 Camila Oliveira de Souza
Camila Oliveira de Souza Xuenan Sun
Xuenan Sun Dayoung Oh
Dayoung Oh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 23 August 2021
Sec. Cellular Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.715877
This article is part of the Research TopicG protein-coupled receptors: Role in metabolic disordersView all 9 articles
Seven transmembrane receptors (7TMRs), often termed G protein-coupled receptors (GPCRs), are the most common target of therapeutic drugs used today. Many studies suggest that distinct members of the GPCR superfamily represent potential targets for the treatment of various metabolic disorders including obesity and type 2 diabetes (T2D). GPCRs typically activate different classes of heterotrimeric G proteins, which can be subgrouped into four major functional types: Gαs, Gαi, Gαq/11, and G12/13, in response to agonist binding. Accumulating evidence suggests that GPCRs can also initiate β-arrestin-dependent, G protein-independent signaling. Thus, the physiological outcome of activating a certain GPCR in a particular tissue may also be modulated by β-arrestin-dependent, but G protein-independent signaling pathways. In this review, we will focus on the role of G protein- and β-arrestin-dependent signaling pathways in the development of obesity and T2D-related metabolic disorders.
Type 2 diabetes (T2D) is a complex, heterogeneous disease afflicting an increasing proportion of the population. In 2018, around 8.2% of the United States population had T2D (1). Insulin resistance is key to the pathogenesis of T2D, and obesity is the most common cause of insulin resistance in humans (2). As the worldwide prevalence of obesity is rising to epidemic proportions, a parallel epidemic of T2D is eminent (3). In most individuals, insulin resistance can be compensated by pancreatic β-cells through hyperinsulinemia. However, eventually β-cell dysfunction emerges and is characterized by a decrease in β-cell mass, as well as poor ability of β-cells to correctly secrete insulin in response to glucose. In this context, hyperinsulinemia is no longer able to compensate resulting in hyperglycemia and the development of T2D (2, 4, 5). Therefore, insulin resistance and lower insulin secretion are the two coexisting pathophysiological markers in most patients with T2D (2, 4, 5).
G protein-coupled receptors (GPCRs) regulate virtually all metabolic processes, including glucose and energy homeostasis. In this review, we focus on GPCRs that function in metabolic disorders, particularly in T2D and obesity-related diseases. Several endogenous ligands such as free fatty acids and their receptors (e.g., GPR40, GPR41, GPR43, GPR84, GPR119, and GPR120) have been extensively studied in the regulation of insulin secretion, insulin sensitization, β-cell expansion, and glucose homeostasis (Figure 1 and Table 1). Concomitantly, drugs that target these GPCRs in metabolic tissues have emerged as attractive T2D therapeutic targets as well (47). Thus, this review will discuss GPCRs and their signaling pathways (G protein-dependent and/or β-arrestin-dependent) that can be targeted pharmacologically to treat T2D by improving insulin sensitivity (Figures 1 and 2).
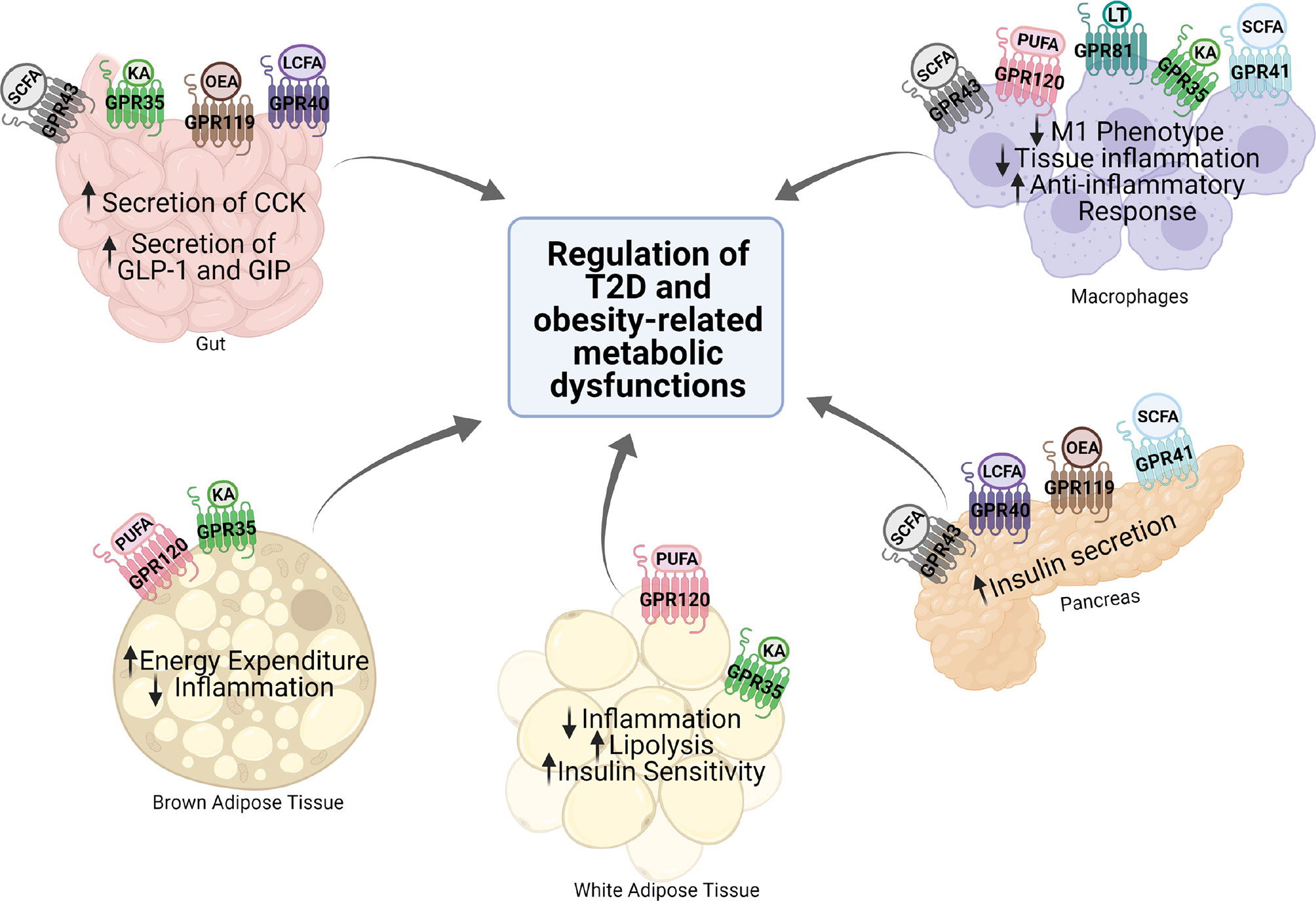
Figure 1 Expression and function of various GPCRs in metabolic tissues. Various GPCRs target insulin resistance, obesity, and T2D-related pathophysiology in the metabolic tissues. KA, kynurenic acid; LCFA, long-chain fatty acid; SCFA, short-chain fatty acid; OEA, oleylethanolamide; PUFA, polyunsaturated fatty acid; CCK, cholecystokinin; GLP-1, glucagon-like peptide 1; GIP, gastric inhibitory polypeptide.
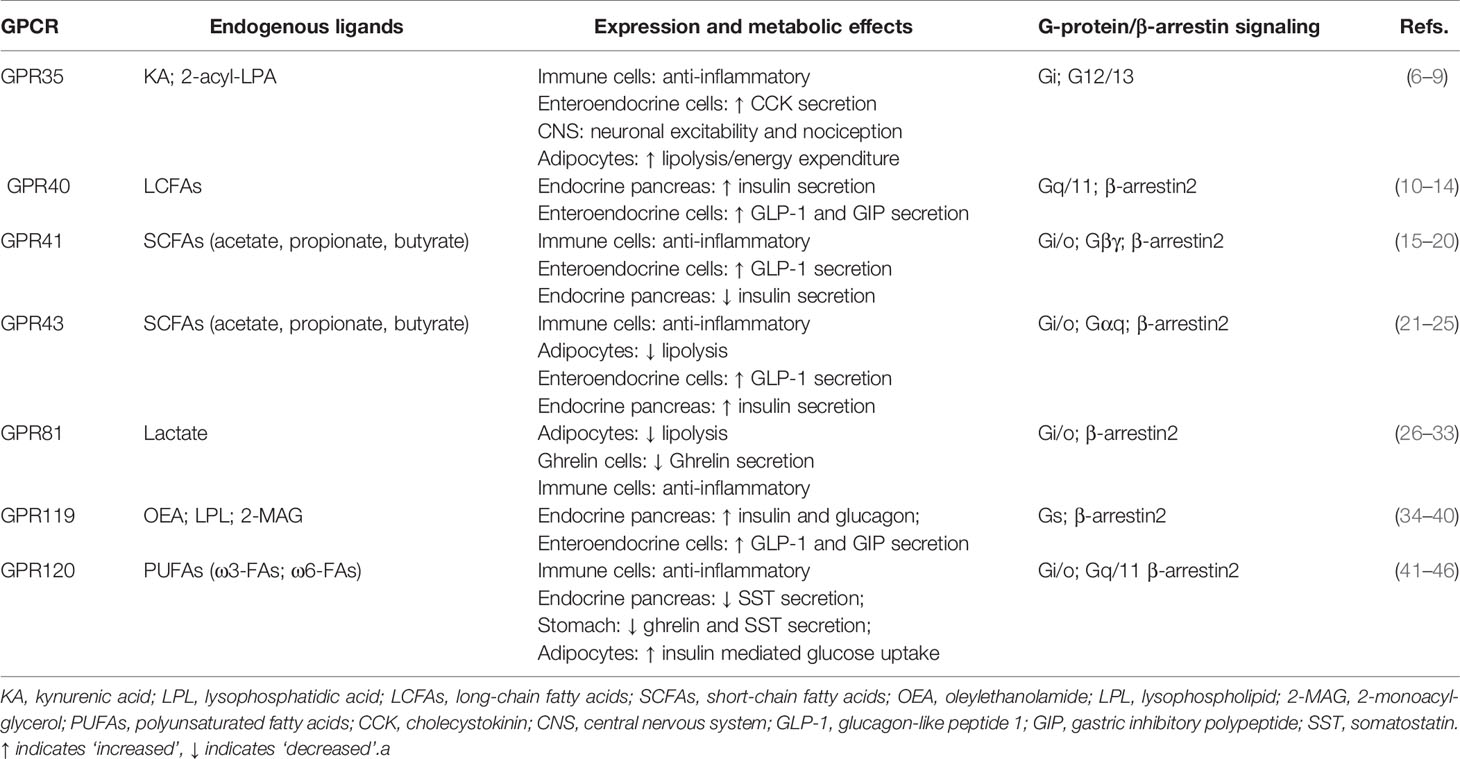
Table 1 Selection of GPCRs and their endogenous agonists act as signaling molecules.
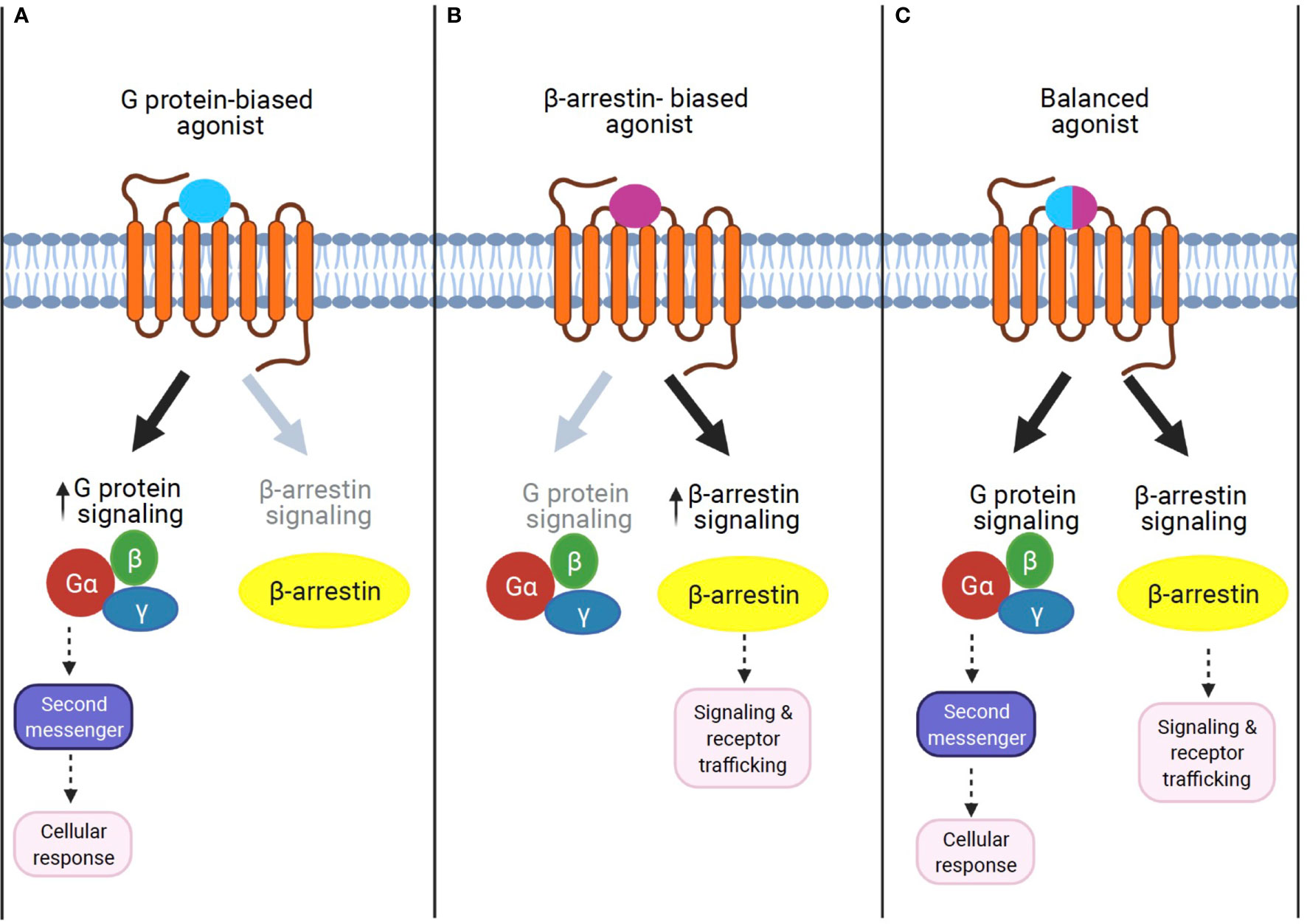
Figure 2 The concept of GPCR signaling: biased signaling. (A) G protein-biased agonist. Biased agonists selectively activate the GPCR-dependent signaling pathway. Previous studies demonstrate that sustained G protein-mediated signaling can affect cellular response through second messenger activation. (B) β-Arrestin biased agonist. Biased agonists selectively activate the β-arrestin-dependent signaling pathway. The β-arrestin-mediated signaling leads to distinct physiological outcomes. (C) Balanced agonist. Balanced agonists activate both the G protein- and β-arrestin-dependent signaling pathway.
GPCRs are the most common target of therapeutic drugs today. These seven transmembrane receptors (7TMRs) are synthesized, folded, and assembled in the endoplasmic reticulum, packed in vesicles, and transported to the plasma membrane (48). Upon binding to its cognitive ligands, GPCRs undergo a conformational change, which is transmitted to the cytoplasmic portion to couple with a heterotrimer (α, β, and γ subunits) of GTP-binding protein (G proteins) (49). GPCRs typically couple into a specific G protein such as Gs, Gi, Gq/11, or G12/13. Coupling to Gs stimulates adenylate cyclase (AC) to increase cAMP levels, while coupling to Gi inhibits adenylate cyclase. Gq/11 activation simulates phospholipase C (PLC) to hydrolyze membrane phospholipids to release inositol 1,4,5,-triphosphate (IP3) and diacylglycerols (DAGs), which then leads to increased intracellular calcium concentrations. Gq/11 can also lead to PI3K and AKT activation (49–51). Following G protein activation, a family of G protein-coupled receptor kinases (GRKs) can phosphorylate the cytoplasmic domain of the GPCR, which recruits β-arrestin adapter molecule. Once recruited to the GPCR, β-arrestin can facilitate internalization of the receptor or propagate a separate signaling cascade mediating distinct biological effects.
β-Arrestin1 (arrestin 2) and β-arrestin2 (arrestin 3) are ubiquitously expressed cytosolic adaptor proteins that were originally discovered for their inhibitory roles in GPCR signaling via the G protein-mediated signaling pathway (52). β-Arrestin1 and 2 share ~80% amino acid sequence identity and highly conserved structural features, but present unique, as well as shared roles in GPCR signaling and regulation (53). In response to receptor activation, β-arrestins can act as adaptor proteins to trigger the removal of activated GPCRs from the cell surface via clathrin-coated pits (54). This ability of β-arrestin1 and β-arrestin2 to terminate GPCR signaling through internalization is known as their classical or canonical actions (54, 55). While receptor desensitization requires β-arrestin interaction with activated GPCRs, β-arrestins can also transduce intracellular signaling as an adaptor protein (56, 57). For example, β-arrestins have been shown to form signaling scaffolds for mitogen-activated protein kinases (MAPKs) such as the extracellular signaling kinases (ERKs) and c-Jun N-terminal kinase 3 (JNK3) on endosomes with internalized GPCRs. Subsequently, β-arrestins have been known to promote some of these pathways even when the G protein activity is disabled. The finding that β-arrestins can mediate the G protein-independent signaling pathway of GPCRs led to the discovery that the two signaling pathways are pharmacologically distinct. In other words, it is possible to identify agonists that can selectively activate either G protein-dependent or β-arrestin-dependent signaling. Such agonists, which can selectively activate one or the other signaling pathway, are termed “biased agonists,” and this phenomenon of selective activation is termed “biased agonism” (58). Although “biased agonism” is often used in a sense to refer to “β-arrestin-biased agonism” in GPCR signaling (59, 60), it generally describes the disparity of the efficacies of agonists in activating signals mediated by different downstream effectors, for example, different G protein isoforms, G protein versus β-arrestin, or biases from many other signaling pathways (61). In some cases, the biased agonist could act as an antagonist or an inverse agonist for G protein-dependent signaling but as an agonist for β-arrestin-dependent signaling in a single GPCR (62, 63). Unlike G protein-mediated signaling pathways, which are transient and rapid, the β-arrestin-mediated pathway is often persistent and slow (58, 64, 65).
A number of GPCRs have been extensively studied in metabolic tissues (i.e., white and brown adipose tissue, gut, liver, and pancreatic β-cells), which are shown to modulate metabolic response such as insulin secretion, glucose homeostasis, as well as energy expenditure and more (41, 47). Certain GPCRs play important roles in improving inflammation and insulin response in adipose tissue. Given their pleiotropic effects, GPCRs in white adipose tissue (WAT) and brown adipose tissue (BAT) are potential targets for the treatment of metabolic diseases (66–68). GPCRs are also involved in the regulation of insulin secretion. Although the molecular mechanisms of islet GPCR remain to be elucidated, functional studies of the β-cells have shown that activation of GPCRs can modulate β-cell signaling through alterations in intracellular levels of cAMP, IP3, and Ca2+, as well as in protein phosphorylation and acylation (23, 48, 69, 70). Such alterations modulate insulin secretion, indicating that β-cell GPCRs are promising targets for the development of antidiabetic therapeutics. Additionally, some GPCRs are abundantly expressed in macrophages and regulate diverse macrophage functions, including cell–cell contact, survival, chemotaxis, and the activation of inflammatory mediator production (42). These macrophage-enriched GPCRs are also implicated in metabolic dysfunction related to obesity. In fact, their agonists may interact with components of multiple pathways in macrophages to modulate signaling crosstalk with metabolic tissues, coordinating a precise and appropriate cellular response in order to improve the insulin signaling and other obesity-related metabolic disorders.
The following sections will discuss the role of GPCR signaling in key metabolic tissues (Table 1).
GPR35 is a class A (rhodopsin-like) GPCR identified in 1998 (71). It is expressed in various tissues, such as central and peripheral nervous tissues, the gastrointestinal tract (GI), and lymphoid tissues (72–74). In the nervous tissues, several investigators have suggested that GPR35 activation regulates neuronal excitability, synaptic release (6), and nociception (73). In the GI tract, GPR35 has been linked to the development of gastric cancer (74), but it also actively modulates energy balance through the secretion of peptide hormones, such as cholecystokinin (CCK). GPR35 is coexpressed with the CCK1 receptor and the proton sensing receptors in GI vagal afferents neurons, suggesting that it may be part of the gut–brain signal axis that regulates energy balance (75).
The endogenous ligand for GPR35 has remained controversial. Kynurenic acid (KYNA), a tryptophan metabolite, was first proposed as a potential ligand candidate (72, 76). However, even at very high concentrations, KYNA seems to be almost inactive on human GPR35 (7). 2-Acyl lysophosphatidic acid is another endogenous ligand for GPR35. Oka et al. described that 2-acyl lysophosphatidic acid induces Ca2+ response, activates RhoA, increases the phosphorylation of ERKs, and also triggers the internalization of GPR35 in GPR35-expressing HEK293 cells (7). Additionally, the chemokine CXCL17 has also been proposed as a GPR35 endogenous agonist (8); however, subsequent studies by other teams (77, 78) have failed to support this study. Thus, despite the significant efforts to identify the real endogenous activator(s) for GPR35, GPR35 remains as a liganded orphan receptor.
Although the search for the selective and sensitive GPR35 ligands is underway, there are a number of studies to characterize the metabolic function of GPR35 in animal models. Agudelo et al. have shown that KYNA alleviates metabolic alterations triggered by high fat diet (HFD)-feeding, as it reduced weight gain, improved glucose tolerance, and remarkably reduced circulating TG levels. These effects of KYNA were associated with increased expression of adipose tissue thermogenic genes, specifically the expression of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) in adipocytes. Interestingly, the effects of KYNA are lost in GPR35 KO mice, which are more susceptible to the effects of HFD-feeding, gaining more weight, developing glucose intolerance, and showing reduced browning of the subcutaneous adipose tissue (9). These findings identify a new metabolic role of GPR35 that can potentially be exploited for the treatment of obesity-related metabolic disorders and T2D.
G protein‐coupled receptor 40 (GPR40), or free fatty acid receptor 1 (FFAR1), is found in pancreatic islets, being particularly enriched in the pancreatic β-cells (Figure 1). GPR40 can be activated by medium‐ to long‐chain free fatty acids (FFAs) (76, 79). Activation of GPR40 by FFAs or synthetic agonists enhances insulin secretion (10–12), partly through the amplification of intracellular calcium signaling in a glucose‐dependent manner (11, 12). GPR40 couples to the Gq/11, leading to the formation of IP3 and increasing intracellular calcium. Although enhancement of glucose-stimulated insulin secretion (GSIS) in β-cells requires extracellular calcium signaling (79), GPR40 stimulation increases intracellular calcium, which is dependent on glucose levels (12) and is mediated through the activation of PLC and an L-type Ca2+ channel (80).
GPR40-mediated signal transduction is known to be primarily through G protein-dependent mechanisms (10, 12). However, GPR40 can also activate the functionally distinct G protein-independent, but β-arrestins-dependent signaling pathway (13). The GPR40–β-arrestin2-mediated signaling axis is functionally linked to insulin secretion (14). Further studies with various GPR40 agonists’ activation indicate that G protein- and β-arrestin-biased signaling can be differentially modulated by different ligands, thus eliciting ligand-specific responses (biased agonism; Figure 2). While GPR40 agonists, palmitic acid, and oleic acid act through Gq/11-mediated mechanisms, the synthetic agonist TAK-875 can act as a β-arrestin2-biased agonist, engaging β-arrestin2-dependent signaling to induce the insulinotropic activity of GPR40 (13). The biased GPR40 activation has shown a promising potential as a therapeutic target to enhance insulin secretion in T2D (79), but phase III clinical trials with TAK-875 were recently terminated due to signs of liver toxicity in patients (81). Therefore, therapies based on GPR40 agonism provide an attractive alternative in the discovery of antidiabetic drugs, but further studies are needed to determine if potential side effects induced by this approach can be avoided.
GPR41, also known as free fatty acid receptor 3 (FFA3), is expressed in adipose tissue, pancreas, spleen, lymph nodes, bone marrow, and peripheral blood mononuclear cells including monocytes (15–17). GPR41 and GPR43 (described in the next section) are activated by short-chain fatty acids (SCFAs) such as acetate, propionate, and butyrate, which are produced during dietary fiber fermentation by gut resident bacteria (76). Despite similarities in the receptor sequence, KO mice studies revealed contradictory results about the effects of GPR41 and/or GPR43 loss-of-function on metabolism (18, 19, 21–23, 69, 82). Those studies indicated opposite effects of the two SCFA sensing GPCRs on insulin secretion. GPR41 was found to inhibit glucose-dependent insulin secretion (69), while GPR43 was reported to potentiate insulin secretion (19, 23). Therefore, the effects of SCFAs on insulin secretion seem to be fine-tuned by the balance between GPR41 and GPR43 expression and activation. However, high selective agonists and antagonists for GPR41 and 43, as well as tissue-specific GPR41 and/or GPR43 KO mice, are required to fully elucidate the involvement of these receptors in SCFA-mediated effects. Since loss of GPR41 caused decreased GPR43 expression (83) and dual GPR41 and GPR43 KO mice exhibited higher insulin secretion and improved glucose tolerance, determination of specific function of each receptor in the regulation of insulin secretion is complex (84).
Both SCFA receptors, GPR41 and GPR43, seem to have a preference for Gi/o signaling, resulting in the inhibition of AC and the reduction in cAMP production (16, 19). In pancreatic islets, the activation of GPR41 by its endogenous ligand, propionate, inhibited the glucose-dependent insulin secretion through the Gαi/o pathway (19). However, other G protein-dependent pathways may also be triggered by GPR41 activation, as it was observed that SCFAs and ketone bodies induced GPR41-mediated activation of sympathetic neurons through Gβγ-PLCβ-MAPK signaling, stimulating body energy expenditure and helping to maintain metabolic homeostasis (18). The role of β-arrestin signaling by GPR41 and GPR43 activation is not clear; however, in monocytes, it was described that GPR41 and GPR43 form a heteromer, which, in addition to enhancing Ca2+ signaling, also induced β-arrestin-2 recruitment (20).
Although promising, together these findings show how essential future studies are in order to more clearly define the role and mechanisms of GPR41 in insulin secretion, as well as the potential druggability of its agonists/antagonists to improve T2D and obesity-related metabolic abnormalities.
G protein‐coupled receptor 43 (GPR43), also known as free fatty acid receptor 2 (FFA2), has been reported to be present in cells of the distal ileum, colon, and adipose tissue, with the highest expression found in immune cells such as monocytes and neutrophils (15, 16, 85). GPR43 appears to play a role during inflammation, as immune challenges such as LPS, TNFα, or granulocyte-macrophage colony stimulating factor (GM-CSF) were found to raise GPR43 transcript levels in human monocytes (86, 87). Although GPR43 was not identified in the human adipose tissue (17), GPR43 inhibits lipolysis in mouse adipocytes (21). Adipocytes treated with GPR43 natural ligands, acetate and propionate, exhibit a reduction in lipolytic activity. Since this reduction in the lipolytic activity was abolished in adipocytes isolated from GPR43 KO mice, it seems to be the result of GPR43 activation (21). In addition to higher lipolysis and higher energy expenditure, deletion of GPR43 was also reported to improve the glucose homeostasis in obesity, as HFD-fed GPR43 KO mice exhibited lower body fat mass and increased insulin sensitivity (22). Despite the beneficial effects of the deletion of GPR43 in adipocytes, HFD-fed GPR43 KO mice exhibited dysfunctional β-cells, which showed reduced cell mass and lower expression of β-cell differentiation genes. Those abnormalities blunted the insulin secretion in GPR43 KO mice (23, 24). On the other hand, the treatment with acetate, endogenous GPR43 agonist, improved insulin secretion in mouse but not in human islets (23, 24).
It has been shown that acetate and synthetic GPR43 agonists can differently modulate GPR43 activation via coupling to multiple G protein pathways in mouse and human islets (69). PA ((S)-2-(4-chlorophenyl)-3,3-dimethyl-N-(5-phenylthiazol-2-yl)butanamide), a synthetic GPR43 agonist, potentiated insulin secretion in isolated murine islets, human islets, and Min6 cells in vitro by increasing intracellular IP3 and Ca2+ levels in a GPR43-, Gq-, and PLC-dependent manner (23). However, another GPR43 synthetic agonist, 4-CMTB (4-Chloro-α-(1-methylethyl)-N-2-thiazolyl-benzeneacetamide), invariably inhibited GSIS in human pseudoislets, contrary to mouse islets, where it augmented GSIS (88). This finding that mouse and human islets responded differently to acetate and GPR43 agonists in GSIS assay will require close attention in future studies, since GPR43 is considered as a potential T2D target.
It has been proposed that β-arrestin2-mediated signaling can also be activated by GPR43 agonists, causing further inhibition of NF-κB and downregulation of its inflammatory gene targets (25). However, this is the only study where GPR43 and β-arrestin2 internalization was triggered by PA (25). Therefore, the idea of GPR43 biased agonism might be plausible, but more studies are necessary to verify its potential druggability for T2D and obesity-related metabolic disorders.
GPR81 is a member of the hydroxyl-carboxylic acid receptor family. GPR81 is highly expressed in adipose tissue but also found in kidney, skeletal muscle, and liver (26, 89). Lactate is an endogenous ligand for GPR81. In adipocytes, the activation of GPR81 inhibits lipolysis by decreased cAMP and phosphorylation of PKA, which consequently reduces the activity of the hormone-sensitive lipase (27–29). GPR81 stimulated by lactate decreases intracellular cAMP and lipolysis, which was also found to work synergistically with insulin (90). In fact, Ahmed et al. observed that lactate and GPR81 unexpectedly functioned in an autocrine and paracrine loop to mediate insulin-induced antilipolytic effects (90). Additionally, GPR81 might also be linked to obesity, since HFD-fed GPR81 KO mice exhibited lower weight gain (90).
Obesity is also associated with increased inflammation, and several studies observed that GPR81 plays a role in inflammation. GPR81 expression in adipocytes and endothelial cells is reduced under inflammatory conditions (30, 31). Similarly, GPR81 expression is significantly decreased in the white adipose tissue of HFD-fed mice (91), as well as in adipocytes of ob/ob mice, an animal model of T2D characterized by high inflammation (30). Despite the clear link between inflammation and GPR81 function in the adipose tissue, the effects of GPR81 in macrophages and other immune cells are not fully understood. There are several reports that the expression of GPR81 in immune cells promote downregulation of the innate immune response (26, 32, 33). Hoque et al. observed that the immunosuppressive function of GPR81 was attributed to the downregulation of TLR- and/or NLRP3-mediated signaling (33). This GPR81-stimulated reduction of the inflammatory responses in macrophages/monocytes seems to be due to downstream signaling of GPR81 and β-arrestin2 and not due to a reduction in cAMP (G protein-dependent) (33). The exact mechanism for how GPR81 and β-arrestin2 counteract the NLRP3 and TLR pathways is currently unknown and will require further studies. Given the strong association between inflammation and obesity-related metabolic dysfunction, GPR81 biased agonism might be a valuable drug therapy for T2D and needs to be further investigated.
GPR119 is a Gs-coupled receptor that is expressed in pancreatic β-cells and gastrointestinal enteroendrocrine cells (34). GPR119 directly leads to an increase in insulin secretion in β-cells and promotes the release of both glucagon-like peptide-1 (GLP1) and gastric inhibitory polypeptide (GIP) in enteroendocrine cells (34–36). Thus, GPR119 stimulation can augment insulin secretion both by direct effects on β-cells and indirectly through GLP1 (34–36). GPR119 couples to Gs in response to several lipid-based agonists, with highly constitutive activity. GPR119 was originally described as a receptor for N-acylethanolamines, such as oleylethanolamide (OEA), which are generated locally in enterocytes (92). Lysophospholipids (LPL), another known class of endogenous GPR119 ligands, are directly absorbed from the diet or generated by catalysis of endogenous phospholipids (93). 2-Monoacylglycerols (2-MAGs), generated in high amounts during intestinal digestion of triglycerides, also bind and activate GPR119 (36). These findings illustrate that the most effective endogenous, fat-derived ligand for GPR119 is still ill defined, with OEA, LPL, or 2-monoacylglycerols being potential candidates (36, 76). Despite the debate about potential agonists, GPR119 signaling through Gs is well established, inducing cAMP accumulation and/or downstream activation of CREB (cAMP response element binding protein) in response to both natural ligands and synthetic GPR119 agonists (34, 37–39). GPR119 activation has also been proposed to induce ligand-mediated calcium release (94).
G protein-independent signaling via β-arrestin recruitment has only been sparsely described in the GPR119-mediated signaling pathway (40, 95). Hassing et al. described that GPR119 activation by OEA can trigger a β-arrestin biased signaling (40). Given the effects of GPR119 in β-cells (34–36), more studies are necessary to understand the effects of GPR119 biased agonism and investigate whether it might be plausible to stimulate the insulin release and treat T2D.
GPR120 (a.k.a. free fatty acid receptor 4 (FFA4)) is the most abundant one among free fatty acid receptors in the mouse adipose tissue (17); however, GPR120 is also found in the pancreas, where expression is suggested to be restricted to the δ-cells (96), in lung (97), and in immune cells, specifically macrophages (41) (Figure 1). GPR120 is highly expressed in different types of macrophages (monocytes, Kupffer cells in the liver, osteoclasts in the bone, resident macrophages in the lung) and plays an important role in the regulation of inflammation (42). GPR120 is described as a receptor for omega-3 polyunsaturated fatty acids (ω3-FAs) (76), the activation of which reduces adipose tissue inflammation and protects against global insulin resistance (41, 43). The pathways coupled to GPR120 stimulation diverge between G protein- and β-arrestin-dependent pathways (98) and are critical for the regulation of metabolic or inflammatory processes (41). We and others have shown that GPR120 effectively responds to ω3-FAs, and the activation of GPR120 stimulates the PI3K/Akt pathway triggering GLUT4 translocation to the cell membrane and increasing glucose uptake in adipocytes by a Gq/11-dependent mechanism, not by the β-arrestin-dependent pathway (41). In addition, Paschoal et al. showed that GPR120 agonist stimulation in adipocytes displayed biphasic ERK phosphorylation with G protein-mediated acute phase of ERK activation followed by a steady-state, β-arrestin-mediated ERK signaling pathway (44).
Although GPR120 stimulation leads to both Gq/11 and β-arrestin-mediated pathway activation, it has been shown that the G protein-independent, β-arrestin2-dependent signaling pathway is responsible for the GPR120-mediated anti-inflammatory effects in macrophages (43). The activation of GPR120 signaling pathways regulate the macrophage phenotypic switch, influencing their response to inflammation and ability to migrate to other tissues (43).
Mechanistically, it has been shown that GPR120 activation and concomitant recruitment of β-arrestin-2 promote further interactions between β-arrestin-2 and TAB-1. This receptor mediated β-arrestin-2/TAB-1 interaction is suggested to prevent the formation of a TAB-1/TAK-1 (transforming growth factor kinase) complex, blocking the subsequent signaling that results in the activation of inflammatory responses (41, 43). β-Arrestin-2 pull-down experiments demonstrated the physical interaction between β-arrestin-2, GPR120, and TAB-1 in macrophages after stimulation with DHA or DHA plus LPS, respectively (41). Similar results have been reported using a synthetic agonist of GPR120 to replace the fatty acid: compound A (cpdA) (41). This molecule produced anti-inflammatory effects in macrophages in both in vitro and in vivo models (43). Treatment of primary macrophages with cpdA in conjunction with LPS inhibited the phosphorylation of many of the previously discussed phospho-regulated kinases that are typically activated in inflammatory processes (e.g., p-IKK, p-JNK, pTAK-1) (43).
The functions of GPR120 in the adipose tissue can be linked to pathological ramifications of obesity (41, 43, 99, 100); thus, GPR120 as a target for the development of novel compounds to treat metabolic syndrome becomes a very promising approach. However, efforts to identify or generate GPR120 biased agonists are still ongoing (101).
The glucagon-like peptide-1 receptor (GLP-1R) is a GPCR predominantly expressed in the β-cells, intestine, heart, breast, and brain (102). GLP-1R mediates a number of physiological effects, and due to its functions, the GLP-1R is a major therapeutic target for treatment of type 2 diabetes and obesity. It increases insulin secretion by direct stimulation of gene expression, synthesis, and secretion of insulin from pancreatic β-cells; it enhances β-cell mass by increasing neogenesis and proliferation, while decreasing apoptosis; it suppresses glucagon secretion and inhibits gastric emptying; and extra-pancreatically GLP-1 also acts at a range of sites such as the nervous system, where it signals satiety, reducing food intake (103–105). A large number of GLP1-based therapeutics are already well established, and the scientific basis underlying these therapeutic approaches is quite advanced; therefore, this subject will only be briefly reviewed.
The GLP-1R physiological effects rely on downstream signaling pathways mediated by GLP-1 interaction. This receptor activates Gαs proteins enhancing the formation of cAMP; however, it can pleiotropically interact with multiple other G proteins including Gi/o and Gq proteins, leading to the activation of downstream signaling pathways that include the mobilization of intracellular calcium and the phosphorylation of mitogen activated protein kinases such as extracellular regulated kinases 1/2 (ERK1/2), protein kinase B (Akt/PKB), phosphoinositide 3 (PI3) kinase, and p38, among others (102, 106). In addition to signaling via G proteins, the GLP-1R can also promote β-arrestin-1 biased, non-G protein-mediated cellular signaling (47, 102).
Several groups have reported GLP-1R biased agonism; for instance, the drugs Oxyntomodulin and exendin are GLP-1R biased agonists that recruit β-arrestin in a higher potency than G protein-mediated cAMP and ERK1/2 activation (102, 107, 108). In addition to direct evidence of biased agonism by different peptide ligands, there is also some evidence that the kinetics of GLP-1R internalization and recycling mediated by distinct peptides may contribute to biased agonism profiles. The potencies of GLP-1 and exendin-induced internalization are 10-fold higher than that of liraglutide (107). With emerging evidence that internalized GLP-1Rs can continue to signal inside the cell and that spatial-temporal control of signaling pathways promotes distinct physiological functions, this ability of different ligands to promote different kinetics of internalization and recycling of the GLP-1R may contribute to the observed ligand-biased agonism (107). Taken together, all these studies indicate that biased signaling occurs at this receptor, and this may have the potential to be exploited in drug development.
Together, these findings highlight the current lack in understanding the role of the biased G protein- and/or β-arrestin-mediated mechanism in GPCR signaling, even for well-known GPCRs. Therefore, future studies that identify and synthesize biased agonists for each GPCR, as well as explore the biased signaling mechanism in target tissues, are imperative (Figure 2). In the next section, we will discuss promising approaches that β-arrestin biased activation may uniquely represent to treat T2D and obesity-related comorbidities.
Among four members of the arrestin family, β-arrestin1 (arrestin 2) and β-arrestin2 (arrestin-3) are widely expressed in different tissues and implicated in many GPCR signaling pathways to regulate cellular responses. β-Arrestin signaling serves multiple purposes; it can modulate the activity of several cellular signaling proteins such as PI3K and AKT (109), c-Src, MAPKs, cAMP phosphodiesterase, calmodulin, protein phosphatases, ubiquitin ligases, deubiquitinating enzymes, and many others (54, 110). It is still unclear whether these noncanonical β-arrestin functions require prior recruitment by GPCRs, as the main function of these proteins is to terminate GPCR cellular pathways. Since β-arrestins can act independently as signaling molecules, therefore, it is important to understand their independent role in metabolic homeostasis (111). An increasing number of studies have focused on the role of β-arrestins in metabolic tissues, such as white and brown adipose tissue, liver, and the pancreatic β cell. Table 2 and the following sections will provide new insights for the role of β-arrestins in metabolic tissues, regardless of G protein-dependent or independent signaling.
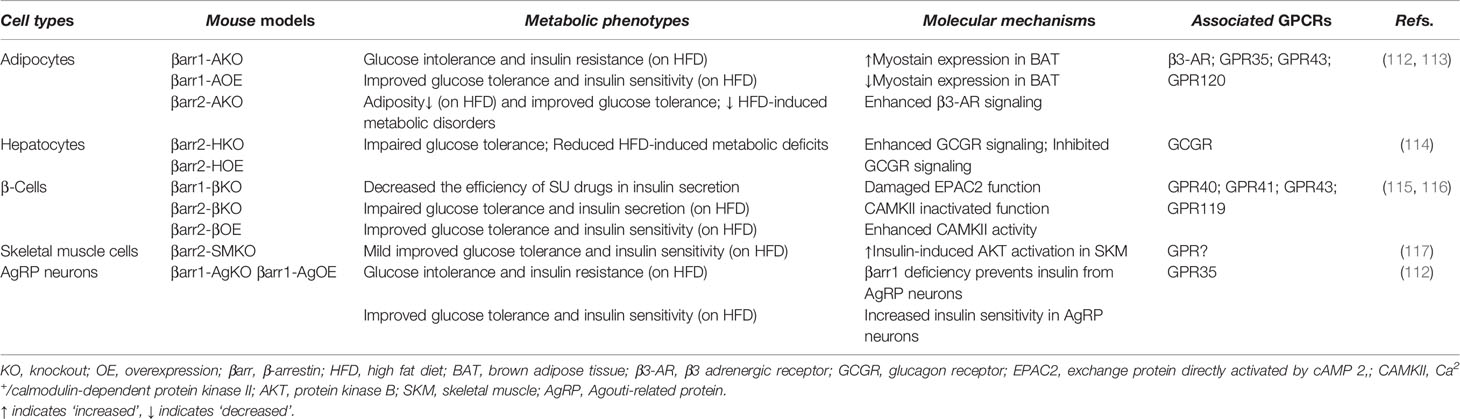
Table 2 Metabolic functions of β-arrestins in distinct cell types and associated GPCRs.
It is known that white adipocytes play essential roles in storing extra lipids as energy and releasing fatty acids as resources. In contrast, mutilocular brown adipocytes oxidize fatty acids and other substrates to produce heat for maintaining body temperature in mammals. During the past few years, many laboratories have used mouse genetics to identify the role of β-arrestin1 and 2 in adipocyte function and whole-body glucose homeostasis (118, 119). Pydi et al. (120) reported that selectively lacked β-arrestin1 in adipocyte (β-arr1 AKO) mice on HFD are glucose intolerance and insulin resistance (Table 2). On the other hand, mice overexpressing β-arrestin1 in adipocytes (β-arr1 AOE) were protected against HFD-induced metabolic deficits (120). In contrast to β-arrestin1, mice lacking β-arrestin2 in adipocytes (βarr2-AKO) display improved metabolic phenotypes (118). These findings convincingly identify that β-arrestin1 and 2 not only are required for the maintenance of glucose homeostasis in their own right but also strongly suggest that strategies aiming to enhance β-arrestin activity in adipocytes may be beneficial for the treatment of T2D and obesity-related metabolic disorders.
Insulin resistance is a key etiological factor in the development of T2D. It can be triggered by desensitization of insulin signaling at several steps (2). Studies have demonstrated that β-arrestin1 can function as a nodal point for heterologous desensitization and crosstalk between receptor tyrosine kinases (RTKs) and GPCR signaling pathways (121, 122). β-Arrestin1 has been showed to modulate ubiquitination and degradation of one major substrate in the insulin cascade, the RTK, and insulin receptor substrate (IRS) (122). β-Arrestin1 competes with IRS proteins for ubiquitination and degradation, such that β-arrestin1 deficiency accelerates insulin-induced IRS degradation, exacerbating cellular insulin resistance, whereas overexpression of β-arrestin1 restrains this process, leading to increased insulin signaling downstream of IRS-1 and improving cellular insulin sensitivity (122).
Pancreatic β-cell specific β-arrestin2 KO mice on HFD (βarr2-βKO mice) showed impaired insulin release and glucose tolerance, whereas the β-cell specific overexpression of β-arrestin2 mice on HFD (βarr2-βOE mice) exhibited improved GSIS and glucose tolerance compared to HFD-fed control mice (116). These data indicate that β-arrestin2 act as an important regulator of β-cell function.
Impaired β-cell function is a major etiological defect underlying T2D. The failure to maintain compensatory hyperinsulinemia and the following decrease in plasma insulin levels is a key cause of hyperglycemia (2, 4, 5). Therapeutic measures to increase endogenous insulin secretion, and, indeed, administration of exogenous insulin itself, have been the cornerstones of T2D treatment for decades. In recent years, several developments have emerged that focus attention on the role of β-arrestins as therapeutic targets to enhance β-cell function and lower glucose levels.
Different areas of brain play key roles in regulating and maintaining euglycemia. Neuronal subpopulations of the arcuate nucleus (ARC) of the hypothalamus, which synthesize and release agouti-related peptide (AgRP), have been studied extensively in relation to metabolic function (123). Numerous studies have shown that AgRP neurons play a key role in regulating food intake and energy homeostasis (124–127). Recent studies have shown that mice lacking β-arrestin1 in AgRP neurons on HFD displayed impaired glucose tolerance and insulin sensitivity accompanied with liver steatosis and increase in the plasma FFA level (112). Interestingly, they found that mice with specific deletion of β-arrestin1 in AgRP neurons (βarr1-AgKO) have increased PKA activity in adipose tissue, resulting in the accumulated lipolysis (112). In contrast, a mouse model where β-arrestin1 was overexpressed in these neurons (βarr1-AgOE) has significantly improved glucose and insulin tolerance (112). Collectively, these metabolic phenotypes may provide a novel way to improve glucose tolerance and insulin sensitivity in the AgRP neurons through the increased activation of β-arrestin1. Further studies are necessary to delineate the importance of GPCR signaling and the association between β-arrestin1 and related GPCRs in AgRP neurons.
Hepatocytes play a significant role in T2D by controlling lipid metabolism and whole body glucose homeostasis (57). Insulin and glucagon are the two major hormones that regulate the metabolic function of hepatocytes (128). Many studies focus on the Gs-coupled glucagon receptor (GCGR) in hepatocytes. The GCGR can trigger the cAMP/PKA-dependent signaling pathway to promote gluconeogenesis and glycogen breakdown, thereby increasing hepatic glucose production (129, 130).
Zhu et al. generated liver (hepatocyte) specific β-arrestin1 and β-arrestin2 KO mice (βarr1-HKO and βarr2-HKO) (131). While βarr1-HKO mice did not show any significant difference in metabolic phenotype compared to the control mice, βarr2-HKO mice displayed impaired glucose tolerance and hyperglycemia (131). When βarr2-HKO mice were treated with anti-GCGR antibody, the blood glucose level of these mice was back to the normal level. However, in mice where hepatocytes overexpressed β-arrestin2 (βarr2-HOE), the opposite metabolic phenotype was observed (131). Consistent with in vivo studies, they also found that glucagon treatment in primary hepatocytes isolated from control mice caused the internalization of the GCGR, whereas this effect was absent in hepatocytes isolated from βarr2-HKO mice (131). Collectively, both in vivo and in vitro data demonstrate that β-arrestin2 plays a negative role in GCGR signaling.
Skeletal muscle is a major insulin target tissue for regulating whole body glucose homeostasis. Skeletal muscle specific β-arrestin2 KO mice (βarr2-SMKO) on HFD displayed slight improvements of glucose tolerance and insulin sensitivity (117). More detailed studies need to be done to illustrate how β-arrestin2 deficiency in skeletal muscle influences insulin signaling. In summary, skeletal muscle expressed GPCRs represent promising therapeutic targets for modulating insulin sensitivity and treating T2D and obesity-related metabolic disorders.
GPCRs are uniquely druggable targets that form the basis of the leading antidiabetic treatments, as evidenced by the large number of GLP1R/GLP1 therapies currently in use. Additionally, GPCRs that can influence insulin resistance, β-cell dysfunction, or both have recently been identified, and preclinical studies hold great promise. In addition, β-arrestins are crucial regulators of GPCR signaling. Although aspects of the GPCR/β-arrestin signaling network had been previously well established, the novelty of the recent studies highlighted in this review is the ability of β-arrestins to orchestrate a complex signaling network with or without GPCR activation that specifically controls metabolic homeostasis. Studies with β-arrestin KO mice have provided several key insights into the physiological implications of β-arrestin-dependent signaling. The multitudes of important metabolic processes that are regulated by β-arrestins offer new perspectives for the development of novel classes of therapeutic agents for the treatment of T2D and obesity-related pathophysiological conditions. Biased agonists for several GPCRs present a unique opportunity to explore the possibilities of developing a novel class of drugs. Such drugs may include G protein- or β-arrestin biased agonists that can interrupt or enhance metabolically relevant interactions of β-arrestin1 and β-arrestin2 with key signaling molecules. In this regard, additional preclinical studies are warranted to further analyze the effect of potential G protein- or β-arrestin biased signaling machinery to instruct novel therapeutic regimes for the treatment of metabolic disease.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by grant from the NIH (R01-DK108773 to DO) and the American Heart Association (14SDG19880020 to DO).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We gratefully acknowledge all members of the Oh laboratory for their constant support and enthusiasm, and Drs. Nai-Wen Chi and Olivia Osborn (Univ. of California San Diego) for critical reading and editorial assistance. The figures were generated in part by using BioRender. We apologize to all authors whose work we could not discuss because of the short length of this review.
1. Control CfD, Prevention. National Diabetes Statistics Report, 2020. Atlanta, GA: Centers for Disease Control and Prevention, US Department of Health and Human Services (2020) p. 12–5.
2. Kahn SE, Hull RL, Utzschneider KM. Mechanisms Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nature (2006) 444(7121):840–6. doi: 10.1038/nature05482
3. Barnes AS. The Epidemic of Obesity and Diabetes: Trends and Treatments. Texas Heart Institute J (2011) 38(2):142–4.
4. Cerf ME. Beta Cell Dysfunction and Insulin Resistance. Front Endocrinol (2013) 4:37. doi: 10.3389/fendo.2013.00037
5. Prentki M, Nolan CJ. Islet Beta Cell Failure in Type 2 Diabetes. J Clin Invest (2006) 116(7):1802–12. doi: 10.1172/JCI29103
6. Guo J, Williams DJ, Puhl HL, Ikeda SR. Inhibition of N-Type Calcium Channels by Activation of GPR35, an Orphan Receptor, Heterologously Expressed in Rat Sympathetic Neurons. J Pharmacol Exp Ther (2008) 324(1):342–51. doi: 10.1124/jpet.107.127266
7. Oka S, Ota R, Shima M, Yamashita A, Sugiura T. GPR35 Is a Novel Lysophosphatidic Acid Receptor. Biochem Biophys Res Commun (2010) 395(2):232–7. doi: 10.1016/j.bbrc.2010.03.169
8. Maravillas-Montero JL, Burkhardt AM, Hevezi PA, Carnevale CD, Smit MJ, Zlotnik A. Cutting Edge: GPR35/CXCR8 I the Receptor of the Mucosal Chemokine CXCL17. J Immunol (Baltimore Md: 1950) (2015) 194(1):29–33. doi: 10.4049/jimmunol.1401704
9. Agudelo LZ, Ferreira DMS, Cervenka I, Bryzgalova G, Dadvar S, Jannig PR, et al. Kynurenic Acid and Gpr35 Regulate Adipose Tissue Energy Homeostasis and Inflammation. Cell Metab (2018) 27(2):378–92.e5. doi: 10.1016/j.cmet.2018.01.004
10. Tan CP, Feng Y, Zhou Y-P, Eiermann GJ, Petrov A, Zhou C, et al. Selective Small-Molecule Agonists of G Protein–Coupled Receptor 40 Promote Glucose-Dependent Insulin Secretion and Reduce Blood Glucose in Mice. Diabetes (2008) 57(8):2211–9. doi: 10.2337/db08-0130
11. Lin DC-H, Zhang J, Zhuang R, Li F, Nguyen K, Chen M, et al. AMG 837: A Novel GPR40/FFA1 Agonist That Enhances Insulin Secretion and Lowers Glucose Levels in Rodents. PloS One (2011) 6(11):e27270. doi: 10.1371/journal.pone.0027270
12. Fujiwara K, Maekawa F, Yada T. Oleic Acid Interacts With GPR40 to Induce Ca2+ Signaling in Rat Islet β-Cells: Mediation by PLC and L-Type Ca2+ Channel and Link to Insulin Release. Am J Physiol Endocrinol Metab (2005) 289(4):E670–7. doi: 10.1152/ajpendo.00035.2005
13. Mancini AD, Bertrand G, Vivot K, Carpentier É, Tremblay C, Ghislain J, et al. β-Arrestin Recruitment and Biased Agonism at Free Fatty Acid Receptor 1. J Biol Chem (2015) 290(34):21131–40. doi: 10.1074/jbc.M115.644450
14. Qian J, Wu C, Chen X, Li X, Ying G, Jin L, et al. Differential Requirements of Arrestin-3 and Clathrin for Ligand-Dependent and -Independent Internalization of Human G Protein-Coupled Receptor 40. Cell Signal (2014) 26(11):2412–23. doi: 10.1016/j.cellsig.2014.07.019
15. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The Orphan G Protein-Coupled Receptors GPR41 and GPR43 Are Activated by Propionate and Other Short Chain Carboxylic Acids. J Biol Chem (2003) 278(13):11312–9. doi: 10.1074/jbc.M211609200
16. Le Poul E, Loison C, Struyf S, Springael J-Y, Lannoy V, Decobecq M-E, et al. Functional Characterization of Human Receptors for Short Chain Fatty Acids and Their Role in Polymorphonuclear Cell Activation. J Biol Chem (2003) 278(28):25481–9. doi: 10.1074/jbc.M301403200
17. Amisten S, Neville M, Hawkes R, Persaud SJ, Karpe F, Salehi A. An Atlas of G-Protein Coupled Receptor Expression and Function in Human Subcutaneous Adipose Tissue. Pharmacol Ther (2015) 146:61–93. doi: 10.1016/j.pharmthera.2014.09.007
18. Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, et al. Short-Chain Fatty Acids and Ketones Directly Regulate Sympathetic Nervous System via G Protein-Coupled Receptor 41 (GPR41). Proc Natl Acad Sci (2011) 108(19):8030–5. doi: 10.1073/pnas.1016088108
19. Priyadarshini M, Layden BT. FFAR3 Modulates Insulin Secretion and Global Gene Expression in Mouse Islets. Islets (2015) 7(2):e1045182. doi: 10.1080/19382014.2015.1045182
20. Ang Z, Xiong D, Wu M, Ding JL. FFAR2-FFAR3 Receptor Heteromerization Modulates Short-Chain Fatty Acid Sensing. FASEB J: Off Publ Fed Am Societies Exp Biol (2018) 32(1):289–303. doi: 10.1096/fj.201700252RR
21. Ge H, Li X, Weiszmann J, Wang P, Baribault H, Chen J-L, et al. Activation of G Protein-Coupled Receptor 43 in Adipocytes Leads to Inhibition of Lipolysis and Suppression of Plasma Free Fatty Acids. Endocrinology (2008) 149(9):4519–26. doi: 10.1210/en.2008-0059
22. Bjursell M, Admyre T, Göransson M, Marley AE, Smith DM, Oscarsson J, et al. Improved Glucose Control and Reduced Body Fat Mass in Free Fatty Acid Receptor 2-Deficient Mice Fed a High-Fat Diet. Am J Physiol Endocrinol Metab (2011) 300(1):E211–20. doi: 10.1152/ajpendo.00229.2010
23. McNelis JC, Lee YS, Mayoral R, van der Kant R, Johnson AM, Wollam J, et al. GPR43 Potentiates β-Cell Function in Obesity. Diabetes (2015) 64(9):3203–17. doi: 10.2337/db14-1938
24. Villa SR, Priyadarshini M, Fuller MH, Bhardwaj T, Brodsky MR, Angueira AR, et al. Loss of Free Fatty Acid Receptor 2 Leads to Impaired Islet Mass and Beta Cell Survival. Sci Rep (2016) 6:28159. doi: 10.1038/srep28159
25. Lee SU, In HJ, Kwon MS, Park BO, Jo M, Kim MO, et al. β-Arrestin 2 Mediates G Protein-Coupled Receptor 43 Signals to Nuclear Factor-κb. Biol Pharm Bull (2013) 36(11):1754–9. doi: 10.1248/bpb.b13-00312
26. Brown TP, Ganapathy V. Lactate/GPR81 Signaling and Proton Motive Force in Cancer: Role in Angiogenesis, Immune Escape, Nutrition, and Warburg Phenomenon. Pharmacol Ther (2020) 206:107451. doi: 10.1016/j.pharmthera.2019.107451
27. Cai T-Q, Ren N, Jin L, Cheng K, Kash S, Chen R, et al. Role of GPR81 in Lactate-Mediated Reduction of Adipose Lipolysis. Biochem Biophys Res Commun (2008) 377(3):987–91. doi: 10.1016/j.bbrc.2008.10.088
28. Ge H, Weiszmann J, Reagan JD, Gupte J, Baribault H, Gyuris T, et al. Elucidation of Signaling and Functional Activities of an Orphan GPCR, Gpr81. J Lipid Res (2008) 49(4):797–803. doi: 10.1194/jlr.M700513-JLR200
29. Liu C, Wu J, Zhu J, Kuei C, Yu J, Shelton J, et al. Lactate Inhibits Lipolysis in Fat Cells Through Activation of an Orphan G-Protein-Coupled Receptor, GPR81. J Biol Chem (2009) 284(5):2811–22. doi: 10.1074/jbc.M806409200
30. Feingold KR, Moser A, Shigenaga JK, Grunfeld C. Inflammation Inhibits GPR81 Expression in Adipose Tissue. Inflammation Research: Off J Eur Histamine Res Soc [et al] (2011) 60(10):991–5. doi: 10.1007/s00011-011-0361-2
31. Boitsova EB, Morgun AV, Osipova ED, Pozhilenkova EA, Martinova GP, Frolova OV, et al. The Inhibitory Effect of LPS on the Expression of GPR81 Lactate Receptor in Blood-Brain Barrier Model In Vitro. J Neuroinflamm (2018) 15(1):196. doi: 10.1186/s12974-018-1233-2
32. Ranganathan P, Shanmugam A, Swafford D, Suryawanshi A, Bhattacharjee P, Hussein MS, et al. GPR81, a Cell-Surface Receptor for Lactate, Regulates Intestinal Homeostasis and Protects Mice From Experimental Colitis. J Immunol (Baltimore Md: 1950) (2018) 200(5):1781–9. doi: 10.4049/jimmunol.1700604
33. Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. Lactate Reduces Liver and Pancreatic Injury in Toll-Like Receptor- and Inflammasome-Mediated Inflammation via GPR81-Mediated Suppression of Innate Immunity. Gastroenterology (2014) 146(7):1763–74. doi: 10.1053/j.gastro.2014.03.014
34. Chu ZL, Carroll C, Alfonso J, Gutierrez V, He H, Lucman A, et al. A Role for Intestinal Endocrine Cell-Expressed G Protein-Coupled Receptor 119 in Glycemic Control by Enhancing Glucagon-Like Peptide-1 and Glucose-Dependent Insulinotropic Peptide Release. Endocrinology (2008) 149(5):2038–47. doi: 10.1210/en.2007-0966
35. Lauffer L, Iakoubov R, Brubaker PL. GPR119: “Double-Dipping” for Better Glycemic Control. Endocrinology (2008) 149(5):2035–7. doi: 10.1210/en.2008-0182
36. Hansen HS, Rosenkilde MM, Holst JJ, Schwartz TW. GPR119 as a Fat Sensor. Trends Pharmacol Sci (2012) 33(7):374–81. doi: 10.1016/j.tips.2012.03.014
37. Hansen KB, Rosenkilde MM, Knop FK, Wellner N, Diep TA, Rehfeld JF, et al. 2-Oleoyl Glycerol Is a GPR119 Agonist and Signals GLP-1 Release in Humans. J Clin Endocrinol Metab (2011) 96(9):E1409–17. doi: 10.1210/jc.2011-0647
38. Yoshida S, Ohishi T, Matsui T, Tanaka H, Oshima H, Yonetoku Y, et al. Novel GPR119 Agonist AS1535907 Contributes to First-Phase Insulin Secretion in Rat Perfused Pancreas and Diabetic Db/Db Mice. Biochem Biophys Res Commun (2010) 402(2):280–5. doi: 10.1016/j.bbrc.2010.10.015
39. Ning Y, O’neill K, Lan H, Pang L, Shan L, Hawes B, et al. Endogenous and Synthetic Agonists of GPR119 Differ in Signalling Pathways and Their Effects on Insulin Secretion in MIN6c4 Insulinoma Cells. Br J Pharmacol (2008) 155(7):1056–65. doi: 10.1038/bjp.2008.337
40. Hassing HA, Fares S, Larsen O, Pad H, Hauge M, Jones RM, et al. Biased Signaling of Lipids and Allosteric Actions of Synthetic Molecules for GPR119. Biochem Pharmacol (2016) 119:66–75. doi: 10.1016/j.bcp.2016.08.018
41. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-Inflammatory and Insulin-Sensitizing Effects. Cell (2010) 142(5):687–98. doi: 10.1016/j.cell.2010.07.041
42. Lattin J, Zidar DA, Schroder K, Kellie S, Hume DA, Sweet MJ. G-Protein-Coupled Receptor Expression, Function, and Signaling in Macrophages. J Leukocyte Biol (2007) 82(1):16–32. doi: 10.1189/jlb.0107051
43. Oh DY, Walenta E, Akiyama TE, Lagakos WS, Lackey D, Pessentheiner AR, et al. A Gpr120-Selective Agonist Improves Insulin Resistance and Chronic Inflammation in Obese Mice. Nat Med (2014) 20(8):942–7. doi: 10.1038/nm.3614
44. Paschoal VA, Walenta E, Talukdar S, Pessentheiner AR, Osborn O, Hah N, et al. Positive Reinforcing Mechanisms Between GPR120 and Pparγ Modulate Insulin Sensitivity. Cell Metab (2020) 31(6):1173–88.e5. doi: 10.1016/j.cmet.2020.04.020
45. Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential Kinetic and Spatial Patterns of β-Arrestin and G Protein-Mediated ERK Activation by the Angiotensin II Receptor*. J Biol Chem (2004) 279(34):35518–25. doi: 10.1074/jbc.M405878200
46. Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. β-Arrestin Scaffolding of the ERK Cascade Enhances Cytosolic ERK Activity But Inhibits ERK-Mediated Transcription Following Angiotensin AT1a Receptor Stimulation*. J Biol Chem (2002) 277(11):9429–36. doi: 10.1074/jbc.M106457200
47. Oh DY, Olefsky JM. G Protein-Coupled Receptors as Targets for Anti-Diabetic Therapeutics. Nat Rev Drug Discov (2016) 15(3):161–72. doi: 10.1038/nrd.2015.4
48. Winzell MS, Ahrén B. G-Protein-Coupled Receptors and Islet Function—Implications for Treatment of Type 2 Diabetes. Pharmacol Ther (2007) 116(3):437–48. doi: 10.1016/j.pharmthera.2007.08.002
49. Kobilka BK. G Protein Coupled Receptor Structure and Activation. Biochim Biophys Acta (2007) 1768(4):794–807. doi: 10.1016/j.bbamem.2006.10.021
50. Gether U. Uncovering Molecular Mechanisms Involved in Activation of G Protein-Coupled Receptors. Endocrine Rev (2000) 21(1):90–113. doi: 10.1210/edrv.21.1.0390
51. Katritch V, Cherezov V, Stevens RC. Structure-Function of the G Protein-Coupled Receptor Superfamily. Annu Rev Pharmacol Toxicol (2013) 53:531–56. doi: 10.1146/annurev-pharmtox-032112-135923
52. Jean-Charles PY, Kaur S, Shenoy SK. G Protein-Coupled Receptor Signaling Through β-Arrestin-Dependent Mechanisms. J Cardiovasc Pharmacol (2017) 70(3):142–58. doi: 10.1097/FJC.0000000000000482
53. Ranjan R, Dwivedi H, Baidya M, Kumar M, Shukla AK. Novel Structural Insights Into GPCR–β-Arrestin Interaction and Signaling. Trends Cell Biol (2017) 27(11):851–62. doi: 10.1016/j.tcb.2017.05.008
54. Pydi SP, Barella LF, Meister J, Wess J. Key Metabolic Functions of β-Arrestins: Studies With Novel Mouse Models. Trends Endocrinol Metab (2020). doi: (2021) 32(2):118–29. doi: 10.1016/j.tem.2020.11.008
55. Pierce KL, Lefkowitz RJ. Classical and New Roles of β-Arrestins in the Regulation of G-PROTEIN-COUPLED Receptors. Nat Rev Neurosci (2001) 2(10):727–33. doi: 10.1038/35094577
56. Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Association of Beta-Arrestin With G Protein-Coupled Receptors During Clathrin-Mediated Endocytosis Dictates the Profile of Receptor Resensitization. J Biol Chem (1999) 274(45):32248–57. doi: 10.1074/jbc.274.45.32248
57. Pydi SP, Barella LF, Meister J, Wess J. Key Metabolic Functions of β-Arrestins: Studies With Novel Mouse Models. Trends Endocrinol Metab (2021) 32(2):118–29. doi: 10.1016/j.tem.2020.11.008
58. Shukla AK, Xiao K, Lefkowitz RJ. Emerging Paradigms of Beta-Arrestin-Dependent Seven Transmembrane Receptor Signaling. Trends Biochem Sci (2011) 36(9):457–69. doi: 10.1016/j.tibs.2011.06.003
59. Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, Lefkowitz RJ. Beta-Arrestin-Biased Agonism at the Beta2-Adrenergic Receptor. J Biol Chem (2008) 283(9):5669–76. doi: 10.1074/jbc.M708118200
60. Violin JD, Lefkowitz RJ. Beta-Arrestin-Biased Ligands at Seven-Transmembrane Receptors. Trends Pharmacol Sci (2007) 28(8):416–22. doi: 10.1016/j.tips.2007.06.006
61. Smith JS, Lefkowitz RJ, Rajagopal S. Biased Signalling: From Simple Switches to Allosteric Microprocessors. Nat Rev Drug Discov (2018) 17(4):243–60. doi: 10.1038/nrd.2017.229
62. van Gastel J, Hendrickx JO, Leysen H, Santos-Otte P, Luttrell LM, Martin B, et al. Beta-Arrestin Based Receptor Signaling Paradigms: Potential Therapeutic Targets for Complex Age-Related Disorders. Front Pharmacol (2018) 9:1369. doi: 10.3389/fphar.2018.01369
63. Woo AY, Ge XY, Pan L, Xing G, Mo YM, Xing RJ, et al. Discovery of Beta-Arrestin-Biased Beta2-Adrenoceptor Agonists From 2-Amino-2-Phenylethanol Derivatives. Acta Pharmacol Sin (2019) 40(8):1095–105. doi: 10.1038/s41401-018-0200-x
64. Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential Kinetic and Spatial Patterns of Beta-Arrestin and G Protein-Mediated ERK Activation by the Angiotensin II Receptor. J Biol Chem (2004) 279(34):35518–25. doi: 10.1074/jbc.M405878200
65. Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB, et al. Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-Coupled Receptor Signaling. J Biol Chem (2016) 291(53):27147–59. doi: 10.1074/jbc.M116.754887
66. Caron A, Reynolds RP, Castorena CM, Michael NJ, Lee CE, Lee S, et al. Adipocyte Gs But Not Gi Signaling Regulates Whole-Body Glucose Homeostasis. Mol Metab (2019) 27:11–21. doi: 10.1016/j.molmet.2019.06.019
67. Långberg EC, Seed Ahmed M, Efendic S, Gu HF, Östenson CG. Genetic Association of Adrenergic Receptor Alpha 2A With Obesity and Type 2 Diabetes. Obesity (2013) 21(8):1720–5. doi: 10.1002/oby.20162
68. Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. Perilipin, a Major Hormonally Regulated Adipocyte-Specific Phosphoprotein Associated With the Periphery of Lipid Storage Droplets. J Biol Chem (1991) 266(17):11341–6. doi: 10.1016/S0021-9258(18)99168-4
69. Priyadarshini M, Villa SR, Fuller M, Wicksteed B, Mackay CR, Alquier T, et al. FFAR2, Regulates Insulin Secretion. Mol Endocrinol (2015) 29(7):1055–66. doi: 10.1210/me.2015-1007
70. Sebastiani G, Ceccarelli E, Castagna MG, Dotta F. G-Protein-Coupled Receptors (GPCRs) in the Treatment of Diabetes: Current View and Future Perspectives. Best Pract Res Clin Endocrinol Metab (2018) 32(2):201–13. doi: 10.1016/j.beem.2018.02.005
71. O’Dowd BF, Nguyen T, Marchese A, Cheng R, Lynch KR, Heng HH, et al. Discovery of Three Novel G-Protein-Coupled Receptor Genes. Genomics (1998) 47(2):310–3. doi: 10.1006/geno.1998.5095
72. Wang J, Simonavicius N, Wu X, Swaminath G, Reagan J, Tian H, et al. Kynurenic Acid as a Ligand for Orphan G Protein-Coupled Receptor GPR35. J Biol Chem (2006) 281(31):22021–8. doi: 10.1074/jbc.M603503200
73. Ohshiro H, Tonai-Kachi H, Ichikawa K. GPR35 Is a Functional Receptor in Rat Dorsal Root Ganglion Neurons. Biochem Biophys Res Commun (2008) 365(2):344–8. doi: 10.1016/j.bbrc.2007.10.197
74. Okumura S, Baba H, Kumada T, Nanmoku K, Nakajima H, Nakane Y, et al. Cloning of a G-Protein-Coupled Receptor That Shows an Activity to Transform NIH3T3 Cells and Is Expressed in Gastric Cancer Cells. Cancer Sci (2004) 95(2):131–5. doi: 10.1111/j.1349-7006.2004.tb03193.x
75. Egerod KL, Petersen N, Timshel PN, Rekling JC, Wang Y, Liu Q, et al. Profiling of G Protein-Coupled Receptors in Vagal Afferents Reveals Novel Gut-to-Brain Sensing Mechanisms. Mol Metab (2018) 12:62–75. doi: 10.1016/j.molmet.2018.03.016
76. Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-Mediated Signaling of Metabolites. Cell Metab (2017) 25(4):777–96. doi: 10.1016/j.cmet.2017.03.008
77. Binti Mohd Amir NAS, Mackenzie AE, Jenkins L, Boustani K, Hillier MC, Tsuchiya T, et al. Evidence for the Existence of a CXCL17 Receptor Distinct From GPR35. J Immunol (Baltimore Md: 1950) (2018) 201(2):714–24. doi: 10.4049/jimmunol.1700884
78. Park SJ, Lee SJ, Nam SY, Im DS. GPR35 Mediates Lodoxamide-Induced Migration Inhibitory Response But Not CXCL17-Induced Migration Stimulatory Response in THP-1 Cells; Is GPR35 a Receptor for CXCL17? Br J Pharmacol (2018) 175(1):154–61. doi: 10.1111/bph.14082
79. Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, et al. Free Fatty Acids Regulate Insulin Secretion From Pancreatic β Cells Through GPR40. Nature (2003) 422(6928):173–6. doi: 10.1038/nature01478
80. Feng DD, Luo Z, S-g R, Hernandez M, Tawadros N, DJ K, et al. Reduction in Voltage-Gated K+ Currents in Primary Cultured Rat Pancreatic β-Cells by Linoleic Acids. Endocrinology (2006) 147(2):674–82. doi: 10.1210/en.2005-0225
81. Kim M, Gu GJ, Koh YS, Lee SH, Na YR, Seok SH, et al. Fasiglifam (TAK-875), a G Protein-Coupled Receptor 40 (GPR40) Agonist, May Induce Hepatotoxicity Through Reactive Oxygen Species Generation in a GPR40-Dependent Manner. Biomolecules Ther (2018) 26(6):599–607. doi: 10.4062/biomolther.2017.225
82. Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, et al. Short-Chain Fatty Acids Stimulate Glucagon-Like Peptide-1 Secretion via the G-Protein–Coupled Receptor Ffar2. Diabetes (2012) 61(2):364–71. doi: 10.2337/db11-1019
83. Zaibi MS, Stocker CJ, O’Dowd J, Davies A, Bellahcene M, Cawthorne MA, et al. Roles of GPR41 and GPR43 in Leptin Secretory Responses of Murine Adipocytes to Short Chain Fatty Acids. FEBS Lett (2010) 584(11):2381–6. doi: 10.1016/j.febslet.2010.04.027
84. Tang C, Ahmed K, Gille A, Lu S, Gröne H-J, Tunaru S, et al. Loss of FFA2 and FFA3 Increases Insulin Secretion and Improves Glucose Tolerance in Type 2 Diabetes. Nat Med (2015) 21(2):173–7. doi: 10.1038/nm.3779
85. Nilsson NE, Kotarsky K, Owman C, Olde B. Identification of a Free Fatty Acid Receptor, FFA2R, Expressed on Leukocytes and Activated by Short-Chain Fatty Acids. Biochem Biophys Res Commun (2003) 303(4):1047–52. doi: 10.1016/S0006-291X(03)00488-1
86. Senga T, Iwamoto S, Yoshida T, Yokota T, Adachi K, Azuma E, et al. LSSIG Is a Novel Murine Leukocyte-Specific GPCR That Is Induced by the Activation of STAT3. Blood J Am Soc Hematol (2003) 101(3):1185–7. doi: 10.1182/blood-2002-06-1881
87. Ang Z, Er JZ, Ding JL. The Short-Chain Fatty Acid Receptor GPR43 Is Transcriptionally Regulated by XBP1 in Human Monocytes. Sci Rep (2015) 5(1):1–9. doi: 10.1038/srep08134
88. Lorza-Gil E, Kaiser G, Rexen Ulven E, König GM, Gerst F, Oquendo MB, et al. FFA2-, But Not FFA3-Agonists Inhibit GSIS of Human Pseudoislets: A Comparative Study With Mouse Islets and Rat INS-1E Cells. Sci Rep (2020) 10(1):16497. doi: 10.1038/s41598-020-73467-5
89. Davidsson Ö, Nilsson K, Brånalt J, Andersson T, Berggren K, Chen Y, et al. Identification of Novel GPR81 Agonist Lead Series for Target Biology Evaluation. Bioorganic Medicinal Chem Lett (2020) 30(4):126953. doi: 10.1016/j.bmcl.2020.126953
90. Ahmed K, Tunaru S, Tang C, Müller M, Gille A, Sassmann A, et al. An Autocrine Lactate Loop Mediates Insulin-Dependent Inhibition of Lipolysis Through GPR81. Cell Metab (2010) 11(4):311–9. doi: 10.1016/j.cmet.2010.02.012
91. Wanders D, Graff EC, Judd RL. Effects of High Fat Diet on GPR109A and GPR81 Gene Expression. Biochem Biophys Res Commun (2012) 425(2):278–83. doi: 10.1016/j.bbrc.2012.07.082
92. Overton HA, Babbs AJ, Doel SM, Fyfe MC, Gardner LS, Griffin G, et al. Deorphanization of a G Protein-Coupled Receptor for Oleoylethanolamide and Its Use in the Discovery of Small-Molecule Hypophagic Agents. Cell Metab (2006) 3(3):167–75. doi: 10.1016/j.cmet.2006.02.004
93. Soga T, Ohishi T, Matsui T, Saito T, Matsumoto M, Takasaki J, et al. Lysophosphatidylcholine Enhances Glucose-Dependent Insulin Secretion via an Orphan G-Protein-Coupled Receptor. Biochem Biophys Res Commun (2005) 326(4):744–51. doi: 10.1016/j.bbrc.2004.11.120
94. Lan H, Lin H, Wang C, Wright M, Xu S, Kang L, et al. Agonists at GPR119 Mediate Secretion of GLP-1 From Mouse Enteroendocrine Cells Through Glucose-Independent Pathways. Br J Pharmacol (2012) 165(8):2799–807. doi: 10.1111/j.1476-5381.2011.01754.x
95. Abdel-Magid AF. GPR119 Modulators for the Treatment of Diabetes, Obesity, and Related Diseases: Patent Highlight. ACS Med Chem Lett (2012) 3(12):955–8. doi: 10.1021/ml300296q
96. Stone VM, Dhayal S, Brocklehurst KJ, Lenaghan C, Winzell MS, Hammar M, et al. GPR120 (FFAR4) Is Preferentially Expressed in Pancreatic Delta Cells and Regulates Somatostatin Secretion From Murine Islets of Langerhans. Diabetologia (2014) 57(6):1182–91. doi: 10.1007/s00125-014-3213-0
97. Moore K, Zhang Q, Murgolo N, Hosted T, Duffy R. Cloning, Expression, and Pharmacological Characterization of the GPR120 Free Fatty Acid Receptor From Cynomolgus Monkey: Comparison With Human GPR120 Splice Variants. Comp Biochem Physiol Part B: Biochem Mol Biol (2009) 154(4):419–26. doi: 10.1016/j.cbpb.2009.08.005
98. Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, et al. GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-Inflammatory and Insulin-Sensitizing Effects. Cell (2010) 142(5):687–98. doi: 10.1016/j.cell.2010.07.041
99. Villegas-Comonfort S, Takei Y, Tsujimoto G, Hirasawa A, García-Sáinz J. Effects of Arachidonic Acid on FFA4 Receptor: Signaling, Phosphorylation and Internalization. Prostaglandins Leukotrienes Essential Fatty Acids (2017) 117:1–10. doi: 10.1016/j.plefa.2017.01.013
100. Milligan G, Alvarez-Curto E, Watterson K, Ulven T, Hudson B. Characterizing Pharmacological Ligands to Study the Long-Chain Fatty Acid Receptors GPR 40/FFA 1 and GPR 120/FFA 4. Br J Pharmacol (2015) 172(13):3254–65. doi: 10.1111/bph.12879
101. Li A, Yang D, Zhu M, Tsai KC, Xiao KH, Yu X, et al. Discovery of Novel FFA4 (GPR120) Receptor Agonists With β-Arrestin2-Biased Characteristics. Future Medicinal Chem (2015) 7(18):2429–37. doi: 10.4155/fmc.15.160
102. Fletcher Madeleine M, Halls Michelle L, Christopoulos A, Sexton Patrick M, Wootten D. The Complexity of Signalling Mediated by the Glucagon-Like Peptide-1 Receptor. Biochem Soc Trans (2016) 44(2):582–8. doi: 10.1042/BST20150244
103. Farilla L, Bulotta A, Hirshberg B, Li Calzi S, Khoury N, Noushmehr H, et al. Glucagon-Like Peptide 1 Inhibits Cell Apoptosis and Improves Glucose Responsiveness of Freshly Isolated Human Islets. Endocrinology (2003) 144(12):5149–58. doi: 10.1210/en.2003-0323
104. Kuna RS, Girada SB, Asalla S, Vallentyne J, Maddika S, Patterson JT, et al. Glucagon-Like Peptide-1 Receptor-Mediated Endosomal cAMP Generation Promotes Glucose-Stimulated Insulin Secretion in Pancreatic β-Cells. Am J Physiol Endocrinol Metab (2013) 305(2):E161–70. doi: 10.1152/ajpendo.00551.2012
105. Rajan S, Dickson LM, Mathew E, Orr CM, Ellenbroek JH, Philipson LH, et al. Chronic Hyperglycemia Downregulates GLP-1 Receptor Signaling in Pancreatic β-Cells via Protein Kinase A. Mol Metab (2015) 4(4):265–76. doi: 10.1016/j.molmet.2015.01.010
106. Pabreja K, Mohd MA, Koole C, Wootten D, Furness SG. Molecular Mechanisms Underlying Physiological and Receptor Pleiotropic Effects Mediated by GLP-1R Activation. Br J Pharmacol (2014) 171(5):1114–28. doi: 10.1111/bph.12313
107. Weston C, Poyner D, Patel V, Dowell S, Ladds G. Investigating G Protein Signalling Bias at the Glucagon-Like Peptide-1 Receptor in Yeast. Br J Pharmacol (2014) 171(15):3651–65. doi: 10.1111/bph.12716
108. Jorgensen R, Kubale V, Vrecl M, Schwartz TW, Elling CE. Oxyntomodulin Differentially Affects Glucagon-Like Peptide-1 Receptor β-Arrestin Recruitment and Signaling Through Gα. J Pharmacol Exp Ther (2007) 322(1):148–54. doi: 10.1124/jpet.107.120006
109. Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, et al. Visualization of Arrestin Recruitment by a G-Protein-Coupled Receptor. Nature (2014) 512(7513):218–22. doi: 10.1038/nature13430
110. Thomsen ARB, Plouffe B, Cahill TJ 3rd, Shukla AK, Tarrasch JT, Dosey AM, et al. GPCR-G Protein-Beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell (2016) 166(4):907–19. doi: 10.1016/j.cell.2016.07.004
111. Zhuang LN, Hu WX, Zhang ML, Xin SM, Jia WP, Zhao J, et al. Beta-Arrestin-1 Protein Represses Diet-Induced Obesity. J Biol Chem (2011) 286(32):28396–402. doi: 10.1074/jbc.M111.223206
112. Pydi SP, Cui Z, He Z, Barella LF, Pham J, Cui Y, et al. Beneficial Metabolic Role of β-Arrestin-1 Expressed by AgRP Neurons. Sci Adv (2020) 6(23):eaaz1341. doi: 10.1126/sciadv.aaz1341
113. Pydi SP, Jain S, Tung W, Cui Y, Zhu L, Sakamoto W, et al. Adipocyte β-Arrestin-2 Is Essential for Maintaining Whole Body Glucose and Energy Homeostasis. Nat Commun (2019) 10(1):2936. doi: 10.1038/s41467-019-11003-4
114. Zhu L, Rossi M, Cui Y, Lee RJ, Sakamoto W, Perry NA, et al. Hepatic β-Arrestin 2 Is Essential for Maintaining Euglycemia. J Clin Invest (2017) 127(8):2941–5. doi: 10.1172/JCI92913
115. Barella LF, Rossi M, Zhu L, Cui Y, Mei FC, Cheng X, et al. β-Cell-Intrinsic β-Arrestin 1 Signaling Enhances Sulfonylurea-Induced Insulin Secretion. J Clin Invest (2019) 129(9):3732–7. doi: 10.1172/JCI126309
116. Zhu L, Almaça J, Dadi PK, Hong H, Sakamoto W, Rossi M, et al. β-Arrestin-2 Is an Essential Regulator of Pancreatic β-Cell Function Under Physiological and Pathophysiological Conditions. Nat Commun (2017) 8(1):14295. doi: 10.1038/ncomms14295
117. Meister J, Bone DBJ, Godlewski G, Liu Z, Lee RJ, Vishnivetskiy SA, et al. Metabolic Effects of Skeletal Muscle-Specific Deletion of Beta-Arrestin-1 and -2 in Mice. PloS Genet (2019) 15(10):e1008424. doi: 10.1371/journal.pgen.1008424
118. Pydi SP, Jain S, Tung W, Cui Y, Zhu L, Sakamoto W, et al. Adipocyte Beta-Arrestin-2 Is Essential for Maintaining Whole Body Glucose and Energy Homeostasis. Nat Commun (2019) 10(1):2936. doi: 10.1038/s41467-019-11003-4
119. Wang L, Pydi SP, Cui Y, Zhu L, Meister J, Gavrilova O, et al. Selective Activation of Gs Signaling in Adipocytes Causes Striking Metabolic Improvements in Mice. Mol Metab (2019) 27:83–91. doi: 10.1016/j.molmet.2019.06.018
120. Pydi SP, Jain S, Barella LF, Zhu L, Sakamoto W, Meister J, et al. β-Arrestin-1 Suppresses Myogenic Reprogramming of Brown Fat to Maintain Euglycemia. Sci Advances (2020) 6(23):eaba1733. doi: 10.1126/sciadv.aba1733
121. Dalle S, Imamura T, Rose DW, Worrall DS, Ugi S, Hupfeld CJ, et al. Insulin Induces Heterologous Desensitization of G-Protein-Coupled Receptor and Insulin-Like Growth Factor I Signaling by Downregulating Beta-Arrestin-1. Mol Cell Biol (2002) 22(17):6272–85. doi: 10.1128/MCB.22.17.6272-6285.2002
122. Usui I, Imamura T, Huang J, Satoh H, Shenoy SK, Lefkowitz RJ, et al. β-Arrestin-1 Competitively Inhibits Insulin-Induced Ubiquitination and Degradation of Insulin Receptor Substrate 1. Mol Cell Biol (2004) 24(20):8929–37. doi: 10.1128/MCB.24.20.8929-8937.2004
123. Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, et al. Hypothalamic K(ATP) Channels Control Hepatic Glucose Production. Nature (2005) 434(7036):1026–31. doi: 10.1038/nature03439
124. Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, et al. Agouti-Related Peptide-Expressing Neurons Are Mandatory for Feeding. Nat Neurosci (2005) 8(10):1289–91. doi: 10.1038/nn1548
125. Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP Neurons Are Essential for Feeding in Adult Mice But can be Ablated in Neonates. Science (2005) 310(5748):683–5. doi: 10.1126/science.1115524
126. Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford ML. Leptin Inhibits Hypothalamic Neurons by Activation of ATP-Sensitive Potassium Channels. Nature (1997) 390(6659):521–5. doi: 10.1038/37379
127. Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML. Insulin Activates ATP-Sensitive K+ Channels in Hypothalamic Neurons of Lean, But Not Obese Rats. Nat Neurosci (2000) 3(8):757–8. doi: 10.1038/77660
128. Lin HV, Accili D. Hormonal Regulation of Hepatic Glucose Production in Health and Disease. Cell Metab (2011) 14(1):9–19. doi: 10.1016/j.cmet.2011.06.003
129. Cho YM, Merchant CE, Kieffer TJ. Targeting the Glucagon Receptor Family for Diabetes and Obesity Therapy. Pharmacol Ther (2012) 135(3):247–78. doi: 10.1016/j.pharmthera.2012.05.009
130. Htike ZZ, Zaccardi F, Papamargaritis D, Webb DR, Khunti K, Davies MJ. Efficacy and Safety of Glucagon-Like Peptide-1 Receptor Agonists in Type 2 Diabetes: A Systematic Review and Mixed-Treatment Comparison Analysis. Diabetes Obes Metab (2017) 19(4):524–36. doi: 10.1111/dom.12849
Keywords: GPCRs, β-arrestins, biased signaling, type 2 diabetes, obesity
Citation: Oliveira de Souza C, Sun X and Oh D (2021) Metabolic Functions of G Protein-Coupled Receptors and β-Arrestin-Mediated Signaling Pathways in the Pathophysiology of Type 2 Diabetes and Obesity. Front. Endocrinol. 12:715877. doi: 10.3389/fendo.2021.715877
Received: 27 May 2021; Accepted: 29 July 2021;
Published: 23 August 2021.
Edited by:
Shanu Jain, National Institutes of Health (NIH), United StatesReviewed by:
Sudha K. Shenoy, Duke University School of Medicine, United StatesCopyright © 2021 Oliveira de Souza, Sun and Oh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dayoung Oh, ZGF5b3VuZy5vaEB1dHNvdXRod2VzdGVybi5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.