 Qian Zhang
Qian Zhang Xinhua Xiao
Xinhua Xiao Jia Zheng
Jia Zheng- Key Laboratory of Endocrinology, Ministry of Health, Department of Endocrinology, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China
The prevalence of obesity has become a threatening global public health issue. The consequence of obesity is abnormal energy metabolism. Unlike white adipose tissue (WAT), brown adipose tissue (BAT) has a unique role in nonshivering thermogenesis. Lipids and glucose are consumed to maintain energy and metabolic homeostasis in BAT. Recently, accumulating evidence has indicated that exposure to excess maternal energy intake affects energy metabolism in offspring throughout their life. However, whether excess intrauterine energy intake influences BAT metabolism in adulthood is not clear. In this study, mouse dams were exposed to excess energy intake by feeding a high-fat diet (HFD) before and during pregnancy and lactation. The histology of BAT was assessed by hematoxylin and eosin staining. The genome-wide methylation profile of BAT was determined by a DNA methylation array, and specific site DNA methylation was quantitatively analyzed by methylated DNA immunoprecipitation (MeDIP) qPCR. We found that intrauterine exposure to a high-energy diet resulted in blood lipid panel disorders and impaired the BAT structure. Higher methylation levels of genes involved in thermogenesis and fatty acid oxidation (FAO) in BAT, such as Acaa2, Acsl1, and Cox7a1, were found in 16-week-old offspring from mothers fed with HFD. Furthermore, the expression of Acaa2, Acsl1, and Cox7a1 was down-regulated by intrauterine exposure to excess energy intake. In summary, our results reveal that excess maternal energy leads to a long-term disorder of BAT in offspring that involves the activation of DNA methylation of BAT-specific genes involved in fatty acid oxidation and thermogenesis.
Introduction
Obesity remains a significant global public health issue (1). Currently, the obesity and overweight occur not only in developed and developing countries but also in undeveloped regions. Obesity is involved in multiple diseases, such as type 2 diabetes, cardiovascular disease, nonalcoholic steatohepatitis, and even cancer (2–5). Obesity is a multi-factorial disease. In addition to genetic factors, environmental changes also contribute to the incidence of obesity, especially during critical developmental periods, such as gestation and lactation (6). Epidemiological data indicates that a maternal high-fat diet is one of the key risk factors for childhood obesity (7, 8). Obesity during childhood is strongly associated with obesity and other unhealthy outcomes in adulthood (9). Moreover, animal research has demonstrated that exposure to excess energy intake during early life stages, such as the fetal and infancy periods, induces obesity and dysfunction in adipose tissue in adulthood (10–13). Increasing evidence supports the developmental origins of a health and diseases (DOHaD) concept; that is, exposure during an early life period is the origin of the development of health and diseases in adulthood (14). This phenomenon is also explained as metabolic programming, which is defined as adverse exposure during gestation and lactation that programs the health and illnesses in adulthood (15, 16). Epigenetic mechanisms, including DNA methylation, chromatin remodel, and noncoding RNAs (17, 18), can influence gene expression chiefly at the level of transcription (19) and play a critical role in metabolic programming (20). Among these epigenetic mechanisms, DNA methylation, which takes place at the 5’ position of cytosine in CpG islands at specific locations in genome, has been proven to be a key mechanism in the progression of obesity and lipid metabolism (20). DNA methylation, which occurs in CpG-rich regions known as CpG islands in the genome, can result in gene expression alteration (21).
In mammals, white adipose tissue (WAT) is at high ratio in the body and is well studied (22). As a primary site of excess energy storage, WAT hypertrophy is closely related to obesity. Another type of adipose tissue is brown adipose tissue (BAT). The main function of BAT is nonshivering thermogenesis. In BAT, fatty acids and glucose are utilized as fuel to produce heat under stimuli (22, 23). In the past, the BAT was considered only to exist rodent animals and newborn babies. However, recent studies have confirmed that active BAT depots also exist in adult humans (24). Recently, scientists discovered the important role of BAT in adults and its potential strategy for obesity treatment (25).
However, the majority of studies focus on the effect of maternal HFD on the morphological and functional disorders of WAT in offspring. These studies reveal that maternal obesity is closely related to increased WAT, and glucose and lipid metabolism disorders during the adult life of offspring (26–30). Several studies have proven that maternal obesity also disturbs the thermogenesis function of BAT in offspring, resulting in obesity in adulthood (31–33).
In this study, we hypothesized that maternal HFD exposure affects offspring BAT morphology, biological function, and DNA methylation modification, which boosts the progression of obesity. Therefore, we focused on whole-genomic methylation in the BAT from offspring exposed to a maternal HFD, as evaluated by a DNA methylation array.
Materials and Methods
Animal Treatments and Diets
This work was designed according to the NIH guidelines for the use of laboratory animals (NIH Publications No.86-23, revised 1996). All experimental protocols were approved by the Animal Care Committee of Peking Union Medical Hospital (Permit Number: XHDW-2015-0051).Thirty-two female C57BL/6J mice (five weeks old) were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China). Sixteen females were randomly assigned to consume a normal control rodent diet (NC, 16% kcal fat, 64% kcal carbohydrate, 20% kcal protein, D11112201, Research Diets, New Brunswick, NJ). Another sixteen mice were fed a high-fat diet (HFD, 45% kcal fat, 35% kcal carbohydrate, 20% kcal protein, D12451, Research Diets, New Brunswick, NJ) to induce obesity. After a 4-week diet scheme, female mice were mated with male mice fed with a normal diet. Female mouse diet protocols were continued during the following gestation and lactation periods. Thus, two different diet-exposed dams were obtained: normal control (NC) mice fed a normal diet and diet-induced obesity (DIO) mice fed a HFD. After birth, litters were reduced to six pups (3 males and 3 females) per mother to avoid competition for milk. After weaning, only male offspring (one male pup from each litter was randomly assigned to the experimental groups) were randomly divided into the following groups to receive a postweaning control diet (CD) or a HFD (n = 8 each group) for 13 weeks. Other surviving mice were kept feeding for other study. Thus, four subgroups were obtained, NC-CD, NC-HFD, DIO-CD, and DIO-HFD, to research the interaction between the maternal and offspring diets on the progression of obesity (Figure 1). The female offspring were not included in this study, because of confounding factors related to their hormone profile and estrus cycle. Food consumption and body weight were monitored. Offspring were sacrificed at 16 weeks old.
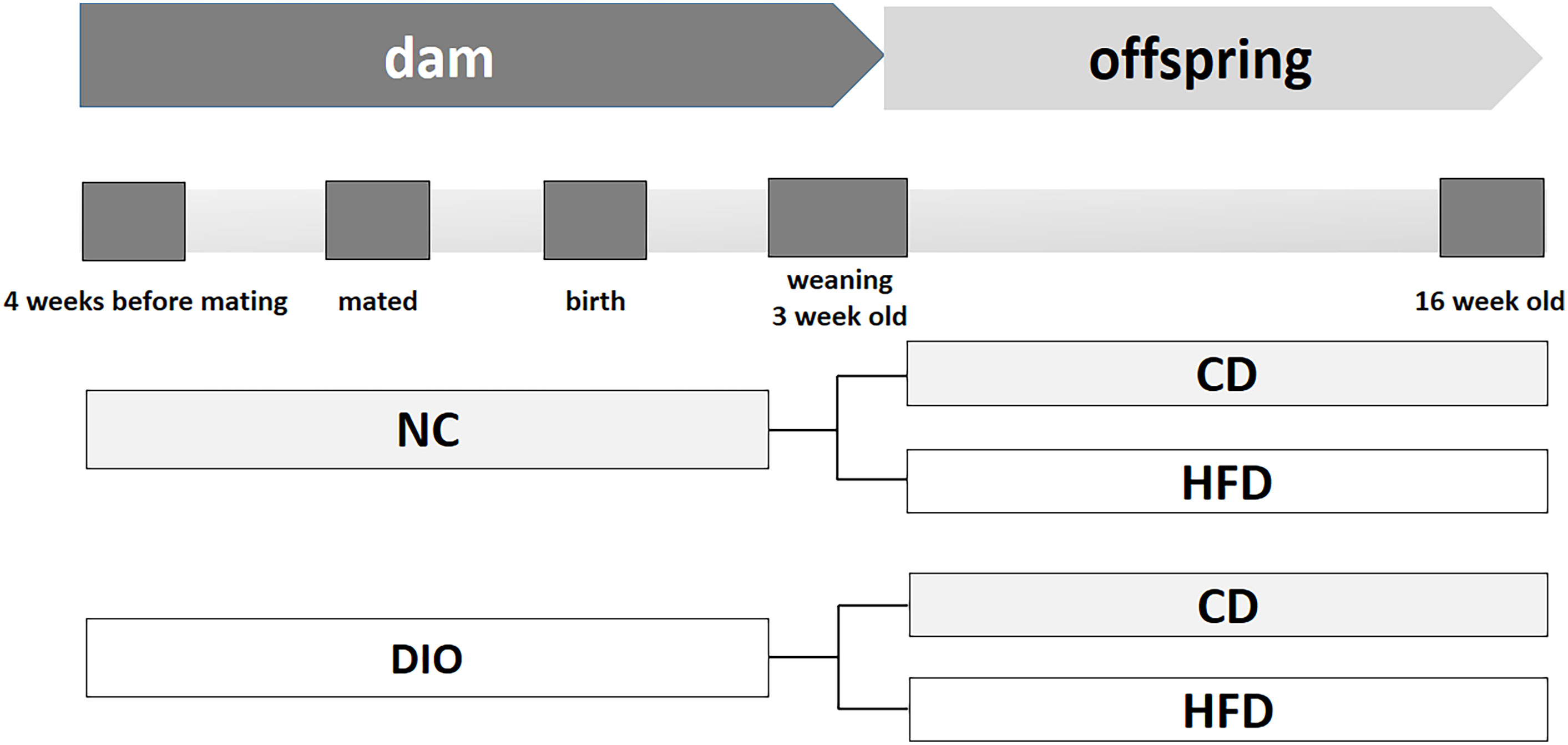
Figure 1 Schema of animal experiment protocol. NC, normal control; DIO, diet induced obesity; CD, control diet; HFD, high-fat diet.
Sample and Tissue Collection
After an overdose injection of pentobarbital sodium (150 mg/kg), mice were sacrificed. Other surviving mice were kept feeding. Then, blood was collected by cardiac puncture. Interscapular BAT was separated and weighed. Part of the BAT was frozen in liquid nitrogen and stored at -80°C for future molecular experiments. The other part of the BAT was fixed in 10% formalin for histological analyses.
Metabolic Analyses of Circulating Factors
Serum was collected from mice that were fasted overnight. Total cholesterol (TC) and triglyceride (TG) levels were assayed by enzyme end-point assays with a commercial kit (Roche Diagnostics, GmbH, Mannheim, Germany) according to the manufacturer’s instructions.
Hematoxylin and Eosin (H&E) Staining
The BAT was fixed in 4% paraformaldehyde. Then, tissues were embedded in paraffin. Five micrometer sections were cut by a microtome (Nickon, Tokyo, Japan). Finally, the section was stained with H&E. Photographs (magnification 400 x) were taken using a Nikon microscope (Nikon, Tokyo, Japan). Lipid area was measured by Image-Pro Plus version 7.0 (Media Cybernetics Inc., Rockville, MD). Lipid droplets in the histopathological image were defined as circular unstained objects, as descripted in previous reference (34).
DNA Preparation and DNA Methylation Microarray
To determine the effect of maternal DIO on DNA methylation in offspring BAT, genomic DNA was extracted from BAT using a DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA). Isolated genomic DNA from the DIO-CD group and NC-CD group (n = 3 for each group) was fragmented to 200 to 1,000 bp by sonication. Through reaction with magnetic beads coupled with mouse monoclonal antibodies against 5-methylcytidine (Diagenode, Liege, Belgium), the methylated DNA was immunoprecipitated. The total input DNA was labeled with Cy3-labeled random 9-mers, while the immunoprecipitated DNA was labelled with Cy5-labeled random 9-mers. Then, labelled DNA was hybridized to an Arraystar Mouse RefSeq Promoter Array (Arraystar Inc., Rockville, MD). This array covers 22,327 well-characterized RefSeq promoter regions with ∼180,000 probes. The promoter regions were defined as ∼-1,300 to +500 bp of the transcription start sites (TSSs). The array was scanned by an Agilent Scanner G2505C (Agilent Technologies, Waldbronn, Germany).
Data Normalization and Analysis
Raw data were generated and normalized to log 2 ratio data and then analyzed to identify enriched peaks by NimbleScan Software v2.5 (Roche NimbleGen Inc., Madison, WI). To calculate the difference in enriched peaks between DIO-CD and NC-CD offspring BAT, the average log2 ratios and M’ were calculated for each probe, according to the following formula,
The NimbleScan sliding-window peak-finding algorithm was used to find the differentially enriched peaks (DEPs). DEPs were defined as follows, (i) at least one of the two groups had a median value of log2(MeDIP/Input) ≥ 0.3 and a median value of M’ > 0 within the peak; and (ii) at least half of the probes in a peak had a median value of the coefficient of variability (CV) ≤ 0.8 for both groups. Then, PeakScores were calculated to identify differentially methylated regions. The DEP was defined as follows: PeakScore ≥ 2 and P ≤ 0.01. The CpG region was categorized according to CpG island density, including high CpG promoters/regions (HCPs), intermediate CpG promoters/regions (ICPs), and low CpG promoters/regions (LCPs) (35). All differentially methylated genes (DMGs) were clustered to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway terms by using DAVID Bioinformatics Resources version 6.7 (http://david.abcc.ncifcrf.gov/) (36). Significant GO functional clusters and KEGG pathways were defined as the follows, at least 2 DMGs in each GO and/or KEGG pathway term, a Benjamini–Hochberg FDR P < 0.05 (KEGG) or 0.01 (GO) and a fold enrichment ≥ 1.50 (KEGG) or 1.10 (GO) within each cluster.
Methylated DNA Immunoprecipitation (MeDIP) qPCR
To detect the methylation status of specific genes, MeDIP qPCR was used. Genomic DNA was fragmented by ultrasonication. DNA fragments of 100∼500 bp on average were obtained. Similar to DNA methylation microarray, denatured DNA containing RNase A was incubated with magnetic beads coupled with mouse monoclonal antibodies against 5-methylcytidine antibody (Zymo Research, Irvine, CA) or mouse anti-IgG overnight at 4°C. Then, the mixture was incubated with protein G magnetic beads at 4°C for 1 h, washed in digestion buffer at 65°C for 3 h, and finally quantified by quantitative PCR (qPCR). CpG island analyses and MeDIP qPCR primer designs were based on the genome-wide epigenetic database of the National Center for Biotechnology Information and MethPrimer 1.0 (37). MeDIP qPCR primers are listed in Table 1.
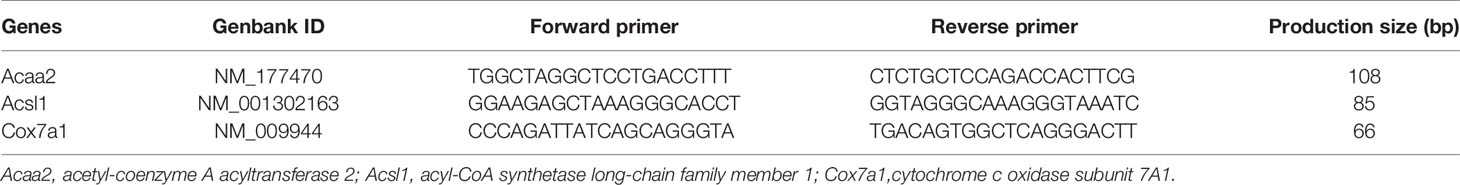
Table 1 Primers for MeDIP qPCR.
RNA Isolation and qPCR Analysis
Total RNA was extracted by using TRIzol reagent (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. The expression of acetyl-coenzyme A acyltransferase 2 (Acaa2), acyl-CoA synthetase long-chain family member 1 (Acsl1) and cytochrome c oxidase subunit 7A1, mitochondrial (Cox7a1) was measured using an ABI Prism 7900 system (Applied Biosystems, Foster City, CA) by the comparative Ct method (2-△△Ct). The relative RNA expression levels were standardized to Gadph. The primer sequences are listed in Table 2.
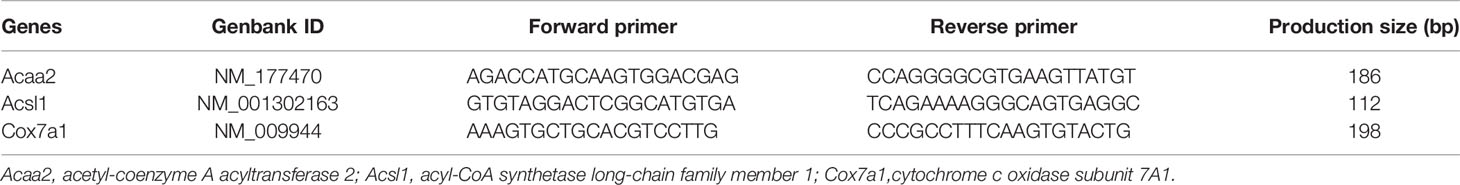
Table 2 Primers for qPCR.
Statistical Analysis
The research data are shown as the mean ± SEM. Differences between the two dam groups were analyzed using Student’s t test. Differences among the four offspring groups were analyzed using two-way ANOVA to analyze the maternal diet effect, offspring diet effect and their interaction. Prism Software version 7 (GraphPad Software Inc., San Diego, CA) was used to perform statistical analyses. Statistical significance was defined as P < 0.05.
Results
The Effect of Maternal HFD on Food Intake and Body Weight
During pregnancy, DIO dams consumed more caloric energy than NC dams (P < 0.01, Figure 2A). After 4-week HFD, DIO mice model was successfully established (Figure 2B). On the deliver day, the weight of DIO group was higher than that of NC group (P < 0.01, Figure 2B). And DIO females had a 1.40-fold increase in gestational body weight gain compared with NC females (P < 0.01, Figure 2C).
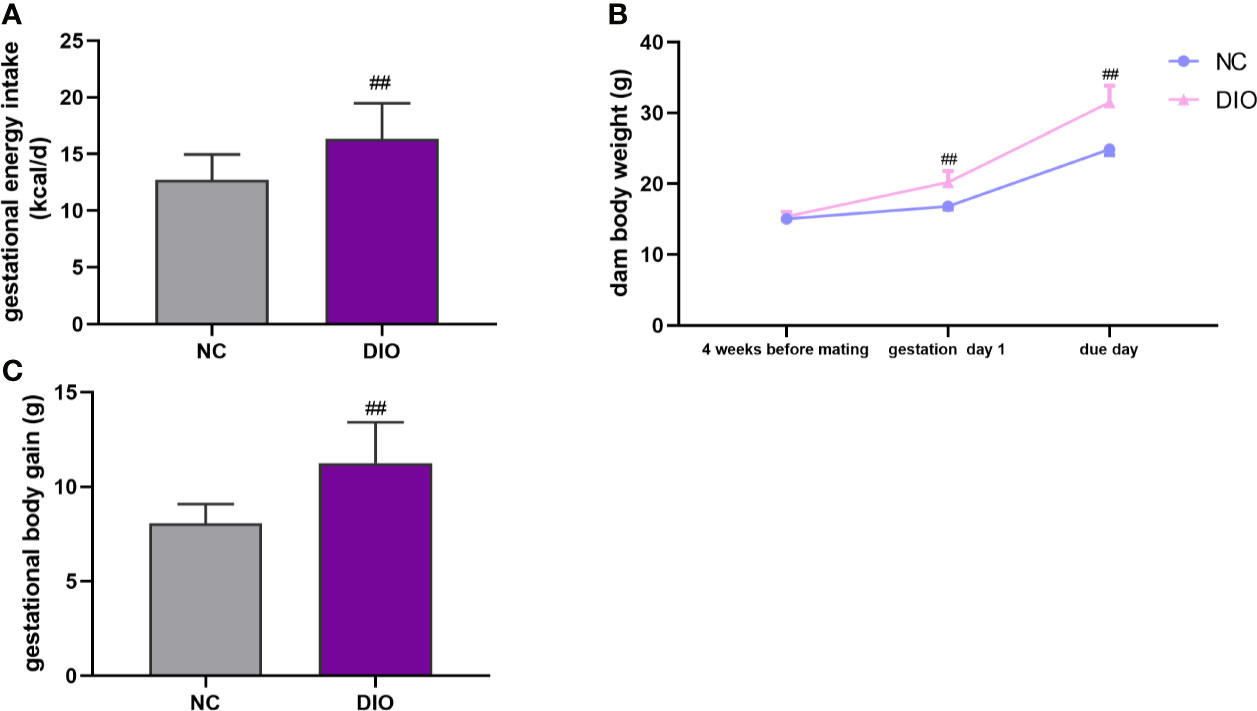
Figure 2 Maternal characteristics. Maternal (A) energy intake during pregnancy, (B) body weight and (C) body weight gain during pregnancy. Values represent the mean ± SEM, n = 16 each group. ##P < 0.01 DIO vs. NC group. NC, normal control; DIO, diet-induced obesity.
The Effect of Maternal HFD on Body Weight and BAT Weight in Male Offspring
There were no differences in litter sizes (7.00 ± 0.73 vs. 7.38 ± 0.62), male: female ratio (0.47 ± 0.06 vs 0.50 ± 0.09), and birth weight (1.33 ± 0.09 g vs 1.27 ± 0.06 g) between offspring from DIO and NC dams (P > 0.05). Offspring fed with HFD had more energy intake at 16 weeks old (diet exposure P < 0.05 or 0.01, Figure 3A). In all groups of offspring, the HFD resulted in an increase in the body weight of male offspring at 8 and 16 weeks old (diet exposure P < 0.01, Figure 3B). Although there is no significant difference of energy intake between DIO-CD and NC-CD group. Maternal DIO induced a significant increase in body weight (prenatal exposure P < 0.01, Figure 3B). Although BAT weight was elevated in NC-HFD offspring compared to NC-CD offspring (diet exposure P < 0.01, Figure 3B), BAT weight in DIO-HFD offspring was similar to that in DIO-CD offspring. Notably, BAT weight in DIO-CD offspring was higher than that in NC-CD offspring (prenatal exposure P < 0.01, Figure 3C).
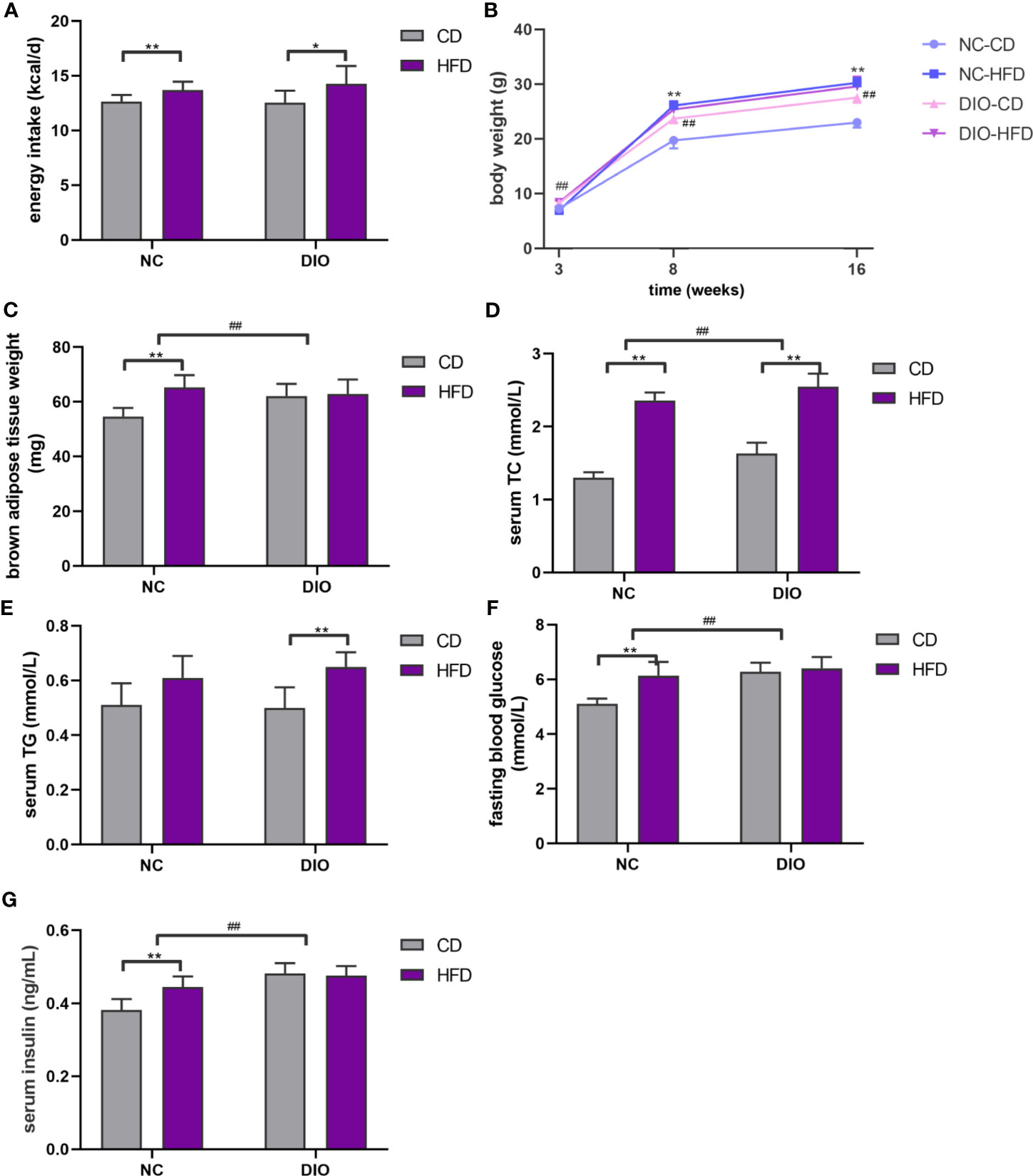
Figure 3 Offspring characteristics. (A) Energy intake, (B) body weight, (C) brown adipose tissue weight (BAT), (D) serum total cholesterol (TC), (E) serum triglyceride (TG), (F) fasting blood glucose (FBG), (G) serum insulin. Values represent the mean ± SEM, n = 8 each group. P values represent significance in the main effect for each source of variation (diet or prenatal exposure) as calculated by two-way ANOVA: *P < 0.05, **P < 0.01 offspring diet effect, ##P < 0.01 maternal diet effect. NC, normal control; DIO, diet-induced obesity; CD, control diet; HFD, high-fat diet.
The Effect of Maternal HFD on the Serum Lipid Profile in Male Offspring
Serum TC levels were 1.82-fold higher in the NC-HFD offspring than in the NC-CD offspring (diet exposure P < 0.01, Figure 3D). Furthermore, DIO-HFD offspring exhibited more hyperlipidemia than NC-CD offspring (1.96-fold, diet exposure P < 0.01, prenatal exposure P < 0.01, Figure 3D). However, elevated serum TG levels were observed only in the DIO-HFD group, compared with those in the DIO-CD group (diet exposure P < 0.01, Figure 3E).
The Effect of Maternal HFD on the Fasting Blood Glucose and Serum Insulin in Male Offspring
Mice in NC-HFD had higher fasting blood glucose (FBG) and serum insulin than those of NC-CD group (diet exposure P < 0.01, Figures 3F, G). And FBG and serum insulin in DIO-CD offspring was also higher than that in NC-CD group (prenatal exposure P < 0.01, Figures 3F, G).
The Effect of Maternal HFD on the Morphology of BAT in Male Offspring
H&E stained slides showed that the NC-HFD, DIO-CD, and DIO-HFD groups had large brown adipocytes and white-like adipocytes with unilocular lipid droplets (Figure 4A). The fat area of the BAT in both HFD groups was 2.09-fold and 1.23-fold higher than that in the CD group (diet exposure P < 0.01, Figure 4B). DIO-HFD offspring had a 2.52-fold increase in the fat area of BAT compared with NC-CD offspring (diet exposure P < 0.01, prenatal exposure P < 0.01, Figure 4B).

Figure 4 The effect of maternal HFD disrupts the morphology of brown adipose tissue (BAT) in male offspring. (A) H&E staining (magnification 400 x), (B) lipid area analysis (lipid area/total adipocyte area, %) of BAT (n = 8 for each group). Values represent the mean ± SEM, n = 8 each group. P values represent significance in the main effect for each source of variation (diet or prenatal exposure) as calculated by two-way ANOVA: **P < 0.01 offspring diet effect, ##P < 0.01 maternal diet effect. NC, normal control; DIO, diet-induced obesity; CD, control diet; HFD, high-fat diet.
The Effect of Maternal HFD on Genome Methylation in BAT of Male Offspring
DNA methylation array data were uploaded to NCBI’s Gene Expression Omnibus repository (GEO) under the series accession number GSE173218 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE173218). An overall comparison of the DNA methylation of BAT in the DIO-CD and NC-CD groups was performed. In total, 937 regions on 21 chromosomes were differentially methylated in the DIO-CD group compared with the NC-CD group, particularly on chromosomes 4, 5, 6, 11, and 17 (Figure 5A). Of these differentially methylated regions (DMRs), 554 were hypermethylated (59.12%) and 383 were hypomethylated (40.88%) of the DMRs (Supplementary Table 1). Five hundred sixty-two DMRs (59.98%) were located in HCP, 247 DMRs (26.36%) were located in ICP, and 128 DMRs (13.66%) were located in LCP (Figure 5B).
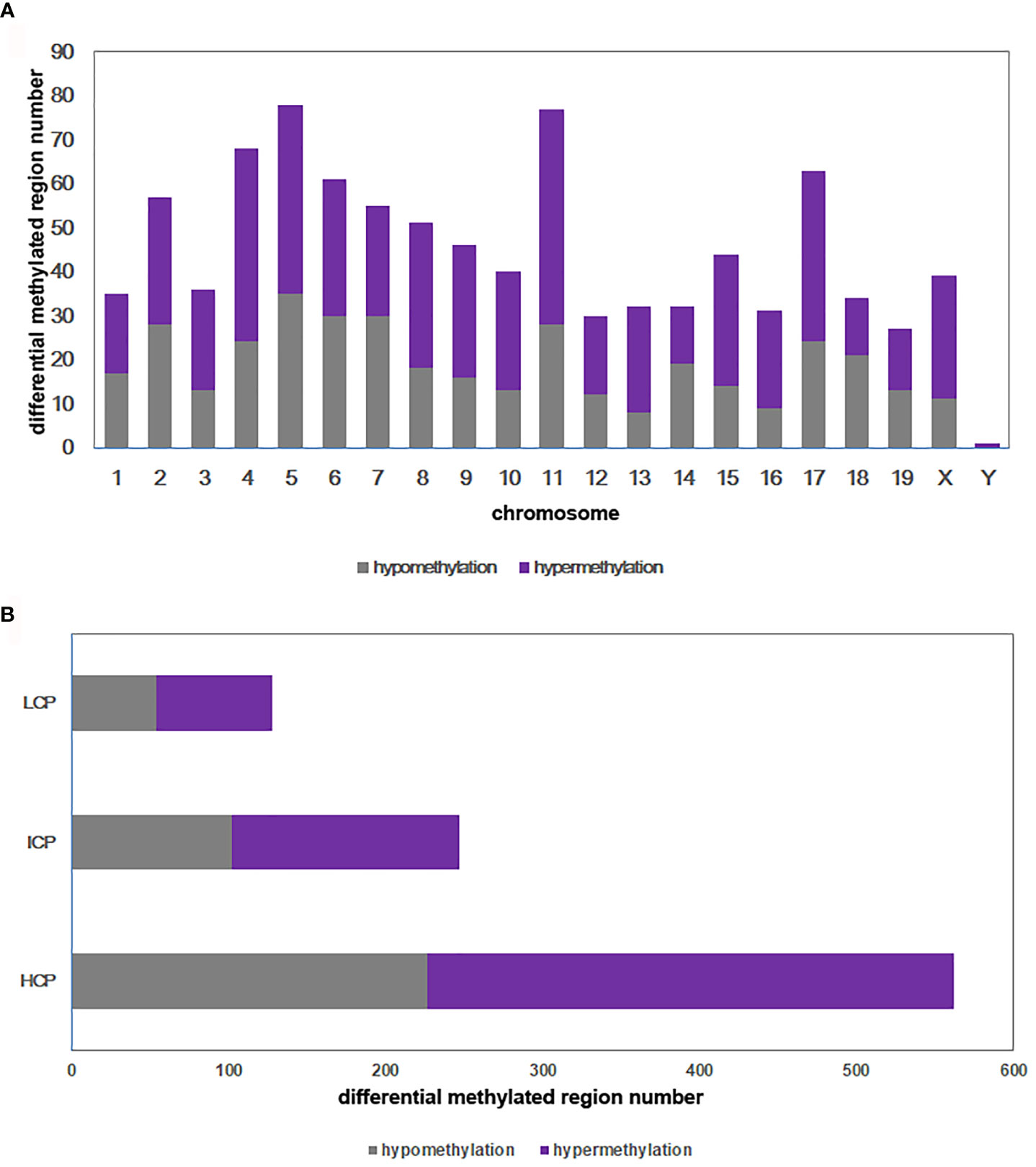
Figure 5 Differential methylated regions between DIO-CD group and NC-CD group. (A) Chromosomal distribution of differentially methylated regions. (B) CpG density of differentially methylated regions. Classification of all regions with high (HCP), intermediated (ICP), and low (LCP) CpG content.
The Effect of Maternal HFD on BAT Methylated Gene Functional Clusters in Male Offspring
To examine the biological function of differentially methylated genes in BAT from offspring exposed to maternal HFD, GO and KEGG enrichment analyses were performed. Table 3 presents the top five prevalent GO terms for differentially methylated genes. The top five significant biological process (BP) GO terms were transcription, negative regulation of cell proliferation, protein ubiquitination, negative regulation of transcription from the RNA polymerase II promoter, and regulation of transcription. Table 4 presents the enriched KEGG pathways for differentially methylated genes. The relationship of this enriched KEGG pathways was shown in Figure 6. The pathway connections in this figure were based on the KEGG database. The differentially methylated genes were most significantly enriched in the Hippo signaling pathway, Jak-Stat signaling pathway, nucleotide excision repair, Wnt signaling pathway, axon guidance, adipocytokine signaling pathway, purine metabolism, basal transcription factors, AMPK signaling pathway, and oocyte meiosis.

Table 3 The enriched GO terms with differentially methylated genes in DIO-CD offspring vs NC-CD offspring (P < 0.01).
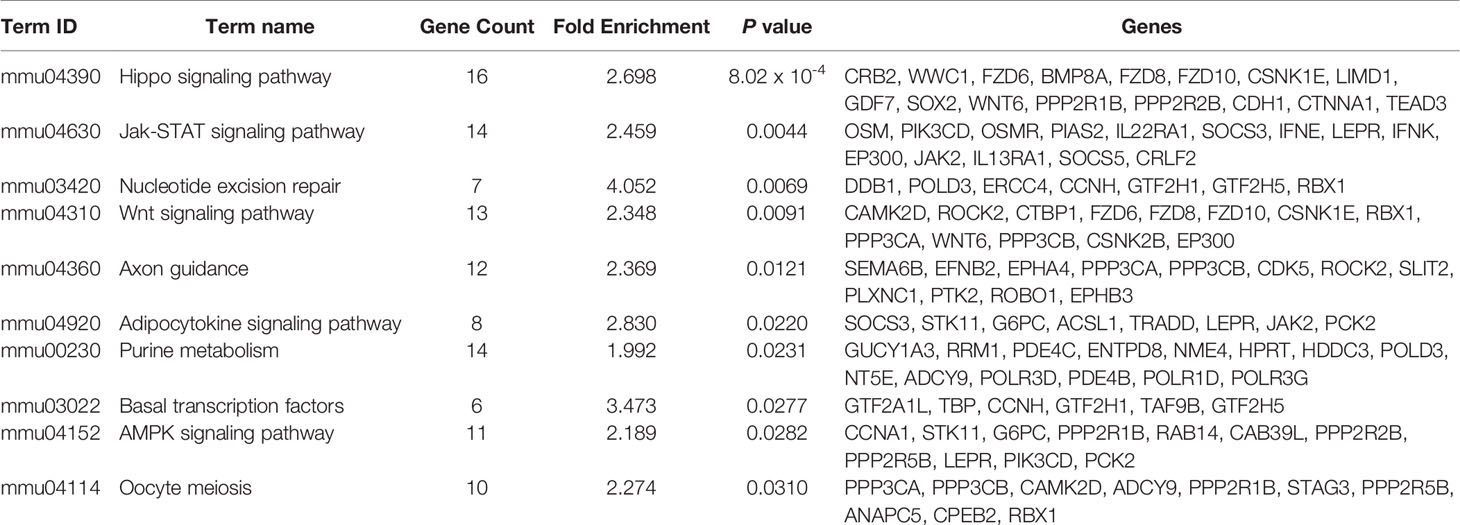
Table 4 The enriched KEGG pathway with differentially methylated genes in DIO-CD offspring vs NC-CD offspring (P < 0.05).
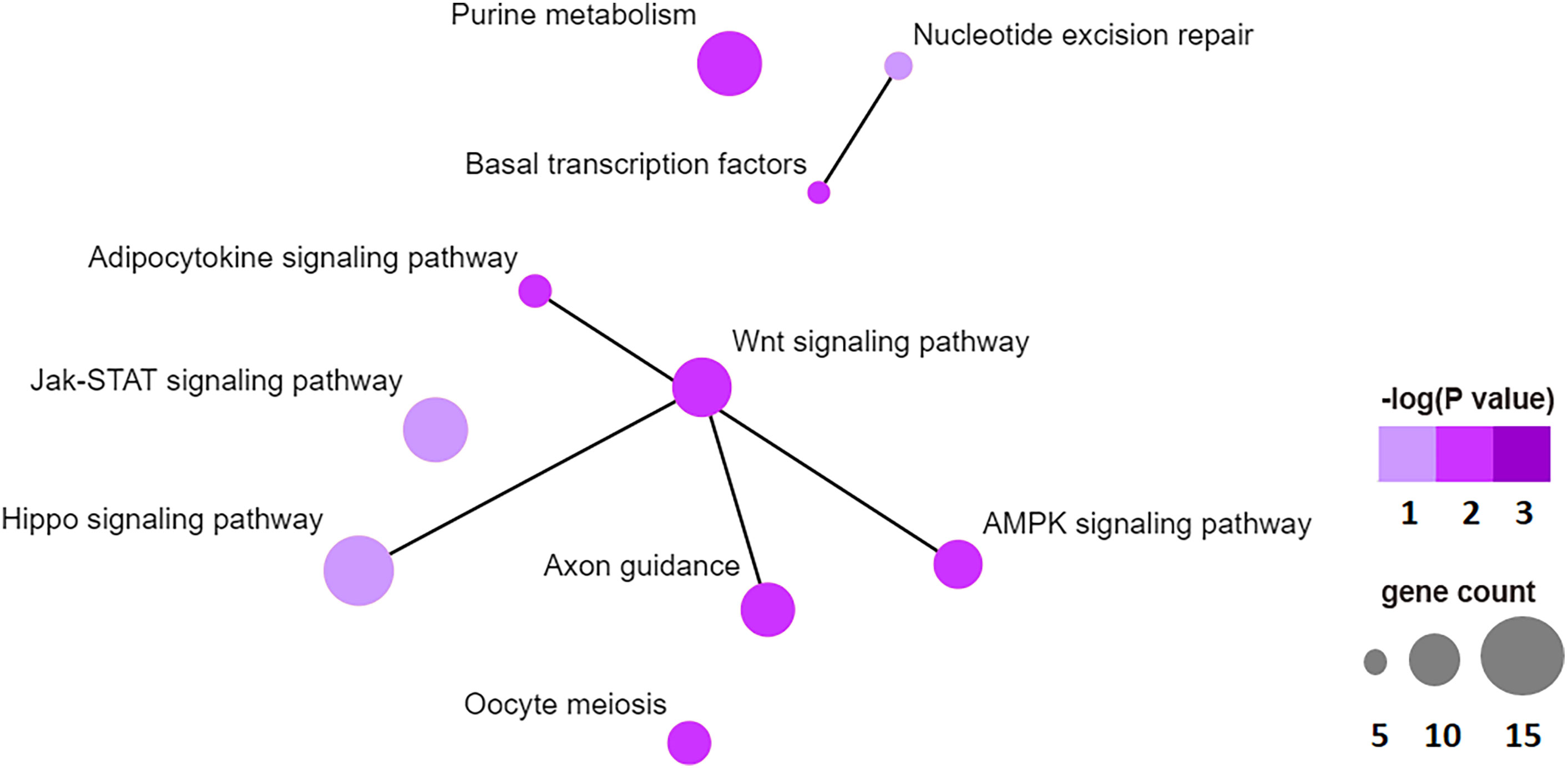
Figure 6 Top 10 significant enriched KEGG pathway network, like hippo signaling pathway, Jak-STAT signaling pathway, nucleotide excision repair, WNT signaling pathway, axon guidance, adipocytokine signaling pathway, purine metabolism, basal transcription factors, AMPK signaling pathway, oocyte meiosis.
The Effect of Maternal HFD on BAT-Specific Gene Methylation in Male Offspring by Using MeDIP qPCR
Because Acaa2, Acsl1 and Cox7a1 are related to energy metabolism, we chose these three differentially methylated genes, which were found in methylation array, to perform MeDIP qPCR analysis. Figures 7A, C, E show CpG islands in Acaa2, Acsl1 and Cox7a1. The gene methylation levels of Acaa2, Acsl1 and Cox7a1 were higher in the NC-HFD group than in the NC-CD group (diet exposure P < 0.01, Figures 7B, D, F). Interestingly, DIO-CD offspring had higher Acaa2, Acsl1 and Cox7a1 gene methylation levels than NC-CD offspring (prenatal exposure, P < 0.01, Figures 7B, D, F).
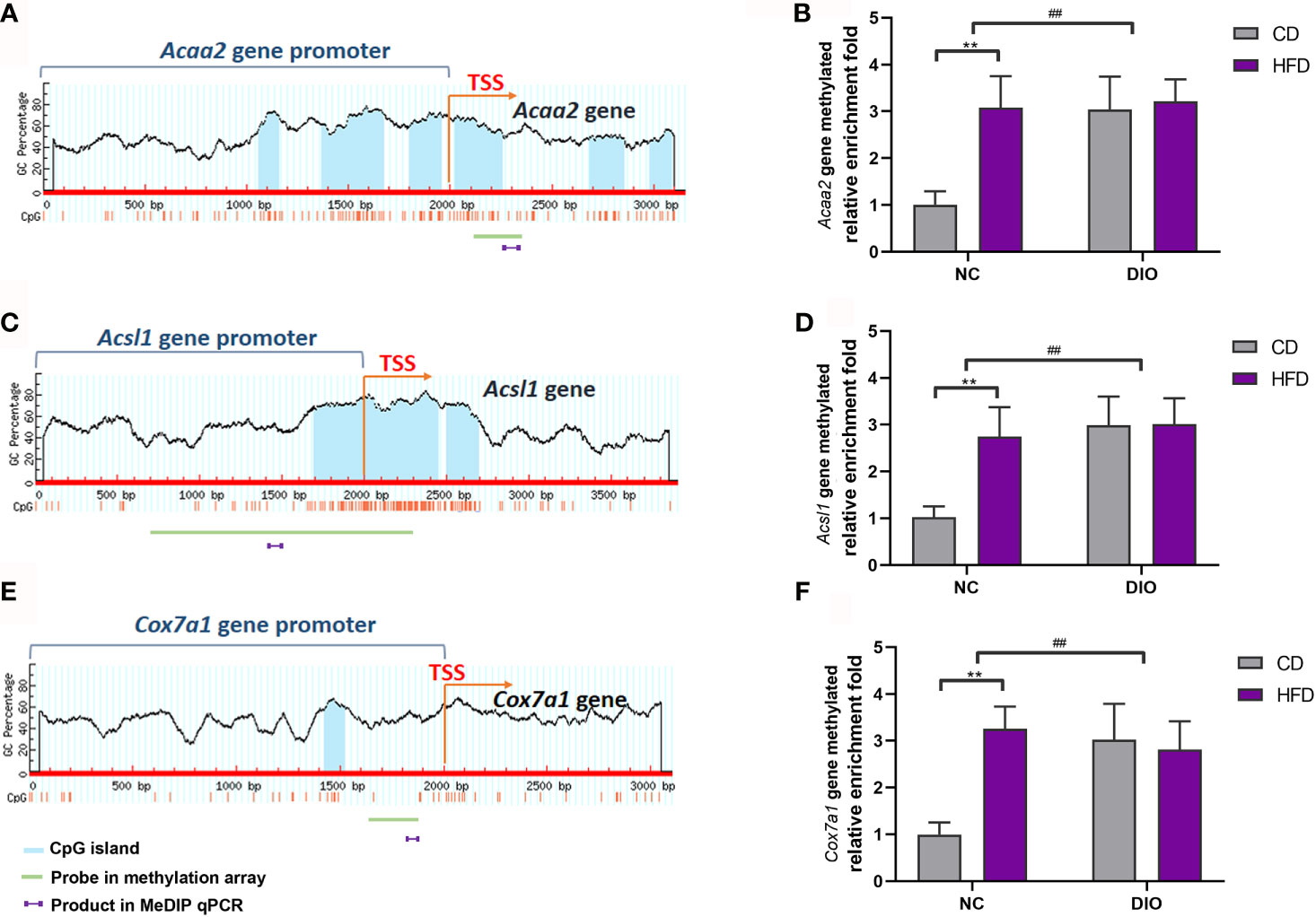
Figure 7 The effect of maternal HFD activated the gene methylation of Acaa2, Acsl1, and Cox7a1 promoter in male offspring BAT. Diagram showed the CpG island region in promoters and transcriptional starting site (TSS) shores of (A) Acaa2, (C) Acsl1, and (E) Cox7a1. The methylation changes of (B) Acaa2, (D) Acsl1, (F) Cox7a1 quantified by MeDIP-qPCR in BAT. Values represent the mean ± SEM, n = 8 each group. P values represent significance in the main effect for each source of variation (diet or prenatal exposure) as calculated by two-way ANOVA: **P < 0.01 offspring diet effect, ##P < 0.01 maternal diet effect. NC, normal control; DIO, diet-induced obesity; CD, control diet; HFD, high-fat diet.
The Effect of Maternal HFD on Gene Expression in Male Offspring BAT
The expression levels of Acaa2, Acsl1 and Cox7a1 were reduced in the NC-HFD offspring (diet exposure, P < 0.01, Figure 8) and offspring from DIO mothers (prenatal exposure, P < 0.01, Figure 8) compared to those in the NC-CD group.
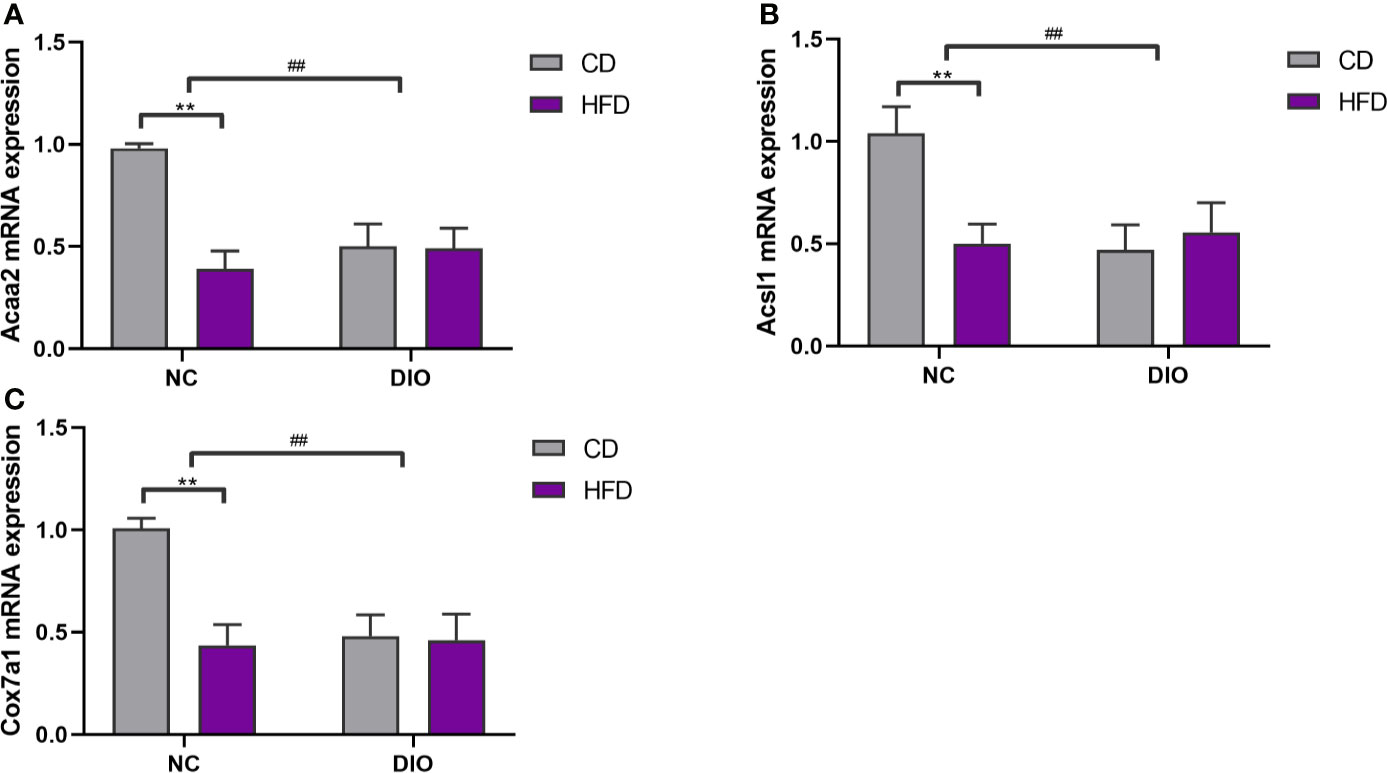
Figure 8 The effect of maternal HFD reduced (A) Acaa2, (B) Acsl1, (C) Cox7a1 expression in male offspring BAT. Values represent the mean ± SEM, n = 8 each group. P values represent significance in the main effect for each source of variation (diet or prenatal exposure) as calculated by two-way ANOVA: **P < 0.01 offspring diet effect, ##P < 0.01 maternal diet effect. NC, normal control; DIO, diet-induced obesity; CD, control diet; HFD, high-fat diet.
Discussion
In our research, although maternal HFD before and during pregnancy and lactation did not affect offspring energy intake, it increased offspring body weight at 8 and 16 weeks old. Previous studies also found that maternal HFD during this critical period elevated offspring body weight from weaning to 20 weeks old in a rodent model (38, 39). These studies highlight the long term effect of the maternal diet model on the offspring metabolic phenotype related to impaired energy metabolism. In addition, we found that pups from mothers fed a HFD had higher serum TC levels at 16 weeks old. Previous studies also found that serum TC levels were persistently higher in offspring from maternal HFD exposure than in those from maternal chow exposure at 3 weeks old (12, 40).
Moreover, our data showed that maternal HFD exposure led to an increase BAT weight in adult male offspring. In addition, the histological results of H&E staining also showed the accumulation of lipid droplets in brown adipocytes in male offspring from mothers fed a HFD. These results reveal that maternal HFD before and during pregnancy and lactation resulted in a male offspring BAT “whitening” phenotype. BAT has an important effect on energy metabolism regulation. BAT “whitening” is related to the progression of obesity. Activation of BAT is a potent target for metabolic diseases, such as diabetes and obesity (40, 41).
A previous study proved that maternal HFD induced early-onset obesity and increased lipid accumulation in BAT in weanling rats (32). The BAT of fetal mice from obese mothers had more lipid droplets, and reduced mitochondrial DNA and mitochondrial biogenesis markers (42). Offspring from mothers fed a HFD during lactation had larger brown adipocytes with the accumulation of fat droplets and increased BAT mass in adult offspring (33). Regarding the function of BAT, mitochondrial DNA mass and mitochondrial biological markers (Ndufs8, Atp5a1, and Nrf2) were reduced in the fetal BAT in mice from obese mothers (42). Moreover, maternal obesity inhibits the expression of Drp1, which is upstream from the transcription factor of energy metabolism regulation Forkhead box O1 (FoxO1) in fetal BAT (43). Interestingly, a normal diet after weaning only partially reverted the mitochondrial dysfunction caused by maternal HFD (43). Thus, our research provides proof that maternal HFD consumption induces BAT “whitening” in adult male offspring.
Some research has addressed the differences in the genomic methylation status between white adipose tissue and brown adipose tissue. One study observed dynamic DNA methylation changes during adipogenesis in white and brown adipose tissues. Their results revealed that there were more hypermethylated sites overall in white adipose tissue than in brown adipose tissue. Hypermethylated sites were mostly located in intronic and intergenic regions in white adipose tissue. However, hypomethylated sites in brown adipose were mostly in exonic regions. These hypomethylated promoters play a key role in the transcription of genes involved in brown fat functions, such as the mitochondrial respiratory chain and fatty acid oxidation (44). Decreased uncoupling protein-1 (UCP-1) enhancer CpG methylation is associated with high BAT-specific expression of UCP-1 (45).
Increasing evidence suggests epigenetic modification is a key mechanism in gestation and lactation programming in offspring (46, 47). Among these epigenetic mechanisms, DNA methylation has been proven in several intrauterine adverse environmental exposure models (47–50). In a rodent intrauterine growth retardation model, DNA methylation alterations were found in several key transcription factors, such as peroxisome proliferator-activated receptor-α (PPARα), glucocorticoid receptor (GR), pancreatic duodenal homeobox-1 (PDX-1), and hepatocyte nuclear factor-α (HNF-α) (46, 49, 50). In addition, maternal HFD affects the DNA methylation status of multiple tissues in offspring, including the pancreas, hypothalamus, muscle and liver (48, 51–54). Moreover, the maternal nutrition status also affects DNA methylation in adipose tissues (55, 56). Maternal obesity alters the DNA methylation of key pro-adipogenic genes, including Zfp423 and C/EBP-β, in the WAT in offspring (57). Yu et al. found increased DNA methylation of BAT-specific genes, including Ucp1, cytochrome c oxidase subunit (Cox5b), and fatty acid elongase 3 (Elovl3), in offspring mice from mothers with streptozotocin-induced diabetes (58). Excessive maternal glucocorticoids during pregnancy inhibited Ppargc1a expression through promoter DNA hypermethylation, which impaired fetal BAT function (59). In our study, we did not found Ucp1, Cox5b, Elovl3, and Ppargc1a methylation status changes in DIO-CD mice, partly because of the different maternal exposure and modeling method.
Some fatty acid oxidation (FAO)-related genes were hypermethylated in BAT from DOI-CD offspring compared to the NC-CD group. BAT mitochondria provide energy basis for thermogenesis. In mitochondria, fatty acids are a source of oxidative fuel. Unlike white adipocytes, brown adipocytes have several specific proteins that participate in FAO, such as Acsl1 and Acaa2. FAO function is important for the activation and maintenance of BAT thermogenic programming (60). Enzymes involved in FAO are the switches of thermogenesis (61). After fatty acids are transported in the mitochondria, they are catalyzed to fatty acyl-CoAs by acyl-CoA synthetases, such as Acsl1. Then, long-chain fatty acids are transported into the mitochondrial matrix by carnitine palmitoyltransferase. In the mitochondrial matrix, acyl-CoAs are activated to acetyl-CoAs by acyl-CoA dehydrogenases. Finally, acetyl-CoA products participate in the citric acid cycle and electron transport chain. A previous study found that short term cold stimulation increased BAT-specific FAO-related gene expression in subcutaneous WAT (62). The Acaa2 and Acsl1 expression levels were lower in BAT of G protein-coupled receptor 120 (GPR120)-deficient mice than in BAT of normal mice (63). GPR120 is highly expressed in the BAT and cold exposure further increases its expression in BAT of mice (64). HFD-fed GPR120-deficient mice are more prone to obesity and fatty liver than wide-type mice (65). Cold exposure increases BAT-specific mitochondrial FAO enzyme expression in subcutaneous WAT, including Acaa2 (62). Acsl1 is also essential for the synthesis of triacylglycerol, mitochondrial function and FAO activity in brown adipocytes (66). Acsl1-deficient mice showed an obese phenotype and severe cold intolerance (67). We found that maternal HFD activated Acaa2 and Acsl1 methylation, and inhibited their expression in BAT from male offspring. This result reflects that maternal HFD-induced obesity in male offspring obesity is regarded to the inhibitory action of BAT fatty acid oxidation.
As a terminal and rate-limiting enzyme of the respiratory chain, COX7A1 is expressed in tissues that are rich in mitochondria and high in aerobic capacity in mammals, such as the heart, skeletal muscle, and BAT (68, 69). Similar to UCP1, COX also mediates BAT thermogenesis function through translocation from coupled protons to uncoupled protons in mitochondria. Cold stimulation strongly increased COX7A1 protein expression in rodent BAT (70). Cox7a1 is a thermogenic gene, especially in a cold-responsive pattern. During cold-exposure, C57BL/6J wild-type mice showed increased COX activity and norepinephrine-induced heat production, but were unaffected in Cox7a1 knockout mice (71). Cox7a1 expression was increased in BAT compared with WAT. It is involved in activating brown adipocytes (72). Depletion of BAT in a rodent model increased the likelihood of HFD-induced obesity (73). Our results showed that male offspring mice from HFD dams have higher Cox7a1 gene methylation levels, and lower Cox7a1 expression. These results indicate that maternal HFD impairs the BAT thermogenic program in offspring.
Conclusion
In summary, our data suggested that maternal HFD consumption before and during pregnancy and lactation disrupts BAT structure and function. This is related to the activated methylation of Acaa2, Acsl1, and Cox7a1 promoters. The hypermethylation of these key BAT-specific genes disturbs BAT FAO and thermogenesis and increases the risks of obesity and metabolic dysfunctions in male offspring (Figure 9). All of these findings uncover the transgenerational effects of maternal HFD in BAT. Specifically targeting BAT intervention during the early stage of life may become a promising strategy for metabolic diseases.
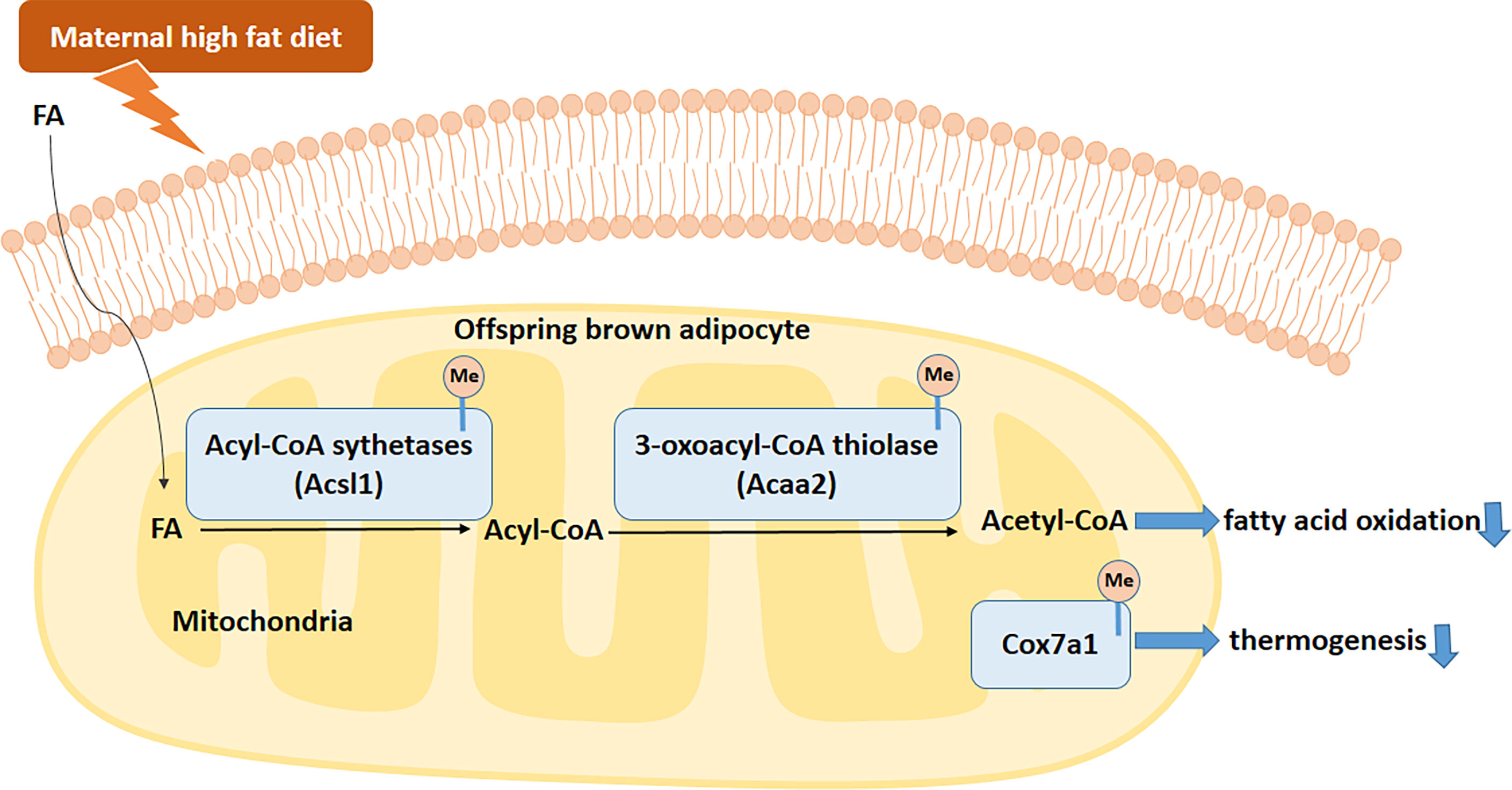
Figure 9 Proposed mechanism by which maternal HFD disrupts brown adipose tissue (BAT) fatty acid oxidation (FAO) and thermogenesis in male offspring. Fatty acids (FAs) enter brown adipocytes and are activated by acyl-CoA sythetases, including Acsl1, to form acyl-CoAs. Then acyl-CoAs were catabolized to acetyl-CoAs by acyl-CoA dehydrogenases, such as Acaa2. Maternal HFD active the methylation of offspring BAT FAO related enzymes, such as Acsl1 and Acaa2. Meanwhile, Cox7a1 gene methylation in offspring BAT was also active by maternal HFD. To sum up, maternal HFD disturbs the BAT FAO and thermogenesis through activated specific gene DNA methylation. FA, fatty acid.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
All experimental protocols were approved by the Animal Care Committee of Peking Union Medical Hospital (Permit Number: XHDW-2015-0051).
Author Contributions
XX designed the experiments, contributed reagents and materials. QZ, JZ, TW, and XW conducted the experiments. MY, ML, and FP analyzed the data. QZ wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the grants from National Natural Science Foundation of China (No. 81870579, 81870545, 81570715, 81170736), Beijing Natural Science Foundation (7202163), Beijing Natural Science Foundation (7202163), Beijing Municipal Science & Technology Commission (Z201100005520011), National Key R&D Program of China (2017YFC1309603), National Key Research and Development Program of China (2016YFA0101002, 2018YFC2001100), Scientific Activities Foundation for Selected Returned Overseas Professionals of Human Resources and Social Security Ministry, Beijing Dongcheng District Outstanding Talent Funding Project (2019DCT-M-05), Medical Epigenetics Research Center, Chinese Academy of Medical Sciences (2017PT31036, 2018PT31021), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (No. 2017PT32020, No. 2018PT32001), Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (CIFMS2017-I2M-1-008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.705827/full#supplementary-material
References
1. Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, Regional, and National Prevalence of Overweight and Obesity in Children and Adults During 1980-2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet (2014) 384(9945):766–81. doi: 10.1016/s0140-6736(14)60460-8
3. Gallagher EJ, LeRoith D. Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality. Physiol Rev (2015) 95(3):727–48. doi: 10.1152/physrev.00030.2014
4. Koppe SW. Obesity and the Liver: Nonalcoholic Fatty Liver Disease. Transl Res (2014) 164(4):312–22. doi: 10.1016/j.trsl.2014.06.008
5. Goodwin PJ, Stambolic V. Impact of the Obesity Epidemic on Cancer. Annu Rev Med (2015) 66:281–96. doi: 10.1146/annurev-med-051613-012328
6. Lisboa PC, de Oliveira E, Manhães AC, Santos-Silva AP, Pinheiro CR, Younes-Rapozo V, et al. Effects of Maternal Nicotine Exposure on Thyroid Hormone Metabolism and Function in Adult Rat Progeny. J Endocrinol (2015) 224(3):315–25. doi: 10.1530/joe-14-0473
7. Weng SF, Redsell SA, Nathan D, Swift JA, Yang M, Glazebrook C. Estimating Overweight Risk in Childhood From Predictors During Infancy. Pediatrics (2013) 132(2):e414–21. doi: 10.1542/peds.2012-3858
8. O'Reilly JR, Reynolds RM. The Risk of Maternal Obesity to the Long-Term Health of the Offspring. Clin Endocrinol (Oxf) (2013) 78(1):9–16. doi: 10.1111/cen.12055
9. Biro FM, Wien M. Childhood Obesity and Adult Morbidities. Am J Clin Nutr (2010) 91(5):1499s–505s. doi: 10.3945/ajcn.2010.28701B
10. Bayol SA, Farrington SJ, Stickland NC. A Maternal 'Junk Food' Diet in Pregnancy and Lactation Promotes an Exacerbated Taste for 'Junk Food' and a Greater Propensity for Obesity in Rat Offspring. Br J Nutr (2007) 98(4):843–51. doi: 10.1017/s0007114507812037
11. Franco JG, Fernandes TP, Rocha CP, Calviño C, Pazos-Moura CC, Lisboa PC, et al. Maternal High-Fat Diet Induces Obesity and Adrenal and Thyroid Dysfunction in Male Rat Offspring at Weaning. J Physiol (2012) 590(21):5503–18. doi: 10.1113/jphysiol.2012.240655
12. Guo Y, Wang Z, Chen L, Tang L, Wen S, Liu Y, et al. Diet Induced Maternal Obesity Affects Offspring Gut Microbiota and Persists Into Young Adulthood. Food Funct (2018) 9(8):4317–27. doi: 10.1039/c8fo00444g
13. Paul HA, Collins KH, Bomhof MR, Vogel HJ, Reimer RA. Potential Impact of Metabolic and Gut Microbial Response to Pregnancy and Lactation in Lean and Diet-Induced Obese Rats on Offspring Obesity Risk. Mol Nutr Food Res (2018) 62(4):1700820. doi: 10.1002/mnfr.201700820
14. Grandjean P, Barouki R, Bellinger DC, Casteleyn L, Chadwick LH, Cordier S, et al. Life-Long Implications of Developmental Exposure to Environmental Stressors: New Perspectives. Endocrinology (2015) 156(10):3408–15. doi: 10.1210/en.2015-1350
15. Gluckman PD, Hanson MA, Buklijas T. A Conceptual Framework for the Developmental Origins of Health and Disease. J Dev Origins Health Dis (2010) 1(1):6–18. doi: 10.1017/s2040174409990171
16. Almeida DL, Pavanello A, Saavedra LP, Pereira TS, de Castro-Prado MAA, de Freitas Mathias PC. Environmental Monitoring and the Developmental Origins of Health and Disease. J Dev origins Health Dis (2019) 10(6):608–15. doi: 10.1017/s2040174419000151
17. Moutinho C, Esteller M. MicroRNAs and Epigenetics. Adv Cancer Res (2017) 135:189–220. doi: 10.1016/bs.acr.2017.06.003
18. Zanger UM, Klein K, Kugler N, Petrikat T, Ryu CS. Epigenetics and MicroRNAs in Pharmacogenetics. Adv Pharmacol (2018) 83:33–64. doi: 10.1016/bs.apha.2018.02.003
19. Gibney ER, Nolan CM. Epigenetics and Gene Expression. Heredity (Edinb) (2010) 105(1):4–13. doi: 10.1038/hdy.2010.54
20. Obri A, Serra D, Herrero L, Mera P. The Role of Epigenetics in the Development of Obesity. Biochem Pharmacol (2020) 177:113973. doi: 10.1016/j.bcp.2020.113973
21. Lee YS. Genetics of Nonsyndromic Obesity. Curr Opin Pediatr (2013) 25(6):666–73. doi: 10.1097/MOP.0b013e3283658fba
22. Balistreri CR, Caruso C, Candore G. The Role of Adipose Tissue and Adipokines in Obesity-Related Inflammatory Diseases. Mediators Inflamm (2010) 2010:802078. doi: 10.1155/2010/802078
23. Gesta S, Tseng YH, Kahn CR. Developmental Origin of Fat: Tracking Obesity to Its Source. Cell (2007) 131(2):242–56. doi: 10.1016/j.cell.2007.10.004
24. Park A, Kim WK, Bae KH. Distinction of White, Beige and Brown Adipocytes Derived From Mesenchymal Stem Cells. World J Stem Cells (2014) 6(1):33–42. doi: 10.4252/wjsc.v6.i1.33
25. Osuna-Prieto FJ, Martinez-Tellez B, Sanchez-Delgado G, Aguilera CM, Lozano-Sánchez J, Arráez-Román D, et al. Activation of Human Brown Adipose Tissue by Capsinoids, Catechins, Ephedrine, and Other Dietary Components: A Systematic Review. Adv Nutr (2019) 10(2):291–302. doi: 10.1093/advances/nmy067
26. Mendes-da-Silva C, Giriko C, Mennitti LV, Hosoume LF, Souto Tdos S, Silva AV. Maternal High-Fat Diet During Pregnancy or Lactation Changes the Somatic and Neurological Development of the Offspring. Arq Neuropsiquiatr (2014) 72(2):136–44. doi: 10.1590/0004-282x20130220
27. Jawerbaum A, White V. Review on Intrauterine Programming: Consequences in Rodent Models of Mild Diabetes and Mild Fat Overfeeding Are Not Mild. Placenta (2017) 52:21–32. doi: 10.1016/j.placenta.2017.02.009
28. Nguyen LT, Saad S, Tan Y, Pollock C, Chen H. Maternal High-Fat Diet Induces Metabolic Stress Response Disorders in Offspring Hypothalamus. J Mol Endocrinol (2017) 59(1):81–92. doi: 10.1530/jme-17-0056
29. de Paula Simino LA, de Fante T, Figueiredo Fontana M, Oliveira Borges F, Torsoni MA, Milanski M, et al. Lipid Overload During Gestation and Lactation Can Independently Alter Lipid Homeostasis in Offspring and Promote Metabolic Impairment After New Challenge to High-Fat Diet. Nutr Metab (Lond) (2017) 14:16. doi: 10.1186/s12986-017-0168-4
30. White CL, Purpera MN, Morrison CD. Maternal Obesity Is Necessary for Programming Effect of High-Fat Diet on Offspring. Am J Physiol Regulatory Integr Comp Physiol (2009) 296(5):R1464–72. doi: 10.1152/ajpregu.91015.2008
31. Xiao XQ, Williams SM, Grayson BE, Glavas MM, Cowley MA, Smith MS, et al. Excess Weight Gain During the Early Postnatal Period Is Associated With Permanent Reprogramming of Brown Adipose Tissue Adaptive Thermogenesis. Endocrinology (2007) 148(9):4150–9. doi: 10.1210/en.2007-0373
32. Almeida MM, Dias-Rocha CP, Souza AS, Muros MF, Mendonca LS, Pazos-Moura CC, et al. Perinatal Maternal High-Fat Diet Induces Early Obesity and Sex-Specific Alterations of the Endocannabinoid System in White and Brown Adipose Tissue of Weanling Rat Offspring. Br J Nutr (2017) 118(10):788–803. doi: 10.1017/s0007114517002884
33. Liang X, Yang Q, Zhang L, Maricelli JW, Rodgers BD, Zhu MJ, et al. Maternal High-Fat Diet During Lactation Impairs Thermogenic Function of Brown Adipose Tissue in Offspring Mice. Sci Rep (2016) 6:34345. doi: 10.1038/srep34345
34. Horai Y, Kakimoto T, Takemoto K, Tanaka M. Quantitative Analysis of Histopathological Findings Using Image Processing Software. J Toxicol Pathol (2017) 30(4):351–8. doi: 10.1293/tox.2017-0031
35. Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, Silencing Potential and Evolutionary Impact of Promoter DNA Methylation in the Human Genome. Nat Genet (2007) 39(4):457–66. doi: 10.1038/ng1990
36. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol (2003) 4(5):P3. doi: 10.1186/gb-2003-4-5-p3
37. Li LC, Dahiya R. MethPrimer: Designing Primers for Methylation PCRs. Bioinf (Oxford England) (2002) 18(11):1427–31. doi: 10.1093/bioinformatics/18.11.1427
38. Gali Ramamoorthy T, Allen TJ, Davies A, Harno E, Sefton C, Murgatroyd C, et al. Maternal Overnutrition Programs Epigenetic Changes in the Regulatory Regions of Hypothalamic Pomc in the Offspring of Rats. Int J Obes (Lond) (2018) 42(8):1431–44. doi: 10.1038/s41366-018-0094-1
39. Zheng J, Xiao X, Zhang Q, Yu M, Xu J, Wang Z. Maternal High-Fat Diet Modulates Hepatic Glucose, Lipid Homeostasis and Gene Expression in the PPAR Pathway in the Early Life of Offspring. Int J Mol Sci (2014) 15(9):14967–83. doi: 10.3390/ijms150914967
40. Lima MS, Perez GS, Morais GL, Santos LS, Cordeiro GS, Couto RD, et al. Effects of Maternal High Fat Intake During Pregnancy and Lactation on Total Cholesterol and Adipose Tissue in Neonatal Rats. Braz J Biol (2018) 78(4):615–8. doi: 10.1590/1519-6984.166788
41. Roman S, Agil A, Peran M, Alvaro-Galue E, Ruiz-Ojeda FJ, Fernández-Vázquez G, et al. Brown Adipose Tissue and Novel Therapeutic Approaches to Treat Metabolic Disorders. Transl Res (2015) 165(4):464–79. doi: 10.1016/j.trsl.2014.11.002
42. Wang H, Chen Y, Mao X, Du M. Maternal Obesity Impairs Fetal Mitochondriogenesis and Brown Adipose Tissue Development Partially via Upregulation of miR-204-5p. Biochim Biophys Acta Mol Basis Dis (2019) 1865(10):2706–15. doi: 10.1016/j.bbadis.2019.07.012
43. Lettieri Barbato D, Tatulli G, Vegliante R, Cannata SM, Bernardini S, Ciriolo MR, et al. Dietary Fat Overload Reprograms Brown Fat Mitochondria. Front Physiol (2015) 6:272. doi: 10.3389/fphys.2015.00272
44. Lim YC, Chia SY, Jin S, Han W, Ding C, Sun L. Dynamic DNA Methylation Landscape Defines Brown and White Cell Specificity During Adipogenesis. Mol Metab (2016) 5(10):1033–41. doi: 10.1016/j.molmet.2016.08.006
45. Shore A, Karamitri A, Kemp P, Speakman JR, Lomax MA. Role of Ucp1 Enhancer Methylation and Chromatin Remodelling in the Control of Ucp1 Expression in Murine Adipose Tissue. Diabetologia (2010) 53(6):1164–73. doi: 10.1007/s00125-010-1701-4
46. Lillycrop KA, Burdge GC. Epigenetic Changes in Early Life and Future Risk of Obesity. Int J Obes (Lond) (2011) 35(1):72–83. doi: 10.1038/ijo.2010.122
47. Heerwagen MJ, Miller MR, Barbour LA, Friedman JE. Maternal Obesity and Fetal Metabolic Programming: A Fertile Epigenetic Soil. Am J Physiol Regulatory Integr Comp Physiol (2010) 299(3):R711–22. doi: 10.1152/ajpregu.00310.2010
48. Aagaard-Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, et al. Developmental Origins of Disease and Determinants of Chromatin Structure: Maternal Diet Modifies the Primate Fetal Epigenome. J Mol Endocrinol (2008) 41(2):91–102. doi: 10.1677/jme-08-0025
49. Sandovici I, Smith NH, Nitert MD, Ackers-Johnson M, Uribe-Lewis S, Ito Y, et al. Maternal Diet and Aging Alter the Epigenetic Control of a Promoter-Enhancer Interaction at the Hnf4a Gene in Rat Pancreatic Islets. Proc Natl Acad Sci U S A (2011) 108(13):5449–54. doi: 10.1073/pnas.1019007108
50. Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of Type 2 Diabetes Following Intrauterine Growth Retardation in Rats Is Associated With Progressive Epigenetic Silencing of Pdx1. J Clin Invest (2008) 118(6):2316–24. doi: 10.1172/jci33655
51. Dudley KJ, Sloboda DM, Connor KL, Beltrand J, Vickers MH. Offspring of Mothers Fed a High Fat Diet Display Hepatic Cell Cycle Inhibition and Associated Changes in Gene Expression and DNA Methylation. PLoS One (2011) 6(7):e21662. doi: 10.1371/journal.pone.0021662
52. Han J, Xu J, Long YS, Epstein PN, Liu YQ. Rat Maternal Diabetes Impairs Pancreatic Beta-Cell Function in the Offspring. Am J Physiol Endocrinol Metab (2007) 293(1):E228–36. doi: 10.1152/ajpendo.00479.2006
53. Steculorum SM, Bouret SG. Maternal Diabetes Compromises the Organization of Hypothalamic Feeding Circuits and Impairs Leptin Sensitivity in Offspring. Endocrinology (2011) 152(11):4171–9. doi: 10.1210/en.2011-1279
54. Tawfik SH, Haiba MM, Saad MI, Abdelkhalek TM, Hanafi MY, Kamel MA. Intrauterine Diabetic Milieu Instigates Dysregulated Adipocytokines Production in F1 Offspring. J Anim Sci Technol (2017) 59:1. doi: 10.1186/s40781-016-0125-1
55. Su R, Yan J, Yang H. Transgenerational Glucose Intolerance of Tumor Necrosis Factor With Epigenetic Alteration in Rat Perirenal Adipose Tissue Induced by Intrauterine Hyperglycemia. J Diabetes Res (2016) 2016:4952801. doi: 10.1155/2016/4952801
56. Dong HP, Tan MZ, Liu QJ, Wang J, Zhong SB. The Study on the Effect of Hyperglycemia on Offspring Fatty Tissue Metabolism During Pregnancy. Eur Rev Med Pharmacol Sci (2017) 21(16):3658–64.
57. Borengasser SJ, Zhong Y, Kang P, Lindsey F, Ronis MJ, Badger TM, et al. Maternal Obesity Enhances White Adipose Tissue Differentiation and Alters Genome-Scale DNA Methylation in Male Rat Offspring. Endocrinology (2013) 154(11):4113–25. doi: 10.1210/en.2012-2255
58. Yu DQ, Lv PP, Yan YS, Xu GX, Sadhukhan A, Dong S, et al. Intrauterine Exposure to Hyperglycemia Retards the Development of Brown Adipose Tissue. FASEB J (2019) 33(4):5425–39. doi: 10.1096/fj.201801818R
59. Chen YT, Hu Y, Yang QY, Son JS, Liu XD, de Avila JM, et al. Excessive Glucocorticoids During Pregnancy Impair Fetal Brown Fat Development and Predispose Offspring to Metabolic Dysfunctions. Diabetes (2020) 69(8):1662–74. doi: 10.2337/db20-0009
60. Gonzalez-Hurtado E, Lee J, Choi J, Wolfgang MJ. Fatty Acid Oxidation Is Required for Active and Quiescent Brown Adipose Tissue Maintenance and Thermogenic Programing. Mol Metab (2018) 7:45–56. doi: 10.1016/j.molmet.2017.11.004
61. Guerra C, Koza RA, Walsh K, Kurtz DM, Wood PA, Kozak LP. Abnormal Nonshivering Thermogenesis in Mice With Inherited Defects of Fatty Acid Oxidation. J Clin Invest (1998) 102(9):1724–31. doi: 10.1172/jci4532
62. Nakamura Y, Sato T, Shiimura Y, Miura Y, Kojima M. FABP3 and Brown Adipocyte-Characteristic Mitochondrial Fatty Acid Oxidation Enzymes Are Induced in Beige Cells in a Different Pathway From UCP1. Biochem Biophys Res Commun (2013) 441(1):42–6. doi: 10.1016/j.bbrc.2013.10.014
63. Schilperoort M, van Dam AD, Hoeke G, Shabalina IG, Okolo A, Hanyaloglu AC, et al. The GPR120 Agonist TUG-891 Promotes Metabolic Health by Stimulating Mitochondrial Respiration in Brown Fat. EMBO Mol Med (2018) 10(3):e8047. doi: 10.15252/emmm.201708047
64. Rosell M, Kaforou M, Frontini A, Okolo A, Chan YW, Nikolopoulou E, et al. Brown and White Adipose Tissues: Intrinsic Differences in Gene Expression and Response to Cold Exposure in Mice. Am J Physiol Endocrinol Metab (2014) 306(8):E945–64. doi: 10.1152/ajpendo.00473.2013
65. Ichimura A, Hirasawa A, Poulain-Godefroy O, Bonnefond A, Hara T, Yengo L, et al. Dysfunction of Lipid Sensor GPR120 Leads to Obesity in Both Mouse and Human. Nature (2012) 483(7389):350–4. doi: 10.1038/nature10798
66. Townsend KL, An D, Lynes MD, Huang TL, Zhang H, Goodyear LJ, et al. Increased Mitochondrial Activity in BMP7-Treated Brown Adipocytes, Due to Increased CPT1- and CD36-Mediated Fatty Acid Uptake. Antioxid Redox Signal (2013) 19(3):243–57. doi: 10.1089/ars.2012.4536
67. Ellis JM, Li LO, Wu PC, Koves TR, Ilkayeva O, Stevens RD, et al. Adipose Acyl-CoA Synthetase-1 Directs Fatty Acids Toward Beta-Oxidation and Is Required for Cold Thermogenesis. Cell Metab (2010) 12(1):53–64. doi: 10.1016/j.cmet.2010.05.012
68. Villani G, Greco M, Papa S, Attardi G. Low Reserve of Cytochrome C Oxidase Capacity In Vivo in the Respiratory Chain of a Variety of Human Cell Types. J Biol Chem (1998) 273(48):31829–36. doi: 10.1074/jbc.273.48.31829
69. Acín-Pérez R, Bayona-Bafaluy MP, Bueno M, Machicado C, Fernández-Silva P, Pérez-Martos A, et al. An Intragenic Suppressor in the Cytochrome C Oxidase I Gene of Mouse Mitochondrial DNA. Hum Mol Genet (2003) 12(3):329–39. doi: 10.1093/hmg/ddg021
70. Forner F, Kumar C, Luber CA, Fromme T, Klingenspor M, Mann M. Proteome Differences Between Brown and White Fat Mitochondria Reveal Specialized Metabolic Functions. Cell Metab (2009) 10(4):324–35. doi: 10.1016/j.cmet.2009.08.014
71. Maurer SF, Fromme T, Grossman LI, Hüttemann M, Klingenspor M. The Brown and Brite Adipocyte Marker Cox7a1 Is Not Required for Non-Shivering Thermogenesis in Mice. Sci Rep (2015) 5:17704. doi: 10.1038/srep17704
72. Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, et al. Transcriptional Control of Brown Fat Determination by PRDM16. Cell Metab (2007) 6(1):38–54. doi: 10.1016/j.cmet.2007.06.001
Keywords: maternal exposure, obesity, developmental programming, DNA methylation, high fat diet
Citation: Zhang Q, Xiao X, Zheng J, Li M, Yu M, Ping F, Wang T and Wang X (2021) Maternal High-Fat Diet Disturbs the DNA Methylation Profile in the Brown Adipose Tissue of Offspring Mice. Front. Endocrinol. 12:705827. doi: 10.3389/fendo.2021.705827
Received: 06 May 2021; Accepted: 30 July 2021;
Published: 08 October 2021.
Edited by:
Adam Gesing, Medical University of Lodz, PolandReviewed by:
Amanda Brandon, The University of Sydney, AustraliaPeter L. Molloy, Commonwealth Scientific and Industrial Research Organisation (CSIRO), Australia
Copyright © 2021 Zhang, Xiao, Zheng, Li, Yu, Ping, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinhua Xiao, eGlhb3hoMjAxNEB2aXAuMTYzLmNvbQ==