 Erick Spears
Erick Spears Ioannis Serafimidis
Ioannis Serafimidis Alvin C. Powers1,3,4*
Alvin C. Powers1,3,4* Anthony Gavalas
Anthony Gavalas- 1Division of Diabetes, Endocrinology and Metabolism, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
- 2Center of Basic Research, Biomedical Research Foundation of the Academy of Athens, Athens, Greece
- 3Department of Molecular Physiology and Biophysics, Vanderbilt University School of Medicine, Nashville, TN, United States
- 4VA Tennessee Valley Healthcare System, Nashville, TN, United States
- 5Paul Langerhans Institute Dresden (PLID) of Helmholtz Center Munich at the University Clinic Carl Gustav Carus of TU Dresden, Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany
- 6German Centre for Diabetes Research (DZD), Neuherberg, Germany
In all forms of diabetes, β cell mass or function is reduced and therefore the capacity of the pancreatic cells for regeneration or replenishment is a critical need. Diverse lines of research have shown the capacity of endocrine as well as acinar, ductal and centroacinar cells to generate new β cells. Several experimental approaches using injury models, pharmacological or genetic interventions, isolation and in vitro expansion of putative progenitors followed by transplantations or a combination thereof have suggested several pathways for β cell neogenesis or regeneration. The experimental results have also generated controversy related to the limitations and interpretation of the experimental approaches and ultimately their physiological relevance, particularly when considering differences between mouse, the primary animal model, and human. As a result, consensus is lacking regarding the relative importance of islet cell proliferation or progenitor differentiation and transdifferentiation of other pancreatic cell types in generating new β cells. In this review we summarize and evaluate recent experimental approaches and findings related to islet regeneration and address their relevance and potential clinical application in the fight against diabetes.
Introduction
Insulin-producing β cells are critical for maintaining systemic glucose homeostasis. In both type 1 diabetes (T1D) and type 2 diabetes (T2D), reduced and/or inadequate β cell mass leads to insufficient insulin secretion and hyperglycemia. T1D is the result of autoimmune destruction of β cells. At clinical T1D onset, there are still remaining β cells, but the β cell mass declines further with increasing T1D duration. However, many individuals with long-standing T1D continue to secrete some insulin, indicating the persistence of some β cells. In T2D, the combination of insulin resistance due to obesity and impaired functional β cell mass leads to hyperglycemia. With increasing duration of T2D, β cell mass and function progressively declines. Thus, in both forms of diabetes, there is a great need to restore or increase β cell mass (1). In this review, we compare and contrast two primary mechanisms by which β cell mass can be increased: 1) differentiation of progenitor/stem cells or transdifferentiation of other pancreatic cell types into new β cells; and 2) stimulation of proliferation of remaining β cells. These two approaches are discussed with respect to their physiological relevance with the Pros and Cons of each discussed below and in Table 1. While presented as two distinct processes, in reality, both may play a critical role in restoring β cell mass. Prior to discussing these mechanisms, we first review important experimental considerations.
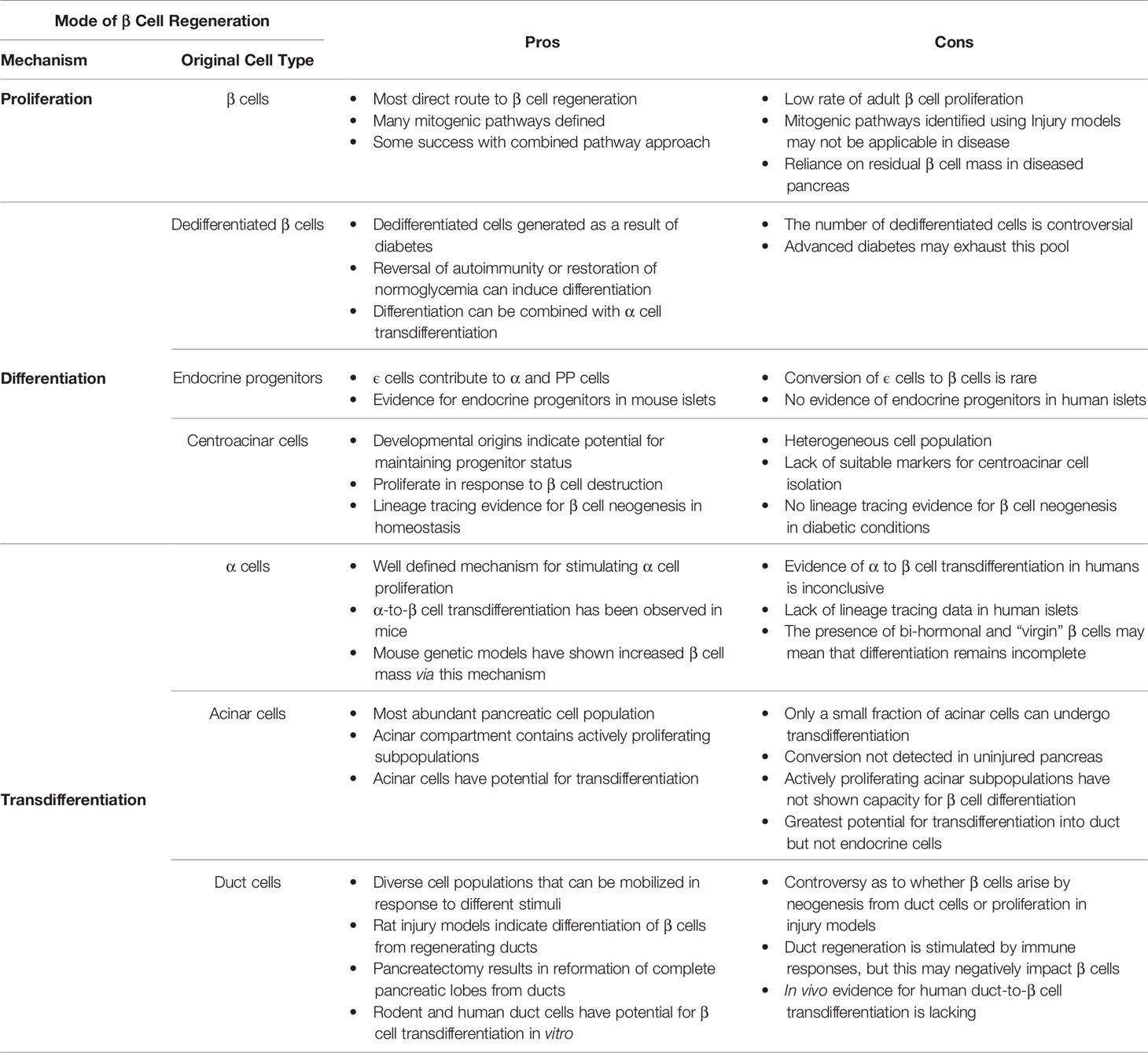
Table 1 Pros and Cons of β cell proliferation versus neogenesis through progenitor differentiation or transdifferentiation.
First, proliferation and differentiation are important processes in establishing and maintaining baseline β cell mass. However, for diabetes-relevant studies, it is desirable to begin with reduced β cell mass that is subsequently restored since what determines restoration may differ from the determinants of baseline β cell mass. Several experimental approaches can be used to reduce β cell mass and the approach taken may influence the experimental outcome of β cell restoration. For example, chemical ablation of β cells with toxins such as streptozotocin or alloxan may impact not only the β cell, but also the progenitor cells responsible for restoration of β cell mass. In contrast, a genetic approach, with deletion of a gene leading to reduced β cell mass, is the product of irreversible genetic alterations that may not be truly representative of the genetic landscape of the natural disease state.
Second, while studying human tissues is important, there are considerable limitations and challenges. Although major strides have been made in the acquisition, processing, and manipulation of human pancreatic islets in recent years, human islets available for research are still relatively scarce. Among tissues that are procured for research, there is wide variability in physiologic responses due to cross-sectional genetic variability in the human population. Efforts to address this scarcity and genetic variability have employed directed differentiation of human pluripotent stem cells into islet cells in culture. These techniques have met with some success, but this is still a relatively new area of research and suffers from the limitations seen in most static culture models.
Third, mice have many strengths but some experimental limitations. Insulin promoter-driven Cre recombinase or fluorescence reporter lines, for β cell targeted gene knockout or β cell-specific reporter expression, have shown variations in β cell specificity with unwanted off-target effects. Furthermore, while mouse β cells show greater regenerative potential than human β cells, their regeneration is still difficult to stimulate. Many of the techniques used to assess β cell regeneration in mice, such as high fat diet or partial pancreatectomy, represent extreme stresses that may not be physiologically relevant to human islets, even those under the physiological stress that result in β cell loss during diabetes.
Fourth, there are profound differences between human and mouse islets that must be considered when relating islet studies performed in mice to humans. Mouse islets have a different architecture than human islets, having α cells on the outside of the islet surrounding a core of β cells. In contrast, human islets have interspersed α and β cells, implying differences in intercellular contacts. Mouse islets have a much higher β:α cell ratio (approximately 7:1) than human islets, in which the β:α ratio is closer to 2:1. Mouse islet cells have shown greater plasticity with respect to proliferation, dedifferentiation and transdifferentiation than do human islets. As discussed below, many of the individual mitogenic pathways in β cells were identified in mouse experiments. Unfortunately, the targeting of individual pathways in human β cells was not sufficient to stimulate significant proliferation as it was in mice. Only when combinatorial approaches were employed did human β cells achieve proliferation to a level that could potentially restore β cell mass in a diabetic setting. All of these differences, among others, indicate that caution is required when translating findings from mice to humans.
Taken together, these experimental considerations represent challenges that must be overcome when assessing the mechanisms of adult β cell regeneration. A consideration of the developmental origin of β cells and their lineage relationship with the other pancreatic cell types is also important as it can illuminate the molecular mechanisms of β cell proliferation and regeneration in the adult. All pancreatic cell types are derived from the same pancreas progenitor population that emerges at the posterior foregut region of the developing embryo. Pancreas progenitors are defined by the expression of the transcription factors PDX1, SOX9 and PTF1A and, as development proceeds, they form a branched epithelium. Acinar progenitors are gradually confined at the tips of this epithelium while trunk cells become bipotent endocrine/duct progenitors. All endocrine progenitors arise in the trunk as NGN3+ cells and migrate into the mesenchyme to form the islets that contain insulin producing β-cells (2, 3). Progenitor differentiation gives way to proliferation which increases β cell mass during late fetal and early neonatal development. The capacity for β cells to proliferate decreases with age. Fetal and neonatal β cells proliferate at the highest rates while adult β cells proliferation, under normal physiological conditions, is very low meaning that β cell mass is fixed in early adolescence.
Taking into account these experimental considerations, the goal of this review will be to detail the current understanding of the molecular and cellular mechanisms involved in β cell regeneration. Previous attempts at β cell regenerative therapies have been extensively reviewed recently (1, 4, 5). Our goal is not to repeat this information, but instead, we hope the reader will gain a comprehensive view of the potential for β cell regeneration and a novel understanding of the underlying molecular and cellular mechanisms. This discussion is accompanied by our assessment of the pros and cons of targeting these different mechanisms of β cell regeneration in hopes of inspiring new and innovative ideas about potential β cell regeneration therapies.
The challenge for adult β cell regeneration lies in reverting pancreatic cell types to an earlier developmental state that will allow them to regain their proliferative capacity and differentiate to fully functional β cells. Resident progenitor or stem cells would be an additional route for the restoration of functional β cell mass.
Endocrine Cell Proliferation
Upon differentiation from progenitors during fetal pancreas development, early β cells expand through proliferation (6). While necessary to increase β cell mass in the fetal and neonatal pancreas, β cell proliferation declines rapidly into early adolescence and is essentially nonexistent in adulthood. The lack of proliferative capacity in adult β cells becomes problematic in individuals who develop diabetes because both T1D and T2D are associated with loss of β cell mass at some point during their progression. For this reason, major emphasis in diabetes research has been placed on understanding β cell proliferation during development and stimulating β cell proliferation in adults.
Fetal and Neonatal β Cell Proliferation
Early studies in the mouse demonstrated a critical role for β cell proliferation in the establishment of adult β cell mass (6, 7). This indicates that it is the systemic environment in young animals that allows for the expansion of β cell mass through proliferation. The proliferative capacity observed in these early β cells is lost in adults due to decreases in stimulatory humoral factors (8), increased cyclin-dependent kinase inhibitor, p16INK4A, expression (9) and decreased responsiveness to PDGF signaling due to decreased expression of the PDGF receptor (10). The upregulation of cell cycle inhibitors and loss of growth factor signaling clamps down the intrinsic proliferative capacity of β cells in adults. Interestingly, parabiosis experiments with young and old mice have shown that the humoral factors which stimulate β cell proliferation in young animals may return adults β cells to a partially proliferation competent state (11). The proliferative capacity of β cells seems to be in direct contrast to their functional capacity. The proliferative fetal and neonatal β cells lack a robust glucose-stimulated insulin response. As these cells mature functionally and become more glucose-responsive, they lose the ability to proliferate (12). The loss of proliferative capacity in adult β cells may be necessary for this functional maturation and is the product of a shift in the nutritional environment after birth (13). These studies have elucidated a great deal about fetal and neonatal β cell proliferation. It is clear from the introduction that there are profound differences between mice and humans and findings in mice must be validated in humans.
Studies in humans are comparatively limited for obvious moral and ethical reasons. Developmental studies of human islets are generally restricted to histological analysis of cadaveric tissues. Nevertheless, studies in human tissues indicate the same dynamics of β cell proliferation as those seen in mice, a peak in β cell proliferation in neonatal stages and a rapid decline through adolescence, though on a time scale that reflects the difference between human and mouse life spans (7, 14). Cadaveric human pancreatic islets may be studied in culture for a relatively short time. These studies indicate that cultured neonatal islets lack the functional maturity that is typically associated with those containing proliferative β cells (15). Unfortunately, the implications of these experiments on β cell proliferation are highly correlative as it is difficult to detect β cell proliferation in cultured human islets. It is also possible to study human islets as mouse kidney xenografts, offering the islets a more dynamic, in vivo environment. While human β cell proliferation has been observed, and even stimulated, in these models (16), the study of neonatal islets in this context has been difficult due to a lack of availability.
Adult β Cell Proliferation
The steep decline in β cell proliferation after the early neonatal period results in fixation of β cell mass during adolescence (7). Human β cell proliferation peaks around 4% during the early neonatal period and, under normal physiological conditions, decreases to approximately 0.2% in adults (14). In mice, the β cell proliferation rate is generally higher, reaching approximately 10% during development and resting at approximately 1% in adults (17). Unlike many other tissues in the body that have the capacity to regenerate, proliferation in adult pancreatic islets is only sufficient to maintain a relatively stable endocrine cell mass. Adult β cells lack the proliferative capacity to compensate for severe loss of β cells, as inflicted during T1D and T2D progression. There are, however, physiological circumstances under which adult β cells are stimulated to proliferate. These circumstances indicate that adult β cells maintain the capacity to proliferate at higher rates than normally seen and offer hope that stimulated β cell proliferation may be a viable option for the restoration β cell mass after diabetic loss.
Pregnancy results in an increased demand for insulin and in the mouse this is accompanied by an adaptive expansion of the β cell population (18). This proliferative response is driven by the placental lactogens and prolactin in mice (19, 20). However, the mechanism of this β cell expansion, whether due to proliferation or de novo differentiation from progenitors, is still in debate. Autopsy studies revealed little increase in proliferation marker Ki67, in the pancreas of pregnant women (21). While human β cell mass typically increases through hyperplasia, the lack of proliferation markers led to the interpretation that the observed increase in β cell mass was the result of β cell neogenesis through progenitor differentiation. Early in vitro studies indicated that placental lactogens, prolactin and human growth hormone can all stimulate β cell proliferation in cultured human islets (19). However, more recent studies have indicated that, while activation of downstream pathway components of prolactin signaling (i.e., STAT5A) could upregulate prolactin signaling targets, they failed to stimulate human β cell proliferation (22). In all, the story of pregnancy-induced β cell expansion is complex and difficult to translate to humans. Many of the lessons learned from the experiments described above appear to be more relevant to studies of gestational diabetes than to generalized β cell proliferation.
Similar to pregnancy, non-diabetic obesity induces insulin resistance and a compensatory increase in β cell mass (23). Although obesity-induced T2D ultimately results in decreased β cell mass, prediabetic obesity causes an initial increase on β cell mass prior to the development of T2D (24–26). Mouse models of obesity and insulin resistance, both genetic and high fat diet, show profound increases in β cell mass (27, 28). These studies have elucidated some molecular pathways for adult β cell proliferation and indicate that intracellular glucose signaling drives β cell proliferation, during hyperglycemia, through glucose metabolism pathways (29–31). Confounding the translatability of these studies, human islets xenograft studies have demonstrated that, despite profound β cell proliferation in native mouse pancreas, human β cells from islets transplanted under the kidney capsules of the same mice did not proliferate in response to high fat diet (32). Again, the differences in β cell plasticity between humans and mice make it difficult to ascertain the mechanisms of stimulated adult β cell proliferation.
Pancreatic injury models in mice suggest that regenerative signals within the whole pancreas could stimulate β cell regeneration (6, 33, 34). However, such regeneration was observed only in young animals and it was not clear whether the β cell regeneration occurred via proliferation pathways or through β cell neogenesis (34). Furthermore, the issue of translatability to the human system was again raised as studies in partial pancreatectomy patients failed to demonstrate increased β cell proliferation, even after removal of up to 50% of the pancreas mass (35). Chemical injury models, such as streptozotocin treatment, have helped to elucidate some of the molecular mechanisms involved in adult β cells proliferation (36).
Extracellular Signals Stimulating Adult β Cell Proliferation
Despite the difficulties outlined above, there has been significant progress made in understanding β cell proliferation under normal physiological conditions and some successes in regeneration of adult β cells through proliferation. Mouse studies, particularly in development, have elucidated many of the molecular components necessary for β cell proliferation. Secreted factors such as incretin hormones (37, 38), adipokines (39–42), interleukins (43, 44), liver-derived factors (45) and secreted growth factors (46–49) have been shown to stimulate β cells proliferation under dynamic physiological conditions. In addition to secreted factors, extracellular matrix components have also been shown to play a critical role in the proliferative capacity of β cells through establishing an islet microenvironment with localized growth factor signaling (50). Of particular recent interest are a family of pleiotropic matricellular proteins known as the cellular communication network (CCN) proteins. Two members of this family have been shown to stimulate adult β cell proliferation after injury and during pregnancy (CCN2/CTGF) (51) and as a secreted factor from pre-weaning mouse serum (CCN4/WISP1 (52). Recently, it has been shown that intraislet endothelial cells and macrophages contribute to β cell proliferation through VEGF signaling and remodeling of the extracellular matrix (53). These studies of extracellular stimulatory factors have paved the way for understanding intracellular signaling pathways that drive β cell proliferation and may be targeted in efforts to stimulate adult β cell proliferation.
Intracellular Pathways Involved in β Cell Proliferation
Extracellular signals transmitted into the β cell stimulate transduction pathways that regulate cellular functions. The challenge is finding the signals that stimulate the necessary signaling pathways to stimulate β cell proliferation, so called mitogenic pathways. Much effort has been placed on elucidating mitogenic signaling pathways in β cells in order to restore a proliferative capacity to adult β cells. There are numerous mitogenic pathways found in proliferating cells throughout the body. It is important to understand the pathways that stimulate β cell proliferation in order to access a physiologically relevant mechanism for β cell regeneration. While there is a broad literature regarding many mitogenic signaling pathways, it is the pathways that have been successfully targeted to stimulate adult β cell proliferation that will be the focus here.
Cyclin dependent kinases (CDKs) drive cell cycle entry and progression and CDK inhibitors are commonly upregulated in cells that have exited regular cell cycle. This is the case in adult β cells where CDK inhibitors such as p16INK4a/CDKN2A, p21/CDKN1A and p18/CDKN2C are known to be key regulators of β cell proliferation (17). A major role of mitogenic pathways in β cells is to overcome the actions of these kinases and allow β cells to progress into active cells cycle.
The most successful identification of β cells mitogens has come through studies designed to identify soluble signals that stimulate β cell proliferation. Using high throughput methods, DYRK1A inhibitors were shown to stimulate adult human β cell proliferation implicating the Calcineurin/NFAT/DYRK1A pathway as a major mitogenic pathway in β cells (54). At the same time, studies demonstrated that osteoprotegerin, a target of prolactin receptor signaling, could stimulate β cell proliferation through the RANKL binding and modulation of the CREB and GSK3 pathways (36). Mouse studies identified a liver-derived mitogen, serpinB1, that stimulates β cells proliferation in zebrafish, mouse, and human adult β cells by stimulating MAP kinase pathway and inhibiting GSK3 (45). A similar effect in mouse and human adult β cells was observed with inhibition of the KRAS signaling pathway and its downstream effector RASSF1A, or by stimulating ERK1/2 phosphorylation with a GLP-1 agonist in combination with inhibition of the tumor suppressor Menin (55). Finally, studies of CDK inhibitors demonstrated that decreased expression of p18/CDKN2C and p21/CDKN1A resulted in adult human β cell proliferation (56). In all, at least four distinct mitogenic pathways and two cell cycle regulators important for adult β cell proliferation were identified during this short time. Given that mouse β cells have greater plasticity with regard to proliferative capacity, mouse studies were sufficient to identify these individual mitogenic pathways. However, none of these pathways, individually, produced enough proliferation in human β cells to be considered a reasonable treatment for diabetic β cell loss.
More success in the human setting has come from studies of multiple mitogenic pathways. Combined inhibition of the Calcineurin/NFAT/DYRK1A pathway with the DYRK1A inhibitor, Harmine, and the TGFβ signaling pathway, with inhibitors such as ALK5, resulted in a synergistic increase in β cell proliferation in primary cultures and human islet xenografts (16). The TGFβ signaling pathway is commonly known as being antagonistic to mitogenic signals in many tissues. This combinatorial approach may produce a robust enough proliferative response in β cells to be of clinical use in stimulating their regeneration.
Proliferation of α Cells
Regeneration of β cells through stimulated proliferation has been, so far, the focus of this section. While this is not as well characterized as for β cells, all of the endocrine cells in the pancreatic islet appear to follow the same dynamic pattern of proliferation as β cells, i.e., highest proliferation in the early neonatal period with a steep decline in proliferation into adulthood (14). There are well defined physiological conditions under which adult pancreatic α cells will be stimulated to proliferate. Interrupted glucagon signaling, either genetically or pharmaceutically, has been shown to result in robust α cell proliferation (57–60). Such interruption inhibited amino acid uptake in the liver resulting in higher serum amino acid levels which ultimately stimulated α cell proliferation (57). This phenomenon is well characterized and has been observed in zebrafish, mice, and human islet xenografts (57, 58, 61). While not a direct stimulation of β cell regeneration, α cells have been shown to transdifferentiate into β cells under certain conditions as discussed below. Stimulated α cell proliferation coupled with α-to-β cell transdifferentiation could be another route to β cell regeneration. This method of stimulating proliferation in cells other than β cells followed by stimulated transdifferentiation may be especially necessary in individuals with long term T1D where β cell mass may be so low that directly stimulating β cell proliferation is not a viable option.
Endocrine Cell Transdifferentiation and Endocrine Progenitors
Diverse lines of research have demonstrated the capacity of endocrine, as well as acinar, ductal and centroacinar cells to generate new β cells via a range of different mechanisms. A number of studies seeking to establish conditions promoting endocrine cell interconversion or identify endocrine progenitors residing in the islet were inspired by the fact that all endocrine cells are derived from dedicated Ngn3+ progenitors and thus bear a close lineage relationship.
Endocrine Transdifferentiation
Genetic studies showed that ectopic expression of Pax4 or inactivation of Arx in α cells resulted in the neogenesis of functional β cells from α cells (62, 63). Similarly, inactivation of the tumor suppressor Men1 in α cells triggered their conversion into β cells albeit with the concurrent development of insulinomas (64). The predisposition of α cells for this transdifferentiation has been attributed to the similar chromatin states of α and β cells (65). This propensity was further confirmed following extreme conditions of β cell ablation using the diphtheria toxin receptor (DTR) approach. When β cells were nearly eliminated in post-pubescent mice, transdifferentiated α cells accounted for a large proportion of newly generated β cells. This transdifferentiation capacity is absent in pre-pubescent mice, however, in these mice, δ cells can transdifferentiate into β cells in a FoxO1 dependent manner. Both processes generated bi-hormonal cells suggesting a mechanism of gradual conversion (66, 67). This conversion was accelerated in mice by the combined inactivation of Arx and DNA methyltransferase 1 (Dnmt1). Interestingly, combined loss of these genes and expression of β cell markers were observed in α cells of T1D human islets suggesting that this transdifferentiation may actually be employed in humans for β cell mass replenishment (68). Moreover, in vitro re-aggregated adult human α or PP cells acquired some aspects of β cell physiology including insulin production and glucose sensing following ectopic expression of PDX1 and MAFA (69).
Pharmacological stimulation of α-to-β cell conversion would provide a possible means of β cell mass restoration in diabetes. It has been suggested that artemisinins, which stimulate GABA signaling, or GABA long term administration trigger this conversion by translocating Arx to the cytoplasm and promoting its subsequent degradation (70, 71). However, these results are still debated as other studies using lineage tracing or in vitro culture of mouse and human islets were not successful in inducing this conversion (72, 73). Other interesting possibilities include two kinase inhibitors which target ribosomal S6 kinase (RSK) and cyclin-dependent kinase-2 (CDK2) and, at least in vitro, upregulate multiple β cell markers including PAX4 in a dose dependent manner (74, 75). In addition, stimulation of the liver receptor homologue-1 (LRH-1/NR5A2), a nuclear receptor that represses inflammation in digestive organs, with a small molecule agonist has been shown to increase β cell (76) mass in pre-clinical mouse models of experimental autoimmune diabetes. An increase in the number of α/β bihormonal cells and the repression of the α cell genetic program, following the administration of the agonist, suggested that the implicated mechanism was an α to β conversion, but this was not shown by lineage tracing.
The forced conversion of α to β cells resulted in a significant number of cells that persisted as bi-hormonal cells suggesting that the conversion proceeded through distinct steps rather than a sudden, all or none, transformation. This interpretation was strengthened by the recent identification of ‘virgin’ β cells that express insulin but lack other key β cell markers, do not sense glucose, and do not support calcium influx. These cells reside in a specialized niche at the periphery of the murine islet, which supports α to β cell interconversion, and correspond to an intermediate cell type between α and β cells constituting a life-long source of fully functional β cells. Interestingly, such immature cells can be identified in the normal human pancreas from donors of different ages and from T1D donors, suggesting that these cells could be a plausible candidate for the replenishment of β cell mass from endogenous sources (77).
It has been shown in rat and mouse studies that stressors such as pancreatectomy induced hyperglycemia (78), loss of FoxO1 expression combined with multiparity or aging (79) and hyperglycemia induced by the ectopic expression of an activated KATP channel (80, 81) resulted in β cell dedifferentiation marked by loss of expression of β cell identity genes, degranulation and upregulation of disallowed genes. Glycemic normalization could either completely or partially reverse this dedifferentiation depending on its timely or delayed implementation, respectively (80–82). Such dedifferentiation has also been seen in diabetic humans and, if substantial, it would provide a possible means for β cell mass recovery. This is still a matter of debate as estimates of the presence of dedifferentiated hormone-/synaptophysin+ islet cells in diabetics vary from as little as 2% (83, 84) to as much as nearly 17% (85). The number of these cells may depend on the time elapsed from the onset of the disease and its severity. Their restoration could be combined with α cell transdifferentiation. A recent study showed that this is precisely what happened when autoimmunity of NOD mice was reversed, and animals were treated with a combination of gastrin and EGF (86).
Endocrine Progentiors
Resident progenitor cells in the islet could provide yet another route for β cell mass restoration. Such a dedicated population, at least in the mouse, appears to be ghrelin producing cells (ϵ cells). They represent 5-10% of the total endocrine cells at birth but their numbers decline significantly postnatally (87, 88). Lineage tracing studies showed that ϵ cells contribute half the population of PP cells, approximately 25% of α cells as well as rare β cells in the islets of adult mice. Intriguingly, the generation of a subset of these cells is Ngn3 independent (89). A fasting mimicking diet in mice could induced the stepwise expression of Sox17 and Pdx1 leading to Ngn3-driven generation of β cells. Repeated cycles of this diet restored normoglycemia in mouse models of T1D and T2D. This regime had similar effects in T1D human pancreatic islets, and those effects could be recapitulated by PKA and mTOR inhibition (90). Whereas the authors suggested that these treatments drove reprogramming of endocrine cells, the possibility of a mobilization of resident endocrine progenitors has not been addressed. Intriguingly, persistent, functionally important expression of Ngn3 at both the mRNA and protein levels has been detected in mouse islets (91) providing a possible mechanistic underpinning for the generation of new β cells from resident islet cells. Importantly, a recent study identified a small cell population in the mouse islets comprising just 1% of the endocrine cells. These cells have a distinct transcriptional signature that is highly reminiscent of Ngn3+ embryonic endocrine progenitors and are lacking markers of terminally differentiated pancreatic cells. They are marked by strong expression of Procr, a cell surface marker enriched in stem cells of other organs. Procr+ cells originate from Ngn3+ cells and clonal analysis in vivo showed that they differentiate into all types of endocrine cells while self-renewing. Furthermore, these could support organoid growth in vitro in the presence of endothelial cells and differentiated into functional β cells (92). The identification of a similar human population would certainly open additional exciting prospects.
Acinar Cell Transdifferentiation
As a candidate source cell population for diabetes cell therapy, acinar cells have the obvious advantages of abundance and demonstrated proliferative capacity and plasticity. In humans, acinar cells constitute more than 50% of the mass of the organ and an abundance of acinar cells are left over from islet isolation procedures. The acinar compartment contains actively proliferating subpopulations such as Bmi1+ cells as well as quiescent progenitors such as Dclk1+ and Stmn+ cells that can mediate complete acinar restoration following cerulein mediated acute pancreatitis (93–95). Acinar cells can undergo transdifferentiation into progenitor, duct-like cells during a process known as acinar to ductal metaplasia (ADM) that facilitates regeneration after injury. This process can also be initiated by oncogenic signalling in which case the transdifferentiated cells become the precursors of pancreatic ductal adenocarcinoma (PDAC) (96). On the other hand, none of these actively proliferating or quiescent acinar subpopulations has demonstrated a capacity for β cell differentiation and ADM does not entail the generation of Ngn3+ endocrine progenitors, let alone β cell neogenesis. Additionally, acinar cells harbour extensive repressive histone modifications of endocrine genes that need to be removed before endocrine conversion (65).
The abundance of acinar cells inspired a number of studies where acinar cells were cultured in suspension under different conditions with the aim to generate functional β cells. Such cultures of rat acinar cells resulted in extensive cell death and transdifferentiation to mostly duct cells but addition of EGF and LIF gave rise to a number of endocrine cells which was increased by additional Notch inhibition. The cells were shown to be of acinar origin by acinar cell specific incorporation of lectin and they were able to rescue diabetes in alloxan-treated mice (97, 98). In a similar approach, suspension culture of mouse acinar cells in the presence of EGF and nicotinamide resulted in β cells comprising 5% of the cultured cells and lineage tracing suggested that these were indeed derived from elastase/amylase expressing cells (99). Such encouraging findings were not restricted to rodents since non-endocrine adult human pancreatic epithelial cells supported endocrine differentiation after in vitro expansion and subsequent transplantation under the mouse kidney capsule (100). In this study, the origin of the cells was not lineage traced but a later, similar, study showed that half of the elastase 2A promoter lineage traced human acinar cells, transduced with STAT3 and activated MAPK, upregulated Ngn3 expression and that 3D culture promoted their conversion to β cells (101).
Acinar cell conversion into β cells has not been detected in the uninjured mouse pancreas but temporal labelling of acinar cells using a Ptf1aCreERT2 driver and pancreatic duct ligation showed a small, long term contribution of acinar cells to endocrine cell generation via a duct-like intermediate. The conversion was enhanced when the number of pre-existing β cells was reduced by streptozotocin treatment (102).
Although not directly addressed in the studies mentioned above, in vitro conversion following relatively long periods of cell culture or conversion in vivo following injury imply that the underlying mechanism would be dedifferentiation of a small number of competent acinar cells, possibly followed by proliferation, and final endocrine differentiation. However, acinar cells also have the capacity for transdifferentiation following transduction by systemically administered viruses that expressed a cocktail of the Pdx1, Ngn3 and MafA transcription factors. Subsequent studies showed that transgenic expression of the same combination of transcription factors or even just Pdx1 through the elastase 2A promoter could induce the transdifferentiation of a few acinar cells into endocrine cells including β cells (103–105). This transdifferentiation appears to be inhibited under hyperglycemic conditions (106).
A common thread in all these studies is that only a small number of acinar cells can undergo transdifferentiation. Single cell RNA Seq analyses may provide the keys to identify these elusive cells and the molecular pathways involved in their transdifferentiation.
Duct Cell Transdifferentiation
The ducts have been postulated as a source of islet cells since the beginning of the 20th century (107, 108). The image of an islet emerging from an adult human duct published in the 1980s (109), the subsequent observations that rodent and human islets or single endocrine cells are very often associated with ducts in adults (110, 111) and the common developmental lineage of duct and endocrine cells strengthened the interest in this hypothesis.
This hypothesis was initially pursued in rat and mouse injury models. Rat pancreatectomy (Px) resulted in duct dedifferentiation and expansion followed by recapitulation of aspects of embryonic development to form apparently new pancreatic lobes containing islets (112). Regeneration of the β cell mass following rat pancreatectomy was enhanced by long-term administration of gastrin (113). Px experiments in mouse lead to contradictory results with some groups reporting both β cell neogenesis and replication of pre-existing β cells (114, 115) whereas others detected only enhanced β cell replication (6, 116). Pancreatic duct ligation (PDL) resulted in the appearance of small islets or isolated Ins+ cells near the ducts as well as in the appearance of Nng3+ cells. The latter were able to give rise to endocrine cells in vitro (117, 118). However, these PDL studies raised controversy due to reproducibility issues and it was also suggested that changes in the composition of the tissue due to the surgical procedure might have led to misinterpretation of the results (119, 120). In any case, these procedures trigger an immune response and cytokines have been shown to stimulate duct proliferation, epithelial to mesenchymal transition and the initiation of the endocrine differentiation program through the upregulation of Ngn3 (121–123). Extensive acinar and endocrine ablation using the DTR system (124) led to duct cell driven regeneration of endocrine cells. This approach induced less inflammation and may therefore have fewer confounding effects (125). Apart from the controversy that some of these experiments generated, none of these approaches included rigorous lineage tracing to rigorously demonstrate the ductal origin of the additional β cells.
This has been attempted in other studies using a large number of diverse Cre driver lines. An early study, using a 1.6 Kb piece of the exclusive duct marker carbonic anhydrase II (CAII) promoter to drive Cre expression, suggested that duct cells contributed to β cell neogenesis, particularly following PDL (126). However, the fidelity of this transgene was questioned and an in vitro study using lineage tracing and human duct cells concluded that it was CAII- but not CAII+ cells that could be converted into β cells (127). Two studies using BAC transgenic lines expressing the inducible form of Cre from the full set of Hnf1b or Sox9 regulatory elements found that, whereas Hnf1b+ and Sox9+ cells generate endocrine cells during development, they stop doing so after birth (128, 129). In contrast, a study using a different Sox9 BAC transgenic line suggested that Sox9+ cells can generate β cells in the context of mild hyperglycemia and long-term administration of EGF and gastrin (130). On the other hand, knock-in approaches using the duct specific Krt19 and Muc1 loci to drive expression of CreERT failed to detect a contribution of duct cells to β cell neogenesis.
Other studies using genetic manipulations combined with lineage tracing demonstrated the ability of some duct cells for β cell neogenesis. Ectopic expression of Pax4 in α cells rescues toxin induced diabetes by converting α to β cells and the former were replenished from Ngn3+ cells of ductal origin (131). In adult, TGF-β receptor mouse mutants, β cells arose from intra-islet ducts following partial pancreatectomy (132) suggesting that suppression of TGFβ signalling may enhance duct cell conversion into β cells. Deletion of the tumor suppressor ubiquitin ligase Fbw7 specifically in duct cells led to the upregulation of Ngn3 expression in a small subset of duct cells and their subsequent conversion into functional β cells (133).
Taken together, and despite reservations for some of the experimental approaches and/or their interpretations, these findings suggested that some duct cells may have the capacity to contribute to β cell neogenesis. A number of in vitro and transplantation studies of both mouse and human duct cells have strengthened this notion.
Human duct cells present in islets proliferated and induced Nkx6.1 expression following transplantation under the kidney capsule provided that the mice carried growth stimulatory traits (134). This was supported by pharmacological treatments using BMP7 (127), a combination of EGF and gastrin (135) or preadipocyte factor 1 (136). These approaches suggested that rodent and human duct cells could proliferate and differentiate into endocrine cells in vitro and they were also used to reverse diabetes in mice. Expansion of human ductal tissue in matrigel resulted in the formation of cysts that contained functional islet-like structures (137). However, the possibility that the initial tissue contained contaminating islet cells was not excluded and a similar approach later showed that removal of endocrine cells by NCAM-mediated magnetic sorting severely depleted the endocrine differentiation potential of these cells (138). To reduce or eliminate contaminating endocrine cells several presumed duct specific markers have been used.
Human duct cells isolated by FACS using the carbohydrate antigen 19-9 expanded in vitro and, when transplanted into NOD-SCID mice, 1% of them were converted into insulin producing cells (139). This suggested that a small percentage of duct cells may retain progenitor properties, and this has been confirmed by several studies. Indeed, only a subset of duct cells has organoid forming capacity. For example, CD24+ duct cells isolated from mouse expand as spheroids and show endocrine differentiation potential (140). CD133 is another duct progenitor marker in both humans and mice. Mouse CD133high/CD71low mouse cells are enriched in tripotent colony forming progenitor cells (141). Human duct cells expanded in vitro showed increased expression of NGN3 with CD133 co-expression and FACS isolated CD133+ cells from these cultures showed endocrine differentiation potential (142). Finally, expression of the key endocrine developmental regulators NGN3, MAFA, PDX1 and PAX6 in expanded CD133 human cells converted duct cells into endocrine cells with β-cell properties. Tripotent progenitor cells have been identified in the human main ducts as P2RY1+/ALK3+ cells that can differentiate into all pancreatic lineages upon transplantation, including functional β cells. Consistent with that, single cell RNA Seq analyses suggested the presence of three distinct differentiation axes (143).
In assessing all the results presented above for their potential in diabetes therapy it is important to bear in mind that the ductal tree is an intricate network containing intercalated ducts, intra-and interlobular ducts as well as main ducts. Molecular analyses at the single cell level identified at least four different duct cell types (144) and different experiments may mobilize different subsets of duct progenitors or induce dedifferentiation of different subsets of duct cells. The challenge will be to identify the cells of origin with unique molecular markers and the distinct signals implicated in their mobilization.
Centroacinar Cells as Progenitor Cells
Pancreatic centroacinar cells (CACs) exist in all vertebrates. CACs are rare, specialized cells situated in the middle of the acinar lobules. They are small, with minimal cytoplasm, numerous mitochondria, and long cytoplasmic extensions, some of which reach the endocrine compartment. They are contiguous with ductal cells and form a fenestrated lining on the luminal surface of each acinus (145, 146). They were first identified in the rodent pancreas in the early 1960s and it has been suggested that they may participate in pancreas regeneration following ethionine mediated ablation of acinar cells (147, 148). It has been postulated that CACs originate developmentally from a domain at the interface of bipotent trunk cells and tip acinar progenitors thus possibly retaining their progenitor status (119, 145). Their expression profile supports this hypothesis as CACs are Notch responsive (149) express Hes1 and Sox9 together with Ptf1a (150) and proliferate in response to injuries such as streptozotocin-induced destruction of β cells, partial pancreatectomy and cerulein induced acute or chronic pancreatitis (151–153). Lineage tracing of Notch responsive cells in the adult zebrafish following β cell ablation or partial pancreatectomy showed that they give rise to endocrine cells. However, lineage tracing in the adult mouse using either a replacement knock-in Hes1CreERT2 allele or a BAC Sox9CreERT2 allele did not detect a contribution of labelled cells in β cell neogenesis in either the resting pancreas or following PDL (128, 154). The latter experiments relied on the assumption that all CACs expressed either of the two markers but given the scarcity of these cells and their small size this was far from certain. The lack of a unique marker for CACs has hampered progress but the finding that mouse CACs can be distinguished and isolated by virtue of their high aldehyde dehydrogenase (ALDH) activity provided plausible candidates among the several Aldh isoforms. Moreover, ALDHhigh cells differentiate into endocrine or acinar cells in vitro either forming pancreatospheres in suspension culture or when injected in embryonic pancreatic buds (150). Aldh1b1, an Aldh isoform, is expressed in all mouse pancreas progenitors during development and its expression is retained specifically in CACs in the adult (155). Lineage tracing using a knock-in Aldh1b1CreERT2 allele that retained all the regulatory elements and the functionality of the targeted Aldh1b1 allele suggested that Aldh1b1 expressing cells are self-renewing and contribute to all three pancreatic lineages under homeostatic conditions. Moreover, mouse Aldh1b1 expressing cells were necessary and sufficient to form pancreatic organoids in vitro (156). Similarly, only ALDHhigh cells from adult human pancreas can form organoids and show endocrine differentiation potential in vitro and in vivo after transplantation (157). Immunofluorescence experiments suggested heterogeneity of the Aldh1b1 expressing mouse CACs and single cell RNA Seq showed that these cells are not all identical but are distributed along two main paths of differentiation and that the earliest progenitors among them are devoid of detectable Sox9 and Hes1 expression (156).
Identification of additional surface markers and elucidation of self-renewing and differentiation requirements may provide the means to isolate, expand and differentiate human CACs into endocrine cells for diabetes treatment.
Discussion - Pros and Cons Towards Clinical β Cell Regeneration
Single cell RNA Seq has revealed that there is significant heterogeneity among pancreas differentiated cells that were once considered homogeneous stable populations (143, 144, 158–160). Terminally differentiated, mitotically inactive cells may exist in a dynamic equilibrium with proliferating differentiated cells and progenitors. This might be the underlying reason for the bewildering variety of pancreatic responses to injury and disease. At the same time, it may provide the opportunity for multiple points of intervention to restore β cells mass.
Accordingly, there are multiple possible approaches to β cell replenishment that have been mainly explored in mice but show some promise of applicability to humans (Figure 1). The most direct route for restoration of β cell mass would stimulate the proliferation of existing β cells. This is happening in vivo, particularly in rodents, even under physiological conditions and several mitogenic pathways have been identified that show promise when manipulated in combination (16, 54, 161, 162). Transferring these to a clinical setting will not be without challenges since the proliferation rate of human β cells is much lower than that in rodents. Additionally, in severe diabetes one will have to rely on a greatly diminished mass of stressed β cells (Table 1).
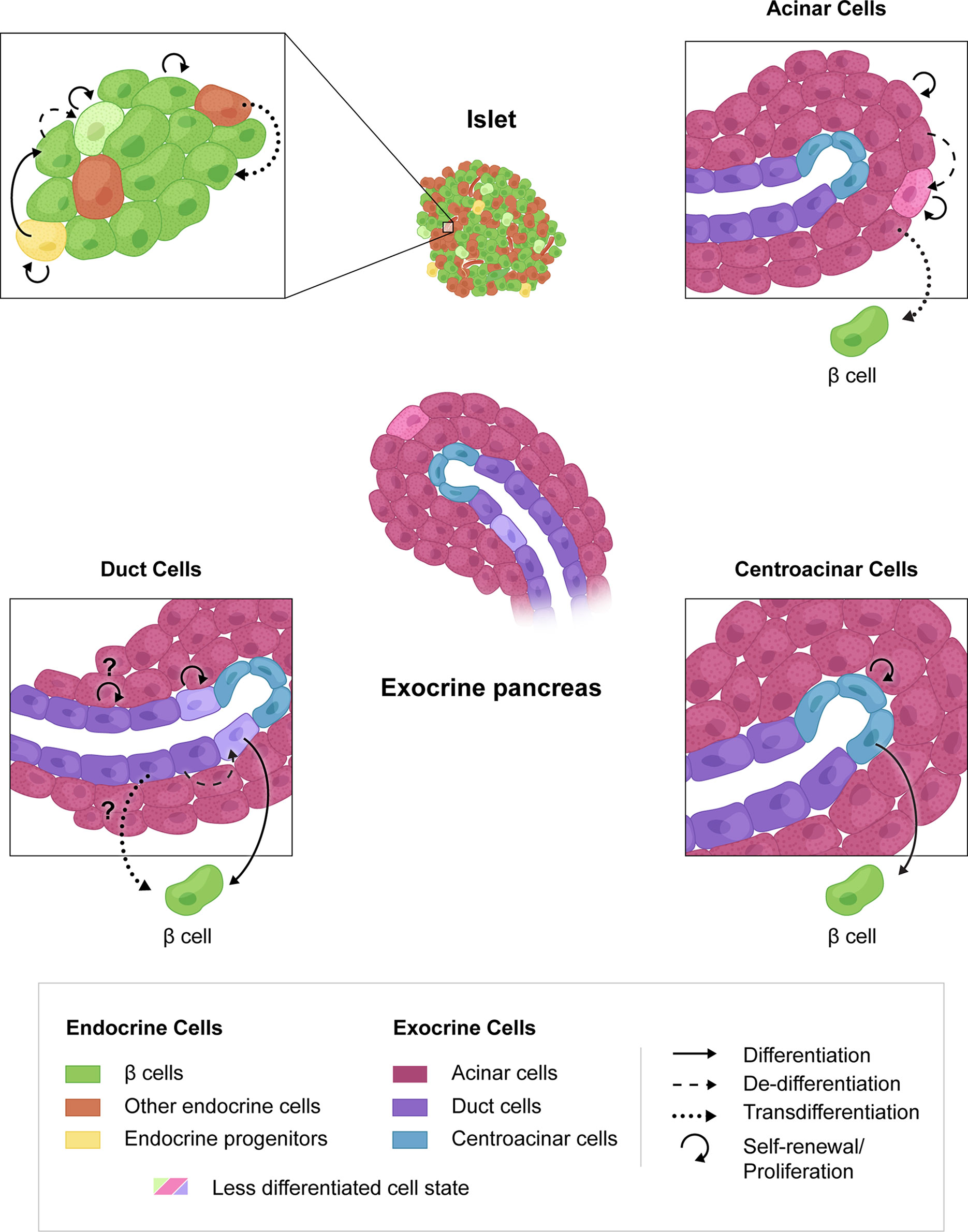
Figure 1 Mechanisms of β cell regeneration through proliferation, progenitor differentiation or transdifferentiation. β cell proliferation is the most direct route to regeneration of β cell mass (upper left, circular arrow). The presence of endocrine progenitors in human islets is unknown but could lend to increased β cell mass. Transdifferentiation from other endocrine cells (upper left), acinar cells (upper right), duct cells (lower left) or centroacinar cells (lower right) may also be targeted for β cell regeneration. Possible cell interconversions that have not been shown experimentally are indicated by question marks.
Dedifferentiated β cells are another likely source for β cell replenishment but the extent of their presence in the diabetic pancreas is controversial (83, 84). It may depend on the stage of the disease as it is possible that advanced, long term diabetes may exhaust this cellular pool. The upregulation of progenitor markers in these cells suggests that they would be more amenable to stimulation of proliferation. This could be combined with treatments that promote β cell differentiation as a consequence of autoimmunity reversal and restoration of normoglycemia (86). Such mobilization of dedifferentiated β cells could be combined with α cell proliferation and transdifferentiation (77) (Table 1).
Progenitor cells are expected to be more proliferative, while remaining malleable towards β cell differentiation. The identification of dedicated progenitor cells in the murine islet (89) has generated excitement but, so far, remains an exclusive murine feature. Murine centroacinar cells, or at least a subpopulation thereof, express progenitor markers, remain proliferative, can contribute to all three pancreatic lineages under homeostatic conditions (89) but it remains unknown if they can be stimulated to generate β cells under diabetic conditions. A possible path would be their isolation, in vitro expansion, and differentiation into β cells followed by transplantation. However, specific surface markers that would mediate their efficient and specific isolation have not been identified (Table 1).
Transdifferentiation is another major route that may be useful in restoring β cell mass. Because of greater similarity and physical proximity, α cells would be the cell of choice and their transdifferentiation has been well documented by lineage tracing in murine models. Additionally, they are more proliferative than β cells, have defined proliferation pathways and their proliferation is further stimulated by β cell destruction (57–60, 67). However, because of the presence of bi-hormonal and virgin β cells, it is questionable whether this differentiation is sufficiently driven to completion, and it remains unclear whether it can occur in human islets. Acinar cells are abundant, contain proliferative subpopulations and it has been established that a small subpopulation is amenable to β cell conversion via transcription factor mediated reprogramming (103–105). However, their tendency is to rather differentiate into duct cells and transdifferentiation into β cells in either the normal or injured pancreas has not been documented. Duct cells as a possible β cell source have remained controversial. Ducts contain diverse subpopulations that can be mobilized using different stimuli and rodent injury models have repeatedly suggested their transdifferentiation capacity which could be recapitulated in vitro (139). However, β cell proliferation has been proposed as the reason for apparent β cell neogenesis and convincing lineage tracing experiments are scarce with the exception of a model where the tumor suppressor gene Fbw7 has been inactivated in duct cells (139). Evidence that this transdifferentiation can happen in vivo is lacking and stimulation of duct regeneration by cytokines may simultaneously adversely affect β cell function.
Irrespective of the relative merits of mechanisms and possible cell sources, there are general concerns to be addressed. Two general routes appear possible for the restoration of β cell mass. The first would be in vivo stimulation of proliferation, differentiation, transdifferentiation or a combination thereof. This would involve delivery of small molecules or transcription factors to the target population and specificity of the delivery would be of major importance to ensure both efficiency and elimination of side effects. There are several promising avenues for targeting β cells (163–166) but knowledge of the receptors present on the surface of other target cells will be essential. This necessitates the unequivocal identification and thorough characterization of the target cells, but this remains elusive even in mouse models. An additional risk to be evaluated is the possibility that stimulation of endocrine cell proliferation may result in the development of insulinomas or glucagonomas. In this respect transdifferentiation may appear a safer, albeit slower, route. The other route would be the isolation of progenitor cells from a biopsy followed by in vitro expansion and differentiation to provide enough β cells for transplantation. This remains a daunting undertaking as it requires the identification of specific surface molecules, to enable efficient isolation, as well as the growth requirements for expansion in vitro. Additionally, transplantation of the differentiated cells will face the same hurdles as hPS cell derived endocrine cells regarding the suitability of candidate transplantation sites, adequate oxygenation, the use of macroencapsulation devices to protect against the escape of undifferentiated cells and the development of fibrotic tissue around the device that would eventually block the function of the transplanted cells.
Beyond these challenges, choosing between the method of β cell regeneration is confounded by the difficulty in evaluating β cell mass in individuals to be treated. As mentioned above, both T1D and T2D are associated with loss of β cell mass with time. The difficulty lies in the heterogeneity of β cell loss among individuals, even those with the same duration of disease. In some individuals with T1D, there may be sufficient residual β cell mass to consider treatment that will stimulate proliferation whereas in other individuals, the loss of β cells may be so severe that a neogenic treatment may be required. Further compounding this problem is a difficulty in assessing β cell mass. Current technologies allow β cell mass calculation in cadaveric pancreas samples, but we lack the imaging technology to assess β cell mass in living patients. This further highlights the challenges to the development of β cell regenerating therapies.
In summary, whereas there are several promising possibilities to restore β cells using adult pancreatic cells significant challenges remain and require additional research.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Author Contributions
All authors reviewed and edited the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
Work in the authors’ laboratories was supported by funding provided by the National Institute of Diabetes and Digestive and Kidney Diseases [the Human Islet Research Network (HIRN; RRID : SCR_014393; https://hirnetwork.org; DK112232, DK123716, DK123743, DK104211, DK108120), and by DK106755, DK117147, DK20593] (AP), The Leona M. and Harry B. Helmsley Charitable Trust, and the Department of Veterans Affairs (BX000666)(AP), the German Center for Diabetes Research (DZD)(82DZD00101) (AG) the German Research Foundation (DFG)(GA 2004/3-2 and IRTG 2251)(AG), and the Greek General Secreteriat of Research and Technology (GSRT) (Code: T1EDK-03532)(IS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhong F, Jiang Y. Endogenous Pancreatic β Cell Regeneration: A Potential Strategy for the Recovery of β Cell Deficiency in Diabetes. Front Endocrinol (2019) 10:101. doi: 10.3389/fendo.2019.00101
2. Bastidas-Ponce A, Scheibner K, Lickert H, Bakhti M. Cellular and Molecular Mechanisms Coordinating Pancreas Development. Development (2017) 144:2873–88. doi: 10.1242/dev.140756
3. Shih HP, Wang A, Sander M. Pancreas Organogenesis: From Lineage Determination to Morphogenesis. Annu Rev Cell Dev Biol (2013) 29:81–105. doi: 10.1146/annurev-cellbio-101512-122405
4. Aguayo-Mazzucato C, Bonner-Weir S. Pancreatic Beta Cell Regeneration as a Possible Therapy for Diabetes. Cell Metab (2018) 27:57–67. doi: 10.1016/j.cmet.2017.08.007
5. Docherty FM, Sussel L. Islet Regeneration: Endogenous and Exogenous Approaches. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22073306
6. Dor Y, Brown J, Martinez OI, Melton DA. Adult Pancreatic Beta-Cells are Formed by Self-Duplication Rather Than Stem-Cell Differentiation. Nature (2004) 429:41–6. doi: 10.1038/nature02520
7. Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, et al. Beta-Cell Replication Is the Primary Mechanism Subserving the Postnatal Expansion of Beta-Cell Mass in Humans. Diabetes (2008) 57:1584–94. doi: 10.2337/db07-1369
8. Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of Aged Progenitor Cells by Exposure to a Young Systemic Environment. Nature (2005) 433:760–4. doi: 10.1038/nature03260
9. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. P16ink4a Induces an Age-Dependent Decline in Islet Regenerative Potential. Nature (2006) 443:453–7. doi: 10.1038/nature05092
10. Chen C, Chai J, Singh L, Kuo CY, Jin L, Feng T, et al. Characterization of an In Vitro Differentiation Assay for Pancreatic-Like Cell Development From Murine Embryonic Stem Cells: Detailed Gene Expression Analysis. Assay Drug Dev Technol (2011) 9:403–19. doi: 10.1089/adt.2010.0314
11. Salpeter SJ, Khalaileh A, Weinberg-Corem N, Ziv O, Glaser B, Dor Y. Systemic Regulation of the Age-Related Decline of Pancreatic β-Cell Replication. Diabetes (2013) 62:2843–8. doi: 10.2337/db13-0160
12. Puri S, Roy N, Russ HA, Leonhardt L, French EK, Roy R, et al. Replication Confers β Cell Immaturity. Nat Commun (2018) 9:485–12. doi: 10.1038/s41467-018-02939-0
13. Helman A, Cangelosi AL, Davis JC, Pham Q, Rothman A, Faust AL, et al. A Nutrient-Sensing Transition at Birth Triggers Glucose-Responsive Insulin Secretion. Cell Metab (2020) 31:1004–16.e5. doi: 10.1016/j.cmet.2020.04.004
14. Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, et al. Formation of a Human β-Cell Population Within Pancreatic Islets is Set Early in Life. J Clin Endocrinol Metab (2012) 97:3197–206. doi: 10.1210/jc.2012-1206
15. Henquin J-C, Nenquin M. Immaturity of Insulin Secretion by Pancreatic Islets Isolated From One Human Neonate. J Diabetes Invest (2018) 9:270–3. doi: 10.1111/jdi.12701
16. Wang P, Karakose E, Liu H, Swartz E, Ackeifi C, Zlatanic V, et al. Combined Inhibition of DYRK1A, SMAD, and Trithorax Pathways Synergizes to Induce Robust Replication in Adult Human Beta Cells. Cell Metab (2019) 29:638–52.e5. doi: 10.1016/j.cmet.2018.12.005
17. Kulkarni RN, Mizrachi E-B, Ocana AG, Stewart AF. Human β-Cell Proliferation and Intracellular Signaling: Driving in the Dark Without a Road Map. Diabetes (2012) 61:2205–13. doi: 10.2337/db12-0018
18. Baeyens L, Hindi S, Sorenson RL, German MS. β-Cell Adaptation in Pregnancy. Diabetes Obes Metab (2016) 18 Suppl 1:63–70. doi: 10.1111/dom.12716
19. Brelje TC, Scharp DW, Lacy PE, Ogren L, Talamantes F, Robertson M, et al. Effect of Homologous Placental Lactogens, Prolactins, and Growth Hormones on Islet B-Cell Division and Insulin Secretion in Rat, Mouse, and Human Islets: Implication for Placental Lactogen Regulation of Islet Function During Pregnancy. Endocrinology (1993) 132:879–87. doi: 10.1210/endo.132.2.8425500
20. Xu Y, Wang X, Gao L, Zhu J, Zhang H, Shi H, et al. Prolactin-Stimulated Survivin Induction Is Required for Beta Cell Mass Expansion During Pregnancy in Mice. Diabetologia (2015) 58:2064–73. doi: 10.1007/s00125-015-3670-0
21. Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, et al. Adaptive Changes in Pancreatic Beta Cell Fractional Area and Beta Cell Turnover in Human Pregnancy. Diabetologia (2010) 53:2167–76. doi: 10.1007/s00125-010-1809-6
22. Chen H, Kleinberger JW, Takane KK, Salim F, Fiaschi-Taesch N, Pappas K, et al. Augmented Stat5 Signaling Bypasses Multiple Impediments to Lactogen-Mediated Proliferation in Human β-Cells. Diabetes (2015) 64:3784–97. doi: 10.2337/db15-0083
23. Linnemann AK, Baan M, Davis DB. Pancreatic β-Cell Proliferation in Obesity. Adv Nutr (Bethesda Md) (2014) 5:278–88. doi: 10.3945/an.113.005488
24. Hanley SC, Austin E, Assouline-Thomas B, Kapeluto J, Blaichman J, Moosavi M, et al. {Beta}-Cell Mass Dynamics and Islet Cell Plasticity in Human Type 2 Diabetes. Endocrinology (2010) 151:1462–72. doi: 10.1210/en.2009-1277
25. Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic Beta-Cell Mass in European Subjects With Type 2 Diabetes. Diabetes Obes Metab (2008) 10 Suppl 4:32–42. doi: 10.1111/j.1463-1326.2008.00969.x
26. Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-Cell Mass and Turnover in Humans: Effects of Obesity and Aging. Diabetes Care (2013) 36:111–7. doi: 10.2337/dc12-0421
27. Hull RL, Kodama K, Utzschneider KM, Carr DB, Prigeon RL, Kahn SE. Dietary-Fat-Induced Obesity in Mice Results in Beta Cell Hyperplasia But Not Increased Insulin Release: Evidence for Specificity of Impaired Beta Cell Adaptation. Diabetologia (2005) 48:1350–8. doi: 10.1007/s00125-005-1772-9
28. Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, et al. A Gene Expression Network Model of Type 2 Diabetes Links Cell Cycle Regulation in Islets With Diabetes Susceptibility. Genome Res (2008) 18:706–16. doi: 10.1101/gr.074914.107
29. Persaud SJ, Liu B, Sampaio HB, Jones PM, Muller DS. Calcium/calmodulin-Dependent Kinase IV Controls Glucose-Induced Irs2 Expression in Mouse Beta Cells Via Activation of cAMP Response Element-Binding Protein. Diabetologia (2011) 54:1109–20. doi: 10.1007/s00125-011-2050-7
30. Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, Stolovich-Rain M, et al. Control of Pancreatic Beta Cell Regeneration by Glucose Metabolism. Cell Metab (2011) 13:440–9. doi: 10.1016/j.cmet.2011.02.012
31. Salpeter SJ, Klochendler A, Weinberg-Corem N, Porat S, Granot Z, Shapiro AMJ, et al. Glucose Regulates Cyclin D2 Expression in Quiescent and Replicating Pancreatic β-Cells Through Glycolysis and Calcium Channels. Endocrinology (2011) 152:2589–98. doi: 10.1210/en.2010-1372
32. Dai C, Kayton NS, Shostak A, Poffenberger G, Cyphert HA, Aramandla R, et al. Stress-Impaired Transcription Factor Expression and Insulin Secretion in Transplanted Human Islets. J Clin Invest (2016) 126:1857–70. doi: 10.1172/JCI83657
33. Cano DA, Rulifson IC, Heiser PW, Swigart LB, Pelengaris S, German M, et al. Regulated Beta-Cell Regeneration in the Adult Mouse Pancreas. Diabetes (2008) 57:958–66. doi: 10.2337/db07-0913
34. Rankin MM, Kushner JA. Adaptive Beta-Cell Proliferation is Severely Restricted With Advanced Age. Diabetes (2009) 58:1365–72. doi: 10.2337/db08-1198
35. Menge BA, Tannapfel A, Belyaev O, Drescher R, Müller C, Uhl W, et al. Partial Pancreatectomy in Adult Humans Does Not Provoke Beta-Cell Regeneration. Diabetes (2008) 57:142–9. doi: 10.2337/db07-1294
36. Kondegowda NG, Fenutria R, Pollack IR, Orthofer M, Garcia-Ocaña A, Penninger JM, et al. Osteoprotegerin and Denosumab Stimulate Human Beta Cell Proliferation Through Inhibition of the Receptor Activator of NF-κB Ligand Pathway. Cell Metab (2015) 22:77–85. doi: 10.1016/j.cmet.2015.05.021
37. Buteau J, Foisy S, Joly E, Prentki M. Glucagon-Like Peptide 1 Induces Pancreatic Beta-Cell Proliferation Via Transactivation of the Epidermal Growth Factor Receptor. Diabetes (2003) 52:124–32. doi: 10.2337/diabetes.52.1.124
38. Fusco J, Xiao X, Prasadan K, Sheng Q, Chen C, Ming Y-C, et al. GLP-1/Exendin-4 Induces β-Cell Proliferation Via the Epidermal Growth Factor Receptor. Sci Rep (2017) 7:9100–6. doi: 10.1038/s41598-017-09898-4
39. Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB, Moede T, et al. Adipsin Is an Adipokine That Improves β Cell Function in Diabetes. Cell (2014) 158:41–53. doi: 10.1016/j.cell.2014.06.005
40. Park S, Hong SM, Sung SR, Jung HK. Long-Term Effects of Central Leptin and Resistin on Body Weight, Insulin Resistance, and Beta-Cell Function and Mass by the Modulation of Hypothalamic Leptin and Insulin Signaling. Endocrinology (2008) 149:445–54. doi: 10.1210/en.2007-0754
41. Pham MN, Kolb H, Mandrup-Poulsen T, Battelino T, Ludvigsson J, Pozzilli P, et al. Serum Adipokines as Biomarkers of Beta-Cell Function in Patients With Type 1 Diabetes: Positive Association With Leptin and Resistin and Negative Association With Adiponectin. Diabetes Metab Res Rev (2013) 29:166–70. doi: 10.1002/dmrr.2378
42. Retnakaran R. Adiponectin and β-Cell Adaptation in Pregnancy. Diabetes (2017) 66:1121–2. doi: 10.2337/dbi17-0001
43. Dauriz M, Trombetta M, Boselli L, Santi L, Brangani C, Pichiri I, et al. Interleukin-6 as a Potential Positive Modulator of Human Beta-Cell Function: An Exploratory Analysis-the Verona Newly Diagnosed Type 2 Diabetes Study (VNDS) 6. Acta Diabetol (2016) 53:393–402. doi: 10.1007/s00592-015-0807-z
44. Dirice E, Kahraman S, Jiang W, El Ouaamari A, De Jesus DF, Teo AKK, et al. Soluble Factors Secreted by T Cells Promote β-Cell Proliferation. Diabetes (2014) 63:188–202. doi: 10.2337/db13-0204
45. El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou J-Y, Shirakawa J, et al. SerpinB1 Promotes Pancreatic β Cell Proliferation. Cell Metab (2016) 23:194–205. doi: 10.1016/j.cmet.2015.12.001
46. Agudo J, Ayuso E, Jimenez V, Salavert A, Casellas A, Tafuro S, et al. IGF-I Mediates Regeneration of Endocrine Pancreas by Increasing Beta Cell Replication Through Cell Cycle Protein Modulation in Mice. Diabetologia (2008) 51:1862–72. doi: 10.1007/s00125-008-1087-8
47. Alvarez-Perez JC, Ernst S, Demirci C, Casinelli GP, Mellado-Gil JMD, Rausell-Palamos F, et al. Hepatocyte Growth Factor/C-Met Signaling Is Required for β-Cell Regeneration. Diabetes (2014) 63:216–23. doi: 10.2337/db13-0333
48. Gaddy DF, Riedel MJ, Pejawar-Gaddy S, Kieffer TJ, Robbins PD. In Vivo Expression of HGF/NK1 and GLP-1 From dsAAV Vectors Enhances Pancreatic ß-Cell Proliferation and Improves Pathology in the Db/Db Mouse Model of Diabetes. Diabetes (2010) 59:3108–16. doi: 10.2337/db09-1886
49. Modi H, Cornu M, Thorens B. Glutamine Stimulates Biosynthesis and Secretion of Insulin-Like Growth Factor 2 (IGF2), an Autocrine Regulator of Beta Cell Mass and Function. J Biol Chem (2014) 289:31972–82. doi: 10.1074/jbc.M114.587733
50. Townsend SE, Gannon M. Extracellular Matrix-Associated Factors Play Critical Roles in Regulating Pancreatic β-Cell Proliferation and Survival. Endocrinology (2019) 160:1885–94. doi: 10.1210/en.2019-00206
51. Pasek RC, Dunn JC, Elsakr JM, Aramandla M, Matta AR, Gannon M. Vascular-Derived Connective Tissue Growth Factor (Ctgf) Is Critical for Pregnancy-Induced β Cell Hyperplasia in Adult Mice. Islets (2017) 9:150–8. doi: 10.1080/19382014.2017.1356963
52. Fernandez-Ruiz R, García-Alamán A, Esteban Y, Mir-Coll J, Serra-Navarro B, Fontcuberta-PiSunyer M, et al. Wisp1 Is a Circulating Factor That Stimulates Proliferation of Adult Mouse and Human Beta Cells. Nat Commun (2020) 11:5982–14. doi: 10.1038/s41467-020-19657-1
53. Saunders DC, Aamodt KI, Richardson TM, Hopkirk AJ, Aramandla R, Poffenberger G, et al. Coordinated Interactions Between Endothelial Cells and Macrophages in the Islet Microenvironment Promote Beta Cell Regeneration. NPJ Regener Med (2021) 6:22. doi: 10.1038/s41536-021-00129-z
54. Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, et al. Inhibition of DYRK1A Stimulates Human β-Cell Proliferation. Diabetes (2016) 65:1660–71. doi: 10.2337/db15-1127
55. Chamberlain CE, Scheel DW, McGlynn K, Kim H, Miyatsuka T, Wang J, et al. Menin Determines K-RAS Proliferative Outputs in Endocrine Cells. J Clin Invest (2014) 124:4093–101. doi: 10.1172/JCI69004
56. Robitaille K, Rourke JL, McBane JE, Fu A, Baird S, Du Q, et al. High-Throughput Functional Genomics Identifies Regulators of Primary Human Beta Cell Proliferation. J Biol Chem (2016) 291:4614–25. doi: 10.1074/jbc.M115.683912
57. Dean ED, Li M, Prasad N, Wisniewski SN, Von Deylen A, Spaeth J, et al. Interrupted Glucagon Signaling Reveals Hepatic α Cell Axis and Role for L-Glutamine in α Cell Proliferation. Cell Metab (2017) 25:1362–73.e5. doi: 10.1016/j.cmet.2017.05.011
58. Kim J, Okamoto H, Huang Z, Anguiano G, Chen S, Liu Q, et al. Amino Acid Transporter Slc38a5 Controls Glucagon Receptor Inhibition-Induced Pancreatic α Cell Hyperplasia in Mice. Cell Metab (2017) 25:1348–61.e8. doi: 10.1016/j.cmet.2017.05.006
59. Longuet C, Robledo AM, Dean ED, Dai C, Ali S, McGuinness I, et al. Liver-Specific Disruption of the Murine Glucagon Receptor Produces α-Cell Hyperplasia: Evidence for a Circulating α-Cell Growth Factor. Diabetes (2013) 62:1196–205. doi: 10.2337/db11-1605
60. Solloway MJ, Madjidi A, Gu C, Eastham-Anderson J, Clarke HJ, Kljavin N, et al. Glucagon Couples Hepatic Amino Acid Catabolism to mTOR-Dependent Regulation of α-Cell Mass. Cell Rep (2015) 12:495–510. doi: 10.1016/j.celrep.2015.06.034
61. Li M, Dean ED, Zhao L, Nicholson WE, Powers AC, Chen W. Glucagon Receptor Inactivation Leads to α-Cell Hyperplasia in Zebrafish. J Endocrinol (2015) 227:93–103. doi: 10.1530/JOE-15-0284
62. Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, et al. The Ectopic Expression of Pax4 in the Mouse Pancreas Converts Progenitor Cells Into Alpha and Subsequently Beta Cells. Cell (2009) 138:449–62. doi: 10.1016/j.cell.2009.05.035
63. Courtney M, Gjernes E, Druelle N, Ravaud C, Vieira A, Ben-Othman N, et al. The Inactivation of Arx in Pancreatic Alpha-Cells Triggers Their Neogenesis and Conversion Into Functional Beta-Like Cells. PloS Genet (2013) 9:e1003934. doi: 10.1371/journal.pgen.1003934
64. Lu J, Herrera PL, Carreira C, Bonnavion R, Seigne C, Calender A, et al. Alpha Cell-Specific Men1 Ablation Triggers the Transdifferentiation of Glucagon-Expressing Cells and Insulinoma Development. Gastroenterology (2010) 138:1954–65. doi: 10.1053/j.gastro.2010.01.046
65. Bramswig NC, Everett LJ, Schug J, Dorrell C, Liu C, Luo Y, et al. Epigenomic Plasticity Enables Human Pancreatic Alpha to Beta Cell Reprogramming. J Clin Invest (2013) 123:1275–84. doi: 10.1172/JCI66514
66. Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, et al. Diabetes Recovery by Age-Dependent Conversion of Pancreatic Delta-Cells Into Insulin Producers. Nature (2014) 514:503–7. doi: 10.1038/nature13633
67. Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, et al. Conversion of Adult Pancreatic Alpha-Cells to Beta-Cells After Extreme Beta-Cell Loss. Nature (2010) 464:1149–54. doi: 10.1038/nature08894
68. Chakravarthy H, Gu X, Enge M, Dai X, Wang Y, Damond N, et al. Converting Adult Pancreatic Islet Alpha Cells Into Beta Cells by Targeting Both Dnmt1 and Arx. Cell Metab (2017) 25:622–34. doi: 10.1016/j.cmet.2017.01.009
69. Furuyama K, Chera S, van Gurp L, Oropeza D, Ghila L, Damond N, et al. Diabetes Relief in Mice by Glucose-Sensing Insulin-Secreting Human Alpha-Cells. Nature (2019) 567:43–8. doi: 10.1038/s41586-019-0942-8
70. Ben-Othman N, Vieira A, Courtney M, Record F, Gjernes E, Avolio F, et al. Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-Like Cell Neogenesis. Cell (2017) 168:73–85 e11. doi: 10.1016/j.cell.2016.11.002
71. Li J, Casteels T, Frogne T, Ingvorsen C, Honore C, Courtney M, et al. Artemisinins Target GABAA Receptor Signaling and Impair Alpha Cell Identity. Cell (2017) 168:86–100.e15. doi: 10.1016/j.cell.2016.11.010
72. Ackermann AM, Moss NG, Kaestner KH. GABA and Artesunate Do Not Induce Pancreatic Alpha-to-Beta Cell Transdifferentiation In Vivo. Cell Metab (2018) 28:787–92.e3. doi: 10.1016/j.cmet.2018.07.002
73. van der Meulen T, Lee S, Noordeloos E, Donaldson CJ, Adams MW, Noguchi GM, et al. Artemether Does Not Turn Alpha Cells Into Beta Cells. Cell Metab (2018) 27:218–225 e4. doi: 10.1016/j.cmet.2017.10.002
74. Fomina-Yadlin D, Kubicek S, Vetere A, He KH, Schreiber SL, Wagner BK. GW8510 Increases Insulin Expression in Pancreatic Alpha Cells Through Activation of P53 Transcriptional Activity. PloS One (2012) 7:e28808. doi: 10.1371/journal.pone.0028808
75. Fomina-Yadlin D, Kubicek S, Walpita D, Dancik V, Hecksher-Sorensen J, Bittker JA, et al. Small-Molecule Inducers of Insulin Expression in Pancreatic Alpha-Cells. Proc Natl Acad Sci U S A (2010) 107:15099–104. doi: 10.1073/pnas.1010018107
76. Cobo-Vuilleumier N, Lorenzo PI, Rodriguez NG, Herrera Gomez IG, Fuente-Martin E, Lopez-Noriega L, et al. LRH-1 Agonism Favours an Immune-Islet Dialogue Which Protects Against Diabetes Mellitus. Nat Commun (2018) 9:1488. doi: 10.1038/s41467-018-03943-0
77. van der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dolleman S, et al. Virgin Beta Cells Persist Throughout Life at a Neogenic Niche Within Pancreatic Islets. Cell Metab (2017) 25:911–26.e6. doi: 10.1016/j.cmet.2017.03.017
78. Jonas JC, Sharma A, Hasenkamp W, Ilkova H, Patane G, Laybutt R, et al. Chronic Hyperglycemia Triggers Loss of Pancreatic Beta Cell Differentiation in an Animal Model of Diabetes. J Biol Chem (1999) 274:14112–21. doi: 10.1074/jbc.274.20.14112
79. Talchai C, Xuan S, Kitamura T, DePinho RA, Accili D. Generation of Functional Insulin-Producing Cells in the Gut by Foxo1 Ablation. Nat Genet (2012) 44:406–12, S1. doi: 10.1038/ng.2215
80. Brereton MF, Iberl M, Shimomura K, Zhang Q, Adriaenssens AE, Proks P, et al. Reversible Changes in Pancreatic Islet Structure and Function Produced by Elevated Blood Glucose. Nat Commun (2014) 5:4639. doi: 10.1038/ncomms5639
81. Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic Beta Cell Dedifferentiation in Diabetes and Redifferentiation Following Insulin Therapy. Cell Metab (2014) 19:872–82. doi: 10.1016/j.cmet.2014.03.010
82. Laybutt DR, Sharma A, Sgroi DC, Gaudet J, Bonner-Weir S, Weir GC. Genetic Regulation of Metabolic Pathways in Beta-Cells Disrupted by Hyperglycemia. J Biol Chem (2002) 277:10912–21. doi: 10.1074/jbc.M111751200
83. Butler AE, Dhawan S, Hoang J, Cory M, Zeng K, Fritsch H, et al. Beta-Cell Deficit in Obese Type 2 Diabetes, a Minor Role of Beta-Cell Dedifferentiation and Degranulation. J Clin Endocrinol Metab (2016) 101:523–32. doi: 10.1210/jc.2015-3566
84. Md Moin AS, Dhawan S, Cory M, Butler PC, Rizza RA, Butler AE. Increased Frequency of Hormone Negative and Polyhormonal Endocrine Cells in Lean Individuals With Type 2 Diabetes. J Clin Endocrinol Metab (2016) 101:3628–36. doi: 10.1210/jc.2016-2496
85. Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, et al. Evidence of Beta-Cell Dedifferentiation in Human Type 2 Diabetes. J Clin Endocrinol Metab (2016) 101:1044–54. doi: 10.1210/jc.2015-2860
86. Tang S, Zhang M, Zeng S, Huang Y, Qin M, Nasri U, et al. Reversal of Autoimmunity by Mixed Chimerism Enables Reactivation of Beta Cells and Transdifferentiation of Alpha Cells in Diabetic NOD Mice. Proc Natl Acad Sci USA (2020) 117:31219–30. doi: 10.1073/pnas.2012389117
87. Andralojc KM, Mercalli A, Nowak KW, Albarello L, Calcagno R, Luzi L, et al. Ghrelin-Producing Epsilon Cells in the Developing and Adult Human Pancreas. Diabetologia (2009) 52:486–93. doi: 10.1007/s00125-008-1238-y
88. Wierup N, Svensson H, Mulder H, Sundler F. The Ghrelin Cell: A Novel Developmentally Regulated Islet Cell in the Human Pancreas. Regul Pept (2002) 107:63–9. doi: 10.1016/S0167-0115(02)00067-8
89. Arnes L, Hill JT, Gross S, Magnuson MA, Sussel L. Ghrelin Expression in the Mouse Pancreas Defines a Unique Multipotent Progenitor Population. PloS One (2012) 7:e52026. doi: 10.1371/journal.pone.0052026
90. Cheng CW, Villani V, Buono R, Wei M, Kumar S, Yilmaz OH, et al. Fasting-Mimicking Diet Promotes Ngn3-Driven Beta-Cell Regeneration to Reverse Diabetes. Cell (2017) 168:775–88.e12. doi: 10.1016/j.cell.2017.01.040
91. Wang S, Jensen JN, Seymour PA, Hsu W, Dor Y, Sander M, et al. Sustained Neurog3 Expression in Hormone-Expressing Islet Cells Is Required for Endocrine Maturation and Function. Proc Natl Acad Sci USA (2009) 106:9715–20. doi: 10.1073/pnas.0904247106
92. Wang D, Wang J, Bai L, Pan H, Feng H, Clevers H, et al. Long-Term Expansion of Pancreatic Islet Organoids From Resident Procr(+) Progenitors. Cell (2020) 180:1198–1211 e19. doi: 10.1016/j.cell.2020.02.048
93. Sangiorgi E, Capecchi MR. Bmi1 Lineage Tracing Identifies a Self-Renewing Pancreatic Acinar Cell Subpopulation Capable of Maintaining Pancreatic Organ Homeostasis. Proc Natl Acad Sci USA (2009) 106:7101–6. doi: 10.1073/pnas.0902508106
94. Westphalen CB, Takemoto Y, Tanaka T, Macchini M, Jiang Z, Renz BW, et al. Dclk1 Defines Quiescent Pancreatic Progenitors That Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell (2016) 18:441–55. doi: 10.1016/j.stem.2016.03.016
95. Wollny D, Zhao S, Everlien I, Lun X, Brunken J, Brune D, et al. Single-Cell Analysis Uncovers Clonal Acinar Cell Heterogeneity in the Adult Pancreas. Dev Cell (2016) 39:289–301. doi: 10.1016/j.devcel.2016.10.002
96. Storz P. Acinar Cell Plasticity and Development of Pancreatic Ductal Adenocarcinoma. Nat Rev Gastroenterol Hepatol (2017) 14:296–304. doi: 10.1038/nrgastro.2017.12
97. Baeyens L, Bonne S, Bos T, Rooman I, Peleman C, Lahoutte T, et al. Notch Signaling as Gatekeeper of Rat Acinar-to-Beta-Cell Conversion In Vitro. Gastroenterology (2009) 136:1750–60.e13. doi: 10.1053/j.gastro.2009.01.047
98. Baeyens L, De Breuck S, Lardon J, Mfopou JK, Rooman I, Bouwens L. In Vitro Generation of Insulin-Producing Beta Cells From Adult Exocrine Pancreatic Cells. Diabetologia (2005) 48:49–57. doi: 10.1007/s00125-004-1606-1
99. Minami K, Okuno M, Miyawaki K, Okumachi A, Ishizaki K, Oyama K, et al. Lineage Tracing and Characterization of Insulin-Secreting Cells Generated From Adult Pancreatic Acinar Cells. Proc Natl Acad Sci USA (2005) 102:15116–21. doi: 10.1073/pnas.0507567102
100. Hao E, Tyrberg B, Itkin-Ansari P, Lakey JR, Geron I, Monosov EZ, et al. Beta-Cell Differentiation From Nonendocrine Epithelial Cells of the Adult Human Pancreas. Nat Med (2006) 12:310–6. doi: 10.1038/nm1367
101. Lemper M, Leuckx G, Heremans Y, German MS, Heimberg H, Bouwens L, et al. Reprogramming of Human Pancreatic Exocrine Cells to Beta-Like Cells. Cell Death Differ (2015) 22:1117–30. doi: 10.1038/cdd.2014.193
102. Pan FC, Bankaitis ED, Boyer D, Xu X, Van de Casteele M, Magnuson MA, et al. Spatiotemporal Patterns of Multipotentiality in Ptf1a-Expressing Cells During Pancreas Organogenesis and Injury-Induced Facultative Restoration. Development (2013) 140:751–64. doi: 10.1242/dev.090159
103. Li W, Nakanishi M, Zumsteg A, Shear M, Wright C, Melton DA, et al. In Vivo Reprogramming of Pancreatic Acinar Cells to Three Islet Endocrine Subtypes. Elife (2014) 3:e01846. doi: 10.7554/eLife.01846
104. Miyazaki S, Tashiro F, Miyazaki J. Transgenic Expression of a Single Transcription Factor Pdx1 Induces Transdifferentiation of Pancreatic Acinar Cells to Endocrine Cells in Adult Mice. PloS One (2016) 11:e0161190. doi: 10.1371/journal.pone.0161190
105. Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In Vivo Reprogramming of Adult Pancreatic Exocrine Cells to Beta-Cells. Nature (2008) 455:627–32. doi: 10.1038/nature07314
106. Cavelti-Weder C, Li W, Zumsteg A, Stemann-Andersen M, Zhang Y, Yamada T, et al. Hyperglycaemia Attenuates In Vivo Reprogramming of Pancreatic Exocrine Cells to Beta Cells in Mice. Diabetologia (2016) 59:522–32. doi: 10.1007/s00125-015-3838-7
107. Bensley RR. Studies on the Pancreas of the Guinea Pig. Am J Anat (1911) 12:297–388. doi: 10.1002/aja.1000120304
109. Swartz FJ, Carstens PH. An Islet of Langerhans Located Within the Epithelium of a Human Pancreatic Duct. Histol Histopathol (1986) 1:111–7.
110. Watanabe T, Yaegashi H, Koizumi M, Toyota T, Takahashi T. Changing Distribution of Islets in the Developing Human Pancreas: A Computer-Assisted Three-Dimensional Reconstruction Study. Pancreas (1999) 18:349–54. doi: 10.1097/00006676-199905000-00004
111. Bertelli E, Bendayan M. Association Between Endocrine Pancreas and Ductal System. More Than an Epiphenomenon of Endocrine Differentiation and Development? J Histochem Cytochem (2005) 53:1071–86. doi: 10.1369/jhc.5R6640.2005
112. Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A Second Pathway for Regeneration of Adult Exocrine and Endocrine Pancreas. A possible recapitulation of embryonic development. Diabetes (1993) 42:1715–20. doi: 10.2337/diab.42.12.1715
113. Tellez N, Montanya E. Gastrin Induces Ductal Cell Dedifferentiation and Beta-Cell Neogenesis After 90% Pancreatectomy. J Endocrinol (2014) 223:67–78. doi: 10.1530/JOE-14-0222
114. Ackermann Misfeldt A, Costa RH, Gannon M. Beta-Cell Proliferation, But Not Neogenesis, Following 60% Partial Pancreatectomy is Impaired in the Absence of Foxm1. Diabetes (2008) 57:3069–77. doi: 10.2337/db08-0878
115. Peshavaria M, Larmie BL, Lausier J, Satish B, Habibovic A, Roskens V, et al. Regulation of Pancreatic Beta-Cell Regeneration in the Normoglycemic 60% Partial-Pancreatectomy Mouse. Diabetes (2006) 55:3289–98. doi: 10.2337/db06-0017
116. Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and Regeneration of Adult Beta Cells Does Not Involve Specialized Progenitors. Dev Cell (2007) 12:817–26. doi: 10.1016/j.devcel.2007.04.011
117. Wang RN, Kloppel G, Bouwens L. Duct- to Islet-Cell Differentiation and Islet Growth in the Pancreas of Duct-Ligated Adult Rats. Diabetologia (1995) 38:1405–11. doi: 10.1007/BF00400600
118. Xu X, D’Hoker J, Stange G, Bonne S, De Leu N, Xiao X, et al. Beta Cells can be Generated From Endogenous Progenitors in Injured Adult Mouse Pancreas. Cell (2008) 132:197–207. doi: 10.1016/j.cell.2007.12.015
119. Kopp JL, Dubois CL, Hao E, Thorel F, Herrera PL, Sander M. Progenitor Cell Domains in the Developing and Adult Pancreas. Cell Cycle (2011) 10:1921–7. doi: 10.4161/cc.10.12.16010
120. Van de Casteele M, Leuckx G, Cai Y, Yuchi Y, Coppens V, De Groef S, et al. Partial Duct Ligation: Beta-Cell Proliferation and Beyond. Diabetes (2014) 63:2567–77. doi: 10.2337/db13-0831
121. Gu D, Sarvetnick N. Epithelial Cell Proliferation and Islet Neogenesis in IFN-G Transgenic Mice. Development (1993) 118:33–46. doi: 10.1242/dev.118.1.33
122. Wang TC, Bonner-Weir S, Oates PS, Chulak M, Simon B, Merlino GT, et al. Pancreatic Gastrin Stimulates Islet Differentiation of Transforming Growth Factor Alpha-Induced Ductular Precursor Cells. J Clin Invest (1993) 92:1349–56. doi: 10.1172/JCI116708
123. Valdez IA, Dirice E, Gupta MK, Shirakawa J, Teo AKK, Kulkarni RN. Proinflammatory Cytokines Induce Endocrine Differentiation in Pancreatic Ductal Cells Via STAT3-Dependent NGN3 Activation. Cell Rep (2016) 15:460–70. doi: 10.1016/j.celrep.2016.03.036
124. Saito M, Iwawaki T, Taya C, Yonekawa H, Noda M, Inui Y, et al. Diphtheria Toxin Receptor-Mediated Conditional and Targeted Cell Ablation in Transgenic Mice. Nat Biotechnol (2001) 19:746–50. doi: 10.1038/90795
125. Criscimanna A, Speicher JA, Houshmand G, Shiota C, Prasadan K, Ji B, et al. Duct Cells Contribute to Regeneration of Endocrine and Acinar Cells Following Pancreatic Damage in Adult Mice. Gastroenterology (2011) 141:1451–62, 1462 e1-6. doi: 10.1053/j.gastro.2011.07.003
126. Inada A, Nienaber C, Katsuta H, Fujitani Y, Levine J, Morita R, et al. Carbonic Anhydrase II-Positive Pancreatic Cells are Progenitors for Both Endocrine and Exocrine Pancreas After Birth. Proc Natl Acad Sci USA (2008) 105:19915–9. doi: 10.1073/pnas.0805803105
127. Klein D, Alvarez-Cubela S, Lanzoni G, Vargas N, Prabakar KR, Boulina M, et al. BMP-7 Induces Adult Human Pancreatic Exocrine-To-Endocrine Conversion. Diabetes (2015) 64:4123–34. doi: 10.2337/db15-0688
128. Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, et al. Sox9+ Ductal Cells are Multipotent Progenitors Throughout Development But do Not Produce New Endocrine Cells in the Normal or Injured Adult Pancreas. Development (2011) 138:653–65. doi: 10.1242/dev.056499
129. Solar M, Cardalda C, Houbracken I, Martin M, Maestro MA, De Medts N, et al. Pancreatic Exocrine Duct Cells Give Rise to Insulin-Producing Beta Cells During Embryogenesis But Not After Birth. Dev Cell (2009) 17:849–60. doi: 10.1016/j.devcel.2009.11.003
130. Zhang M, Lin Q, Qi T, Wang T, Chen CC, Riggs AD, et al. Growth Factors and Medium Hyperglycemia Induce Sox9+ Ductal Cell Differentiation Into Beta Cells in Mice With Reversal of Diabetes. Proc Natl Acad Sci U S A (2016) 113:650–5. doi: 10.1073/pnas.1524200113
131. Al-Hasani K, Pfeifer A, Courtney M, Ben-Othman N, Gjernes E, Vieira A, et al. Adult Duct-Lining Cells Can Reprogram Into Beta-Like Cells Able to Counter Repeated Cycles of Toxin-Induced Diabetes. Dev Cell (2013) 26:86–100. doi: 10.1016/j.devcel.2013.05.018
132. El-Gohary Y, Wiersch J, Tulachan S, Xiao X, Guo P, Rymer C, et al. Intraislet Pancreatic Ducts Can Give Rise to Insulin-Positive Cells. Endocrinology (2016) 157:166–75. doi: 10.1210/en.2015-1175
133. Sancho R, Gruber R, Gu G, Behrens A. Loss of Fbw7 Reprograms Adult Pancreatic Ductal Cells Into Alpha, Delta, and Beta Cells. Cell Stem Cell (2014) 15:139–53. doi: 10.1016/j.stem.2014.06.019
134. Tyrberg B, Ustinov J, Otonkoski T, Andersson A. Stimulated Endocrine Cell Proliferation and Differentiation in Transplanted Human Pancreatic Islets: Effects of the Ob Gene and Compensatory Growth of the Implantation Organ. Diabetes (2001) 50:301–7. doi: 10.2337/diabetes.50.2.301
135. Suarez-Pinzon WL, Lakey JR, Brand SJ, Rabinovitch A. Combination Therapy With Epidermal Growth Factor and Gastrin Induces Neogenesis of Human Islet {Beta}-Cells From Pancreatic Duct Cells and an Increase in Functional {Beta}-Cell Mass. J Clin Endocrinol Metab (2005) 90:3401–9. doi: 10.1210/jc.2004-0761
136. Rhee M, Lee SH, Kim JW, Ham DS, Park HS, Yang HK, et al. Preadipocyte Factor 1 Induces Pancreatic Ductal Cell Differentiation Into Insulin-Producing Cells. Sci Rep (2016) 6:23960. doi: 10.1038/srep23960
137. Bonner-Weir S, Taneja M, Weir GC, Tatarkiewicz K, Song KH, Sharma A, et al. In Vitro Cultivation of Human Islets From Expanded Ductal Tissue. Proc Natl Acad Sci U S A (2000) 97:7999–8004. doi: 10.1073/pnas.97.14.7999
138. Gao R, Ustinov J, Korsgren O, Otonkoski T. In Vitro Neogenesis of Human Islets Reflects the Plasticity of Differentiated Human Pancreatic Cells. Diabetologia (2005) 48:2296–304. doi: 10.1007/s00125-005-1935-8
139. Yatoh S, Dodge R, Akashi T, Omer A, Sharma A, Weir GC, et al. Differentiation of Affinity-Purified Human Pancreatic Duct Cells to Beta-Cells. Diabetes (2007) 56:1802–9. doi: 10.2337/db06-1670
140. Rezanejad H, Ouziel-Yahalom L, Keyzer CA, Sullivan BA, Hollister-Lock J, Li WC, et al. Heterogeneity of SOX9 and HNF1beta in Pancreatic Ducts Is Dynamic. Stem Cell Rep (2018) 10:725–38. doi: 10.1016/j.stemcr.2018.01.028
141. Jin L, Gao D, Feng T, Tremblay JR, Ghazalli N, Luo A, et al. Cells With Surface Expression of CD133highCD71low Are Enriched for Tripotent Colony-Forming Progenitor Cells in the Adult Murine Pancreas. Stem Cell Res (2016) 16:40–53. doi: 10.1016/j.scr.2015.11.015
142. Gomez DL, O’Driscoll M, Sheets TP, Hruban RH, Oberholzer J, McGarrigle JJ, et al. Neurogenin 3 Expressing Cells in the Human Exocrine Pancreas Have the Capacity for Endocrine Cell Fate. PloS One (2015) 10:e0133862. doi: 10.1371/journal.pone.0133862
143. Qadir MMF, Alvarez-Cubela S, Klein D, van Dijk J, Muniz-Anquela R, Moreno-Hernandez YB, et al. Single-Cell Resolution Analysis of the Human Pancreatic Ductal Progenitor Cell Niche. Proc Natl Acad Sci USA (2020) 117:10876–87. doi: 10.1073/pnas.1918314117
144. Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-Cell Population Structure. Cell Syst (2016) 3:346–360 e4. doi: 10.1016/j.cels.2016.08.011
145. Cleveland MH, Sawyer JM, Afelik S, Jensen J, Leach SD. Exocrine Ontogenies: On the Development of Pancreatic Acinar, Ductal and Centroacinar Cells. Semin Cell Dev Biol (2012) 23:711–9. doi: 10.1016/j.semcdb.2012.06.008
146. Beer RL, Parsons MJ, Rovira M. Centroacinar Cells: At the Center of Pancreas Regeneration. Dev Biol (2016) 413:8–15. doi: 10.1016/j.ydbio.2016.02.027
147. Warshawsky H, Leblond CP, Droz B. Synthesis and Migration of Proteins in the Cells of the Exocrine Pancreas as Revealed by Specific Activity Determination From Radioautographs. J Cell Biol (1963) 16:1–24. doi: 10.1083/jcb.16.1.1
148. Herman L, Fitzgerald PJ. Restitution of Pancreatic Acinar Cells Following Ethionine. J Cell Biol (1962) 12:297–312. doi: 10.1083/jcb.12.2.297
149. Nakano Y, Negishi N, Gocho S, Mine T, Sakurai Y, Yazawa M, et al. Disappearance of Centroacinar Cells in the Notch Ligand-Deficient Pancreas. Genes Cells (2015) 20:500–11. doi: 10.1111/gtc.12243
150. Rovira M, Scott SG, Liss AS, Jensen J, Thayer SP, Leach SD. Isolation and Characterization of Centroacinar/Terminal Ductal Progenitor Cells in Adult Mouse Pancreas. Proc Natl Acad Sci U S A (2010) 107:75–80. doi: 10.1073/pnas.0912589107
151. Nagasao J, Yoshioka K, Amasaki H, Tsujio M, Ogawa M, Taniguchi K, et al. Morphological Changes in the Rat Endocrine Pancreas Within 12 H of Intravenous Streptozotocin Administration. Anat Histol Embryol (2005) 34:42–7. doi: 10.1111/j.1439-0264.2004.00566.x
152. Hayashi KY, Tamaki H, Handa K, Takahashi T, Kakita A, Yamashina S. Differentiation and Proliferation of Endocrine Cells in the Regenerating Rat Pancreas After 90% Pancreatectomy. Arch Histol Cytol (2003) 66:163–74. doi: 10.1679/aohc.66.163
153. Gasslander T, Smeds S, Blomqvist L, Ihse I. Proliferative Response of Different Exocrine Pancreatic Cell Types to Hormonal Stimuli. I. Effects of Long-Term Cerulein Administration. Scand J Gastroenterol (1990) 25:1103–10. doi: 10.3109/00365529008998541
154. Kopinke D, Brailsford M, Shea JE, Leavitt R, Scaife CL, Murtaugh LC. Lineage Tracing Reveals the Dynamic Contribution of Hes1+ Cells to the Developing and Adult Pancreas. Development (2011) 138:431–41. doi: 10.1242/dev.053843
155. Ioannou M, Serafimidis I, Arnes L, Sussel L, Singh S, Vasiliou V, et al. ALDH1B1 Is a Potential Stem/Progenitor Marker for Multiple Pancreas Progenitor Pools. Dev Biol (2013) 374:153–63. doi: 10.1016/j.ydbio.2012.10.030
156. Mameishvili E, Serafimidis I, Iwaszkiewicz S, Lesche M, Reinhardt S, Bolicke N, et al. Aldh1b1 Expression Defines Progenitor Cells in the Adult Pancreas and Is Required for Kras-Induced Pancreatic Cancer. Proc Natl Acad Sci USA (2019) 116:20679–88. doi: 10.1073/pnas.1901075116
157. Loomans CJM, Williams Giuliani N, Balak J, Ringnalda F, van Gurp L, Huch M, et al. Expansion of Adult Human Pancreatic Tissue Yields Organoids Harboring Progenitor Cells With Endocrine Differentiation Potential. Stem Cell Rep (2018) 10:712–24. doi: 10.1016/j.stemcr.2018.02.005
158. Muraro MJ, Dharmadhikari G, Grun D, Groen N, Dielen T, Jansen E, et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst (2016) 3:385–94.e3. doi: 10.1016/j.cels.2016.09.002
159. Segerstolpe A, Palasantza A, Eliasson P, Andersson EM, Andreasson AC, Sun X, et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab (2016) 24:593–607. doi: 10.1016/j.cmet.2016.08.020
160. Wollny D, Zhao S, Everlien I, Weichert W, Marciniak-Czochra A, Martin-Villalba A. Single-Cell Analysis Uncovers Clonal Acinar Cell Heterogeneity in the Adult Pancreas. Dev Cell (2016) 39:1–13. doi: 10.1016/j.devcel.2016.10.002
161. Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang Y-Q, et al. Inhibition of DYRK1A and GSK3B Induces Human β-Cell Proliferation. Nat Commun (2015) 6:8372–11. doi: 10.1038/ncomms9372
162. Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, Bender A, et al. A High-Throughput Chemical Screen Reveals That Harmine-Mediated Inhibition of DYRK1A Increases Human Pancreatic Beta Cell Replication. Nat Med (2015) 21:383–8. doi: 10.1038/nm.3820
163. Bhattacharya P, Fan J, Haddad C, Essani A, Gopisetty A, Elshabrawy HA, et al. A Novel Pancreatic Beta-Cell Targeting Bispecific-Antibody (BsAb) can Prevent the Development of Type 1 Diabetes in NOD Mice. Clin Immunol (2014) 153:187–98. doi: 10.1016/j.clim.2014.04.014
164. Sachs S, Bastidas-Ponce A, Tritschler S, Bakhti M, Bottcher A, Sanchez-Garrido MA, et al. Author Correction: Targeted Pharmacological Therapy Restores Beta-Cell Function for Diabetes Remission. Nat Metab (2020) 2:380. doi: 10.1038/s42255-020-0201-1
165. Saunders DC, Brissova M, Phillips N, Shrestha S, Walker JT, Aramandla R, et al. Ectonucleoside Triphosphate Diphosphohydrolase-3 Antibody Targets Adult Human Pancreatic Beta Cells for In Vitro and In Vivo Analysis. Cell Metab (2019) 29:745–54.e4. doi: 10.1016/j.cmet.2018.10.007
Keywords: β cells, β cell proliferation, differentiation, transdifferentiation, acinar cells, duct cells, centroacinar cells
Citation: Spears E, Serafimidis I, Powers AC and Gavalas A (2021) Debates in Pancreatic Beta Cell Biology: Proliferation Versus Progenitor Differentiation and Transdifferentiation in Restoring β Cell Mass. Front. Endocrinol. 12:722250. doi: 10.3389/fendo.2021.722250
Received: 08 June 2021; Accepted: 14 July 2021;
Published: 06 August 2021.
Edited by:
Eckhard Lammert, Heinrich Heine University of Düsseldorf, GermanyReviewed by:
Benoit Gauthier, Andalusian Center of Molecular Biology and Regenerative Medicine (CABIMER), SpainSimona Chera, University of Bergen, Norway
Copyright © 2021 Spears, Serafimidis, Powers and Gavalas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony Gavalas, YW50aG9ueS5nYXZhbGFzQHR1LWRyZXNkZW4uZGU=; Alvin C. Powers, YWwucG93ZXJzQHZ1bWMub3Jn
†These authors share first authorship