
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Endocrinol. , 01 October 2021
Sec. Pituitary Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.735655
 Jianyu Zhu1,2†‡
Jianyu Zhu1,2†‡ Zhicheng Wang2†‡
Zhicheng Wang2†‡ Wenze Wang3Jinghua Fan3Yi Zhang1
Wenze Wang3Jinghua Fan3Yi Zhang1 Xiaoxu Li1†
Xiaoxu Li1† Jie Liu1†Shenzhong Jiang1Kan Deng1Lian Duan1
Jie Liu1†Shenzhong Jiang1Kan Deng1Lian Duan1 Yong Yao1*†
Yong Yao1*† Huijuan Zhu4*†
Huijuan Zhu4*†Purpose: Xanthomatous hypophysitis (XHP) is an extremely rare form of primary hypophysitis for which there is a lack of clinical experience. A comprehensive understanding of its clinical characteristics, diagnosis and treatment is needed.
Methods: Here, we report a case study and conduct a systematic review of XHP. Thirty-six cases were included, and their clinical manifestations, endocrine assessment, imaging features, treatment and follow-up data were collected and analyzed.
Results: The mean age at diagnosis was 39.1 years, and females were predominant (75.0%). The most common symptom was headache (68.6%), and 66.7% of female patients presented menstrual disorders. The most common pituitary dysfunction was growth hormone (GH) deficiency. More than half of patients exhibited central diabetes insipidus (CDI). The majority of patients had an imaging presentation of a cystic lesion with peripheral enhancement. Pituitary stalk thickening was observed in half of the patients. Total lesion resection was achieved in 57.1% of cases. The recurrence rate after partial resection and biopsy was significantly higher than that after total lesion resection (57.1% vs. 0.0%, P = 0.0147). The most common pituitary hormone abnormalities to resolve after surgery were hyperprolactinemia (100.0%) and GH deficiency (91.7%). The typical pathological feature was inflammatory infiltration of foamy histiocytes, which showed positivity for CD68.
Conclusion: Diagnosis of XHP is difficult when relying on clinical symptoms and imaging features. Therefore, surgical histopathology is necessary. Based on the available evidence, total lesion resection is recommended for treatment. However, the long-term prognosis for this rare disease remains unclear.
Hypophysitis is a rare disease characterized by lymphocytic infiltration of the pituitary gland, which can be divided into primary and secondary types (1, 2). Primary hypophysitis is the most common type, including several heterogeneous forms (1, 2). XHP is thought to be the rarest subtype of primary hypophysitis, and there is a lack of clinical experience with this disease (2). In this study, we report a case of XHP in our center and review all of the reported cases in previous literature. The objective of this study is to summarize the clinical characteristics and to bring an up-to-date understanding of XHP to provide evidence for the diagnosis, differential diagnosis and treatment of this rare disease.
A review of the literature was performed in accordance with PRISMA guidelines. The terms “xanthomatous hypophysitis” and “xanthomatous AND hypophysitis” were searched using the online database PubMed until May 2021. Cases whose diagnosis of XHP was confirmed by histological examination were included. Review articles without new cases were excluded. The relevant adequate cases mentioned in the references of these articles were also considered. The search flowchart is shown in Figure 1. lf total of 35 applicable cases reported in 20 articles were finally included.
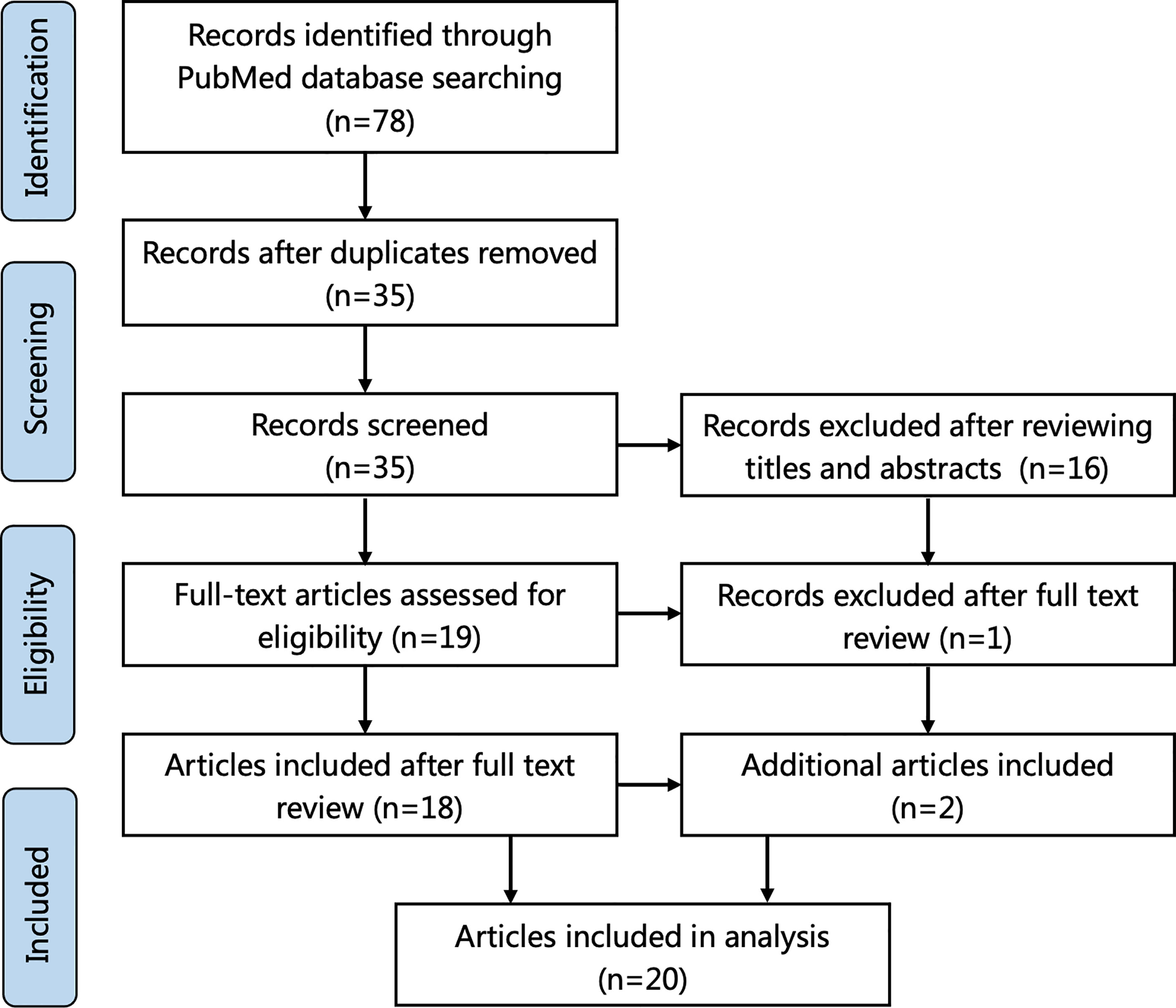
Figure 1 Flowchart of literature selection using the PRISMA guidelines.
Information on clinical symptoms, laboratory results, radiological features, surgery and postoperative conditions was extracted from these articles. Continuous data are expressed as the means ± standard deviations; categorical variables are summarized as frequencies and percentages and were analyzed using the χ2 test or Fisher’s exact test. The data were analyzed using GraphPad Prism, version 8 (GraphPad Software, San Diego, California, USA).
A 36-year-old woman presented with a 5-month history of headaches, polyuria and polydipsia and a 2-month history of amenorrhea. Her daily water intake was 3500-5000 ml. A water deprivation test indicated central diabetes insipidus (CDI). The results of the pituitary hormone test are shown in Table 1. Her anti-peroxidase antibody (>600 IU/mL, normal range<34 IU/mL) and anti-thyroglobulin antibody (730 IU/mL, normal range<115 IU/mL) were high. Thyroid gland ultrasonography showed a normal volume with diffuse heterogeneous background echotexture of both thyroid glands. A visual field test showed partial bilateral temporal defects. A pituitary magnetic resonance imaging (MRI) scan revealed a 1.7×1.2 cm cystic lesion occupying the sella turcica with mainly peripheral enhancement (Figures 2A–D). Desmopressin (0.05 mg) was administered orally three times per day for DI as hormone replacement therapy. Endoscopic transsphenoidal surgery was performed and displayed a soft yellowish lesion with pus-like discharge (Figures 2E, F). The lesion was totally removed. The pathologic examination showed inflammatory infiltration of normal pituicytes by foamy histiocytes and lymphocytes, with fibrosis, local cysts and necrosis. Immunohistochemistry of the foamy histiocytes indicated strong staining for CD68 (Figures 2G–J).
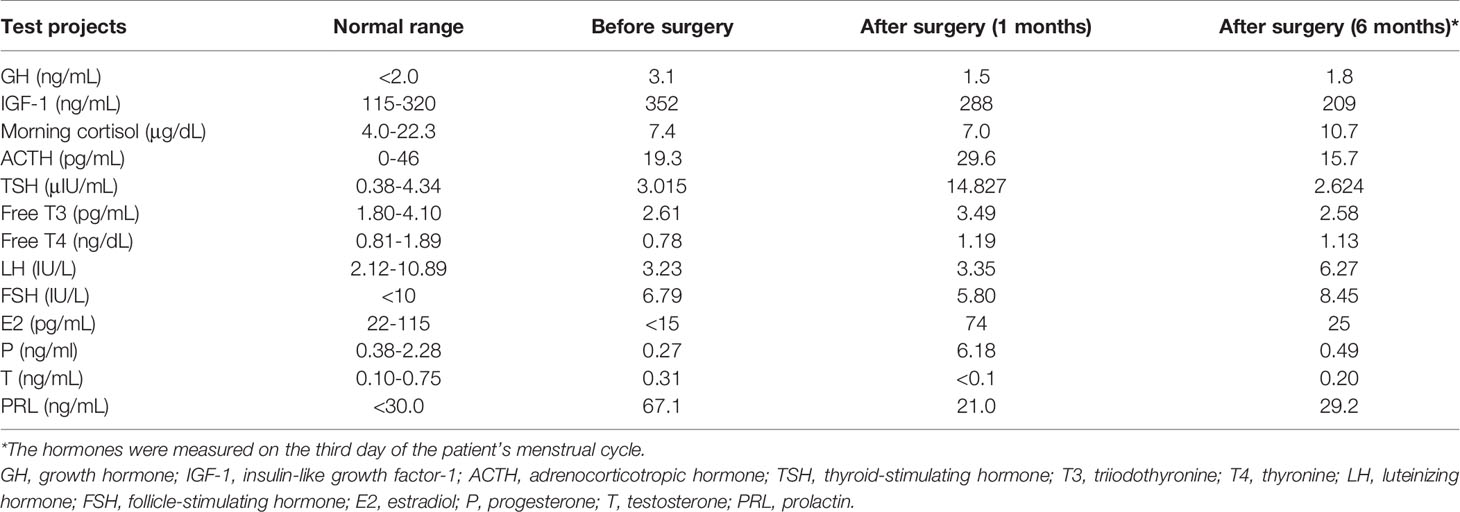
Table 1 The results of the pituitary hormone test in our case.
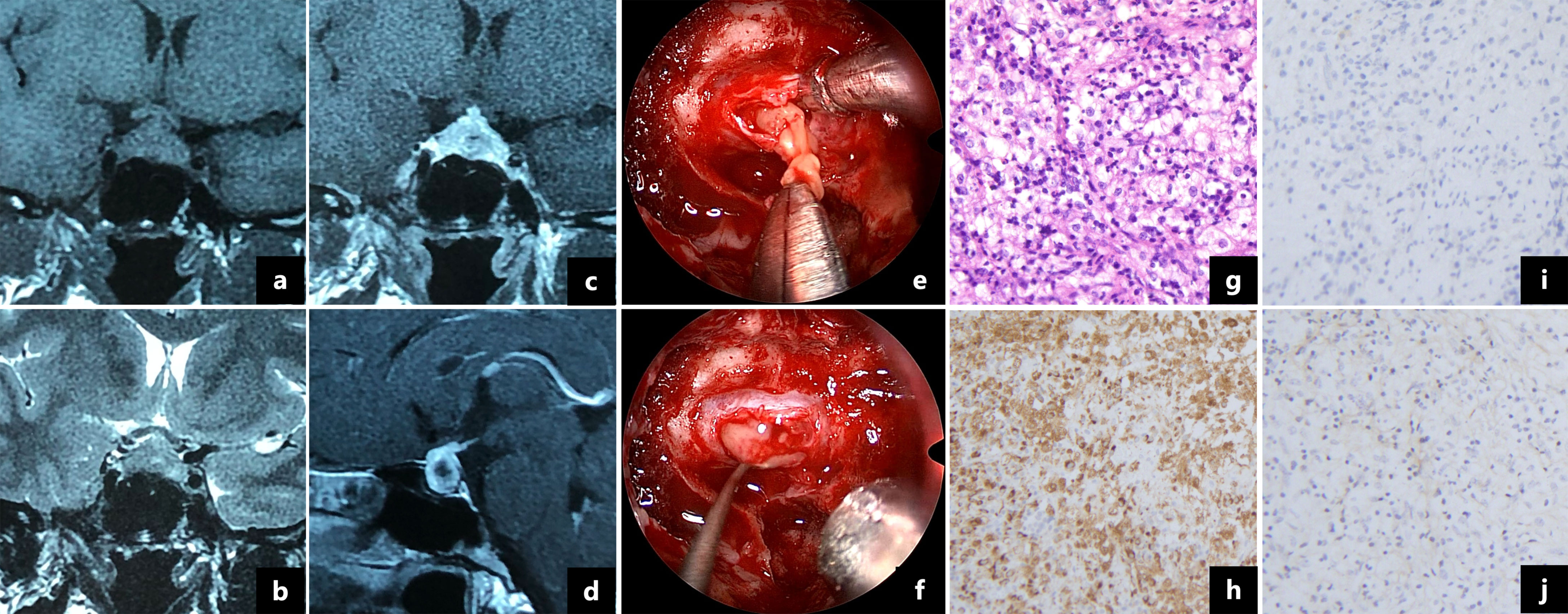
Figure 2 Magnetic resonance imaging showed a 1.7×1.2 cm cystic lesion with a thickened pituitary stalk and mainly peripheral enhancement (A–D). Intraoperative pictures revealed a soft yellow lesion (E) with pus-like fluid (F). Pathologic examination showed that the pituitary gland was infiltrated by foamy histocytes and lymphocytes (G, ×200). The foamy cells were immunopositive for CD68 (H, ×200) and immunonegative for CD1a (I, ×200) and S-100 protein (J, ×200).
The patient was discharged with no surgical complications. Postoperatively, her headaches and visual field defects resolved. The dose of desmopressin decreased during follow-up, and levothyroxine was administered because of autoimmune thyroiditis. The patient’s menstruation resumed three months after surgery. At the 6-month follow-up, desmopressin was discontinued, and the pituitary hormones were within the normal range (Table 1).
There were 36 cases of XHP, including 35 in previous literature (3–22) and one in our center (summarized in Table 2). The mean age at diagnosis was 39.1 years (range 12-72), and 27 (75.0%) patients were female. The most common symptoms were headache (68.6%), visual impairment (34.3%) and diabetes insipidus (DI) (36.1%). A total of 66.7% of female patients presented menstrual disorders, and three (8.3%) patients presented fever (Figure 3A). In addition, apoplectic events occurred in two patients (10, 13). Interestingly, five patients had autoimmune disease, including two who had autoimmune thyroiditis (12), one who had rheumatoid arthritis (13), one who had ulcerative colitis (9) and one who had both rheumatoid arthritis and Sjogren’s syndrome (16).
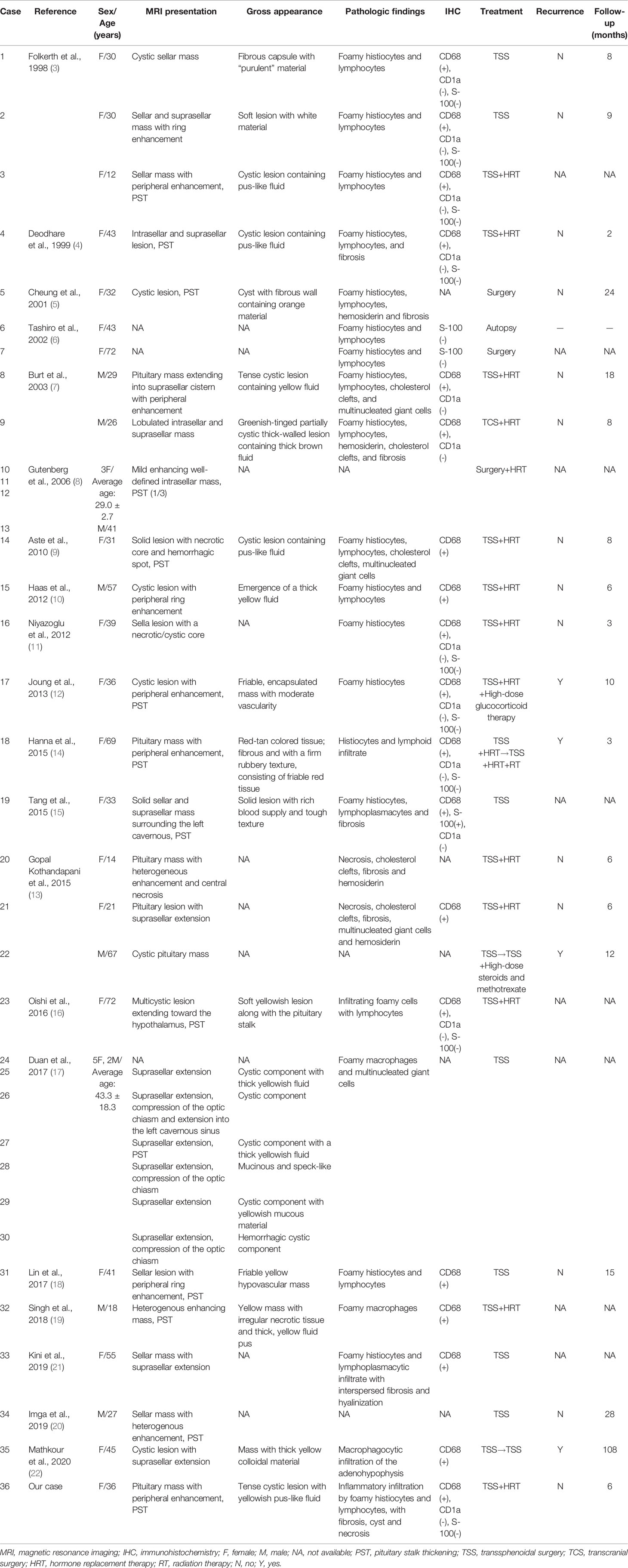
Table 2 Review of 36 cases of xanthomatous hypophysitis.
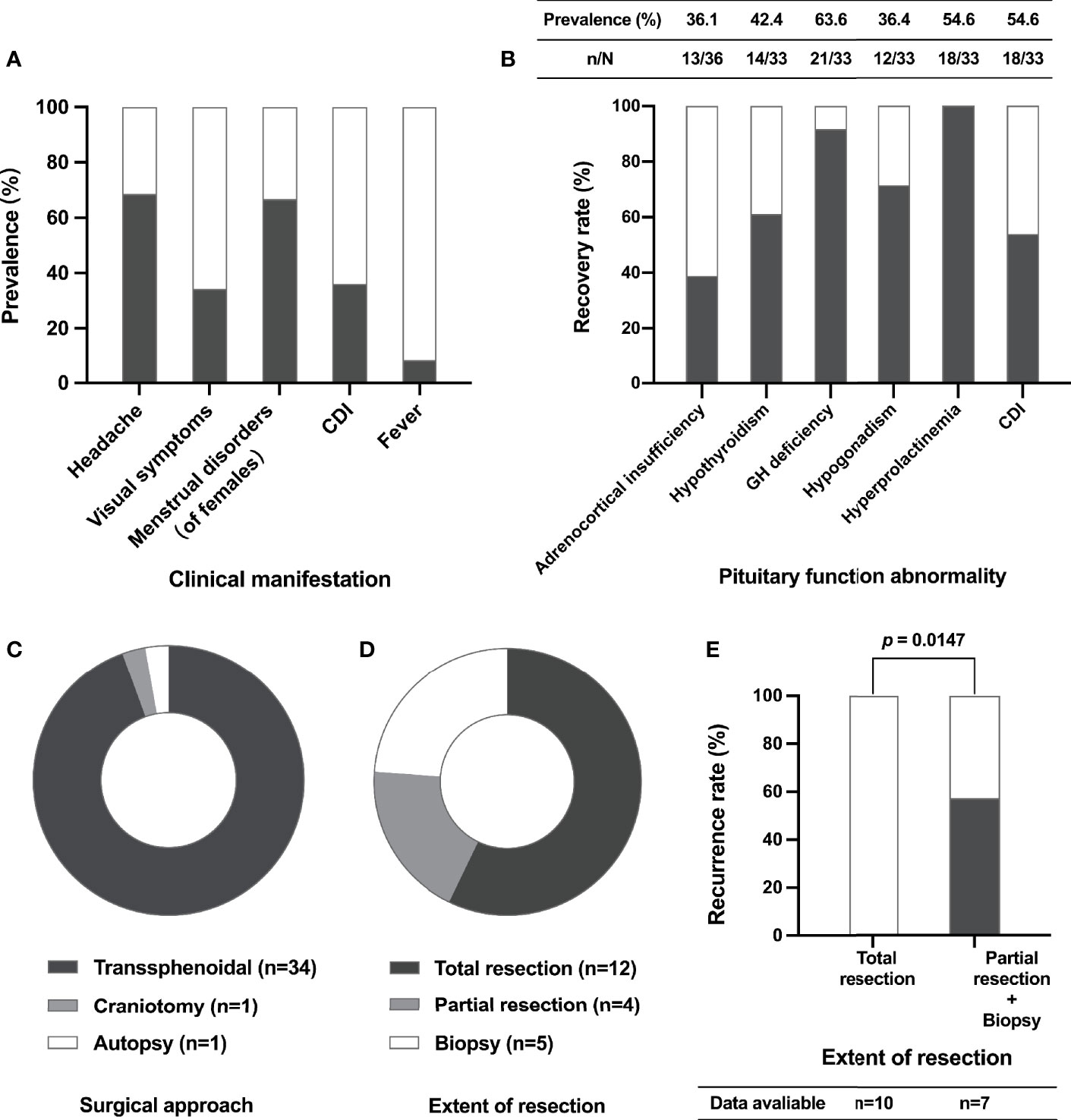
Figure 3 Clinical characteristics, surgical aspects and recurrence of XHP cases. The prevalence of different clinical manifestations of XHP patients (A); the prevalence of pituitary function abnormalities before surgery (table) and the recovery rate of pituitary function after surgery (histogram) (B); the composition of different surgical approaches (C); the composition of different extents of lesion resection (D); the relationship between recurrence and extent of resection (E). CDI, central diabetes insipidus; GH, growth hormone.
Data on anterior pituitary function before surgery were available in 33 cases (Figure 3B). Hypogonadotropic hypogonadism, adrenocortical insufficiency, central hypothyroidism and growth hormone (GH) deficiency occurred in 63.6%, 54.6%, 54.6% and 36.4% of patients, respectively. In addition, 42.4% of patients had hyperprolactinemia.
The MRI presentation of patients with XHP is shown in Table 2. Based on the available data, the MRI scan showed a cystic mass with ring or peripheral enhancement in almost all of these patients. The mean maximum diameter of these lesions was 16.3 ± 4.8 mm (data available in 20 patients). In addition, pituitary stalk thickening (PST) was shown in 50.0% of patients.
A total of 35 patients underwent surgery, including one who received craniotomy and 34 (97.14%) who received transsphenoidal surgery (Figure 3C). Based on the available data, total lesion resection was achieved in 57.14% of patients (Figure 3D). New-onset transient DI was recorded in 3 patients, and rhinorrhea appeared in one patient. Follow-up data were available in 17 patients. The median follow-up time was 8.0 (4.5, 16.5) months, ranging from 2.0 to 108.0 months. Notably, the recurrence rate of patients who underwent partial lesion resection and biopsy was significantly higher than that of patients who received total resection (57.1% vs. 0.0%, P = 0.0147) (Figure 3E). Of the four patients who developed recurrence after surgery, one received high-dose glucocorticoid therapy, and the mass showed reduction three months later (12); one patient suffered recurrence again after repeat surgery, and radiation therapy was given (14); one patient was treated with high-dose steroids, but the prognosis was unknown (13); and one patient underwent reoperation and achieved total tumor resection with no recurrence during the 9-year follow-up (22).
The most common pituitary hormone abnormality to recover is hyperprolactinemia (100.0%), followed by GH deficiency (91.7%), hypogonadism (71.4%) and hypothyroidism (61.1%) postoperatively. Recovery of adrenocortical insufficiency was only noted in 38.9% of patients. In addition, 53.8% of patients with CDI were cured through surgery (Figure 3B).
The pathological results were available for 30 cases. The descriptions of “foamy histocytes”, “lymphocytes”, “fibrosis”, “hemosiderin” and “necrosis” appeared in 20 (66.7%), 17 (56.7%), 8 (26.7%), 4 (13.3%), 4 (13.3%), and 3 (10.0%) cases, respectively. The immunohistochemical results were available for 26 cases. CD68 status results were reported in 19 cases, and all showed positivity. Of the 11 cases reporting CD1a results and 12 cases reporting S-100 results, 100.0% and 91.7% showed negativity, respectively (Table 2).
Based on the histopathologic features, primary hypophysitis can be divided into five categories: lymphocytic hypophysitis (LHP), granulomatous hypophysitis (GHP), IgG-4 hypophysitis (IgG4HP), XH and necrotizing hypophysitis (NHP) (1, 23). The first two are main forms, and the last three are rare variants (1). As the most common form of hypophysitis, LHP predominantly occurs in females, especially during pregnancy (2, 24). Approximately 20-50% of cases with LHP have been shown to have other autoimmune diseases (25). Similar to patients with XHP, headaches (48.0%) and visual defects (40.0%) are the most frequent complaints in patients with LHP, and 70.0% have anterior pituitary dysfunction (26). In addition, CDI occurs in up to 70.0% of cases with LHP (26) but is relatively uncommon (36.1%) in XHP. GHP is the second most common subtype of primary hypophysitis and shows a female predilection (23, 27). Headaches and visual changes present in 61.0% and 40.2% of cases with GHP, but CDI only occurs in 26.8% of these patients (27). Unlike other subtypes of primary hypophysitis, IgG4HP is usually found in the context of IgG4-related disease and mainly occurs in males and older patients (mean age: 64.2 years) (23, 28). A total of 52.4%, 26.2% and 17.9% of patients with IgG4HP have panhypopituitarism, anterior hypopituitarism and CDI, respectively (28). In addition, NHP is the rarest subtype of primary hypophysitis and is thought to be a severe form of one of the other subtypes of hypophysitis (23). Because XHP has similar clinical presentations to other subtypes of hypophysitis, it is difficult to differentiate these disease types by symptoms alone.
Interestingly, five patients with XHP had other autoimmune diseases. Thus, there are some views that an autoimmune phenomenon may participate in the pathogenesis, and XHP is thought to be part of a systemic autoimmune condition (12, 13). In addition, XHP may develop as a local reaction to hemorrhage, necrosis and inflammation, but the pathogenesis remains unclear (13, 23).
MRI is an important tool in the diagnosis of pituitary diseases that can provide some clues for differential diagnosis. A finding in our study was that PST occurred in half of cases with GHP. PST can be seen in a variety of diseases (29, 30). A retrospective study that summarized 321 patients with PST in our center, showed that the etiologies of PST included neoplastic (75.2%), inflammatory (13.1%), and congenital diseases (11.7%) (30). It is not difficult to identify benign tumors such as craniopharyngioma, pituitary adenoma and meningioma and Rathke cleft cyst by MRI because these diseases usually have typical imaging features (31). And age, medical history, and laboratory examination help identify germ cell tumors and metastatic tumors (32). Besides, histiocytosis such as Langerhans cell histiocytosis (LCH) and Erdheim-Chester disease (ECD) can also affect the pituitary stalk, and diabetes insipidus is found in a quarter of patients with LCH and ECD (33, 34). However, both LCH and ECD can affect multiple systems (33, 34) and long-bone involvement is considered to be a hallmark of ECD (34). Therefore, multi-organ radiological screening and appropriate histology are sufficient for differential diagnosis (33, 34).
In our study, we found that most XHPs manifest as cystic sellar lesions with peripheral enhancement on MRI. However, this performance is also seen in both LHP and NHP (24). Thus, the differential diagnosis of hypophysitis by radiological examination is still a challenge. In this situation, biopsy seems to be unavoidable to make a definitive diagnosis. As one of the largest pituitary centers in China, we have performed hundreds of biopsy procedures on patients with PST or CDI in the last few years (30, 32). According to our experience, transsphenoidal biopsy is a safe and effective way to diagnose these indistinguishable sellar lesions (32). Therefore, for sellar lesions that are difficult to identify by clinical manifestations, laboratory examinations and radiological features, biopsy surgery should be considered, especially in experienced pituitary centers. According to previous descriptions in the literature, the typical gross presentation of XHP is a cystic lesion with yellowish pus-like fluid (Table 2). In addition, based on our intraoperative findings, the lesion had a thick wall, and the texture was tough.
The common feature of primary hypophysitis is inflammatory infiltration of lymphocytes and plasma cells into the pituitary gland, but the individual features among different subtypes differ except LHP (1). The diagnosis of GHP can be confirmed by the presence of multinucleated giant cells, and IgG4HP is characterized by IgG4-positive plasma cells (1, 2, 23). The typical pathological feature of XHP is foamy histiocyte infiltration, which can be marked by CD68 glycoprotein on immunohistochemistry (Table 2). Since Langerhans cells stain positive for CD1a, S-100 and CD207 (33), the absence of CD1a and S-100 helps to rule out LCH. Thus, histopathology remains the gold-standard diagnostic criterion for different forms of primary hypophysitis and histiocytosis.
Unlike LHP, which usually presents as a self-resolving process and has a good response to glucocorticoid therapy (35), clinical experience in the treatment of XHP is extremely limited. Although Joung et al. (12) reported a patient with recurrent XHP whose mass decreased after high-dose glucocorticoid therapy, and another patient showed less response to steroid therapy (14). In addition, tentative bromocriptine therapy failed to reduce the volume of the mass (5). Our study found that the recurrence rate was significantly lower in patients who underwent total lesion resection than in those who underwent partial resection or biopsy. In addition to the adrenocortical axis, anterior pituitary function can improve obviously, and more than half of patients with CDI can be cured through surgery. Therefore, surgical resection should be considered, especially for patients suffering from mass effects and pituitary dysfunction, and total mass resection is recommended. In addition, close follow-up and routine assessment of pituitary function are essential, and hormone replacement therapy should be given postoperatively when necessary (2, 23). Nevertheless, there were still two problems in the treatment of XHP. On the one hand, the lack of follow-up data does not allow for an accurate assessment of the long-term recurrence of XHP after surgery, resulting in a lack of best evidence for the treatment. On the other hand, the gross appearance of XHP—as well as the tough texture of the mass— is unfamiliar to surgeons, which leads some patients to undergo only partial resection or biopsy. Therefore, we have summarized the characteristics of XHP to increase surgeons’ awareness of this rare disease.
There were some limitations in our study. First, due to the rarity of XHP, selection bias was intrinsic to this investigation. Second, the majority of data were extracted from descriptions in previous literature, and this may have influenced the accuracy and integrity of the results. Third, due to the heterogeneity of surgical techniques in different centers and the incompleteness of follow-up data, the optimal treatment and long-term prognosis for XHP is still unclear. Therefore, the establishment of a database of rare diseases and close follow-up should be emphasized to provide evidence for guiding the diagnosis and treatment of these rare diseases.
XHP is an extremely rare form of primary hypophysitis. Due to the similarity of its clinical manifestations and radiological findings to those of other forms of primary hypophysitis, the diagnosis and differential diagnosis of XHP are difficult to reach without a pathological examination. According to our study, pituitary dysfunction can be improved through surgery, and patients who undergo total lesion resection have a relatively low recurrence rate. Thus, surgery is both a diagnostic and a therapeutic method. However, because of the limited data, the real prognosis of this rare disease is still unclear.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
YY, HZ, and JZ: conception and design. JZ, ZW, and YZ: development of methodology. JZ, ZW, XL, JL, and SJ: acquisition and analysis of data. JZ, ZW, and YY: writing, review, and revision of manuscript. WW and JF: technical and material support. YY, HZ, LD, and KD: study supervision. All authors have read and approved the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the Chinese Academy of Medical Sciences Innovation Fund (CAMS-2016-I2M-1-002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fukuoka H. Hypophysitis. Endocrinol Metab Clin North Am (2015) 44(1):143–9. doi: 10.1016/j.ecl.2014.10.011
2. Joshi MN, Whitelaw BC, Carroll PV. Mechanisms in Endocrinology: Hypophysitis: Diagnosis and Treatment. Eur J Endocrinol (2018) 179(3):R151–r163. doi: 10.1530/eje-17-0009
3. Folkerth RD, Price DL Jr., Schwartz M, Black PM, De Girolami U. Xanthomatous Hypophysitis. Am J Surg Pathol (1998) 22(6):736–41. doi: 10.1097/00000478-199806000-00011
4. Deodhare SS, Bilbao JM, Kovacs K, Horvath E, Nomikos P, Buchfelder M, et al. Xanthomatous Hypophysitis: A Novel Entity of Obscure Etiology. Endocr Pathol (1999) 10(3):237–41. doi: 10.1007/bf02738885
5. Cheung CC, Ezzat S, Smyth HS, Asa SL. The Spectrum and Significance of Primary Hypophysitis. J Clin Endocrinol Metab (2001) 86(3):1048–53. doi: 10.1210/jcem.86.3.7265
6. Tashiro T, Sano T, Xu B, Wakatsuki S, Kagawa N, Nishioka H, et al. Spectrum of Different Types of Hypophysitis: A Clinicopathologic Study of Hypophysitis in 31 Cases. Endocr Pathol (2002) 13(3):183–95. doi: 10.1385/ep:13:3:183
7. Burt MG, Morey AL, Turner JJ, Pell M, Sheehy JP, Ho KK. Xanthomatous Pituitary Lesions: A Report of Two Cases and Review of the Literature. Pituitary (2003) 6(3):161–8. doi: 10.1023/b:pitu.0000011177.43408.56
8. Gutenberg A, Hans V, Puchner MJ, Kreutzer J, Brück W, Caturegli P, et al. Primary Hypophysitis: Clinical-Pathological Correlations. Eur J Endocrinol (2006) 155(1):101–7. doi: 10.1530/eje.1.02183
9. Aste L, Bellinzona M, Meleddu V, Farci G, Manieli C, Godano U. Xanthomatous Hypophysitis Mimicking a Pituitary Adenoma: Case Report and Review of the Literature. J Oncol (2010) 2010:1–5. doi: 10.1155/2010/195323
10. Haas AV, Smith TW. Xanthomatous Hypophysitis: An Unusual Case of a Sellar Mass. Neurological Bulletin. (2012) 4(1):41–4. doi: 10.7191/neurol_bull.2012.1034
11. Niyazoglu M, Celik O, Bakkaloglu DV, Oz B, Tanriöver N, Gazioglu N, et al. Xanthomatous Hypophysitis. J Clin Neurosci (2012) 19(12):1742–4. doi: 10.1016/j.jocn.2011.08.041
12. Joung JY, Jeong H, Cho YY, Huh K, Suh YL, Kim KW, et al. Steroid Responsive Xanthomatous Hypophysitis Associated With Autoimmune Thyroiditis: A Case Report. Endocrinol Metab (Seoul). (2013) 28(1):65–9. doi: 10.3803/EnM.2013.28.1.65
13. Gopal-Kothandapani JS, Bagga V, Wharton SB, Connolly DJ, Sinha S, Dimitri PJ. Xanthogranulomatous Hypophysitis: A Rare and Often Mistaken Pituitary Lesion. Endocrinol Diabetes Metab Case Rep (2015) 25(1):1–9. doi: 10.1530/edm-14-0089
14. Hanna B, Li YM, Beutler T, Goyal P, Hall WA. Xanthomatous Hypophysitis. J Clin Neurosci (2015) 22(7):1091–7. doi: 10.1016/j.jocn.2015.01.019
15. Tang F, Liu H, Zhou S, Liu J, Xiao E, Tan C. MRI Manifestation of Xanthomatous Hypophysitis: A Case Report and Review of the Literature. Zhong Nan Da Xue Xue Bao Yi Xue Ban. (2015) 40(2):228–32. doi: 10.11817/j.issn.1672-7347.2015.02.019
16. Oishi M, Hayashi Y, Fukui I, Kita D, Miyamori T, Nakada M. Xanthomatous Hypophysitis Associated With Autoimmune Disease in an Elderly Patient: A Rare Case Report. Surg Neurol Int (2016) 7(Suppl 16):S449–53. doi: 10.4103/2152-7806.185773
17. Duan K, Asa SL, Winer D, Gelareh Z, Gentili F, Mete O. Xanthomatous Hypophysitis Is Associated With Ruptured Rathke’s Cleft Cyst. Endocr Pathol (2017) 28(1):83–90. doi: 10.1007/s12022-017-9471-x
18. Lin W, Gao L, Guo X, Wang W, Xing B. Xanthomatous Hypophysitis Presenting With Diabetes Insipidus Completely Cured Through Transsphenoidal Surgery: Case Report and Literature Review. World Neurosurg. (2017) 1041051:e7–1051.e13. doi: 10.1016/j.wneu.2017.05.156
19. Singh K, Kanodia AK, Ross P, Torgersen A, Maclean J, Leese G, et al. Xanthomatous Hypophysitis Causing Hypogonadotropic Hypogonadism Resulting in Delayed Presentation of Slipped Capital Femoral Epiphysis. Br J Neurosurg. (2018) 19:1–4. doi: 10.1080/02688697.2018.1525482
20. Imga NN, Yildirim AE, Baser OO, Berker D. Clinical and Hormonal Characteristics of Patients With Different Types of Hypophysitis: A Single-Center Experience. Arch Endocrinol Metab (2019) 63(1):47–52. doi: 10.20945/2359-3997000000102
21. Kini H, Rao R, Pai M. Xanthomatous Hypophysitis: A Rare Case Report With Review of Literature. Indian J Pathol Microbiol (2019) 62(3):448–50. doi: 10.4103/ijpm.Ijpm_319_18
22. Mathkour M, Zeoli T, Werner C, Scullen T, Garces J, Keen J, et al. Recurring Primary Xanthomatous Hypophysitis Behaving Like Pituitary Adenoma: Additional Case and Literature Review. World Neurosurg (2020) 138:27–34. doi: 10.1016/j.wneu.2020.02.055
23. Gubbi S, Hannah-Shmouni F, Verbalis JG, Koch CA. Hypophysitis: An Update on the Novel Forms, Diagnosis and Management of Disorders of Pituitary Inflammation. Best Pract Res Clin Endocrinol Metab (2019) 33(6):101371. doi: 10.1016/j.beem.2019.101371
24. Caranci F, Leone G, Ponsiglione A, Muto M, Tortora F, Muto M, et al. Imaging Findings in Hypophysitis: A Review. Radiol Med (2020) 125(3):319–28. doi: 10.1007/s11547-019-01120-x
25. Naran J, Can AS. Lymphocytic Hypophysitis. In: StatPearls. Treasure Island (FL: StatPearls Publishing LLC (2021). StatPearls Publishing Copyright © 2021.
26. Wang S, Wang L, Yao Y, Feng F, Yang H, Liang Z, et al. Primary Lymphocytic Hypophysitis: Clinical Characteristics and Treatment of 50 Cases in a Single Centre in China Over 18 Years. Clin Endocrinol (Oxf) (2017) 87(2):177–84. doi: 10.1111/cen.13354
27. Hunn BH, Martin WG, Simpson S Jr., McLean CA. Idiopathic Granulomatous Hypophysitis: A Systematic Review of 82 Cases in the Literature. Pituitary (2014) 17(4):357–65. doi: 10.1007/s11102-013-0510-4
28. Shikuma J, Kan K, Ito R, Hara K, Sakai H, Miwa T, et al. Critical Review of IgG4-Related Hypophysitis. Pituitary (2017) 20(2):282–91. doi: 10.1007/s11102-016-0773-7
29. Salvatori R. The Differential Diagnosis of Pituitary Stalk Thickening. Endocr Pract (2019) 25(6):616–8. doi: 10.4158/ep-2019-0070
30. Zhou X, Zhu H, Yao Y, Lian X, Feng F, Wang L, et al. Etiological Spectrum and Pattern of Change in Pituitary Stalk Thickening: Experience in 321 Patients. J Clin Endocrinol Metab (2019) 104(8):3419–27. doi: 10.1210/jc.2018-02297
31. Adams NC, Farrell TP, O’Shea A, O’Hare A, Thornton J, Power S, et al. Neuroimaging of Central Diabetes Insipidus-When, How and Findings. Neuroradiology (2018) 60(10):995–1012. doi: 10.1007/s00234-018-2072-7
32. Wang Z, Zhu J, Yao Y, Zhu H, Deng K, Lu L, et al. Clinical and Pathological Features of 124 Patients With Indistinguishable Sellar Lesions and Central Diabetes Insipidus. J Clin Neurosci (2020) 80:215–22. doi: 10.1016/j.jocn.2020.08.001
33. Monsereenusorn C, Rodriguez-Galindo C. Clinical Characteristics and Treatment of Langerhans Cell Histiocytosis. Hematol Oncol Clin North Am (2015) 29(5):853–73. doi: 10.1016/j.hoc.2015.06.005
34. Haroche J, Cohen-Aubart F, Amoura Z. Erdheim-Chester Disease. Blood (2020) 135(16):1311–8. doi: 10.1182/blood.2019002766
Keywords: xanthomatous hypophysitis, clinical characteristics, pathological examination, surgery, recurrence
Citation: Zhu J, Wang Z, Wang W, Fan J, Zhang Y, Li X, Liu J, Jiang S, Deng K, Duan L, Yao Y and Zhu H (2021) Xanthomatous Hypophysitis: A Case Report and Comprehensive Literature Review. Front. Endocrinol. 12:735655. doi: 10.3389/fendo.2021.735655
Received: 03 July 2021; Accepted: 15 September 2021;
Published: 01 October 2021.
Edited by:
Mônica Gadelha, Federal University of Rio de Janeiro, BrazilReviewed by:
Krystallenia I. Alexandraki, National and Kapodistrian University of Athens, GreeceCopyright © 2021 Zhu, Wang, Wang, Fan, Zhang, Li, Liu, Jiang, Deng, Duan, Yao and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Yao, ZnJlZXRpZ2VyeWFvQDE2My5jb20=; Huijuan Zhu, c2hlbmd4aW4yMDA0QDE2My5jb20=
†ORCID: Jianyu Zhu, orcid.org/0000-0002-0484-699X
Zhicheng Wang, orcid.org/0000-0002-8835-2208
Xiaoxu Li, orcid.org/0000-0003-2662-1385
Jie Liu, orcid.org/0000-0003-2740-1923
Yong Yao, orcid.org/0000-0001-6982-8832
Huijuan Zhu, orcid.org/0000-0001-5172-6870
‡These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.