
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 23 December 2021
Sec. Pediatric Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.758723
 Chaoyan Tang1†
Chaoyan Tang1† Liheng Meng2†
Liheng Meng2† Ping Zhang1Xinghuan Liang2Chaozhi Dang1Hui Liang1Junfeng Wu1Haiyun Lan1*
Ping Zhang1Xinghuan Liang2Chaozhi Dang1Hui Liang1Junfeng Wu1Haiyun Lan1* Yingfen Qin2*
Yingfen Qin2*Background: We aimed to analyze a novel ABCC8 variant of a Chinese patient with suspected maturity-onset diabetes of the young (MODY) and to provide evidence for precise diagnosis and appropriate treatment.
Method: A Chinese family with suspected MODY was recruited in this study, which included a 15-year-old female patient with diabetes. Clinical data and blood samples were collected from the proband and other family members. All of the living relatives were given an oral glucose tolerance test. Next-generation sequencing was performed to identify the mutated genes in the proband. Sanger sequencing was utilized to confirm the location of the pathogenic variant in all subjects. Further treatment was referred to targeted family members according to genetic testing.
Results: The proband was found to have a random blood glucose level of 244.8 mg/dl and an HbA1c level of 9.2%. Before this investigation, her grandparents had been diagnosed with diabetes. The second uncle, two aunts, mother, and cousin of the proband were diagnosed with diabetes by abnormal HbA1C (6.5–12.1%) and fasting blood glucose (FBG, 91.4–189.7 mg/dl). The second aunt of the proband had impaired glucose homeostasis (HbA1C = 6.4% and FBG = 88.0 mg/dl). One novel missense variant c.1432G>A (p.A478T) in exon 9 of the ABCC8 gene was detected in the proband with suspected MODY. The variant was also found in six family members with diabetes or impaired glucose homeostasis, including her second uncle, two aunts, mother, and cousin. After the treatment was switched to glimepiride, the fasting blood glucose was adjusted to 99.54 mg/dl, the 2-h postprandial blood glucose was 153.54 mg/dl, serum fructosamine was 259 μmol/l, and HbA1c was 5.8%. The glycemic control remained optimal, and no hypoglycemic episodes were observed in the living relatives.
Conclusion: This study revealed one novel missense variant of the ABCC8 gene in Chinese families. The present findings indicated that the members of this family responded to treatment with sulfonylureas as previously seen in ABCC8 MODY.
Maturity-onset diabetes of the young (MODY) is a rare type of diabetes with a defect in pancreatic β-cell function, which is predominantly inherited as a typically autosomal dominant and rarely autosomal recessive trait (1, 2). It has been reported that MODY accounts for approximately 1 to 2% of all cases of diabetes in Europe and North American countries (3, 4). Monogenic diabetes is usually misdiagnosed as type 1 or type 2 diabetes mellitus (DM) as the clinical features are similar (5), but the treatment strategies and prognosis differ from other types of DM (6). The most common forms of monogenic diabetes share residual insulin secretion, leading to the detection of C-peptide, which distinguishes MODY from severe type 1 diabetes. To date, at least 14 MODY subtypes have been identified depending on specific variants, including GCK, HNF4A, HNF1A, and others (7). In patients with MODY, the most frequently mutated gene is GCK (8).
MODY12 is caused by ABCC8-activating variants, which are associated with the pancreatic ATP-sensitive potassium channel (KATP-channel). Bowman et al. (9) first discovered a pedigree of MODY12 with an ABCC8 missense variant. There are more than 30 families reported worldwide to carry the heterozygous variants of ABCC8 in MODY. A shift from insulin to normal- or high-dose sulfonylurea drugs is the main treatment strategy for MODY12, and the response to this treatment is similar to the low doses of sulfonylurea treatment of HNF1A/HNF4A MODY. The ABCC8 gene, a member of the ABCC subfamily and 100 kb in length, is located at 11p15.1 and encodes the SUR1 protein (10). SUR1 is an ATP-binding cassette transporter and a regulatory subunit and modulates KATP-channel and insulin release. Variants in the ABCC8 gene are the most frequent cause of hyperinsulinemic hypoglycemia in infants (11). There are 2,318 common and rare single-nucleotide polymorphisms found in the ABCC8 gene, of which rs757110, rs1799854, rs1799859, and rs1801261 are the most frequently reported variants in DM (12). Nevertheless, the role and mechanisms of ABCC8 in MODY12 are not yet clear and require more research.
This study reports a novel missense variant of the ABCC8 gene in a Chinese family with suspected MODY. Further treatment was modified according to genetic testing.
A 15-year-old female with increased blood glucose was recruited in this study on December 18, 2019. Clinical information and blood samples were obtained from 11 living family members.
Peripheral blood (5 ml) was collected from the included family members. Blood cells were isolated after permeation and ruptured in the blood cell lysis solution. A high-salt solution was added to the lysis solution to precipitate proteins and impurities. Centrifugation removed the precipitate to obtain a supernatant containing only DNA. Isopropanol was added to the precipitate and to recover the DNA, which was washed with 70% ethanol to remove the salt and finally mixed with 1 X Buffer TE to dissolve the DNA. Human genomic DNA was extracted for library preparation. The genomic DNA was broken into main bands of <500 bp, with peak values at 350-bp small DNA fragments. The ends of the broken DNA fragments were then filled in; an “A” base was added at the 3′ end so that the DNA fragment was connected to a special linker with a “T” base at the 5′ end. Pre-Capture LM-PCR amplified the library with adapters. A total of 206 known diabetes-related genes were collected for capture (Supplementary File 1). After the library was combined, the human exon library was added for hybridization, the exon capture area was captured, and the product was enriched by PCR amplification. Agarose gel electrophoresis was used to determine the fragment size of the library, while Qubit3.0 and real-time PCR were used to determine the concentration of the library. The qualified library was diluted to the concentration indicated by the computer, and a NextSeq500 sequencer (Illumina, San Diego, CA, USA) was used for sequencing analysis.
Sanger sequencing was performed to verify the variant. The primers were designed according to the sequence of the verification site of the ABCC8 gene through the UCSC website and Primer3 as follows: forward: 5′-GAGACCTGCTGCTGTCGAG-3′ and reverse: 5′-CCAAGGCTTGTCCCACTCTA-3′. PCR was used for amplification. According to high-throughput sequencing, positive variant sites were identified, specific primers were designed at both ends of the mutant gene site fragments to be detected for amplification, and then analysis was done by agarose gel electrophoresis. The amplified product was sequenced by Sanger sequencing to obtain gene fragment sequences, and the variants and short fragment deletions or insertions of the positive site were further analyzed and verified by Sanger sequencing to determine the specific variant of the site.
All participants underwent an oral glucose tolerance test (OGTT). The parameters for diabetes were tested concurrently, including fasting blood glucose (FBG), postprandial blood glucose (PBG), 0-h C-peptide, and 2-h C-peptide. HbA1c was determined using immunoturbidimetric hemolytic automated analysis. Abnormal glucose homeostasis and diabetes-related antibodies were tested by islet autoantibody assay: glutamic acid decarboxylase autoantibody (GADA), insulin autoantibody (IAA), islet antigen-2 antibody (IA-2Ab), zinc transporter-8 autoantibody (ZnT8A), and islet cell antibody (ICA-IgG).
A 15-year-old girl was admitted to our hospital. She was suffering from a fever and underwent a random blood glucose test with a result of 266.22 mg/dl on December 5, 2019. She returned to our hospital because of increased blood glucose on December 18, 2019. Her random blood glucose was 244.8 mg/dl and her HbA1c was 9.2%. There was no obvious polydipsia, polyuria, hunger, or weight loss. The physical examination on admission revealed a body mass index (BMI) of 21.72 kg/m2, a regular pulse of 85 BPM, and blood pressure of 125/65 mmHg. The laboratory findings revealed that urine glucose was 1+ and urine ketones were negative. FBG was 196.2 mg/dl; the next-day FBG was 151.56 mg/dl, and the 2-h PBG was 279 mg/dl. The serum HbA1c level was 9.3%, glycated albumin was 374 μmol/l, the fasting C-peptide was 2.05 ng/ml, and the 2-h postprandial C-peptide was 4.7 ng/ml. The islet GADA, IA-2Ab, IAA, and ZnT8A were negative, and the ICA-IgG was positive (Table 1). The abdominal ultrasound and fundus photography showed no abnormalities. The ultrasonic Doppler examination indicated that the brachiocephalic arteries were normal and not atherosclerotic.
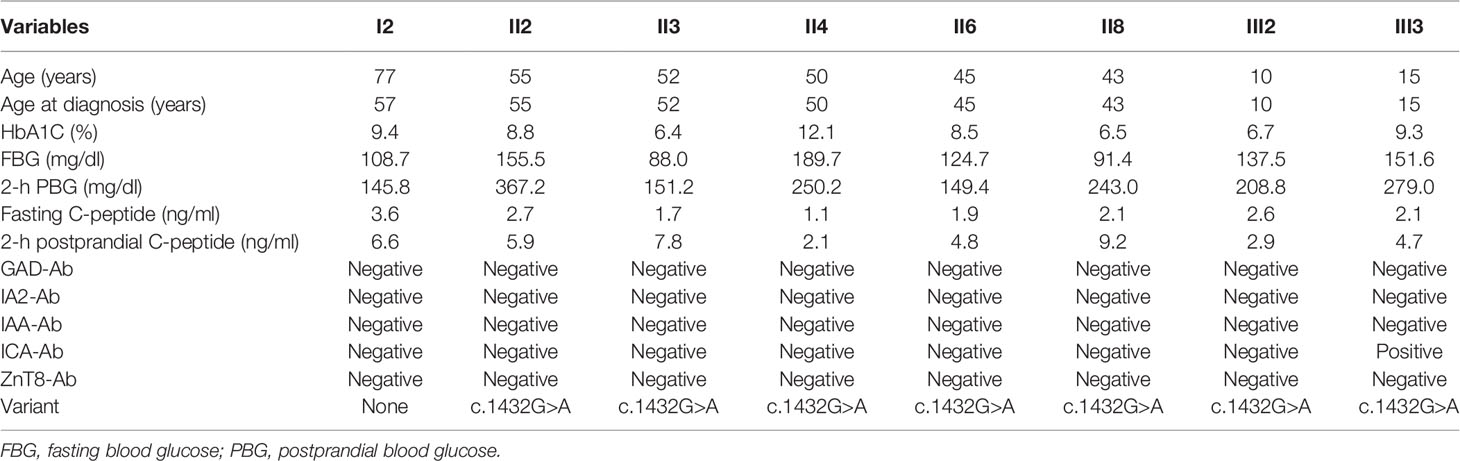
Table 1 Clinical characteristics of each patient with MODY12.
Through family history, we learned that the grandfather (I1) and grandmother (I2) of the proband had been diagnosed with diabetes. The grandfather (I1) of the proband was born in 1940, diagnosed with diabetes in 1996, and died in 2000. The grandmother (I2) of the proband was born in 1943 and diagnosed with diabetes in 2003. We then recruited all the living relatives for an OGTT test. A total of nine members in the three-generation family were diagnosed with diabetes or abnormal glucose homeostasis. Based on the OGTT test, the first aunt (II2), uncle (II4), fourth aunt (II6), mother (II8), and cousin (III2) of the proband were diagnosed with diabetes by abnormal HbA1C (6.5–12.1%) and fasting blood glucose (FBG, 91.4–189.7 mg/dl). The second aunt (II3) of the proband had impaired glucose homeostasis (HbA1C = 6.4% and FBG = 88.0 mg/dl). The fasting C-peptide ranged from 1.1 to 3.6 ng/ml, and the 2-h postprandial C-peptide was from 2.1 to 6.6 in these relatives (Table 1). No relatives presented with typical symptoms of diabetes or received any treatment. The family pedigree is shown in Figure 1.
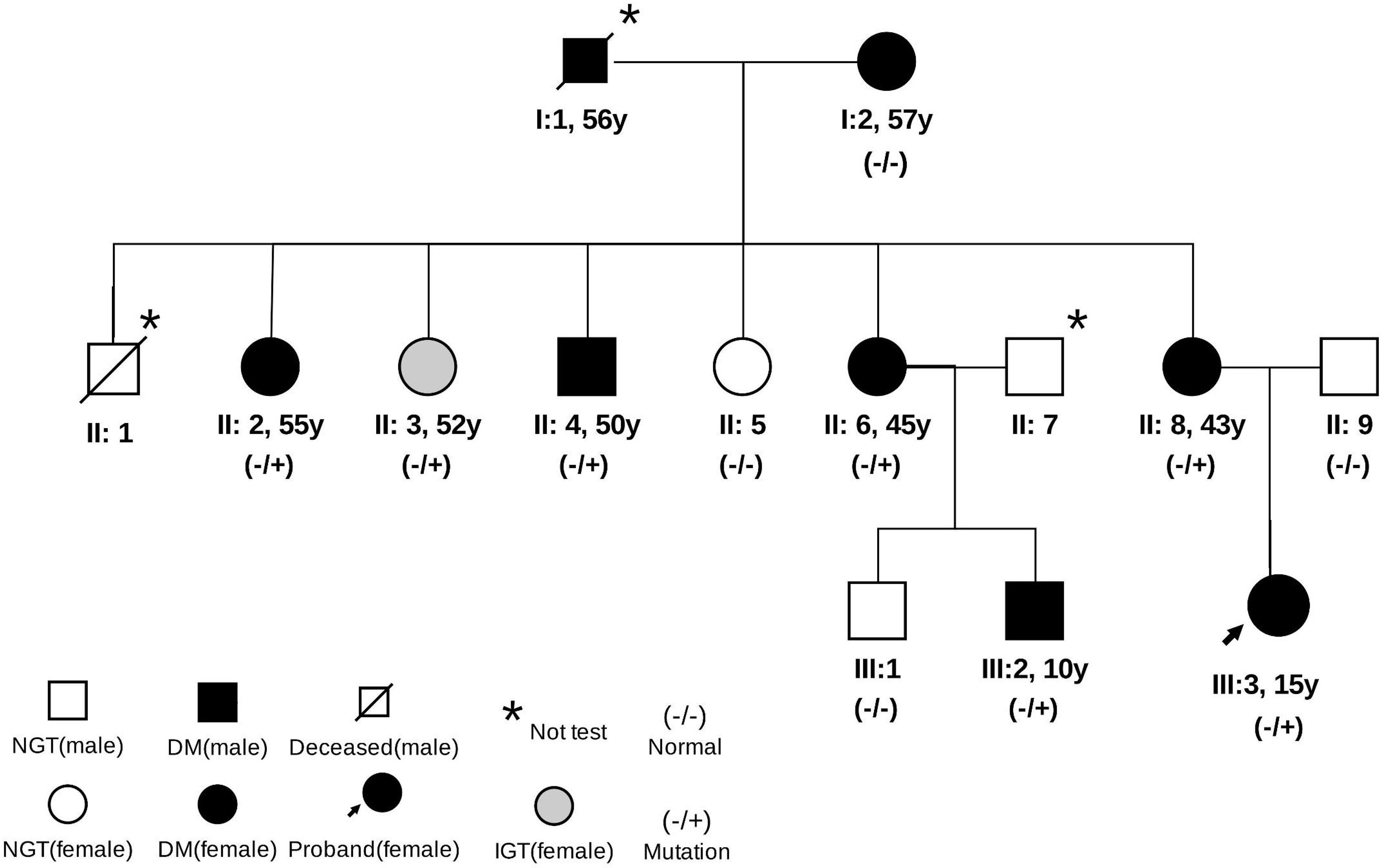
Figure 1 Pedigree of the family. Data under the symbols: age at diabetes diagnosis. NGT, normal glucose tolerance; IGT, impaired glucose tolerance.
Thus, we suspected that this family might be suffering MODY and performed target capture and sequencing for 206 known diabetes-related genes in the participants. In this genetic testing, a novel heterozygous variant, c.1432G>A (p.A478T), of the ABCC8 gene was detected in the proband. Sanger sequencing revealed the missense variant c.1432G>A (p.A478T) in exon 9 of the ABCC8 gene in the proband and her mother, but not her father (Figure 2). These results verified that the heterozygous variant in the proband might have originated from the mother (heterozygous status). Furthermore, by copy number and SNP analysis, no copy number variants that could be associated with clinical manifestations were detected. This variant was also found in the family members with diabetes or impaired glucose homeostasis, except for the grandmother: her aunt (II2), second aunt (II3), fourth aunt (II6), and cousin (III2). The father (II9), third aunt (II5), and cousin (III1) of the proband had normal glucose homeostasis and no missense variants. The missense variant showed a co-segregation of diabetes in the family members. However, the variant was not tested in the grandfather (I2) of the proband because he died in 2000. According to the history of the grandfather, it is presumed that he was diabetic with the ABCC8 MODY missense variant. Considering the ages at diagnosis and that three consecutive generations were affected, we speculated that the ABCC8 variant was causal (13–15).
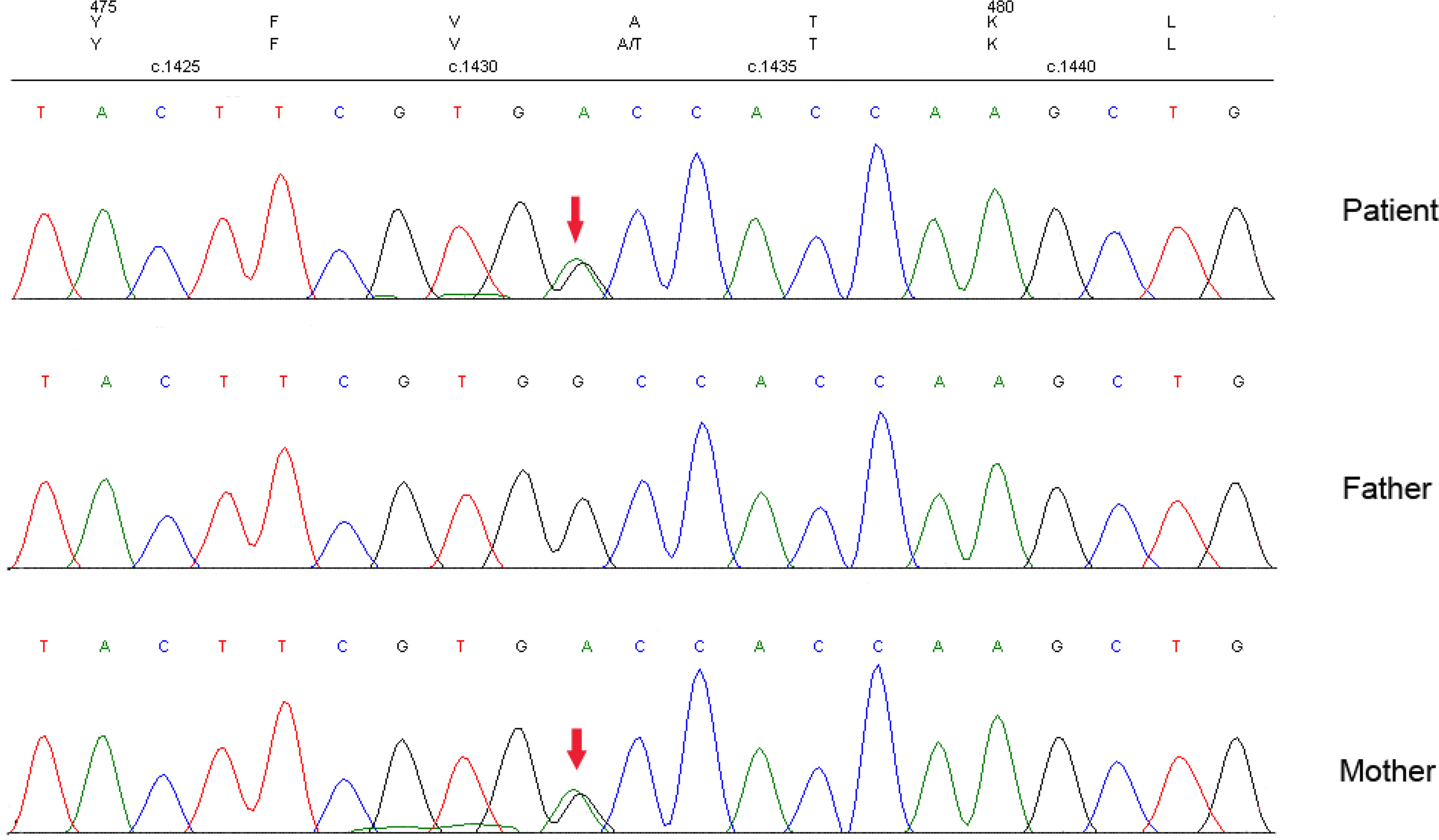
Figure 2 Gene sequencing results of the proband and her parents.
The timeline for the episode of care for the proband (III3) is shown in Figure 3A. Before the genetic test, the proband (III3) was treated subcutaneously with 4 U of short-acting insulin three times daily before meals starting on December 22, 2019. Blood glucose was controlled at FPG fluctuations of 72–126 mg/dl and PBG fluctuations of 108–234 mg/dl. Occasionally, she experienced hypoglycemia after conscious exercise or missed insulin injections while at boarding school. Given the results of genotyping, her treatments were modified. The proband (III3) switched from insulin to 1 mg glimepiride per day on March 19, 2020. After 1 month of treatment, FBG was 99.54 mg/dl, 2-h PBG was 153.54 mg/dl, and serum fructosamine was 259 μmol/l. The HbA1c levels ranged from 5.8 to 6.5% over a 1-year follow-up after the glimepiride treatment. There were no instances of hypoglycemia, and self-monitored fasting glucose was maintained in the range of 81–115.2 mg/dl. The glycemic control at different time points is presented in Figures 3B–D. Advice on health education, diet control, and strengthening exercises was provided to the affected family members. For the mother (II8) and second aunt (II3) of the proband, mild hyperglycemia was corrected successfully by diet alone. The uncle (II4) of the proband switched from insulin to 2 mg glimepiride per day and 150 mg acarbose per day. The aunt (II2) of the proband was given 1 mg glimepiride and 150 mg acarbose per day. The fourth aunt (II6) and cousin (III2) of the proband were given 1 mg glimepiride per day. Glycemic control remained optimal, and no hypoglycemic episodes were observed in the relatives.
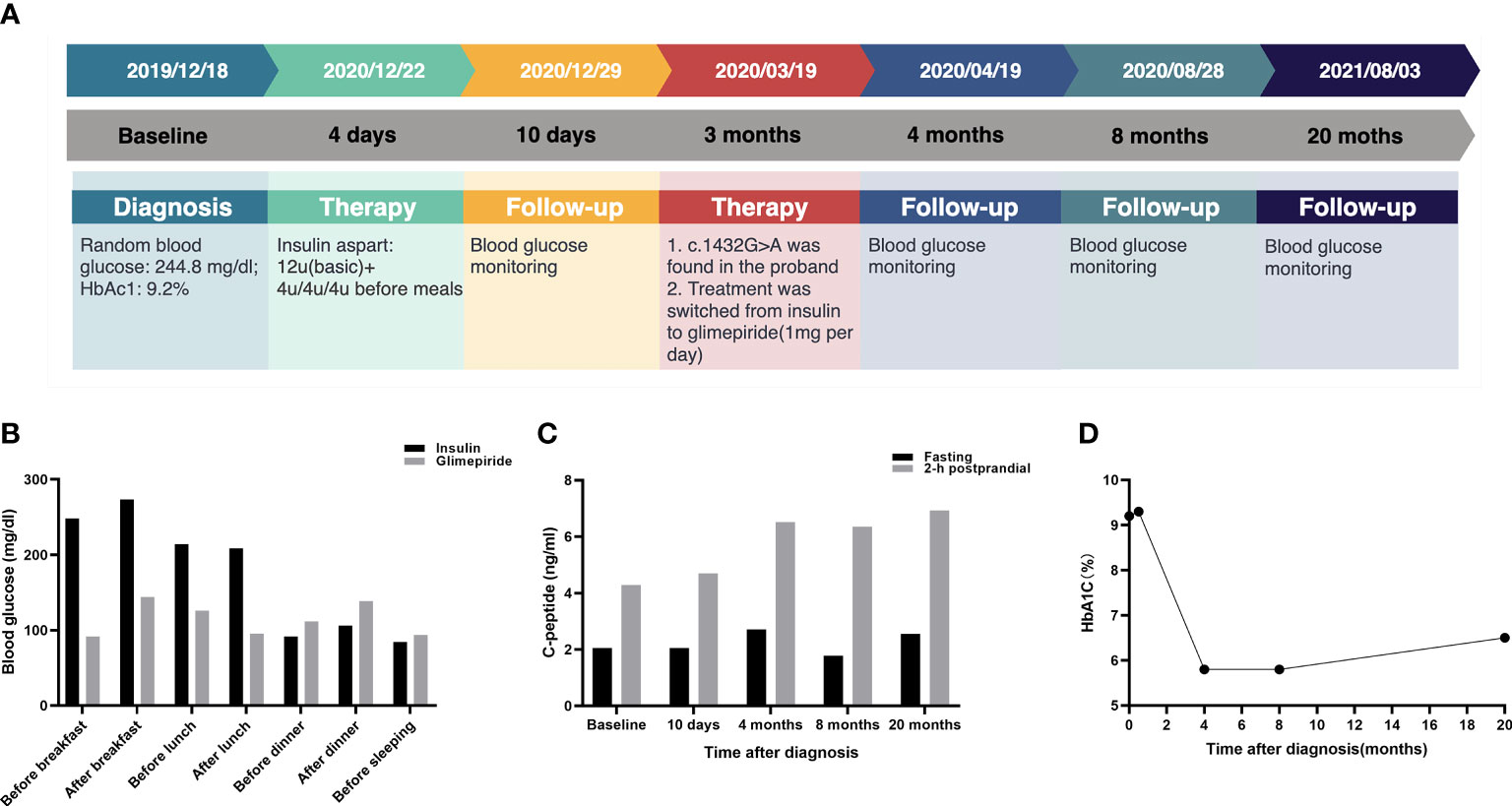
Figure 3 Glycemic control for the proband at follow-up. (A) Timeline for the episode of care. The top panel indicates the date of intervention. The middle panel shows the time period after the diagnosis. The bottom panel presents the intervention for the proband. (B) Comparison of glycemic control between insulin and glimepiride treatment for proband. (C) The C-peptide and 2-h postprandial C-peptide at different time points. (D) HbA1C levels at different time points.
In this study, we reported on a MODY family with a new missense variant in exon 9 of the ABCC8 gene (c.1432G>A, p.A478T), detected for the first time in the Chinese population. The assessment of MODY in the study was supported by the presence of hyperglycemia in three generations of the same family and the diagnosis of diabetes for the proband (III3) and cousin (III2) at ages <25 years. Family segregation was confirmed by diagnoses of the remaining family members (13–15).
To date, more than 30 ABCC8-MODY families with missense variants have been reported worldwide (Supplementary File 2). Studies in animal models indicate that activating ABCC8 variants may lead to diabetes (16), and since 2006, multiple studies have linked it to a wide range of clinical types of diabetes, including transient neonatal diabetes mellitus, and permanent neonatal diabetes mellitus (17, 18). In addition, congenital hyperinsulinism caused by ABCC8 variants can develop into MODY (19). The family member with abnormal glucose homeostasis had no history of hypoglycemia or hyperglycemia in the neonatal period. ABCC8 MODY is characterized by other clinical characteristics, including a lack of progression to ketosis, detectable C-peptide, negative pancreatic islet autoantibodies, and sensitivity to sulfonylureas. The relative of the patient who presented with abnormal glucose homeostasis was not ketosis-prone. The three-generation family was affected with diabetes, and the proband had early-onset hyperglycemia (≤25 years of age), was not ketosis-prone, and had acceptable C-peptide insulin levels, which allowed her condition to be differentiated from type 1 diabetes. However, the proband had a BMI of 21.72 kg/m2 and did not have acanthosis nigricans or insulin resistance, which also ruled out a diagnosis of type 2 diabetes. Interestingly, the proband was found to be positive for ICA-IgG; however, the islet autoantibodies in the other seven variant carriers were negative. Although there was a low frequency of seropositivity for beta-cell antibodies in MODY, investigations conducted outside of China have discovered that islet autoimmune antibodies can be positive in some MODY patients (17, 20–22). There was also a patient with a history of T1DM, T2DM, and HNF1A diabetes (23). A prior study found that, after a few months, the titer of GADA and ICA in a MODY patient changed from negative to positive. Another MODY patient who was ICA- and GADA-negative acquired autoimmunity, and after a 1-year follow-up, a moderate positive was discovered (24). We hypothesized that mutations in MODY genes altered the structural adjustments of β-cell enzymes or regulated the unknown genes in the autoimmune pathways, therefore tying MODY to autoimmunity. It is worth noting that a prior study found that the C-peptide levels increased with each year of age at diagnosis (25). The HbA1C levels were inversely related to C-peptide (26). The diversity in both C-peptide preservations for the families in this investigation could be related to the combined effect of HbA1C and age at diagnosis. On the islet cell membrane of chromosome 11, ABCC8 encodes a portion of the KATP channel. The KATP channel controls the electrical activity of pancreatic islet cells and links cell homeostasis with the electrical activity of the plasma membrane, thereby regulating the secretion of insulin. Because of the key role of the KATP channel in insulin secretion, the pathogenic variants in ABCC8 genes are associated with severe disorders of glucose homeostasis. The ABCC8 gene variants include inactivating variants and activating variants. Inactivating variants are recognized owing to insulin over-secretion, resulting in hyperinsulinemic hypoglycemia in infants (12, 27). This heterozygous variant of the ABCC8 gene c.1432G>A (p.A478T) has not been reported in relevant clinical cases. This variant is a pathogenic mutation at the same amino acid position, p.A478D, that was reported previously (28). Studies show that sulfonylureas bind to the SUR in islet cells with high affinity (29, 30) and that the channel closes to promote insulin secretion. Sulfonylureas have been used to effectively treat individuals with ABCC8 MODY by acting on KATP channels. In one study, four families of non-obese patients were found to have diabetes before the age of 10 and were genetically diagnosed with ABCC8 MODY. Compared with patients with type 2 diabetes, patients with ABCC8 MODY are more sensitive to sulfonylureas. A group of researchers reported a 27-year-old patient with non-obese diabetes with epilepsy and his mother who were genetically diagnosed with ABCC8 MODY. Their blood glucose levels improved without hypoglycemia after their treatment was changed from insulin to sulfonylureas and SGLT-2 (31).
Prior to genetic testing, the proband (III3) was treated subcutaneously with 4 U short-acting insulin three times daily before meals and occasionally had hypoglycemia after conscious exercise at boarding school. It was inconvenient to administer subcutaneous injections after three meals, and she occasionally missed insulin injections, which also indicated psychological pressure. After genetic diagnosis, treatment was switched to 1 mg glimepiride per day; the patient was able to adhere to her medication regimen without hypoglycemia, and her personality was brighter than before. Furthermore, the HbA1c level of the proband decreased from 9.3 to 6.8%. In the living relatives, diagnosis by genetic testing guided the discontinuation of insulin therapy. The family members with the ABCC8 variant were responsive to treatment with sulfonylurea. In this pedigree, microvascular diseases such as diabetic retinopathy had not been found. In future follow-up, more attention will be given to the prevention and treatment of microvascular diseases.
Several strengths of the current study should be noted. Firstly, our study was the first to report the novel missense variant c.1432G>A (p.A478T) in ABCC8 MODY. This novel heterozygous variant (c.1432G>A) is predicted to be damaging by PROVEAN, MutationTaster, and SIFT. According to the standards and guidelines developed by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (32), one pathogenic supporting_strong and three pathogenic supporting criteria also support this variation being a “likely pathogenic” mutation, including (1) PP1_Strong: the variant cosegregated with diabetes in six non-proband family members and the reference allele segregated with normal glucose homeostasis in two family members, for a total of eight meioses (probability 1/256), meeting the criteria for applying PP1 at the strong level as proposed by Jarvik and Browning (33); (2) PP3: there were more than two lines of computational evidence supporting a deleterious effect on this variant, such as PROVEAN, SIFT, and MutationTaster; (3) PP4: the family history is highly specific for MODY; (4) PM2_Supporting: the variant was absent from controls in the database like Exome Sequencing Project, 1000 Genomes, and ExAC. Secondly, the results of the follow-up indicated that sulfonylureas were effective in treating this Chinese family who expressed clinical MODY. Thirdly, both Sanger sequencing and next-generation sequencing were utilized, which improved the reliability of the results. Limitations, on the other hand, should be mentioned. Importantly, one drawback was that the DNA test of the grandfather was lacking because he died before the study began. Furthermore, a longer follow-up period is required to evaluate the effectiveness of treatment. However, because the variant (c.1432G>A) in the ABCC8 gene has not been previously reported, functional studies are needed to uncover the underlying mechanism and confirm the role of the variant in diabetes of this family.
In conclusion, a novel missense variant of the ABCC8 gene was discovered in a Chinese ABCC8 MODY family for the first time. Genetic diagnosis is of great importance to the clinical typing and treatment of ABCC8 MODY patients. Sulfonylureas were effective for treating the family as previously seen in ABCC8 MODY.
The proband: “Insulin injections before three meals are a little bit troublesome. I occasionally felt hypoglycemia after conscious exercise and missed insulin injections in boarding school. It is convenient for me to take 1 mg glimepiride per day, and I was pleased for the glycemic control remains optimal after the treatment modification.”
Her mother: “She felt pressured for insulin injections and occasionally felt hypoglycemia in boarding school. After the treatment modification, she did not miss any medications and had no hypoglycemia, and her disposition was more cheerful than before. We feel very happy that she was admitted to a key high school with excellent results after the sulfonylurea treatment.”
The datasets generated and analyzed during the current study are not publicly available but are available from the corresponding authors on reasonable request.
The studies involving human participants were reviewed and approved by The Ethics Committee of the First People’s Hospital of Yulin. Written informed consent to participate in this study was provided by the legal guardian/next of kin of the participants.
CT and LM conceived the research and wrote the manuscript. PZ, XL, and CD performed data analyses. HuL and JW provided study materials. HaL and YQ designed this study. All the authors read and approved the final manuscript.
This study was funded by the Scientific Research and Technology Development Program of Yulin City, China (no. 201934047).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all the participants who participated in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.758723/full#supplementary-material
1. Stanik J, Dusatkova P, Cinek O, Valentinova L, Huckova M, Skopkova M, et al. De Novo Mutations of GCK, HNF1A and HNF4A May be More Frequent in MODY Than Previously Assumed. Diabetologia (2014) 57(3):480–4. doi: 10.1007/s00125-013-3119-2
2. Yorifuji T, Kurokawa K, Mamada M, Imai T, Kawai M, Nishi Y, et al. Neonatal Diabetes Mellitus and Neonatal Polycystic, Dysplastic Kidneys: Phenotypically Discordant Recurrence of a Mutation in the Hepatocyte Nuclear Factor-1beta Gene Due to Germline Mosaicism. J Clin Endocrinol Metab (2004) 89(6):2905–8. doi: 10.1210/jc.2003-031828
3. Thomas ER, Brackenridge A, Kidd J, Kariyawasam D, Carroll P, Colclough K, et al. Diagnosis of Monogenic Diabetes: 10-Year Experience in a Large Multi-Ethnic Diabetes Center. J Diabetes Investig (2016) 7(3):332–7. doi: 10.1111/jdi.12432
4. Brahm AJ, Wang G, Wang J, McIntyre AD, Cao H, Ban MR, et al. Genetic Confirmation Rate in Clinically Suspected Maturity-Onset Diabetes of the Young. Can J Diabetes (2016) 40(6):555–60. doi: 10.1016/j.jcjd.2016.05.010
5. Thanabalasingham G, Owen KR. Diagnosis and Management of Maturity Onset Diabetes of the Young (MODY). Bmj (2011) 343:d6044. doi: 10.1136/bmj.d6044
6. Thanabalasingham G, Pal A, Selwood MP, Dudley C, Fisher K, Bingley PJ, et al. Systematic Assessment of Etiology in Adults With a Clinical Diagnosis of Young-Onset Type 2 Diabetes Is a Successful Strategy for Identifying Maturity-Onset Diabetes of the Young. Diabetes Care (2012) 35(6):1206–12. doi: 10.2337/dc11-1243
7. Ivanoshchuk DE, Shakhtshneider EV, Rymar OD, Ovsyannikova AK, Mikhailova SV, Fishman VS, et al. The Mutation Spectrum of Maturity Onset Diabetes of the Young (MODY)-Associated Genes Among Western Siberia Patients. J Pers Med (2021) 11(1):57. doi: 10.3390/jpm11010057
8. Fajans SS, Bell GI. MODY: History, Genetics, Pathophysiology, and Clinical Decision Making. Diabetes Care (2011) 34(8):1878–84. doi: 10.2337/dc11-0035
9. Bowman P, Flanagan SE, Edghill EL, Damhuis A, Shepherd MH, Paisey R, et al. Heterozygous ABCC8 Mutations Are a Cause of MODY. Diabetologia (2012) 55(1):123–7. doi: 10.1007/s00125-011-2319-x
10. Kapoor R. Defining Genotype-Phenotype Correlations in Children With Congenital Hyperinsulinism. London: UCL (University College London) (2010).
11. De Franco E, Saint-Martin C, Brusgaard K, Knight Johnson AE, Aguilar-Bryan L, Bowman P, et al. Update of Variants Identified in the Pancreatic β-Cell KATP Channel Genes KCNJ11 and ABCC8 in Individuals With Congenital Hyperinsulinism and Diabetes. Hum Mutat (2020) 41(5):884–905. doi: 10.1002/humu.23995
12. Haghvirdizadeh P, Sadat Haerian M, Haghvirdizadeh P, Sadat Haerian B. ABCC8 Genetic Variants and Risk of Diabetes Mellitus. Gene (2014) 545(2):198–204. doi: 10.1016/j.gene.2014.04.040
13. American Diabetes Association; 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2021. Diabetes Care (2021) 44(Suppl 1):S15–33. doi: 10.2337/dc21-S002
14. Hattersley AT, Greeley SA, Polak M, Rubio-Cabezas O, Njølstad PR, Mlynarski W, et al. ISPAD Clinical Practice Consensus Guidelines 2018: The Diagnosis and Management of Monogenic Diabetes in Children and Adolescents. Pediatr Diabetes (2018) 19(Suppl 27):47–63. doi: 10.1111/pedi.12772
15. Chinese Diabetes Society. Guideline for the Prevention and Treatment of Type 2 Diabetes Mellitus in China (2020 Edition). Chin J Pract Internal Med (2020) 41(8):681–95. doi: 10.19538/j.nk2021080106
16. Koster JC, Marshall BA, Ensor N, Corbett JA, Nichols CG. Targeted Overactivity of Beta Cell K(ATP) Channels Induces Profound Neonatal Diabetes. Cell (2000) 100(6):645–54. doi: 10.1016/S0092-8674(00)80701-1
17. Gonsorcikova L, Vaxillaire M, Pruhova S, Dechaume A, Dusatkova P, Cinek O, et al. Familial Mild Hyperglycemia Associated With a Novel ABCC8-V84I Mutation Within Three Generations. Pediatr Diabetes (2011) 12(3 Pt 2):266–9. doi: 10.1111/j.1399-5448.2010.00719.x
18. Busiah K, Drunat S, Vaivre-Douret L, Bonnefond A, Simon A, Flechtner I, et al. Neuropsychological Dysfunction and Developmental Defects Associated With Genetic Changes in Infants With Neonatal Diabetes Mellitus: A Prospective Cohort Study. Lancet Diabetes Endocrinol (2013) 1(3):199–207. doi: 10.1016/S2213-8587(13)70059-7
19. Hartemann-Heurtier A, Simon A, Bellanné-Chantelot C, Reynaud R, Cavé H, Polak M, et al. Mutations in the ABCC8 Gene Can Cause Autoantibody-Negative Insulin-Dependent Diabetes. Diabetes Metab (2009) 35(3):233–5. doi: 10.1016/j.diabet.2009.01.003
20. Urbanová J, Rypáčková B, Procházková Z, Kučera P, Cerná M, Anděl M, et al. Positivity for Islet Cell Autoantibodies in Patients With Monogenic Diabetes Is Associated With Later Diabetes Onset and Higher HbA1c Level. Diabetes Med (2014) 31(4):466–71. doi: 10.1111/dme.12314
21. Karaoglan M, Nacarkahya G. Clinical and Laboratory Clues of Maturity-Onset Diabetes of the Young and Determination of Association With Molecular Diagnosis. J Diabetes (2021) 13(2):154–63. doi: 10.1111/1753-0407.13097
22. McDonald TJ, Colclough K, Brown R, Shields B, Shepherd M, Bingley P, et al. Islet Autoantibodies Can Discriminate Maturity-Onset Diabetes of the Young (MODY) From Type 1 Diabetes. Diabetes Med (2011) 28(9):1028–33. doi: 10.1111/j.1464-5491.2011.03287.x
23. Bowden SA, Hoffman RP. Triple Diabetes: Coexistence of Type 1 Diabetes Mellitus and a Novel Mutation in the Gene Responsible for MODY3 in an Overweight Adolescent. Pediatr Diabetes (2008) 9(2):162–4. doi: 10.1111/j.1399-5448.2007.00335.x
24. Maltoni G, Zucchini S, Scipione M, Mantovani V, Salardi S, Cicognani A. Onset of Type 1 Diabetes Mellitus in Two Patients With Maturity Onset Diabetes of the Young. Pediatr Diabetes (2012) 13(2):208–12. doi: 10.1111/j.1399-5448.2011.00788.x
25. Thunander M, Törn C, Petersson C, Ossiansson B, Fornander J, Landin-Olsson M. Levels of C-Peptide, Body Mass Index and Age, and Their Usefulness in Classification of Diabetes in Relation to Autoimmunity, in Adults With Newly Diagnosed Diabetes in Kronoberg, Sweden. Eur J Endocrinol (2012) 166(6):1021–9. doi: 10.1530/EJE-11-0797
26. Hao W, Gitelman S, DiMeglio LA, Boulware D, Greenbaum CJ. Fall in C-Peptide During First 4 Years From Diagnosis of Type 1 Diabetes: Variable Relation to Age, HbA1c, and Insulin Dose. Diabetes Care (2016) 39(10):1664–70. doi: 10.2337/dc16-0360
27. Baier LJ, Muller YL, Remedi MS, Traurig M, Piaggi P, Wiessner G, et al. ABCC8 R1420H Loss-Of-Function Variant in a Southwest American Indian Community: Association With Increased Birth Weight and Doubled Risk of Type 2 Diabetes. Diabetes (2015) 64(12):4322–32. doi: 10.2337/db15-0459
28. Macmullen CM, Zhou Q, Snider KE, Tewson PH, Becker SA, Aziz AR, et al. Diazoxide-Unresponsive Congenital Hyperinsulinism in Children With Dominant Mutations of the β-Cell Sulfonylurea Receptor SUR1. Diabetes (2011) 60(6):1797–804. doi: 10.2337/db10-1631
29. Mohan V, Radha V, Nguyen TT, Stawiski EW, Pahuja KB, Goldstein LD, et al. Comprehensive Genomic Analysis Identifies Pathogenic Variants in Maturity-Onset Diabetes of the Young (MODY) Patients in South India. BMC Med Genet (2018) 19(1):22. doi: 10.1186/s12881-018-0528-6
30. Nichols CG. KATP Channels as Molecular Sensors of Cellular Metabolism. Nature (2006) 440(7083):470–6. doi: 10.1038/nature04711
31. Ovsyannikova AK, Rymar OD, Shakhtshneider EV, Klimontov VV, Koroleva EA, Myakina NE, et al. ABCC8-Related Maturity-Onset Diabetes of the Young (MODY12): Clinical Features and Treatment Perspective. Diabetes Ther (2016) 7(3):591–600. doi: 10.1007/s13300-016-0192-9
32. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–23. doi: 10.1038/gim.2015.30
Keywords: maturity-onset diabetes of the young (MODY), ABCC8, variant, sulfonylureas, treatment
Citation: Tang C, Meng L, Zhang P, Liang X, Dang C, Liang H, Wu J, Lan H and Qin Y (2021) Case Report: A Novel ABCC8 Variant in a Chinese Pedigree of Maturity-Onset Diabetes of the Young. Front. Endocrinol. 12:758723. doi: 10.3389/fendo.2021.758723
Received: 14 August 2021; Accepted: 30 November 2021;
Published: 23 December 2021.
Edited by:
Ronald Cohen, University of Chicago, United StatesReviewed by:
Stephen Stone, Washington University School of Medicine in St. Louis, United StatesCopyright © 2021 Tang, Meng, Zhang, Liang, Dang, Liang, Wu, Lan and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiyun Lan, ZG9jdG9ybGh5QDEyNi5jb20=; Yingfen Qin, eWluZ2ZlbnFAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.