 Leiming Zhang
Leiming Zhang Xiaoli Li
Xiaoli Li Tianhao Ying
Tianhao Ying Tian Wang
Tian Wang Fenghua Fu*
Fenghua Fu*- Key Laboratory of Molecular Pharmacology and Drug Evaluation, Ministry of Education, School of Pharmacy, Yantai University, Yantai, China
Glucocorticoids are drugs that are widely used to suppress inflammation and the activation of the immune system. However, the prolonged use or at high doses of glucocorticoid can result in adverse side effects including osteoporosis, bone loss, and an increased risk of fracture. A number of compounds derived from natural plant sources have been reported to exert anti-inflammatory activity by interacting with the glucocorticoid receptor (GR), likely owing to their chemical similarity to glucocorticoids, or by regulating GR, without a concomitant risk of treatment-related side effects such as osteoporosis. Other herbal compounds can counteract the pathogenic processes underlying glucocorticoid-induced osteoporosis (GIOP) by regulating homeostatic bone metabolic processes. Herein, we systematically searched the PubMed, Embase, and Cochrane library databases to identify articles discussing such compounds published as of May 01, 2021. Compounds reported to exert anti-inflammatory glucocorticoid-like activity without inducing GIOP include escin, ginsenosides, and glycyrrhizic acid, while compounds reported to alleviate GIOP by improving osteoblast function or modulating steroid hormone synthesis include tanshinol and icariin.
Introduction
Glucocorticoids are drugs that modulate a diverse array of signaling pathways, modifying cognitive signaling, exerting immunosuppressive and anti-inflammatory activity, and preserving normal organ homeostasis and function (1). Since their first clinical deployment in the 1950s, glucocorticoids have been widely adopted and are the most commonly utilized immunosuppressive drug class in the world (2). The prolonged use of glucocorticoids, however, particularly at higher doses, can result in a variety of adverse side effects including arterial hypertension, Cushing’s syndrome, type 2 diabetes mellitus, osteoporosis, and increased susceptibility to infection (3).
Endogenous glucocorticoids regulate key processes including calcium homeostasis in the intestines and kidneys, bone development, and mesenchymal cell differentiation at physiological concentrations. By stimulating mature osteoblasts to increase canonical Wnt protein production, glucocorticoids can promote the activation of β-catenin signaling in mesenchymal progenitor cells such that they differentiate into osteoblasts rather than chondrocytes or adipocytes, thus favoring osteogenesis. In osteoblasts, Wnt signaling also leads to the expression of osteoprotegerin (OPG), which suppresses osteoclastogenesis to maintain bone homeostasis (4). At very high doses, however, glucocorticoids can negatively impact bone integrity through a range of mechanisms, with GIOP having first been described in individuals with Cushing’s disease expressing excess endogenous glucocorticoid levels (5).
Owing to their potent anti-inflammatory and immunomodulatory activity, glucocorticoids are widely used in clinical contexts. However, their prolonged use can lead to adverse outcomes including glucocorticoid-induced osteoporosis (GIOP), which is the most common secondary cause of osteoporosis and an important iatrogenic risk to patients in many contexts (6). Such osteoporosis has been reported in patients with chronic inflammatory diseases including inflammatory bowel disease, chronic obstructive pulmonary disease, and systemic lupus erythematosus (SLE) (7). Most SLE patients undergo chronic glucocorticoid treatment, and one Dutch study with a 6-year follow-up period detected a dose-dependent association between the use of glucocorticoids and lumbar spine bone loss (8). Similarly, a cohort study of individuals between the ages of 18 and 64 undergoing glucocorticoid treatment for a range of disorders found that higher doses, longer treatment durations, and continuous use were associated with the highest fracture risk (9). Sustained treatment with prednisone (10 mg/d) for over 90 days was associated with 7- and 17-fold increases in the risk of hip and vertebral fractures (9).
Glucocorticoids can modulate bone biology via a number of different mechanisms (Figure 1), suppressing osteogenesis and promoting the apoptotic death of osteoblasts and osteocytes (10). Additionally, these drugs can increase the number of osteoclasts and enhance their function, resulting in an overall increase in osteoclast lifespan (11).
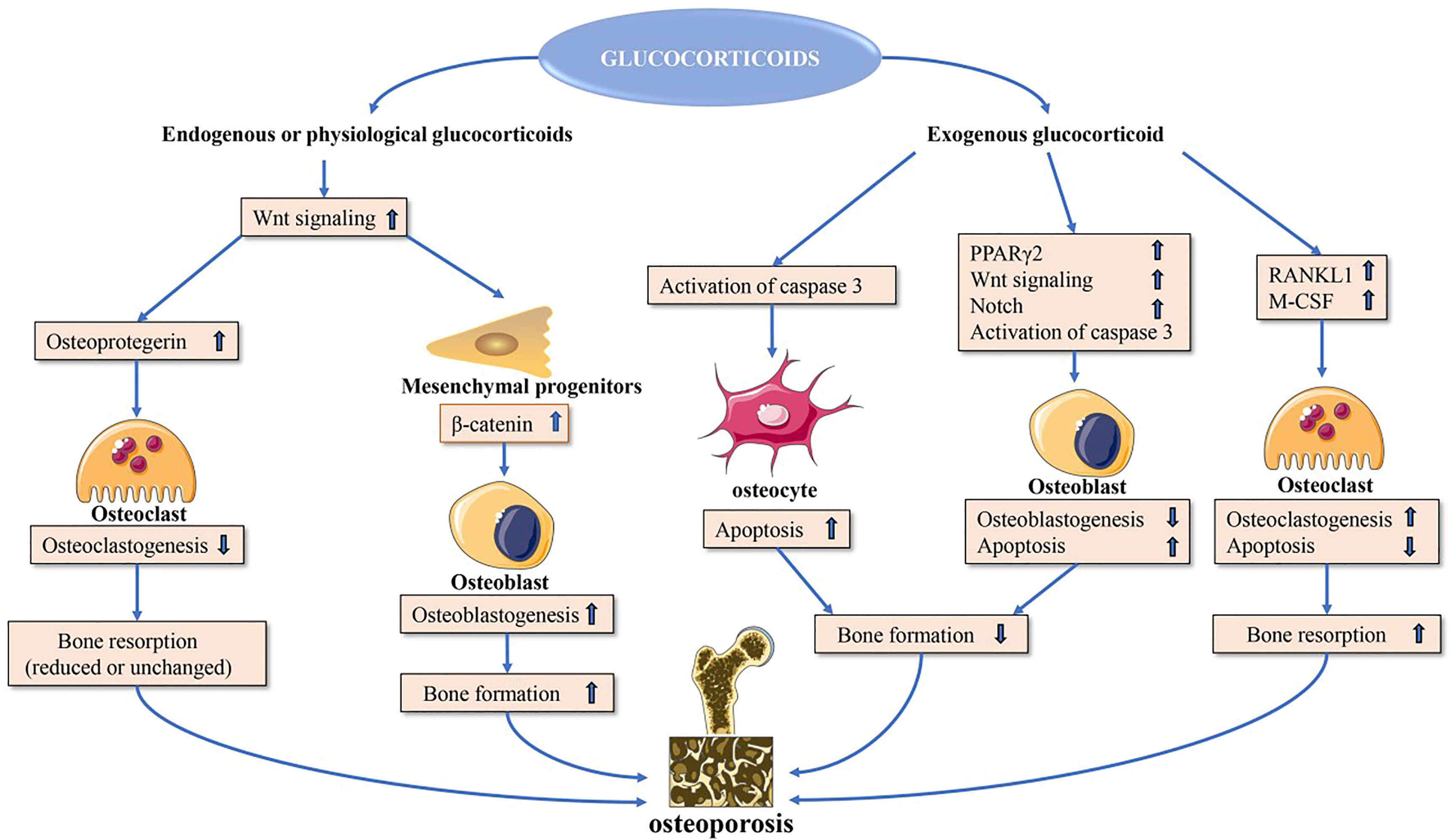
Figure 1 Direct glucocorticoid effects on bone. Endogenous or physiological glucocorticoids stimulate mature osteoblasts to produce canonical Wnt proteins, which activate the β-catenin signaling cascade in mesenchymal progenitor cells and promote them to differentiate towards osteoblasts. These actions favor bone formation. Additionally, Wnt signaling in osteoblasts and osteocytes promotes osteoprotegerin expression, which in turn inhibits osteoclast formation resulting in decreased or unchanged bone resorption. Exogenous glucocorticoids negatively affect osteoblast and osteocyte function. In osteoblasts and osteocytes, increased PPARγ2 and Notch target gene expression and decreased wnt signaling contribute to decreased osteoblastogenesis, and activation of caspase 3 results in increased osteoblast and osteocyte apoptosis. Glucocorticoids induce upregulation of expression of RANKL and M-CSF, which leads to increased osteoclastogenesis and osteoclast lifespan. PPARγ2, peroxisome proliferator-activated receptor-γ2; RANKL, receptor activator of nuclear factor-κB ligand; M-CSF, macrophage colony-stimulating fact.
Osteoblast signaling pathways that can be directly impacted by glucocorticoid exposure include the peroxisome proliferator-activated receptor γ2 (PPARγ2) (12), CCAAT/enhancer-binding protein-α (C/EBPα) (13), adipocyte protein 2 (aP2) (14), and canonical WNT signaling pathways (15). Glucocorticoids promote PPARγ2, C/EBPα, and aP2 upregulation, leading precursor cells to preferentially differentiate into adipocytes instead of osteoblasts, thereby leading to a decrease in overall osteoblast numbers (12–15). Glucocorticoids also increase the expression of inhibitory molecules including sclerostin in the WNT-β-catenin signaling pathway while simultaneously inhibiting the expression of WNT16 in a dose- and time-dependent fashion, further contributing to reduced osteoblastogenesis and bone loss (16, 17).
The receptor activator of nuclear factor-κB ligand (RANKL)-osteoprotegerin (OPG) pathway is also amenable to modulation by glucocorticoids, which increase RANKL production and suppress OPG mRNA expression (18–20). Glucocorticoids can also enhance Notch signaling in osteoblasts and osteocytes, leading to increased Notch target gene expression including hairy and enhancer of split (Hes) and Hes-related with YRPW motif (Hey), which are repressive transcription factors that have the potential to mediate the impairment of osteoblast functionality and consequent reductions in osteogenesis (21, 22).
Glucocorticoid-induced apoptosis is linked to the enhanced activity of effector proteins including caspase 3, 7, and 8 downstream of the pro-apoptotic Bim and Fas/FasL death receptor pathways (10). Glucocorticoids can also stabilize GSK-3β activity to induce osteoblast apoptosis.
Glucocorticoids can impact osteoblasts to increase the RANKL : OPG ratio, thereby promoting osteoclast differentiation and maturation such that the overall rate of bone resorption increases. This effect can be further exacerbated by the ability of glucocorticoid treatment to induce the production of macrophage colony stimulating factor (M-CSF), which is released from osteoblasts and enhances the differentiation and activity of osteoclasts (23). The long-term impact of glucocorticoids on osteoclast function, however, is less certain, with multiple reports indicating that these compounds can interfere with the osteoclast cytoskeleton such that the activity of these cells may be increasingly impaired even as their longevity increases (24–26).
The Impact of Herbal Medicines on Glucocorticoid- Induced Osteoporosis
Many studies have shown that herbal medicines can significantly increase bone density and improve clinical findings in GIOP patients, thus serving as novel tools for the treatment and/or prevention of this debilitating glucocorticoid-related complication (27–29).
Herbal Medicines Exert Glucocorticoid-Like Anti-Inflammatory Activity Without Inducing GIOP
A range of herbal compounds have been suggested to mediate anti-inflammatory activity by signaling through the glucocorticoid receptor, likely owing to their structural similarity to glucocorticoids. Notably, these compounds seem to be able to mediate these effects without a significant risk of negative glucocorticoid-related side effects such as GIOP.
Escin
Escin is a natural mixture of triterpene saponins extracted from the seeds of Aesculus chinensis Bge. or Aesculus wilsonii Rehd. Escin has been reported to exhibit pharmacological effects similar to those associated with glucocorticoid administration. For example, oral escin (5 and 10 mg/kg, p.o.) intake has been found to suppress carrageenan-induced paw edema and to inhibit prostaglandin E2 (PGE2) production (30). Notably, when compared with glucocorticoids, escin (2 mg/kg, i.v.) has been shown not to induce thymic or splenic immune cell apoptosis in mice, nor does it promote the enhanced secretion of endogenous corticosterone (31). Zhang et al. found that the sustained administration of escin (0.45 and 0.9 mg/kg for a period of 10 days, i.v.) in the context of post-surgical bone fracture healing has no adverse impact on wound or bone healing processes (32). There is also evidence that glucocorticoids and escin (2 mg/kg, i.v.) exhibit synergistic anti-inflammatory activity when administered in vitro and in vivo at low doses, suggesting at least partial overlap in the pharmacological pathways impacted by these compounds (33). Combination glucocorticoid and escin(5 and 10 mg/kg for a period of 16 days, i.g.) treatment can significantly decrease synovial inflammatory infiltration, synovial hyperplasia, and bone erosion in a rat model of adjuvant-induced arthritis (AIA) rats while reversing some of the adverse effects of glucocorticoid treatment alone such as reductions in boy weight and increases in the spleen index relative to untreated rats (34). Administering escin (10 mg/kg for a period of 14 days, p.o.) together with a low dose of dexamethasone (Dex) has been shown to markedly suppress paw swelling, joint pathology, arthritic index scores, and immune organ pathology in an animal model, all while reducing the necessary Dex dose and thus decreasing the rate of adverse effects associated with Dex administration (35). The anti-edema and anti-inflammatory properties of escin may be attributable to its ability to bind to the glucocorticoid receptor (GR), consistent with glucocorticoid-like activity (36). Escin (1.8 and 3.6 mg/kg, i.v.) may additionally augment the antioxidant capacity of tissue in the context of lipopolysaccharide (LPS)-induced acute lung injury (ALI) and endotoxin-induced liver injury by suppressing the production of inflammatory compounds including NO, TNF-α, and IL-1β while simultaneously promoting GR upregulation in the liver and lungs (37, 38).
Ginsenosides
Ginsenosides are the primary active ingredients isolated from ginseng, and they have been reported to exhibit anti-inflammatory activity in vitro and in vivo owing to their structural similarity to steroid hormones. Compound K is a ginsenoside that has, in vitro, been shown to suppress TNF-α-induced fibroblast-like synoviocyte (FLS) migration, proliferation, and secretion, consistent with joint-protective activity (80 mg/kg for a period of 14 days, i.g.) (39). In a rat model of myocardial infarction (MI), ginsenoside Rg3 (30 mg/kg for a period of 7 days, i.g.) reduces inflammation via the inhibition of the NF-κB pathway (40). Combining the ginsenosides Rh1 (20 mg/kg, i.p.) and Rg2 (20 mg/kg, i.p.) can suppress LPS-induced tissue damage and inflammation by interfering with the ability of LPS to bind to and trigger the activation of TLR4 (41). Ginsenoside Rb1 (10 and 20 mg/kg, i.p.) markedly alleviates LPS- or cantharidin-induced acute kidney injury, LPS-induced septicemia, and dimethyl benzene-induced ear edema in mice (42). Ginsenoside treatment is also not associated with any significant adverse reactions. In mice overexpressing TNF-α, ginsenoside Rg1 (20 mg/kg, i.g.) can prevent bone erosion, inhibit synovial inflammation, and reduce serum levels of both IL-6 and TNF-α, and treatment for 12 weeks with ginsenoside Rg1 was not associated with any liver or kidney damage (43). Ginsenoside Rd (10 mg/kg, i.p.) can suppress ischemia-induced microglial activation and inhibit proinflammatory cytokine production while inducing fewer severe side effects as compared to glucocorticoids (44). Importantly, these ginsenosides can also work in synergy with glucocorticoids to inhibit inflammation. For example, combining corticosterone with low concentrations of Rg1 can suppress the LPS-induced production of NO and TNF-α by RAW264.7 macrophages while simultaneously promoting GR upregulation (45). Ginsenosides can also shape cellular responses in a GR-mediated manner, as in the case of Rg1 (12.5 mg/kg, i.p.), which suppresses LPS-induced NF-κB nuclear translocation and inflammatory cytokine production in a GR-dependent fashion. Notably, Rg1 (20 mg/kg for a period of 21 days, i.g.) has no adverse impact on murine osteoblast differentiation or proliferation (46). In a murine collagen-induced arthritis (CIA) model system, the ginsenoside Rh1 (10 mg/kg for a period of 10 days, i.p.) was also able to augment the anti-inflammatory activity of Dex by enhancing GR expression and binding without inducing hyperglycemia (47). Ginsenoside CK (112 mg/kg for a period of 24 days, i.g.) can also activate GR to suppress β-arrestin2 expression, thereby inhibiting inflammation (48).
Glycyrrhizic Acid and Glycyrrhetinic Acid
Glycyrrhizic acid (also known as glycyrrhizin) is the primary glycoside derivative of licorice, and it has been ascribed a range of anti-inflammatory activities. By suppressing signaling through the Smad3 and MAPK pathways, for example, glycyrrhizin (30 and 100 mg/kg for a period of 28 days, i.g.) has been shown to reduce the severity of bleomycin-induced inflammation and pulmonary fibrosis in mice (49). Glycyrrhizin (10 mg/kg for once every day in the first 3 weeks following by given once every 3 days until the twelfth week, intra-articular knee injection) treatment can also alleviate inflammation and the degeneration of cartilage tissue in a rat model of osteoarthritis via regulating the TLR4/NF-κB and HMGB1 pathways (50). In vivo, glycyrrhizic acid undergoes hydrolysis to yield glycyrrhetinic acid, which is structurally similar to steroid hormones such that it is able to exert a range of biological effects including glucocorticoid-like anti-inflammatory activity through interactions with steroid hormone receptors and metabolic enzymes. For example, in a murine ALI model system, glycyrrhetinic acid (10, 20 and 40 mg/kg for a period of 7 days, i.g.) was able to reduce injury severity by suppressing NLRP3 inflammasome activation through the ROS-PI3K/AKT pathway (51). Glycyrrhetinic acid (40 μM) may also be hepatoprotective in the context of chronic liver inflammation, functioning by suppressing the phosphorylation of IκBα phosphorylation and the nuclear translocation of p65 so as to reduce iNOS expression, thus alleviating inflammation (52). Glycyrrhetinic acid and glycyrrhizic acid can interact with GR as ligands, modulating glucocorticoid resistance can also prevent inflammation by disrupting the GR-HSP90 (53, 54). As a relatively weak glucocorticoid-like drug, glycyrrhizic acid can enhance the effects of glucocorticoids while antagonizing the adverse effects associated with high-dose glucocorticoid treatment. Licorice (75 mg/kg for a period of 5 days) can also suppress 11 beta-hydroxysteroid dehydrogenase mRNA expression while potentiating glucocorticoid activity (55). Therefore, glycyrrhizic acid and glycyrrhetinic acid seem to be able to exert glucocorticoid-like anti-inflammatory activity without a significant risk of negative glucocorticoid-related side effects such as GIOP.
Herbal Medicines Capable of Inhibiting or Treating GIOP
Icariin
Icariin is the main active ingredient of epimedium, which is a natural compound that has been increasingly studied in the context of osteoporosis treatment and prevention, as it has been shown to simultaneously suppress bone resorption and expedite bone formation (56). Icariin (125 mg/kg for a period of 14 days, i.g.) can promote primary osteoblast maturation and associated bone remodeling, inducing osteoblast mineralization and the expression of key markers of terminal differentiation such as alkaline phosphatase (ALP) and type I collagen (57–60). Icariin(0.1 μM) also exhibits robust anti-apoptotic activity, promoting BMSC proliferation and osteogenic differentiation via Wnt/β-catenin pathway activation (61).
With respect to the symptoms of GIOP, icariin (5 μM for a period of 48 h) can enhance trabecular bone density in the context of glucocorticoid exposure, promoting osteogenic differentiation via the suppression of Notch signaling (62). Through the enhancement of autophagic activity, icariin (50 mg/kg for a period of 30 days, i.p.) can reduce OVX-induced bone loss in animal model systems (63), in addition to disrupting the Dex-induced apoptotic death of osteocytes (58). Icariin can also activate the ERK and ER pathways to control bone homeostasis, promoting OPG expression and Wnt pathway activation. Inhibiting osteoclastogenesis is at least partially responsible for the anti-osteoporotic activity of icariin and compounds derived therefrom. The levels of the osteoclast differentiation marker tartrate-resistant acid phosphatase (TRAP) are reduced in a dose-dependent manner when osteoclast precursor cells are treated with icariin (10 nM, every 3 days) (64). Icariin (10 μM) is also able to directly suppress RANKL-induced hemopoietic cell differentiation into osteoclasts (65). In addition to regulating osteoclastogenesis, icariin (50 and 100 μM) can arrest cell cycle progression in osteoclast precursors, thereby inducing their apoptotic death (66). It can further reverse deleterious Dex-induced trabecular phenotypes while stimulating bone remodeling, increasing bone calcium, OCN, and FGF-23 levels while reducing the levels of bone resorption markers including CTX and TRAP-5b. Indeed, in GIOP model mice, icariin (100 mg/kg for a period of 6 or 12 weeks, p.o.) treatment has been shown to protect against bone degeneration, hypercalciuria, and hypocalcemia (67). As such, icariin may be a valuable tool for use in the induction of bone regeneration owing to its potent osteogenic bioactivity.
Many clinical studies have shown that Chinese medicine containing epimedium has achieved good clinical effects in the treatment of GIOP patients. Hugu Capsules (comprised of epimedium, polygonum multiflorum, rehmannia glutinosa and other traditional chinese medicines) can significantly increase the bone mass of 51 patients with GIOP, improve bone turnover, and relieve pain (68). Through observation of 50 GIOP patients, Wu et al. found that taking Xianling Gubao capsule while using glucocorticoids treatment can increase the BMD of the patient’s lumbar spine and proximal femur, thereby reducing the incidence of osteoporotic fractures and having fewer adverse reactions (69). Through clinical observation of 66 patients with GIOP, Shi et al. found that Bugu Capsules (including epimedium) can significantly reduce the impact of OP caused by glucocorticoids, reduce blood calcium, parathyroid hormone levels, and increase bone density (70).
Tanshinones
Tanshinone IIA, extracted from Salvia miltiorrhiza Bunge, is a perennial herbal plant widely used as a folk remedy in Asian countries. Several studies have proved that Tanshinone IIA possesses many biological activities, such as anti-inflammatory, free-radical scavenging abilities, antioxidant properties, liver protection, and anti-cancer properties. Tanshinones are compounds that can also simultaneously inhibit osteoclastogenesis and bone resorption while promoting more robust bone formation with concomitant osteoblastogenesis. These tanshinones (2, 5 μg/ml) suppress osteoclast development through the disruption of RANKL-mediated NF-κB, MAPK, Akt, and M-CSF/c-Src signaling pathway activation (71, 72). Tanshinone IIA, for example, can inhibit osteoclastogenesis through the inhibition of RANKL-induced c-Fos and NFAT c1 (71), with Tanshinone IIA (20 μg/mL for a period of 30 min) pretreatment reportedly reducing the fusion, actin ring formation, and resorptive activity of osteoclasts in a co-culture system containing M-CSF and RANKL-treated calvarial osteoblasts and BMCs (73). Mechanistically, Tanshinone IIA (10 μg/mL) can function as a selective COX-2 inhibitor to suppress PGE2 and to thereby modulate OPG and RANKL expression (74), all of which are related to osteoclast function. Tanshinones (1 μM for a period of 24 h) can also disrupt the apoptotic death of osteoblasts and consequent osteoporosis observed upon glucocorticoid treatment by inactivating Nox4 (75). In osteoporosis model mice, Tanshinone IIA (10 mg/kg for a period of 6 weeks, p.o.) was able to decrease the incidence of fractures and severe osteopenia while augmenting bone strength, mineral levels, and collagen in the bone matrix (76). Tanshinone (10 mg/kg for a period of 21 days, i.v.) was also able to upregulate phosphoglycerate dehydrogenase and to thereby suppress OVX-induced osteoporosis and BMSC senescence (77). Tanshinone can alleviate the adverse effects of Dex treatment and consequence cellular injuries such as caspase-9-dependent apoptosis, increased cytosolic cytochrome c and Nox levels, and increased ROS generation (75). Current preclinical evidence suggests that these Tanshinones preserve skeletal integrity primarily by suppressing bone resorption and osteoclast formation, underscoring their potential value for the treatment of GIOP.
Many other herbal medicines have also been found to reduce GIOP incidence or severity through a range of mechanisms. For example, celastrol can suppress GIOP incidence in rats by modulating the Wnt and PI3K/AKT signaling pathways (78), while KRG can induce the apoptotic death of osteoblasts, highlighting its potential therapeutic utility as a tool to delay the onset of osteoporosis (79). Osthole has been shown to prevent Dex-induced osteoporosis in female rats, potentially by normalizing hormone and cytokine homeostasis through increases in TGF-β1 production (80) (Table 1).
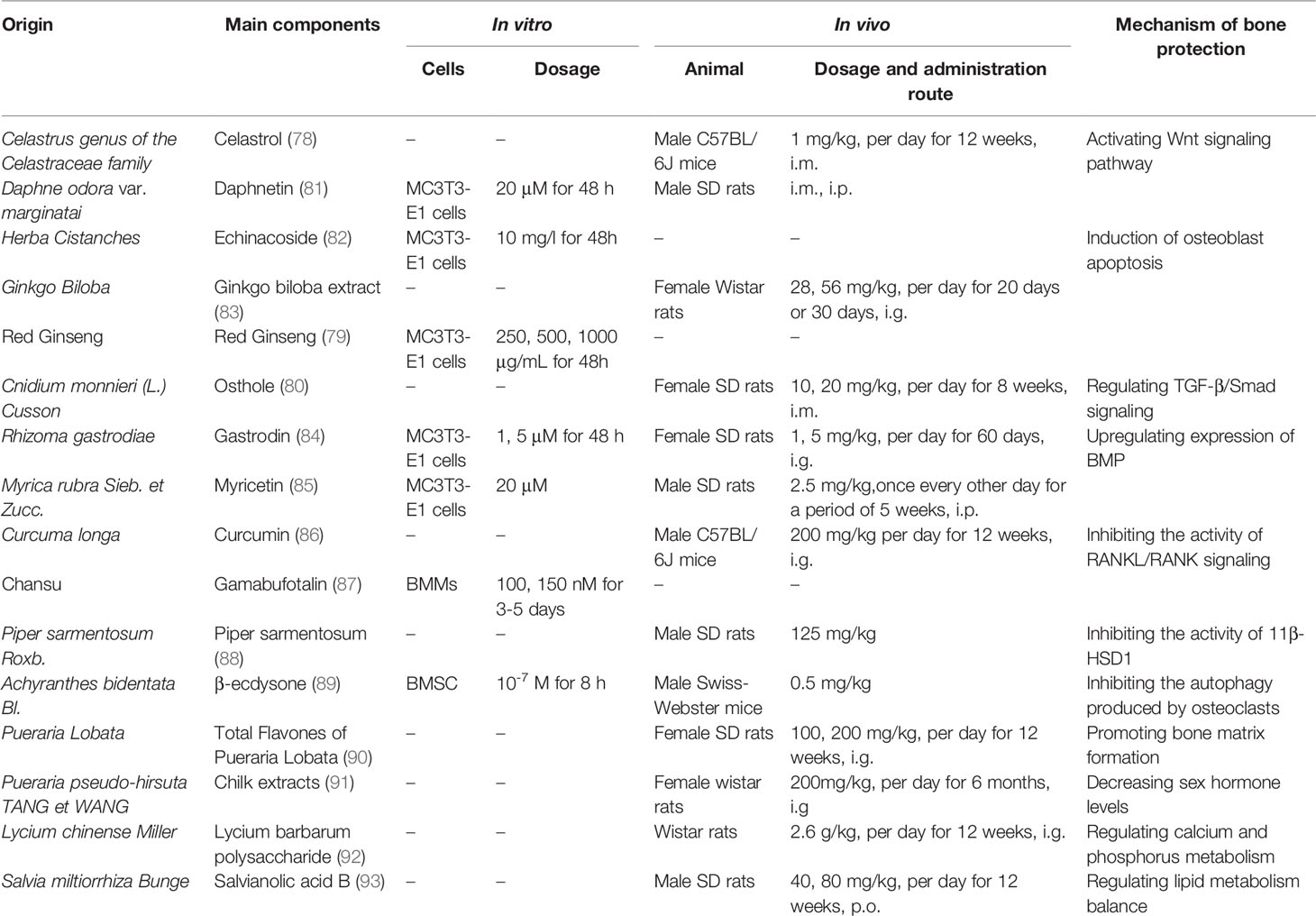
Table 1 Herbal medicines capable of inhibiting or treating GIOP.
Conclusion
Much like other hormone molecules, glucocorticoids can exert a range of effects on tissues and organs when employed at physiological and pharmacological doses. While awareness of osteoporosis and other risks associated with prolonged or high doses glucocorticoid use is growing, GIOP remains underdiagnosed and inadequately treated. Herbal medicines characterized to date have been shown to treat GIOP through two primary mechanisms, with some exerting glucocorticoid-like activity without a risk of adverse reactions, and the others treating GIOP through mechanisms including the regulation of Wnt signaling pathway, the induction of osteoblast apoptosis, and the inhibition of RANKL/RANK signaling.
However, Further clinical studies of these herbal medicines are needed to demonstrate prevention properties in GIOP patients. For example, sodium aescinate has been widely used in clinic to treat traumatic and inflammatory edema, etc. A randomized, parallel, controlled clinical trial can be conducted to evaluate the anti-inflammatory efficacy combined with glucocorticoids, as well as the side effects, GIOP.
Author Contributions
LZ and FF conceived the idea. XL and TY drafted the manuscript. LZ, TW, and FF supervised the process and contributed to editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Science Foundation of China (No. 81973547).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.744647/full#supplementary-material
References
1. Scherholz ML, Schlesinger N, Androulakis IP. Chronopharmacology of Glucocorticoids. Adv Drug Delivery Rev (2019) 151-152:245–61. doi: 10.1016/j.addr.2019.02.004
2. Buttgereit F. Views on Glucocorticoid Therapy in Rheumatology: The Age of Convergence. Nat Rev Rheumatol (2020) 16(4):239–46. doi: 10.1038/s41584-020-0370-z
3. Strehl C, Bijlsma JW, de Wit M, Boers M, Caeyers N, Cutolo M, et al. Defining Conditions Where Long-Term Glucocorticoid Treatment has an Acceptably Low Level of Harm to Facilitate Implementation of Existing Recommendations: Viewpoints From an EULAR Task Force. Ann Rheum Dis (2016) 75(6):952–7. doi: 10.1136/annrheumdis-2015-208916
4. Seibel MJ, Cooper MS, Zhou H. Glucocorticoid-Induced Osteoporosis: Mechanisms, Management, and Future Perspectives. Lancet Diabetes Endocrinol (2013) 1(1):59–70. doi: 10.1016/S2213-8587(13)70045-7
5. Cushing H. The Basophil Adenomas of the Pituitary Body and Their Clinical Manifestations (Pituitary Basophilism). 1932. Obes Res (1994) 2(5):486–508. doi: 10.1002/j.1550-8528.1994.tb00097.x
6. Chotiyarnwong P, McCloskey EV. Pathogenesis of Glucocorticoid-Induced Osteoporosis and Options for Treatment. Nat Rev Endocrinol (2020) 16(8):437–47. doi: 10.1038/s41574-020-0341-0
7. Briot K, Geusens P, Em Bultink I, Lems WF, Roux C. Inflammatory Diseases and Bone Fragility. Osteoporos Int (2017) 28(12):3301–14. doi: 10.1007/s00198-017-4189-7
8. Jacobs J, Korswagen LA, Schilder AM, van Tuyl LH, Dijkmans BA, Lems WF, et al. Six-Year Follow-Up Study of Bone Mineral Density in Patients With Systemic Lupus Erythematosus. Osteoporos Int (2013) 24(6):1827–33. doi: 10.1007/s00198-012-2157-9
9. Steinbuch M, Youket TE, Cohen S. Oral Glucocorticoid Use Is Associated With an Increased Risk of Fracture. Osteoporos Int (2004) 15(4):323–8. doi: 10.1007/s00198-003-1548-3
10. Hofbauer LC, Rauner M. Minireview: Live and Let Die: Molecular Effects of Glucocorticoids on Bone Cells. Mol Endocrinol (2009) 23(10):1525–31. doi: 10.1210/me.2009-0069
11. den Uyl D, Bultink IE, Lems WF. Advances in Glucocorticoid-Induced Osteoporosis. Curr Rheumatol Rep (2011) 13(3):233–40. doi: 10.1007/s11926-011-0173-y
12. Ito S, Suzuki N, Kato S, Takahashi T, Takagi M. Glucocorticoids Induce the Differentiation of a Mesenchymal Progenitor Cell Line, ROB-C26 Into Adipocytes and Osteoblasts, But Fail to Induce Terminal Osteoblast Differentiation. Bone (2007) 40(1):84–92. doi: 10.1016/j.bone.2006.07.012
13. Li J, Zhang N, Huang X, Xu J, Fernandes JC, Dai K, et al. Dexamethasone Shifts Bone Marrow Stromal Cells From Osteoblasts to Adipocytes by C/EBPalpha Promoter Methylation. Cell Death Dis (2013) 4(10):e832. doi: 10.1038/cddis.2013.348
14. Ohlsson C, Nilsson KH, Henning P, Wu J, Gustafsson KL, Poutanen M, et al. WNT16 Overexpression Partly Protects Against Glucocorticoid-Induced Bone Loss. Am J Physiol Endocrinol Metab (2018) 314(6):E597–604. doi: 10.1152/ajpendo.00292.2017
15. Hildebrandt S, Baschant U, Thiele S, Tuckermann J, Hofbauer LC, Rauner M. Glucocorticoids Suppress Wnt16 Expression in Osteoblasts in vitro and in vivo. Sci Rep (2018) 8(1):8711. doi: 10.1038/s41598-018-26300-z
16. Yang YJ, Zhu Z, Wang DT, Zhang XL, Liu YY, Lai WX, et al. Tanshinol Alleviates Impaired Bone Formation by Inhibiting Adipogenesis via KLF15/Pparγ2 Signaling in GIO Rats. Acta Pharmacol Sin (2018) 39(4):633–41. doi: 10.1038/aps.2017.134
17. Ohnaka K, Tanabe M, Kawate H, Nawata H, Takayanagi R. Glucocorticoid Suppresses the Canonical Wnt Signal in Cultured Human Osteoblasts. Biochem Biophys Res Commun (2005) 329(1):177–81. doi: 10.1016/j.bbrc.2005.01.117
18. Swanson C, Lorentzon M, Conaway HH, Lerner UH. Glucocorticoid Regulation of Osteoclast Differentiation and Expression of Receptor Activator of Nuclear factor-kappaB (NF-Kappab) Ligand, Osteoprotegerin, and Receptor Activator of NF-KappaB in Mouse Calvarial Bones. Endocrinology (2006) 147(7):3613–22. doi: 10.1210/en.2005-0717
19. Piemontese M, Xiong J, Fujiwara Y, Thostenson JD, O’Brien CA. Cortical Bone Loss Caused by Glucocorticoid Excess Requires RANKL Production by Osteocytes and is Associated With Reduced OPG Expression in Mice. Am J Physiol Endocrinol Metab (2016) 311(3):E587–93. doi: 10.1152/ajpendo.00219.2016
20. Jing Z, Wang C, Yang Q, Wei X, Jin Y, Meng Q, et al. Luteolin Attenuates Glucocorticoid-Induced Osteoporosis by Regulating ERK/Lrp-5/GSK-3β Signaling Pathway In Vivo and In Vitro. J Cell Physiol (2019) 234(4):4472–90. doi: 10.1002/jcp.27252
21. Zanotti S, Yu J, Adhikari S, Canalis E. Glucocorticoids Inhibit Notch Target Gene Expression in Osteoblasts. J Cell Biochem (2018) 119(7):6016–23. doi: 10.1002/jcb.26798
22. Zanotti S, Canalis E. Notch Signaling and the Skeleton. Endocr Rev (2016) 37(3):223–53. doi: 10.1210/er.2016-1002
23. Achuthan A, Aslam ASM, Nguyen Q, Lam PY, Fleetwood AJ, Frye AT, et al. Glucocorticoids Promote Apoptosis of Proinflammatory Monocytes by Inhibiting ERK Activity. Cell Death Dis (2018) 9(3):267. doi: 10.1038/s41419-018-0332-4
24. Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ, et al. Glucocorticoids Suppress Bone Formation via the Osteoclast. J Clin Invest (2006) 116(8):2152–60. doi: 10.1172/JCI28084
25. Teitelbaum SL. Glucocorticoids and the Osteoclast. Clin Exp Rheumatol (2015) 33(4 Suppl 92):S37–9.
26. Jia D, O’Brien CA, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids Act Directly on Osteoclasts to Increase Their Life Span and Reduce Bone Density. Endocrinology (2006) 147(12):5592–9. doi: 10.1210/en.2006-0459
27. Suvarna V, Sarkar M, Chaubey P, Khan T, Sherje A, Patel K, et al. Bone Health and Natural Products- An Insight. Front Pharmacol (2018) 9:981. doi: 10.3389/fphar.2018.00981
28. Lin J, Zhu J, Wang Y, Zhang N, Gober HJ, Qiu X, et al. Chinese Single Herbs and Active Ingredients for Postmenopausal Osteoporosis: From Preclinical Evidence to Action Mechanism. Biosci Trends (2017) 11(5):496–506. doi: 10.5582/bst.2017.01216
29. Zhang ND, Han T, Huang BK, Rahman K, Jiang YP, Xu HT, et al. Traditional Chinese Medicine Formulas for the Treatment of Osteoporosis: Implication for Antiosteoporotic Drug Discovery. J Ethnopharmacol (2016) 189:61–80. doi: 10.1016/j.jep.2016.05.025
30. Li M, Lu C, Zhang L, Zhang J, Du Y, Duan S, et al. Oral Administration of Escin Inhibits Acute Inflammation and Reduces Intestinal Mucosal Injury in Animal Models. Evid Based Complement Alternat Med (2015) 2015:503617. doi: 10.1155/2015/503617
31. Wang T, Fu F, Zhang L, Han B, Zhu M, Zhang X. Effects of Escin on Acute Inflammation and the Immune System in Mice. Pharmacol Rep (2009) 61(4):697–704. doi: 10.1016/s1734-1140(09)70122-7
32. Zhang L, Wang H, Wang T, Jiang N, Yu P, Liu F, et al. Potent Anti-Inflammatory Agent Escin Does Not Affect the Healing of Tibia Fracture and Abdominal Wound in an Animal Model. Exp Ther Med (2012) 3(4):735–39. doi: 10.3892/etm.2012.467
33. Xin W, Zhang L, Sun F, Jiang N, Fan H, Wang T, et al. Escin Exerts Synergistic Anti-Inflammatory Effects With Low Doses of Glucocorticoids In Vivo and In Vitro. Phytomedicine (2011) 18(4):272–7. doi: 10.1016/j.phymed.2010.08.013
34. Du Y, Song Y, Zhang L, Zhang M, Fu F. Combined Treatment With Low Dose Prednisone and Escin Improves the Anti-Arthritic Effect in Experimental Arthritis. Int Immunopharmacol (2016) 31:257–65. doi: 10.1016/j.intimp.2016.01.006
35. Zhang L, Huang Y, Wu C, Du Y, Li P, Wang M, et al. Network Pharmacology Based Research on the Combination Mechanism Between Escin and Low Dose Glucocorticoids in Anti-Rheumatoid Arthritis. Front Pharmacol (2019) 10:280. doi: 10.3389/fphar.2019.00280
36. Zhang L, Wang H, Fan H, Wang T, Jiang N, Yu P, et al. The Potent Anti-Inflammatory Agent Escin Does Not Increase Corticosterone Secretion and Immune Cell Apoptosis in Mice. Fitoterapia (2011) 82(6):861–7. doi: 10.1016/j.fitote.2011.04.004
37. Jiang N, Xin W, Wang T, Zhang L, Fan H, Du Y, et al. Protective Effect of Aescin From the Seeds of Aesculus Hippocastanum on Liver Injury Induced by Endotoxin in Mice. Phytomedicine (2011) 18(14):1276–84. doi: 10.1016/j.phymed.2011.06.011
38. Xin W, Zhang L, Fan H, Jiang N, Wang T, Fu F. Escin Attenuates Acute Lung Injury Induced by Endotoxin in Mice. Eur J Pharm Sci (2011) 42(1-2):73–80. doi: 10.1016/j.ejps.2010.10.008
39. Wang Y, Chen J, Luo X, Zhang Y, Si M, Wu H, et al. Ginsenoside Metabolite Compound K Exerts Joint-Protective Effect by Interfering With Synoviocyte Function Mediated by TNF-α and Tumor Necrosis Factor Receptor Type 2. Eur J Pharmacol (2016) 771:48–55. doi: 10.1016/j.ejphar.2015.12.019
40. Tu C, Wan B, Zeng Y. Ginsenoside Rg3 Alleviates Inflammation in a Rat Model of Myocardial Infarction via the SIRT1/NF-κb Pathway. Exp Ther Med (2020) 20(6):238. doi: 10.3892/etm.2020.9368
41. Huynh DTN, Baek N, Sim S, Myung CS, Heo KS. Minor Ginsenoside Rg2 and Rh1 Attenuates LPS-Induced Acute Liver and Kidney Damages via Downregulating Activation of TLR4-STAT1 and Inflammatory Cytokine Production in Macrophages. Int J Mol Sci (2020) 21(18):6656. doi: 10.3390/ijms21186656
42. Gao H, Kang N, Hu C, Zhang Z, Xu Q, Liu Y, et al. Ginsenoside Rb1 Exerts Anti-Inflammatory Effects In Vitro and In Vivo by Modulating Toll-Like Receptor 4 Dimerization and NF-Kb/MAPKs Signaling Pathways. Phytomedicine (2020) 69:153197. doi: 10.1016/j.phymed.2020.153197
43. Hou T, Liu Y, Wang X, Jiao D, Xu H, Shi Q, et al. Ginsenoside Rg1 Promotes Lymphatic Drainage and Improves Chronic Inflammatory Arthritis. J Musculoskelet Neuronal Interact (2020) 20(4):526–34.
44. Zhang G, Xia F, Zhang Y, Zhang X, Cao Y, Wang L, et al. Ginsenoside Rd Is Efficacious Against Acute Ischemic Stroke by Suppressing Microglial Proteasome-Mediated Inflammation. Mol Neurobiol (2016) 53(4):2529–40. doi: 10.1007/s12035-015-9261-8
45. Song Y, Zhao F, Zhang L, Du Y, Wang T, Fu F. Ginsenoside Rg1 Exerts Synergistic Anti-Inflammatory Effects With Low Doses of Glucocorticoids in vitro. Fitoterapia (2013) 91:173–79. doi: 10.1016/j.fitote.2013.09.001
46. Du J, Cheng B, Zhu X, Ling C. Ginsenoside Rg1, a Novel Glucocorticoid Receptor Agonist of Plant Origin, Maintains Glucocorticoid Efficacy With Reduced Side Effects. J Immunol (2011) 187(2):942–50. doi: 10.4049/jimmunol.1002579
47. Li J, Du J, Liu D, Cheng B, Fang F, Weng L, et al. Ginsenoside Rh1 Potentiates Dexamethasone’s Anti-Inflammatory Effects for Chronic Inflammatory Disease by Reversing Dexamethasone-Induced Resistance. Arthritis Res Ther (2014) 16(3):R106. doi: 10.1186/ar4556
48. Wang R, Zhang M, Hu S, Liu K, Tai Y, Tao J, et al. Ginsenoside Metabolite Compound-K Regulates Macrophage Function Through Inhibition of β-Arrestin2. BioMed Pharmacother (2019) 115:108909. doi: 10.1016/j.biopha.2019.108909
49. Zhu ZH, Li X, He LF, Cai HF, Ye B, Wu ZM. Glycyrrhizic Acid, as an Inhibitor of HMGB1, Alleviates Bleomycin-Induced Pulmonary Toxicity in Mice Through the MAPK and Smad3 Pathways. Immunopharmacol Immunotoxicol (2021) 18:1–10. doi: 10.1080/08923973.2021.1939371
50. Luo Y, Li J, Wang B, Zhang Q, Bian Y, Wang R. Protective Effect of Glycyrrhizin on Osteoarthritis Cartilage Degeneration and Inflammation Response in a Rat Model. J Bioenerg Biomembr (2021) 53(3):285–93. doi: 10.1007/s10863-021-09889-1
51. Wang K, Zhang Y, Cao Y, Shi Z, Lin Y, Chen Y, et al. Glycyrrhetinic Acid Alleviates Acute Lung Injury by PI3K/AKT Suppressing Macrophagic Nlrp3 Inflammasome Activation. Biochem Biophys Res Commun (2020) 532(4):555–62. doi: 10.1016/j.bbrc.2020.08.044
52. Chen HJ, Kang SP, Lee IJ, Lin YL. Glycyrrhetinic Acid Suppressed NF-κb Activation in TNF-α-Induced Hepatocytes. J Agric Food Chem (2014) 62(3):618–25. doi: 10.1021/jf405352g
53. Kao TC, Wu CH, Yen GC. Glycyrrhizic Acid and 18β-Glycyrrhetinic Acid Recover Glucocorticoid Resistance via PI3K-Induced AP1, CRE and NFAT Activation. Phytomedicine (2013) 20(3-4):295–302. doi: 10.1016/j.phymed.2012.10.013
54. Kao TC, Shyu MH, Yen GC. Glycyrrhizic Acid and 18beta-Glycyrrhetinic Acid Inhibit Inflammation via PI3K/Akt/GSK3beta Signaling and Glucocorticoid Receptor Activation. J Agric Food Chem (2010) 58(15):8623–9. doi: 10.1021/jf101841r
55. Whorwood CB, Sheppard MC, Stewart PM. Licorice Inhibits 11 Beta-Hydroxysteroid Dehydrogenase Messenger Ribonucleic Acid Levels and Potentiates Glucocorticoid Hormone Action. Endocrinology (1993) 132(6):2287–92. doi: 10.1210/endo.132.6.8504732
56. Ming LG, Chen KM, Xian CJ. Functions and Action Mechanisms of Flavonoids Genistein and Icariin in Regulating Bone Remodeling. J Cell Physiol (2013) 228(3):513–21. doi: 10.1002/jcp.24158
57. Song L, Zhao J, Zhang X, Li H, Zhou Y. Icariin Induces Osteoblast Proliferation, Differentiation and Mineralization Through Estrogen Receptor-Mediated ERK and JNK Signal Activation. Eur J Pharmacol (2013) 714(1-3):15–22. doi: 10.1016/j.ejphar.2013.05.039
58. Feng R, Feng L, Yuan Z, Wang D, Wang F, Tan B, et al. Icariin Protects Against Glucocorticoid-Induced Osteoporosis In Vitro and Prevents Glucocorticoid-Induced Osteocyte Apoptosis In Vivo. Cell Biochem Biophys (2013) 67(1):189–97. doi: 10.1007/s12013-013-9533-8
59. Ma XN, Zhou J, Ge BF, Zhen P, Ma HP, Shi WG, et al. Icariin Induces Osteoblast Differentiation and Mineralization Without Dexamethasone In Vitro. Planta Med (2013) 79(16):1501–8. doi: 10.1055/s-0033-1350802
60. Ma HP, Ma XN, Ge BF, Zhen P, Zhou J, Gao YH, et al. Icariin Attenuates Hypoxia-Induced Oxidative Stress and Apoptosis in Osteoblasts and Preserves Their Osteogenic Differentiation Potential In Vitro. Cell Prolif (2014) 47(6):527–39. doi: 10.1111/cpr.12147
61. Gao J, Xiang S, Wei X, Yadav RI, Han M, Zheng W, et al. Icariin Promotes the Osteogenesis of Bone Marrow Mesenchymal Stem Cells Through Regulating Sclerostin and Activating the Wnt/β-Catenin Signaling Pathway. BioMed Res Int (2021) 2021:6666836. doi: 10.1155/2021/6666836
62. Xu Y, Li L, Tang Y, Yang J, Jin Y, Ma C. Icariin Promotes Osteogenic Differentiation by Suppressing Notch Signaling. Eur J Pharmacol (2019) 865:172794. doi: 10.1016/j.ejphar.2019.172794
63. Liang X, Hou Z, Xie Y, Yan F, Li S, Zhu X, et al. Icariin Promotes Osteogenic Differentiation of Bone Marrow Stromal Cells and Prevents Bone Loss in OVX Mice via Activating Autophagy. J Cell Biochem (2019) 120(8):13121–32. doi: 10.1002/jcb.28585
64. Hsieh TP, Sheu SY, Sun JS, Chen MH. Icariin Inhibits Osteoclast Differentiation and Bone Resorption by Suppression of MAPKs/NF-κb Regulated HIF-1α and PGE(2) Synthesis. Phytomedicine (2011) 18(2-3):176–85. doi: 10.1016/j.phymed.2010.04.003
65. Chen KM, Ge BF, Liu XY, Ma PH, Lu MB, Bai MH, et al. Icariin Inhibits the Osteoclast Formation Induced by RANKL and Macrophage-Colony Stimulating Factor in Mouse Bone Marrow Culture. Pharmazie (2007) 62(5):388–91.
66. Zhang D, Zhang J, Fong C, Yao X, Yang M. Herba Epimedii Flavonoids Suppress Osteoclastic Differentiation and Bone Resorption by Inducing G2/M Arrest and Apoptosis. Biochimie (2012) 94(12):2514–22. doi: 10.1016/j.biochi.2012.06.033
67. Zhang J, Song J, Shao J. Icariin Attenuates Glucocorticoid-Induced Bone Deteriorations, Hypocalcemia and Hypercalciuria in Mice. Int J Clin Exp Med (2015) 8(5):7306–14.
68. Sun P, Wang JZ, Liu F, Xu L, Hu LP, Wang XD, et al. Clinical Study of Hugu Capsule in Treatment for Glucocorticoid-Induced Osteoporosis. J Pract Med (2016) 32(2):4122–4. doi: 10.3969/j.issn.1006-5725.2016.24.041
69. Wu JJ, Wen LP, Wu YG, Shen Q, Han Y. Effects of Xianling Gubao Capsules for the Treatment Bone Loss Induced by Glucocorticoid. China J Orthopaedics Traumatol (2009) 22(03):193–5. doi: 10.3969/j.issn.1003-0034.2009.03.013
70. Shi JQ, Chen LJ, Xu ZY, Chen QX, Li D, Lin GP. The Effect of Bugu Capsule on Osteoporosis of Systemic Lupus Erythematosus by Glucocorticoid Therapy and Expxession of Cytokin. Chin J Dermatovenereol (2010) 24(02):169–71.
71. Kwak HB, Yang D, Ha H, Lee JH, Kim HN, Woo ER, et al. Tanshinone IIA Inhibits Osteoclast Differentiation Through Down-Regulation of C-Fos and Nfatc1. Exp Mol Med (2006) 38(3):256–64. doi: 10.1038/emm.2006.31
72. Cheng L, Zhou S, Zhao Y, Sun Y, Xu Z, Yuan B, et al. Tanshinone IIA Attenuates Osteoclastogenesis in Ovariectomized Mice by Inactivating NF-kB and Akt Signaling Pathways. Am J Transl Res (2018) 10(5):1457–68.
73. Kim HH, Kim JH, Kwak HB, Huang H, Han SH, Ha H, et al. Inhibition of Osteoclast Differentiation and Bone Resorption by Tanshinone IIA Isolated From Salvia Miltiorrhiza Bunge. Biochem Pharmacol (2004) 67(9):1647–56. doi: 10.1016/j.bcp.2003.12.031
74. Kwak HB, Sun HM, Ha H, Kim HN, Lee JH, Kim HH, et al. Tanshinone IIA Suppresses Inflammatory Bone Loss by Inhibiting the Synthesis of Prostaglandin E2 in Osteoblasts. Eur J Pharmacol (2008) 601(1-3):30–7. doi: 10.1016/j.ejphar.2008.10.034
75. Li J, He C, Tong W, Zou Y, Li D, Zhang C, et al. Tanshinone IIA Blocks Dexamethasone-Induced Apoptosis in Osteoblasts Through Inhibiting Nox4-Derived ROS Production. Int J Clin Exp Pathol (2015) 8(10):13695–706.
76. Zhu S, Wei W, Liu Z, Yang Y, Jia H. Tanshinone−IIA Attenuates the Deleterious Effects of Oxidative Stress in Osteoporosis Through the NF−κb Signaling Pathway. Mol Med Rep (2018) 17(5):6969–76. doi: 10.3892/mmr.2018.8741
77. Wang L, Cheng L, Zhang B, Wang N, Wang F. Tanshinone Prevents Alveolar Bone Loss in Ovariectomized Osteoporosis Rats by Up-Regulating Phosphoglycerate Dehydrogenase. Toxicol Appl Pharmacol (2019) 376:9–16. doi: 10.1016/j.taap.2019.05.014
78. Xi J, Li Q, Luo X, Wang Y, Li J, Guo L, et al. Celastrol Inhibits Glucocorticoid−Induced Osteoporosis in Rat via the PI3K/AKT and Wnt Signaling Pathways. Mol Med Rep (2018) 18(5):4753–9. doi: 10.3892/mmr.2018.9436
79. Kim J, Lee H, Kang KS, Chun KH, Hwang GS. Protective Effect of Korean Red Ginseng Against Glucocorticoid-Induced Osteoporosis In Vitro and In Vivo. J Ginseng Res (2015) 39(1):46–53. doi: 10.1016/j.jgr.2014.06.001
80. Tang Q. Experimental Study on Inhibitory Effect of Osthole on Rat Osteoporosis Induced by Dexamethasone and Its Mechanism. [Dissertation/Master’s Thesis]. Suzhou (2006).
81. Wang YK, Chen J, Chen J, Dong C, Yan X, Zhu ZP, et al. Daphnetin Ameliorates Glucocorticoid-Induced Osteoporosis via Activation of Wnt/GSK-3β/β-Catenin Signaling. Toxicol Appl Pharmacol (2020) 409:115333. doi: 10.1016/j.taap.2020.115333
82. Li S, Jiang H, Gu X. Echinacoside Suppresses Dexamethasone-Induced Growth Inhibition and Apoptosis in Osteoblastic MC3T3-E1 Cells. Exp Ther Med (2018) 16(2):643–8. doi: 10.3892/etm.2018.6199
83. Lucinda LMF, Aarestrup BJV, Brandão JS, Peters VM, de Reis JEP, . de Guerra MO. The Effect of Ginkgo Biloba Extract Treatment in the Bcl-2 Expression by Osteoblasts in the Femoral Trabecular Bone of Wistar Rats With Glucocorticoid-Induced Osteoporosis. Rev Bras Farmacognosia (2014) 24(3):363–66. doi: 10.1016/j.bjp.2014.07.015
84. Liu S, Fang T, Yang L, Chen Z, Mu S, Fu Q. Gastrodin Protects MC3T3-E1 Osteoblasts From Dexamethasone-Induced Cellular Dysfunction and Promotes Bone Formation via Induction of the NRF2 Signaling Pathway. Int J Mol Med (2018) 41(4):2059–69. doi: 10.3892/ijmm.2018.3414
85. Fan S, Gao X, Chen P, Li X. Myricetin Ameliorates Glucocorticoid-Induced Osteoporosis Through the ERK Signaling Pathway. Life Sci (2018) 207:205–11. doi: 10.1016/j.lfs.2018.06.006
86. Li G, Bu J, Zhu Y, Xiao X, Liang Z, Zhang R. Curcumin Improves Bone Microarchitecture in Glucocorticoid-Induced Secondary Osteoporosis Mice Through the Activation of microRNA-365 via Regulating MMP-9. Int J Clin Exp Pathol (2015) 8(12):15684–95.
87. Sun K, Zhu J, Deng Y, Xu X, Kong F, Sun X, et al. Gamabufotalin Inhibits Osteoclastgenesis and Counteracts Estrogen-Deficient Bone Loss in Mice by Suppressing RANKL-Induced NF-κb and ERK/MAPK Pathways. Front Pharmacol (2021) 12:629968. doi: 10.3389/fphar.2021.629968
88. Suhana MR, Farihah HS, Faizah O, Nazrun SA, Norazlina M, Norliza M, et al. Piper Sarmentosum Prevents Glucocorticoid-Induced Osteoporotic Bone Resorption by Increasing 11β-Hydroxysteroid Dehydrogenase Type 1 Activity. Clin Ter (2011) 162(4):313–8.
89. Dai W, Jiang L, Lay YA, Chen H, Jin G, Zhang H, et al. Prevention of Glucocorticoid Induced Bone Changes With Beta-Ecdysone. Bone (2015) 74:48–57. doi: 10.1016/j.bone.2015.01.001
90. Chen G. Effects of Total Flavones of Pueraria Lobata on Osteoporosis and the Possible Mechanisms. [Dissertation/Master’s Thesis]. Hefei (2014).
91. Zhang M, Sun FJ, Yu HJ, Lin X, Sheng GL, Li GH. The Effect of Chilk Extracts on Dexamethasone-Induced Osteoporosis Rat Model. Chin J Osteoporosis (2011) 17(7):589–92. doi: 10.3969/j.issn.1006-7108.2011.07.009
92. Wang XM, Zhao LK, Liu XX, Qi JY. Effects of LBP on Calcium Absorption and Biochemical Indicators in Glucocorticoid-Induced Osteoporosis Rats. Chin Prev Med (2011) 12(12):1004–7. doi: 10.16506/j.1009-6639.2011.12.009
Keywords: glucocorticoid-induced osteoporosis, herb medicine, escin, ginsenoside, glycyrrhizic acid, icariin
Citation: Zhang L, Li X, Ying T, Wang T and Fu F (2021) The Use of Herbal Medicines for the Prevention of Glucocorticoid-Induced Osteoporosis. Front. Endocrinol. 12:744647. doi: 10.3389/fendo.2021.744647
Received: 20 July 2021; Accepted: 20 October 2021;
Published: 12 November 2021.
Edited by:
Ulrike Baschant, University Hospital Carl Gustav Carus, GermanyReviewed by:
Connie M Weaver, Purdue University, United StatesPaula Goes, Federal University of Ceara, Brazil
Copyright © 2021 Zhang, Li, Ying, Wang and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fenghua Fu, ZnVmaEB5dHUuZWR1LmNu