 Can Thi Bich Ngoc1†
Can Thi Bich Ngoc1† Tran Minh Dien1†
Tran Minh Dien1† Elisa De Franco2Sian Ellard2,3Jayne A. L. Houghton3
Elisa De Franco2Sian Ellard2,3Jayne A. L. Houghton3 Nguyen Ngoc Lan4Bui Phuong Thao1
Nguyen Ngoc Lan4Bui Phuong Thao1 Nguyen Ngoc Khanh1
Nguyen Ngoc Khanh1 Sarah E. Flanagan2Maria E. Craig5,6
Sarah E. Flanagan2Maria E. Craig5,6 Vu Chi Dung1*
Vu Chi Dung1*- 1Department of Endocrinology, Metabolism and Genetics, Vietnam National Children’s Hospital, Hanoi, Vietnam
- 2Institute of Biomedical and Clinical Science, College of Medicine and Health, University of Exeter, Exeter, United Kingdom
- 3Exeter Genomics Laboratory, Royal Devon & Exeter NHS Foundation Trust, Exeter, United Kingdom
- 4Institute of Genome Research, Vietnam Academy of Science and Technology (VAST), Hanoi, Vietnam
- 5Institute of Endocrinology and Diabetes, The Children’s Hospital at Westmead/Discipline of Child and Adolescent Health, University of Sydney, Sydney, NSW, Australia
- 6School of Women’s and Children’s Health, University of New South Wales Medicine, Sydney, NSW, Australia
Background: Neonatal diabetes mellitus (NDM) is defined as insulin-requiring persistent hyperglycemia occurring within the first 6 months of life, which can result from mutations in at least 25 different genes. Activating heterozygous mutations in genes encoding either of the subunits of the ATP-sensitive K+ channel (KATP channel; KCNJ11 or ABCC8) of the pancreatic beta cell are the most common cause of permanent NDM and the second most common cause of transient NDM. Patients with NDM caused by KATP channel mutations are sensitive to sulfonylurea (SU) treatment; therefore, their clinical management can be improved by replacing insulin with oral agents.
Patients and Methods: Seventy patients were diagnosed with NDM between May 2008 and May 2021 at Vietnam National Children’s Hospital, and molecular genetic testing for all genes known to cause NDM was performed at the Exeter Genomic Laboratory, UK. Patients with ABCC8 or KCNJ11 mutations were transferred from insulin to oral SU. Clinical characteristics, molecular genetics, and annual data relating to glycemic control, SU dose, severe hypoglycemia, and side effects were collected. The main outcomes of interest were SU dose, SU failure (defined as permanent reintroduction of daily insulin), and glycemic control (HbA1c).
Results: Fifty-four of 70 patients (77%) with NDM harbored a genetic mutation and of these; 27 (50%) had activating heterozygous mutations in ABCC8 or KCNJ11. A total of 21 pathogenic mutations were identified in the 27 patients, including 13 mutations in ABCC8 and 8 mutations in KCNJ11. Overall, 51% had low birth weight (below 3rd percentile), 23 (85%) were diagnosed before 3 months of age, and 23 (85%) presented with diabetic ketoacidosis. At diagnosis, clinical and biochemical findings (mean ± SD) were pH 7.16 ± 0.16; , 7.9 ± 7.4 mmol/L; BE, −17.9 ± 9.1 mmol/L; HbA1C, 7.98% ± 2.93%; blood glucose, 36.2 ± 12.3 mmol/L; and C-peptide median, 0.09 (range, 0–1.61 nmol/l). Twenty-six patients were successfully transferred from insulin to SU therapy. In the remaining case, remission of diabetes occurred prior to transfer. Glycemic control on SU treatment was better than on insulin treatment: HbA1c and blood glucose level decreased from 7.58% ± 4.63% and 19.04 ± 14.09 mmol/L when treated with insulin to 5.8 ± 0.94% and 6.87 ± 3.46 mmol/L when treated with SU, respectively.
Conclusions: This is the first case series of NDM patients with ABCC8/KCNJ11 mutations reported in Vietnam. SU is safe in the short term for these patients and more effective than insulin therapy, consistent with all studies to date. This is relevant for populations where access to and cost of insulin are problematic, reinforcing the importance of genetic testing for NDM.
Introduction
Neonatal diabetes mellitus (NDM) is defined as uncontrolled hyperglycemia with onset in the first 6 months of life. It is estimated to affect one in 90,000 newborns (1). NDM can be divided into three forms based on phenotypic characteristics; transient NDM (TNDM), permanent NDM (PNDM), and syndromic NDM (2). TNDM and PNDM account for 90% of NDM cases. NDM can result from mutations in at least 25 different genes (2). Most cases of TNDM are caused by imprinting defects on chromosome 6q24, with presentation in infancy, remission, and subsequent relapse in later life. Activating heterozygous mutations in the genes encoding either of the subunits of the ATP-sensitive K+ channel (KATP channel; KCNJ11 or ABCC8) of the pancreatic beta cell are the second most common cause of TNDM (26% of TNDM cases) and the most common causes of PNDM (44% of PNDM cases) (2). The KCNJ11 (MIM # 600937) and ABCC8 (MIM # 60059) genes are located on the short arm of chromosome 11 (11p15.1) and encode the Kir6.2 subunit and the SU receptor 1 (SUR1) regulatory subunit of the KATP channel, respectively. In the normal pancreatic beta-cell, increased glucose enters the cell via a glucose transporter and is metabolized by the enzyme glucokinase, resulting in increased production of ATP. This causes closure of the KATP channel, which, in turn, depolarizes the cell membrane, activating the influx of calcium through voltage-gated calcium channels that subsequently allows exocytosis of insulin granules. Activating KCNJ11 and ABCC8 mutations cause the KATP channels to remain inappropriately open even in the presence of hyperglycemia. Without channel closure, the cell membrane is not able to depolarize effectively; thus, insulin cannot be released from the beta-cell (3).
In the clinical setting, insulin is the immediate choice for establishing glycemic control in NDM patients because it will be effective in all cases where an insulin deficit is involved. If a diagnosis of diabetes is made before 6 months of age and genetic screening is undertaken, the identification of mutations in KCNJ11 or ABCC8 provides an alternative therapeutic strategy. This is because if NDM results from overactive KATP channels, closing these channels is a key step to suppress insulin release, and sulfonylureas (SU) are well-studied KATP channel inhibitors (4). Moreover, SU have proven to be an effective treatment for individuals with NDM resulting from KCNJ11 or ABCC8 mutations (5, 6).
In 2020, De Franco et al. (7) collected and summarized a total of 748 ABCC8 and 205 KCNJ11 pathogenic and likely pathogenic mutations associated with congenital hyperinsulinism and NDM from various countries. In the present study, we report KCNJ11/ABCC8 pathogenic mutations in Vietnamese patients with NDM diagnosed at Vietnam National Children’s Hospital between May 2008 and May 2021, and the outcomes of SU therapy transfer.
Research Design and Methods
Study Design and Individuals
The patients were diagnosed with NDM and were admitted to Vietnam National Children’s Hospital from May 2008 to May 2021. Inclusion criteria were hyperglycemia onset before 6 months of age and fasting blood glucose ≥126 mg/dl (7.0 mmol/L). Fasting was defined as no caloric intake for at least 4 h in children aged 0–1 years or random plasma glucose concentration ≥11.1 mmol/L (200 mg/dl). Hyperglycemia lasting at least 2 weeks required insulin for treatment. All patients with KCNJ11 or ABCC8 mutations and their parents agreed to participate in the study. Exclusion criteria were hyperglycemia due to glucose infusion, infection, stress, drugs, and other factors.
Data Collection and Biochemical Analyses
Clinical phenotype and biochemical tests were performed at Vietnam National Children’s Hospital. Data included pedigree, sex, date of birth, gestational age, birth weight, date of diabetes diagnosis, natural history, and examination at diagnosis such as weight, height, and symptoms of diabetic ketoacidosis (DKA) including tachypnea, dehydration, lethargy, coma, and other symptoms such as convulsion. Blood glucose and HbA1C were measured by the automated Beckman Coulter AU2700/AU680 system. The specimen was collected in the early morning at schedule visits. Hexokinase technology with ORS 6221 reagent of OLYMPUS and ORS6192 reagent were used for blood glucose and HbA1c testing, respectively. Insulin and C-peptide were measured using immunoassay chemiluminiscent technology by automated biochemistry Hitachi 704. Arterial blood gas was measured by spectrometry, using GEM primer 300. Capillary glucose levels were measured at home by One Touch Ultrain in all patients five times/day (before breakfast, lunch, dinner, 22 h and 2 h) or whenever there was an abnormality. Continuous glucose monitoring (CGM) over 7 days was monitored by using the Medtronic Ipro™2 Professional. HbA1C was checked at 3-month intervals. Blood glucose level and HbA1C targets were determined according to the International Society of Pediatrics and Adolescent Diabetes (ISPAD) 2018 guideline (8).
Mutation Analysis
Mutation analysis was performed at the Exeter genomic laboratory, UK. Blood samples were taken with informed consent obtained from the patients and their parents. Genomic DNA was extracted from peripheral blood using phenol/chloroform methods at Vietnam National Children’s Hospital.
The single exon of the KCNJ11 gene was amplified in three overlapping fragments, as previously described (9). The ABCC8 gene was analyzed at the same time as KCNJ11. The 39 exons of ABCC8 were amplified in 38 fragments using previously described primers (10). PCR products were sequenced on an ABI 3100 or ABI 3730 (Applied Biosystems, Warrington, UK). Sequences were compared with the reference sequences (KCNJ11, NM_000525.3; ABCC8, NM_001287174.1) using Staden or Mutation Survey or software version 2.61.
The identified mutations were checked in common databases such as dbSNP154 database, ClinVar database, Leiden open variation database (LOVD), human gene mutation database (HGMD), and the genome aggregation database (gnomAD). In silico analysis was performed using Alamut Visual. Pathogenic variants identified after 2018 were classified using the ACMG best practice guidelines (11). Protein visualization was generated using the Protter website (http://wlab.ethz.ch/protter/start/)
Transfer to Sulfonylureas From Insulin
For this study, clinicians were provided with two recommended protocols for the transfer to the SU glyburide (also known as glibenclamide) for Kir6.2 and SUR1 patients [see www.diabetesgenes.org and (12)]. One was for a rapid inpatient transfer, where the glyburide dose was increased by 0.2 mg/kg/day every day and the other for a slower outpatient transfer, where the glyburide dose was increased by 0.2 mg/kg/day every week. Both involved the gradual withdrawal of insulin, as the SU was introduced depending on blood glucose levels. These protocols were modified by the treating clinicians (5).
Statistical Analysis
Data analyses were performed using SPSS version 12.0 (SPSS Inc., Chicago, IL). Data are expressed as frequency (%), mean ± SD, or median and range. Continuous variables were analyzed using ANOVA or Kruskal–Wallis tests. Proportions were compared using Fisher’s exact tests. Statistical significance was defined as p < 0.05 or the corresponding Bonferroni-adjusted p value for multiple comparisons.
Results
A total of 70 patients were diagnosed with NDM before 6 months of age. Pathogenic variants were identified in 54 (77%). The most common genetic causes were mutations in ABCC8 (26%) and KCNJ11 (24%). The current study focused on the analysis of the 27 patients with ABCC8/KCNJ11 mutations.
Clinical Features of NDM Patients With KCNJ11/ABCC8 Mutations
Of the 27 patients with a KATP channel mutation, 14 had a mutation in ABCC8, and 13 had a KCNJ11 mutations. Clinical characteristics of these patients are provided in Table 1. Five of the 27 patients had TNDM, which was diagnosed before 3 months of age and onset with severe/moderate DKA. The average birth weight was 2758.3 ± 419.0 g with gestational age of 39.2 ± 1.28 weeks. Fifty-two percent (14/27) of the subjects were small for gestational age with a birth weight below the third centile. The average onset age in 27 cases was 60 ± 36.4 days, of which 85% were diagnosed before 3 months of age. At diagnosis, biochemical studies showed blood glucose, 36.2 ± 12.3 mmol/L; HbA1C, 7.16 ± 0.16% (normal range, 4–6.5%); , 7.9 ± 7.4 mmol/L; BE, −17.9 ± 9.1 mmol/L; and C-peptide, median of 0.09 (range, 0–1.61 nmol/L; normal range, 0.26–0.62). Twenty-three patients (85.2%) presented with DKA. Nine patients (33.3%) had a convulsion at diagnosis.

Table 1 Clinical features of 27 NDM Vietnamese patients with KCNJ11/ABCC8 mutations.
Molecular Genetic Analysis
A total of 21 pathogenic variants were identified, including 13 ABCC8 and 8 KCNJ11 mutations (Table 2 and Figure 1). These mutations were predicted to be disease causing by the Mutation Taster tool and evaluated as pathogenic variants in the ClinVar database. Four mutations, namely, ABCC8 (p.E747X), ABCC8 (p.R1183W), KCNJ11 (p.R201C), and KCNJ11 (p.R201H), were found in multiple patients. Except for the ABCC8 (p.E747X) mutation, which was homozygous in patient 4 (13), the remaining mutations were heterozygous.
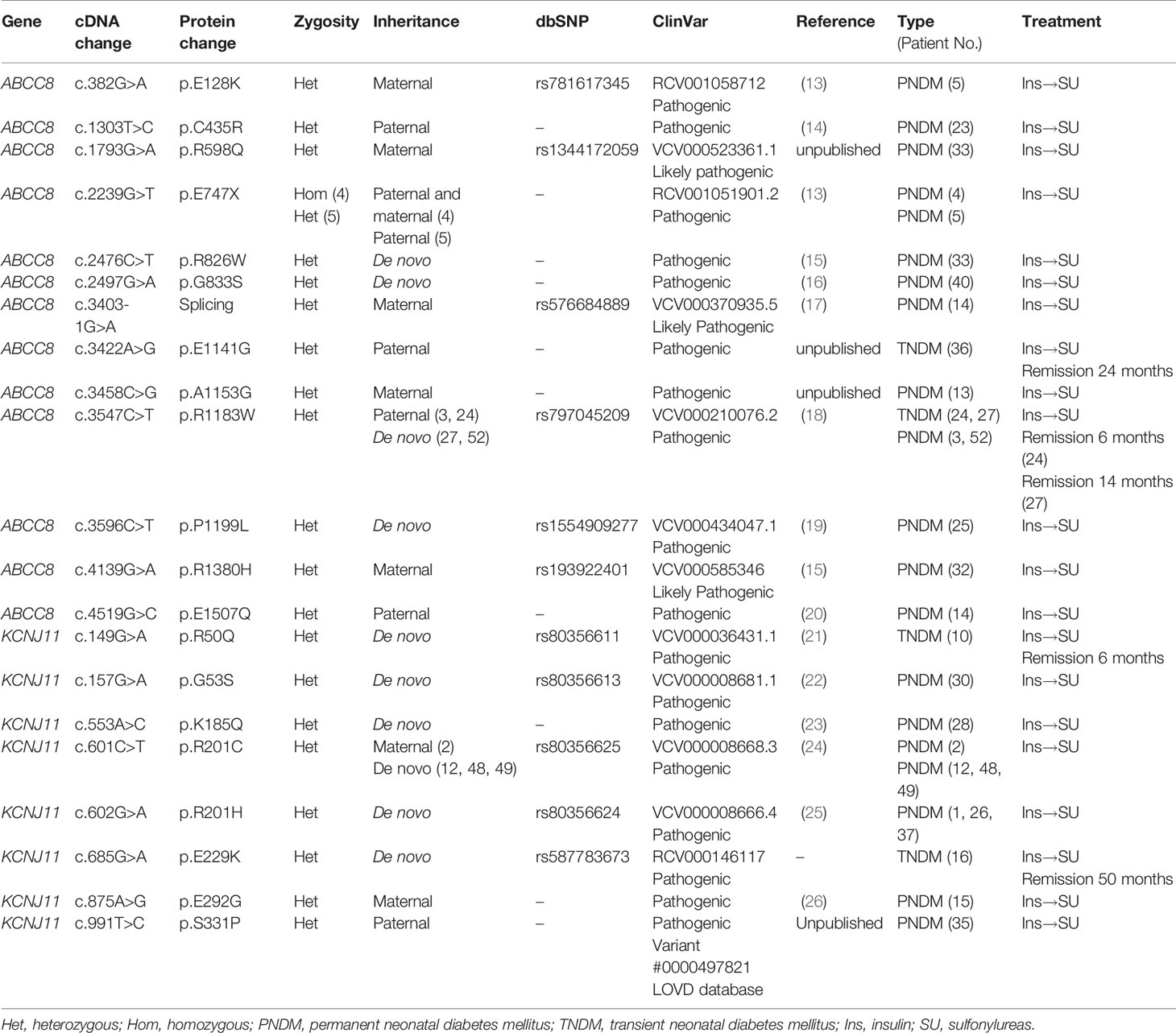
Table 2 Molecular analyses of 27 Vietnamese patients with NDM.
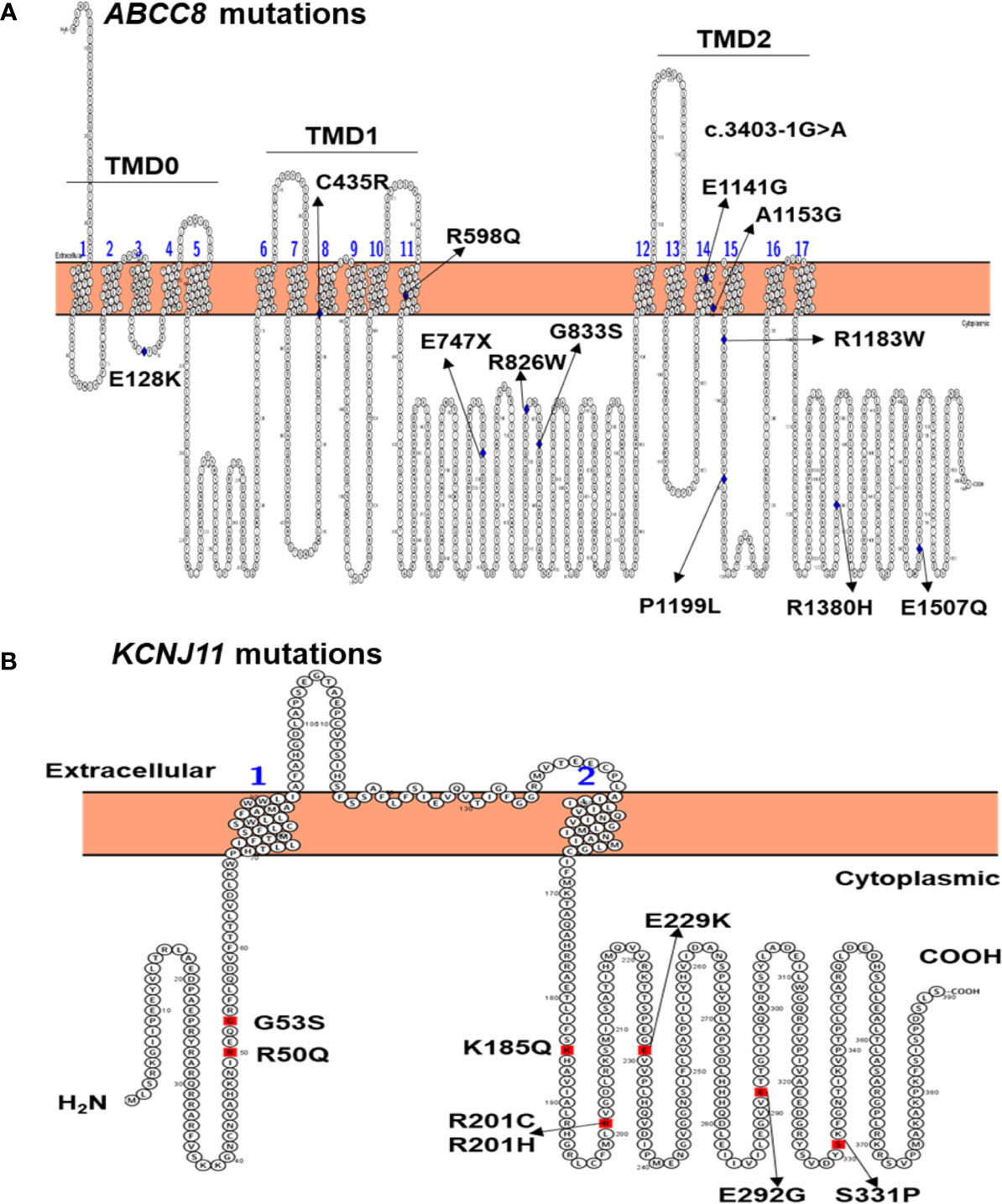
Figure 1 Location of ABCC8 mutations in the SUR1 protein (A) and KCNJ11 mutations in the Kir6.2 protein (B) identified in 27 Vietnamese patients with NDM. TMD, transmembrane domain.
Twelve patients inherited mutations from their parents, while in 14 patients, the mutation had arisen de novo. Patient 33 inherited the ABCC8 (p.R598Q) mutation from his mother and also had a de novo mutation ABCC8 (p.R826W). Three patients 5, 14, and 33 had compound heterozygous mutations in ABCC8. Among 21 pathogenic mutations, four were unpublished, including KCNJ11 (p.S331P), ABCC8 (p.E1141G), and ABCC8 (p.A1153G) (Table 2).
Transfer to Sulfonylureas
Of the 27 patients with mutations in KCNJ11or ABCC8, 26 were successfully transferred from insulin to SU treatment. Patient 24 remitted after 5 months of insulin treatment and before transfer to SU. Glycemic fluctuations reduced when the patients were on SU treatment as compared with insulin treatment (Figure 2A, p = 0.000001). The mean HbA1C level dropped from 7.75% ± 3.60% on insulin treatment (13.3 ± 25 months) to 5.69% ± 1.02% on SU treatment (86.0 ± 56.9 months, p = 0.0000038) (Figure 2B). While on insulin treatment, there was one case of DKA and one case with convulsion due to severe hypoglycemia. After transfer to SU (86.0 ± 56.9 months), there were no patients with DKA or hypoglycemia.
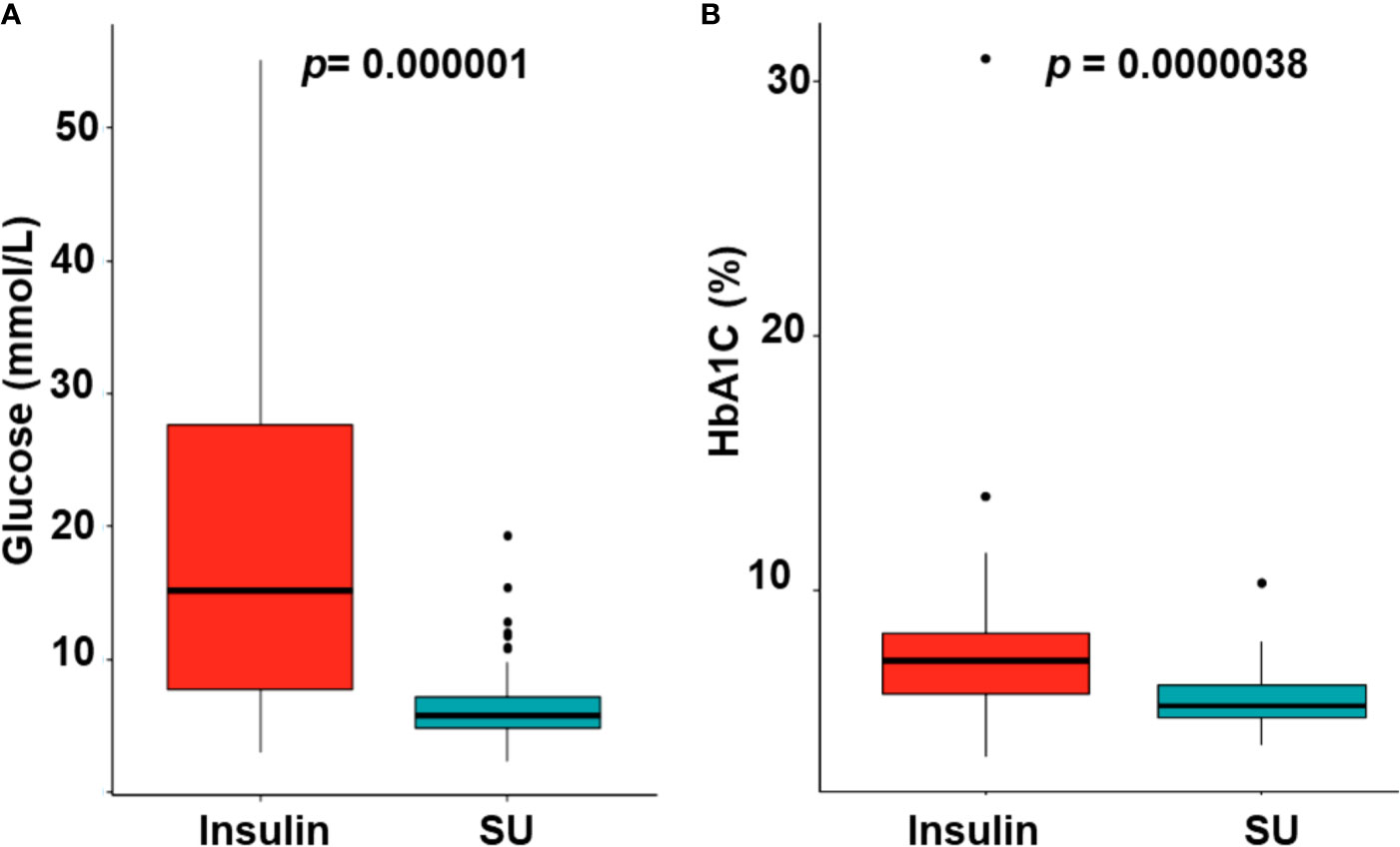
Figure 2 The glycemic control (A) and reduction of HbA1C levels (B) with switching from insulin to sulfonylurea therapy. SU, sulfonylurea.
Two of the 27 patients (patients 2 and 4) were diagnosed with developmental delay, epilepsy, and neonatal diabetes (DEND) syndrome (Table 1). Both individuals successfully transferred to SU, which resulted in improved mental and motor development. Patient 2, who is heterozygous for the KCNJ11 p.R201C mutation, is currently 14 years of age and can understand simple sentences and has good eye contact. Patient 2 showed a decrease in HbA1c from 8.25% ± 0.20% (insulin treatment) to 6.65% ± 0.07% (SU treatment). During insulin treatment, patient 2 was admitted to the hospital twice (due to DKA and convulsion due to severe hypoglycemia). After transfer to SU, there were no further episodes of severe hypoglycemia or DKA. Patient 4 with a homozygous nonsense mutation in ABCC8 p.(E747X) is currently 16 years of age and can walk, speak in short sentences, and understand and answer some simple questions. When patient 4 was treated with insulin, HbA1C was 8.5% ± 0.4%, and blood glucose ranged from 4 to 20 mmol/L; however, HbA1c decreased to 5.3% ± 0.2%, and blood glucose fluctuation ranged from 5 to 10 mmol/L when he was treated with SU.
Two TNDM patients with KCNJ11 mutations (p.(R50Q) and p.(E229K)) and three TNDM patients with ABCC8 mutations [two patients with p.(R1183W) and one patient with p.(E1141G)] remitted at 6, 50, 6, 14, and 24 months of ages, respectively (Table 2).
Treatment requirements (insulin or SU dose) did not show significant differences between the patients with KCNJ11 mutations and those with ABCC8 mutations (data not shown).
Side Effects
None of the patients reported side effects during SU treatment such as diarrhea, nausea, or vomiting. Renal and liver function tests of all patients were checked every 6–12 months; all were within the normal range. No severe hypoglycemic episodes were reported while on SU treatment, and no other side effects were noted.
Discussion
In our comprehensive mutation analysis of a large cohort of 70 patients with NDM enrolled at Vietnam National Children’s Hospital, we identified gene mutations in 54 cases (77%). In these 54 patients with a confirmed genetic diagnosis, mutations in the KATP channel genes was the most common cause of NDM with a rate of 50%. Overall, 51% had low birth weight (below third percentile), 23 (85%) were diagnosed before 3 months of age, and 23 (85%) presented with DKA. Twenty-six patients were successfully transferred from insulin to SU therapy, and glycemic control subsequently improved.
The mutation rate of 77% in our study was lower than that in Ukrainian (12) and Chinese studies but similar to the University of Chicago Monogenic Diabetes Registry (n = 88) (27). In contrast, Russo et al. (28) found the most common genes causing NDM diagnosed during the first 6 months of life were KCNJ11 and ABCC8 (70%), but mutations in KCNJ11 were more common than ABCC8. These differences may be due to ethnicity, race, or size of the study cohort. In our study, we only investigated patients with NDM onset before 6 months of age who have mutations in the genes encoding the KATP channel.
The rate of patients with low birth weight (under third percentile) was 52%, which is similar to the results reported by Besser et al. (29). In the study reported by Russo et al. (28), the patients diagnosed with PNDM before 6 months of age but without mutations in KCNJ11, ABCC8, or INS had higher birth weight than those with mutations in these genes.
The majority of our cohort (85%) presented in DKA. Similarly, Letourneau et al. (27) reported that 66% of patients with neonatal diabetes (and 79% of patients with KCNJ11/ABCC8 mutations) presented with DKA. While this is slightly less than observed in our cohort, there may have been a delay in diagnosis in some of our cases, which is reflected in the later age of diagnosis. This delay may be related to the challenge of diagnosing diabetes in infants who cannot communicate symptoms and in whom polydipsia and polyuria may not be readily apparent—indeed, this could even be reassuring to clinicians.
The numbers of Vietnamese patients with ABCC8 and KCNJ11 mutations were similar (14 versus 13), which is consistent with the findings in the Indian population (30). In contrast, Hashimoto et al. (31) reported more patients with KCNJ11 than ABCC8 mutations (16 versus 8). Recurrent mutations KCNJ11 p.(R201C), KCNJ11 p.(R201H), and ABCC8 p.(R1183W) in Vietnamese patients have been reported in NDM patients in Jordan (32), India (30), Ukraine (12), The United States (33), Japan (34), and China (35). Therefore, these mutations may be considered as common mutations in different ethnic groups. The high rate of de novo mutations, which can arise either during gametogenesis or embryogenesis in NDM patients (15/27), identified in the current study, is consistent with the findings of Edghill et al. (36).
None of the 12 mutations identified in ABCC8 are located in the glibenclamide binding pocket of SUR1. However, four mutations [ABCC8 p.(C435R) and p.(R598Q), p.(E1141G), and p.(A1153G)] are located in the transmembrane domains TMD1 and TMD2, respectively (Figure 1A). Interestingly, p.(C435R) was previously reported in two TNDM patients (15, 37), while patient 23 in our study had PNDM after treatment on SU 72 months (Table 1). Three ABCC8 mutations p.(R598Q), p.(E1141G), and p.(A1153G) are unpublished (Table 2). Patient 33 had a compound heterozygous ABCC8 mutation, p.(R598Q) and p.(R826W) (Tables 1, 2).
Eight KCNJ11 mutations identified in this study are located in the N- and C-terminal regions (Figure 1B), which form the ATP binding pocket of Kir6.2 (38). The p.(R50Q), p.(K185Q), p.(R201C), and p.(R201H) mutations may reduce the response of the channel to ATP, as they lie at the main binding pocket (23, 24, 39). The KCNJ11 p.R50Q mutation has been reported to cause both TNDM (34) and PNDM (9); therefore, patients with this mutation can present with different phenotypes as suggested by Suzuki et al. (34). The KCNJ11 p.(K185Q) mutation was identified in PNDM patient 28 (Table 1). This mutation have been reported previously in a 3-year-old girl with PNDM, and after treatment with insulin, her HbA1C was between 6.8% and 7.8% (23). In our study, patient 28 was also treated with insulin; however, she was transferred to SU, resulting in an HbA1C of 7.6% and blood glucose level of 7.9 mmol/L. Functional studies indicated that this mutation reduced ATP binding to Kir6.2, resulting in a reduction in ATP sensitivity of the KATP channel, leading to PNDM in the patient (23).
SU therapy is effective in the treatment of hyperglycemia in patients with NDM who have a mutation in KCNJ11 or ABCC8. Up to 90–95% of patients with NDM caused by mutations in these genes can cease insulin therapy after initiation of SU therapy (3, 5). In our study, the rate was higher, at 96.3%. The one remaining case (3.7%) had a remission of diabetes at 5 months of age before transfer to SU could be initiated. SU acts on the KATP channel to promote closure, allowing insulin to be released from the beta cells. Since SU therapy increases insulin release, there is a risk for hypoglycemia to occur. However, there was no severe hypoglycemia reported in our study. Excellent glycemic control was maintained after commencing SU therapy (Figure 2A), which is similar to other reports (6, 40). In the study of Bowman et al. (40), there were no reports of severe hypoglycemia in 809 patient-year follow up for the whole KCNJ11 cohort, and 93% of the participants remained on SU therapy for the 10-year duration; however, a high rate (14%) of patients presented with mild and transient side effects of SU such as diarrhea, nausea, weight loss due to reduced appetite, and abdominal pain (40). Whereas, in our study, there was no side effects of SU, continued follow-up will be required to determine the long-term outcome of SU therapy in this group of patients. Interestingly, two patients (patients 2 and 4) with DEND syndrome were successfully transferred to SU treatment, which is consistent with previous studies (41, 42).
In conclusion, we found that all patients in our cohort with ABCC8 and KCNJ11 mutations could be successfully treated with oral SU treatment even if they had previously been treated with insulin. It is essential to perform rapid genetic testing for ABCC8/KCNJ11 in any patient diagnosed with diabetes before 6 months of age, particularly given issues regarding access to and cost of insulin in some populations. On SU treatment, we observed that this therapy is safe in the short term for patients with KATP channel NDM.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Vietnam National Children’s Hospital IRB#1, 18/879 Lathanh, Dongda, Hanoi, Vietnam. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
CN, TD, SE, and VD conceptualized, designed the study, and wrote and reviewed the manuscript. VD, CN, BT, and NNK provided patients’ clinical information, and MC reviewed/edited the manuscript. EF, SF, JH, and NNL analyzed data and wrote and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This project was supported by grants from the Wellcome Trust and the Royal Society (Grant Number: 105636/Z/14/Z). EDF is a Diabetes UK RD Lawrence fellow. SF has a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (105636/Z/14/Z). MC supported by a NHMEC practitioner fellowship (#1136735).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the patients and their family members for their time and support.
References
1. Iafusco D, Massa O, Pasquino B, Colombo C, Iughetti L, Bizzarri C, et al. Minimal Incidence of Neonatal/Infancy Onset Diabetes in Italy Is 1:90,000 Live Births. Acta Diabetologica (2012) 49:405. doi: 10.1007/s00592-011-0331-8
2. Dahl A, Kumar S. Recent Advances in Neonatal Diabetes. Diabetes Metab Syndr Obes (2020) 13:355–64. doi: 10.2147/DMSO.S198932
3. Lemelman MB, Letourneau L, Greeley SAW. Neonatal Diabetes Mellitus: An Update on Diagnosis and Management. Clin Perinatol (2018) 45:41–59. doi: 10.1016/j.clp.2017.10.006
4. Aguilar-Bryan L, Bryan J. Neonatal Diabetes Mellitus. Endocr Rev (2008) 29:265–91. doi: 10.1210/er.2007-0029
5. Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE, Larkin B, et al. Switching From Insulin to Oral Sulfonylureas in Patients With Diabetes Due to Kir6.2 Mutations. N Engl J Med (2006) 355:467–77. doi: 10.1056/NEJMoa061759
6. Rafiq M, Flanagan SE, Patch A-M, Shields BM, Ellard S, Hattersley AT. Neonatal Diabetes International Collaborative Group. Effective Treatment With Oral Sulfonylureas in Patients With Diabetes Due to Sulfonylurea Receptor 1 (SUR1) Mutations. Diabetes Care (2008) 31:204–9. doi: 10.2337/dc07-1785
7. De Franco E, Saint-Martin C, Brusgaard K, Knight Johnson AE, Aguilar-Bryan L, Bowman P, et al. Update of Variants Identified in the Pancreatic β-Cell KATP Channel Genes KCNJ11 and ABCC8 in Individuals With Congenital Hyperinsulinism and Diabetes. Hum Mutat (2020) 41:884–905. doi: 10.1002/humu.23995
8. DiMeglio LA, Acerini CL, Codner E, Craig ME, Hofer SE, Pillay K, et al. ISPAD Clinical Practice Consensus Guidelines 2018: Glycemic Control Targets and Glucose Monitoring for Children, Adolescents, and Young Adults With Diabetes. Pediatr Diabetes (2018) 19:105–14. doi: 10.1111/pedi.12737
9. Flanagan SE, Edghill EL, Gloyn AL, Ellard S, Hattersley AT. Mutations in KCNJ11, Which Encodes Kir6.2, Are a Common Cause of Diabetes Diagnosed in the First 6 Months of Life, With the Phenotype Determined by Genotype. Diabetologia (2006) 49:1190–7. doi: 10.1007/s00125-006-0246-z
10. Ellard S, Flanagan SE, Girard CA, Patch A-M, Harries LW, Parrish A, et al. Permanent Neonatal Diabetes Caused by Dominant, Recessive, or Compound Heterozygous SUR1 Mutations With Opposite Functional Effects. Am J Hum Genet (2007) 81:375–82. doi: 10.1086/519174
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Flanagan SE, Dũng V, Houghton J, De Francov E, Ngoc CTB, Damhuis A, et al. An ABCC8 Nonsense Mutation Causing Neonatal Diabetes Through Altered Transcript Expression. J Clin Res Pediatr Endocrinol (2017) 9:260–4. doi: 10.4274/jcrpe.4624
13. Babenko AP, Polak M, Cavé H, Busiah K, Czernichow P, Scharfmann R, et al. Activating Mutations in the ABCC8 Gene in Neonatal Diabetes Mellitus. N Engl J Med (2006) 355:456–66. doi: 10.1056/NEJMoa055068
14. Patch AM, Flanagan SE, Boustred C, Hattersley AT, Ellard S. Mutations in the ABCC8 Gene Encoding the SUR1 Subunit of the KATP Channel Cause Transient Neonatal Diabetes, Permanent Neonatal Diabetes or Permanent Diabetes Diagnosed Outside the Neonatal Period. Diabetes Obes Metab (2007) 9:28–39. doi: 10.1111/j.1463-1326.2007.00772.x
15. Reilly F, Sanchez-Lechuga B, Clinton S, Crowe G, Burke M, Ng N, et al. Phenotype, Genotype and Glycaemic Variability in People With Activating Mutations in the ABCC8 Gene: Response to Appropriate Therapy. Diabetic Med (2020) 37:876–84. doi: 10.1111/dme.14145
16. Snider KE, Becker S, Boyajian L, Shyng S-L, MacMullen C, Hughes N, et al. Genotype and Phenotype Correlations in 417 Children With Congenital Hyperinsulinism. J Clin Endocrinol Metab (2013) 98:E355–363. doi: 10.1210/jc.2012-2169
17. Kong JH, Kim JB. Transient Neonatal Diabetes Mellitus Caused by a De Novo ABCC8 Gene Mutation. Korean J Pediatr (2011) 54:179–82. doi: 10.3345/kjp.2011.54.4.179
18. Oztekin O, Durmaz E, Kalay S, Flanagan SE, Ellard S, Bircan I. Successful Sulfonylurea Treatment of a Neonate With Neonatal Diabetes Mellitus Due to a Novel Missense Mutation, P.P1199L, in the ABCC8 Gene. J Perinatol (2012) 32:645–7. doi: 10.1038/jp.2012.46
19. Ortiz D, Bryan J. Neonatal Diabetes and Congenital Hyperinsulinism Caused by Mutations in ABCC8/SUR1 Are Associated With Altered and Opposite Affinities for ATP and ADP. Front Endocrinol (Lausanne) (2015) 6:48. doi: 10.3389/fendo.2015.00048
20. Alves C, Flanagan SE, Ellard S, Mackay DJ. Clinical and Molecular Basis of Transient Neonatal Diabetes Mellitus in Brazilian Children. Diabetes Res Clin Pract (2012) 97:e41–44. doi: 10.1016/j.diabres.2012.05.025
21. Gloyn AL, Reimann F, Girard C, Edghill EL, Proks P, Pearson ER, et al. Relapsing Diabetes can Result From Moderately Activating Mutations in KCNJ11. Hum Mol Genet (2005) 14:925–34. doi: 10.1093/hmg/ddi086
22. Shimomura K, de Nanclares GP, Foutinou C, Caimari M, Castaño L, Ashcroft FM. The First Clinical Case of a Mutation at Residue K185 of Kir6.2 (KCNJ11): A Major ATP-Binding Residue. Diabetes Med (2010) 27:225–9. doi: 10.1111/j.1464-5491.2009.02901.x
23. Hattersley AT, Ashcroft FM. Activating Mutations in Kir6.2 and Neonatal Diabetes: New Clinical Syndromes, New Scientific Insights, and New Therapy. Diabetes (2005) 54:2503–13. doi: 10.2337/diabetes.54.9.2503
24. Cho JH, Kang E, Lee BH, Kim GH, Choi JH, Yoo HW. DEND Syndrome With Heterozygous KCNJ11 Mutation Successfully Treated With Sulfonylurea. J Korean Med Sci (2017) 32:1042–5. doi: 10.3346/jkms.2017.32.6.1042
25. Rica I, Luzuriaga C, Pérez de Nanclares G, Estalella I, Aragonés A, Barrio R, et al. The Majority of Cases of Neonatal Diabetes in Spain can be Explained by Known Genetic Abnormalities. Diabetes Med (2007) 24:707–13. doi: 10.1111/j.1464-5491.2007.02140.x
26. Globa E, Zelinska N, Mackay DJG, Temple KI, Houghton JAL, Hattersley AT, et al. Neonatal Diabetes in Ukraine: Incidence, Genetics, Clinical Phenotype and Treatment. J Pediatr Endocrinol Metab (2015) 28:1279–86. doi: 10.1515/jpem-2015-0170
27. Letourneau LR, Carmody D, Wroblewski K, Denson AM, Sanyoura M, Naylor RN, et al. Diabetes Presentation in Infancy: High Risk of Diabetic Ketoacidosis. Diabetes Care (2017) 40:e147–8. doi: 10.2337/dc17-1145
28. Russo L, Iafusco D, Brescianini S, Nocerino V, Bizzarri C, Toni S, et al. Permanent Diabetes During the First Year of Life: Multiple Gene Screening in 54 Patients. Diabetologia (2011) 54:1693–701. doi: 10.1007/s00125-011-2094-8
29. Besser REJ, Flanagan SE, Mackay DGJ, Temple IK, Shepherd MH, Shields BM, et al. Prematurity and Genetic Testing for Neonatal Diabetes. Pediatrics (2016) 138: e20153926.1–7. doi: 10.1542/peds.2015-3926
30. Gopi S, Kavitha B, Kanthimathi S, Kannan A, Kumar R, Joshi R, et al. Genotype-Phenotype Correlation of KATP Channel Gene Defects Causing Permanent Neonatal Diabetes in Indian Patients. Pediatr Diabetes (2021) 22:82–92. doi: 10.1111/pedi.13109
31. Hashimoto Y, Dateki S, Hirose M, Satomura K, Sawada H, Mizuno H, et al. Molecular and Clinical Features of KATP-Channel Neonatal Diabetes Mellitus in Japan. Pediatr Diabetes (2017) 18:532–9. doi: 10.1111/pedi.12447
32. Abujbara MA, Liswi MI, El-Khateeb MS, Flanagan SE, Ellard S, Ajlouni KM. Permanent Neonatal Diabetes Mellitus in Jordan. J Pediatr Endocrinol Metab (2014) 27:879–83. doi: 10.1515/jpem-2014-0069
33. Støy J, Greeley SAW, Paz VP, Ye H, Pastore AN, Skowron KB, et al. Diagnosis and Treatment of Neonatal Diabetes: An United States Experience†. Pediatr Diabetes (2008) 9:450–9. doi: 10.1111/j.1399-5448.2008.00433.x
34. Suzuki S, Makita Y, Mukai T, Matsuo K, Ueda O, Fujieda K. Molecular Basis of Neonatal Diabetes in Japanese Patients. J Clin Endocrinol Metab (2007) 92:3979–85. doi: 10.1210/jc.2007-0486
35. Cao B, Gong C, Wu D, Lu C, Liu F, Liu X, et al. Genetic Analysis and Follow-Up of 25 Neonatal Diabetes Mellitus Patients in China. J Diabetes Res (2016) 2016:6314368. doi: 10.1155/2016/6314368
36. Edghill EL, Gloyn AL, Goriely A, Harries LW, Flanagan SE, Rankin J, et al. Origin of De Novo KCNJ11 Mutations and Risk of Neonatal Diabetes for Subsequent Siblings. J Clin Endocrinol Metab (2007) 92:1773–7. doi: 10.1210/jc.2006-2817
37. Hartemann-Heurtier A, Simon A, Bellanné-Chantelot C, Reynaud R, Cavé H, Polak M, et al. Mutations in the ABCC8 Gene can Cause Autoantibody-Negative Insulin-Dependent Diabetes. Diabetes Metab (2009) 35:233–5. doi: 10.1016/j.diabet.2009.01.003
38. Martin GM, Kandasamy B, DiMaio F, Yoshioka C, Shyng S-L. Anti-Diabetic Drug Binding Site in a Mammalian KATP Channel Revealed by Cryo-EM. Elife (2017) 6:e31054.1–27. doi: 10.7554/eLife.31054
39. Shimomura K, Girard CAJ, Proks P, Nazim J, Lippiat JD, Cerutti F, et al. Mutations at the Same Residue (R50) of Kir6.2 (KCNJ11) That Cause Neonatal Diabetes Produce Different Functional Effects. Diabetes (2006) 55:1705–12. doi: 10.2337/db05-1640
40. Bowman P, Sulen Å, Barbetti F, Beltrand J, Svalastoga P, Codner E, et al. Effectiveness and Safety of Long-Term Treatment With Sulfonylureas in Patients With Neonatal Diabetes Due to KCNJ11 Mutations: An International Cohort Study. Lancet Diabetes Endocrinol (2018) 6:637–46. doi: 10.1016/S2213-8587(18)30106-2
41. Mlynarski W, Tarasov AI, Gach A, Girard CA, Pietrzak I, Zubcevic L, et al. Sulfonylurea Improves CNS Function in a Case of Intermediate DEND Syndrome Caused by a Mutation in KCNJ11. Nat Clin Pract Neurol (2007) 3:640–5. doi: 10.1038/ncpneuro0640
42. Zwaveling-Soonawala N, Hagebeuk EE, Slingerland AS, Ris-Stalpers C, Vulsma T, van Trotsenburg AS. Successful Transfer to Sulfonylurea Therapy in an Infant With Developmental Delay, Epilepsy and Neonatal Diabetes (DEND) Syndrome and a Novel ABCC8 Gene Mutation. Diabetologia (2011) 54:469–71. doi: 10.1007/s00125-010-1981-8
Keywords: neonatal diabetes mellitus, ABCC8 mutations, KCNJ11 mutations, sulfonylureas treatment in neonatal diabetes mellitus, diabetes mellitus in infants
Citation: Ngoc CTB, Dien TM, De Franco E, Ellard S, Houghton JAL, Lan NN, Thao BP, Khanh NN, Flanagan SE, Craig ME and Dung VC (2021) Molecular Genetics, Clinical Characteristics, and Treatment Outcomes of KATP-Channel Neonatal Diabetes Mellitus in Vietnam National Children’s Hospital. Front. Endocrinol. 12:727083. doi: 10.3389/fendo.2021.727083
Received: 18 June 2021; Accepted: 16 August 2021;
Published: 09 September 2021.
Edited by:
Giulio Maltoni, Sant’Orsola-Malpighi Polyclinic, ItalyReviewed by:
Dario Iafusco, University of Campania Luigi Vanvitelli, ItalyPeter Light, University of Alberta, Canada
Copyright © 2021 Ngoc, Dien, De Franco, Ellard, Houghton, Lan, Thao, Khanh, Flanagan, Craig and Dung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vu Chi Dung, ZHVuZ3Z1QG5jaC5vcmcudm4=
†These authors have contributed equally to this work