Jia C, Ma P, et al. PAX8 Regulon in Human Ovarian Cancer Links Lineage Dependency With Epigenetic Vulnerability to HDAC Inhibitors. Elife (2019) 8. doi: 10.7554/eLife.44306
Keywords: pituitary, granular cell, sequencing, SETD2, PAX8, case report
Citation: Hong CS, Elsamadicy AA, Fisayo A, Inzucchi SE, Gopal PP, Vining EM, Erson-Omay EZ and Bulent Omay S (2021) Comprehensive Genomic Characterization of A Case of Granular Cell Tumor of the Posterior Pituitary Gland: A Case Report. Front. Endocrinol. 12:762095. doi: 10.3389/fendo.2021.762095
Received: 20 August 2021; Accepted: 25 October 2021;
Published: 01 December 2021.
Edited by:
Wang Haijun, The First Affiliated Hospital of Sun Yat-Sen University, ChinaReviewed by:
Sergei I. Bannykh, Cedars Sinai Medical Center, United StatesCopyright © 2021 Hong, Elsamadicy, Fisayo, Inzucchi, Gopal, Vining, Erson-Omay and Bulent Omay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: E. Zeynep Erson-Omay, emV5bmVwLmVyc29uQHlhbGUuZWR1; Sacit Bulent Omay, c2FjaXQub21heUB5YWxlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.