
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 12 November 2021
Sec. Pediatric Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.729056
 Fiorenza Ulgiati1
Fiorenza Ulgiati1 Sophie Lhoir2Irina Balikova2Sylvie Tenoutasse1Emese Boros1Catheline Vilain3Claudine Heinrichs1Cécile Brachet1*
Sophie Lhoir2Irina Balikova2Sylvie Tenoutasse1Emese Boros1Catheline Vilain3Claudine Heinrichs1Cécile Brachet1*Objective: Experimental evidence suggests that the clinical manifestations of Triple A syndrome result from oxidative stress. Several conditions caused by oxidative stress display retinal involvement. Our objective was to assess the retina and optic nerve involvement in children with Triple A syndrome.
Methods: Eleven patients with genetically proven Triple A syndrome followed-up in our centre were approached for study participation. The main outcome was the measurement of the thicknesses of the different retinal layers by Optical Coherence Tomography (OCT).
Results: 9 patients with triple A syndrome had OCT measurements. 7 patients were children and 2 were adults; 4 were females and 5 were males. The 7 paediatric patients had at least two OCT measured at a mean interval of 7.9 months after the first one. The average Retinal Nerve Fibre Layer thickness was 74 ± 10 µm in patients compared to the paediatric reference range of 100 ± 2 µm (p<0.05).
Conclusions and Relevance: This is the first study to document retinal layer thicknesses in a series of patients with Triple A syndrome. Nearly all retinal thickness and peripapillary RNFL measurements were very significantly inferior to the reference range in Triple A patients, whatever their age. RNFL thinning was more marked at the temporal part of the optic nerve. OCT being non-invasive, it represents a promising tool to assess the severity of neurodegeneration in patients with Triple A syndrome.
Triple A syndrome or Allgrove syndrome is characterised by ACTH-resistant Adrenal insufficiency, Alacrima and Achalasia plus diverse neurological deficits. It was first described by Allgrove in 1978 (1). This autosomal recessive condition results from biallelic inactivation of the AAAS gene, which encodes a nuclear pore protein called ALADIN (alacrima–achalasia–adrenal insufficiency neurologic disorder) (2, 3). Pathogenic AAAS mutations lead either to absent ALADIN or to aberrant subcellular localisation of the ALADIN protein and nuclear pore complex malfunction. The nuclear pore complex regulates molecular trafficking between the nucleus and the cytoplasm (4).
In patients with triple A syndrome, alacrima is usually present since birth but the other features of the syndrome develop over time, pointing towards a degenerative process damaging the adrenal glands and the neuromuscular control of the lower esophageal sphincter along with other components of the nervous system. Increased oxidative stress has been shown to lead to increased apoptosis and to evolving tissue damage in patients with triple A syndrome. In vitro studies have documented oxidative stress in cultured adrenal cells, neuronal cells and fibroblasts harbouring AAAS mutations (5, 6). In patients’ fibroblasts, altered induction or downregulation of genes associated with oxidative stress and antioxidant defence have been shown (7). Furthermore, ALADIN interacts with the ferritin heavy chain protein (FTH1), which, in addition to its iron storage role, protects the nucleus from oxidative damage (8). Fibroblasts from triple A patients lack nuclear FTH1, suggesting that the nuclear translocation of FTH1 is defective. ALADIN has also been shown to interact directly with progesterone receptor membrane component 2 (PGRMC2), a microsomal protein known to regulate cytochrome P450 hydroxylases and oxidoreductases, which are critical for maintaining the cellular oxidative state and for generating precursors for hormones like cortisol (9). In vivo, a study in one patient revealed oxidative stress, evidenced by lipid peroxidation and LDL oxidation measurements (10).
Optical coherence tomography (OCT) is an imaging technique that allows quantitative imaging of the ten layers of the retina (11). Furthermore, it is particularly suitable for the paediatric population because of its non-invasiveness and short duration. The potential role of OCT measurements as biomarkers of disease progression in neurodegenerative disorders such as multiple sclerosis, Alzheimer’s disease, neuromyelitis optica and Parkinson’s disease, has been widely investigated (12–15).
Following the observation that one young patient followed for triple A syndrome presented poor colour vision, optic nerve atrophy was diagnosed. Some case reports in the literature had reported the findings of optic nerve paleness or colour vision loss in patients with triple A syndrome (16–19). We therefore decided to measure optic nerve thickness with OCT in all the patients followed-up for triple A syndrome by our team.
The aim of our study is to assess retinal involvement by OCT in patients with Triple A syndrome and to discuss the role of these measurements as markers of disease severity and progression.
The study was approved by the ethical committee of Hôpital Universitaire des Enfants Reine Fabiola.
The study is a single-centre retrospective study. The patients or their caregiver consented to publication of medical information about them or their relative.
The study included all patients with a genetically confirmed Triple A syndrome followed-up in HUDERF or previously followed-up in this Hospital between 1988 and mars 2021 and still in contact with the same medical team because of family relationship to an affected patient.
All patients underwent a thorough ophthalmic examination, in order to exclude ocular pathology that could potentially confound OCT analysis. This included measurement of best-corrected visual acuity with age-adapted optotype, colour vision assessment with Ishihara colour plates (colour vision was said to be abnormal if one or more Ishihara plate was incorrectly identified), pupil assessment, automated visual field screening, slit lamp bio microscopy, stereoscopic fundus examination with pupil dilation and digital retinal photography. The Heidelberg Spectralis (Heidelberg Engineering, Heidelberg, Germany) spectral domain (SD) OCT was used to obtain three high-resolution Macula-Fast scans and peripapillary retinal nerve fibre layer (RNFL) scans and retinal map for both eyes of each participant. The RNFL scan images the retinal nerve fibre layer in a 3.4 mm diameter circle centred on the optic nerve head. The Macula-Fast scan is used to assess the thickness and volume in the macular region. This scan images a 6 mm circle centred on the fovea. The area is divided into three circles, measuring 1, 3 and 6 mm in diameter: the two outer circles are divided into four quadrants (superior, nasal, inferior and temporal) and the central circle corresponds to the foveal region. The scans were obtained by one examiner in a single session. Care was taken to ensure that all scans were well centred. Both eyes were examined for each patient.
Continuous variables are presented as mean ± standard deviation (SD). Normative reference ranges as established by Perez-Garcia and colleagues in 2016 are used to compare to the measurements of the RNFL and macular thickness of our pædiatric study population (20). Normative reference ranges as established by Lu Cheng and colleagues are used to compare to the measurements of the ganglion cell layer and the inner plexiform layer of our pædiatric study population (21). Normative reference ranges as published in the Heidelberg Spectralis RNFL thickness database (including 330 eyes enrolled in Canada, the United States, and Germany and enrolling diverse study populations) is used to compare to the measurements of the RNFL and macular thickness of the two adult patients studied (Heidelberg Engineering GmbH 2016). Comparison of the mean ± SD of the varied measurements in our study population with the mean of the reference population was done using the one sample mean Z-test, the variance of the control sample being known. A p value below 0.05 was considered significant.
Eleven patients with Triple A syndrome currently or previously followed in our Paediatric Endocrinology and Ophthalmology Units were identified (Table 1).
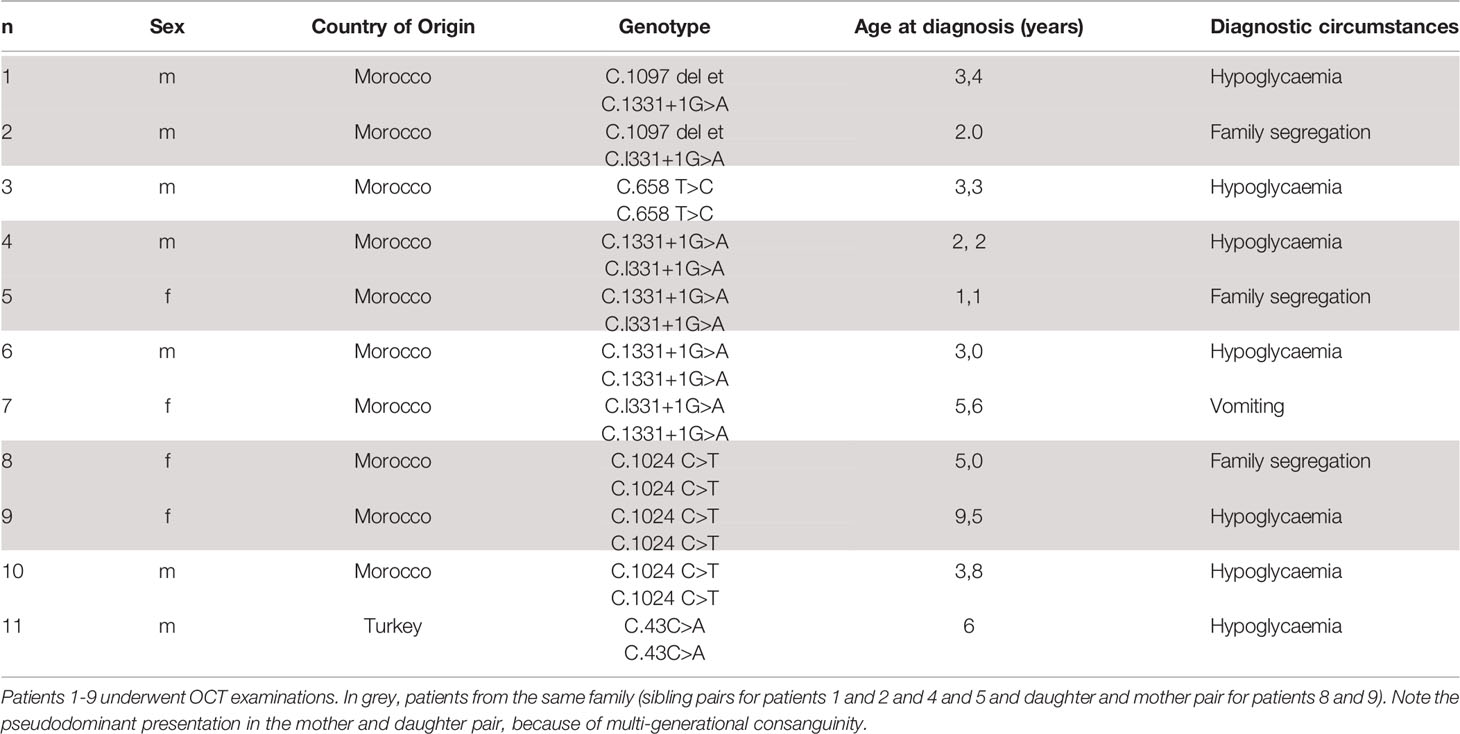
Table 1 Characteristics of the study series of patients with Triple A syndrome.
The 11 patients come from 6 families, 5 of Moroccan origin and one of Turkish origin. Most of the patients (7/11) have been diagnosed after an acute adrenal crisis with severe hypoglycaemia, whereas one patient presented with achalasia, and the remaining were identified after family segregation study. All the patients were treated for ACTH-resistant adrenal insufficiency with hydrocortisone and 6/11 patients were also treated for mineralocorticoid deficiency with 9-αFludrocortisone with good treatment adherence. Alacrima was treated with artificial tears with moderate adherence. Six patients had had surgical treatment for achalasia. OCT measurement could not be performed in two patients because of loss of follow up in one case and severe neurological sequelae in the second case. Of the patients who participated, 7 patients were children and 2 were adults; 4 were females and 5 were males. Age (range) at first OCT analysis was 4 to 39 years. All 7 pædiatric patients had a second OCT measured at a mean interval of 7.9 months after the first one and 6/7 pædiatric patients had a third OCT measured at a mean interval of 9.3 months after the second one.
Table 2 shows results from the ophthalmological evaluation in relation with the OCT findings. Eight patients out of 9 had temporal papillary pallor at fundus examination (not performed in one patient because of neurologic sequelae). Visual acuity was abnormal in all patients ranging from 0.5 to 0.85. Ishihara’s Test for Colour Deficiency could be reliably performed in 6 out of 9 patients. Colour vision was normal in one patient. It could not be performed in 4 patients because of loss of follow-up, severe neurological sequelae, or poor cooperation. Automated visual field screening was performed in 5 patients out of 9. Central scotoma was suspected in 4 patients. It was considered unreliable in 3 patients because of misunderstanding of the test. All patients had superficial punctuated keratitis at Slit lamp examination.
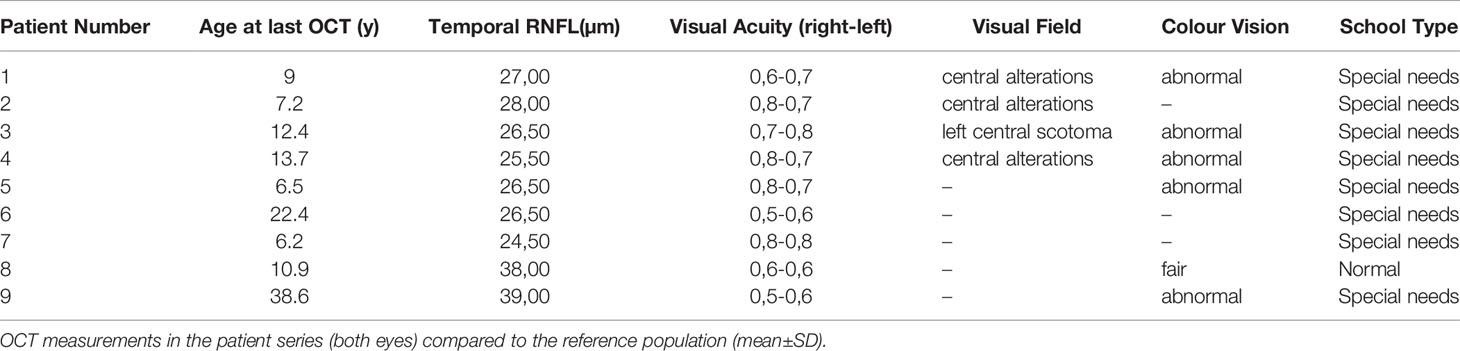
Table 2 Ophthalmological data and school type of the 9 patients with Triple A syndrome whose neurological status allowed an in-depth investigation.
Three out of 108 measurements were excluded from RNFL analysis at first OCT and 2 out of 108 measurements were excluded at second OCT, due to poor scan quality secondary to poor fixation. RNFL thickness was significantly lower in the patients compared to the mean of the reference paediatric population in 5 out of the 6 quadrants and markedly lower in the temporal quadrants (p<0.01) (Table 3). Figure 1 shows the temporal RNFL according to age at first OCT in the patients.
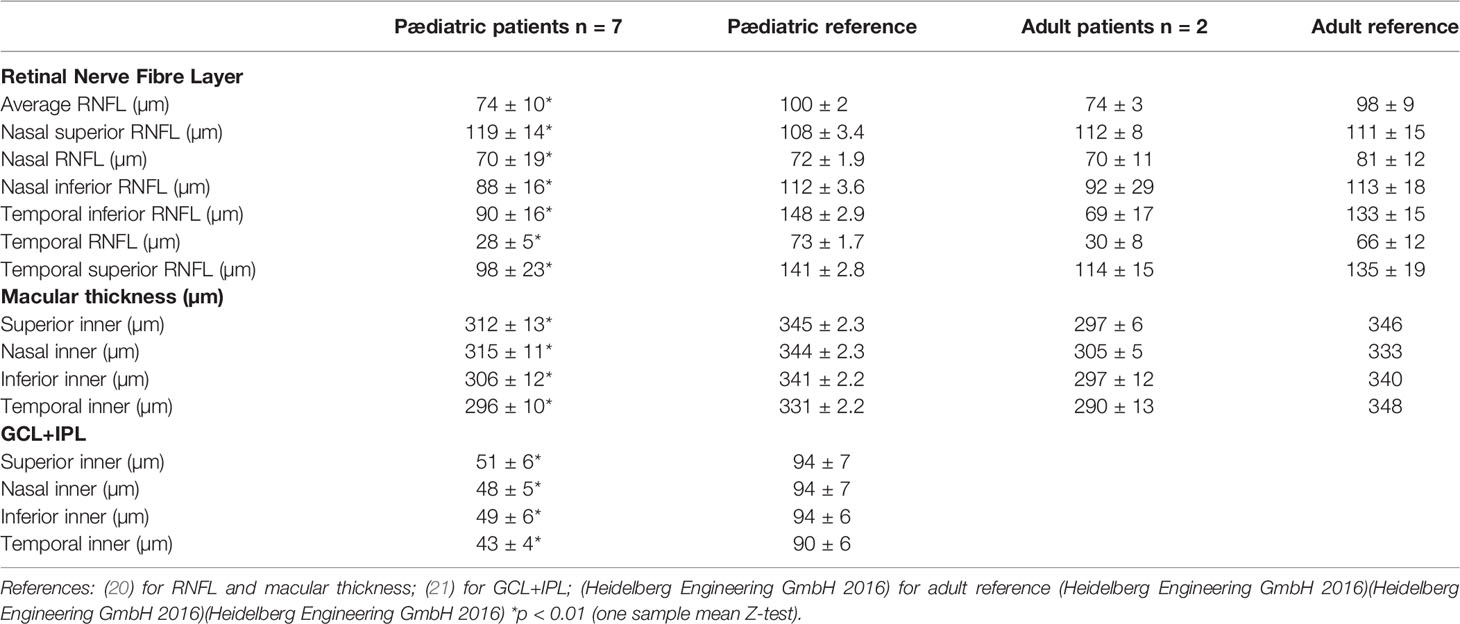
Table 3 Retinal Nerve Fibre Layer thickness (RNFL); macular ganglion cell layer plus macular inner plexiform layer thickness (GCL+IPL).
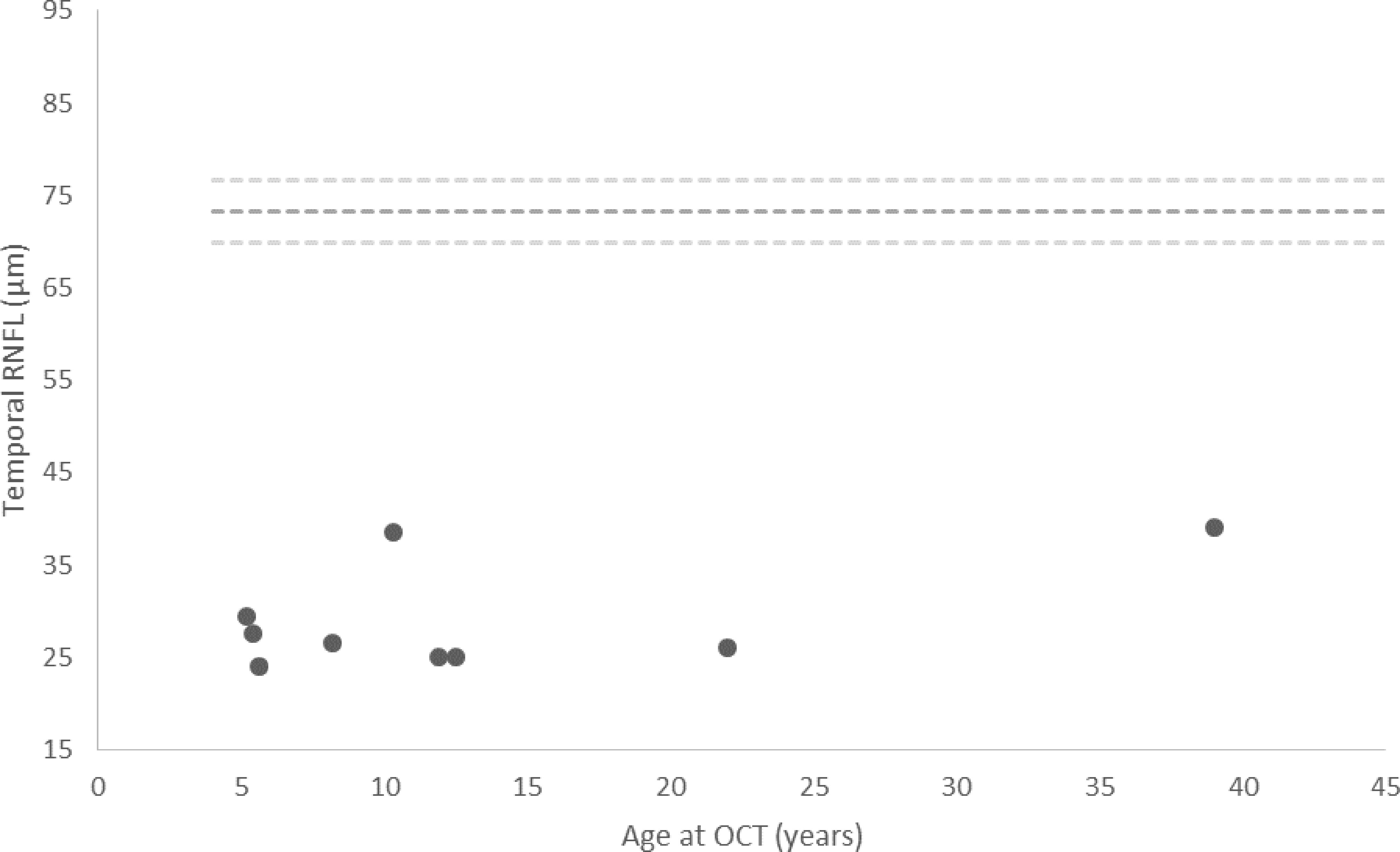
Figure 1 Temporal RNFL thickness measured at initial OCT in patients with Triple A syndrome. Each value is the mean between two eyes. The dashed lines represent the mean ± 2SD of the reference range (20). Of note, the outliers that display a temporal RNFL of nearly 40 µm are the mother and daughter pair.
Macular thickness was significantly reduced in the patients compared to the mean of the reference paediatric population in the four quadrants of the macula (Table 3).
Macular ganglion cell layer (GCL) thickness and macular inner plexiform layer (IPL) thickness was significantly reduced in the patients compared to the mean of the reference paediatric population in the four quadrants of the macula (Table 3).
OCT measurements were repeated in the paediatric patients after a mean interval of 7.9 months between the first and the second OCT and 9.3 months between the second and the third OCT (Figure 2).
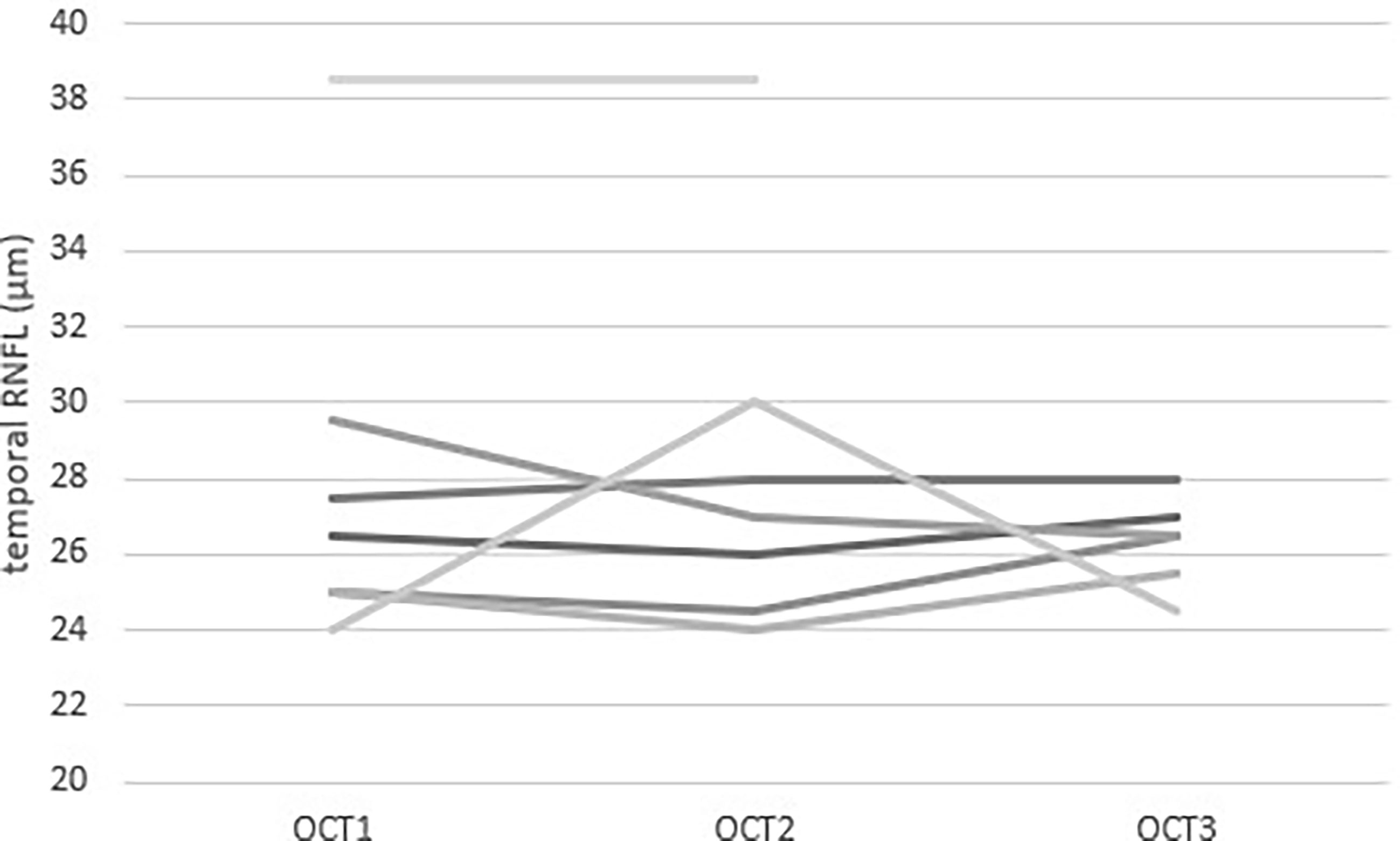
Figure 2 Repeated measurements of Temporal RNFL thickness measured by OCT in 7 paediatric patients with Triple A syndrome. Each value is the mean of the two eyes of one patient. Each line represents a patient. Three patients had three measurements during their follow-up. The mean time between OCT1 and OCT2 was 7.9 months and the mean time between OCT2 and OCT3 was 9.3 months.
This is the first study to document retinal layer thicknesses in patients with Triple A syndrome. Nearly all retinal thickness measurements are significantly lower in patients with Triple A syndrome than in the control cohorts, regardless of the age of the patients. The retinal thinning is more marked at the temporal region of the retina. The retinal thinning does not seem to be a rapidly progressive process.
Optic atrophy is rare in children. In patients with Triple A syndrome, it has been previously described in two teenage siblings (18) earlier reports mention optic disk paleness in a 21 year-old male (19), two 13 and 16 year-old boys (20) and two 3 and 6 year-old children (21).
Triple A syndrome is thought to result from an increased oxidative stress, as documented by in vitro and in vivo studies. Deficiency of ALADIN Impairs Redox Homeostasis in vitro in Adrenal and neuronal cells. (22) and markers of oxidative stress have been measured in a child with Triple A syndrome (10).
The sensitivity of adrenal glands to oxidative damage, is illustrated by three different rare conditions having in common adrenal insufficiency and oxidative stress: mutations in NNT, TXNRD2, or AAAS (22). Steroidogenesis is affected by oxidative stress through different mechanisms (23). The nervous system is also very sensitive to Redox disequilibrium. Mitochondrial dysfunction leading to oxidative stress and increased apoptosis is a feature of several inherited neurodegenerative disorders and OCT has been used to monitor disease progression in these disorders (24). Optic nerve involvement, particularly the preferential loss of the temporal RNFL, the nerve fibres which transmit central and colour vision, is a recognised feature of many mitochondrial disorders, including mitochondrial optic atrophy (25). This loss of temporal retinal ganglion cells and their axons in mitochondrial disease is attributed to the disadvantageous energy conditions of the small parvocellular axons (26). The small retinal ganglion cells in the macula may atrophy as the disease progresses and their axons, which emanate from the macula, run to the temporal aspect of the optic nerve (the papillo-macular bundle is either directly affected by the disease or becomes atrophic as a result of damage to the cell bodies in the macula) (25).
The preferential loss of temporal RNFL observed in our case series of patients with Triple A syndrome is similar to what is observed in mitochondrial optic neuropathies.
This finding, added to the documented oxidative damage observed in AAAS knock-down of adrenal and neuronal cell lines, are strong arguments for the implication of a redox disequilibrium in the retinal thinning we observed.
Our study’s has limitations: first, the small sample size (18 eyes included in OCT analysis, repeated twice in most patients, three times in some of them) due to the extreme rarity of the disease. To address this, it would be interesting to expand these measurements to a larger cohort of patients through an EndoERN collaboration. Another limitation regards the assessment of retinal thinning progression. The timeframe of our study is probably too short given that most neurological deficits progress rather slowly in patients with Triple A syndrome.
In conclusion, OCT being a rapid, objective, reproducible and non-invasive investigation method to document neural degeneration, it might have a clinical usefulness in children with Triple A syndrome to monitor neurodegeneration, as in other neurodegenerative diseases.
In addition, it could allow to assess the efficacy of potential therapeutic agents. In patients with triple A syndrome, the only publication about a disease-modifying agent regards N-acetylcysteine, a well-known antioxidant agent. In vitro, after knock-down of the AAAS gene, adrenal and neuronal cells displayed an increased viability after N-acetylcysteine treatment (5). In vivo, N-acetylcysteine (600 mg twice daily) has been administered to one paediatric patient in order to document its effect on oxidative stress measurements. The oxidative stress was assessed by measurements of a lipid peroxidation product, thiobarbituric acid reactive substances in plasma and measurement of reduced glutathione in whole blood. N-acetylcysteine was shown to lower lipid peroxidation products and increase reduced glutathione levels in the patient (5, 10). Our study documenting severe thinning of the retinal layers in children and adults with Triple A syndrome points towards OCT as a useful follow-up tool in these patients.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Ethics Committee of Queen Fabiola Children’s University Hospital. Written informed consent from the participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Jill Dedobbeleer who studied the cohort of triple A children as part of her master thesis in 2019. We thank the patients and their families for their trust and collaboration. We are grateful to both Frédérique Schwilden and Lambert Leenders, endocrine nurses, for their knowledge-sharing work aiming at the empowerment of the patients with adrenal insufficiency.
1. Allgrove J, Clayden GS, Grant DB, Macaulay JC. Familial Glucocorticoid Deficiency With Achalasia of the Cardia and Deficient Tear Production. Lancet (1978) 311(8077):1284–6. doi: 10.1016/S0140-6736(78)91268-0
2. Tullio-Pelet A, Salomon R, Hadj-Rabia S, Mugnier C, de Laet M-H, Chaouachi B, et al. Mutant WD-Repeat Protein in Triple-A Syndrome. Nat Genet (2000) 26(3):332–5. doi: 10.1038/81642
3. Handschug K, Sperling S, Yoon SJ, Hennig S, Clark AJ, Huebner A. Triple A Syndrome Is Caused by Mutations in AAAS, a New WD-Repeat Protein Gene. Hum Mol Genet (2001) 10(3):283–90. doi: 10.1093/hmg/10.3.283
4. Cronshaw JM, Matunis MJ. The Nuclear Pore Complex Protein ALADIN Is Mislocalized in Triple A Syndrome. Proc Natl Acad Sci USA (2003) 100(10):5823–7. doi: 10.1073/pnas.1031047100
5. Prasad R, Metherell LA, Clark AJ, Storr HL. Deficiency of ALADIN Impairs Redox Homeostasis in Human Adrenal Cells and Inhibits Steroidogenesis. Endocrinology (2013) 154(9):3209–18. doi: 10.1210/en.2013-1241
6. Hirano M, Furiya Y, Asai H, Yasui A, Ueno S. ALADINI482S Causes Selective Failure of Nuclear Protein Import and Hypersensitivity to Oxidative Stress in Triple A Syndrome. Proc Natl Acad Sci USA (2006) 103(7):2298–303. doi: 10.1073/pnas.0505598103
7. Koehler K, End K, Kind B, Landgraf D, Mitzscherling P, Huebner A. Changes in Differential Gene Expression in Fibroblast Cells From Patients With Triple A Syndrome Under Oxidative Stress. Horm Metab Res Horm Stoffwechselforschung Horm Metab (2013) 45(2):102–8. doi: 10.1055/s-0032-1331196
8. Storr HL, Kind B, Parfitt DA, Chapple JP, Lorenz M, Koehler K, et al. Deficiency of Ferritin Heavy-Chain Nuclear Import in Triple a Syndrome Implies Nuclear Oxidative Damage as the Primary Disease Mechanism. Mol Endocrinol Baltim Md (2009) 23(12):2086–94. doi: 10.1210/me.2009-0056
9. Jühlen R, Landgraf D, Huebner A, Koehler K. Identification of a Novel Putative Interaction Partner of the Nucleoporin ALADIN. Biol Open (2016) 5(11):1697–705. doi: 10.1242/bio.021162
10. Fragoso MCBV, Albuquerque EV de A, Cardoso AL de A, da Rosa PWL, de Paulo RB, Schimizu MHM, et al. Triple A Syndrome: Preliminary Response to the Antioxidant N-Acetylcysteine Treatment in a Child. Horm Res Paediatr (2017) 88(2):167–71. doi: 10.1159/000465520
11. Huang D, Swanson E, Lin C, Schuman J, Stinson W, Chang W, et al. Optical Coherence Tomography. Science (1991) 254(5035):1178–81. doi: 10.1126/science.1957169
12. Altintaş O, Işeri P, Ozkan B, Cağlar Y. Correlation Between Retinal Morphological and Functional Findings and Clinical Severity in Parkinson’s Disease. Doc Ophthalmol Adv Ophthalmol (2008) 116(2):137–46. doi: 10.1007/s10633-007-9091-8
13. Fisher JB, Jacobs DA, Markowitz CE, Galetta SL, Volpe NJ, Nano-Schiavi ML, et al. Relation of Visual Function to Retinal Nerve Fiber Layer Thickness in Multiple Sclerosis. Ophthalmology (2006) 113(2):324–32. doi: 10.1016/j.ophtha.2005.10.040
14. Greenberg BM, Frohman E. Optical Coherence Tomography as a Potential Readout in Clinical Trials. Ther Adv Neurol Disord (2010) 3(3):153–60. doi: 10.1177/1756285610368890
15. Iseri PK, Altinaş O, Tokay T, Yüksel N. Relationship Between Cognitive Impairment and Retinal Morphological and Visual Functional Abnormalities in Alzheimer Disease. J Neuroophthalmol Off J North Am Neuroophthalmol Soc (2006) 26(1):18–24. doi: 10.1097/01.wno.0000204645.56873.26
16. Moschos MM, Margetis I, Koehler K, Gatzioufas Z, Huebner A. New Ophthalmic Features in a Family With Triple A Syndrome. Int Ophthalmol (2011) 31(3):239–43. doi: 10.1007/s10792-011-9450-z
17. Tsilou E, Stratakis CA, Rubin BI, Hay BN, Patronas N, Kaiser-Kupfer MI. Ophthalmic Manifestations of Allgrove Syndrome: Report of a Case. Clin Dysmorphol (2001) 10(3):231–3. doi: 10.1097/00019605-200107000-00016
18. Chu ML, Berlin D, Axelrod FB. Allgrove Syndrome: Documenting Cholinergic Dysfunction by Autonomic Tests. J Pediatr (1996) 129(1):156–9. doi: 10.1016/S0022-3476(96)70205-6
19. Ehrich E, Aranoff G, Johnson WG. Familial Achalasia Associated With Adrenocortical Insufficiency, Alacrima, and Neurological Abnormalities. Am J Med Genet (1987) 26(3):637–44. doi: 10.1002/ajmg.1320260319
20. Pérez-García D, Ibañez-Alperte J, Remón L, Cristóbal JA, Sanchez-Cano A, Pinilla I. Study of Spectral-Domain Optical Coherence Tomography in Children: Normal Values and Influence of Age, Sex, and Refractive Status. Eur J Ophthalmol (2016) 26(2):135–41. doi: 10.5301/ejo.5000665
21. Cheng L, Wang M, Deng J, Lv M, Jiang W, Xiong S, et al. Macular Ganglion Cell-Inner Plexiform Layer, Ganglion Cell Complex, and Outer Retinal Layer Thicknesses in a Large Cohort of Chinese Children. Invest Ophthalmol Vis Sci (2019) 60(14):4792–802. doi: 10.1167/iovs.18-26300
22. Prasad R, Kowalczyk JC, Meimaridou E, Storr HL, Metherell LA. Oxidative Stress and Adrenocortical Insufficiency. J Endocrinol (2014) 221(3):R63–73. doi: 10.1530/JOE-13-0346
23. Kil IS, Lee SK, Ryu KW, Woo HA, Hu M-C, Bae SH, et al. Feedback Control of Adrenal Steroidogenesis via H2O2-Dependent, Reversible Inactivation of Peroxiredoxin III in Mitochondria. Mol Cell (2012) 46(5):584–94. doi: 10.1016/j.molcel.2012.05.030
24. Kersten HM, Roxburgh RH, Danesh-Meyer HV. Ophthalmic Manifestations of Inherited Neurodegenerative Disorders. Nat Rev Neurol (2014) 10(6):349–62. doi: 10.1038/nrneurol.2014.79
25. Sitarz KS, Chinnery PF, Yu-Wai-Man P. Disorders of the Optic Nerve in Mitochondrial Cytopathies: New Ideas on Pathogenesis and Therapeutic Targets. Curr Neurol Neurosci Rep (2012) 12(3):308–17. doi: 10.1007/s11910-012-0260-0
Keywords: AAAS, ALADIN, Triple A syndrome, OCT, retina, neurodegeneration, Redox, RNFL
Citation: Ulgiati F, Lhoir S, Balikova I, Tenoutasse S, Boros E, Vilain C, Heinrichs C and Brachet C (2021) The Retina in Patients With Triple A Syndrome: A Window Into Neurodegeneration? Front. Endocrinol. 12:729056. doi: 10.3389/fendo.2021.729056
Received: 22 June 2021; Accepted: 15 October 2021;
Published: 12 November 2021.
Edited by:
George Paltoglou, National and Kapodistrian University of Athens, GreeceReviewed by:
Alfonso Maria Lechuga-Sancho, University of Cádiz, SpainCopyright © 2021 Ulgiati, Lhoir, Balikova, Tenoutasse, Boros, Vilain, Heinrichs and Brachet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cécile Brachet, Y2VjaWxlLmJyYWNoZXRAaHVkZXJmLmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.