 Lijiao Zhang
Lijiao Zhang Yongchang Sun
Yongchang Sun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 28 September 2021
Sec. Bone Research
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.724911
This article is part of the Research Topic Vascular and Skeletal Crosstalk in Health and Disease View all 5 articles
Sarcopenia and osteoporosis are common musculoskeletal comorbidities of chronic obstructive pulmonary disease (COPD) that seriously affect the quality of life and prognosis of the patient. In addition to spatially mechanical interactions, muscle and bone can also serve as endocrine organs by producing myokines and osteokines to regulate muscle and bone functions, respectively. As positive and negative regulators of skeletal muscles, the myokines irisin and myostatin not only promote/inhibit the differentiation and growth of skeletal muscles, but also regulate bone metabolism. Both irisin and myostatin have been shown to be dysregulated and associated with exercise and skeletal muscle dysfunction in COPD. During exercise, skeletal muscles produce a large amount of IL-6 which acts as a myokine, exerting at least two different conflicting functions depending on physiological or pathological conditions. Remarkably, IL-6 is highly expressed in COPD, and considered to be a biomarker of systemic inflammation, which is associated with both sarcopenia and bone loss. For osteokines, receptor activator of nuclear factor kappa-B ligand (RANKL), a classical regulator of bone metabolism, was recently found to play a critical role in skeletal muscle atrophy induced by chronic cigarette smoke (CS) exposure. In this focused review, we described evidence for myokines and osteokines in the pathogenesis of skeletal muscle dysfunction/sarcopenia and osteoporosis in COPD, and proposed muscle-bone crosstalk as an important mechanism underlying the coexistence of muscle and bone diseases in COPD.
Chronic obstructive pulmonary disease (COPD) is a prevalent chronic airway disease characterized by persistent airflow limitation and varying respiratory symptoms including chronic cough with or without sputum production, and exertional dyspnea which limits physical activity of the patient. The major risk factor for COPD is cigarette smoking, which leads to airway inflammation and alveolar destruction (emphysema), the pathological hallmark of the disease (1). COPD is now viewed as a systemic disease with significant comorbidities, such as sarcopenia, osteoporosis, cardiovascular diseases, lung cancer, gastroesophageal reflux, metabolic syndrome, anxiety and depression. These comorbidities contribute markedly to the mortality of COPD, and therefore their assessment and management are an integral part of COPD (1).
Skeletal muscle dysfunction/sarcopenia and osteoporosis are common in COPD, and their pathogenic mechanisms are believed to be associated with systemic inflammation (2). Muscle and bone are closely linked spatially. In addition to mechanical interactions, they also serve as endocrine organs to secrete myokines and osteokines to regulate bone metabolism and skeletal muscle growth and functions, respectively. In recent years, the role of muscle-bone crosstalk in the development of skeletal muscle dysfunction/sarcopenia or osteoporosis has attracted great attention (3), but its involvement in musculoskeletal comorbidities of COPD still awaits investigation. In this focused review, we searched for the studies of osteoporosis and skeletal muscle dysfunction/sarcopenia in COPD, and for muscle-bone crosstalk in general in the past 20 years. We described evidence for myokines and osteokines in the pathogenesis of skeletal muscle dysfunction/sarcopenia and osteoporosis in COPD, and put forward muscle-bone crosstalk as an important mechanism underlying the coexistence of muscle and bone diseases in COPD, aiming to promote research in this fields.
Sarcopenia is an age-related progressive and systemic skeletal muscle disease associated with increased likelihood of adverse consequences such as falls, fractures, physical disability, hospitalizations and mortality (4). The European Working Group on Sarcopenia in Older People (EWGSOP) developed a practical clinical definition and diagnostic criteria for sarcopenia based on three criteria: muscle quantity or mass, muscle strength, and physical performance (4), which has been adopted by many studies. A meta-analysis reported that the overall pooled prevalence estimate of sarcopenia in people with COPD was 27.5% (95%CI, 18.4%–36.5%) (5), and the prevalence increased with age, degree of airflow limitation and severity of disease (6). Moreover, with the same degree of airway obstruction, sarcopenia is more prevalent in individuals with emphysema than in those with airway-type COPD (7). Skeletal muscle dysfunction occurs in patients with COPD and affects both respiratory and nonrespiratory muscles. Compared with upper extremities, skeletal muscle dysfunction is more significant in lower extremities (such as quadriceps femoris), which compromises the ambulatory capacity of COPD patients and has devastating effects on their daily lives (8). Until present, most studies on skeletal muscle dysfunctions in COPD have mainly focused on certain skeletal muscle groups, which often cannot accurately reflect the real prevalence and severity of the disease. For example, one study showed that there was no difference in the prevalence of sarcopenia between stable COPD patients with or without quadriceps weakness, and nearly 1/3 of those with sarcopenia had preserved quadriceps strength (6). Therefore, it is recommended that patients with COPD should be evaluated comprehensively based on the diagnostic criteria for sarcopenia. Currently, multiple studies have shown that sarcopenia/skeletal muscle dysfunction in COPD is associated with more severe airflow obstruction, emphysema, dyspnea score (modified British medical research council, mMRC), decreased quality of life and exercise capacity, frequent exacerbations and increased mortality (9–11). Exercise training is currently the most effective non-pharmacological intervention to improve skeletal muscle function in COPD. COPD patients with sarcopenia respond well to pulmonary rehabilitation, similar to those without sarcopenia (6), with improvement in exercise ability, upper and lower extremity strength, functional performance and health status.
Osteoporosis, a systemic skeletal disease characterized by low bone mineral density(BMD) and/or microarchitectural deterioration of bone, is another common comorbidity of COPD, which is associated with increased bone fragility/fracture, lower mobility and alteration in postural balance (12). A latest meta-analysis reported that the overall pooled prevalence of osteoporosis in patients with COPD was 38% (95%CI, 34%-43%), and BMI<18.5 kg/m2 and the presence of sarcopenia were significant risk factors for osteoporosis in COPD (13). Osteoporosis in COPD patients is usually asymptomatic, and often diagnosed until bone fractures occur, and therefore the prevalence of osteoporosis in COPD is often under-estimated. The risk of bone fracture depends on bone strength, which is comprised of BMD and bone quality, and BMD accounts for about 70% of bone strength (14). Due to the lack of precise evaluation of bone quality, the diagnosis of osteoporosis mainly depends on BMD, however, simply measuring BMD cannot fully reflect the risk of bone fracture. Thus, although BMD is decreased as Global Initiative for Chronic Obstructive Lung Disease(GOLD) stage advances in COPD patients, the prevalence of bone fracture appears to be independent of the GOLD stage, and the actual prevalence is higher than that predicted by BMD (15). It was reported that about 40% of patients with COPD suffered at least one event of vertebral fracture (16), with thoracic fracture as the most common (17). Vertebral fracture can cause back pain, thoracic deformities, kyphosis and height loss, leading to impaired pulmonary function (18). In addition, low BMD/osteoporosis and related fractures are also associated with emphysema, frequent exacerbations (19), increased hospitalization rates and mortality (20), and decreased quality of life in patients with COPD (21).
Skeletal muscle dysfunction/sarcopenia can be caused by an imbalance of muscle protein synthesis and degradation, inflammation, mitochondrial dysfunction, myosatellite cell injury and disturbed calcium homeostasis (8, 22). There are many common risk factors between sarcopenia and osteoporosis in COPD, including systemic inflammation (such as enhanced tumor necrosis factor (TNF)-α and IL-6), cigarette smoking, hypoxemia and/or hypercapnia, malnutrition, oxidative stress and reduced level of physical activity (8, 23). Up to 70% of COPD patients with osteoporosis also show signs of muscle wasting (24), and sarcopenia is also an independent risk factor for osteoporosis in patients with COPD (13). Therefore, we hypothesize that there are interactions between muscle and bone comorbidities in COPD.
Skeletal muscle and bone are inextricably linked genetically, molecularly, and mechanically, and they are closely connected spatially (25). They have various functions in common, such as maintaining posture, locomotion, promoting breathing, protecting internal organs and coordinating overall energy consumption. Studies have also shown that muscle and bone can serve as endocrine organs in producing myokines and osteokines, respectively, which can interact within the so-called muscle-bone unit (26).
There is a close mechanical relationship between skeletal muscle and bone, with the bone providing the muscle with a point of attachment and acting as a lever, and the muscle acting as a pulley to move the body (25). The coordination of muscle mass and bone mass is achieved by mechanical signals produced by muscle strength that transmits anabolic activity in adjacent bones (27). When the stimulus exceeds a certain threshold of mechanical strain, it can induce bone anabolism and enhance bone hardness, while preventing bone damage caused by increased mechanical load. Conversely, if the stimulus is below this threshold of mechanical strain, bone resorption will occur in related bone tissues (27). In daily activities, the mechanical relationship between muscle and bone occurs all the time, and exercise training is just one of the manifestations of the mechanical connection between muscle and bone. Exercise can produce mechanical signals through the physical connection between muscle and bone to affect bone remodeling. Although exercise training was generally considered to be beneficial for improving the loss of muscle mass and bone mass in most cases, some studies indicated that the role of exercise on improving bone loss was controversial. For example, studies showed that resistance training was associated with the improvement of femur bone mass (28), while endurance training might not improve bone mass (29). One study even showed that transient endurance exercise promoted bone resorption and stimulated upregulation of sclerostin, a bone formation inhibiting protein (30). From the biomechanical view, the decrease of muscle mass and function will lead to the reduction of mechanical load on the bone, and eventually lead to bone loss, which is consistent with the coexistence of sarcopenia and osteoporosis in many pathological conditions. However, the loss of bone mass cannot fully explain sarcopenia, nor does muscle atrophy fully explain osteoporosis (31). This suggests that there are other mechanisms of muscle-bone crosstalk besides biomechanical action.
Muscle and bone are two major components of the musculoskeletal system, and they are closely related in embryonic development, postnatal growth and development, and aging (31). In addition to their mechanical connection, the chemical crosstalk between muscle and bone has become a hot topic of research in recent years. Accumulating evidence indicates that muscle and bone can secrete cytokines to act on each other in autocrine, paracrine or endocrine manners. A variety of myokines and osteokines have been found to play critical roles in this muscle-bone crosstalk, including myokines such as myostatin, irisin, IL-6, and osteokines such as RANKL, OPG and osteocalcin.
Myostatin, also known as growth differentiation factor 8 (GDF-8), belongs to the transforming growth factor (TGF)-β superfamily. Myostatin can inhibit the proliferation and differentiation of myoblasts and promote muscle atrophy, and is a potent negative regulator of skeletal muscle growth and development (32). Myostatin signals through the activin type IIB receptor (ACVR2B or ActRIIB), which is widely expressed and forms a heterodimer with activin-like kinase 4 (ALK4, also known as ACVR1B) or ALK5 (also known as TGF-βR1) (33). The intracellular serine/threonine kinase domain of ALK4 and ALK5 phosphorylates SMAD Family Member 2 (Smad2) and Smad3, and then forms a complex with Smad4. The complex translocats to the nucleus to regulate the transcription of genes involved in the proliferation and differentiation of skeletal muscle precursor cells (34, 35), and in protein degradation pathways in mature myofibers (36). The activation of Smad2 and Smad3 by myostatin can also inhibit the Akt/mammalian target of rapamycin (mTOR) pathway in response to pro-growth signals, and therefore, suppresses protein synthesis (37) (Figure 1). Earlier studies also confirmed that human and animals with a mutation in the myostatin genes showed a remarkable growth in muscle mass. A decoy receptor (ACVR2B/Fc) was found to be a potent inhibitor of myostatin and as a result, could induce marked muscle growth when given systemically to wild type mice and microgravity-exposed mice respectively (38). Furthermore, studies showed that the expression of myostatin mRNA was downregulated in skeletal muscles after acute exercise and was even further decreased over long-term physical activity in sedentary men, and the expression of myostatin in muscles was associated with an impaired insulin sensitivity (39), suggesting that downregulation of myostatin in response to exercise may improve muscle performance.
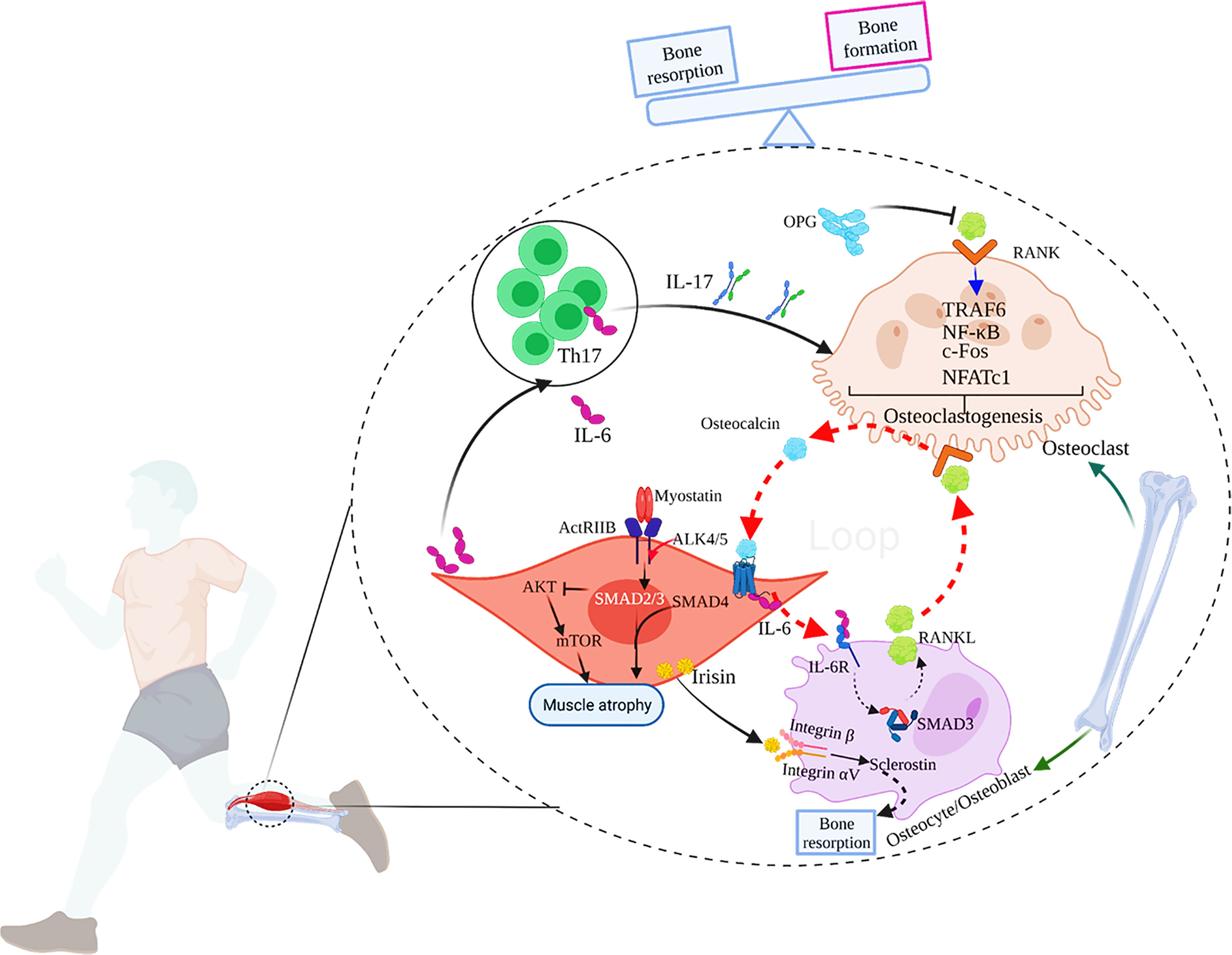
Figure 1 The main mechanisms of muscle-bone crosstalk. 1) IL-17 secreted by Th17 cells induces RANKL expression in mice, which combines with RANK on the surface of osteoclasts to activate TRAF6 and c-Fos to induce osteoclastogenesis, and eventually, bone resorption occurs. OPG, the decoy receptor of RANKL, can prevent RANKL from activating RANK in the extracellular environment, thereby inhibiting osteoclast formation and bone loss; 2) During exercise, the muscle-derived IL-6 (mIL-6) acts on the IL-6 receptor on the surface of osteoblasts and increases the production of RANKL, subsequently promoting osteoclast differentiation and the secretion of bioactive osteocalcin through RANKL/RANK pathway in osteoclasts. In turn, osteocalcin also enhances the production of mIL-6 during exercise. Thus, a feed-forward loop between muscle and bone favors exercise adaptations. 3) Myostatin signals through ActRIIB, which forms a heterodimer with ALK 4/5. The intracellular ALK4/5 phosphorylates SMAD2 and SMAD3, and then forms a complex with SMAD4. The complex translocates to the nucleus to regulate the transcription of genes involved in the proliferation and differentiation of skeletal muscle precursor cells. The activation of SMAD2 and 3 by myostatin also inhibits the Akt/mTOR pathway in response to pro-growth signals, and therefore, suppresses protein synthesis. Ultimately, myostatin contributes to muscle atrophy. 4) Irisin produced by skeletal muscles during exercise acts on integrin αV/β on the surface of osteocytes to promote sclerostin production, leading to bone resorption. 5) IL-6 can also promote the generation of IL-17.
Myostatin also promotes osteoclast development and inhibits bone formation (40). Myostatin alone cannot induce bone marrow-derived macrophages to form osteoclasts, but the presence of myostatin with classical signals [RANKL and macrophage colony-stimulating factor (M-CSF)] dramatically improved the ability of osteoclast precursors to differentiate into mature osteoclasts in vitro (40). On the gene expression level, myostatin can increase the nuclear translocation of Smad2-dependent nuclear factor of activated T cells, cytoplasmic 1 (NFATc1) and subsequently up-regulate the expression of osteoclast differentiation genes (40). The role of myostatin in osteoclast differentiation has also been confirmed in animal models. For instance, deficiency or pharmacological inhibition of myostatin prominently diminished osteoclast formation and promoted bone destruction in the human TNF-α transgenic (hTNFtg) mouse model of rheumatoid arthritis as well as in the serum-transfer-induced arthritis model (40). Furthermore, myostatin may also influence the expression of other cytokines in muscle-bone crosstalk. Knockout of myostatin genes in mice, for example, resulted in enhanced expression of irisin in muscles (41).
Irisin is a newly discovered myokine, mainly derived from skeletal muscles (42). Irisin is proteolytically cleaved from the fibronectin type III domain-containing protein 5 (FNDC5) gene products and secreted into the blood (42). Irisin is regulated by exercise, and its plasma concentration increases in a dose-dependent manner with increase in aerobic exercise intensity (43). In a meta-analysis, Fox et al. evaluated the effect of an acute bout of exercise (including aerobic and/or resistance exercise) on circulating irisin levels, and found that the circulating irisin levels boosted by 15% (95%CI: 10.8%-19.3%) after exercise compared to baseline (44). The expression of irisin is regulated by peroxisome proliferator-activated receptor γ-coactivator 1α (PGC-1α) and Smad3. In contrast to wild type mice, Smad3-/- mice showed higher serum and skeletal muscle FNDC5 protein levels with the same exercise protocol (45). TGF-β binds to TGF-βR1/TGF-βR2 complex, and Smad3 is phosphorylated and translocated into the nucleus to bind the promoters of PGC-1α and FNDC5 to suppress their transcription, and suppression of FNDC5 transcription and protein leads to decrease in circulating irisin in vitro (45) (Figure 2).
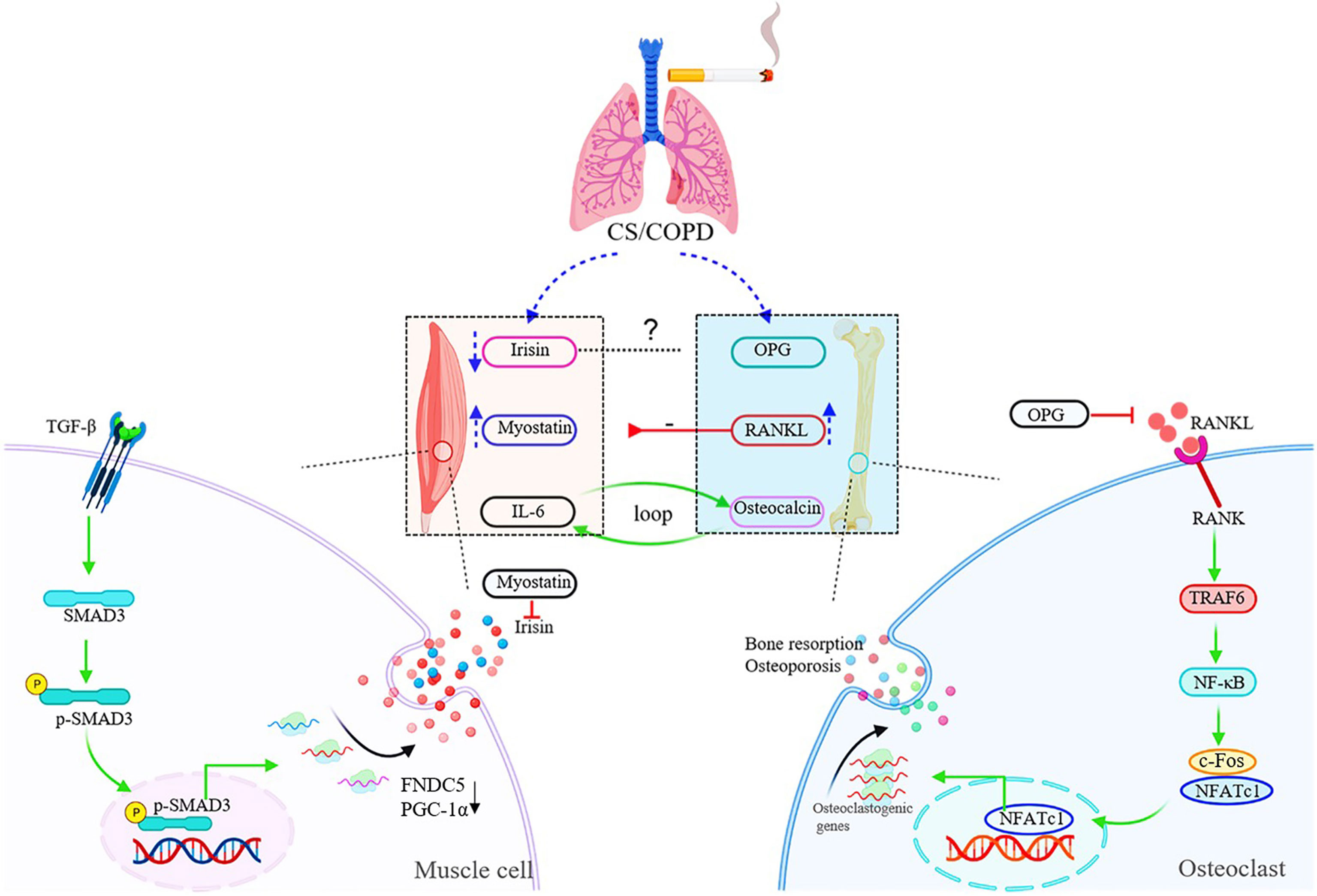
Figure 2 Schematic diagram of muscle-bone crosstalk in COPD. In COPD, the serum levels of irisin are decreased, while myostatin and RANKL are up-regulated in both circulation and skeletal muscles. The raised expression of RANKL induced by chronic cigarette smoke exposure contributes to muscle atrophy/skeletal muscle dysfunction through the RANKL/RANK pathway. The figure also shows the specific signaling pathway of irisin and RANKL. TGF-β binds to TGF-βR1/TGF-βR2 complex, and Smad3 is phosphorylated and translocated into the nucleus to bind the promoters of PGC-1α and FNDC5 to suppress their transcription, and suppression of FNDC5 transcription and protein leads to decrease in circulating irisin in vitro. RANKL activates the homologous receptor RANK on the surface of osteoclasts and osteoclast precursors, and activated RANK causes the recruitment of the adapter protein TRAF6, leading to NF-κB activation and translation of NF-κB to the nucleus. NF-κB increases the expression of c-Fos, and c-Fos interacts with NFATc1 to trigger the transcription of osteoclastogenic genes.
Irisin plays a positive role in regulating muscle mass by promoting differentiation of myoblasts (46). In last few years, several important studies have found that irisin was also a pivotal regulator of bone metabolism. Colaianni et al. found that male C57BL/6 mice injected with recombinant irisin (100μg/kg, once a week) for a month showed increased cortical tissue BMD of femora and tibia, accompanied by increased osteoblast numbers (47). Irisin promoted the differentiation of bone marrow stem cell-derived osteoblasts in vitro (47). Up till now, most studies support the role of irisin in promoting bone formation, such as in inflammatory bowel disease, hind-limb unloading, and microgravity (weightlessness) induced bone loss models, and exogenous administration of recombinant irisin alleviated bone loss (48–50). Nevertheless, a more recent study, firstly identified integrin αV/β5 as the receptor of irisin, found that genetic deletion of FNDC5/irisin completely blocked trabecular bone loss in mouse models of osteoporosis caused by ovariectomy (51). Irisin induced up-regulated expression of sclerostin and receptor activator of nuclear factor kappa-B ligand (RANKL) through integrin αV/β5 in the osteocyte membrane, thereby promoting bone resorption (51) (Figure 1). This discovery prompted discussion to re-examine the role of irisin in muscle-bone crosstalk (52).
IL-6 is a classical pro-inflammatory cytokine mainly produced by T lymphocytes and macrophages. Skeletal muscles can also produce a large amount of IL-6 in response to muscle contraction, which is released into blood stream during exercise, and therefore IL-6 is also regarded as a myokine. In skeletal muscles, IL-6 acts as a pleiotropic factor and appears to have at least two different conflicting functions (53). In exercise and physiological conditions, IL-6 produced by skeletal muscles is increased for several hours in the circulation, which promotes the activation of the signal transducer and activator of transcription 3-suppressor of cytokine signaling 3 (Stat3-SOCS3) signaling pathway through autocrine effects and plays a positive role in regulating the myogenic differentiation (54, 55). However, in pathological conditions, including muscular dystrophy and chronic inflammatory diseases, there is a chronic and lasting elevation of the circulating IL-6. Although Stat3-SOCS3 signaling is also activated in the target skeletal muscle under these pathological conditions, the autocrine loop of regulation in the action of IL-6 is totally lost (53). Besides, IL-6 combined with other molecules negatively regulates muscle differentiation and promotes muscle atrophy (53). Although IL-6 is a well-known pro-inflammatory cytokine, it can also exert anti-inflammatory effects under certain conditions. For instance, the exercise-induced rise in IL-6 promoted production of IL-1 receptor antagonist (IL-1ra) and IL-10 (54), which stimulated an anti-inflammatory systemic environment during exercise. A recent study confirmed through genetic means that the vast majority of circulating IL-6 detectable during exercise originated from muscles (56), and the magnitude of exercise-induced elevation of plasma IL-6 depended on exercise mode, intensity and duration, as well as energy availability (54, 57). Furthermore, it’s reported that the amount of regular physical activity was negatively associated with the basal circulating IL-6 levels (54), that is to say, the less physical activity, the higher basal plasma IL-6. This regulation was attributed to the effects of training adaptations. However, it also has been demonstrated that acute exercise upregulated IL-6 receptor (IL-6R) gene expression in skeletal muscles, and long-term training increased basal IL-6R mRNA levels (57). Therefore, the downregulation of systemic levels of IL-6 induced by exercise training may be compensated for the enhanced production of IL-6R, suggesting a sensitization of skeletal muscles to IL-6 at rest. Meanwhile, it is worth noting that IL-6 signals in osteoblasts to promote cell differentiation and the release of bioactive osteocalcin into circulation, and the muscle-derived IL-6 (mIL-6) promotes nutrient uptake and catabolism in myofibers during exercise in an osteocalcin-dependent manner, thus enhancing exercise capacity (56). In addition to regulating muscle growth and enhancing exercise capacity, IL-6 appears to induce bone resorption through RANKL-dependent enhanced osteoclastogenesis/osteoblast differentiation (58) (Figure 1) as well as via osteoblast-derived prostaglandia E2 (PGE2)-dependent osteoclast activation (59, 60).
RANKL is produced by osteoblasts, stromal cells, and other types of cells under the regulation of various hormones and cytokines. Osteoblasts and stromal cells also produce OPG-the decoy receptor of RANKL. Both RANKL and OPG belong to the TNF family. RANKL activates the homologous receptor RANK on the surface of osteoclasts and osteoclast precursors, and activated RANK causes the recruitment of the adapter protein TNF receptor associated factor 6 (TRAF6), leading to NF-κB activation and translation of NF-κB to the nucleus. NF-κB increases the expression of c-Fos and c-Fos interacts with NFATc1 to trigger the transcription of osteoclastogenic genes (61), which contributes to bone resorption and osteoporosis. OPG can prevent RANKL from activating RANK in the extracellular environment, thereby inhibiting osteoclast formation and bone loss (61) (Figure 2). In wild-type mouse models, RANKL and RANK are expressed in both bones and muscles, and RANKL is particularly highly expressed in oxidative muscles like the soleus dominated by type I fibers (62). In huRANKL transgenic mice, the overexpression of huRANKL not only induced bone loss, but also resulted in decreased leg muscle, lower maximal speed and limb force, decreased number of both type I and II myofibers, and impaired glucose metabolism (62). These changes were associated with the increased levels of antimyogenic and inflammatory gene expression in muscles, such as myostatin and protein tyrosine phosphatase receptor-γ (62). In mouse models, intervention with OPG-Fc (an inhibitor of both RANKL and TNF-related apoptosis-inducing ligand) and denosumab (a selective RANKL inhibitor) rescued muscle weakness to the similar levels, suggesting that the effect of RANKL on muscle is mainly mediated by RANK signals (62). In postmenopausal women, the elevated expression of RANKL plays a pivotal role in the development of osteoporosis (63). Denosumab treatment not only improved BMD and reduced the risk of fracture, but also, interestingly, increased the appendicular lean mass and grip strength (62, 64).
Osteocalcin is a small γ-carboxyglutamate protein expressed by osteoblasts, and is only expressed in the late differentiation stage of osteoblasts and after the arrest of proliferation, under the regulation of Runx2/Cbfa1 transcription factor (65). γ-carboxylation occurs on three glutamate residues of osteocalcin before osteocalcin is secreted by osteoblasts. The post-translational modification increases the affinity of osteocalcin to hydroxyapatite. Therefore, the majority of the osteocalcin secreted by osteoblasts is stored in bone matrix, which constitutes the most abundant non-collagenous polypeptides (66). Osteocalcin in circulation exists in two forms: γ-carboxylated osteocalcin and uncarboxylated osteocalcin, and only uncarboxylated osteocalcin plays the role of endocrine hormone (67). In the process of bone resorption under acidic conditions, osteoclasts decarboxylate γ-carboxylated osteocalcin in bone matrix, activate and release bioactive osteocalcin into blood (68). The effects of osteocalcin on bone metabolism remain controversial from studies in vivo and in vitro. It is speculated that osteocalcin plays a dual role. On the one hand, osteocalcin is a biochemical marker of bone turnover and can regulate bone remodeling by regulating the activity of osteoblasts and osteoclasts. On the other hand, it is a regulator of bone mineralization (69).
Previous studies explored the role of osteocalcin in regulating glucose homeostasis, insulin sensitivity and energy metabolism (68, 70). In recent years, more studies have found that osteocalcin plays an important role in regulating muscle quantity, muscle function, and movement adaptability (71). It has been identified that G-protein coupled receptor C family 6a (Gprc6a) is the osteocalcin receptor on cells, which is highly expressed in skeletal muscles, especially in the oxidative muscles that require long-term work, such as the soleus muscle (71). Uncarboxylated osteocalcin could promote myoblast proliferation through the PI3K/Akt/p38 MAPK pathway, or through the Gprc6a-ERK1/2 signaling pathway in vitro (72). Mera et al. demonstrated a notable decrease in muscle mass in Gprc6a-deficient mice, and the administration of osteocalcin ameliorated muscle mass in wild type 9-month-old mice (73), suggesting that osteocalcin signaling in myofibers is necessary for maintaining muscle quantity in elderly mice, partly due to the promotion of protein synthesis in the myotubes. In early adulthood, circulating osteocalcin levels begin to decline sharply, especially during aging; Circulating osteocalcin levels in women and men reach their lowest levels before the age of 30 and 50, respectively (71). Interestingly, exercise has been shown to increase circulating osteocalcin levels. For example, a bout endurance aerobic-based exercise in 3-month-old mice (74) and a 45-minute exercise in young women (71) significantly increased circulating osteocalcin levels. Osteocalcin not only favors the uptake and catabolism of glucose and fatty acids in myofibers, but also enhances the production of mIL-6 during exercise, a myokine that promotes the secretion of bioactive osteocalcin (Figure 1), while exogenous osteocalcin administration improved the exercise capacity of mice (71). Hence, it’s possible that osteocalcin signaling in myofibers may be a novel and promising means to attenuate the age-related decline in muscle function.
In addition to the molecules described above, muscle and bone also produce other myokines and osteokines (75, 76), such as insulin-like growth factor (IGF)-1, fibroblast growth factor (FGF)-2, PGE2, IL-7, IL-15, beta-aminoisobutyric acid (BAIBA), TGF-β, and sclerostin, which form a delicate network in muscle-bone crosstalk. Whether one or more of these molecules are involved in musculoskeletal comorbidities of COPD still needs further investigation, so we will not introduce these cytokines in more details here. We mostly focused on those involved in bone and muscle comorbidities in COPD, and those believed to play key roles in muscle-bone crosstalk.
Aging, chronic inflammation, inactivity, bed rest and other pathological conditions in COPD result in the loss of muscle and bone mass and functional impairment. Although the clinical significance of sarcopenia and osteoporosis is well recognized, the molecular mechanisms under these pathological conditions remain to be investigated. However, myokines and osteokines potentially involved in muscle-bone crosstalk in COPD have newly been revealed, including IL-6, irisin, myostatin, RANKL, osteocalcin, etc.
It is well known that COPD is characterized by both local and systemic inflammation, and IL-6, as a key pro-inflammatory cytokine, is highly expressed in patients with COPD. Elevated circulating IL-6 levels are associated with decreased forced expiratory volume in one second (FEV1), strength of quadriceps femoris and exercise capacity in COPD patients (77). In a 3-year study of inflammatory markers in patients with COPD, elevated serum IL-6 was found to predict increased mortality (78). However, as described in the previous section, IL-6 is also a myokine that plays both anti-inflammatory and pro-inflammatory roles in skeletal muscles. It can regulate the differentiation of osteoclasts and enhance exercise adaptability. From the perspective of muscle-bone crosstalk, the role of IL-6 in COPD should be re-examined.
Some studies have examined the expression of irisin in COPD, and its association with emphysema and exercise. Compared with healthy controls, COPD patients showed decreased level of serum irisin which was positively correlated with physical activity (79). Exercise training raised circulating irisin in a chronic cigarette smoke (CS)-exposure induced mouse model of COPD (80). In patients with COPD, serum irisin levels were significantly correlated with diffusing capacity of lung for carbon monoxide/alveolar volume (DLCO/VA) and percentage of low-attenuation area (LAA%) (81). Interestingly, an experimental study showed that irisin ameliorated emphysema in a CS-induced model of COPD in mice, in which irisin played an anti-oxidative role through nuclear factor erythroid 2-related factor 2 (Nrf2, a transcription factor with antioxidant properties) and heme oxygenase-1 (HO-1, an antioxidant in the antioxidant pathway) (80). In vitro, irisin was found to promote the expression of Nrf2 and decrease apoptosis induced by CS exposure in A549 cells (81). These findings suggest that irisin, in addition to its role as a myokine, may be also involved in the pathogenesis of emphysema induced by epithelial apoptosis in COPD.
Myostatin is a negative regulator of skeletal muscle growth and development, and some studies have reported dysregulated expression of myostatin in COPD and its association with skeletal muscle dysfunction. Serum myostatin levels were elevated, and negatively correlated with total muscle mass in male COPD patients (82), and myostatin mRNA expression was also elevated in skeletal muscles from COPD patients (83). Myostatin maintains the satellite cells (SCs) in quiescent state, and its absence can trigger the activation of SCs (84). In addition, inhibition of myostatin alleviated muscle atrophy by upregulating muscle regeneration markers in limb muscles of rats (85). A recent study found that the markers of muscle regeneration (Pax-7, Myf-5, MyoD and myogenia) and the numbers of Pax-7+/Myf-5- SCs in the vastus lateralis of sarcopenic COPD patients were decreased, while markers of muscle injury and the myostatin level were increased (86). In a clinical study of sarcopenic COPD patients, Bimagrumab (an ACVR2 inhibitor that blocks the myostatin pathway) was found to safely increase skeletal muscle mass in patients with COPD, but no improvement in muscle function or physical performance was observed (87). Clinically, pulmonary rehabilitation is the most effective non-pharmacological treatment for COPD patients at present. Pulmonary rehabilitation was found to improve quadriceps femoris strength and physical performance (88) and decrease myostatin expression in quadriceps femoris (89). Moreover, resistance training can not only attenuate the expression of myostatin in skeletal muscles, but also up-regulate the proportion of myogenin/MyoD, and safely and effectively counteract skeletal muscle dysfunction in acute exacerbation of COPD (90).
Latest studies also indicate that osteokines play critical roles in muscle-bone crosstalk in COPD. BMD was found to be associated with the imbalance of RANKL/OPG and systemic inflammation in peripheral blood of patients with COPD (91). In addition, in IL-17A knockout mice, bone loss induced by chronic CS exposure was attenuated, with downregulation of RANKL, suggesting that IL-17 promoted bone resorption by inducing RANKL expression in this model (92). Our most recent study found that chronic CS exposure induced skeletal muscle atrophy and muscle weakness in mice, with up-regulated expression of RANKL and its receptor RANK in skeletal muscles. RANKL neutrolization attenuated skeletal muscle dysfunction and reduced the expression of myostatin and MuFR1/Atrogin1. In vitro, CS extract induced up-regulation of RANKL/RANK in skeletal muscle cells, and blocking RANKL down-regulated myostatin expression (93). These results indicate that the RANKL/RANK pathway plays a key role in skeletal muscle atrophy induced by chronic CS exposure.
To our knowledge, there are few studies on osteocalcin in COPD, and it is only used as a marker of bone turnover or bone formation (94, 95), without further investigation on the mechanism of osteocalcin in the muscle-bone crosstalk of COPD. As mentioned earlier, osteocalcin does play a role in muscle-bone crosstalk during exercise. During exercise, a feedback loop between bone (via osteocalcin) and muscle (via IL-6) enhances exercise adaptation: osteocalcin promotes nutrient absorption and catabolism of myofibers, and osteocalcin can trigger IL-6 expression and secretion in skeletal muscles. IL-6 also enhances the production of bioactive osteocalcin (56). The roles of osteocalcin and IL-6 in muscle-bone comorbidities of COPD deserve further investigation. Based on the evidence available to date, we propose a muscle-bone crosstalk mechanism for skeletal muscle dysfunction/sarcopenia and osteoporosis in COPD, as shown in Figure 2.
COPD is a major cause of chronic morbidity and mortality worldwide, with many people dying prematurely from the disease or its complications. Sarcopenia and osteoporosis, as important comorbidities of COPD, are associated with COPD severity and prognosis. Currently, the mechanisms underlying the muscle-bone relationship in COPD is still unclear, and there is still a lack of comprehensive and systematic measures to improve the condition and prognosis of COPD and its comorbidities in clinical practice. Fortunately, studies have revealed the potential role of muscle-bone crosstalk involving myokines and osteokines, such as IL-6, irisin, myostatin, RANKL/RANK and osteocalcin, in the development of sarcopenia and osteoporosis in COPD. Identification of key molecules in the pathogenesis of musculoskeletal diseases holds promise for precision therapy of comorbidities in COPD. Importantly, rehabilitation is now the most effective non-pharmacological therapy for improving outcomes of COPD (1), but its impact on the delicate network of myokines and osteokines is underappreciated. Further understanding of muscle-bone crosstalk in physical exercise in COPD may shed light on implementation of better modalities of non-pharcological management for a disease currently without a pharmalogical cure.
YS conceived and revised this manuscript. LZ collected materials, wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81770040, 81970041) and Natural Science Foundation of Beijing Municipality (7192224).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Vogelmeier C, Agusti A, Anzueto A, Barnes P, Bourbeau J, Criner G, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. In: 2021 Report. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) (2019). Available online at: http://www.goldcopd.org..
2. Clynes MA, Gregson CL, Bruyere O, Cooper C, Dennison EM. Osteosarcopenia: Where Osteoporosis and Sarcopenia Collide. Rheumatol (Oxford) (2021) 60(2):529–37. doi: 10.1093/rheumatology/keaa755
3. Kirk B, Zanker J, Duque G. Osteosarcopenia: Epidemiology, Diagnosis, and Treatment-Facts and Numbers. J Cachexia Sarcopenia Muscle (2020) 11(3):609–18. doi: 10.1002/jcsm.12567
4. Cruz-Jentoft AJ, Bahat G, Bauer J, Boirie Y, Bruyere O, Cederholm T, et al. Sarcopenia: Revised European Consensus on Definition and Diagnosis. Age Ageing (2019) 48(4):601. doi: 10.1093/ageing/afz046
5. Sepulveda-Loyola W, Osadnik C, Phu S, Morita AA, Duque G, Probst VS. Diagnosis, Prevalence, and Clinical Impact of Sarcopenia in COPD: A Systematic Review and Meta-Analysis. J Cachexia Sarcopenia Muscle (2020) 11(5):1164–76. doi: 10.1002/jcsm.12600
6. Jones SE, Maddocks M, Kon SS, Canavan JL, Nolan CM, Clark AL, et al. Sarcopenia in COPD: Prevalence, Clinical Correlates and Response to Pulmonary Rehabilitation. Thorax (2015) 70(3):213–8. doi: 10.1136/thoraxjnl-2014-206440
7. Vanfleteren LE, Spruit MA, Groenen M, Gaffron S, van Empel VP, Bruijnzeel PL, et al. Clusters of Comorbidities Based on Validated Objective Measurements and Systemic Inflammation in Patients With Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2013) 187(7):728–35. doi: 10.1164/rccm.201209-1665OC
8. Jaitovich A, Barreiro E. Skeletal Muscle Dysfunction in Chronic Obstructive Pulmonary Disease. What We Know and Can Do for Our Patients. Am J Respir Crit Care Med (2018) 198(2):175–86. doi: 10.1164/rccm.201710-2140CI
9. Ju S, Lee SJ, Park MJ, Cho YJ, Jeong YY, Jeon KN, et al. Clinical Importance of Cross-Sectional Area of Intercostal Muscles in Patients With Chronic Obstructive Pulmonary Disease. Clin Respir J (2018) 12(3):939–47. doi: 10.1111/crj.12609
10. Bak SH, Kwon SO, Han SS, Kim WJ. Computed Tomography-Derived Area and Density of Pectoralis Muscle Associated Disease Severity and Longitudinal Changes in Chronic Obstructive Pulmonary Disease: A Case Control Study. Respir Res (2019) 20(1):226. doi: 10.1186/s12931-019-1191-y
11. Tanimura K, Sato S, Fuseya Y, Hasegawa K, Uemasu K, Sato A, et al. Quantitative Assessment of Erector Spinae Muscles in Patients With Chronic Obstructive Pulmonary Disease. Novel Chest Computed Tomography-Derived Index for Prognosis. Ann Am Thorac Soc (2016) 13(3):334–41. doi: 10.1513/AnnalsATS.201507-446OC
12. Gazzotti MR, Roco CM, Pradella CO, Nascimento OA, Porto EF, Adas M, et al. Frequency of Osteoporosis and Vertebral Fractures in Chronic Obstructive Pulmonary Disease (COPD) Patients. Arch Bronconeumol (2019) 55(5):252–7. doi: 10.1016/j.arbres.2018.10.010
13. Chen YW, Ramsook AH, Coxson HO, Bon J, Reid WD. Prevalence and Risk Factors for Osteoporosis in Individuals With COPD: A Systematic Review and Meta-Analysis. Chest (2019) 156(6):1092–110. doi: 10.1016/j.chest.2019.06.036
14. Nih Consensus Development Panel on Osteoporosis Prevention D. Therapy. Osteoporosis Prevention, Diagnosis, and Therapy. JAMA (2001) 285(6):785–95. doi: 10.1001/jama.285.6.785
15. Watanabe R, Tanaka T, Aita K, Hagiya M, Homma T, Yokosuka K, et al. Osteoporosis is Highly Prevalent in Japanese Males With Chronic Obstructive Pulmonary Disease and is Associated With Deteriorated Pulmonary Function. J Bone Miner Metab (2015) 33(4):392–400. doi: 10.1007/s00774-014-0605-7
16. Nuti R, Siviero P, Maggi S, Guglielmi G, Caffarelli C, Crepaldi G, et al. Vertebral Fractures in Patients With Chronic Obstructive Pulmonary Disease: The EOLO Study. Osteoporos Int (2009) 20(6):989–98. doi: 10.1007/s00198-008-0770-4
17. Kjensli A, Falch JA, Ryg M, Blenk T, Armbrecht G, Diep LM, et al. High Prevalence of Vertebral Deformities in COPD Patients: Relationship to Disease Severity. Eur Respir J (2009) 33(5):1018–24. doi: 10.1183/09031936.00073908
18. Inoue D, Watanabe R, Okazaki R. COPD and Osteoporosis: Links, Risks, and Treatment Challenges. Int J Chron Obstruct Pulmon Dis (2016) 11:637–48. doi: 10.2147/COPD.S79638
19. Romme EA, Murchison JT, Edwards LD, van Beek E Jr., Murchison DM, Rutten EP, et al. CT-Measured Bone Attenuation in Patients With Chronic Obstructive Pulmonary Disease: Relation to Clinical Features and Outcomes. J Bone Miner Res (2013) 28(6):1369–77. doi: 10.1002/jbmr.1873
20. Pascual-Guardia S, Badenes-Bonet D, Martin-Ontiyuelo C, Zuccarino F, Marin-Corral J, Rodriguez A, et al. Hospital Admissions and Mortality in Patients With COPD Exacerbations and Vertebral Body Compression Fractures. Int J Chron Obstruct Pulmon Dis (2017) 12:1837–45. doi: 10.2147/COPD.S129213
21. Adachi JD, Ioannidis G, Pickard L, Berger C, Prior JC, Joseph L, et al. The Association Between Osteoporotic Fractures and Health-Related Quality of Life as Measured by the Health Utilities Index in the Canadian Multicentre Osteoporosis Study (CaMos). Osteoporos Int (2003) 14(11):895–904. doi: 10.1007/s00198-003-1483-3
22. Thoma A, Lightfoot AP. NF-kB and Inflammatory Cytokine Signalling: Role in Skeletal Muscle Atrophy. Adv Exp Med Biol (2018) 1088:267–79. doi: 10.1007/978-981-13-1435-3_12
24. Chua JR, Tee ML. Association of Sarcopenia With Osteoporosis in Patients With Chronic Obstructive Pulmonary Disease. Osteoporos Sarcopenia (2020) 6(3):129–32. doi: 10.1016/j.afos.2020.07.004
25. Avin KG, Bloomfield SA, Gross TS, Warden SJ. Biomechanical Aspects of the Muscle-Bone Interaction. Curr Osteoporos Rep (2015) 13(1):1–8. doi: 10.1007/s11914-014-0244-x
26. Maurel DB, Jahn K, Lara-Castillo N. Muscle-Bone Crosstalk: Emerging Opportunities for Novel Therapeutic Approaches to Treat Musculoskeletal Pathologies. Biomedicines (2017) 5(4):62. doi: 10.3390/biomedicines5040062
27. Frost HM. A 2003 Update of Bone Physiology and Wolff’s Law for Clinicians. Angle Orthod (2004) 74(1):3–15. doi: 10.1043/0003-3219(2004)074<0003:AUOBPA>2.0.CO;2
28. Eatemadololama A, Karimi MT, Rahnama N, Rasolzadegan MH. Resistance Exercise Training Restores Bone Mineral Density in Renal Transplant Recipients. Clin cases Miner Bone Metab (2017) 14(2):157–60. doi: 10.11138/ccmbm/2017.14.1.157
29. Piasecki J, McPhee JS, Hannam K, Deere KC, Elhakeem A, Piasecki M, et al. Hip and Spine Bone Mineral Density are Greater in Master Sprinters, But Not Endurance Runners Compared With non-Athletic Controls. Arch Osteoporos (2018) 13(1):72. doi: 10.1007/s11657-018-0486-9
30. Pickering ME, Simon M, Sornay-Rendu E, Chikh K, Carlier MC, Raby AL, et al. Serum Sclerostin Increases After Acute Physical Activity. Calcif Tissue Int (2017) 101(2):170–3. doi: 10.1007/s00223-017-0272-5
31. Brotto M, Bonewald L. Bone and Muscle: Interactions Beyond Mechanical. Bone (2015) 80:109–14. doi: 10.1016/j.bone.2015.02.010
32. Lee SJ. Regulation of Muscle Mass by Myostatin. Annu Rev Cell Dev Biol (2004) 20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836
33. Lee SJ, McPherron AC. Regulation of Myostatin Activity and Muscle Growth. Proc Natl Acad Sci USA (2001) 98(16):9306–11. doi: 10.1073/pnas.151270098
34. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin Inhibits Myoblast Differentiation by Down-Regulating MyoD Expression. J Biol Chem (2002) 277(51):49831–40. doi: 10.1074/jbc.M204291200
35. Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin Signals Through a Transforming Growth Factor Beta-Like Signaling Pathway to Block Adipogenesis. Mol Cell Biol (2003) 23(20):7230–42. doi: 10.1128/MCB.23.20.7230-7242.2003
36. Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, et al. Smad2 and 3 Transcription Factors Control Muscle Mass in Adulthood. Am J Physiol Cell Physiol (2009) 296(6):C1248–57. doi: 10.1152/ajpcell.00104.2009
37. Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin Reduces Akt/TORC1/p70S6K Signaling, Inhibiting Myoblast Differentiation and Myotube Size. Am J Physiol Cell Physiol (2009) 296(6):C1258–70. doi: 10.1152/ajpcell.00105.2009
38. Lee SJ, Lehar A, Meir JU, Koch C, Morgan A, Warren LE, et al. Targeting Myostatin/Activin A Protects Against Skeletal Muscle and Bone Loss During Spaceflight. Proc Natl Acad Sci USA (2020) 117(38):23942–51. doi: 10.1073/pnas.2014716117
39. Hjorth M, Pourteymour S, Gorgens SW, Langleite TM, Lee S, Holen T, et al. Myostatin in Relation to Physical Activity and Dysglycaemia and its Effect on Energy Metabolism in Human Skeletal Muscle Cells. Acta Physiol (Oxf) (2016) 217(1):45–60. doi: 10.1111/apha.12631
40. Dankbar B, Fennen M, Brunert D, Hayer S, Frank S, Wehmeyer C, et al. Myostatin is a Direct Regulator of Osteoclast Differentiation and Its Inhibition Reduces Inflammatory Joint Destruction in Mice. Nat Med (2015) 21(9):1085–90. doi: 10.1038/nm.3917
41. Shan T, Liang X, Bi P, Kuang S. Myostatin Knockout Drives Browning of White Adipose Tissue Through Activating the AMPK-PGC1alpha-Fndc5 Pathway in Muscle. FASEB J (2013) 27(5):1981–9. doi: 10.1096/fj.12-225755
42. Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-α-Dependent Myokine That Drives Brown-Fat-Like Development of White Fat and Thermogenesis. Nature (2012) 481(7382):463–8. doi: 10.1038/nature10777
43. Daskalopoulou SS, Cooke AB, Gomez YH, Mutter AF, Filippaios A, Mesfum ET, et al. Plasma Irisin Levels Progressively Increase in Response to Increasing Exercise Workloads in Young, Healthy, Active Subjects. Eur J Endocrinol (2014) 171(3):343–52. doi: 10.1530/EJE-14-0204
44. Fox J, Rioux BV, Goulet EDB, Johanssen NM, Swift DL, Bouchard DR, et al. Effect of an Acute Exercise Bout on Immediate Post-Exercise Irisin Concentration in Adults: A Meta-Analysis. Scand J Med Sci Sports (2018) 28(1):16–28. doi: 10.1111/sms.12904
45. Tiano JP, Springer DA, Rane SG. SMAD3 Negatively Regulates Serum Irisin and Skeletal Muscle FNDC5 and Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha (PGC-1alpha) During Exercise. J Biol Chem (2015) 290(18):11431. doi: 10.1074/jbc.A114.617399
46. Reza MM, Subramaniyam N, Sim CM, Ge X, Sathiakumar D, McFarlane C, et al. Irisin Is a Pro-Myogenic Factor That Induces Skeletal Muscle Hypertrophy and Rescues Denervation-Induced Atrophy. Nat Commun (2017) 8(1):1104. doi: 10.1038/s41467-017-01131-0
47. Colaianni G, Cuscito C, Mongelli T, Pignataro P, Buccoliero C, Liu P, et al. The Myokine Irisin Increases Cortical Bone Mass. Proc Natl Acad Sci USA (2015) 112(39):12157–62. doi: 10.1073/pnas.1516622112
48. Narayanan SA, Metzger CE, Bloomfield SA, Zawieja DC. Inflammation-Induced Lymphatic Architecture and Bone Turnover Changes are Ameliorated by Irisin Treatment in Chronic Inflammatory Bowel Disease. FASEB J (2018) 32(9):4848–61. doi: 10.1096/fj.201800178R
49. Colaianni G, Mongelli T, Cuscito C, Pignataro P, Lippo L, Spiro G, et al. Irisin Prevents and Restores Bone Loss and Muscle Atrophy in Hind-Limb Suspended Mice. Sci Rep (2017) 7(1):2811. doi: 10.1038/s41598-017-02557-8
50. Colucci S, Colaianni G, Brunetti G, Ferranti F, Mascetti G, Mori G, et al. Irisin Prevents Microgravity-Induced Impairment of Osteoblast Differentiation In Vitro During the Space Flight CRS-14 Mission. FASEB J (2020) 34(8):10096–106. doi: 10.1096/fj.202000216R
51. Kim H, Wrann CD, Jedrychowski M, Vidoni S, Kitase Y, Nagano K, et al. Irisin Mediates Effects on Bone and Fat via αv Integrin Receptors. Cell (2018) 175(7):1756–68.e17. doi: 10.1016/j.cell.2018.10.025
52. Farmer SR. Boning Up on Irisin. N Engl J Med (2019) 380(15):1480–2. doi: 10.1056/NEJMcibr1900041
53. Pelosi M, De Rossi M, Barberi L, Musaro A. IL-6 Impairs Myogenic Differentiation by Downmodulation of P90rsk/Eef2 and mTOR/P70s6k Axes, Without Affecting AKT Activity. BioMed Res Int (2014) 2014:206026. doi: 10.1155/2014/206026
54. Pedersen BK, Febbraio MA. Muscle as an Endocrine Organ: Focus on Muscle-Derived Interleukin-6. Physiol Rev (2008) 88(4):1379–406. doi: 10.1152/physrev.90100.2007
55. Carson JA, Baltgalvis KA. Interleukin 6 as a Key Regulator of Muscle Mass During Cachexia. Exerc Sport Sci Rev (2010) 38(4):168–76. doi: 10.1097/JES.0b013e3181f44f11
56. Chowdhury S, Schulz L, Palmisano B, Singh P, Berger JM, Yadav VK, et al. Muscle-Derived Interleukin 6 Increases Exercise Capacity by Signaling in Osteoblasts. J Clin Invest (2020) 130(6):2888–902. doi: 10.1172/JCI133572
57. Keller C, Steensberg A, Hansen AK, Fischer CP, Plomgaard P, Pedersen BK. Effect of Exercise, Training, and Glycogen Availability on IL-6 Receptor Expression in Human Skeletal Muscle. J Appl Physiol (1985) (2005) 99(6):2075–9. doi: 10.1152/japplphysiol.00590.2005
58. Le Goff B, Blanchard F, Berthelot JM, Heymann D, Maugars Y. Role for Interleukin-6 in Structural Joint Damage and Systemic Bone Loss in Rheumatoid Arthritis. Joint Bone Spine (2010) 77(3):201–5. doi: 10.1016/j.jbspin.2010.03.002
59. Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, Leukemia Inhibitory Factor, and Oncostatin M Stimulate Bone Resorption and Regulate the Expression of Receptor Activator of NF-Kappa B Ligand, Osteoprotegerin, and Receptor Activator of NF-Kappa B in Mouse Calvariae. J Immunol (2002) 169(6):3353–62. doi: 10.4049/jimmunol.169.6.3353
60. Axmann R, Bohm C, Kronke G, Zwerina J, Smolen J, Schett G. Inhibition of Interleukin-6 Receptor Directly Blocks Osteoclast Formation In Vitro and In Vivo. Arthritis Rheum (2009) 60(9):2747–56. doi: 10.1002/art.24781
61. Kearns AE, Khosla S, Kostenuik PJ. Receptor Activator of Nuclear Factor kappaB Ligand and Osteoprotegerin Regulation of Bone Remodeling in Health and Disease. Endocr Rev (2008) 29(2):155–92. doi: 10.1210/er.2007-0014
62. Bonnet N, Bourgoin L, Biver E, Douni E, Ferrari S. RANKL Inhibition Improves Muscle Strength and Insulin Sensitivity and Restores Bone Mass. J Clin Invest (2020) 130(6):3329. doi: 10.1172/JCI138278
63. Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK Ligand in Mediating Increased Bone Resorption in Early Postmenopausal Women. J Clin Invest (2003) 111(8):1221–30. doi: 10.1172/JCI17215
64. McCloskey EV, Johansson H, Oden A, Austin M, Siris E, Wang A, et al. Denosumab Reduces the Risk of Osteoporotic Fractures in Postmenopausal Women, Particularly in Those With Moderate to High Fracture Risk as Assessed With FRAX. J Bone Miner Res (2012) 27(7):1480–6. doi: 10.1002/jbmr.1606
65. Carvallo L, Henriquez B, Paredes R, Olate J, Onate S, van Wijnen AJ, et al. 1alpha,25-Dihydroxy Vitamin D3-Enhanced Expression of the Osteocalcin Gene Involves Increased Promoter Occupancy of Basal Transcription Regulators and Gradual Recruitment of the 1alpha,25-Dihydroxy Vitamin D3 Receptor-SRC-1 Coactivator Complex. J Cell Physiol (2008) 214(3):740–9. doi: 10.1002/jcp.21267
66. Hauschka PV, Lian JB, Cole DE, Gundberg CM. Osteocalcin and Matrix Gla Protein: Vitamin K-Dependent Proteins in Bone. Physiol Rev (1989) 69(3):990–1047. doi: 10.1152/physrev.1989.69.3.990
67. Zoch ML, Clemens TL, Riddle RC. New Insights Into the Biology of Osteocalcin. Bone (2016) 82:42–9. doi: 10.1016/j.bone.2015.05.046
68. Ferron M, Wei J, Yoshizawa T, Del Fattore A, DePinho RA, Teti A, et al. Insulin Signaling in Osteoblasts Integrates Bone Remodeling and Energy Metabolism. Cell (2010) 142(2):296–308. doi: 10.1016/j.cell.2010.06.003
69. Neve A, Corrado A, Cantatore FP. Osteocalcin: Skeletal and Extra-Skeletal Effects. J Cell Physiol (2013) 228(6):1149–53. doi: 10.1002/jcp.24278
70. Lacombe J, Karsenty G, Ferron M. In Vivo Analysis of the Contribution of Bone Resorption to the Control of Glucose Metabolism in Mice. Mol Metab (2013) 2(4):498–504. doi: 10.1016/j.molmet.2013.08.004
71. Mera P, Laue K, Ferron M, Confavreux C, Wei J, Galan-Diez M, et al. Osteocalcin Signaling in Myofibers Is Necessary and Sufficient for Optimum Adaptation to Exercise. Cell Metab (2017) 25(1):218. doi: 10.1016/j.cmet.2016.12.003
72. Liu S, Gao F, Wen L, Ouyang M, Wang Y, Wang Q, et al. Osteocalcin Induces Proliferation via Positive Activation of the PI3K/Akt, P38 MAPK Pathways and Promotes Differentiation Through Activation of the GPRC6A-ERK1/2 Pathway in C2C12 Myoblast Cells. Cell Physiol Biochem (2017) 43(3):1100–12. doi: 10.1159/000481752
73. Mera P, Laue K, Wei J, Berger JM, Karsenty G. Osteocalcin is Necessary and Sufficient to Maintain Muscle Mass in Older Mice. Mol Metab (2016) 5(10):1042–7. doi: 10.1016/j.molmet.2016.07.002
74. Karsenty G, Olson EN. Bone and Muscle Endocrine Functions: Unexpected Paradigms of Inter-Organ Communication. Cell (2016) 164(6):1248–56. doi: 10.1016/j.cell.2016.02.043
75. Severinsen MCK, Pedersen BK. Muscle-Organ Crosstalk: The Emerging Roles of Myokines. Endocr Rev (2020) 41(4):594–609. doi: 10.1210/endrev/bnaa016
76. Bonewald L. Use it or Lose it to Age: A Review of Bone and Muscle Communication. Bone (2019) 120:212–8. doi: 10.1016/j.bone.2018.11.002
77. Yende S, Waterer GW, Tolley EA, Newman AB, Bauer DC, Taaffe DR, et al. Inflammatory Markers are Associated With Ventilatory Limitation and Muscle Dysfunction in Obstructive Lung Disease in Well Functioning Elderly Subjects. Thorax (2006) 61(1):10–6. doi: 10.1136/thx.2004.034181
78. Celli BR, Locantore N, Yates J, Tal-Singer R, Miller BE, Bakke P, et al. Inflammatory Biomarkers Improve Clinical Prediction of Mortality in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2012) 185(10):1065–72. doi: 10.1164/rccm.201110-1792OC
79. Ijiri N, Kanazawa H, Asai K, Watanabe T, Hirata K. Irisin, a Newly Discovered Myokine, Is a Novel Biomarker Associated With Physical Activity in Patients With Chronic Obstructive Pulmonary Disease. Respirology (2015) 20(4):612–7. doi: 10.1111/resp.12513
80. Kubo H, Asai K, Kojima K, Sugitani A, Kyomoto Y, Okamoto A, et al. Exercise Ameliorates Emphysema Of Cigarette Smoke-Induced COPD In Mice Through The Exercise-Irisin-Nrf2 Axis. Int J Chron Obstruct Pulmon Dis (2019) 14:2507–16. doi: 10.2147/COPD.S226623
81. Sugiyama Y, Asai K, Yamada K, Kureya Y, Ijiri N, Watanabe T, et al. Decreased Levels of Irisin, a Skeletal Muscle Cell-Derived Myokine, are Related to Emphysema Associated With Chronic Obstructive Pulmonary Disease. Int J Chron Obstruct Pulmon Dis (2017) 12:765–72. doi: 10.2147/COPD.S126233
82. Ju CR, Chen RC. Serum Myostatin Levels and Skeletal Muscle Wasting in Chronic Obstructive Pulmonary Disease. Respir Med (2012) 106(1):102–8. doi: 10.1016/j.rmed.2011.07.016
83. Plant PJ, Brooks D, Faughnan M, Bayley T, Bain J, Singer L, et al. Cellular Markers of Muscle Atrophy in Chronic Obstructive Pulmonary Disease. Am J Respir Cell Mol Biol (2010) 42(4):461–71. doi: 10.1165/rcmb.2008-0382OC
84. Ohno Y, Matsuba Y, Hashimoto N, Sugiura T, Ohira Y, Yoshioka T, et al. Suppression of Myostatin Stimulates Regenerative Potential of Injured Antigravitational Soleus Muscle in Mice Under Unloading Condition. Int J Med Sci (2016) 13(9):680–5. doi: 10.7150/ijms.16267
85. Wurtzel CN, Gumucio JP, Grekin JA, Khouri RK Jr., Russell AJ, Bedi A, et al. Pharmacological Inhibition of Myostatin Protects Against Skeletal Muscle Atrophy and Weakness After Anterior Cruciate Ligament Tear. J Orthop Res (2017) 35(11):2499–505. doi: 10.1002/jor.23537
86. Sancho-Munoz A, Guitart M, Rodriguez DA, Gea J, Martinez-Llorens J, Barreiro E. Deficient Muscle Regeneration Potential in Sarcopenic COPD Patients: Role of Satellite Cells. J Cell Physiol (2021) 236(4):3083–98. doi: 10.1002/jcp.30073
87. Polkey MI, Praestgaard J, Berwick A, Franssen FME, Singh D, Steiner MC, et al. Activin Type II Receptor Blockade for Treatment of Muscle Depletion in Chronic Obstructive Pulmonary Disease. A Randomized Trial. Am J Respir Crit Care Med (2019) 199(3):313–20. doi: 10.1164/rccm.201802-0286OC
88. Troosters T, Gosselink R, Decramer M. Short- and Long-Term Effects of Outpatient Rehabilitation in Patients With Chronic Obstructive Pulmonary Disease: A Randomized Trial. Am J Med (2000) 109(3):207–12. doi: 10.1016/s0002-9343(00)00472-1
89. Vogiatzis I, Simoes DC, Stratakos G, Kourepini E, Terzis G, Manta P, et al. Effect of Pulmonary Rehabilitation on Muscle Remodelling in Cachectic Patients With COPD. Eur Respir J (2010) 36(2):301–10. doi: 10.1183/09031936.00112909
90. Troosters T, Probst VS, Crul T, Pitta F, Gayan-Ramirez G, Decramer M, et al. Resistance Training Prevents Deterioration in Quadriceps Muscle Function During Acute Exacerbations of Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2010) 181(10):1072–7. doi: 10.1164/rccm.200908-1203OC
91. Bai P, Sun Y, Jin J, Hou J, Li R, Zhang Q, et al. Disturbance of the OPG/RANK/RANKL Pathway and Systemic Inflammation in COPD Patients With Emphysema and Osteoporosis. Respir Res (2011) 12:157. doi: 10.1186/1465-9921-12-157
92. Xiong J, Tian J, Zhou L, Le Y, Sun Y. Interleukin-17a Deficiency Attenuated Emphysema and Bone Loss in Mice Exposed to Cigarette Smoke. Int J Chron Obstruct Pulmon Dis (2020) 15:301–10. doi: 10.2147/COPD.S235384
93. Xiong J, Le Y, Rao Y, Zhou L, Hu Y, Guo S, et al. RANKL Mediates Muscle Atrophy and Dysfunction in a Cigarette Smoke-Induced Model of Chronic Obstructive Pulmonary Disease. Am J Respir Cell Mol Biol (2021) 64(5):617–28. doi: 10.1165/rcmb.2020-0449OC
94. Duckers JM, Evans BA, Fraser WD, Stone MD, Bolton CE, Shale DJ. Low Bone Mineral Density in Men With Chronic Obstructive Pulmonary Disease. Respir Res (2011) 12:101. doi: 10.1186/1465-9921-12-101
Keywords: COPD, sarcopenia, osteoporosis, myokines, osteokines, crosstalk
Citation: Zhang L and Sun Y (2021) Muscle-Bone Crosstalk in Chronic Obstructive Pulmonary Disease. Front. Endocrinol. 12:724911. doi: 10.3389/fendo.2021.724911
Received: 15 June 2021; Accepted: 13 September 2021;
Published: 28 September 2021.
Edited by:
Giacomina Brunetti, University of Bari Aldo Moro, ItalyReviewed by:
Anna Piotrowska, University School of Physical Education in Krakow, PolandCopyright © 2021 Zhang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongchang Sun, c3VueUBiam11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.