 Seonghwan Hwang
Seonghwan Hwang Hwayoung Yun
Hwayoung Yun Sungwon Moon1†
Sungwon Moon1† Bin Gao
Bin Gao- 1College of Pharmacy and Research Institute for Drug Development, Pusan National University, Busan, South Korea
- 2Laboratory of Liver Diseases, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD, United States
Nonalcoholic fatty liver disease (NAFLD) includes a spectrum of liver disorders, from fatty liver to nonalcoholic steatohepatitis (NASH), cirrhosis, and hepatocellular carcinoma. Compared with fatty liver, NASH is characterized by increased liver injury and inflammation, in which liver-infiltrating immune cells, with neutrophil infiltration as a hallmark of NASH, play a critical role in promoting the progression of fatty liver to NASH. Neutrophils are the first responders to injury and infection in various tissues, establishing the first line of defense through multiple mechanisms such as phagocytosis, cytokine secretion, reactive oxygen species production, and neutrophil extracellular trap formation; however, their roles in the pathogenesis of NASH remain obscure. The current review summarizes the roles of neutrophils that facilitate the progression of fatty liver to NASH and their involvement in inflammation resolution during NASH pathogenesis. The notion that neutrophils are potential therapeutic targets for the treatment of NASH is also discussed.
1 Introduction
Neutrophils are the most populous subset of leukocytes in the circulation and participate in various processes of immune reactions and inflammation (1). Neutrophils act as an effector of innate immunity to handle microorganism infection and execute a series of reactions to maintain homeostasis during tissue injury (2–4). Neutrophils are equipped with a variety of protective mechanisms against infection. These include phagocytosis, reactive oxygen species (ROS) production, oxidative burst, release of proteolytic enzymes via degranulation, and formation of web-like structures called neutrophil extracellular traps (NETs) that extrude genomic DNA and enzymes to fight against microorganisms (5). With regard to sterile inflammation, neutrophils are also activated and recruited to the site of injury as one of the first responders and participate in the inflammatory response to restore the physiological function of the tissue (3, 4). Because of their versatile functions, neutrophils have been highlighted as a critical mediator of diseases in multiple organs, including the liver.
Although hepatitis C virus (HCV) and hepatitis B virus (HBV) infections have traditionally been the leading causes of chronic liver disease that requires liver transplantation, the position of HCV and HBV infection has recently been challenged by several other etiologies, among which is nonalcoholic steatohepatitis (NASH) whose cases are rapidly increasing (6). NASH has recently become prevalent: fatty liver is observed in approximately 25% of the adult population, and 25% of the individuals with fatty liver are estimated to proceed to NASH (7). In the USA, NASH has become the second most common indication for liver transplantation (8, 9). NASH is marked by liver inflammation and damage caused by fat accumulation in the liver (10). The inflammatory properties of NASH are closely associated with the recruitment of innate immune cells, including neutrophils and monocytes; thus, investigators have recently focused on the role of neutrophils in exacerbating inflammation and tissue damage during NASH development (11). In this review, we summarize the recent advances in our understanding of the pathological role of neutrophils in the development of NASH (Figure 1), as well as their potential as therapeutic targets. The current review also discusses the protective function of neutrophils in NASH pathogenesis, which has recently gained attention (12).
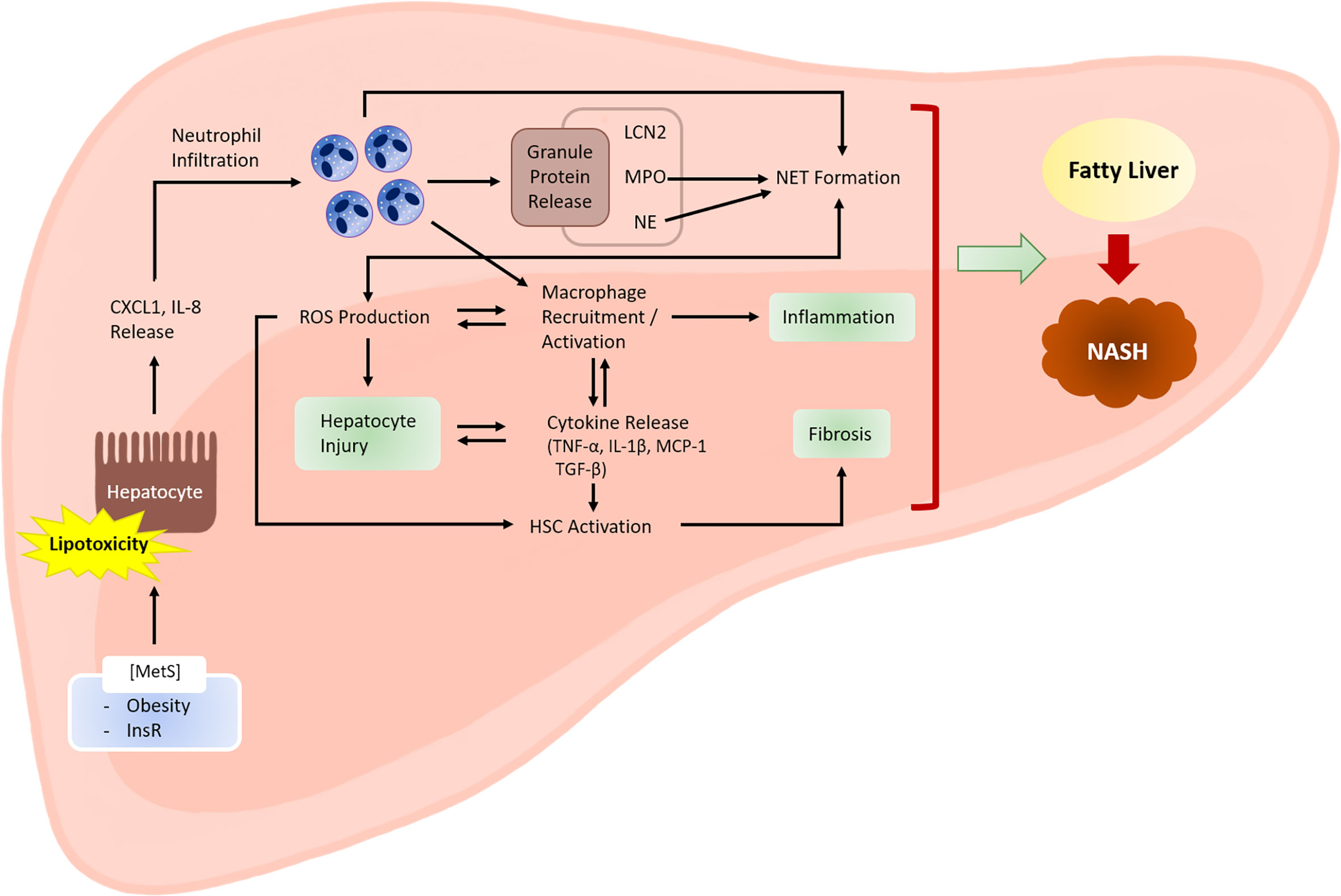
Figure 1 Role of neutrophils in the development of NASH. Metabolic syndrome is often associated with excessive lipid accumulation in the liver. Under these conditions, the probability of hepatocytes being exposed to lipotoxic lipid species, such as fatty acids, ceramides, cholesterol, and sphingolipids, is high. Lipotoxic hepatocytes release neutrophil-recruiting chemokines, including CXCL1 and IL-8. Infiltrating neutrophils exert various actions that facilitate NASH development. Activated neutrophils produce ROS via an oxidative burst that involves the activity of enzymes, such as NADPH oxidase 2. ROS may directly cause hepatocyte injury. ROS also activate and recruit macrophages, which further enhance hepatocyte injury and inflammation by releasing inflammatory cytokines. Cytokines released by macrophages (e.g., transforming growth factor-β) activate hepatic stellate cells (HSCs) and promote fibrosis. Neutrophil-derived ROS also contribute to HSC activation. Neutrophil granule proteins, such as LCN2, MPO, and NE, have increasingly been recognized to contribute to sterile inflammation, although the exact mechanisms through which they contribute to NASH are unclear. NETs mediate inflammation during NASH development through mechanisms that are not yet fully understood. Neutrophil-derived factors such as ROS and granule proteins (e.g., MPO and NE) contribute to NET formation. Hepatocyte injury, inflammation, and fibrosis are the three hallmarks of NASH. MetS, metabolic syndrome; InsR, insulin resistance.
2 Pathogenesis of NASH
Nonalcoholic fatty liver disease (NAFLD) encompasses a disease spectrum that ranges from fatty liver to NASH, cirrhosis, and hepatocellular carcinoma (13). Fatty liver is the benign and reversible stage of NAFLD and is caused by excessive fat accumulation in hepatocytes (defined as the presence of lipid droplets in >5% of hepatocytes) (14). NASH is the more severe form of fatty liver and is characterized by the presence of liver injury, inflammation, and fibrosis (15), which are not remarkably observed in the fatty liver of obese humans and mice fed with a high-fat diet (HFD) (16). The mechanism underlying the progression of fatty liver to NASH has been extensively investigated to identify a therapeutic target for the pharmacological intervention of NASH; however, no medications have been approved for use in the treatment of NASH (17). Because this review focuses on the role of neutrophils in NASH pathogenesis, this section highlights the inflammatory basis of the pathogenic events in NASH development and the link between lipotoxicity, hepatocyte injury, and inflammation.
2.1 Hepatic Fat Accumulation and Lipotoxicity
Fat accumulation in the liver is a priming factor in the development of NAFLD (18). This event is accompanied by the conversion of surplus energy sources such as carbohydrates and fatty acids into triglycerides, which are stored in the liver as lipid droplets (19). Hepatic fatty acids can come from a variety of sources: i) adipocyte-derived fatty acids generated by lipolysis of triglycerides, ii) de novo lipogenesis from sugars (e.g., glucose and fructose), and iii) intake of dietary sugar and lipid species (17). Fatty acid is relatively toxic and thus is preferred to be converted into triglycerides for storage, and mitochondrial fatty acid beta oxidation is another way to dispose of fatty acids (20). However, when there is excessive accumulation of fat in the liver, these protective mechanisms normally fail to completely eliminate fatty acids that can cause cellular stresses in a variety of ways that are discussed in more detail below. In addition, fatty acids serve as a source of several lipotoxic lipid classes (e.g., ceramides and sphingolipids), and they increase in amount during NAFLD progression (21). Free cholesterol is another type of toxic lipid that is elevated in the liver of patients with NASH (22). It is increasingly regarded as a contributing factor to NASH pathogenesis and is widely used as a supplement with NASH-inducing diets in experimental models.
2.2 Cellular Stress and Cell Death
Accumulation of lipotoxic lipid species in the liver causes cellular stress in hepatocytes, eventually leading to hepatocyte death (23). At the subcellular organelle level, lipotoxicity induces different types of stress in distinct locations such as the endoplasmic reticulum (ER), lysosomes, and mitochondria (24–26). Among the multiple types of lipotoxic lipids, palmitic acid has been particularly well documented as an inducer of hepatocyte stress. Palmitic acid is increased in the liver of NASH patients and experimental NASH models (27–29), and it triggers ER stress (30). Under the conditions with chronic ER stress, the unfolded protein response may facilitate inflammation and the death of hepatocytes (31). Exposure of hepatocytes to palmitic acid also disrupts mitochondrial and lysosomal functions (32). Severe injury in hepatocytes upon exposure to palmitic acid readily provokes hepatocyte death through the mechanisms involving molecular players that mediate organelle stress. For example, palmitic acid stimulates apoptosis of hepatocytes via the extrinsic and intrinsic pathways. Mitochondrial dysfunction and cytochrome c release are critical elements that activate the intrinsic apoptosis pathway (33, 34). Hepatocytes express death receptors such as tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptors that mediate apoptosis, especially via the extrinsic pathway, and the expression of these receptors is increased in NASH patients and mouse models (35). There are also other types of cell death, such as necrosis and pyroptosis, that can be observed in lipotoxic hepatocytes (36–38).
Lipotoxic hepatocytes undergoing cell death release chemokines and cytokines, which recruit and activate the innate immune cells, such as neutrophils and macrophages, for the initiation and amplification of inflammation (39–41). These inflammatory cells also release factors that signal through death receptors on hepatocytes (e.g., TRAIL-R1, TRAIL-R2, TNFR1, and Fas), which may further stimulate hepatocyte death and inflammation, thereby creating a positive feedback loop (36).
2.3 Gut Dysfunction
In addition to hepatocyte stress and death, other factors may also enhance inflammation in the pathogenesis of NASH. For example, gut barrier dysfunction is profound in NASH, which is believed to cause the translocation of gut bacteria to the circulation and further to the liver via portal vein (42). The compromise in the integrity of intestinal epithelial barrier may also promote the migration of the proinflammatory bacterial products such as endotoxin to the circulation and cause endotoxemia (43). Under endotoxic conditions, the influx of the proinflammatory bacterial products such as lipopolysaccharides to the liver is enhanced and trigger hepatic inflammation and NASH development.
Mouries et al., demonstrated that intestinal epithelial barrier and gut vascular barrier were disrupted in mice with diet-induced NASH (44). Mice with defective intestinal epithelial barrier develop more severe NASH when fed a diet high in fat, fructose, and cholesterol (45). The intestines of NAFLD patients are characterized by the disruption of intestinal tight junction and an increase in cytokine secretion and intestinal inflammation (46).
The diversity of microbial species become altered in NASH patients. Jiang et al. demonstrated that Escherichia, Anaerobacter, Lactobacillus, and Streptococcus were more abundant in the gut microbiota of NAFLD patients, while Alistipes and Prevotella were less abundant in NAFLD patients compared to healthy individuals (46). The diversity of microbial species is important for the integrity of the intestinal epithelial barrier and the regulation of metabolic functions; thus the change in their diversity may also contribute to the development of NAFLD and NASH (46, 47).
2.4 Inflammation
The progression of fatty liver to NASH is driven by innate immunity, where liver-resident macrophages (also known as Kupffer cells), infiltrating monocytes, and neutrophils play a critical role (48, 49). In the early phase of liver injury, Kupffer cells are activated and release chemokines and proinflammatory cytokines, including C-C motif chemokine ligand 2 and TNF-α, which result in the recruitment of monocytes and neutrophils into the injured liver (50, 51). Interleukin (IL)-1β and IL-6 released by Kupffer cells can further enhance NASH pathogenesis by increasing the lipogenesis and insulin sensitivity in hepatocytes (52–54). The recruited monocytes express high levels of lymphocyte antigen 6C (defined as LY6Chi monocytes) and differentiate into M1 type macrophages that exacerbate inflammation by releasing cytokines and ROS (55–59). Some of these cytokines, including transforming growth factor-β, may also directly act on hepatic stellate cells (HSCs), causing their transformation into myofibroblasts that promote fibrogenesis (60, 61). Different types of macrophages also play an important role in inflammation resolution, which are referred to as either alternatively-activated macrophages or M2 type macrophages. M2 macrophages are known to counteract the proinflammatory environment and participate in tissue repair (62); however, they also promote fibrotic changes in the liver that occur during the pathogenesis of NASH (63, 64).
The advent of single-cell analysis has expanded our understanding of the diverse subsets of infiltrating monocytes and macrophages in the liver, which can be characterized by the expression of distinct surface marker proteins. More recent studies reported that a specific subset of macrophages called NASH-associated macrophages, which express unique marker proteins, such as triggering receptor expressed on myeloid cells 2 (TREM2) and CD9, are enriched in the livers of NASH mouse models and patients (65, 66). Remmerie et al. demonstrated a reduction in Kupffer cells and an increase in a unique population of macrophages expressing SPP1 (also known as osteopontin) in the liver during NASH progression (67). The group of Philippe Gual reported that CD44 which is expressed in macrophages is upregulated during NASH progression and enhances NASH progression by controlling macrophage polarization and infiltration (68). However, whether and how these specific macrophage subsets affect the development of NASH are not fully understood.
Other types of immune cells that contribute to the pathogenesis of NASH include cytotoxic T cells, B cells, T-helper cells (e.g., Th1, Th2, and Th17), dendritic cells, natural killer cells, and innate lymphoid cells, and there is accumulating evidence supporting the pathogenic roles of these immune cells (69, 70). However, the overall pathogenic mechanisms of NASH encompass a broad range of biological events and molecular players, and thus are beyond the scope of the current review. Building upon the information stated in this section regarding the inflammatory aspects of NASH pathogenesis, primarily involving the action of macrophages, the following section will elaborate on the role of neutrophils in NASH development.
3 Involvement of Neutrophils in NASH Pathogenesis
Neutrophils are the first type of immune cells that respond to inflammatory changes in various tissues, including the liver, and execute a program that eventually induces a chronic inflammatory state by promoting macrophage recruitment and interacting with antigen-presenting cells (71–73). Thus, neutrophils have been studied as crucial players in the development of inflammatory liver diseases, including NASH. Treatment with a monoclonal antibody against Ly6G, which is present on neutrophils, results in a partial depletion of neutrophils in mice and thus has been used to investigate the role of neutrophils in the development of a variety of diseases (74, 75). Depletion of neutrophils in HFD-fed mice using Ly6G monoclonal antibody treatment significantly reduced the body weight gain, blood glucose levels, and hepatic triglyceride accumulation (76). It also repressed the expression of inflammatory and fibrotic proteins in the liver and decreased the activity of transaminases (e.g., alanine aminotransferase and aspartate aminotransferase), indicating that neutrophils are implicated in metabolic dysregulation, inflammation, and fibrosis during NASH development (76). The neutrophil-to-lymphocyte ratio (NLR) is closely related to the severity of NAFLD. In particular, NLR was correlated with the degree of hepatocyte ballooning, lobular inflammation, and fibrosis in a study of NASH patients (77). A more recent study also reported that NLR was associated with NAFLD activity score, hepatocyte degeneration, steatosis, inflammation, and fibrosis, indicating the possibility that NLR could function as a marker for histological grade and fibrosis stage of NASH (78). Although these studies emphasize the correlation between the abundance of neutrophils and the severity of NASH, there has been a recent increase in our understanding of the mechanism by which hepatic neutrophil infiltration accelerates NAFLD progression, which is further discussed below.
Neutrophil infiltration in the liver is a salient feature of alcoholic steatohepatitis (ASH) and NASH (79), both of which are regarded as the leading causes of end-stage liver disease and liver transplantation (6). In the context of ASH pathogenesis, the mechanisms by which neutrophil population is increased in the circulation, how neutrophils undergo hepatic infiltration, and how these neutrophils are implicated in the exacerbation of liver injury and inflammation have been well-documented (79, 80). In particular, multiple studies have investigated the pathogenic roles of neutrophils in murine models of ASH, such as chronic and binge ethanol feeding (the National Institute on Alcohol Abuse and Alcoholism model) (81–85). Neutrophils produce and release ROS, proteases, and inflammatory mediators to enhance hepatocyte injury and subsequent inflammation and fibrosis (80).
Although the histopathological features of NASH and ASH are similar to each other, albeit not identical, their immunological pathogenic mechanisms are different (86). For example, lipotoxicity-induced immune cell activation is regarded important to NASH pathogenesis, while the concept of lipotoxicity has not been highlighted in the field of ASH research although fatty liver stage is involved in the spectrum of alcoholic liver disease. Thus, it is not recommended to simply apply the knowledge obtained from ASH research to the understanding of NASH pathogenesis and the identification of NASH therapeutic targets. Compared with the well-documented role of neutrophil infiltration in the development of ASH, the contribution of neutrophils to NASH development has been relatively obscure, in part because of the lack of appropriate animal models that can recapitulate the NASH-associated elevation in hepatic neutrophil population as well as molecular features corresponding to the activation and recruitment of neutrophils.
3.1 HFD+CXCL1-Induced NASH Mouse Model With Increased Hepatic Neutrophil Infiltration
Diet-induced obesity is a widely used model to study fatty liver in mice; however, it is generally difficult to embody a NASH-like environment in the liver of mice by simply feeding them with an HFD chronically. Obesity, diabetes, and hyperlipidemia are major risk factors for NAFLD in humans, and long-term HFD feeding in mice gives rise to these risk factors. However, long-term HFD feeding seldom induces hepatic inflammation or hepatic infiltration of neutrophils (82).
Inflammation is a key event that promotes the progression of fatty liver to NASH, and the inflammation observed in NASH patients is characterized by a significant infiltration of neutrophils in the liver and other immune cells such as macrophages (61, 87). NASH-associated neutrophil infiltration in the liver is often accompanied by elevated expression of chemokines for neutrophil chemotaxis. Bertola et al. reported that NASH patients had a higher hepatic expression of chemokines that recruit neutrophils, such as C-X-C motif chemokine ligand 1 (CXCL1) and IL-8, than obese individuals with fatty liver (88).
Recent studies have revealed several differences in neutrophil biology between mice and humans, which may confer resistance to fatty liver to NASH progression in mice. First, mice have relatively fewer neutrophils in the circulation (~1 × 109/L) than humans (~4 × 109/L) (1, 85). In addition, inflamed mouse hepatocytes have less ability to attract neutrophils than inflamed human hepatocytes as the human IL-8 gene has no counterpart in mice, and mouse hepatocytes induce CXCL1 less effectively than human hepatocytes when exposed to inflammatory mediators (89). Accordingly, metabolic dysregulation and injury in the mouse liver less effectively recruit neutrophils, which could partially explain why HFD-fed mice were more likely to develop fatty liver than NASH.
Although there is no experimental model that reflects the full spectrum of NAFLD progression in mice (90), several dietary NASH models have recently been established by adding lipotoxic materials (e.g., cholesterol) and carbohydrates to a regular HFD (90). Some of these NASH models have been reported to show increased neutrophil infiltration in the liver (91–93).
Experimental NASH mouse models have been mostly induced by dietary compositions that provide overnutrition and contribute to liver injury; however, recent studies have reported new approaches that exploit the molecular mechanisms that mediate hepatic neutrophil infiltration and thus accelerate the progression of fatty liver to NASH. Adenovirus-driven overexpression of Cxcl1 in 3-month HFD-fed mice enhanced the expression of neutrophil-recruiting chemokines in hepatocytes and promoted the recruitment of neutrophils in the liver (16, 89). CXCL1-driven neutrophil infiltration promotes ROS production and activates stress kinases, including apoptosis signal-regulating kinase 1 and p38 mitogen-activated protein kinase (p38MAPK), which relay oxidative stress to cell death signaling (94). ROS also impair the proper folding of proteins, resulting in ER stress, which is often observed in NASH patients (31, 95, 96). CXCL1-induced, neutrophil-driven liver damage further led to inflammatory and fibrogenic processes and facilitated the progression of NASH; the expression profiles of the genes involved in inflammation and fibrosis were similar to those found in NASH patients (16). The overexpression of the human IL-8 gene in mice could also increase hepatic neutrophil infiltration and facilitate the progression of fatty liver to NASH; the concomitant overexpression of Cxcl1 and IL-8 further amplified the effect of the single overexpression of either Cxcl1 or IL-8 (89).
Neutrophilic oxidative bursts are crucial for ROS production by activated neutrophils (5). CXCL1-induced liver injury in HFD-fed mice was found to be dependent on neutrophil cytosolic factor 1 (also known as p47phox), which is one of the components of the NADPH oxidase 2 complex that mediates oxidative burst (97, 98), corroborating the importance of neutrophilic ROS production in neutrophil-driven NASH development (16). In addition, CXCL1-induced NASH was ameliorated by treatment with IL-22, which is a cytokine produced by immune cells such as Th17, Th22, and type 3 innate lymphoid cells (ILC3s) (99) but acts on epithelial cells, including hepatocytes, by upregulating various genes with cytoprotective and anti-oxidative properties (100, 101). In particular, the protective effect of IL-22 against neutrophil-driven NASH was reversed in mice lacking the genes encoding the two antioxidant enzymes, namely, metallothionein-1 and metallothionein-2, which implies the important function of ROS in neutrophil-driven NASH development. Interestingly, neutrophil elastase (NE) or NET was not crucial in CXCL1-induced NASH development because deletion of the gene encoding NE or peptidyl arginine deiminase-4 (PAD4) failed to reverse the NASH-inducing ability of CXCL1 overexpression in HFD-fed mice (16).
CXCL6 is another chemokine that recruits neutrophils by binding CXCR1 and CXCR2 (102). Due to the similar function of CXCL6 to that of CXCL1 and IL-8, it is reasonable to speculate that CXCL6 might be involved in the pathogenesis of NASH. However, the role of CXCL6 in NASH pathogenesis is still obscure. For neutrophils to be recruited from the blood to the affected sites, neutrophils interact with adhesion molecules such as E-selectin expressed on endothelial cells. E-selectin has been reported to be upregulated in the liver of NASH patients compared with the fatty liver (88). In CXCL1-induced NASH model, the expression of E-selectin was also highly elevated in the liver, indicating the involvement of E-selectin in the development of NASH in mice (16).
3.2 Role of Neutrophil-Specific MicroRNA-223 in NASH Development
miRs have emerged as important regulators of various genes involved in metabolism and inflammation in the liver (103–105). Several miRs, including miR-122, miR-192, miR-223, miR-21, and miR-29, affect the pathogenesis of NAFLD in vitro and in experimental animal models (106–113). These miRs usually have specific cell types, where they show the highest expression. For example, miR-122 and miR-192 are highly expressed in hepatocytes, miR-21 is commonly found in the circulation, and miR-29 is enriched in HSCs (114). MiR-223 is particularly interesting and is expressed at the highest levels in neutrophils, whose activation and maturation are repressed by miR-223 (115–117). Although miR-223 is also expressed in macrophages and is involved in macrophage polarization, its expression levels in macrophages are approximately 10% that in neutrophils (118); thus, it is regarded as a neutrophil-specific miR (119). miR-223 is upregulated in hepatocytes in HFD-fed mice and NASH patients (107, 109). miR-223 targets and inhibits several genes in mice that are involved in inflammation and tumorigenesis, such as C-X-C motif chemokine ligand 10 (CXCL10) and transcriptional coactivator with PDZ-binding motif (TAZ), and deletion of the miR-223 gene in HFD-fed mice promoted the development of NASH and NASH-associated hepatocellular carcinoma (109). Mechanistically, He et al. reported that miR-223, which is abundantly expressed in neutrophils, was transported to hepatocytes through the extracellular vesicles (EVs) derived from neutrophils (118). In particular, the transfer of miR-223-enriched EVs is mediated by the interaction between low-density lipoprotein receptor (LDLR) in hepatocytes and apolipoprotein E (APOE) in EVs (118). In addition, miR-223 protects against liver fibrosis by targeting multiple genes in hepatocytes and HSCs (120), which may contribute to the role of miR-223 in preventing NASH progression. The IL-6 signaling in myeloid cells, such as neutrophils, is important for the generation and release of miR-223-enriched EVs, which may inhibit the progression of fibrosis in NASH-associated fibrosis (121, 122). As miR-223 is expressed by both hepatocytes and neutrophils, specific deletion of miR-223 in each cell population by crossing miR-223-floxed mice with either Albumin Cre or Lysozyme M Cre mice will help better understand its role in the pathogenesis of NASH.
Elevation of miR-223 levels in the circulation and liver in NASH patients may be a compensatory mechanism to modulate the proinflammatory environment that prevails during NAFLD progression; thus, the deletion of miR-223 gene could accelerate NASH development in HFD-fed mice. Along with the model that is characterized by an increase in neutrophil infiltration in the liver, inhibition of the anti-inflammatory function of neutrophilic miR-223 could be another method to develop a genetic NASH model (85, 116, 117, 123).
3.3 Role of Neutrophil-Derived Specific Molecules in NASH Development
Neutrophil granules are sites where a large amount of protein is expressed, which mediates various neutrophil functions (124). Neutrophil granule proteins are released into the phagosome or extracellular space by degranulation and execute their functions (125). The different types of granules include azurophilic granules, specific granules, gelatinase granules, and secretory granules (Table 1) (126, 127). In particular, emerging evidence has demonstrated that the proteins expressed by azurophil granules [e.g., myeloperoxidase (MPO) and NE] and specific granules [e.g., lipocalin 2 (LCN2)] play a key role in various inflammatory processes during NASH pathogenesis. S100 calcium-binding protein A8 (S100A8, also known as MRP8) and S100 calcium-binding protein A9 (S100A9, also known as MRP14) are Ca2+-binding proteins that constitute 40% of neutrophil cytosolic protein weight. Their roles in NASH have also been discussed (128). In addition, neutrophils can produce a large number of inflammatory cytokines, chemokines, and inflammatory mediators, which likely play an important role in controlling NASH development and progression (129). Here, we primarily discussed the role of neutrophil-derived specific molecules in NASH development and progression. The changes in the levels of these molecules in experimental NASH as well as in clinical settings and the consequences of genetic or pharmacologic modulation of these factors in experimental NASH were also summarized (Tables 2 and 3).

Table 1 Types of neutrophil granules and their contents.
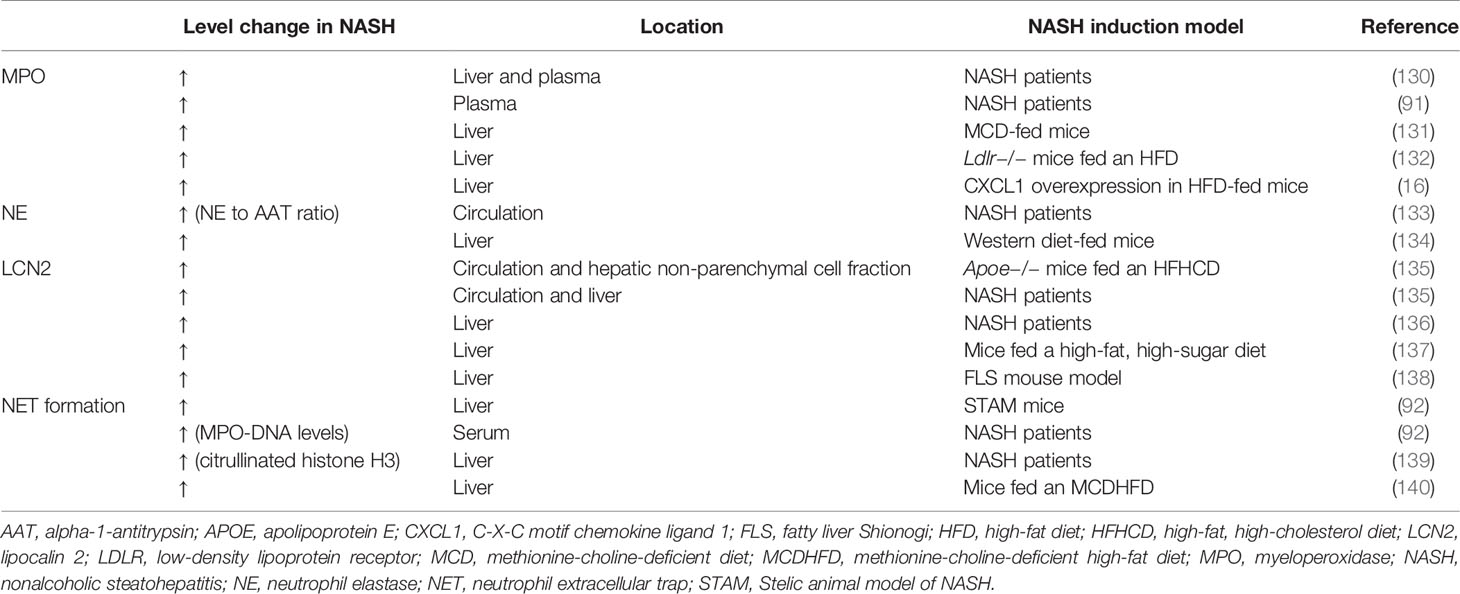
Table 2 Elevation of neutrophil-related factors in NASH patients and experimental NASH models.
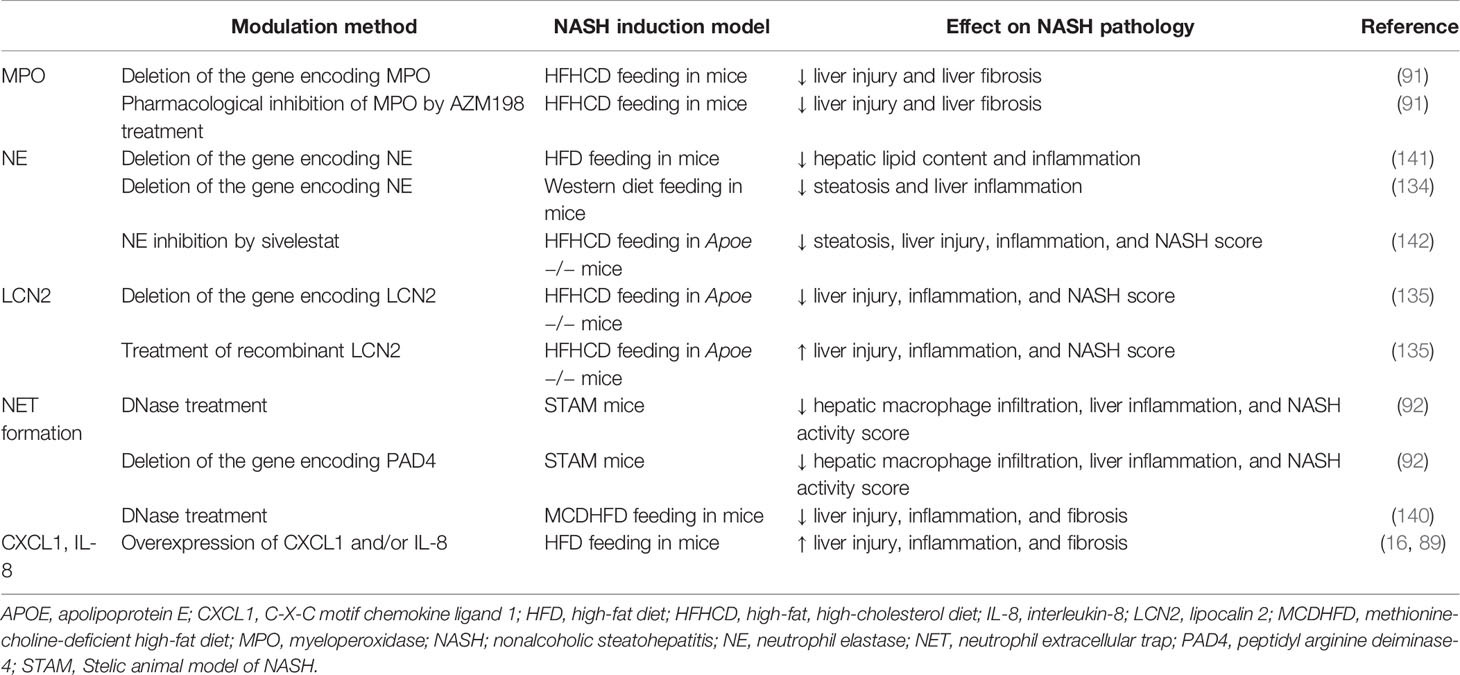
Table 3 Modulation of neutrophil-related factors that affect the degree of experimental NASH.
3.3.1 Myeloperoxidase
Myeloperoxidase (MPO) catalyzes the generation of ROS, which is crucial for the ability of neutrophils to kill microorganisms (5). ROS production by MPO is also involved in the occurrence of tissue damage and inflammation in chronic inflammatory diseases (143). Rensen et al. reported that the number of neutrophils was increased in the liver of NASH patients than in those with fatty liver, and the enhanced inflammation observed in NASH patients was associated with increased expression and activity of MPO (130). The plasma MPO levels and number of MPO-positive cells in the liver were increased in patients with NASH (130). In addition, NASH patients showed an increase in the accumulation of proteins modified by hypochlorite and nitrates which can be formed by the MPO-H2O2 system (130). An increased population of MPO-positive cells was also associated with the upregulation of CXC chemokines and hepatic neutrophil infiltration in the liver of NASH patients (130).
The contribution of MPO to fatty liver to NASH progression was further clarified by another study conducted by Rensen et al. (132). In this study, NASH was induced by feeding LDLR-deficient mice with an HFD. LDLR-null mice with MPO deficiency in the hematopoietic system showed reduced inflammation and fibrosis in the liver, indicating that neutrophil-derived MPO plays a crucial role in the development of NASH in mice. Furthermore, MPO-deficient mice showed a reduction in hepatic cholesterol, which is known to exacerbate NASH progression.
Feeding mice with a methionine and choline-deficient diet (MCD) is a classic method of inducing NASH in mice (90, 144). MCD-induced NASH is accompanied by an increase in MPO expression by neutrophils in the liver (131). Whole-body deletion of the MPO gene attenuates hepatocyte death and the severity of NASH and fibrosis (131). Mechanistically, MPO-derived oxidative stress causes hepatocyte injury through mitochondrial permeability transition and activates HSCs, which results from the crosstalk between neutrophilic MPO, hepatocytes, and HSCs (131).
MPO deficiency could also prevent the development of NASH in mice by attenuating liver injury and fibrosis induced by feeding a high-fat, high-cholesterol, high-carbohydrate diet (91), a widely used dietary model of NASH with a combination of different types of toxic substances (145). This study also reported that pharmacological inhibition of MPO through treatment with AZM198, an MPO inhibitor, could repress NASH progression and liver fibrosis induced by feeding a high-fat, high-cholesterol diet (HFHCD) (91).
3.3.2 Neutrophil Elastase
Neutrophil elastase (NE) is a serine protease that is released by neutrophils during inflammation through the degranulation process (146). NE is also important in NET formation because it contributes to the histone degradation and chromatin decondensation during NET formation (147, 148). NE is not only implicated in pathogen infection but also in sterile inflammation, which commonly develops in the liver of NASH patients (126).
NE that is released into the extracellular space is bound to alpha-1-antitrypsin (AAT), which inhibits the activity of NE; thus, the ratio of NE to AAT in the serum is used to predict the activity of NE (149–151). In a study that recruited NAFLD patients and healthy controls, the ratio of NE to AAT was higher in NAFLD patients and was closely associated with liver inflammation in patients with NASH, indicating that it can be used as a marker to evaluate the severity of NASH in humans (133). Another clinical study reported that the plasma concentration of NE was associated with the severity of NAFLD. The advanced stages of NAFLD with NASH and fibrosis features are characterized by higher levels of hepatic NE (152). This study also showed that the hepatic levels of proteinase 3, another type of neutrophil serine protease, were correlated with the severity of NAFLD (152, 153).
Talukdar et al. demonstrated that HFD feeding increased the infiltration of neutrophils in the liver of mice, and liver-infiltrating neutrophils inhibited the insulin signaling by degradation of insulin receptor substrate 1 (141). They also showed that NE possibly mediated these effects of neutrophils on liver metabolism because deletion of the Elane gene, which encodes NE, increased the hepatic insulin sensitivity and reduced the expression of hepatic inflammatory genes in HFD-fed mice (141). Elane−/− mice were also resistant to western diet-induced NASH, with decreased hepatic expression of lipogenic and inflammatory genes and reduced hepatic levels of ceramides that promote inflammation through a variety of signaling pathways (134). The pharmacological inhibition of NE by treatment with sivelestat reduced the serum transaminase activity, expression of inflammatory mediators, and NASH score in Apoe-deficient mice fed with an HFHCD that showed NASH phenotypes (142). This study also showed that the MCD-induced NASH was prevented by the Ly6G monoclonal antibody-induced depletion of neutrophils (142).
Although previous studies have suggested that NE contributes to the pathogenesis of NAFLD, our recent data revealed that deletion of the Elane gene did not affect the serum ALT levels in HFD+CXCL1-fed mice (16). Conflicts among these data may be because of the different models and wild-type control mice used (littermate controls were used in our studies).
3.3.3 Lipocalin 2
Lipocalin 2 (LCN2) is a proinflammatory cytokine found in specific granules of neutrophils. LCN2 has biological functions in various processes, ranging from innate immunity to cell death, cell proliferation, and metabolism (154–158). LCN2 has been implicated in the development of inflammatory and metabolic diseases (159, 160) and is generally induced by injury and inflammatory conditions in the liver (161). The LCN2 levels are elevated in the experimental models of NASH and promote inflammation by attracting neutrophils and inducing the expression of proinflammatory cytokines (e.g., TNF-α, IL-1β, MCP-1) (135, 162). In addition, LCN2 was found to be elevated in the livers of NASH patients. The mRNA and protein levels of LCN2 in the liver were higher in NASH patients than in those with fatty liver (136). This increase is thought to be because of the proinflammatory environment in NASH, as evidenced by the upregulation of LCN2 in the HepG2 hepatoma cell line upon exposure to proinflammatory mediators, such as TNF-α and IL-6 (136). The expression of LCN2 in the liver was highly increased in fatty liver Shionogi (FLS) mouse model (138) which features spontaneous development of fatty liver that progresses to NASH and eventually hepatocellular carcinoma (163, 164). In the FLS model, CXCL1 was found to be elevated, which might further exacerbate the progression of NASH in concert with LCN2 (138). In line with these studies, Xu et al. reported that LCN2 mRNA levels were increased by more than 28-folds in a high-fat, high-sugar-induced NASH mouse model; their protein-protein interaction analysis using Search Tool for the Retrieval of Interacting Genes (STRING, http://string-db.org) database supported the notion that LCN2 might physically interact with various inflammatory mediators (e.g., IL-1R2, IL-3, IL-6, IL-18, and IL-17A) or might be involved in the inflammatory cascades mediated by inflammatory mediators (137). HFHCD-induced NASH was accompanied by an upregulation of LCN2 expression in the liver, in which the major LCN2-enriched cell type was neutrophils (135). Genetic ablation of LCN2 in Apoe-null mice significantly suppressed the severity of hepatic injury and inflammation, whereas chronic administration of recombinant LCN2 enhanced the severity of HFHCD-induced NASH (135). Mechanistically, LCN2 promoted the activation of C-X-C motif chemokine receptor 2-mediated extracellular signal-regulated kinase (ERK), which enhances the crosstalk between neutrophils and macrophages in the liver and induces inflammatory responses (135).
3.3.4 Potential Roles of Neutrophil-Derived S100A8/A9 in NASH Development
S100A8 and S100A9 are Ca2+-binding proteins that belong to the S100 family and account for a substantial proportion of the neutrophil cytosolic protein population (165–167). S100A8 and S100A9 preferentially form heterodimers; the S100A8/A9 heterodimer is released by activated neutrophils and is implicated in the pathogenesis of various diseases as an innate immune mediator (168–170). The expression of S100A8 and S100A9 is elevated in the visceral adipose tissues of obese patients with diabetes (171). However, the contribution of S100A8/A9 to NASH development is not well understood although the serum levels of S100A8 and S100A9 are elevated in NASH patients (172). As it has been increasingly recognized that the crosstalk between the liver and adipose tissue plays a crucial role in NASH development, it is reasonable to further examine the role of adipose tissue proteins S100A8/A9 in NASH development. Indeed, S100A8 and S100A9 are upregulated in the adipose tissue samples of NASH patients and CXCL1-induced experimental NASH models, and the administration of paquinimod, a S100A9 inhibitor, attenuated the CXCL1-induced NASH in mice (128). This finding further supports the possibility that neutrophil-derived specific molecules might be implicated in the crosstalk between organs that exacerbates NASH.
3.4 Role of NET in NASH Development
NET is a web-like structure of DNA fibers composed of histones and granule proteins (173). NETs are formed by expulsion of the nuclear materials of neutrophils into the extracellular space (147). NET was originally reported to be a mechanism through which neutrophils participate in host defense by capturing and killing microorganisms and preventing their dissemination (174); however, increasing evidence has supported the role of NET as a critical mediator of sterile inflammation, which is critically implicated in the development of various diseases including cancer (175–177). NET also potentiates neutrophil function through a positive feedback loop and thus has attracted attention as a therapeutic target for inflammatory diseases (178).
NET formation is stimulated in experimental NASH models. In the Stelic animal model of NASH (STAM), neonatal streptozotocin treatment and HFD induces NASH and hepatocellular carcinoma in mice by creating a diabetic and obesogenic environment (179). STAM mice showed elevated neutrophil infiltration and NET formation in the liver (92). Inhibition of NET formation through deoxyribonuclease (DNase) treatment or the deletion of the gene encoding PAD4 attenuated the monocyte infiltration and inflammation in the liver of STAM mice, although steatosis was not significantly improved (92). In addition, DNase treatment or PAD4 deletion inhibited the NASH-associated hepatocellular carcinoma development in STAM mice. Moreover, the serum levels of MPO-DNA, which is a marker of NET, were elevated in NASH patients than in healthy individuals (92). A more recent study revealed that not only the serum marker but also the hepatic marker of NET formation (e.g., citrullinated histone H3) was increased in the liver of NASH patients and that NET was correlated with NAFLD severity (139).
NET formation was also promoted in a methionine-choline-deficient high-fat diet (MCDHFD)-induced NASH model. In mice fed with an MCDHFD, NETs were detected in the early stages of NASH, and the depletion of NETs by intraperitoneal injection of DNase I alleviated the MCDHFD-induced liver injury, inflammation, and fibrosis (140). During the development of MCDHFD-induced NASH, hepatic sphingosine 1-phosphate (S1P) levels were correlated with NET formation. S1P activated p38MAPK- and ERK-mediated ROS production in neutrophils via the S1P receptor 2 signaling pathway, which switched apoptosis to NETosis and promoted NASH development (140).
4 Potential Beneficial Functions of Neutrophils in NASH
Damaged or infected tissues undergo a series of inflammatory processes, where neutrophil infiltration is one of the first events to remove pathogenic microorganisms or overcome injuries. These inflammatory processes are usually followed by tissue repair; however, excessive and prolonged inflammation that is not properly resolved by various repair mechanisms may lead to chronic destructive inflammation and fibrosis, which are critically implicated in the development of various inflammatory diseases.
Resolution of inflammation is a coordinated and active process that is designed to maintain tissue homeostasis (180, 181). Timely resolution of inflammation is important because tissue integrity and function are impaired during inflammation; thus, prolonged inflammation may lead to collateral tissue damage (182, 183). Neutrophil infiltration is correlated with the severity of tissue damage and inflammation (184). Thus, neutrophil apoptosis and the clearance of apoptotic neutrophils are tightly regulated in order to resolve the inflammation (185, 186). Apoptotic neutrophils are normally cleared from the affected site by the macrophages through the process of efferocytosis (187).
Neutrophils are generally thought to initiate and aggravate inflammation; the current review has so far discussed the deleterious function of neutrophils, that is, exacerbation of tissue injury and inflammation during the pathogenesis of liver diseases. However, recent publications have increasingly described the pro-resolving and tissue-restorative functions of neutrophils (12, 188). A study by Wang et al. reported the cooperation of neutrophils with macrophages which promotes the conversion of proinflammatory monocytes to pro-resolving macrophages to orchestrate the resolution of tissue inflammation and repair (189). Proteases derived from neutrophils, such as NE, inhibit the production of IL-1β and TNF-α and cause the degradation of these cytokines (190). Neutrophils also inhibit the production of cytokines induced by lipopolysaccharides or the fragment of the cell wall of gram-positive bacteria (191). Although NETs promote inflammation, they capture and degrade proinflammatory mediators, especially in sites where the density of neutrophils is high, which is thought to be associated with the action of neutrophilic proteases that are components of NETs (192).
Formyl peptide receptor 2 binds multiple pro-resolving ligands, including annexin A1, lipoxin A4, 15-epi-lipoxin A5, and 17-epi-resolvin D1, which contribute to multiple processes that mediate the maintenance of tissue homeostasis under inflammatory conditions (193). Some of these processes include inhibition of neutrophil infiltration and attachment and stimulation of neutrophil apoptosis, thereby attenuating tissue injury and accelerating inflammation resolution (193). Interestingly, neutrophils produce and release some of these pro-resolving ligands (e.g., annexin A1) via the membrane-derived microvesicles, through which neutrophils can actively participate in the resolution of inflammation (194–197).
Researchers in the field of hepatology have also recently demonstrated the pro-resolving role of neutrophils in several models of liver diseases, including NASH. Calvente et al. reported that depletion of neutrophils in mice recovering from MCD-induced NASH or CCl4-induced fibrosis prolonged the period of tissue damage, inflammation, and fibrosis (198). Mechanistically, neutrophils participate in the resolution of inflammation by transferring miR-223 from neutrophils to macrophages via the neutrophil-derived EVs or as a complex with lipoproteins or argonaut 2 (198). In macrophages, miR-223 inhibited the activity of NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasomes and induced the polarization of restorative macrophages, which release cytokines such as IL-10, leading to the resolution of inflammation and fibrosis. The anti-inflammatory role of miR-223 was also observed in a study by He et al.; they showed that miR-223-deficient mice were more vulnerable to diet-induced NASH and hepatocellular carcinoma (109). It was later demonstrated that neutrophil-derived EVs deliver miR-223 to hepatocytes where they exert their anti-inflammatory and anti-tumorigenic functions (109, 118).
Advancements in single-cell analysis have made it possible to study the function of different subsets of immune cells in the development of various diseases (199). In particular, recent publications have elucidated several distinct subsets of macrophages that are enriched in the livers of humans and mice with NASH (65, 67). However, neutrophils have been traditionally thought to be homogeneous, and little is known about the different subsets of neutrophils that may function distinctively during the development of NASH. The notion that neutrophils are both proinflammatory and pro-resolving warrants further investigation of the heterogeneity of neutrophils in the context of NASH development.
5 Neutrophils as Potential Therapeutic Targets for the Treatment of NASH
The involvement of neutrophils in multiple processes in the pathogenesis of liver diseases has made them attractive targets for therapeutic intervention. As discussed in this review, neutrophils generally exacerbate inflammation and liver injury, which have established strategies to deplete neutrophils or inhibit the activity of neutrophils. Blockade of granulocyte colony-stimulating factor 3 receptor with monoclonal antibodies inhibits the production and activation of neutrophils and has been suggested to ameliorate several diseases, including arthritis (200, 201). Neutrophils are short lived and susceptible to apoptosis, and deletion of the Foxo3a gene that is needed for neutrophils to survive prevented inflammatory diseases in mice due to excessive neutrophil apoptosis (202). However, whether pharmacological interventions to reduce the neutrophil population in the circulation or at the site of inflammation are useful for treating NASH should be investigated further.
Because proteins expressed by neutrophil granules significantly contribute to the pathogenic functions of neutrophils, researchers have attempted to develop pharmacological modulators that can control the activities of granule proteins, such as NE, MPO, and LCN2. Elafin and AAT are endogenous factors that have the ability to inhibit NE, and emerging evidence has demonstrated that they may attenuate several diseases in experimental models (203, 204). Elafin is an antimicrobial and anti-inflammatory protein with a molecular weight of approximately 6 kDa (205). Lentiviral overexpression of elafin inhibits HFD-induced steatosis in mice (206). MPO has also been studied as a target of anti-neutrophil therapies because it mediates multiple steps in neutrophil-induced inflammation and tissue damage (207). It has been attempted to inhibit the activity of MPO using chemicals that block the active site of MPO, remove MPO from the chlorination reactions, or scavenge HOCl; however, whether these strategies are valid for the treatment of NASH in experimental animal models remain unknown (208–214). The therapeutic potential of LCN2 inhibition has been examined for the treatment of various diseases, including cancer (215, 216), which has led to the investigation of biological therapeutics such as monoclonal antibodies, RNA interference technology, and nanoparticle-based LCN2 modulators (158, 161, 217–219).
NET formation was originally highlighted as an immune response of neutrophils against microorganism infection; however, accumulating evidence has suggested the involvement of NETs in sterile inflammation, which has facilitated the investigation of the potential application of anti-NETosis therapy in the treatment of inflammatory diseases (147). PAD4 is regarded as one of the most important enzymes involved in the formation of NETs (220), and deletion of the gene that encodes PAD4 or pharmacological inhibition of PAD4 has been widely implemented to study the pathogenic role and therapeutic potential of NETs (221). The study by Tsung et al. on the role of NET in the development of NASH-associated hepatocellular carcinoma corroborates the applicability of anti-NETosis therapy in the treatment of full-spectrum NAFLD (92).
Neutrophils have been actively studied as targets for pharmacological intervention in multiple diseases including autoimmune diseases, atherosclerosis, asthma, infectious diseases, psoriasis, and sepsis (222). Either enhancement or inhibition of the function of neutrophils has been studied as the strategic approach against these diseases. For example, blocking neutrophil recruitment, blocking neutrophil-derived mediators, and targeting NETs have been attempted to inhibit the excessive tissue damage caused by neutrophils in pulmonary diseases, atherosclerosis, and psoriasis (125). However, little is known about whether these strategies can produce positive outcomes for the treatment of NASH. Considering the involvement of neutrophils in the multiple processes of NASH pathogenesis that have been discussed in this review, it is justifiable to further explore the feasibility of anti-neutrophil therapy as a therapeutic strategy for NASH.
6 Conclusions
Knowledge on the role of neutrophils has rapidly expanded in recent years, which has not only enhanced our understanding of the pathogenic mechanism of NASH, but also laid the foundation for its application in establishing an experimental NASH model that may be utilized for drug screening. Although the clinical data to support the applicability of the neutrophil-modulating approach have yet to be presented, the potential benefit of this strategy has been increasingly supported by studies conducted in NASH animal models. Moreover, the population of neutrophils can be more heterogeneous than we currently recognize. Advances in analytical techniques, such as the advent of single-cell RNA sequencing, have further raised questions regarding the different subsets of neutrophils that may distinctively participate in NASH pathogenesis. Elucidation of these remaining questions will help us better elucidate the role of neutrophils in NASH development and open new avenues for the therapeutic intervention of NASH.
Author Contributions
SH, HY, and SM wrote the manuscript. YC edited the manuscript and figure. BG supervised the writing and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Pusan National University Research Grant, 2021 (SH). This work was also supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2021R1F1A1056033) (SH) and by the intramural program of NIAAA, NIH (BG).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. von Vietinghoff S, Ley K. Homeostatic Regulation of Blood Neutrophil Counts. J Immunol (2008) 181(8):5183–8. doi: 10.4049/jimmunol.181.8.5183
2. Segal AW. How Neutrophils Kill Microbes. Annu Rev Immunol (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
3. Chen GY, Nuñez G. Sterile Inflammation: Sensing and Reacting to Damage. Nat Rev Immunol (2010) 10(12):826–37. doi: 10.1038/nri2873
4. Wang J. Neutrophils in Tissue Injury and Repair. Cell Tissue Res (2018) 371(3):531–9. doi: 10.1007/s00441-017-2785-7
5. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect Microbiol (2017) 7:373. doi: 10.3389/fcimb.2017.00373
6. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic Steatohepatitis is the Second Leading Etiology of Liver Disease Among Adults Awaiting Liver Transplantation in the United States. Gastroenterology (2015) 148(3):547–55. doi: 10.1053/j.gastro.2014.11.039
7. Peng C, Stewart AG, Woodman OL, Ritchie RH, Qin CX. Non-Alcoholic Steatohepatitis: A Review of Its Mechanism, Models and Medical Treatments. Front Pharmacol (2020) 11:603926. doi: 10.3389/fphar.2020.603926
8. Anstee QM, Targher G, Day CP. Progression of NAFLD to Diabetes Mellitus, Cardiovascular Disease or Cirrhosis. Nat Rev Gastroenterol Hepatol (2013) 10(6):330–44. doi: 10.1038/nrgastro.2013.41
9. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat Rev Gastroenterol Hepatol (2018) 15(1):11–20. doi: 10.1038/nrgastro.2017.109
10. Cotter TG, Rinella M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology (2020) 158(7):1851–64. doi: 10.1053/j.gastro.2020.01.052
11. Chen Y, Tian Z. Roles of Hepatic Innate and Innate-Like Lymphocytes in Nonalcoholic Steatohepatitis. Front Immunol (2020) 11:1500. doi: 10.3389/fimmu.2020.01500
12. Peiseler M, Kubes P. More Friend Than Foe: The Emerging Role of Neutrophils in Tissue Repair. J Clin Invest (2019) 129(7):2629–39. doi: 10.1172/jci124616
13. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic Fatty Liver Disease and Hepatocellular Carcinoma: A Weighty Connection. Hepatology (2010) 51(5):1820–32. doi: 10.1002/hep.23594
14. Brunt EM, Tiniakos DG. Histopathology of Nonalcoholic Fatty Liver Disease. World J Gastroenterol (2010) 16(42):5286–96. doi: 10.3748/wjg.v16.i42.5286
15. Luedde T, Kaplowitz N, Schwabe RF. Cell Death and Cell Death Responses in Liver Disease: Mechanisms and Clinical Relevance. Gastroenterology (2014) 147(4):765–83.e4. doi: 10.1053/j.gastro.2014.07.018
16. Hwang S, He Y, Xiang X, Seo W, Kim SJ, Ma J, et al. Interleukin-22 Ameliorates Neutrophil-Driven Nonalcoholic Steatohepatitis Through Multiple Targets. Hepatology (2020) 72(2):412–29. doi: 10.1002/hep.31031
17. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat Med (2018) 24(7):908–22. doi: 10.1038/s41591-018-0104-9
18. Buzzetti E, Pinzani M, Tsochatzis EA. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism (2016) 65(8):1038–48. doi: 10.1016/j.metabol.2015.12.012
19. Reddy JK, Rao MS. Lipid Metabolism and Liver Inflammation. II. Fatty Liver Disease and Fatty Acid Oxidation. Am J Physiol Gastrointest Liver Physiol (2006) 290(5):G852–8. doi: 10.1152/ajpgi.00521.2005
20. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular Mechanisms of Hepatic Lipid Accumulation in Non-Alcoholic Fatty Liver Disease. Cell Mol Life Sci (2018) 75(18):3313–27. doi: 10.1007/s00018-018-2860-6
21. Pagadala M, Kasumov T, McCullough AJ, Zein NN, Kirwan JP. Role of Ceramides in Nonalcoholic Fatty Liver Disease. Trends Endocrinol Metab (2012) 23(8):365–71. doi: 10.1016/j.tem.2012.04.005
22. Ioannou GN. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol Metab (2016) 27(2):84–95. doi: 10.1016/j.tem.2015.11.008
23. Marra F, Svegliati-Baroni G. Lipotoxicity and the Gut-Liver Axis in NASH Pathogenesis. J Hepatol (2018) 68(2):280–95. doi: 10.1016/j.jhep.2017.11.014
24. Su Q, Baker C, Christian P, Naples M, Tong X, Zhang K, et al. Hepatic Mitochondrial and ER Stress Induced by Defective Pparα Signaling in the Pathogenesis of Hepatic Steatosis. Am J Physiol Endocrinol Metab (2014) 306(11):E1264–73. doi: 10.1152/ajpendo.00438.2013
25. Han J, Kaufman RJ. The Role of ER Stress in Lipid Metabolism and Lipotoxicity. J Lipid Res (2016) 57(8):1329–38. doi: 10.1194/jlr.R067595
26. Zámbó V, Simon-Szabó L, Szelényi P, Kereszturi E, Bánhegyi G, Csala M. Lipotoxicity in the Liver. World J Hepatol (2013) 5(10):550–7. doi: 10.4254/wjh.v5.i10.550
27. Allard JP, Aghdassi E, Mohammed S, Raman M, Avand G, Arendt BM, et al. Nutritional Assessment and Hepatic Fatty Acid Composition in Non-Alcoholic Fatty Liver Disease (NAFLD): A Cross-Sectional Study. J Hepatol (2008) 48(2):300–7. doi: 10.1016/j.jhep.2007.09.009
28. Malhi H, Gores GJ. Molecular Mechanisms of Lipotoxicity in Nonalcoholic Fatty Liver Disease. Semin Liver Dis (2008) 28(4):360–9. doi: 10.1055/s-0028-1091980
29. Garcia-Jaramillo M, Spooner MH, Löhr CV, Wong CP, Zhang W, Jump DB. Lipidomic and Transcriptomic Analysis of Western Diet-Induced Nonalcoholic Steatohepatitis (NASH) in Female Ldlr -/- Mice. PloS One (2019) 14(4):e0214387. doi: 10.1371/journal.pone.0214387
30. Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated Fatty Acids Induce Endoplasmic Reticulum Stress and Apoptosis Independently of Ceramide in Liver Cells. Am J Physiol Endocrinol Metab (2006) 291(2):E275–81. doi: 10.1152/ajpendo.00644.2005
31. Lebeaupin C, Vallée D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic Reticulum Stress Signalling and the Pathogenesis of Non-Alcoholic Fatty Liver Disease. J Hepatol (2018) 69(4):927–47. doi: 10.1016/j.jhep.2018.06.008
32. Li Z, Berk M, McIntyre TM, Gores GJ, Feldstein AE. The Lysosomal-Mitochondrial Axis in Free Fatty Acid-Induced Hepatic Lipotoxicity. Hepatology (2008) 47(5):1495–503. doi: 10.1002/hep.22183
33. Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free Fatty Acids Induce JNK-Dependent Hepatocyte Lipoapoptosis. J Biol Chem (2006) 281(17):12093–101. doi: 10.1074/jbc.M510660200
34. Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The Release of Cytochrome C From Mitochondria: A Primary Site for Bcl-2 Regulation of Apoptosis. Science (1997) 275(5303):1132–6. doi: 10.1126/science.275.5303.1132
35. Hirsova P, Ibrahim SH, Bronk SF, Yagita H, Gores GJ. Vismodegib Suppresses TRAIL-Mediated Liver Injury in a Mouse Model of Nonalcoholic Steatohepatitis. PloS One (2013) 8(7):e70599. doi: 10.1371/journal.pone.0070599
36. Hirsova P, Gores GJ. Death Receptor-Mediated Cell Death and Proinflammatory Signaling in Nonalcoholic Steatohepatitis. Cell Mol Gastroenterol Hepatol (2015) 1(1):17–27. doi: 10.1016/j.jcmgh.2014.11.005
37. Farrell GC, Haczeyni F, Chitturi S. Pathogenesis of NASH: How Metabolic Complications of Overnutrition Favour Lipotoxicity and Pro-Inflammatory Fatty Liver Disease. Adv Exp Med Biol (2018) 1061:19–44. doi: 10.1007/978-981-10-8684-7_3
38. Geng Y, Faber KN, de Meijer VE, Blokzijl H, Moshage H. How Does Hepatic Lipid Accumulation Lead to Lipotoxicity in Non-Alcoholic Fatty Liver Disease? Hepatol Int (2021) 15(1):21–35. doi: 10.1007/s12072-020-10121-2
39. Rada P, González-Rodríguez Á, García-Monzón C, Valverde ÁM. Understanding Lipotoxicity in NAFLD Pathogenesis: Is CD36 a Key Driver? Cell Death Dis (2020) 11(9):802. doi: 10.1038/s41419-020-03003-w
40. Koyama Y, Brenner DA. Liver Inflammation and Fibrosis. J Clin Invest (2017) 127(1):55–64. doi: 10.1172/jci88881
41. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology (2016) 150(4):956–67. doi: 10.1053/j.gastro.2015.12.037
42. Saltzman ET, Palacios T, Thomsen M, Vitetta L. Intestinal Microbiome Shifts, Dysbiosis, Inflammation, and Non-Alcoholic Fatty Liver Disease. Front Microbiol (2018) 9:61. doi: 10.3389/fmicb.2018.00061
43. Kitabatake H, Tanaka N, Fujimori N, Komatsu M, Okubo A, Kakegawa K, et al. Association Between Endotoxemia and Histological Features of Nonalcoholic Fatty Liver Disease. World J Gastroenterol (2017) 23(4):712–22. doi: 10.3748/wjg.v23.i4.712
44. Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, Wiest R, et al. Microbiota-Driven Gut Vascular Barrier Disruption is a Prerequisite for Non-Alcoholic Steatohepatitis Development. J Hepatol (2019) 71(6):1216–28. doi: 10.1016/j.jhep.2019.08.005
45. Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y, et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology (2016) 151(4):733–46.e12. doi: 10.1053/j.gastro.2016.06.022
46. Jiang W, Wu N, Wang X, Chi Y, Zhang Y, Qiu X, et al. Dysbiosis Gut Microbiota Associated With Inflammation and Impaired Mucosal Immune Function in Intestine of Humans With Non-Alcoholic Fatty Liver Disease. Sci Rep (2015) 5:8096. doi: 10.1038/srep08096
47. Grabherr F, Grander C, Effenberger M, Adolph TE, Tilg H. Gut Dysfunction and Non-Alcoholic Fatty Liver Disease. Front Endocrinol (Lausanne) (2019) 10:611. doi: 10.3389/fendo.2019.00611
48. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate Immunity and Inflammation in NAFLD/NASH. Dig Dis Sci (2016) 61(5):1294–303. doi: 10.1007/s10620-016-4049-x
49. Cai J, Zhang XJ, Li H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology (2019) 70(3):1026–37. doi: 10.1002/hep.30506
50. Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer Cells Trigger Nonalcoholic Steatohepatitis Development in Diet-Induced Mouse Model Through Tumor Necrosis Factor-α Production. J Biol Chem (2012) 287(48):40161–72. doi: 10.1074/jbc.M112.417014
51. Marra F, Tacke F. Roles for Chemokines in Liver Disease. Gastroenterology (2014) 147(3):577–94.e1. doi: 10.1053/j.gastro.2014.06.043
52. Negrin KA, Roth Flach RJ, DiStefano MT, Matevossian A, Friedline RH, Jung D, et al. IL-1 Signaling in Obesity-Induced Hepatic Lipogenesis and Steatosis. PloS One (2014) 9(9):e107265. doi: 10.1371/journal.pone.0107265
53. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, et al. Kupffer Cells Promote Hepatic Steatosis via Interleukin-1beta-Dependent Suppression of Peroxisome Proliferator-Activated Receptor Alpha Activity. Hepatology (2010) 51(2):511–22. doi: 10.1002/hep.23337
54. Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 Induces Cellular Insulin Resistance in Hepatocytes. Diabetes (2002) 51(12):3391–9. doi: 10.2337/diabetes.51.12.3391
55. Tacke F, Zimmermann HW. Macrophage Heterogeneity in Liver Injury and Fibrosis. J Hepatol (2014) 60(5):1090–6. doi: 10.1016/j.jhep.2013.12.025
56. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic Recruitment of the Inflammatory Gr1+ Monocyte Subset Upon Liver Injury Promotes Hepatic Fibrosis. Hepatology (2009) 50(1):261–74. doi: 10.1002/hep.22950
57. Mehal WZ, Schuppan D. Antifibrotic Therapies in the Liver. Semin Liver Dis (2015) 35(2):184–98. doi: 10.1055/s-0035-1550055
58. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver Fibrosis and Repair: Immune Regulation of Wound Healing in a Solid Organ. Nat Rev Immunol (2014) 14(3):181–94. doi: 10.1038/nri3623
59. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C Expression Identifies the Recruited Macrophage Phenotype, Which Orchestrates the Regression of Murine Liver Fibrosis. Proc Natl Acad Sci USA (2012) 109(46):E3186–95. doi: 10.1073/pnas.1119964109
60. Tsuchida T, Friedman SL. Mechanisms of Hepatic Stellate Cell Activation. Nat Rev Gastroenterol Hepatol (2017) 14(7):397–411. doi: 10.1038/nrgastro.2017.38
61. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The Role of Macrophages in Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Nat Rev Gastroenterol Hepatol (2019) 16(3):145–59. doi: 10.1038/s41575-018-0082-x
62. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, et al. M2 Kupffer Cells Promote M1 Kupffer Cell Apoptosis: A Protective Mechanism Against Alcoholic and Nonalcoholic Fatty Liver Disease. Hepatology (2014) 59(1):130–42. doi: 10.1002/hep.26607
63. van der Heide D, Weiskirchen R, Bansal R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front Immunol (2019) 10:2852. doi: 10.3389/fimmu.2019.02852
64. Sun YY, Li XF, Meng XM, Huang C, Zhang L, Li J. Macrophage Phenotype in Liver Injury and Repair. Scand J Immunol (2017) 85(3):166–74. doi: 10.1111/sji.12468
65. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol Cell (2019) 75(3):644–60.e5. doi: 10.1016/j.molcel.2019.07.028
66. Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity (2020) 52(6):1057–74.e7. doi: 10.1016/j.immuni.2020.04.001
67. Remmerie A, Martens L, Thoné T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct From Kupffer Cells in the Fatty Liver. Immunity (2020) 53(3):641–57.e14. doi: 10.1016/j.immuni.2020.08.004
68. Patouraux S, Rousseau D, Bonnafous S, Lebeaupin C, Luci C, Canivet CM, et al. CD44 is a Key Player in Non-Alcoholic Steatohepatitis. J Hepatol (2017) 67(2):328–38. doi: 10.1016/j.jhep.2017.03.003
69. Van Herck MA, Weyler J, Kwanten WJ, Dirinck EL, De Winter BY, Francque SM, et al. The Differential Roles of T Cells in Non-Alcoholic Fatty Liver Disease and Obesity. Front Immunol (2019) 10:82. doi: 10.3389/fimmu.2019.00082
70. Luci C, Vieira E, Perchet T, Gual P, Golub R. Natural Killer Cells and Type 1 Innate Lymphoid Cells Are New Actors in Non-Alcoholic Fatty Liver Disease. Front Immunol (2019) 10:1192. doi: 10.3389/fimmu.2019.01192
71. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the Activation and Regulation of Innate and Adaptive Immunity. Nat Rev Immunol (2011) 11(8):519–31. doi: 10.1038/nri3024
72. Nathan C. Neutrophils and Immunity: Challenges and Opportunities. Nat Rev Immunol (2006) 6(3):173–82. doi: 10.1038/nri1785
73. Borregaard N. Neutrophils, From Marrow to Microbes. Immunity (2010) 33(5):657–70. doi: 10.1016/j.immuni.2010.11.011
74. Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-Specific Monoclonal Antibody to Deplete Neutrophils in Mice. J Leukoc Biol (2008) 83(1):64–70. doi: 10.1189/jlb.0407247
75. Boivin G, Faget J, Ancey PB, Gkasti A, Mussard J, Engblom C, et al. Durable and Controlled Depletion of Neutrophils in Mice. Nat Commun (2020) 11(1):2762. doi: 10.1038/s41467-020-16596-9
76. Ou R, Liu J, Lv M, Wang J, Wang J, Zhu L, et al. Neutrophil Depletion Improves Diet-Induced Non-Alcoholic Fatty Liver Disease in Mice. Endocrine (2017) 57(1):72–82. doi: 10.1007/s12020-017-1323-4
77. Alkhouri N, Morris-Stiff G, Campbell C, Lopez R, Tamimi TA, Yerian L, et al. Neutrophil to Lymphocyte Ratio: A New Marker for Predicting Steatohepatitis and Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Liver Int (2012) 32(2):297–302. doi: 10.1111/j.1478-3231.2011.02639.x
78. Yilmaz H, Yalcin KS, Namuslu M, Celik HT, Sozen M, Inan O, et al. Neutrophil-Lymphocyte Ratio (NLR) Could Be Better Predictor Than C-Reactive Protein (CRP) for Liver Fibrosis in Non-Alcoholic Steatohepatitis(NASH). Ann Clin Lab Sci (2015) 45(3):278–86.
79. Gao B, Tsukamoto H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology (2016) 150(8):1704–9. doi: 10.1053/j.gastro.2016.01.025
80. Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory Pathways in Alcoholic Steatohepatitis. J Hepatol (2019) 70(2):249–59. doi: 10.1016/j.jhep.2018.10.023
81. Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse Model of Chronic and Binge Ethanol Feeding (the NIAAA Model). Nat Protoc (2013) 8(3):627–37. doi: 10.1038/nprot.2013.032
82. Chang B, Xu MJ, Zhou Z, Cai Y, Li M, Wang W, et al. Short- or Long-Term High-Fat Diet Feeding Plus Acute Ethanol Binge Synergistically Induce Acute Liver Injury in Mice: An Important Role for CXCL1. Hepatology (2015) 62(4):1070–85. doi: 10.1002/hep.27921
83. Mathews S, Feng D, Maricic I, Ju C, Kumar V, Gao B. Invariant Natural Killer T Cells Contribute to Chronic-Plus-Binge Ethanol-Mediated Liver Injury by Promoting Hepatic Neutrophil Infiltration. Cell Mol Immunol (2016) 13(2):206–16. doi: 10.1038/cmi.2015.06
84. Bertola A, Park O, Gao B. Chronic Plus Binge Ethanol Feeding Synergistically Induces Neutrophil Infiltration and Liver Injury in Mice: A Critical Role for E-Selectin. Hepatology (2013) 58(5):1814–23. doi: 10.1002/hep.26419
85. Li M, He Y, Zhou Z, Ramirez T, Gao Y, Gao Y, et al. MicroRNA-223 Ameliorates Alcoholic Liver Injury by Inhibiting the IL-6-P47(Phox)-Oxidative Stress Pathway in Neutrophils. Gut (2017) 66(4):705–15. doi: 10.1136/gutjnl-2016-311861
86. Wang H, Mehal W, Nagy LE, Rotman Y. Immunological Mechanisms and Therapeutic Targets of Fatty Liver Diseases. Cell Mol Immunol (2021) 18(1):73–91. doi: 10.1038/s41423-020-00579-3
87. Grunhut J, Wang W, Aykut B, Gakhal I, Torres-Hernandez A, Miller G. Macrophages in Nonalcoholic Steatohepatitis: Friend or Foe? Eur Med J Hepatol (2018) 6(1):100–9.
88. Bertola A, Bonnafous S, Anty R, Patouraux S, Saint-Paul MC, Iannelli A, et al. Hepatic Expression Patterns of Inflammatory and Immune Response Genes Associated With Obesity and NASH in Morbidly Obese Patients. PloS One (2010) 5(10):e13577. doi: 10.1371/journal.pone.0013577
89. Hwang S, Wang X, Rodrigues RM, Ma J, He Y, Seo W, et al. Protective and Detrimental Roles of P38α Mitogen-Activated Protein Kinase in Different Stages of Nonalcoholic Fatty Liver Disease. Hepatology (2020) 72(3):873–91. doi: 10.1002/hep.31390
90. Farrell G, Schattenberg JM, Leclercq I, Yeh MM, Goldin R, Teoh N, et al. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology (2019) 69(5):2241–57. doi: 10.1002/hep.30333
91. Koop AC, Thiele ND, Steins D, Michaëlsson E, Wehmeyer M, Scheja L, et al. Therapeutic Targeting of Myeloperoxidase Attenuates NASH in Mice. Hepatol Commun (2020) 4(10):1441–58. doi: 10.1002/hep4.1566
92. van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO, et al. Neutrophil Extracellular Traps Promote Inflammation and Development of Hepatocellular Carcinoma in Nonalcoholic Steatohepatitis. Hepatology (2018) 68(4):1347–60. doi: 10.1002/hep.29914
93. Liang W, Lindeman JH, Menke AL, Koonen DP, Morrison M, Havekes LM, et al. Metabolically Induced Liver Inflammation Leads to NASH and Differs From LPS- or IL-1β-Induced Chronic Inflammation. Lab Invest (2014) 94(5):491–502. doi: 10.1038/labinvest.2014.11
94. Son Y, Kim S, Chung HT, Pae HO. Reactive Oxygen Species in the Activation of MAP Kinases. Methods Enzymol (2013) 528:27–48. doi: 10.1016/b978-0-12-405881-1.00002-1
95. Maiers JL, Malhi H. Endoplasmic Reticulum Stress in Metabolic Liver Diseases and Hepatic Fibrosis. Semin Liver Dis (2019) 39(2):235–48. doi: 10.1055/s-0039-1681032
96. Zhang XQ, Xu CF, Yu CH, Chen WX, Li YM. Role of Endoplasmic Reticulum Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease. World J Gastroenterol (2014) 20(7):1768–76. doi: 10.3748/wjg.v20.i7.1768
97. El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. P47phox, the Phagocyte NADPH Oxidase/NOX2 Organizer: Structure, Phosphorylation and Implication in Diseases. Exp Mol Med (2009) 41(4):217–25. doi: 10.3858/emm.2009.41.4.058
98. Belambri SA, Rolas L, Raad H, Hurtado-Nedelec M, Dang PM, El-Benna J. NADPH Oxidase Activation in Neutrophils: Role of the Phosphorylation of its Subunits. Eur J Clin Invest (2018) 48 Suppl 2:e12951. doi: 10.1111/eci.12951
99. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: Immunobiology and Pathology. Annu Rev Immunol (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
100. Hwang S, Feng D, Gao B. Interleukin-22 Acts as a Mitochondrial Protector. Theranostics (2020) 10(17):7836–40. doi: 10.7150/thno.48022
101. Kong X, Feng D, Mathews S, Gao B. Hepatoprotective and Anti-Fibrotic Functions of Interleukin-22: Therapeutic Potential for the Treatment of Alcoholic Liver Disease. J Gastroenterol Hepatol (2013) 28(Suppl 1):56–60. doi: 10.1111/jgh.12032
102. Proost P, De Wolf-Peeters C, Conings R, Opdenakker G, Billiau A, Van Damme J. Identification of a Novel Granulocyte Chemotactic Protein (GCP-2) From Human Tumor Cells. In Vitro and In Vivo Comparison With Natural Forms of GRO, IP-10, and IL-8. J Immunol (1993) 150(3):1000–10.
103. Rottiers V, Näär AM. MicroRNAs in Metabolism and Metabolic Disorders. Nat Rev Mol Cell Biol (2012) 13(4):239–50. doi: 10.1038/nrm3313
104. Deiuliis JA. MicroRNAs as Regulators of Metabolic Disease: Pathophysiologic Significance and Emerging Role as Biomarkers and Therapeutics. Int J Obes (Lond) (2016) 40(1):88–101. doi: 10.1038/ijo.2015.170
105. Landrier JF, Derghal A, Mounien L. MicroRNAs in Obesity and Related Metabolic Disorders. Cells (2019) 8(8):859. doi: 10.3390/cells8080859
106. Liu CH, Ampuero J, Gil-Gómez A, Montero-Vallejo R, Rojas Á, Muñoz-Hernández R, et al. miRNAs in Patients With Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. J Hepatol (2018) 69(6):1335–48. doi: 10.1016/j.jhep.2018.08.008
107. Pirola CJ, Fernández Gianotti T, Castaño GO, Mallardi P, San Martino J, Mora Gonzalez Lopez Ledesma M, et al. Circulating microRNA Signature in Non-Alcoholic Fatty Liver Disease: From Serum Non-Coding RNAs to Liver Histology and Disease Pathogenesis. Gut (2015) 64(5):800–12. doi: 10.1136/gutjnl-2014-306996
108. Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW, et al. Nonalcoholic Steatohepatitis is Associated With Altered Hepatic MicroRNA Expression. Hepatology (2008) 48(6):1810–20. doi: 10.1002/hep.22569
109. He Y, Hwang S, Cai Y, Kim SJ, Xu M, Yang D, et al. MicroRNA-223 Ameliorates Nonalcoholic Steatohepatitis and Cancer by Targeting Multiple Inflammatory and Oncogenic Genes in Hepatocytes. Hepatology (2019) 70(4):1150–67. doi: 10.1002/hep.30645
110. Wu H, Ng R, Chen X, Steer CJ, Song G. MicroRNA-21 is a Potential Link Between Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma via Modulation of the HBP1-P53-Srebp1c Pathway. Gut (2016) 65(11):1850–60. doi: 10.1136/gutjnl-2014-308430
111. Loyer X, Paradis V, Hénique C, Vion AC, Colnot N, Guerin CL, et al. Liver microRNA-21 is Overexpressed in Non-Alcoholic Steatohepatitis and Contributes to the Disease in Experimental Models by Inhibiting Pparα Expression. Gut (2016) 65(11):1882–94. doi: 10.1136/gutjnl-2014-308883
112. Vinciguerra M, Sgroi A, Veyrat-Durebex C, Rubbia-Brandt L, Buhler LH, Foti M. Unsaturated Fatty Acids Inhibit the Expression of Tumor Suppressor Phosphatase and Tensin Homolog (PTEN) via microRNA-21 Up-Regulation in Hepatocytes. Hepatology (2009) 49(4):1176–84. doi: 10.1002/hep.22737
113. Calo N, Ramadori P, Sobolewski C, Romero Y, Maeder C, Fournier M, et al. Stress-Activated miR-21/miR-21* in Hepatocytes Promotes Lipid and Glucose Metabolic Disorders Associated With High-Fat Diet Consumption. Gut (2016) 65(11):1871–81. doi: 10.1136/gutjnl-2015-310822
114. Wang X, He Y, Mackowiak B, Gao B. MicroRNAs as Regulators, Biomarkers and Therapeutic Targets in Liver Diseases. Gut (2021) 70(4):784–95. doi: 10.1136/gutjnl-2020-322526
115. Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of Progenitor Cell Proliferation and Granulocyte Function by microRNA-223. Nature (2008) 451(7182):1125–9. doi: 10.1038/nature06607
116. Yuan X, Berg N, Lee JW, Le TT, Neudecker V, Jing N, et al. MicroRNA miR-223 as Regulator of Innate Immunity. J Leukoc Biol (2018) 104(3):515–24. doi: 10.1002/jlb.3mr0218-079r
117. Ye D, Zhang T, Lou G, Liu Y. Role of miR-223 in the Pathophysiology of Liver Diseases. Exp Mol Med (2018) 50(9):1–12. doi: 10.1038/s12276-018-0153-7
118. He Y, Rodrigues RM, Wang X, Seo W, Ma J, Hwang S, et al. Neutrophil-To-Hepatocyte Communication via LDLR-Dependent miR-223-Enriched Extracellular Vesicle Transfer Ameliorates Nonalcoholic Steatohepatitis. J Clin Invest (2021) 131(3):e141513. doi: 10.1172/jci141513
119. Zhuang G, Meng C, Guo X, Cheruku PS, Shi L, Xu H, et al. A Novel Regulator of Macrophage Activation: miR-223 in Obesity-Associated Adipose Tissue Inflammation. Circulation (2012) 125(23):2892–903. doi: 10.1161/circulationaha.111.087817
120. Wang X, Seo W, Park SH, Fu Y, Hwang S, Rodrigues RM, et al. MicroRNA-223 Restricts Liver Fibrosis by Inhibiting the TAZ-IHH-GLI2 and PDGF Signaling Pathways via the Crosstalk of Multiple Liver Cell Types. Int J Biol Sci (2021) 17(4):1153–67. doi: 10.7150/ijbs.58365
121. Hou X, Yin S, Ren R, Liu S, Yong L, Liu Y, et al. Myeloid-Cell-Specific IL-6 Signaling Promotes MicroRNA-223-Enriched Exosome Production to Attenuate NAFLD-Associated Fibrosis. Hepatology (2020) 74(1):116–32. doi: 10.1002/Hep.31658
122. Szabo G. Exosomes and MicroRNA-223 at the Intersection of Inflammation and Fibrosis in NAFLD. Hepatology (2021) 74(1):5–8. doi: 10.1002/hep.31805
123. He Y, Feng D, Li M, Gao Y, Ramirez T, Cao H, et al. Hepatic Mitochondrial DNA/Toll-Like Receptor 9/MicroRNA-223 Forms a Negative Feedback Loop to Limit Neutrophil Overactivation and Acetaminophen Hepatotoxicity in Mice. Hepatology (2017) 66(1):220–34. doi: 10.1002/hep.29153
124. Cassatella MA, Östberg NK, Tamassia N, Soehnlein O. Biological Roles of Neutrophil-Derived Granule Proteins and Cytokines. Trends Immunol (2019) 40(7):648–64. doi: 10.1016/j.it.2019.05.003
125. Németh T, Sperandio M, Mócsai A. Neutrophils as Emerging Therapeutic Targets. Nat Rev Drug Discov (2020) 19(4):253–75. doi: 10.1038/s41573-019-0054-z
126. Pham CT. Neutrophil Serine Proteases: Specific Regulators of Inflammation. Nat Rev Immunol (2006) 6(7):541–50. doi: 10.1038/nri1841
127. Sheshachalam A, Srivastava N, Mitchell T, Lacy P, Eitzen G. Granule Protein Processing and Regulated Secretion in Neutrophils. Front Immunol (2014) 5:448. doi: 10.3389/fimmu.2014.00448
128. Rodrigues RM, He Y, Hwang S, Bertola A, Mackowiak B, Ait-Ahmed Y, et al. E-Selectin-Dependent Inflammation and Lipolysis in Adipose Tissue Exacerbate Steatosis-To-NASH Progression via S100A8/9. Cell Mol Gastroenterol Hepatol (2021). doi: 10.1016/j.jcmgh.2021.08.002
129. He Y, Hwang S, Ahmed YA, Feng D, Li N, Ribeiro M, et al. Immunopathobiology and Therapeutic Targets Related to Cytokines in Liver Diseases. Cell Mol Immunol (2021) 18(1):18–37. doi: 10.1038/s41423-020-00580-w
130. Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased Hepatic Myeloperoxidase Activity in Obese Subjects With Nonalcoholic Steatohepatitis. Am J Pathol (2009) 175(4):1473–82. doi: 10.2353/ajpath.2009.080999
131. Pulli B, Ali M, Iwamoto Y, Zeller MW, Schob S, Linnoila JJ, et al. Myeloperoxidase-Hepatocyte-Stellate Cell Cross Talk Promotes Hepatocyte Injury and Fibrosis in Experimental Nonalcoholic Steatohepatitis. Antioxid Redox Signal (2015) 23(16):1255–69. doi: 10.1089/ars.2014.6108
132. Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri-Sverdlov R, et al. Neutrophil-Derived Myeloperoxidase Aggravates Non-Alcoholic Steatohepatitis in Low-Density Lipoprotein Receptor-Deficient Mice. PloS One (2012) 7(12):e52411. doi: 10.1371/journal.pone.0052411
133. Zang S, Ma X, Zhuang Z, Liu J, Bian D, Xun Y, et al. Increased Ratio of Neutrophil Elastase to α1-Antitrypsin is Closely Associated With Liver Inflammation in Patients With Nonalcoholic Steatohepatitis. Clin Exp Pharmacol Physiol (2016) 43(1):13–21. doi: 10.1111/1440-1681.12499
134. Chen J, Liang B, Bian D, Luo Y, Yang J, Li Z, et al. Knockout of Neutrophil Elastase Protects Against Western Diet Induced Nonalcoholic Steatohepatitis in Mice by Regulating Hepatic Ceramides Metabolism. Biochem Biophys Res Commun (2019) 518(4):691–7. doi: 10.1016/j.bbrc.2019.08.111
135. Ye D, Yang K, Zang S, Lin Z, Chau HT, Wang Y, et al. Lipocalin-2 Mediates non-Alcoholic Steatohepatitis by Promoting Neutrophil-Macrophage Crosstalk via the Induction of CXCR2. J Hepatol (2016) 65(5):988–97. doi: 10.1016/j.jhep.2016.05.041
136. Auguet T, Terra X, Quintero Y, Martínez S, Manresa N, Porras JA, et al. Liver Lipocalin 2 Expression in Severely Obese Women With non Alcoholic Fatty Liver Disease. Exp Clin Endocrinol Diabetes (2013) 121(2):119–24. doi: 10.1055/s-0032-1331696
137. Xu G, Wang YM, Ying MM, Chen SD, Li ZR, Ma HL, et al. Serum Lipocalin-2 is a Potential Biomarker for the Clinical Diagnosis of Nonalcoholic Steatohepatitis. Clin Mol Hepatol (2021) 27(2):329–45. doi: 10.3350/cmh.2020.0261
138. Semba T, Nishimura M, Nishimura S, Ohara O, Ishige T, Ohno S, et al. The FLS (Fatty Liver Shionogi) Mouse Reveals Local Expressions of Lipocalin-2, CXCL1 and CXCL9 in the Liver With non-Alcoholic Steatohepatitis. BMC Gastroenterol (2013) 13:120. doi: 10.1186/1471-230x-13-120
139. Miele L, Alberelli MA, Martini M, Liguori A, Marrone G, Cocomazzi A, et al. Nonalcoholic Fatty Liver Disease (NAFLD) Severity is Associated to a Nonhemostatic Contribution and Proinflammatory Phenotype of Platelets. Transl Res (2021) 231:24–38. doi: 10.1016/j.trsl.2020.11.003
140. Zhao X, Yang L, Chang N, Hou L, Zhou X, Yang L, et al. Neutrophils Undergo Switch of Apoptosis to NETosis During Murine Fatty Liver Injury via S1P Receptor 2 Signaling. Cell Death Dis (2020) 11(5):379. doi: 10.1038/s41419-020-2582-1
141. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils Mediate Insulin Resistance in Mice Fed a High-Fat Diet Through Secreted Elastase. Nat Med (2012) 18(9):1407–12. doi: 10.1038/nm.2885
142. Zang S, Wang L, Ma X, Zhu G, Zhuang Z, Xun Y, et al. Neutrophils Play a Crucial Role in the Early Stage of Nonalcoholic Steatohepatitis via Neutrophil Elastase in Mice. Cell Biochem Biophys (2015) 73(2):479–87. doi: 10.1007/s12013-015-0682-9
143. Aratani Y. Myeloperoxidase: Its Role for Host Defense, Inflammation, and Neutrophil Function. Arch Biochem Biophys (2018) 640:47–52. doi: 10.1016/j.abb.2018.01.004
144. Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as Microsomal Catalysts of Lipid Peroxides in Murine Nonalcoholic Steatohepatitis. J Clin Invest (2000) 105(8):1067–75. doi: 10.1172/jci8814
145. Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, Fosgerau K. Mouse Models of Nonalcoholic Steatohepatitis in Preclinical Drug Development. Drug Discov Today (2017) 22(11):1707–18. doi: 10.1016/j.drudis.2017.06.007
146. Kessenbrock K, Dau T, Jenne DE. Tailor-Made Inflammation: How Neutrophil Serine Proteases Modulate the Inflammatory Response. J Mol Med (Berl) (2011) 89(1):23–8. doi: 10.1007/s00109-010-0677-3
147. Papayannopoulos V. Neutrophil Extracellular Traps in Immunity and Disease. Nat Rev Immunol (2018) 18(2):134–47. doi: 10.1038/nri.2017.105
148. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J Cell Biol (2010) 191(3):677–91. doi: 10.1083/jcb.201006052
149. Jonigk D, Al-Omari M, Maegel L, Müller M, Izykowski N, Hong J, et al. Anti-Inflammatory and Immunomodulatory Properties of α1-Antitrypsin Without Inhibition of Elastase. Proc Natl Acad Sci USA (2013) 110(37):15007–12. doi: 10.1073/pnas.1309648110
150. Dau T, Sarker RS, Yildirim AO, Eickelberg O, Jenne DE. Autoprocessing of Neutrophil Elastase Near its Active Site Reduces the Efficiency of Natural and Synthetic Elastase Inhibitors. Nat Commun (2015) 6:6722. doi: 10.1038/ncomms7722
151. Janciauskiene S, Wrenger S, Immenschuh S, Olejnicka B, Greulich T, Welte T, et al. The Multifaceted Effects of Alpha1-Antitrypsin on Neutrophil Functions. Front Pharmacol (2018) 9:341. doi: 10.3389/fphar.2018.00341
152. Mirea AM, Toonen EJM, van den Munckhof I, Munsterman ID, Tjwa E, Jaeger M, et al. Increased Proteinase 3 and Neutrophil Elastase Plasma Concentrations are Associated With Non-Alcoholic Fatty Liver Disease (NAFLD) and Type 2 Diabetes. Mol Med (2019) 25(1):16. doi: 10.1186/s10020-019-0084-3
153. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil Elastase, Proteinase 3, and Cathepsin G as Therapeutic Targets in Human Diseases. Pharmacol Rev (2010) 62(4):726–59. doi: 10.1124/pr.110.002733
154. Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, et al. Lipocalin 2 Mediates an Innate Immune Response to Bacterial Infection by Sequestrating Iron. Nature (2004) 432(7019):917–21. doi: 10.1038/nature03104
155. Viau A, El Karoui K, Laouari D, Burtin M, Nguyen C, Mori K, et al. Lipocalin 2 is Essential for Chronic Kidney Disease Progression in Mice and Humans. J Clin Invest (2010) 120(11):4065–76. doi: 10.1172/jci42004
156. Devireddy LR, Gazin C, Zhu X, Green MR. A Cell-Surface Receptor for Lipocalin 24p3 Selectively Mediates Apoptosis and Iron Uptake. Cell (2005) 123(7):1293–305. doi: 10.1016/j.cell.2005.10.027
157. Mosialou I, Shikhel S, Liu JM, Maurizi A, Luo N, He Z, et al. MC4R-Dependent Suppression of Appetite by Bone-Derived Lipocalin 2. Nature (2017) 543(7645):385–90. doi: 10.1038/nature21697
158. Guo H, Jin D, Zhang Y, Wright W, Bazuine M, Brockman DA, et al. Lipocalin-2 Deficiency Impairs Thermogenesis and Potentiates Diet-Induced Insulin Resistance in Mice. Diabetes (2010) 59(6):1376–85. doi: 10.2337/db09-1735
159. Abella V, Scotece M, Conde J, Gómez R, Lois A, Pino J, et al. The Potential of Lipocalin-2/NGAL as Biomarker for Inflammatory and Metabolic Diseases. Biomarkers (2015) 20(8):565–71. doi: 10.3109/1354750x.2015.1123354
160. Bhusal A, Rahman MH, Lee WH, Bae YC, Lee IK, Suk K. Paradoxical Role of Lipocalin-2 in Metabolic Disorders and Neurological Complications. Biochem Pharmacol (2019) 169:113626. doi: 10.1016/j.bcp.2019.113626
161. Asimakopoulou A, Weiskirchen S, Weiskirchen R. Lipocalin 2 (LCN2) Expression in Hepatic Malfunction and Therapy. Front Physiol (2016) 7:430. doi: 10.3389/fphys.2016.00430
162. Moschen AR, Adolph TE, Gerner RR, Wieser V, Tilg H. Lipocalin-2: A Master Mediator of Intestinal and Metabolic Inflammation. Trends Endocrinol Metab (2017) 28(5):388–97. doi: 10.1016/j.tem.2017.01.003
163. Soga M, Kishimoto Y, Kawaguchi J, Nakai Y, Kawamura Y, Inagaki S, et al. The FLS Mouse: A New Inbred Strain With Spontaneous Fatty Liver. Lab Anim Sci (1999) 49(3):269–75.
164. Soga M, Kishimoto Y, Kawamura Y, Inagaki S, Makino S, Saibara T. Spontaneous Development of Hepatocellular Carcinomas in the FLS Mice With Hereditary Fatty Liver. Cancer Lett (2003) 196(1):43–8. doi: 10.1016/s0304-3835(03)00213-1
165. Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in Inflammation. Front Immunol (2018) 9:1298. doi: 10.3389/fimmu.2018.01298
166. Sroussi HY, Lu Y, Zhang QL, Villines D, Marucha PT. S100A8 and S100A9 Inhibit Neutrophil Oxidative Metabolism in-Vitro: Involvement of Adenosine Metabolites. Free Radic Res (2010) 44(4):389–96. doi: 10.3109/10715760903431434
167. Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identification of P8,14 as a Highly Abundant Heterodimeric Calcium Binding Protein Complex of Myeloid Cells. J Biol Chem (1991) 266(12):7706–13. doi: 10.1016/S0021-9258(20)89506-4
168. Kraakman MJ, Lee MK, Al-Sharea A, Dragoljevic D, Barrett TJ, Montenont E, et al. Neutrophil-Derived S100 Calcium-Binding Proteins A8/A9 Promote Reticulated Thrombocytosis and Atherogenesis in Diabetes. J Clin Invest (2017) 127(6):2133–47. doi: 10.1172/jci92450
169. Nacken W, Roth J, Sorg C, Kerkhoff C. S100A9/S100A8: Myeloid Representatives of the S100 Protein Family as Prominent Players in Innate Immunity. Microsc Res Tech (2003) 60(6):569–80. doi: 10.1002/jemt.10299
170. Schiopu A, Cotoi OS. S100A8 and S100A9: DAMPs at the Crossroads Between Innate Immunity, Traditional Risk Factors, and Cardiovascular Disease. Mediators Inflamm (2013) 2013:828354. doi: 10.1155/2013/828354
171. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valentí V, et al. Increased Levels of Calprotectin in Obesity are Related to Macrophage Content: Impact on Inflammation and Effect of Weight Loss. Mol Med (2011) 17(11-12):1157–67. doi: 10.2119/molmed.2011.00144
172. Serhal R, Hilal G, Boutros G, Sidaoui J, Wardi L, Ezzeddine S, et al. Nonalcoholic Steatohepatitis: Involvement of the Telomerase and Proinflammatory Mediators. BioMed Res Int (2015) 2015:850246. doi: 10.1155/2015/850246
173. Li T, Zhang Z, Li X, Dong G, Zhang M, Xu Z, et al. Neutrophil Extracellular Traps: Signaling Properties and Disease Relevance. Mediators Inflamm (2020) 2020:9254087. doi: 10.1155/2020/9254087
174. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
175. Jorch SK, Kubes P. An Emerging Role for Neutrophil Extracellular Traps in Noninfectious Disease. Nat Med (2017) 23(3):279–87. doi: 10.1038/nm.4294
176. Shen H, Kreisel D, Goldstein DR. Processes of Sterile Inflammation. J Immunol (2013) 191(6):2857–63. doi: 10.4049/jimmunol.1301539
177. Erpenbeck L, Schön MP. Neutrophil Extracellular Traps: Protagonists of Cancer Progression? Oncogene (2017) 36(18):2483–90. doi: 10.1038/onc.2016.406
178. Németh T, Mócsai A. Feedback Amplification of Neutrophil Function. Trends Immunol (2016) 37(6):412–24. doi: 10.1016/j.it.2016.04.002
179. Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, et al. A Murine Model for Non-Alcoholic Steatohepatitis Showing Evidence of Association Between Diabetes and Hepatocellular Carcinoma. Med Mol Morphol (2013) 46(3):141–52. doi: 10.1007/s00795-013-0016-1
180. Serhan CN. Pro-Resolving Lipid Mediators are Leads for Resolution Physiology. Nature (2014) 510(7503):92–101. doi: 10.1038/nature13479
181. Serhan CN, Levy BD. Resolvins in Inflammation: Emergence of the Pro-Resolving Superfamily of Mediators. J Clin Invest (2018) 128(7):2657–69. doi: 10.1172/jci97943
182. Serhan CN. The Resolution of Inflammation: The Devil in the Flask and in the Details. FASEB J (2011) 25(5):1441–8. doi: 10.1096/fj.11-0502ufm
183. Lawrence T, Gilroy DW. Chronic Inflammation: A Failure of Resolution? Int J Exp Pathol (2007) 88(2):85–94. doi: 10.1111/j.1365-2613.2006.00507.x
184. de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil Migration in Infection and Wound Repair: Going Forward in Reverse. Nat Rev Immunol (2016) 16(6):378–91. doi: 10.1038/nri.2016.49
185. Brostjan C, Oehler R. The Role of Neutrophil Death in Chronic Inflammation and Cancer. Cell Death Discov (2020) 6:26. doi: 10.1038/s41420-020-0255-6
186. El Kebir D, Filep JG. Modulation of Neutrophil Apoptosis and the Resolution of Inflammation Through β2 Integrins. Front Immunol (2013) 4:60. doi: 10.3389/fimmu.2013.00060
187. Kourtzelis I, Hajishengallis G, Chavakis T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front Immunol (2020) 11:553. doi: 10.3389/fimmu.2020.00553
188. Phillipson M, Kubes P. The Healing Power of Neutrophils. Trends Immunol (2019) 40(7):635–47. doi: 10.1016/j.it.2019.05.001
189. Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, et al. Neutrophils Promote the Development of Reparative Macrophages Mediated by ROS to Orchestrate Liver Repair. Nat Commun (2019) 10(1):1076. doi: 10.1038/s41467-019-09046-8
190. Gresnigt MS, Joosten LA, Verschueren I, van der Meer JW, Netea MG, Dinarello CA, et al. Neutrophil-Mediated Inhibition of Proinflammatory Cytokine Responses. J Immunol (2012) 189(10):4806–15. doi: 10.4049/jimmunol.1103551
191. van der Meer JH, Netea MG, Dinarello CA. Modulation of Muramyl Dipeptide Stimulation of Cytokine Production by Blood Components. Clin Exp Immunol (2009) 156(3):428–33. doi: 10.1111/j.1365-2249.2009.03926.x
192. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated Neutrophil Extracellular Traps Limit Inflammation by Degrading Cytokines and Chemokines. Nat Med (2014) 20(5):511–7. doi: 10.1038/nm.3547
193. Filep JG, Ariel A. Neutrophil Heterogeneity and Fate in Inflamed Tissues: Implications for the Resolution of Inflammation. Am J Physiol Cell Physiol (2020) 319(3):C510–C32. doi: 10.1152/ajpcell.00181.2020
194. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of Inflammation: What Controls Its Onset? Front Immunol (2016) 7:160. doi: 10.3389/fimmu.2016.00160
195. Headland SE, Jones HR, Norling LV, Kim A, Souza PR, Corsiero E, et al. Neutrophil-Derived Microvesicles Enter Cartilage and Protect the Joint in Inflammatory Arthritis. Sci Transl Med (2015) 7(315):315ra190. doi: 10.1126/scitranslmed.aac5608
196. Leoni G, Nusrat A. Annexin A1: Shifting the Balance Towards Resolution and Repair. Biol Chem (2016) 397(10):971–9. doi: 10.1515/hsz-2016-0180
197. Sugimoto MA, Vago JP, Teixeira MM, Sousa LP. Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J Immunol Res (2016) 2016:8239258. doi: 10.1155/2016/8239258
198. Calvente CJ, Tameda M, Johnson CD, Del Pilar H, Lin YC, Adronikou N, et al. Neutrophils Contribute to Spontaneous Resolution of Liver Inflammation and Fibrosis via microRNA-223. J Clin Invest (2019) 129(10):4091–109. doi: 10.1172/jci122258
199. Stuart T, Satija R. Integrative Single-Cell Analysis. Nat Rev Genet (2019) 20(5):257–72. doi: 10.1038/s41576-019-0093-7
200. Campbell IK, Leong D, Edwards KM, Rayzman V, Ng M, Goldberg GL, et al. Therapeutic Targeting of the G-CSF Receptor Reduces Neutrophil Trafficking and Joint Inflammation in Antibody-Mediated Inflammatory Arthritis. J Immunol (2016) 197(11):4392–402. doi: 10.4049/jimmunol.1600121
201. Lee MC, McCubbin JA, Christensen AD, Poole DP, Rajasekhar P, Lieu T, et al. G-CSF Receptor Blockade Ameliorates Arthritic Pain and Disease. J Immunol (2017) 198(9):3565–75. doi: 10.4049/jimmunol.1602127
202. Jonsson H, Allen P, Peng SL. Inflammatory Arthritis Requires Foxo3a to Prevent Fas Ligand-Induced Neutrophil Apoptosis. Nat Med (2005) 11(6):666–71. doi: 10.1038/nm1248
203. Wu L, Gao X, Guo Q, Li J, Yao J, Yan K, et al. The Role of Neutrophils in Innate Immunity-Driven Nonalcoholic Steatohepatitis: Lessons Learned and Future Promise. Hepatol Int (2020) 14(5):652–66. doi: 10.1007/s12072-020-10081-7
204. Bronze-da-Rocha E, Santos-Silva A. Neutrophil Elastase Inhibitors and Chronic Kidney Disease. Int J Biol Sci (2018) 14(10):1343–60. doi: 10.7150/ijbs.26111
205. Guyot N, Butler MW, McNally P, Weldon S, Greene CM, Levine RL, et al. Elafin, an Elastase-Specific Inhibitor, is Cleaved by its Cognate Enzyme Neutrophil Elastase in Sputum From Individuals With Cystic Fibrosis. J Biol Chem (2008) 283(47):32377–85. doi: 10.1074/jbc.M803707200
206. Wang J, Ortiz C, Fontenot L, Mukhopadhyay R, Xie Y, Law IKM, et al. Elafin Inhibits Obesity, Hyperglycemia, and Liver Steatosis in High-Fat Diet-Treated Male Mice. Sci Rep (2020) 10(1):12785. doi: 10.1038/s41598-020-69634-3
207. Soubhye J, Aldib I, Delporte C, Prévost M, Dufrasne F, Antwerpen PV. Myeloperoxidase as a Target for the Treatment of Inflammatory Syndromes: Mechanisms and Structure Activity Relationships of Inhibitors. Curr Med Chem (2016) 23(35):3975–4008. doi: 10.2174/0929867323666160607111806
208. Malle E, Furtmüller PG, Sattler W, Obinger C. Myeloperoxidase: A Target for New Drug Development? Br J Pharmacol (2007) 152(6):838–54. doi: 10.1038/sj.bjp.0707358
209. Burner U, Obinger C, Paumann M, Furtmüller PG, Kettle AJ. Transient and Steady-State Kinetics of the Oxidation of Substituted Benzoic Acid Hydrazides by Myeloperoxidase. J Biol Chem (1999) 274(14):9494–502. doi: 10.1074/jbc.274.14.9494
210. van Zyl JM, Basson K, Uebel RA, van der Walt BJ. Isoniazid-Mediated Irreversible Inhibition of the Myeloperoxidase Antimicrobial System of the Human Neutrophil and the Effect of Thyronines. Biochem Pharmacol (1989) 38(14):2363–73. doi: 10.1016/0006-2952(89)90477-2
211. Soubhye J, Aldib I, Prévost M, Elfving B, Gelbcke M, Podrecca M, et al. Hybrid Molecules Inhibiting Myeloperoxidase Activity and Serotonin Reuptake: A Possible New Approach of Major Depressive Disorders With Inflammatory Syndrome. J Pharm Pharmacol (2014) 66(8):1122–32. doi: 10.1111/jphp.12236
212. Tidén AK, Sjögren T, Svensson M, Bernlind A, Senthilmohan R, Auchère F, et al. 2-Thioxanthines are Mechanism-Based Inactivators of Myeloperoxidase That Block Oxidative Stress During Inflammation. J Biol Chem (2011) 286(43):37578–89. doi: 10.1074/jbc.M111.266981
213. Malvezzi A, Queiroz RF, de Rezende L, Augusto O, Amaral AT. MPO Inhibitors Selected by Virtual Screening. Mol Inform (2011) 30(6-7):605–13. doi: 10.1002/minf.201100016
214. Kettle AJ, Gedye CA, Hampton MB, Winterbourn CC. Inhibition of Myeloperoxidase by Benzoic Acid Hydrazides. Biochem J (1995) 308( Pt 2):559–63. doi: 10.1042/bj3080559
215. Hu C, Yang K, Li M, Huang W, Zhang F, Wang H. Lipocalin 2: A Potential Therapeutic Target for Breast Cancer Metastasis. Oncol Targets Ther (2018) 11:8099–106. doi: 10.2147/ott.S181223
216. Santiago-Sánchez GS, Pita-Grisanti V, Quiñones-Díaz B, Gumpper K, Cruz-Monserrate Z, Vivas-Mejía PE. Biological Functions and Therapeutic Potential of Lipocalin 2 in Cancer. Int J Mol Sci (2020) 21(12):4365. doi: 10.3390/ijms21124365
217. Tarín C, Fernandez-Garcia CE, Burillo E, Pastor-Vargas C, Llamas-Granda P, Castejón B, et al. Lipocalin-2 Deficiency or Blockade Protects Against Aortic Abdominal Aneurysm Development in Mice. Cardiovasc Res (2016) 111(3):262–73. doi: 10.1093/cvr/cvw112
218. Volpe V, Raia Z, Sanguigno L, Somma D, Mastrovito P, Moscato F, et al. NGAL Controls the Metastatic Potential of Anaplastic Thyroid Carcinoma Cells. J Clin Endocrinol Metab (2013) 98(1):228–35. doi: 10.1210/jc.2012-2528
219. Miyamoto T, Kashima H, Yamada Y, Kobara H, Asaka R, Ando H, et al. Lipocalin 2 Enhances Migration and Resistance Against Cisplatin in Endometrial Carcinoma Cells. PloS One (2016) 11(5):e0155220. doi: 10.1371/journal.pone.0155220
220. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone Hypercitrullination Mediates Chromatin Decondensation and Neutrophil Extracellular Trap Formation. J Cell Biol (2009) 184(2):205–13. doi: 10.1083/jcb.200806072
221. Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of PAD4 in NET Formation. Front Immunol (2012) 3:360. doi: 10.3389/fimmu.2012.00360
Keywords: neutrophil, nonalcoholic steatohepatitis, nonalcoholic fatty liver disease, inflammation, fibrosis
Citation: Hwang S, Yun H, Moon S, Cho YE and Gao B (2021) Role of Neutrophils in the Pathogenesis of Nonalcoholic Steatohepatitis. Front. Endocrinol. 12:751802. doi: 10.3389/fendo.2021.751802
Received: 02 August 2021; Accepted: 23 September 2021;
Published: 11 October 2021.
Edited by:
Petra Hirsova, Mayo Clinic, United StatesReviewed by:
Yankai Wen, University of Texas Health Science Center at Houston, United StatesHua Wang, Anhui Medical University, China
Carmelo Luci, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2021 Hwang, Yun, Moon, Cho and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seonghwan Hwang, c2hod2FuZ0BwdXNhbi5hYy5rcg==
†These authors have contributed equally to this work