 Lijun Yin
Lijun Yin Man Luo
Man Luo Ru Wang
Ru Wang Jianping Ye
Jianping Ye Xiaohui Wang
Xiaohui Wang- 1School of Kinesiology, Shanghai University of Sport, Shanghai, China
- 2Metabolism Research Center, Zhengzhou University Affiliated Zhengzhou Central Hospital, Zhengzhou, China
- 3Center for Advanced Medicine, College of Medicine, Zhengzhou University, Zhengzhou, China
- 4Shanghai Diabetes Institute, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai, China
Androgens have a complex role in the regulation of insulin sensitivity in the pathogenesis of type 2 diabetes. In male subjects, a reduction in androgens increases the risk for insulin resistance, which is improved by androgen injections. However, in female subjects with polycystic ovary syndrome (PCOS), androgen excess becomes a risk factor for insulin resistance. The exact mechanism underlying the complex activities of androgens remains unknown. In this review, a hormone synergy-based view is proposed for understanding this complexity. Mitochondrial overactivation by substrate influx is a mechanism of insulin resistance in obesity. This concept may apply to the androgen-induced insulin resistance in PCOS. Androgens and estrogens both exhibit activities in the induction of mitochondrial oxidative phosphorylation. The two hormones may synergize in mitochondria to induce overproduction of ATP. ATP surplus in the pancreatic β-cells and α-cells causes excess secretion of insulin and glucagon, respectively, leading to peripheral insulin resistance in the early phase of type 2 diabetes. In the skeletal muscle and liver, the ATP surplus contributes to insulin resistance through suppression of AMPK and activation of mTOR. Consistent ATP surplus leads to mitochondrial dysfunction as a consequence of mitophagy inhibition, which provides a potential mechanism for mitochondrial dysfunction in β-cells and brown adipocytes in PCOS. The hormone synergy-based view provides a basis for the overactivation and dysfunction of mitochondria in PCOS-associated type 2 diabetes. The molecular mechanism for the synergy is discussed in this review with a focus on transcriptional regulation. This view suggests a unifying mechanism for the distinct metabolic roles of androgens in the control of insulin action in men with hypogonadism and women with PCOS.
Introduction
Testosterone is the primary sex hormone in regulating male sex organ development and reproduction activities. It is produced by the testis in males and ovarian interstitial cells in females. Testosterone is converted into the more active form, dihydrotestosterone (DHT), in the cell cytoplasm by 5α-reductase. DHT directly activates the androgen receptor (AR) for induction of target gene transcription. After DHT binds to its receptor, the AR undergoes a conformational change to disassociate from the heat shock protein and travels to the nucleus. In the nucleus, AR binds to the androgen response elements (AREs) of target genes to modulate gene transcription for the control of male organ development, reproductive cell differentiation, muscle growth, bone strength, and acceleration of energy (glucose, fatty acids, and amino acids) metabolism. Additionally, androgens have receptor-independent activity in the regulation of endothelial cell proliferation (1). Testosterone-driven energy metabolism favors energy expenditure to prevent metabolic disorders, such as obesity and type 2 diabetes in male subjects (2, 3). Testosterone deficiency (hypogonadism) increases the risk of metabolic disorders in male subjects and animals (4–6). In some cases, however, hypogonadism can be the result of obesity due to hypothalamus–pituitary axis inhibition by aromatase conversion of androgen into estradiol in peripheral tissues [such as white adipose tissue (WAT)] (7). Injection of male hormones is a treatment strategy for obesity and type 2 diabetes in male patients with hypogonadism. Although the hormone treatment is effective in most studies, there are reports of inefficacy in some studies (8, 9).
In female subjects, testosterone excess (hypergonadism) is a risk factor for type 2 diabetes due to testosterone’s role in the induction of insulin resistance (10). Elevated plasma testosterone levels are often associated with insulin resistance in obese girls (11) and women with polycystic ovary syndrome (PCOS) (12, 13). Inhibition of testosterone production and antagonism of its activity are clinical strategies to control insulin resistance in PCOS patients (14–16). Treatment often leads to visceral fat reduction and improved insulin sensitivity. Studies have shown that testosterone exhibits opposite effects on the regulation of insulin sensitivity in male versus female patients (17, 18); however, the exact mechanism remains unknown. Here, we propose a potential mechanism by integration of multidisciplinary information with a focus on mitochondria to explain the androgen activity. Estrogen protects insulin sensitivity in female subjects (19). Reduced estrogen levels in postmenopausal patients lead to an increased risk of insulin resistance, which can be improved by estrogen supplementation (19). Both estrogens and androgens regulate metabolism through nuclear receptors. Activation of these receptors involves transcription-mediated reprogramming of the neuroendocrine system according to current studies; however, there is no unifying mechanism of how sex hormones regulate insulin sensitivity in PCOS. Mitochondria are the targets of both hormones (more discussion below). Interestingly, mitochondrial genes that play a role in oxidative phosphorylation and antioxidant properties have been observed to be sexually dimorphic in the skeletal muscle and liver of female rats (20). Therefore, we propose that mitochondria may be the key targets for androgens and estrogens in the control of insulin sensitivity, which may hold an answer for the gender-dependent effects of androgens. This review will integrate information from several fields, such as obesity, diabetes, cardiovascular diseases, endocrinology, and mitochondrial biology to explore the mechanism. The aim is to propose a mitochondrion-centered mechanism for how androgens together with estrogen regulate insulin sensitivity, with energy metabolism in mitochondria as the primary focus.
Mitochondria in Energy Expenditure
Mitochondria are the center of energy metabolism in cells, as mitochondria carry out both the catabolism and anabolism of substrates for fuel (21). In the catabolism process, substrates such as glucose, fatty acids, and amino acids are broken down to generate ATP or heat through OXPHOS, which is used as energy for cellular activities. Heat production (thermogenesis) is required for the maintenance of body temperature in mammalians (22). Induction of thermogenesis is an ideal approach in the control of obesity. Thermogenesis includes UCP1-dependent and UCP1-independent mechanisms (23). The UCP1-dependent mechanism is dominant in brown and beige adipocytes (24). The UCP1-independent system comprises the adenine nucleotide transporters 1/2 (ANT1/2) and UCP3, which play a major role in non-adipocytes (23). Mammalian ANT is a “new” uncoupling protein found in the mitochondria of several tissues including the muscle, kidney, liver, and brown fat (25) and is equivalent to the ADP/ATP carrier (AAC) in yeast. The reduction of energy output by dysfunctional mitochondrial may lead to a buildup of intermediate metabolites, which plays a role in the pathogenesis of insulin resistance.
Mitochondria export intermediate metabolites, such as acetyl-CoA and oxaloacetate to use for anabolism. In the cytoplasm, acetyl-CoA is a substrate for de novo lipogenesis of fatty acids and cholesterols. In hepatocytes, oxaloacetate is a substrate for gluconeogenesis. Insulin plays a role in regulating these metabolite levels by stimulating lipogenesis and inhibiting gluconeogenesis. However, these metabolites may also feedback to impact insulin sensitivity. In the lipotoxicity model of insulin resistance, acetyl-CoA buildup inhibits insulin-induced glucose utilization in the skeletal muscle through substrate competition (26). Oxaloacetate buildup in liver hepatocytes promotes gluconeogenesis, contributing to insulin resistance of the liver. In obesity, overproduction of these metabolites in tissues contributes to systemic insulin resistance, and mitochondrial dysfunction in OXPHOS results in insulin resistance through the buildup of the metabolites (27).
Mitochondrial function is regulated by multiple factors, such as biogenesis and mitophagy. Each mitochondrion contains about 1,200 different types of proteins, of which 13 proteins are encoded by mitochondrial genome DNA (mtDNA), and the rest are encoded by nuclear DNA (28). Crosstalk of the nuclear and mitochondrial genomes is required for mitochondrial biogenesis. In the mechanism, the biogenesis is controlled by a network of transcription factors, such as peroxisome proliferator-activated receptor γ (PPARγ), PPARα (29, 30), estrogen-related receptors (ERRs) (31), cAMP response element-binding protein (CREB), and Forkhead box transcription factor (FOXO) (32, 33). In addition, these transcription factors require peroxisome proliferator-activated receptor γ coactivator 1 alpha (PGC-1α) as a primary cofactor. They induce expression of other transcription factors, such as nuclear respiratory factor-1 (NRF-1), which in turn activates expression of mitochondrial DNA transcription factor A (TFAM) in the nucleus (34–36). TFAM is transferred into mitochondria to induce expression and duplication of mtDNA (37, 38). TFAM also requires PGC-1α in the induction of mitochondrion-related gene expression (39). NRF-2, an isoform of NRF-1, is required for expression of the cytochrome c oxidase (COX) in complex IV of the respiratory chain, in which NRF-2 interacts with PGC-1α (30). Therefore, PGC-1α is a major coactivator in the transcription network for mitochondrial biogenesis. Hormones that induce the PGC-1α activity may induce mitochondrial biogenesis through this transcription network.
Mitophagy is the process by which mitochondria are recycled. Mitochondrial components are frequently damaged by high levels of reactive oxygen species (ROS). ROS levels are increased upon active ATP production and have been shown to be coupled with heat production (40, 41). Irreversibly damaged mitochondria are removed by the process of mitophagy (42), a specific form of autophagy in the quality control system of mitochondria. Mitophagy is regulated by PTEN-induced kinase 1 (PINK1), which is on the outer mitochondrial membrane (OMM). In the damaged mitochondria, PINK1 recruits Parkin through phosphorylation. Parkin promotes mitophagy through ubiquitination of proteins on the mitochondrial membrane in the formation of autophagosomes (43). Other mitochondrial proteins, such as NIX, BNIP3, and FUNDC1, are also involved in mitophagy, and a defect in any of those molecules may contribute to impaired mitophagy. A defect in mitophagy has been reported in the pathogenesis of insulin resistance in several studies (44–46). Therefore, dysregulation of the mitochondria quality control process may lead to mitochondrial dysfunction in the pathogenesis of insulin resistance.
Energy Surplus Leads to Mitochondrial Dysfunction in Obesity
Obesity and type 2 diabetes represent the body’s compensatory responses to energy surplus conditions. In these conditions, mitochondria suffer from an oversupply of fuel substrates such as lipids, glucose, amino acids, and their derivatives. A chronic state of fuel surplus may lead to mitochondrial dysfunction and reduction in ATP production capacity (47). Mitochondrial inflexibility is a type of mitochondrial dysfunction in which substrate switch between fatty acids and glucose is disordered (26). This concept, however, is challenged by a new study (48). The signaling mechanism of mitochondrial inflexibility remains unclear (27). We propose that mitochondrial inflexibility is a mitochondrial compensatory response to fuel surplus, where signaling pathways are involved in the suppression of AMPK and Sirtuin-1 (SIRT1) and the activation of mammalian target of rapamycin (mTOR). This mitochondrial dysfunction worsens metabolic disorders by reducing energy expenditure, which leads to further accumulation of fuels in the insulin-sensitive cells (49). These mitochondrial dysfunctions are improved by lifestyle modifications such as physical exercise, calorie restriction, and weight loss, which are established strategies in the control of obesity, type 2 diabetes, and metabolic disorders (27). These practices induce activation of AMPK and SIRT1, while reducing mTOR activity at the molecular level (50). These molecular pathways provide a mechanism for the correction of mitochondrial dysfunction in the practices, suggesting that energy surplus is a major cause of mitochondrial dysfunction.
The activities of AMPK and SIRT1 are reduced in obesity and type 2 diabetes as a result of energy surplus. Energy status in cells is sensed by molecules including AMPK, SIRT1, and mTOR (51). AMPK is activated in energy-deficient states in order to restore energy supply (51, 52). AMPK activity is reduced when there is a rise in ATP or ATP/AMP ratio, which occurs during states of overfeeding, obesity, type 2 diabetes, and lack of physical exercise (51). Inhibition of AMPK leads to mitochondrial degeneration or dysfunction in the energy surplus condition (51). In addition, AMPK inhibition limits the mobilization of energy substrates, leading to an accumulation of glycogen and fatty acids in the cytoplasm (51). AMPK inhibition decreases the phosphorylation of a mitochondrial scaffold protein, a kinase anchor protein 1 (AKAP1), therefore decreasing mitochondrial ATP/heat production through suppression of the respiratory chain (53). AMPK inhibition also induces a reduction in SIRT1 activity (54). A rise in NADH/NAD+ ratio, another indicator of energy status, reduces SIRT1 activity as well. In diabetes and obesity, excess insulin, branch chain amino acids, and ATP activate mTOR. This mTOR activation induces downregulation of mitochondrial biogenesis- and autophagy-related genes (50). Taken together, alteration in AMPK, SIRT1, and mTOR activity occur in energy surplus states such as obesity and type 2 diabetes to compensate for mitochondrial dysfunction and metabolic disorders.
Metabolic Organs Targeted by Testosterone
Testosterone controls energy metabolism through the actions in several organs, including the brain (55), skeletal muscle, adipose tissue (56), liver, and pancreatic islet cells (4, 13, 57, 58). Here, the discussion focuses on the liver for the availability of transcriptomic data. The testosterone activity is mediated by AR activity, which is regulated by post-translational modifications including phosphorylation, acetylation, methylation, SUMOylation, and ubiquitination [more detail in review (59)]. Testosterone controls energy metabolism through the induction of gene transcription, which has been investigated in the liver of castrated male pigs using the RNA-seq strategy. Testosterone deficiency led to reduced gene expression in multiple metabolic pathways, such as fatty acid oxidation, steroid biosynthesis, cholesterol and bile acid metabolism, and glucose metabolism (60). Analysis conducted with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database suggested that energy output by mitochondria is reduced when fatty acid synthesis and tricarboxylic acid (TCA) cycle activity is decreased (60). Decreased mitochondrial energy output led to an upregulation of inflammatory proteins, oxidative stress, and apoptotic responses. This study suggests that testosterone deficiency causes decreased catabolism of energy substrates (glucose and fatty acids) in mitochondria during ATP and heat production. The effects of testosterone may apply to other tissues as well, such as the skeletal muscle, fat, and pancreatic islet cells (61). In testosterone-deficient patients and animals, decreased catabolism of energy substrates increased the risk of obesity. For example, castration of male rats induced expression of fatty acid synthesis-related genes, thereby leading to fat accumulation in the skeletal muscle and increased subcutaneous fat deposition in normal diet-fed rats (58). In the skeletal muscle, androgens promote myogenesis in male mice and humans by enhancing the expression of glycogen synthase, GLUT4, and insulin receptor substrate 1 (IRS1) (19). In β-cell, testosterone has a protective effect because β-cell-specific AR knockout (ARKO) male mice experience glucose intolerance and β-cell failure similar to hypogonadal men (62, 63). Together, these results illustrate the important effects of androgens on energy metabolism. However, the transcriptome of other tissues in testosterone-deficient animals and patients remains to be revealed.
Testosterone Induces Mitochondrial Biogenesis
Testosterone affects mitochondrial function in several ways, including mitochondrial structure. Mitochondria have two layers of membranes, OMM and inner mitochondrial membrane (IMM). The IMM holds the respiratory chain and maintains the mitochondrial membrane potential by pumping protons into the intermembrane space. The cristae of the IMM provide structural support to the respiratory chain. Under pathological conditions, the orderly arrangement of the tubular and lamellar mitochondrial cristae may be disrupted (64). In androgen-deficient rats, crista number and length are reduced in the cardiomyocytes, but these pathologic changes are reversed by androgen supplementation (65). Similar changes are observed in ARKO mice (66). The mechanism behind androgen’s role in the regulation of cristae has yet to be elucidated and may be related to gene expression. These studies suggest that androgen is required in the maintenance of mitochondrial structure in male animals.
Androgen stimulates mitochondrial biogenesis through activation of the AR/PGC-1α/TFAM pathway (67). PGC-α is a crucial positive regulator of mitochondrial biogenesis, and TFAM plays a vital role in the transcription and replication of mtDNA in mitochondrial biogenesis. When androgen activity is blocked by AR gene knockout, the mitochondrial number is reduced together with a fall in PGC-1α expression in the muscle of ARKO mice (68). The expression of PGC-1α and TFAM in the muscle of the castrated rats (65), mice (69), and pigs (67) was reduced, but these effects were reversed by the administration of exogenous androgen. In the cellular model, testosterone induces the expression of PGC-1α and TFAM in the C2C12 cells (68). AR may mediate the androgen signal through direct binding to the target gene promoter. AREs have been identified in the promoter of TFAM gene (67). However, TFAM seems to exhibit different activities in the muscle cells vs. adipocytes. It was reported that the inactivation of adipocyte TFAM gene protected the knockout mice from obesity and insulin resistance in the dietary obesity model (70). It remains unknown how testosterone regulates PGC-1α expression. Non-genomic pathways may play a role in the regulation, given that no ARE has been identified in PGC-1α gene so far.
Androgens increase mitochondrial content through induction of transcription and duplication of mtDNA, which encodes 13 crucial components of the respiratory chain. Those include the seven subunits (ND1–6 and ND4L) of complex I, CytB of complex III, COX1-3 subunits of complex IV, and two subunits (ATP6 and ATP8) of complex V. Mutations in mtDNA or changes in their copy number are a risk factor for mitochondrial dysfunction, excessive ROS production, and ATP production deficiency, which are often observed in the inherited metabolic diseases (71–73). Castration leads to a reduction in mtDNA copy number (by almost 38%) in the muscle of male pigs (67), suggesting that testosterone is required for the maintenance of mtDNA copy number. Emerging evidence suggests that AR acts in mitochondria to induce gene transcription. AR is found in mitochondria (74), and AREs are identified in the mitochondrial genome (75, 76). AR contains a mitochondrial localization sequence (MLS) for its translocation into mitochondria. Deletion of MLS through gene mutation abolishes AR import into mitochondria (74). Mitochondrial AR is reported in the C2C12 skeletal muscle cell line, with a function similar to the nuclear Ars (77). In prostate cancer cell lines, AR was reported to inhibit respiration chain complex activity in an overexpression study. AR overexpression in PC-3 cells decreases the activity of complex I, complex II, and complex III in the respiratory chain. Inhibition of AR activity by gene knockdown and pharmacological agents increases the complex III activity by 22% and 10%, respectively (74). These results suggest that androgens may regulate the expression and duplication of mtDNA in the control of mitochondrial biogenesis. AR may act directly in mitochondria in addition to its activity in the nucleus. Overactivation of mitochondrial AR may lead to suppression of mitochondrial respiration.
Testosterone Regulates Mitophagy
Mitophagy determines mitochondrial number and mass. Testosterone inhibits mitophagy as demonstrated by the accelerated mitophagy in the androgen-deficient mice (78). Inhibition of mitophagy was observed by the decreased expression of fusion-control proteins including OPA1 and MFN2 in the castrated rats, which was reversed by androgen supplementation (65, 69). Androgens induced expression of OPA1 in cultured C2C12 cells to promote mitochondria fusion activity (75). In contrast, androgen deficiency increases mitochondrial fission. In the mechanism, an increase in the activity of fission protein DRP1 is observed in cardiomyocytes of castrated rats, which is reversed by androgen supplementation (65). Clearance of damaged mitochondria is enhanced by elevation of LC3 II/I ratio in the castrated mice (79, 80). Digestion of recycled mitochondria requires the fusion of mitophagosome with lysosome, which is promoted by conversion of the inactivated form (LC3-I) into the activated form (LC3-II). An increase in the ratio of LC3 II/I enhances fusion of mitophagosome with lysosome in the skeletal muscle of castrated mice (79, 80), which is observed with reduction in the mitochondrial mass and OXPHOS function. These studies suggest that androgens may raise mitochondrial mass through induction of fusion and inhibition of fission in physiological conditions. In androgen-deficient conditions, this effect is gone, leading to mitochondrial mass reduction through elevated fission and mitophagy.
Testosterone Affects Mitochondrial ATP Production
Mitochondria are the “powerhouse” in eukaryotic cells to provide energy for cellular activities. ATP production through OXPHOS reactions accounts for about 90% of ATP production in cells (81). Preclinical studies revealed that decreased complex I and II activity was closely associated with decreased ATP production in the cardiomyocytes of obese male rats with insulin resistance (82). Clinical studies revealed that a deficiency of COX in complex IV was associated with decreased ATP production and induced cell apoptosis in the skeletal muscle of humans (83). Additionally, the activity of the ATP synthase β subunit was greatly reduced in the vastus lateralis muscle of obese patients (84). This evidence confirms that deficiency in mitochondrial respiration is closely associated with metabolic disorders.
Androgens are responsible for maintaining the structural integrity of the mitochondrial respiratory chain. A study in vitro showed that the AR antagonist, flutamide, decreased complex I activity, mitochondrial membrane potential, and ATP production (by almost 51.2%) in cultured hepatocytes (46, 85). This fall in mitochondrial membrane potential leads to an upregulation of permeability transition pore (PTP) openings, resulting in loss of mitochondrial content and cristae (86). In the hippocampus and substantia nigra of castrated male rats, a lack of androgens is associated with a reduction in ATP synthesis and complex I and complex III activity and a decrease in MFN2 and OPA1 levels in cardiomyocytes (87, 88). Interestingly, mitochondrial dysfunction was reversed by the administration of exogenous testosterone (65, 89). These studies suggest that androgens play a crucial role in the regulation of ATP production through an impact on the mitochondrial respiratory chain. Several mechanisms are discussed below.
Androgens may protect the respiratory chain of mitochondria by alleviating oxidative damage. At physiological levels, ROS serve as a redox messenger in the regulation of multiple cellular processes, including cell growth, differentiation, proliferation, and apoptosis (90). However, excessive ROS cause damage to several biological molecules, including DNA-repair enzymes. Damage to these enzymes consequently causes OXPHOS malfunction (91). Mitochondria are the main site of ROS production; therefore, mtDNA is more susceptible to oxidative damage than nuclear DNA (92). In the skeletal muscle of testosterone-deficient rats, ROS production by mitochondria is increased, as indicated by the increase in plasma malondialdehyde (MDA) concentration (93). ROS increases cell apoptosis in testosterone-deprived men and male rats (94), which were attenuated by exogenous testosterone supplementation (95). Inhibition of AR activity by flutamide also increased ROS (H2O2) levels and damaged mitochondria, as indicated by a drop in the mitochondrial membrane potential and ATP production in cultured hepatocytes (46). Therefore, these results suggest that androgen/AR may reduce ROS and protect the mitochondrial respiratory chain.
Androgens may also exert their effects through the regulation of cardiolipin, a phospholipid in stabilizing the mitochondrial respiratory chain and IMM structure for mitochondrial function (96). Chemical-induced reduction in testosterone was associated with a significant loss of cardiolipin in the brains of mice (97). However, the connection between cardiolipin and androgens remains to be elucidated.
It should be noted that at least partial effects of testosterone on mitochondria are exerted through its intratissue aromatization, especially in the WAT and brain, leading to the increase in estrogen (98). Aromatase, which is encoded by CYP19A1 gene, actively converts testosterone into estradiol in granulosa cells located in the ovaries (99). Estrogen has a broad impact on energy metabolism through the regulation of mitochondria.
Effects of Estrogen on Mitochondria
Estrogen is secreted by the ovaries as well as the adrenal gland in females. The active form of estrogen is 17β-estradiol (E2). Estrogen typically acts through the nuclear estrogen receptors (ERs) ERα and ERβ. Like androgens, estrogens exert their activity through ER by transcriptional control of gene expression at the estrogen response elements (EREs) on the target genes (100). ERs are activated when a ligand binds to the receptor in the cytoplasm, which is followed by nuclear translocation. ER is also activated by a G protein-coupled receptor, G protein-coupled ER (GPER), which is mainly responsible for rapid non-genomic responses (101).
Estrogen has a profound impact on glucose and lipid metabolism in females. Reduction of estrogen or its receptor is closely associated with impaired energy metabolism, which includes a reduction in lipolysis and glucose uptake and an increased risk of obesity (102). In genetic studies, the inactivation of ERs in the skeletal muscle or the whole body of female mice by ERα gene knockout increased the risk of obesity and insulin resistance (103, 104). In an epidemiology study, the administration of exogenous estrogen reduced the risk of metabolic disorders in postmenopausal women (105). In randomized clinical trials, estrogen hormone therapy greatly reduced fasting glucose, insulin resistance, and the risk of diabetes in postmenopausal women (102). A study done in female mice also confirmed the protective effects of estrogen against obesity and insulin resistance, which disappeared in ovariectomized female mice (106). These studies suggest that estrogen/ERs play a role in the regulation of energy metabolism through effects on glucose and lipid metabolism.
Mitochondria play an important role in estrogen’s effects on energy metabolism (31, 107). In preclinical and clinical studies, reduction in mitochondrial mass and impairment in mitochondrial structure and ATP production are observed in the skeletal muscle of ovariectomized animals or menopausal women (31). Those changes were reversed by the administration of exogenous estrogen. Estrogens affect mitochondria in multiple aspects including protein content and activity, phospholipid content of membranes, oxidant and antioxidant capacities, oxidative phosphorylation, and calcium retention capacities (31). Inactivation of ERα by gene deletion leads to mitochondrial dysfunction and impaired fission–fusion dynamics of mitochondria in females (103). Thus, it is obvious that estrogen has a broad impact on mitochondrial structure and function.
Although less clear than that of ERα in the regulation of energy metabolism, ERβ has also been reported to regulate mitochondrial function (108). ERβ-selective ligands prevented high fat diet-induced lipid accumulation and promoted the expression of mitochondrial biogenesis-related indicators in brown adipose tissue (BAT) and WAT in male and female mice (109, 110). The estrogen-regulated mitochondrial biogenesis markers include PGC-1α (111, 112) and NRFs. Although no ERE has been identified in the PGC-1α gene promoter, it is available in the promoter DNA of NRF-1 gene (113). Estrogen deficiency leads to decreased expression of genes involved in the mitochondrial respiratory chain, oxidative phosphorylation, and metabolic pathways of glucose and lipid in the ovariectomized rats (114–116). This reduction is observed in the transcription factors including NRF-1, TFAM, and PGC-1α in the skeletal muscle of estrogen-deficient female rats (117). These alterations are reversed by the administration of estrogen in ovariectomized rats (118, 119). In addition, estrogen regulates the expression of the COX subunit 7a-related polypeptide (COX7RP), which acts as an assembly-promoting factor for the mitochondrial respiratory chain super complex in the muscle cells (120). COX activity and mitochondrial ATP content are reduced by COX7RP gene knockdown (121). These results suggest that estrogen stimulates mitochondrial biogenesis through the promotion of NRF-1, TFAM, and PGC-1α gene expression, and assembly of the mitochondrial respiratory chain through COX7RP.
Like testosterones, estrogens may act through ERs in mitochondria. The presence of ERs in mitochondria was reported in various cell types by multiple methods, including proteomics analysis of human heart mitochondria, fluorescence probe analysis of human tumor cells, immunoprecipitation of mtDNA, and Western blotting in MCF-7 cells, as recently reviewed (122, 123). ERβ seems to be the main ER present in mitochondria. ERβ exerts several functions by increasing key regulators of mitochondrial function and respiratory chain proteins in cardiomyocytes of female mice, as well as increasing anti-apoptosis effects after pressure overload in the heart tissue of female mice (31).
Activation of ERs may trigger several signaling pathways including extracellular signal-regulated kinase 1 and 2 (ERK1/2), p38 mitogen-activated protein kinases (MAPKs), phosphoinositide 3-kinase (PI3K), c-Jun-NH2-terminal protein kinase (JNK), protein kinase B (PKB), glycogen synthase kinase 3β (GSK3β), β-catenin, calcineurin, and mTOR (124, 125). These activities are reported in the study of ischemia–reperfusion of cardiac remodulating models (126). The ERK, p38 MAPK, and PI3K/Akt signaling pathways are reported to protect mitochondria from H2O2-induced damage in C2C12 myoblasts (127). In ovariectomized mice, estrogen therapy improved mitochondrial function in the skeletal muscle by correcting membrane viscosity, bioenergetic function, respiration (complex I, III activities), and antioxidant activities (128). Taken together, estrogen plays vital roles in the regulation of energy metabolism through its positive impacts on mitochondrial biogenesis and function. The presence of ERs in mitochondria also makes the effects possible. AR and ERs share location and activities in the mitochondria and nucleus, which suggests a synergy between estrogens and androgens in the regulation of mitochondria (Figure 1).
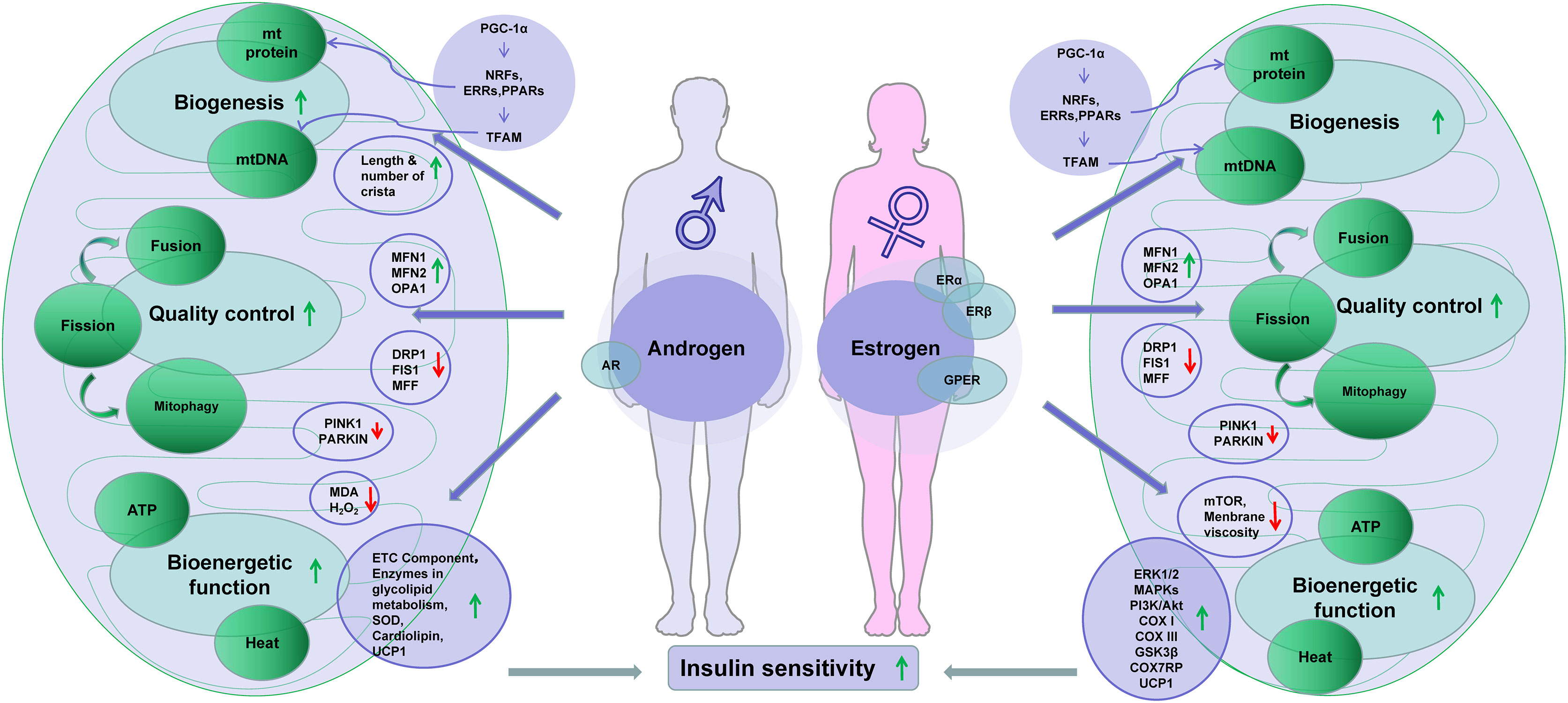
Figure 1 Sex hormones improve insulin sensitivity through regulation of mitochondria. Note: downward arrowheads stand for downregulation, while the upward arrowheads indicate upregulation. Some elements of this figure were produced using Servier Medical Art, https://smart.servier.com.
Excessive Androgen Induces Metabolic Disorders in Female
Androgen and estrogen both promote mitochondrial functions according to the discussion above. Therefore, one may predict that the two hormones may synergize in the regulation of mitochondrial function to control obesity and insulin resistance. However, the fact is opposite to the prediction. Androgen excess is a feature of PCOS in female patients, in which androgen increases the risk for insulin resistance and metabolic syndrome (129, 130). Androgen is elevated in the blood of PCOS patients, which is coupled with higher susceptibility to muscle insulin resistance and obesity (129–131). The mechanism of androgen-related insulin resistance is related to a decrease in expression of adipokines, including adiponectin and omentin-1, which are beneficial to insulin sensitivity in the mouse model of PCOS (132, 133). Other changes associated with androgen excess include an increase in visceral adiposity, reduction of BAT function, and impairment of glucose-stimulated insulin secretion by pancreatic β-cells in PCOS models (61). These factors may partly explain insulin resistance in PCOS, but the basis of those factors remains unknown. We propose that through hormone synergy, androgen may overactivate mitochondria in the presence of estrogen in the female body, leading to insulin resistance.
Androgen and Estrogen Synergy in Mitochondrial Overactivation
ATP production is a primary indicator of mitochondrial function. This function is induced by substrates, hormones, and energy demand as discussed above. In a recent review, excessive substrate availability in obesity has been proposed as a major factor contributing to mitochondrial overproduction of ATP in the mechanism of insulin resistance (134). The insulin resistance occurs following ATP surplus in multiple tissues, in which ATP production exceeds demand (134). ATP surplus in the pancreatic β-cells leads to more secretion of insulin for hyperinsulinemia. ATP surplus in the pancreatic α-cells leads to excessive secretion of glucagon for hyperglucagonemia. These hormone disorders are well-known risk factors for insulin resistance in the muscle, liver, and adipose tissues in obesity. In insulin-sensitive tissues, ATP surplus inhibits the AMPK signaling pathway and activates the mTOR signaling pathway to directly inhibit insulin sensitivity. Additionally, the fall in AMPK activity contributes to mitochondrial degeneration and dysfunction through suppression of mitophagy, which also contributes to insulin resistance. The insulin-sensitizing medicine, metformin, inhibits mitochondrial ATP production in the liver, thereby pharmacologically inducing insulin sensitization. Therefore, ATP surplus due to mitochondrial overactivation is a promising mechanism for insulin resistance. In PCOS, excessive androgen levels may synergize with estrogen to cause mitochondrial overactivation, which in turn leads to mitochondrial dysfunction through the mechanisms discussed above. This possibility is supported by mitochondrial changes in the skeletal muscle, BAT, and β-cells in PCOS models (Figure 2).
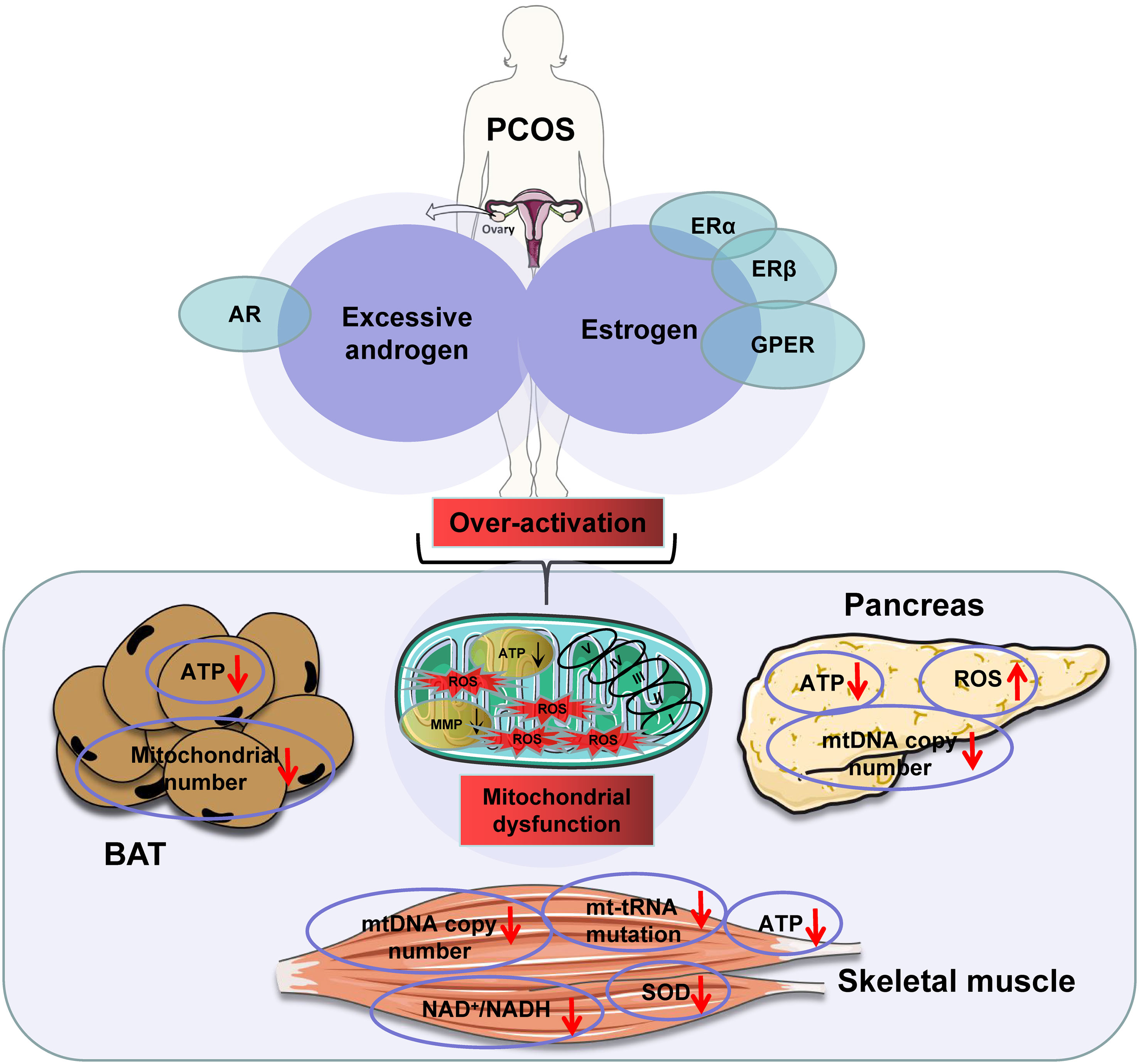
Figure 2 The excessive androgens in the presence of estrogens induce mitochondrial dysfunction in polycystic ovary syndrome (PCOS) patients. Note: downward arrowheads indicate a decrease, upward arrowheads mean an increase. Some elements of this figure were produced using Servier Medical Art, https://smart.servier.com.
Mitochondrial dysfunction is a mechanism for muscle insulin resistance in the PCOS models. Mitochondrial dysfunction is associated with insulin resistance in the skeletal muscle in PCOS (135) and in the mouse model of hyperandrogenemia induced by dehydroepiandrosterone (DHEA) injection (136). Mitochondrial dysfunction is reflected by a significant increase in the NAD+/NADH ratio and decrease in ATP contents in the PCOS mouse model (137). In patients with PCOS, mitochondrial dysfunction was reported by a reduction in superoxide dismutase levels, mtDNA copy number, mitochondrial membrane potential, and ATP levels (138). However, mitochondrial dysfunction was not observed in the study of PCOS patients using the primary myotubes (139). This dysfunction is likely the result of mitochondrial overactivation by androgens in the presence of estrogens.
The mitochondrial dysfunction is reported in BAT of PCOS models. BAT mitochondria produce heat through UCP1 (140), whose activity is induced by cold temperatures or adrenergic stimulation mimics. Interestingly, BAT mass and function are both decreased in PCOS patients with elevated circulating androgen levels (141). In cellular models, differentiation of brown adipocytes is inhibited by androgen in a dose-dependent manner, leading to decreased expression of UCP1. These changes correspond to a reduction in the mitochondrial number and “whitening” of the interscapular BAT in androgen-induced PCOS models (140). This mitochondrial dysfunction is supported by a decrease in other mitochondrial proteins including PGC-1α and Cidea (cell death-inducing DNA fragmentation factor-like effector A). In addition to direct effects on adipocytes, androgens may affect BAT through rewiring neurons in the central nervous system, which is a proposed mechanism for the central obesity seen in PCOS patients (142).
Androgen effect on β-cells supports that mitochondrion overactivation precedes mitochondrial dysfunction. In a study of the acute effect of androgen on β-cells, androgen was found to induce hypersecretion of insulin through activation of the cAMP/PKA pathway, which was followed by β-cell dysfunction in the female mice (13). In DHT-treated female rats, androgen induced mitochondrial dysfunction in pancreatic β-cells by inhibiting oxygen consumption and ATP production and reducing mtDNA copy number (143). Expression of transcription factors for mitochondrial biogenesis (TFAM, NRF-1, and PGC-1α) was all decreased in the model as well. These observations were confirmed in a later study where higher ADP/ATP ratio, decreased mtDNA copy number, increased ROS production, and downregulation of mitochondrial biogenesis were seen in the β-cells of rat models (144). The exact mechanism behind mitochondrial dysfunction remains unknown. However, overproduction of ROS by mitochondria is a promising theory, as studies have shown that DHT can induce mitochondrial ROS production by altering the balance between oxidative and anti-oxidative arms of mitochondria in PCOS rats (145). As a consequence of ROS elevation, mutations in mtDNA, decreases in mitochondrial membrane potential, and abnormal expression of the respiratory chain complexes were reported in the PCOS rats (146). ROS elevation is associated with a rise in ATP production.
Mitochondrial Dysfunction Explains Central Obesity in Polycystic Ovary Syndrome Patients
PCOS patients have a high prevalence of central obesity due to increased visceral fat. Accumulation of visceral fat raises the risk of insulin resistance in both men and women (147), especially in postmenopausal female subjects (148–150). Androgen excess is a risk factor for central obesity in females. Inhibition of androgen effects with the receptor antagonist flutamide decreases visceral fat in PCOS patients (151), suggesting a significant role of androgens in central obesity. There are several hypotheses for the effects of androgens on central obesity in literature.
The first is the failure of leptin actions. In PCOS patients, central obesity is associated with a failure of leptin signaling within the central nervous system (142). Leptin conveys the signal of the body’s energy reserves to the central nervous system in the control of food intake and the promotion of energy expenditure. Defects in leptin action may promote central obesity by increasing energy intake and decreasing energy expenditure (152). Exogenous testosterone treatment was reported to reduce serum leptin in humans (153), confirming the regulatory effects of androgen on leptin.
The second is the impairment of new adipocyte generation in the subcutaneous fat. Adipose tissue expansion involves adipogenesis and adipocyte hypertrophy (154). In PCOS patients, subcutaneous fat pads produce androgen, leading to androgen excess (147). Androgens inhibit differentiation of the mesenchymal stem cells into preadipocytes in the subcutaneous fat. This contributes to fat deposition into the visceral fat pads (147).
The third is inhibition of adipocyte lipolysis, thereby promoting hypertrophy of mature adipocytes by androgen. Androgen downregulates signaling pathway proteins important for lipolysis, such as expression of catecholamine receptor and hormone-sensitive lipase (HSL), in white adipocytes (155–157). This decreases fatty acid release from the adipocytes and causes adipocyte hypertrophy. Adipocyte hypertrophy is a risk factor for adipose tissue hypoxia, adipocyte dysfunction, adipose inflammation, and insulin resistance (158, 159).
The fourth is whitening of beige or brown adipocytes following mitochondrial dysfunction to reduce energy expenditure by thermogenesis as discussed above. These studies suggest that androgen excess may promote visceral adiposity through several mechanisms in PCOS patients. Mitochondrial dysfunction appears to be a common player in all four of these mechanisms.
Conclusion and Perspectives
Sex hormones play a key role in the regulation of energy metabolism. Androgens are required for the maintenance of energy balance in male subjects through the promotion of mitochondrial function. Estrogens have a similar activity in females in the control of energy metabolism through their effects on mitochondria. The activities of these two sex hormones overlap in mitochondria, which suggests a possibility of synergy to induce mitochondrial overactivation in PCOS. These two hormones work through their receptors to induce the expression of nuclear DNA and mitochondrial DNA to promote mitochondrial biogenesis, which lay the foundation for the synergy concept in the induction of mitochondrial function. When the synergistic effect leads to ATP overproduction in cells, it can cause insulin resistance through multiple mechanisms, such as excess secretion of insulin in β-cells and excess glucagon secretion in α-cells. In addition to the endocrine disorders, ATP surplus inhibits AMPK, activates mTOR, and induces ROS production, which contribute to insulin resistance in the skeletal muscle and liver. Mitochondrial overactivation may lead to mitochondrial dysfunction through the alterations of these signaling molecules. Mitochondrial dysfunction in β-cells impairs insulin secretion, which contributes to hyperglycemia in type 2 diabetes. Therefore, mitochondrial overactivation from androgen and estrogen synergism may be a cause of insulin resistance in PCOS.
In the molecular mechanism, the androgen and estrogen synergy may super-induce the activity of PGC-1α because they both upregulate the activity and expression of PGC-1α. Overexpression of PGC-1α is known to induce insulin resistance and impair glucose metabolism in the liver (160). Overexpression of PGC-1α also inhibits glucose-induced insulin secretion in β-cells (161). In addition, the overexpression of PGC-1α induces insulin resistance in the skeletal muscle through the expression of mammalian tribbles homolog (TRB-3) (162), a direct negative regulator of Akt activity in the insulin signaling pathway (163). In type 2 diabetes, chronic hyperglycemia also contributes to the overactivation of mitochondria through the substrate effects (164). This hormone synergy-based view is supported by existing literature on PCOS models and is perfect to fill the gap in the mechanism of metabolic disorder in the PCOS syndrome. However, the possibility remains to be verified by studies in vivo. This view represents a unifying mechanism for the distinct roles of androgens in the control of insulin sensitivity in hypogonadal men and PCOS women, which may shed light on a mitochondrion-targeted strategy for the treatment of PCOS in the future.
Author Contributions
XW and JY contributed to the conception and design of the study. LY wrote the first draft of the manuscript. ML and RW wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) with a fund (No. 31872801).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We highly appreciate the efforts of Ms. Kara Ye and Sarah Ye at the Medical College of Georgia at Augusta University (Georgia State, USA) in editing the English of this manuscript.
References
1. Torres-Estay V, Carreño DV, Fuenzalida P, Watts A, San Francisco IF, Montecinos VP, et al. Androgens Modulate Male-Derived Endothelial Cell Homeostasis Using Androgen Receptor-Dependent and Receptor-Independent Mechanisms. Angiogenesis (2017) 20(1):25–38. doi: 10.1007/s10456-016-9525-6
2. Corona G, Giagulli VA, Maseroli E, Vignozzi L, Aversa A, Zitzmann M, et al. Testosterone Supplementation and Body Composition: Results From a Meta-Analysis of Observational Studies. J Endocrinol Invest (2016) 39(9):967–81. doi: 10.1007/s40618-016-0480-2
3. Yu IC, Lin HY, Sparks JD, Yeh S, Chang C. Androgen Receptor Roles in Insulin Resistance and Obesity in Males: The Linkage of Androgen-Deprivation Therapy to Metabolic Syndrome. Diabetes (2014) 63(10):3180–8. doi: 10.2337/db13-1505
4. Chang C, Yeh S, Lee SO, Chang TM. Androgen Receptor (AR) Pathophysiological Roles in Androgen-Related Diseases in Skin, Bone/Muscle, Metabolic Syndrome and Neuron/Immune Systems: Lessons Learned From Mice Lacking AR in Specific Cells. Nucl Recept Signaling (2013) 11:e001. doi: 10.1621/nrs.11001
5. Zhang L, Cai Y, Wei S, Ling Y, Zhu L, Li D, et al. Testosterone Deficiency Induces Changes of the Transcriptomes of Visceral Adipose Tissue in Miniature Pigs Fed a High-Fat and High-Cholesterol Diet. Int J Mol Sci (2016) 17(12):2125. doi: 10.3390/ijms17122125
6. Hermoso DAM, Bizerra PFV, Constantin RP, Ishii-Iwamoto EL, Gilglioni EH. Association Between Metabolic Syndrome, Hepatic Steatosis, and Testosterone Deficiency: Evidences From Studies With Men and Rodents. Aging Male (2020) 23:1296–315. doi: 10.1080/13685538.2020.1764927
7. Carrageta DF, Oliveira PF, Alves MG, Monteiro MP. Obesity and Male Hypogonadism: Tales of a Vicious Cycle. Obes Rev: Off J Int Assoc Study Obes (2019) 20(8):1148–58. doi: 10.1111/obr.12863
8. Elliott J, Kelly SE, Millar AC, Peterson J, Chen L, Johnston A, et al. Testosterone Therapy in Hypogonadal Men: A Systematic Review and Network Meta-Analysis. BMJ Open (2017) 7(11):e015284. doi: 10.1136/bmjopen-2016-015284
9. Corona G, Maseroli E, Rastrelli G, Isidori AM, Sforza A, Mannucci E, et al. Cardiovascular Risk Associated With Testosterone-Boosting Medications: A Systematic Review and Meta-Analysis. Expert Opin Drug Saf (2014) 13(10):1327–51. doi: 10.1517/14740338.2014.950653
10. Morford JJ, Wu S, Mauvais-Jarvis F. The Impact of Androgen Actions in Neurons on Metabolic Health and Disease. Mol Cell Endocrinol (2018) 465:92–102. doi: 10.1016/j.mce.2017.09.001
11. Chynoweth J, Hosking J, Jeffery A, Pinkney J. Contrasting Impact of Androgens on Male and Female Adiposity, Fat Distribution and Insulin Resistance in Childhood and Adolescence (EarlyBird 75). Pediatr Obes (2020) 15:e12685. doi: 10.1111/ijpo.12685
12. Rubin KH, Glintborg D, Nybo M, Abrahamsen B, Andersen M. Development and Risk Factors of Type 2 Diabetes in a Nationwide Population of Women With Polycystic Ovary Syndrome. J Clin Endocrinol Metab (2017) 102(10):3848–57. doi: 10.1210/jc.2017-01354
13. Navarro G, Allard C, Morford Jamie J, Xu W, Liu S, Molinas AJ, et al. Androgen Excess in Pancreatic β Cells and Neurons Predisposes Female Mice to Type 2 Diabetes. JCI Insight (2018) 3(12):1–17. doi: 10.1172/jci.insight.98607
14. Pal M, Khan J, Kumar R, Surolia A, Gupta S. Testosterone Supplementation Improves Insulin Responsiveness in HFD Fed Male T2DM Mice and Potentiates Insulin Signaling in the Skeletal Muscle and C2C12 Myocyte Cell Line. PloS One (2019) 14(11):e0224162. doi: 10.1371/journal.pone.0224162
15. Lee OD, Tillman K. An Overview of Testosterone Therapy. Am J Men’s Health (2016) 10(1):68–72. doi: 10.1177/1557988314556671
16. Corona G, Giagulli VA, Maseroli E, Vignozzi L, Aversa A, Zitzmann M, et al. Therapy Of Endocrine Disease: Testosterone Supplementation and Body Composition: Results From a Meta-Analysis Study. Eur J Endocrinol (2016) 174(3):R99–116. doi: 10.1530/EJE-15-0262
17. Cameron JL, Jain R, Rais M, White AE, Beer TM, Kievit P, et al. Perpetuating Effects of Androgen Deficiency on Insulin Resistance. Int J Obes (2016) 40(12):1856–63. doi: 10.1038/ijo.2016.148
18. Diamanti-Kandarakis E, Pappalou O, Kandaraki EA. The Role of Androgen Excess on Insulin Sensitivity in Women. Front Horm Res (2019) 53:50–64. doi: 10.1159/000494902
19. Bianchi VE, Locatelli V. Testosterone a Key Factor in Gender Related Metabolic Syndrome. Obes Rev (2018) 19(4):557–75. doi: 10.1111/obr.12633
20. Ventura-Clapier R, Moulin M, Piquereau J, Lemaire C, Mericskay M, Veksler V, et al. Mitochondria: A Central Target for Sex Differences in Pathologies. Clin Sci (2017) 131(9):803–22. doi: 10.1042/CS20160485
21. Spinelli JB, Haigis MC. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat Cell Biol (2018) 20(7):745–54. doi: 10.1038/s41556-018-0124-1
22. Roesler A, Kazak L. UCP1-Independent Thermogenesis. Biochem J (2020) 477(3):709–25. doi: 10.1042/BCJ20190463
23. Chouchani ET, Kazak L, Spiegelman BM. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab (2019) 29(1):27–37. doi: 10.1016/j.cmet.2018.11.002
24. Chang SH, Song NJ, Choi JH, Yun UJ, Park KW, et al. Mechanisms Underlying UCP1 Dependent and Independent Adipocyte Thermogenesis. Obes Rev (2019) 20(2):241–51. doi: 10.1111/obr.12796
25. Bertholet AM, Chouchani ET, Kazak L, Angelin A, Fedorenko A, Long JZ, et al. H Transport Is an Integral Function of the Mitochondrial ADP/ATP Carrier. Nature (2019) 571(7766):515–20. doi: 10.1038/s41586-019-1400-3
26. Muoio DM. Metabolic Inflexibility: When Mitochondrial Indecision Leads to Metabolic Gridlock. Cell (2014) 159(6):1253–62. doi: 10.1016/j.cell.2014.11.034
27. Hesselink MK, Schrauwen-Hinderling V, Schrauwen P. Skeletal Muscle Mitochondria as a Target to Prevent or Treat Type 2 Diabetes Mellitus. Nat Rev Endocrinol (2016) 12(11):633–45. doi: 10.1038/nrendo.2016.104
28. Calvo SE, Mootha VK. The Mitochondrial Proteome and Human Disease. Annu Rev Genomics Hum Genet (2010) 11:25–44. doi: 10.1146/annurev-genom-082509-141720
29. Dominy JE, Puigserver P. Mitochondrial Biogenesis Through Activation of Nuclear Signaling Proteins. Cold Spring Harb Perspect Biol (2013) 5(7):341–50. doi: 10.1101/cshperspect.a015008
30. Scarpulla RC. Metabolic Control of Mitochondrial Biogenesis Through the PGC-1 Family Regulatory Network. Biochim Biophys Acta (2011) 1813(7):1269–78. doi: 10.1016/j.bbamcr.2010.09.019
31. Ventura-Clapier R, Piquereau J, Veksler V, Garnier A. Estrogens, Estrogen Receptors Effects on Cardiac and Skeletal Muscle Mitochondria. Front Endocrinol (2019) 10:557. doi: 10.3389/fendo.2019.00557
32. Kim S, Koh H. Role of FOXO Transcription Factors in Crosstalk Between Mitochondria and the Nucleus. J Bioenerg Biomembr (2017) 49(4):335–41. doi: 10.1007/s10863-017-9705-0
33. Calissi G, Lam EW, Link W. Therapeutic Strategies Targeting FOXO Transcription Factors. Nat Rev Drug Discovery (2020) 20:21–38. doi: 10.1038/s41573-020-0088-2
34. de Oliveira Bristot VJ, de Bem Alves AC, Cardoso LR, da Luz Scheffer D, Aguiar AS Jr. The Role of PGC-1α/UCP2 Signaling in the Beneficial Effects of Physical Exercise on the Brain. Front Neurosci (2019) 13:292. doi: 10.3389/fnins.2019.00292
35. Gureev AP, Shaforostova EA, Popov VN. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front Genet (2019) 10:435. doi: 10.3389/fgene.2019.00435
36. Zhao Z, Pu Y. Lixisenatide Enhances Mitochondrial Biogenesis and Function Through Regulating the CREB/PGC-1α Pathway. Biochem Biophys Res Commun (2019) 508(4):1120–5. doi: 10.1016/j.bbrc.2018.11.135
37. Taherzadeh-Fard E, Saft C, Akkad DA, et al. PGC-1alpha Downstream Transcription Factors NRF-1 and TFAM Are Genetic Modifiers of Huntington Disease. Mol Neurodegeneration (2011) 6(1):32. doi: 10.1186/1750-1326-6-32
38. Cameron RB, Beeson CC, Schnellmann RG. Development of Therapeutics That Induce Mitochondrial Biogenesis for the Treatment of Acute and Chronic Degenerative Diseases. J Med Chem (2016) 59(23):10411–34. doi: 10.1021/acs.jmedchem.6b00669
39. Theilen NT, Kunkel GH, Tyagi SC. The Role of Exercise and TFAM in Preventing Skeletal Muscle Atrophy. J Cell Physiol (2017) 232(9):2348–58. doi: 10.1002/jcp.25737
40. Qiao J, Chen C, Shangguan D, Mu X, Wang S, Jiang L, et al. Simultaneous Monitoring of Mitochondrial Temperature and ATP Fluctuation Using Fluorescent Probes in Living Cells. Anal Chem (2018) 90(21):12553–8. doi: 10.1021/acs.analchem.8b02496
41. Okabe K, Inada N, Gota C, Harada Y, Funatsu T, Uchiyama S. Intracellular Temperature Mapping With a Fluorescent Polymeric Thermometer and Fluorescence Lifetime Imaging Microscopy. Nat Commun (2012) 3:705. doi: 10.1038/ncomms1714
42. Youle RJ, Narendra DP. Mechanisms of Mitophagy. Nat Rev Mol Cell Biol (2011) 12(1):9–14. doi: 10.1038/nrm3028
43. Leduc-Gaudet JP, Hussain SNA, Barreiro E, Gouspillou G. Mitochondrial Dynamics and Mitophagy in Skeletal Muscle Health and Aging. Int J Mol Sci (2021) 22(15):1–19. doi: 10.3390/ijms22158179
44. Yu R, Lendahl U, Nistér M, Zhao J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front Endocrinol (2020) 11:374. doi: 10.3389/fendo.2020.00374
45. Montgomery MK, Turner N. Mitochondrial Dysfunction and Insulin Resistance: An Update. Endocr Connect (2015) 4(1):R1–R15. doi: 10.1530/EC-14-0092
46. Zhang L, Guo J, Zhang Q, Zhou W, Li J, Yin J, et al. Flutamide Induces Hepatic Cell Death and Mitochondrial Dysfunction via Inhibition of Nrf2-Mediated Heme Oxygenase-1. Oxid Med Cell Longev (2018) 2018:8017073. doi: 10.1155/2018/8017073
47. Lowell Bradford B, Shulman Gerald I. Mitochondrial Dysfunction and Type 2 Diabetes. Science (2005) 307(5708):384–7. doi: 10.1126/science.1104343
48. Song JD, Alves TC, Befroy DE, Perry RJ, Mason GF, Zhang XM, et al. Dissociation of Muscle Insulin Resistance From Alterations in Mitochondrial Substrate Preference. Cell Metab (2020) 32(5):726–735 e725. doi: 10.1016/j.cmet.2020.09.008
49. Tsilingiris D, Tzeravini E, Koliaki C, Dalamaga M, Kokkinos A. The Role of Mitochondrial Adaptation and Metabolic Flexibility in the Pathophysiology of Obesity and Insulin Resistance: An Updated Overview. Curr Obes Rep (2021) 10(3):191–213. doi: 10.1007/s13679-021-00434-0
50. Huwatibieke B, Yin WZ, Liu LC, Jin Y, Xiang X, Han J, et al. Mammalian Target of Rapamycin Signaling Pathway Regulates Mitochondrial Quality Control of Brown Adipocytes in Mice. Front Physiol (2021) 12:638352. doi: 10.3389/fphys.2021.638352
51. Herzig S, Shaw RJ. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat Rev Mol Cell Biol (2018) 19(2):121–35. doi: 10.1038/nrm.2017.95
52. González A, Hall MN, Lin SC, Hardie DG. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab (2020) 31(3):472–92. doi: 10.1016/j.cmet.2020.01.015
53. Hoffman NJ, Parker BL, Chaudhuri R, Fisher-Wellman KH, Kleinert M, Humphrey SJ, et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab (2015) 22(5):922–35. doi: 10.1016/j.cmet.2015.09.001
54. Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK Regulates Energy Expenditure by Modulating NAD+ Metabolism and SIRT1 Activity. Nature (2009) 458(7241):1056–60. doi: 10.1038/nature07813
55. Yu IC, Lin HY, Liu NC, Sparks JD, Yeh S, Fang LY, et al. Neuronal Androgen Receptor Regulates Insulin Sensitivity via Suppression of Hypothalamic NF-κb-Mediated PTP1B Expression. Diabetes (2013) 62(2):411–23. doi: 10.2337/db12-0135
56. Ghanim H, Dhindsa S, Batra M, Green K, Abuaysheh S, Kuhadiya ND, et al. Testosterone Increases the Expression and Phosphorylation of AMP Kinase α in Men With Hypogonadism and Type 2 Diabetes. J Clin Endocrinol Metab (2020) 105(4):1169–75. doi: 10.1210/clinem/dgz288
57. McInnes KJ, Smith LB, Hunger NI, Saunders PT, Andrew R, Walker BR. Deletion of the Androgen Receptor in Adipose Tissue in Male Mice Elevates Retinol Binding Protein 4 and Reveals Independent Effects on Visceral Fat Mass and on Glucose Homeostasis. Diabetes (2012) 61(5):1072–81. doi: 10.2337/db11-1136
58. Baik M, Jeong JY, Park SJ, Yoo SP, Lee JO, Lee JS, et al. Testosterone Deficiency Caused by Castration Increases Adiposity in Male Rats in a Tissue-Specific and Diet-Dependent Manner. Genes Nutr (2020) 15(1):14. doi: 10.1186/s12263-020-00673-1
59. Gioeli D, Paschal BM. Post-Translational Modification of the Androgen Receptor. Mol Cell Endocrinol (2012) 352(1-2):70–8. doi: 10.1016/j.mce.2011.07.004
60. Cai ZW, Jiang XL, Pan YM, Chen L, Zhang L, Zhu K, et al. Transcriptomic Analysis of Hepatic Responses to Testosterone Deficiency in Miniature Pigs Fed a High-Cholesterol Diet. BMC Genomics (2015) 16:59. doi: 10.1186/s12864-015-1283-0
61. Navarro G, Allard C, Xu WW, Mauvais-Jarvis F. The Role of Androgens in Metabolism, Obesity, and Diabetes in Males and Females. Obesity (2015) 23(4):713–9. doi: 10.1002/oby.21033
62. Navarro G, Xu WW, Jacobson DA, Wicksteed B, Allard C, Zhang G, et al. Extranuclear Actions of the Androgen Receptor Enhance Glucose-Stimulated Insulin Secretion in the Male. Cell Metab (2016) 23(5):837–51. doi: 10.1016/j.cmet.2016.03.015
63. Yin LJ, Wang XH. Research Advance in the Effects of Androgen and its Receptor on the Development of Obesity, Obesity-Related Diseases and Disorders of Glucose and Lipid Metabolism. Sheng Li Xue Bao: Acta Physiol Sin (2018) 70(03):319–28.
64. Hu C, Shu L, Huang XS, Yu J, Li L, Gong L, et al. OPA1 and MICOS Regulate Mitochondrial Crista Dynamics and Formation. Cell Death Dis (2020) 11(10):940. doi: 10.1038/s41419-020-03152-y
65. Wang F, Yang J, Sun J, Dong Y, Zhao H, Shi H, et al. Testosterone Replacement Attenuates Mitochondrial Damage in a Rat Model of Myocardial Infarction. J Endocrinol (2015) 225(2):101–11. doi: 10.1530/JOE-14-0638
66. Ikeda Y, Aihara K, Akaike M, Sato T, Ishikawa K, Ise T, et al. Androgen Receptor Counteracts Doxorubicin-Induced Cardiotoxicity in Male Mice. Mol Endocrinol (2010) 24(7):1338–48. doi: 10.1210/me.2009-0402
67. Liu C, Ma JD, Zhang JW, Zhao H, Zhu Y, Qi J, et al. Testosterone Deficiency Caused by Castration Modulates Mitochondria Biogenesis Through the AR/PGC1 Alpha/TFAM Pathway. Front Genet (2019) 10:505. doi: 10.3389/fgene.2019.00505
68. Usui T, Kajita K, Kajita T, Mori I, Hanamoto T, Ikeda T, et al. Elevated Mitochondrial Biogenesis in Skeletal Muscle Is Associated With Testosterone-Induced Body Weight Loss in Male Mice. FEBS Lett (2014) 588(10):1935–41. doi: 10.1016/j.febslet.2014.03.051
69. Rossetti ML, Esser KA, Lee C, Tomko RJ Jr, Eroshkin AM, Gordon BS. Disruptions to the Limb Muscle Core Molecular Clock Coincide With Changes in Mitochondrial Quality Control Following Androgen Depletion. Am J Physiol Endocrinol Metab (2019) 317(4):E631–45. doi: 10.1152/ajpendo.00177.2019
70. Masand R, Paulo E, Wu D, Wang Y, Swaney DL, Jimenez-Morales D, et al. Proteome Imbalance of Mitochondrial Electron Transport Chain in Brown Adipocytes Leads to Metabolic Benefits. Cell Metab (2018) 27(3):616–629.e614. doi: 10.1016/j.cmet.2018.01.018
71. Kwak SH, Park KS. Role of Mitochondrial DNA Variation in the Pathogenesis of Diabetes Mellitus. Front Biosci Landmrk (2016) 21:1151–67. doi: 10.2741/4447
72. Hahn A, Zuryn S. Mitochondrial Genome (mtDNA) Mutations That Generate Reactive Oxygen Species. Antioxidants-Basel (2019) 8(9):1–19. doi: 10.3390/antiox8090392
73. Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, et al. Impaired Mitochondrial Biogenesis Contributes to Mitochondrial Dysfunction in Alzheimer’s Disease. J Neurochem (2012) 120(3):419–29. doi: 10.1111/j.1471-4159.2011.07581.x
74. Bajpai P, Koc E, Sonpavde G, Singh R, Singh KK. Mitochondrial Localization, Import, and Mitochondrial Function of the Androgen Receptor. J Biol Chem (2019) 294(16):6621–34. doi: 10.1074/jbc.RA118.006727
75. Pronsato L, Milanesi L, Vasconsuelo A. Testosterone Induces Up-Regulation of Mitochondrial Gene Expression in Murine C2C12 Skeletal Muscle Cells Accompanied by an Increase of Nuclear Respiratory Factor-1 and Its Downstream Effectors. Mol Cell Endocrinol (2020) 500:110631. doi: 10.1016/j.mce.2019.110631
76. Vasconsuelo A, Milanesi L, Boland R. Actions of 17beta-Estradiol and Testosterone in the Mitochondria and Their Implications in Aging. Ageing Res Rev (2013) 12(4):907–17. doi: 10.1016/j.arr.2013.09.001
77. Pronsato L, Boland R, Milanesi L. Non-Classical Localization of Androgen Receptor in the C2C12 Skeletal Muscle Cell Line. Arch Biochem Biophys (2013) 530(1):13–22. doi: 10.1016/j.abb.2012.12.011
78. Rossetti ML, Gordon BS. The Role of Androgens in the Regulation of Muscle Oxidative Capacity Following Aerobic Exercise Training. Appl Physiol Nutr Me (2017) 42(9):1001–7. doi: 10.1139/apnm-2017-0230
79. Rossetti ML, Steiner JL, Gordon BS. Increased Mitochondrial Turnover in the Skeletal Muscle of Fasted, Castrated Mice Is Related to the Magnitude of Autophagy Activation and Muscle Atrophy. Mol Cell Endocrinol (2018) 473:178–85. doi: 10.1016/j.mce.2018.01.017
80. Steiner JL, Fukuda DH, Rossetti ML, Hoffman JR, Gordon BS. Castration Alters Protein Balance After High-Frequency Muscle Contraction. J Appl Physiol (Bethesda Md: 1985) (2017) 122(2):264–72. doi: 10.1152/japplphysiol.00740.2016
81. Prasun P. Mitochondrial Dysfunction in Metabolic Syndrome. Bba Mol Basis Dis (2020) 1866:165838. doi: 10.1016/j.bbadis.2020.165838
82. Apaiajai N, Chunchai T, Jaiwongkam T, Kerdphoo S, Chattipakorn SC, Chattipakorn N. Testosterone Deprivation Aggravates Left-Ventricular Dysfunction in Male Obese Insulin-Resistant Rats via Impairing Cardiac Mitochondrial Function and Dynamics Proteins. Gerontology (2018) 64(4):333–43. doi: 10.1159/000487188
83. Hatakeyama H, Goto Y. Respiratory Chain Complex Disorganization Impairs Mitochondrial and Cellular Integrity Phenotypic Variation in Cytochrome C Oxidase Deficiency. Am J Pathol (2017) 187(1):110–21. doi: 10.1016/j.ajpath.2016.09.003
84. Tran L, Langlais PR, Hoffman N, Roust L, Katsanos CS. Mitochondrial ATP Synthase Beta-Subunit Production Rate and ATP Synthase Specific Activity Are Reduced in Skeletal Muscle of Humans With Obesity. Exp Physiol (2019) 104(1):126–35. doi: 10.1113/EP087278
85. Al Maruf A, O’Brien P. Flutamide-Induced Cytotoxicity and Oxidative Stress in an In Vitro Rat Hepatocyte System. Oxid Med Cell Longev (2014) 2014:398285. doi: 10.1155/2014/398285
86. Morciano G, Bonora M, Campo G, Aquila G, Rizzo P, Giorgi C, et al. Mechanistic Role of mPTP in Ischemia-Reperfusion Injury. Adv Exp Med Biol (2017) 982:169–89. doi: 10.1007/978-3-319-55330-6_9
87. Hioki T, Suzuki S, Morimoto M, Masaki T, Tozawa R, Morita S, et al. Brain Testosterone Deficiency Leads to Down-Regulation of Mitochondrial Gene Expression in Rat Hippocampus Accompanied by a Decline in Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α Expression. J Mol Neurosci: MN (2014) 52(4):531–7. doi: 10.1007/s12031-013-0108-3
88. Zhang TY, Wang Y, Kang YX, Wang L, Zhao H, Ji X, et al. Testosterone Enhances Mitochondrial Complex V Function in the Substantia Nigra of Aged Male Rats. Aging-Us (2020) 12(11):10398–414. doi: 10.18632/aging.103265
89. Yan WS, Kang YX, Ji XM, Li S, Li Y, Zhang G, et al. Testosterone Upregulates the Expression of Mitochondrial ND1 and ND4 and Alleviates the Oxidative Damage to the Nigrostriatal Dopaminergic System in Orchiectomized Rats. Oxid Med Cell Longev (2017) 2017:1202459. doi: 10.1155/2017/1202459
90. Tauffenberger A, Magistretti PJ. Reactive Oxygen Species: Beyond Their Reactive Behavior. Neurochem Res (2021) 46(1):77–87. doi: 10.1007/s11064-020-03208-7
91. Rottenberg H, Hoek JB. The Mitochondrial Permeability Transition: Nexus of Aging, Disease and Longevity. Cells-Basel (2021) 10(1):1–23. doi: 10.3390/cells10010079
92. Mustafa MF, Fakurazi S, Abdullah MA, Maniam S. Pathogenic Mitochondria DNA Mutations: Current Detection Tools and Interventions. Genes-Basel (2020) 11(2):1–13. doi: 10.3390/genes11020192
93. Rovira-Llopis S, Banuls C, de Maranon AM, Diaz-Morales N, Jover A, Garzon S, et al. Low Testosterone Levels Are Related to Oxidative Stress, Mitochondrial Dysfunction and Altered Subclinical Atherosclerotic Markers in Type 2 Diabetic Male Patients. Free Radical Bio Med (2017) 108:155–62. doi: 10.1016/j.freeradbiomed.2017.03.029
94. Pongkan W, Pintana H, Sivasinprasasn S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Testosterone Deprivation Accelerates Cardiac Dysfunction in Obese Male Rats. J Endocrinol (2016) 229(3):209–20. doi: 10.1530/JOE-16-0002
95. Pintana H, Pongkan W, Pratchayasakul W, Chattipakorn N, Chattipakorn SC. Testosterone Replacement Attenuates Cognitive Decline in Testosterone-Deprived Lean Rats, But Not in Obese Rats, by Mitigating Brain Oxidative Stress. Age (Dordrecht Netherlands) (2015) 37(5):84. doi: 10.1007/s11357-015-9827-4
96. Acaz-Fonseca E, Ortiz-Rodriguez A, Garcia-Segura LM, Astiz M. Sex Differences and Gonadal Hormone Regulation of Brain Cardiolipin, A Key Mitochondrial Phospholipid. J Neuroendocrinol (2019) 32:e12774. doi: 10.1111/jne.12774.
97. Acaz-Fonseca E, Ortiz-Rodriguez A, Lopez-Rodriguez AB, Garcia-Segura LM, Astiz M. Developmental Sex Differences in the Metabolism of Cardiolipin in Mouse Cerebral Cortex Mitochondria. Sci Rep-Uk (2017) 7:43878. doi: 10.1038/srep43878
98. Stocco C. Tissue Physiology and Pathology of Aromatase. Steroids (2012) 77(1-2):27–35. doi: 10.1016/j.steroids.2011.10.013
99. Hammes SR, Levin ER. Impact of Estrogens in Males and Androgens in Females. J Clin Invest (2019) 129(5):1818–26. doi: 10.1172/JCI125755
100. Mauvais-Jarvis F, Clegg DJ, Hevener AL. The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocr Rev (2013) 34(3):309–38. doi: 10.1210/er.2012-1055
101. Zimmerman MA, Budish RA, Kashyap S, Lindsey SH. GPER - Novel Membrane Oestrogen Receptor. Clin Sci (2016) 130(12):1005–16. doi: 10.1042/CS20160114
102. Mauvais-Jarvis F, Manson JE, Stevenson JC, Fonseca VA. Menopausal Hormone Therapy and Type 2 Diabetes Prevention: Evidence, Mechanisms, and Clinical Implications. Endocr Rev (2017) 38(3):173–88. doi: 10.1210/er.2016-1146
103. Ribas V, Drew BG, Zhou Z, Phun J, Kalajian NY, Soleymani T, et al. Skeletal Muscle Action of Estrogen Receptor α Is Critical for the Maintenance of Mitochondrial Function and Metabolic Homeostasis in Females. Sci Trans Med (2016) 8(334):334ra54. doi: 10.1126/scitranslmed.aad3815
104. Ribas V, Nguyen MT, Henstridge DC, Nguyen AK, Beaven SW, Watt MJ, et al. Impaired Oxidative Metabolism and Inflammation Are Associated With Insulin Resistance in ERalpha-Deficient Mice. Am J Physiol Endocrinol Metab (2010) 298(2):E304–319. doi: 10.1152/ajpendo.00504.2009
105. Pentti K, Tuppurainen MT, Honkanen R, Núñez NP, et al. Hormone Therapy Protects From Diabetes: The Kuopio Osteoporosis Risk Factor and Prevention Study. Eur J Endocrinol (2009) 160(6):979–83. doi: 10.1530/EJE-09-0151
106. Stubbins RE, Holcomb VB, Hong J, Zhou Z. Estrogen Modulates Abdominal Adiposity and Protects Female Mice From Obesity and Impaired Glucose Tolerance. Eur J Nutr (2012) 51(7):861–70. doi: 10.1007/s00394-011-0266-4
107. Hevener AL, Ribas V, Moore TM, Zhou Z. The Impact of Skeletal Muscle Erα on Mitochondrial Function and Metabolic Health. Endocrinology (2020) 161(2):1–16. doi: 10.1210/endocr/bqz017
108. Simpkins JW, Yang SH, Sarkar SN, Pearce V. Estrogen Actions on Mitochondria–Physiological and Pathological Implications. Mol Cell Endocrinol (2008) 290:51–9. doi: 10.1016/j.mce.2008.04.013
109. Gonzalez-Granillo M, Savva C, Li XD, Fitch M, Pedrelli M, Hellerstein M, et al. ER Beta Activation in Obesity Improves Whole Body Metabolism via Adipose Tissue Function and Enhanced Mitochondria Biogenesis. Mol Cell Endocrinol (2019) 479:147–58. doi: 10.1016/j.mce.2018.10.007
110. Ponnusamy S, Tran QT, Harvey I, Smallwood HS, Thiyagarajan T, Banerjee S, et al. Pharmacologic Activation of Estrogen Receptor Beta Increases Mitochondrial Function, Energy Expenditure, and Brown Adipose Tissue. FASEB J (2017) 31(1):266–81. doi: 10.1096/fj.201600787rr
111. Witt H, Schubert C, Jaekel J, Fliegner D, Penkalla A, Tiemann K, et al. Sex-Specific Pathways in Early Cardiac Response to Pressure Overload in Mice. J Mol Med (Berlin Germany) (2008) 86(9):1013–24. doi: 10.1007/s00109-008-0385-4
112. Hsieh YC, Yang S, Choudhry MA, Yu HP, Rue LW 3rd, Bland KI, et al. PGC-1 Upregulation via Estrogen Receptors: A Common Mechanism of Salutary Effects of Estrogen and Flutamide on Heart Function After Trauma-Hemorrhage. Am J Physiol Heart Circulatory Physiol (2005) 289(6):H2665–2672. doi: 10.1152/ajpheart.00682.2005
113. Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM. Estradiol Stimulates Transcription of Nuclear Respiratory Factor-1 and Increases Mitochondrial Biogenesis. Mol Endocrinol (Baltimore Md) (2008) 22(3):609–22. doi: 10.1210/me.2007-0029
114. Lancaster TS, Jefferson SJ, Hunter JC, Lopez V, Van Eyk JE, Lakatta EG, et al. Quantitative Proteomic Analysis Reveals Novel Mitochondrial Targets of Estrogen Deficiency in the Aged Female Rat Heart. Physiol Genomics (2012) 44(20):957–69. doi: 10.1152/physiolgenomics.00184.2011
115. Meng Z, Jing H, Gan L, Li H, Luo B. Resveratrol Attenuated Estrogen-Deficient-Induced Cardiac Dysfunction: Role of AMPK, SIRT1, and Mitochondrial Function. Am J Trans Res (2016) 8(6):2641–9.
116. Sbert-Roig M, Bauzá-Thorbrügge M, Galmés-Pascual BM, Capllonch-Amer G, García-Palmer FJ, Lladó I, et al. GPER Mediates the Effects of 17β-Estradiol in Cardiac Mitochondrial Biogenesis and Function. Mol Cell Endocrinol (2016) 420:116–24. doi: 10.1016/j.mce.2015.11.027
117. Capllonch-Amer G, Sbert-Roig M, Galmés-Pascual BM, Proenza AM, Lladó I, Gianotti M, et al. Estradiol Stimulates Mitochondrial Biogenesis and Adiponectin Expression in Skeletal Muscle. J Endocrinol (2014) 221(3):391–403. doi: 10.1530/JOE-14-0008
118. Chen Y, Zhang Z, Hu F, Yang W, Yuan J, Cui J, et al. 17β-Estradiol Prevents Cardiac Diastolic Dysfunction by Stimulating Mitochondrial Function: A Preclinical Study in a Mouse Model of a Human Hypertrophic Cardiomyopathy Mutation. J Steroid Biochem Mol Biol (2015) 147:92–102. doi: 10.1016/j.jsbmb.2014.12.011
119. Torres MJ, Ryan TE, Lin CT, Zeczycki TN, Neufer PD. Impact of 17β-Estradiol on Complex I Kinetics and HO Production in Liver and Skeletal Muscle Mitochondria. J Biol Chem (2018) 293(43):16889–98. doi: 10.1074/jbc.RA118.005148
120. Ikeda K, Shiba S, Horie-Inoue K, Shimokata K, Inoue S. A Stabilizing Factor for Mitochondrial Respiratory Supercomplex Assembly Regulates Energy Metabolism in Muscle. Nat Commun (2013) 4:2147. doi: 10.1038/ncomms3147
121. Ikeda K, Horie-Inoue K, Suzuki T, Hobo R, Nakasato N, Takeda S, et al. Mitochondrial Supercomplex Assembly Promotes Breast and Endometrial Tumorigenesis by Metabolic Alterations and Enhanced Hypoxia Tolerance. Nat Commun (2019) 10(1):4108. doi: 10.1038/s41467-019-12124-6
122. Kobayashi A, Azuma K, Ikeda K, Inoue S. Mechanisms Underlying the Regulation of Mitochondrial Respiratory Chain Complexes by Nuclear Steroid Receptors. Int J Mol Sci (2020) 21(18):1–17. doi: 10.3390/ijms21186683
123. Klinge CM. Estrogens Regulate Life and Death in Mitochondria. J Bioenerg Biomembr (2017) 49(4):307–24. doi: 10.1007/s10863-017-9704-1
124. Regitz-Zagrosek V, Kararigas G. Mechanistic Pathways of Sex Differences in Cardiovascular Disease. Physiol Rev (2017) 97(1):1–37. doi: 10.1152/physrev.00021.2015
125. Ueda K, Karas RH. Emerging Evidence of the Importance of Rapid, Non-Nuclear Estrogen Receptor Signaling in the Cardiovascular System. Steroids (2013) 78(6):589–96. doi: 10.1016/j.steroids.2012.12.006
126. Bopassa JC, Eghbali M, Toro L, Stefani E. A Novel Estrogen Receptor GPER Inhibits Mitochondria Permeability Transition Pore Opening and Protects the Heart Against Ischemia-Reperfusion Injury. Am J Physiol Heart Circulatory Physiol (2010) 298(1):H16–23. doi: 10.1152/ajpheart.00588.2009
127. Boland R, Vasconsuelo A, Milanesi L, Ronda AC, de Boland AR. 17beta-Estradiol Signaling in Skeletal Muscle Cells and Its Relationship to Apoptosis. Steroids (2008) 73:859–63. doi: 10.1016/j.steroids.2007.12.027
128. Torres MJ, Kew KA, Ryan TE, Pennington ER, Lin CT, Buddo KA. 17β-Estradiol Directly Lowers Mitochondrial Membrane Microviscosity and Improves Bioenergetic Function in Skeletal Muscle. Cell Metab (2018) 27(1):167–179.e167. doi: 10.1016/j.cmet.2017.10.003.
129. Azziz R, Carmina E, Chen Z, Dunaif A, Laven JS, Legro RS. Polycystic Ovary Syndrome. Nat Rev Dis Primers (2016) 2:16057. doi: 10.1038/nrdp.2016.57
130. Ollila MM, Piltonen T, Puukka K, Ruokonen A, Järvelin MR, Tapanainen JS, et al. Weight Gain and Dyslipidemia in Early Adulthood Associate With Polycystic Ovary Syndrome: Prospective Cohort Study. J Clin Endocrinol Metab (2016) 101(2):739–47. doi: 10.1210/jc.2015-3543
131. O’Reilly MW, Taylor AE, Crabtree NJ, Hughes BA, Capper F, Crowley RK, et al. Hyperandrogenemia Predicts Metabolic Phenotype in Polycystic Ovary Syndrome: The Utility of Serum Androstenedione. J Clin Endocrinol Metab (2014) 99(3):1027–36. doi: 10.1210/jc.2013-3399
132. Benrick A, Chanclon B, Micallef P, Wu Y, Hadi L, Shelton JM, et al. Adiponectin Protects Against Development of Metabolic Disturbances in a PCOS Mouse Model. Proc Natl Acad Sci USA (2017) 114(34):E7187–96. doi: 10.1073/pnas.1708854114
133. Ozgen IT, Oruclu S, Selek S, Kutlu E, Guzel G, Cesur Y. Omentin-1 Level in Adolescents With Polycystic Ovarian Syndrome. Pediatr Int (2019) 61(2):147–51. doi: 10.1111/ped.13761
134. Ye JP. Mechanism of Insulin Resistance in Obesity: A Role of ATP. Front Med-Prc (2021) 15(3):372–82. doi: 10.1007/s11684-021-0862-5
135. Dantas WS, Gualano B, Rocha MP, Barcellos CR, dos Reis Vieira Yance V, Marcondes JA. Metabolic Disturbance in PCOS: Clinical and Molecular Effects on Skeletal Muscle Tissue. Sci World J (2013) 2013:178364. doi: 10.1155/2013/178364
136. Song X, Shen Q, Fan L, Yu Q, Jia X, Sun Y, et al. Dehydroepiandrosterone-Induced Activation of Mtorc1 and Inhibition of Autophagy Contribute to Skeletal Muscle Insulin Resistance in a Mouse Model of Polycystic Ovary Syndrome. Oncotarget (2018) 9(15):11905–21. doi: 10.18632/oncotarget.24190
137. Shen Q, Bi H, Yu F, Fan L, Zhu M, Jia X, et al. Nontargeted Metabolomic Analysis of Skeletal Muscle in a Dehydroepiandrosterone-Induced Mouse Model of Polycystic Ovary Syndrome. Mol Reprod Dev (2019) 86(4):370–8. doi: 10.1002/mrd.23111
138. Ding Y, Xia BH, Zhang CJ, Zhuo GC. Mutations in Mitochondrial tRNA Genes May be Related to Insulin Resistance in Women With Polycystic Ovary Syndrome. Am J Transl Res (2017) 9(6):2984–96.
139. Eriksen MB, Minet AD, Glintborg D, Gaster M. Intact Primary Mitochondrial Function in Myotubes Established From Women With PCOS. J Clin Endocr Metab (2011) 96(8):E1298–302. doi: 10.1210/jc.2011-0278
140. Lerner A, Kewada D, Ahmed A, Hardy K, Christian M, Franks S. Androgen Reduces Mitochondrial Respiration in Mouse Brown Adipocytes: A Model for Disordered Energy Balance in Polycystic Ovary Syndrome. Int J Mol Sci (2021) 22(1):243. doi: 10.3390/ijms22010243
141. Shorakae S, Jona E, de Courten B, Lambert GW, Lambert EA, Phillips SE, et al. Brown Adipose Tissue Thermogenesis in Polycystic Ovary Syndrome. Clin Endocrinol (2019) 90(3):425–32. doi: 10.1111/cen.13913
142. Nohara K, Laque A, Allard C, Münzberg H, Mauvais-Jarvis F. Central Mechanisms of Adiposity in Adult Female Mice With Androgen Excess. Obes (Silver Spring Md) (2014) 22(6):1477–84. doi: 10.1002/oby.20719
143. Wang HD, Wang XP, Zhu YX, Chen F, Sun Y, Han X. Increased Androgen Levels in Rats Impair Glucose-Stimulated Insulin Secretion Through Disruption of Pancreatic Beta Cell Mitochondrial Function. J Steroid Biochem (2015) 154:254–66. doi: 10.1016/j.jsbmb.2015.09.003
144. Mishra JS, More AS, Kumar S. Elevated Androgen Levels Induce Hyperinsulinemia Through Increase in Ins1 Transcription in Pancreatic Beta Cells in Female Rats. Biol Reprod (2018) 98(4):520–31. doi: 10.1093/biolre/ioy017
145. Cui P, Hu W, Ma T, Hu M, Tong X, Zhang F. Long-Term Androgen Excess Induces Insulin Resistance and Non-Alcoholic Fatty Liver Disease in PCOS-Like Rats. J Steroid Biochem Mol Biol (2021) 208:105829. doi: 10.1016/j.jsbmb.2021.105829
146. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial Reactive Oxygen Species (Ros) and Ros-Induced Ros Release. Physiol Rev (2014) 94(3):909–50. doi: 10.1152/physrev.00026.2013
147. Schiffer L, Arlt W, O’Reilly MW. Understanding the Role of Androgen Action in Female Adipose Tissue. Front Horm Res (2019) 53:33–49. doi: 10.1159/000494901
148. Ezeh U, Pall M, Mathur R, Azziz R. Association of Fat to Lean Mass Ratio With Metabolic Dysfunction in Women With Polycystic Ovary Syndrome. Hum Reprod (Oxford England) (2014) 29(7):1508–17. doi: 10.1093/humrep/deu096
149. Borruel S, Fernández-Durán E, Alpañés M, Martí D, Alvarez-Blasco F, Luque-Ramírez M, et al. Global Adiposity and Thickness of Intraperitoneal and Mesenteric Adipose Tissue Depots Are Increased in Women With Polycystic Ovary Syndrome (PCOS). J Clin Endocrinol Metab (2013) 98(3):1254–63. doi: 10.1210/jc.2012-3698
150. Ofori EK, Alonso SC, Correas-Gomez L, Carnero EA, Zwygart K, Hugues H, et al. Thigh and Abdominal Adipose Tissue Depot Associations With Testosterone Levels in Postmenopausal Females. Clin Endocrinol (2019) 90(3):433–9. doi: 10.1111/cen.13921
151. Ryan GE, Malik S, Mellon PL. Antiandrogen Treatment Ameliorates Reproductive and Metabolic Phenotypes in the Letrozole-Induced Mouse Model of PCOS. Endocrinology (2018) 159(4):1734–47. doi: 10.1210/en.2017-03218
152. Andreoli MF, Donato J, Cakir I, Perello M. Leptin Resensitisation: A Reversion of Leptin-Resistant States. J Endocrinol (2019) 241(3):R81–96. doi: 10.1530/JOE-18-0606
153. Dudek P, Kozakowski J, Zgliczynski W. The Effects of Testosterone Replacement Therapy in Men With Age-Dependent Hypogonadism on Body Composition, and Serum Levels of Leptin, Adiponectin, and C-Reactive Protein. Endokrynol Pol (2020) 71(5):382–7. doi: 10.5603/EP.a2020.0048
154. Schiffer L, Kempegowda P, Arlt W, O'Reilly MW. MECHANISMS IN ENDOCRINOLOGY The Sexually Dimorphic Role of Androgens in Human Metabolic Disease. Eur J Endocrinol (2017) 177(3):R125–43. doi: 10.1530/EJE-17-0124
155. Varlamov O, Chu MP, McGee WK, Cameron JL, O'Rourke RW, Meyer KA, et al. Ovarian Cycle-Specific Regulation of Adipose Tissue Lipid Storage by Testosterone in Female Nonhuman Primates. Endocrinology (2013) 154(11):4126–35. doi: 10.1210/en.2013-1428
156. Varlamov O, Bishop CV, Handu M, Takahashi D, Srinivasan S, White A, et al. Combined Androgen Excess and Western-Style Diet Accelerates Adipose Tissue Dysfunction in Young Adult, Female Nonhuman Primates. Hum Reprod (Oxford England) (2017) 32(9):1892–902. doi: 10.1093/humrep/dex244
157. Zang H, Rydén M, Wåhlen K, Dahlman-Wright K, Arner P, Lindén Hirschberg A. Effects of Testosterone and Estrogen Treatment on Lipolysis Signaling Pathways in Subcutaneous Adipose Tissue of Postmenopausal Women. Fertil Steril (2007) 88(1):100–6. doi: 10.1016/j.fertnstert.2006.11.088
158. Ye J. Emerging Role of Adipose Tissue Hypoxia in Obesity and Insulin Resistance. Int J Obes (Lond) (2009) 33(1):54–66. doi: 10.1038/ijo.2008.229
159. Kloting N, Bluher M. Adipocyte Dysfunction, Inflammation and Metabolic Syndrome. Rev Endocr Metab Dis (2014) 15(4):277–87. doi: 10.1007/s11154-014-9301-0
160. Chen S, Zhang W, Tang C, Tang X, Liu L, Liu C. Vanin-1 Is a Key Activator for Hepatic Gluconeogenesis. Diabetes (2014) 63(6):2073–85. doi: 10.2337/db13-0788
161. Rowe GC, Arany Z. Genetic Models of PGC-1 and Glucose Metabolism and Homeostasis. Rev Endocr Metab Dis (2014) 15(1):21–9. doi: 10.1007/s11154-013-9273-5
162. Choi CS, Befroy DE, Codella R, Kim S, Reznick RM, Hwang YJ, et al. Paradoxical Effects of Increased Expression of PGC-1alpha on Muscle Mitochondrial Function and Insulin-Stimulated Muscle Glucose Metabolism. Proc Natl Acad Sci USA (2008) 105(50):19926–31. doi: 10.1073/pnas.0810339105
163. Marinho R, Mekary RA, Muñoz VR, Gomes RJ, Pauli JR, de Moura LP. Regulation of Hepatic TRB3/Akt Interaction Induced by Physical Exercise and Its Effect on the Hepatic Glucose Production in an Insulin Resistance State. Diabetol Metab Syndr (2015) 7:67. doi: 10.1186/s13098-015-0064-x
Keywords: mitochondria, sex hormone, energy metabolism, insulin sensitivity, sex dimorphism
Citation: Yin L, Luo M, Wang R, Ye J and Wang X (2021) Mitochondria in Sex Hormone-Induced Disorder of Energy Metabolism in Males and Females. Front. Endocrinol. 12:749451. doi: 10.3389/fendo.2021.749451
Received: 29 July 2021; Accepted: 24 November 2021;
Published: 20 December 2021.
Edited by:
Paula Mariana Maloberti, University of Buenos Aires, ArgentinaReviewed by:
Sridhar Muthusami, Karpagam Academy of Higher Education, IndiaAshu Johri, Independent Researcher, New York, United States
Copyright © 2021 Yin, Luo, Wang, Ye and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianping Ye, eWVqaWFucGluZ0B6enUuZWR1LmNu; Xiaohui Wang, V2FuZ3Bhbjk2QDEyNi5jb20=