 Wei Long
Wei Long Fang Guo
Fang Guo Ruen Yao
Ruen Yao Ying Wang
Ying Wang Huaiyan Wang
Huaiyan Wang Bin Yu
Bin Yu Peng Xue
Peng Xue- 1Department of Medical Genetics, Affiliated Changzhou Maternal and Child Health Care Hospital, Nanjing Medical University, Changzhou, China
- 2Department of Medical Genetics and Molecular Diagnostic Laboratory, Shanghai Children’s Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 3Department of Pediatrics, Affiliated Changzhou Maternal and Child Health Care Hospital, Nanjing Medical University, Changzhou, China
- 4Department of Pediatrics, Affiliated Changzhou Children’s Hospital of Nantong University, Changzhou, China
Background: The molecular etiology and the genotype–phenotype correlation of congenital hypothyroidism (CH) remain unclear.
Methods: We performed genetic analysis in 42 newborns with CH using whole-exome sequencing. Patients were divided into a single-gene group and a multi-gene group according to the number of affected genes, or divided into a monoallelic group, a biallelic group, and an oligogenic group according to the pattern of the detected variants. The clinical characteristics were compared between groups.
Results: Thyroid dysgenesis (TD) was observed in 10 patients and goiter in 5 patients, whereas 27 patients had normal-sized gland-in-situ (GIS). We identified 58 variants in five genes in 29 patients. The genes with the most frequent variants were DUOX2 (70.7%), followed by TSHR (12.1%), DUOXA2 (10.3%), and TPO (5.2%). Variants in the genes causing dyshormonogenesis (DH) were more common than those in the genes causing TD (87.9% versus 12.1%). Among the patients with detected variants, 26 (89.7%) were harboring a single gene variant (single-gene group), which include 22 patients harboring biallelic variants (biallelic group) and four patients harboring monoallelic variants (monoallelic group). Three (10.3%) patients harbored variants in two or three genes (multi-gene group or oligogenic group). Compared with the single-gene group, the levothyroxine (L-T4) dose at 1 year of age was higher in the multi-gene group (p = 0.018). A controllable reduction in the L-T4 dose was observed in 25% of patients in the monoallelic group and 59.1% of patients in the biallelic group; however, no patients with such reduction in the L-T4 dose were observed in the oligogenic group.
Conclusions: Patients with normal-sized GIS accounted for the majority of our cohort. Genetic defects in the genes causing DH were more common than those in the genes causing TD, with biallelic variants in DUOX2 being dominant. DH might be the leading pathophysiology of CH in Chinese individuals.
Introduction
Congenital hypothyroidism (CH) is the most common neonatal endocrine disorder with an incidence in newborns from approximately 1/2,000 to 1/4,000 (1). Delayed treatment of CH might result in profound neurodevelopmental delay. A newborn screening (NBS) program has been implemented to facilitate the prompt diagnosis of CH, and levothyroxine (L-T4) has also been used in the treatment of CH. However, the etiology of CH remains unclear.
Numerous studies have identified genetic defects in patients with CH (2). According to the pathophysiology, CH is traditionally subdivided into thyroid dysgenesis (TD) and dyshormonogenesis (DH). TD, which is caused by abnormal development of the thyroid gland, has been reported to be associated with mutations in the following genes: thyroid-stimulating hormone receptor (TSHR), NK2 homeobox 1 (NKX2-1), NK2 homeobox 5 (NKX2-5), forkhead box E1 (FOXE1), paired box 8 (PAX8), and GLIS family zinc finger 3 (GLIS3) (3). DH has been associated with mutations in genes encoding known components of the thyroid hormone biosynthesis machinery (4), such as dual oxidase 2 (DUOX2), dual-oxidase maturation factor 2 (DUOXA2), thyroglobulin (TG), thyroid peroxidase (TPO), solute carrier family 5 member 5 (SLC5A5), solute carrier family 26 member 4 (SLC26A4), and iodotyrosine deiodinase (IYD) (5, 6).
To date, more than 20 genes have been reported to be involved in the pathogenesis of primary CH (2), and new genetic defects continue to be identified due to the use of efficient genetic approaches, such as target region sequencing and whole-exome sequencing (WES) (6). However, these reported genes cannot fully explain the molecular etiology of CH. In the present study, we performed screening of variants using WES and collected clinical data in a cohort of Chinese patients with CH in order to analyze the relationship between the genotype and the phenotype in CH. This is a comprehensive study that evaluates genotypic and phenotypic correlations through implementation of WES detecting genetic defects in a cohort.
Methods
Patients
Changzhou Maternal and Child Health Care Hospital is the only NBS institution in Changzhou. Participants were patients who were diagnosed and treated at this hospital. They all volunteered to participate in this study after informed consent. CH was diagnosed based on findings of elevated levels of serum thyroid-stimulating hormone (TSH; ≥9 mIU/L) and low levels of free thyroxine (FT4; <7.77 pmol/L) for newborns who were positive in the NBS program (heel blood TSH, ≥9.0 mIU/L). The levels of serum TSH and FT4 were determined by electrochemistry immunoassay using the COBAS e601 analyzer (Roche Diagnostics, Mannheim, Germany). The levels of heel blood TSH were detected by time-resolved fluoroimmunoassay using the Wallac 1235 AutoDELFIA (Perkin Elmer, Waltham, MA, USA). Following confirmation of diagnosis, the patients were immediately administered L-T4 at an initial dose of 10–15 μg kg−1 day−1. Patients with other congenital diseases or those whose mothers were diagnosed with Graves’ disease were excluded from the study. The study design and protocol were approved by the Ethics Committee of the Nanjing Medical University (approval no. 2019-258). Written informed consent for participation in this study was provided by the participants’ legal guardians.
Collection of Clinical Data and Blood Samples
All newborns with CH were treated and followed up at the Department of Medical Genetics of Changzhou Maternal and Child Health Care Hospital. The levels of heel blood TSH of newborns at screening and those of serum TSH and FT4 at diagnosis were recorded in the electronic NBS information system. All participants were followed up until the conclusion of the present study. The administered dose of L-T4 during treatment was managed and recorded by a clinician. Patients with controllable reduction of L-T4 dose were defined as patients whose levels of thyroid hormones were normal within 2 months of treatment and whose L-T4 doses were reduced or equal compare to the previous follow-up. The reference ranges for the treatment of CH are shown in Table 1. Thyroid morphology was determined using ultrasound scanning. The methods and reference values of thyroid volumes were based on a published study on Chinese newborns (7). Venous blood was sampled from the proband and the proband’s parents and then stored in an ultra-low-temperature refrigerator.
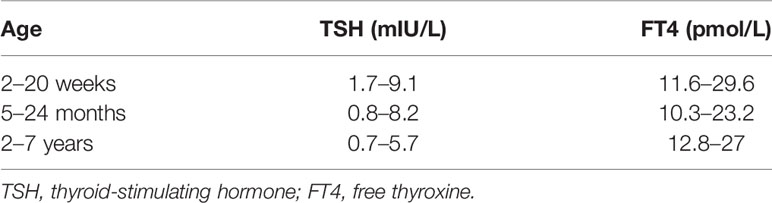
Table 1 Reference ranges in the treatment of patients with congenital hypothyroidism (CH).
Whole-Exome Sequencing
WES and variant analysis were performed in the Department of Medical Genetics and Molecular Diagnostic Laboratory of Shanghai Children’s Medical Center. Briefly, genomic DNA was extracted from blood samples of patients and their parents using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Whole-exome capture was performed using an Agilent SureSelect V6 enrichment capture kit (Agilent Technologies, Inc., Woburn, MA, USA) according to the manufacturer’s instructions. The captured library was sequenced using an Illumina HiSeq 2500 System (Illumina, Inc., San Diego, CA, USA). Original sequencing data were assessed using FastQC (version 0.11.2) for quality control. The Burrows–Wheeler Alignment (BWA) tool (v.0.2.10) was employed for sequencing data alignment to the Genome Reference Consortium Human Build 37 (GRCh37/hg19). Single nucleotide variants and small indels were identified using the Genome Analysis Toolkit (GATK). All variants were saved in VCF format and uploaded to the Ingenuity Variant Analysis (Ingenuity Systems, Redwood City, CA, USA) and the TGex (Translational Genomics Expert) platform for biological analysis and interpretation. Variants were classified based on the guidelines of the American College of Medical Genetics and Genomics (ACMG) (8), excluding benign and likely benign, but including DUOX2 p.His678Arg, which has been reported as a functional SNP (9). Variants detected by WES were confirmed by Sanger sequencing in each patient and the parents.
Patient Classification and Comparison of Characteristics
Patients were assigned into groups using two different strategies: 1) based on the number of affected gene—the single-gene group comprised cases with variants in a single gene, while the multi-gene group comprised cases with variants in multiple genes; 2) based on the pattern of the detected variants—the monoallelic group comprised cases with a single variant in a single gene, the biallelic group comprised cases with homozygous variants or compound heterozygous variants in a single gene determined by analysis of the parents, and the oligogenic group included cases with two or more variants in different genes. The clinical characteristics, such as heel blood TSH at NBS, serum TSH and FT4 at diagnosis, the initial L-T4 dose, the L-T4 dose at 1 year of age, current L-T4 dose, and thyroid morphology, were analyzed among the groups.
Statistical Analysis
The summary statistics for non-normally distributed quantitative variables were expressed as the median plus interquartile range (IQR). Categorical data were summarized as number and percentages. The Mann–Whitney U test or the Kruskal–Wallis test was performed for comparisons between two or more groups, and Dunn’s test was used for comparisons between subgroups. The chi-square test was used for categorical variables, whereas Fisher’s exact test was used if the expected cell count was less than five. Differences were considered statistically significant at a two-sided p-value of 0.05. All statistical tests were performed using R version 3.6.3.
Results
Variant Frequencies in Participants
Following acquisition of consent forms from guardians, 42 newborns with CH (21 males and 21 females) were enrolled in our study, including 40 non-consanguineous individuals and two siblings. The birth years of these participants ranged from 2011 to 2019. During this period, there were a total of 295,650 newborns, and 133 of them were diagnosed with CH. The average birth weight of the enrolled newborns was 3,346 g (range from 2,350 to 4,350 g), while the average gestational age was 39 weeks (from 34 + 6 to 42 weeks). We observed the occurrence of TD in 10 patients and goiter in five patients, whereas 27 patients had normal-sized gland-in-situ (GIS).
In total, we identified 58 variants in five genes—namely, DUOX2, TSHR, DUOXA2, TPO, and SLC26A4—in 29 of the participants (29/42, 69.0%). The positive rate of the variants was 75.0% (24/32) in patients with DH, whereas it was 50% (5/10) in patients with TD. The clinical characteristics and all variants are shown in Supplementary Table S1. Most of the identified variants were heterozygous, except for two homozygous DUOX2 mutations (i.e., p.Lys530* and p.Arg1110Gln) detected in two patients. Most variants were included in the databases or reported in previous studies, except for one heterozygous variant in TSHR (i.e., p. Ala579Val) that was novel. Interestingly, we found that variants in the genes causing DH were more common than those in the genes causing TD (87.9% versus 12.1%). We further observed that, among the 58 identified variants, the genes with the most frequent variants were DUOX2 (70.7%), followed by TSHR (12.1%), DUOXA2 (10.3%), and TPO (5.2%) (Figure 1). In addition, seven of these variants were detected in more than one patient: five DUOX2 variants, one DUOXA2 variant, and one TSHR variant (Table 2). We found that these variants accounted for 53.4% (31/58) of the total variants, with the p.Lys530* and p.Arg1110Gln mutations in DUOX2 constituting the predominant sites in the present cohort.
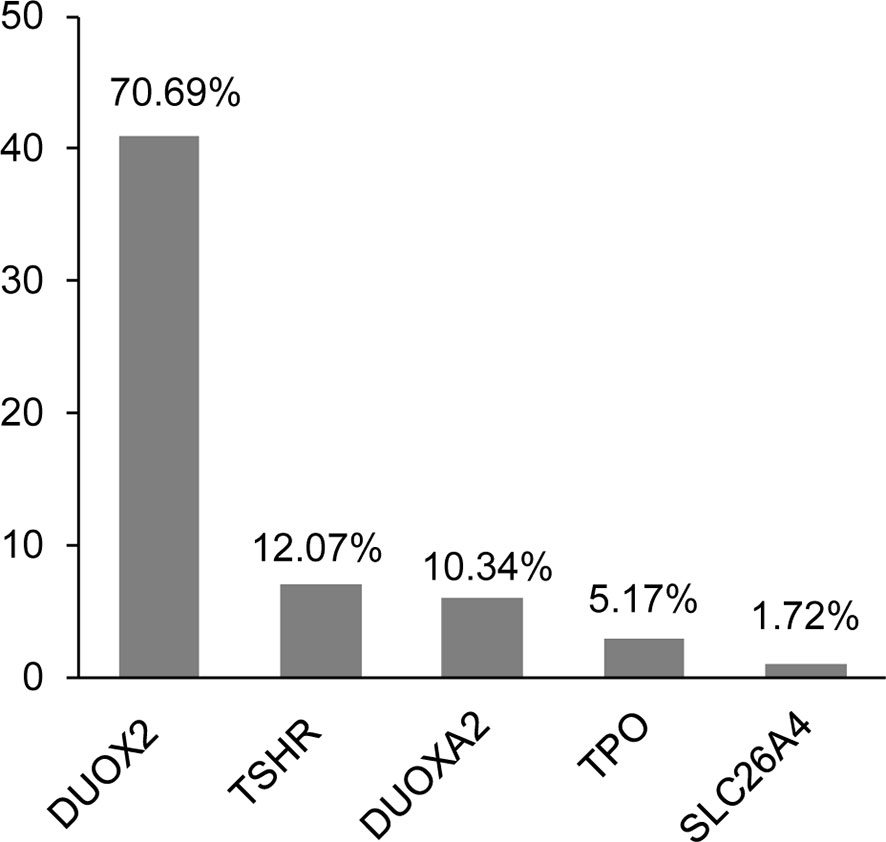
Figure 1 Distribution of the 58 detected variants in five genes.

Table 2 Variants detected in multiple participants.
Characteristics in the Single-Gene and Multi-Gene Groups
We specifically observed that, among the variants detected in patients, 65.5% (19/29) harbored two variants, whereas 20.7% (6/29) harbored three variants. Although multisite variants are common in patients with CH, we noticed that 87.5% (21/24) of our patients harbored variants in a single gene, whereas only two patients and one patient harbored variants in two and three genes, respectively. Based on the number of mutated genes, we classified the patients into two distinct groups. The single-gene group comprised 26 cases (18 cases of GIS, 4 cases of goiter, and 4 cases of TD) with variants in a single gene (Figure 2A and Table 3). More specifically, we noticed that, in the single-gene group, 20 cases had DUOX2 variants, three cases had DUOXA2 variants, and three cases had TSHR variants. The multi-gene group comprised three GIS cases, namely, two cases that harbored variants in DUOX2 and TSHR, and SLC26A4 and TPO, respectively, and one case that harbored variants in DUOXA2, TPO, and TSHR (Table 3).
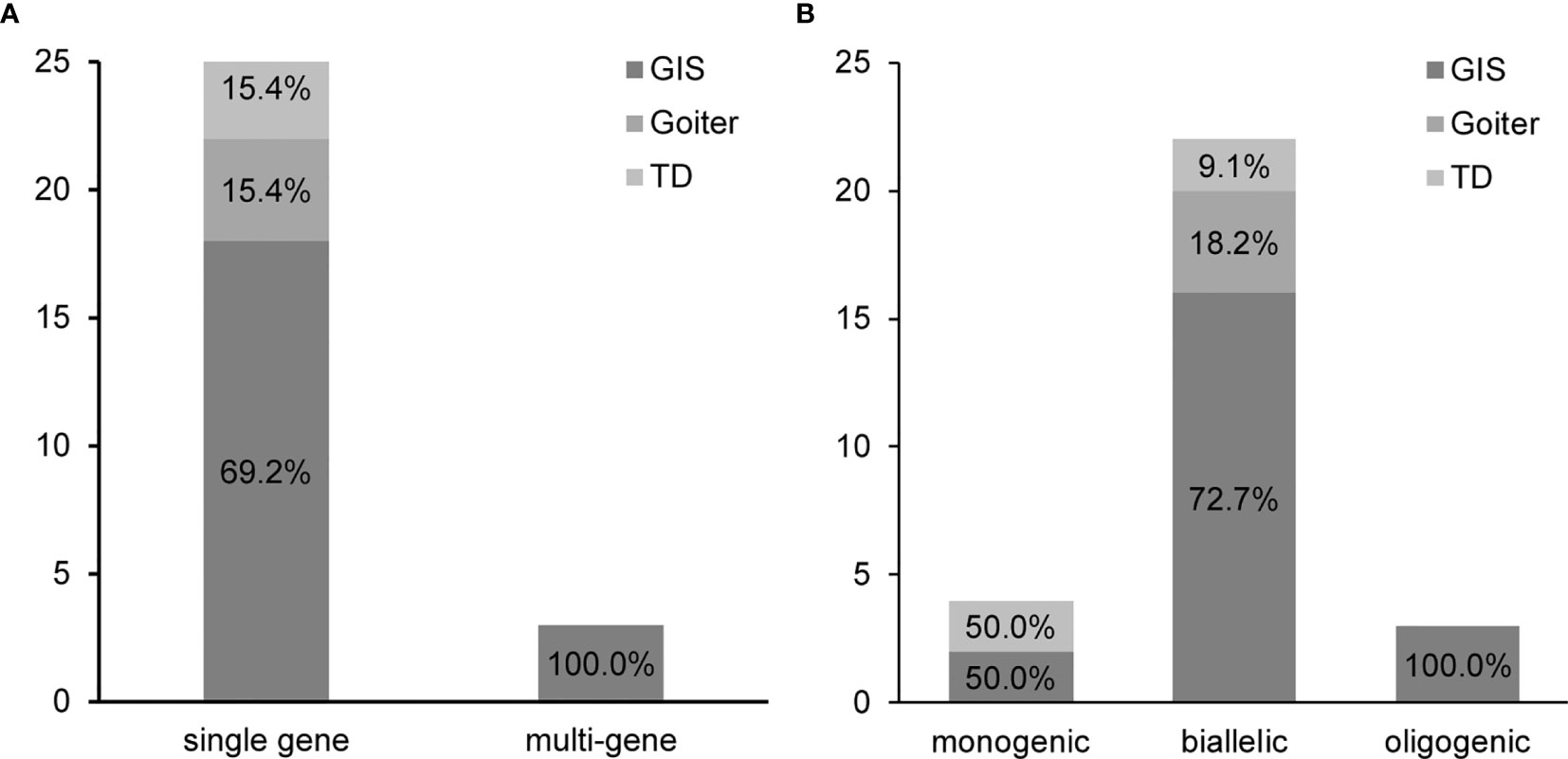
Figure 2 Grouping of 29 patients with congenital hypothyroidism (CH) causative variants. (A) Patients were divided into a single-gene and a multi-gene group. (B) Patients were classified into a monoallelic, a biallelic, and an oligogenic group. GIS, gland-in-situ; TD, thyroid dysgenesis.
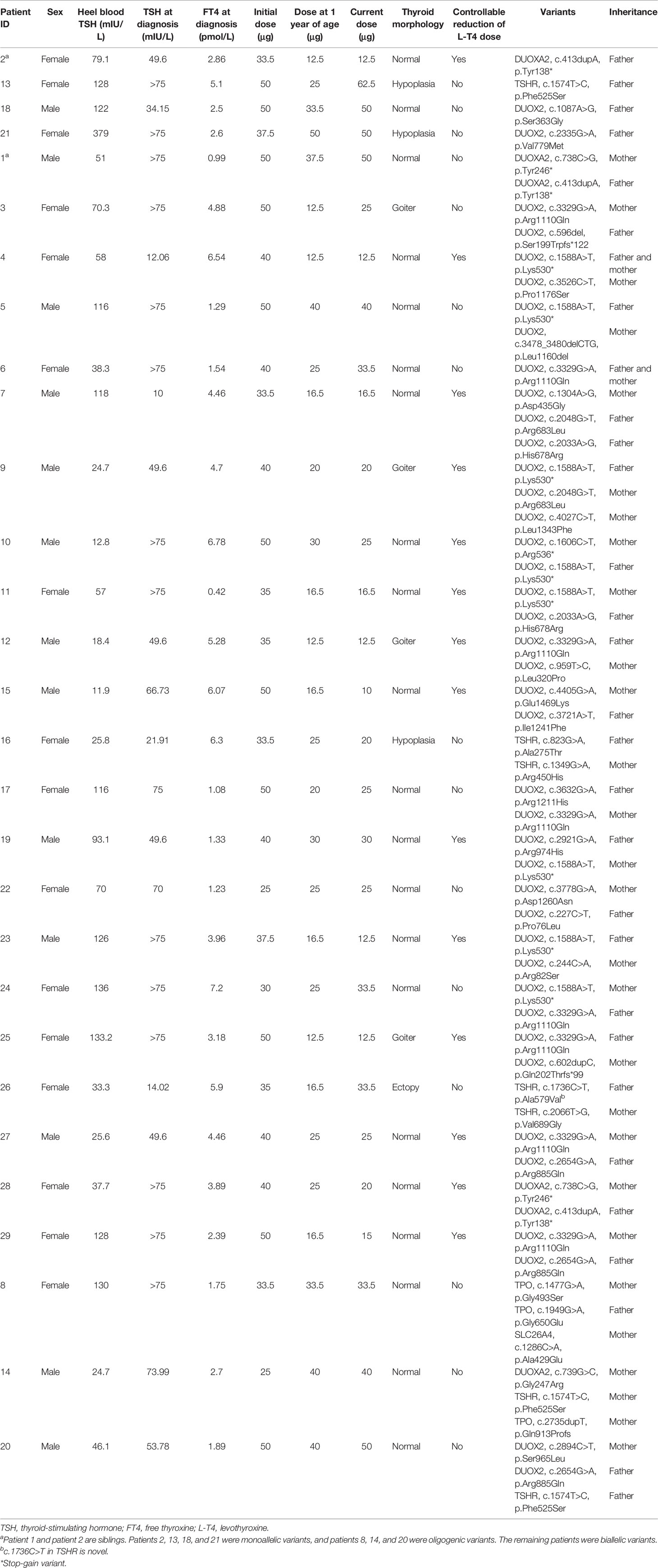
Table 3 Clinical description and variants in patients with monoallelic, biallelic, and oligogenic variants.
We subsequently analyzed the differences in the clinical data between the single-gene and multi-gene groups (Table 4). We did not observe any differences in thyroid morphology, heel blood TSH at screening, and TSH or FT4 at diagnosis (p > 0.05). We further collected data on the L-T4 dose, including the initial dose, dose at 1 years of age, and current dose. Interestingly, we found that the dose at 1 year of age was significantly higher in the multi-gene group than that in the single-gene group (p = 0.018). However, we did not detect a significant difference between the initial and current doses. In addition, we identified a controllable reduction in the L-T4 dose in 53.8% of the patients in the single-gene group; however, no patients with such reduction in the L-T4 dose were observed in the multi-gene group.
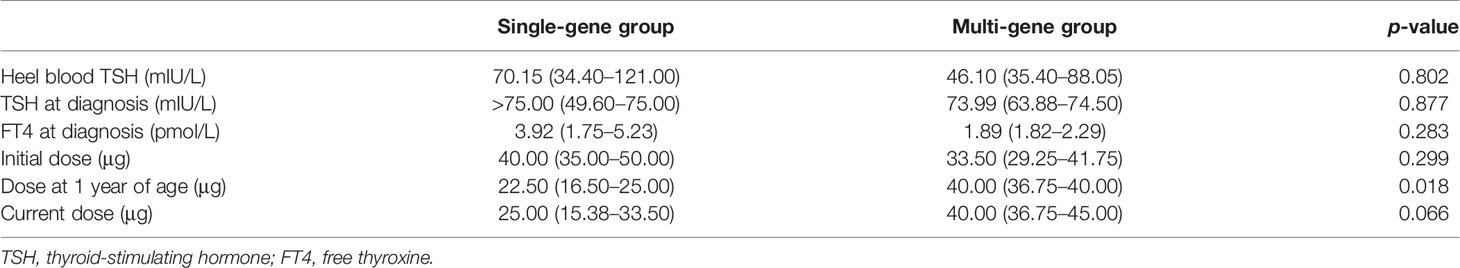
Table 4 Comparison of the clinical characteristics in the single-gene and multi-gene groups.
Characteristics in the Monoallelic, Biallelic, and Oligogenic Groups
Our aforementioned results suggested that multisite variants in a single gene prevailed in patients with CH. Therefore, we analyzed the pattern of the detected variants in their parents using Sanger sequencing. Based on the pattern of the detected variants, we classified the patients into three groups. Patients with variants in a single gene were subdivided into a monoallelic and a biallelic group. As shown in Figure 2B, the biallelic group comprised 22 cases with several variants in a single gene, including homozygous or compound heterozygous variants. The monoallelic group included four cases with a single variant in a single gene. In the biallelic group, we found that 81.8% (18/22) of cases (16 cases of GIS, 4 cases of goiter, and 2 cases of TD) harbored variants in DUOX2, two GIS cases had DUOXA2 variants, and two TD cases had TSHR variants. In the monoallelic group, we detected two cases with DUOX2, one GIS case with DUOXA2, and one TD case with TSHR. Of note is that the components of the cases in the oligogenic group were the same as those in the multi-gene group (Table 3).
The levels of heel blood TSH at screening, TSH and FT4 at diagnosis, and the initial, 1 year, and current L-T4 doses in the biallelic, oligogenic, and monoallelic groups are shown in Table 5. Considering the small sample size of each group, we did not perform comparisons among the three groups. We observed a controllable reduction in the L-T4 dose in 25% (1/4) of patients in the monoallelic group and in 59.1% (13/22) of patients in the biallelic group; however, we did not observe such a reduction in patients in the oligogenic group.
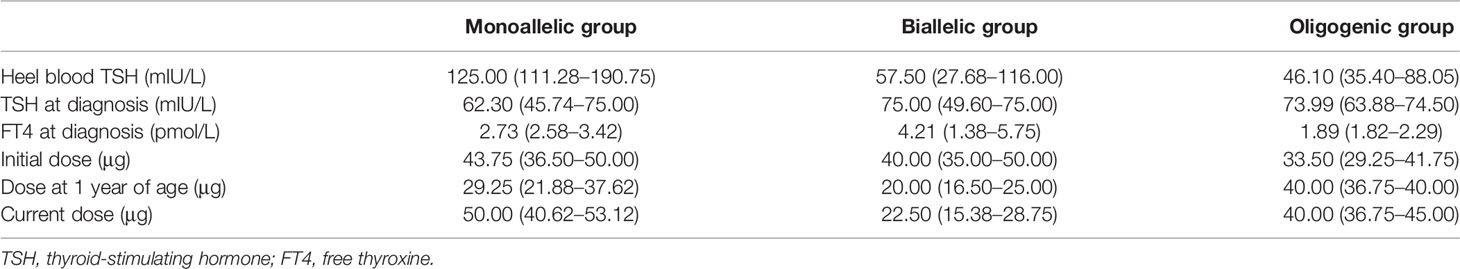
Table 5 Comparison of the clinical characteristics in the monoallelic, biallelic, and oligogenic groups.
Discussion
Our study demonstrated the existence of distinctive characteristics of genetic defects in different patients with CH. Historically, 75%–85% of CH cases have been attributed to TD, with the remainder occurring due to DH (10). However, patients with normal-sized GIS accounted for the majority of the cases in our cohort. Meanwhile, the frequency of genetic defects in the genes causing DH was higher than that in the genes causing TD, which was in agreement with previous studies in China and other Asian countries (11–13). These studies suggested that DH might be the leading pathophysiology of CH in Chinese populations, in contrast to what has been reported in other populations. We assumed that this difference might be a result of the genetic backgrounds of different ethnic populations.
In the present cohort, variants were most frequently identified in DUOX2, followed by TSHR and DUOXA2. The higher frequency of variants in DUOX2 was consistent with the observation of a higher proportion of DH cases in our participants. Variants in DUOX2 have been frequently reported, especially in East Asia (11–14). Several studies have suggested that the most reported variants among Chinese, Japanese, and Thai patients with CH have been identified in DUOX2 (12, 13, 15), suggesting that DUOX2 variants are an even more frequent causative factor for CH than previously recognized. In particular, the p.Lys530* and p.Arg1110Gln variants have been reported in Asian populations, including Chinese (16–18), Japanese (19, 20), Korean (21), and Malaysian (22) patients. The p.Lys530* and p.Arg1110Gln variants in DUOX2 were also identified as the most frequent sites with a rate of 19% in this study population.
Although multisite variants are known to be common in patients with CH, in our cohort, most multisite variants were detected in a single gene, i.e., DUOX2. By detecting the respective variants in the parents of probands, we identified both monoallelic and biallelic DUOX2 variants; the proportion of biallelic DUOX2 variants was overwhelmingly higher. Monoallelic mutations are thought to confer phenotypes due to haploinsufficiency (23). Monoallelic DUOX2 mutations have been reported to result in mild transient CH (16, 24, 25), whereas biallelic DUOX2 mutations cause severe permanent CH. However, subsequent studies have reported biallelic mutations in patients with transient CH (19, 26) and monoallelic mutations in patients with permanent CH (17, 27). Moreover, patients with DUOX2 mutations have been gradually recognized as exhibiting phenotypic heterogeneity (28, 29). In our previous study, we confirmed that the number of DUOX2 variants could not be used to predict the transient or permanent outcomes (30). However, the correlation between variants and hormone characteristics or L-T4 dose remains unclear. Therefore, in the present study, the levels of hormones and the dose of L-T4 were used to analyze the correlation between variants and the phenotypic characteristics.
Application of next-generation sequencing (NGS) revealed that a significant proportion of patients harbored variants in more than a single gene (31–33). Based on these findings, the oligogenic model and the viewpoint of combinational mutant genes resulting in the pathogenesis of CH were proposed. In particular, oligogenicity implied that the pathogenesis of CH could be attributed to the sum of mutations. We detected multi-gene variants, namely, oligogenic variants, in 7.1% of patients in our cohort. Among these patients, patient 14 harbored oligogenic variants on one allele. Considering that the mother of patient 14 has the same variants showing symptoms of hypothyroidism, we kept patient 14 in the follow-up analysis. To date, data on the detection rates of oligogenic variants have been rarely reported. In a study in the Japanese population, oligogenic variants were identified in 18.0% of patients with CH using targeted NGS of 24 causative genes (15). Another study in the Italian population reported that oligogenic defects were found in 26.2% of the cohort using targeted NGS of 11 causative genes (34). Given that differences in ethnic populations have been shown to result in differences in the mutational spectrum, we speculated that the oligogenicity characteristics might be distinctive among different populations.
The oligogenic model has been suggested as the genetic etiology of CH, on the basis that it provides a suitable explanation for the complex forms of inheritance and the variable expressivity of mutations in CH (31, 35). More specifically, the coexistence of mutations in multiple genes might contribute to the severity of the hypothyroid condition and lead to great genotype–phenotype variability (15, 36). However, this hypothesis has not been fully clarified yet. To address this, we compared the clinical data between the single-gene and multi-gene groups. However, we did not observe any differences in the levels of heel blood TSH at NBS or those of serum TSH and FT4 at diagnosis. We also analyzed the differences in the L-T4 treatment doses at different stages (initial, 1 year of age, and current). Our results demonstrated that only the L-T4 treatment dose at 1 year of age was significantly higher in the multi-gene group. Furthermore, we divided the single-gene group into monoallelic and biallelic groups according to the number of variants. Considering the small sample size of each group, we did not perform statistical analysis among the three groups. In the treatment, the L-T4 doses of some patients could be easily controlled in order to keep the thyroid hormones in the normal range; however, this was not easy in some patients. We wondered whether it is related to the genotype. Regarding the change in the treatment dose, we noticed that fewer patients in the oligogenic group had a controllable reduction. However, it is still uncertain whether relatively high doses of L-T4 were administered to patients with oligogenic variants due to the limitation of the small sample size in this study. We will continue to focus on the study of CH.
Previous studies have suggested a detection rate of approximately 20% for CH candidate genes in Japanese and Czech cohorts with CH (15, 37). Using efficient sequencing methods, such as multiplex PCR and targeted NGS, recent studies have reported an increased detection rate of CH candidate genes (14, 16, 33), especially in patients with DH (38). However, the detection rate of variants in TD cases has been shown to be only approximately 5% (3, 39). Unlike TD, DH appears to have a detectable genetic basis in many cases (40, 41). Using WES, the variant detection rate was 69.0% in our cohort. This was consistent with the results of previous studies in the Chinese population using targeted NGS (ranging from 52% to 65%) (14, 33). Efficient sequencing methods and ethnic differences might have contributed to the high detection rates in Chinese cohorts (41). In addition, some ambiguous variants were confirmed as disease-causing variants. Advances in the study of CH candidate genes help to increase the detection rate of variants in later research.
Our results might help elucidate the mechanisms underlying the pathogenesis of CH. However, certain limitations were noted in our study. Firstly, the small sample size of the oligogenic and monoallelic groups might have limited the statistical performance. Secondly, we were unable to detect intronic variants and verify the function of the variants of uncertain significance. Therefore, further studies are required to clarify the molecular etiology and genotype–phenotype correlations in CH.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the ethics committee of Nanjing Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
WL, FG, and YW participated in the diagnostic workup. HW, YW, and PX participated in the interpretation of clinical and biochemical data. RY, WL, and FG performed and interpreted the genetic analysis. WL and FG performed the statistical analysis.WL and BY wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 81903400), Young Talent Plan of Changzhou Health Commission (grant no. CZQM2020096), Changzhou Key Laboratory of High-tech Research (grant no. CM20193009), and Changzhou Social Development Science and Technology Support Project (grant no. CE20205035).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We gratefully thank RY and his team for the help in interpreting the variants found in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.705773/full#supplementary-material
References
1. Rastogi MV, LaFranchi SH. Congenital Hypothyroidism. Orphanet J Rare Dis (2010) 5:17. doi: 10.1186/1750-1172-5-17
2. Kostopoulou E, Miliordos K, Spiliotis B. Genetics of Primary Congenital Hypothyroidism-a Review. Horm (Athens) (2021) 20:225–36. doi: 10.1007/s42000-020-00267-x
3. Mio C, Grani G, Durante C, Damante G. Molecular Defects in Thyroid Dysgenesis. Clin Genet (2020) 97:222–31. doi: 10.1111/cge.13627
4. Peters C, van Trotsenburg ASP, Schoenmakers N. Diagnosis of Endocrine Disease: Congenital Hypothyroidism: Update and Perspectives. Eur J Endocrinol (2018) 179:R297–317. doi: 10.1530/EJE-18-0383
5. Targovnik HM, Citterio C, Siffo S, Rivolta CM. Advances and Perspectives in Genetics of Congenital Thyroid Disorders Associated With Thyroglobulin Gene Mutations. Open J Biol Sci (2016) 1:62–70. doi: 10.17352/pjbrd.000006
6. Stoupa A, Kariyawasam D, Muzza M, de Filippis T, Fugazzola L, Polak M, et al. New Genetics in Congenital Hypothyroidism. Endocrine (2021) 71:696–705. doi: 10.1007/s12020-021-02646-9
7. Yao D, He X, Yang RL, Jiang GP, Xu YH, Zou CC, et al. Sonographic Measurement of Thyroid Volumes in Healthy Chinese Infants Aged 0 to 12 Months. J Ultrasound Med (2011) 30:895–8. doi: 10.7863/jum.2011.30.7.895
8. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
9. Narumi S, Muroya K, Asakura Y, Aachi M, Hasegawa T. Molecular Basis of Thyroid Dyshormonogenesis: Genetic Screening in Population-Based Japanese Patients. J Clin Endocrinol Metab (2011) 96:E1838–42. doi: 10.1210/jc.2011-1573
10. Nettore IC, Cacace V, De Fusco C, Colao A, Macchia PE. The Molecular Causes of Thyroid Dysgenesis: A Systematic Review. J Endocrinol Invest (2013) 36:654–64. doi: 10.3275/8973
11. Zheng Z, Yang L, Sun C, Wu J, Luo F, Zhou W, et al. Genotype and Phenotype Correlation in a Cohort of Chinese Congenital Hypothyroidism Patients With DUOX2 Mutations. Ann Transl Med (2020) 8:1649. doi: 10.21037/atm-20-7165
12. Wang F, Xiaole L, Ma R, Zhao D, Liu S. Dual Oxidase (DUOX) System Genes Defects in Children With Congenital Hypothyroidism. Endocrinology (2021) 162:bqab043. doi: 10.1210/endocr/bqab043
13. Sorapipatcharoen K, Tim-Aroon T, Mahachoklertwattana P, Chantratita W, Iemwimangsa N, Sensorn I, et al. DUOX2 Variants Are a Frequent Cause of Congenital Primary Hypothyroidism in Thai Patients. Endocr Connect (2020) 9:1121–34. doi: 10.1530/EC-20-0411
14. Sun F, Zhang JX, Yang CY, Gao GQ, Zhu WB, Han B, et al. The Genetic Characteristics of Congenital Hypothyroidism in China by Comprehensive Screening of 21 Candidate Genes. Eur J Endocrinol (2018) 178:623–33. doi: 10.1530/EJE-17-1017
15. Yamaguchi T, Nakamura A, Nakayama K, Hishimura N, Morikawa S, Ishizu K, et al. Targeted Next-Generation Sequencing for Congenital Hypothyroidism With Positive Neonatal TSH Screening. J Clin Endocrinol Metab (2020) 105:dgaa308. doi: 10.1210/clinem/dgaa308
16. Fu C, Luo S, Zhang S, Wang J, Zheng H, Yang Q, et al. Next-Generation Sequencing Analysis of DUOX2 in 192 Chinese Subclinical Congenital Hypothyroidism (SCH) and CH Patients. Clin Chim Acta (2016) 458:30–4. doi: 10.1016/j.cca.2016.04.019
17. Tan M, Huang Y, Jiang X, Li P, Tang C, Jia X, et al. Clinical, and Molecular Characteristics of Congenital Hypothyroidism Caused by DUOX2 Mutations: A Population-Based Cohort Study in Guangzhou. Horm Metab Res (2016) 48:581–8. doi: 10.1055/s-0042-112224
18. Shin JH, Kim HY, Kim YM, Lee H, Bae MH, Park KH, et al. Genetic Evaluation of Congenital Hypothyroidism With Gland in Situ Using Targeted Exome Sequencing. Ann Clin Lab Sci (2021) 51:73–81.
19. Maruo Y, Takahashi H, Soeda I, Nishikura N, Matsui K, Ota Y, et al. Transient Congenital Hypothyroidism Caused by Biallelic Mutations of the Dual Oxidase 2 Gene in Japanese Patients Detected by a Neonatal Screening Program. J Clin Endocrinol Metab (2008) 93:4261–7. doi: 10.1210/jc.2008-0856
20. Maruo Y, Nagasaki K, Matsui K, Mimura Y, Mori A, Fukami M, et al. Natural Course of Congenital Hypothyroidism by Dual Oxidase 2 Mutations From the Neonatal Period Through Puberty. Eur J Endocrinol (2016) 174:453–63. doi: 10.1530/EJE-15-0959
21. Park KJ, Park HK, Kim YJ, Lee KR, Park JH, Park JH, et al. DUOX2 Mutations Are Frequently Associated With Congenital Hypothyroidism in the Korean Population. Ann Lab Med (2016) 36:145–53. doi: 10.3343/alm.2016.36.2.145
22. Chow YP, Abdul Murad NA, Mohd Rani Z, Khoo JS, Chong PS, Wu LL, et al. Exome Sequencing Identifies SLC26A4, GJB2, SCARB2 and DUOX2 Mutations in 2 Siblings With Pendred Syndrome in a Malaysian Family. Orphanet J Rare Dis (2017) 12:40. doi: 10.1186/s13023-017-0575-7
23. Grasberger H, De Deken X, Miot F, Pohlenz J, Refetoff S. Missense Mutations of Dual Oxidase 2 (DUOX2) Implicated in Congenital Hypothyroidism Have Impaired Trafficking in Cells Reconstituted With DUOX2 Maturation Factor. Mol Endocrinol (2007) 21:1408–21. doi: 10.1210/me.2007-0018
24. Moreno JC, Bikker H, Kempers MJ, van Trotsenburg AS, Baas F, de Vijlder JJ, et al. Inactivating Mutations in the Gene for Thyroid Oxidase 2 (THOX2) and Congenital Hypothyroidism. N Engl J Med (2002) 347:95–102. doi: 10.1056/NEJMoa012752
25. Grasberger H. Defects of Thyroidal Hydrogen Peroxide Generation in Congenital Hypothyroidism. Mol Cell Endocrinol (2010) 322:99–106. doi: 10.1016/j.mce.2010.01.029
26. Enacan RE, Masnata ME, Belforte F, Papendieck P, Olcese MC, Siffo S, et al. Transient Congenital Hypothyroidism Due to Biallelic Defects of DUOX2 Gene. Two Clinical Cases. Arch Argent Pediatr (2017) 115:E162–5. doi: 10.5546/aap.2017.e162
27. Wang F, Lu K, Yang Z, Zhang S, Lu W, Zhang L, et al. Genotypes and Phenotypes of Congenital Goitre and Hypothyroidism Caused by Mutations in Dual Oxidase 2 Genes. Clin Endocrinol (2014) 81:452–7. doi: 10.1111/cen.12469
28. Srichomkwun P, Takamatsu J, Nickerson D, Bamshad M, Chong J, Refetoff S. DUOX2 Gene Mutation Manifesting as Resistance to Thyrotropin Phenotype. Thyroid (2017) 27:129–31. doi: 10.1089/thy.2016.0469
29. Kizys MML, Louzada RA, Mitne-Neto M, Jara JR, Furuzawa GK, de Carvalho DP, et al. DUOX2 Mutations Are Associated With Congenital Hypothyroidism With Ectopic Thyroid Gland. J Clin Endocrinol Metab (2017) 102:4060–71. doi: 10.1210/jc.2017-00832
30. Long W, Zhou L, Wang Y, Liu J, Wang H, Yu B. Complicated Relationship Between Genetic Mutations and Phenotypic Characteristics in Transient and Permanent Congenital Hypothyroidism: Analysis of Pooled Literature Data. Int J Endocrinol (2020) 2020:6808517. doi: 10.1155/2020/6808517
31. de Filippis T, Gelmini G, Paraboschi E, Vigone MC, Di Frenna M, Marelli F, et al. A Frequent Oligogenic Involvement in Congenital Hypothyroidism. Hum Mol Genet (2017) 26:2507–14. doi: 10.1093/hmg/ddx145
32. Nicholas AK, Serra EG, Cangul H, Alyaarubi S, Ullah I, Schoenmakers E, et al. Comprehensive Screening of Eight Known Causative Genes in Congenital Hypothyroidism With Gland-In-Situ. J Clin Endocrinol Metab (2016) 101:4521–31. doi: 10.1210/jc.2016-1879
33. Long W, Lu G, Zhou W, Yang Y, Zhang B, Zhou H, et al. Targeted Next-Generation Sequencing of Thirteen Causative Genes in Chinese Patients With Congenital Hypothyroidism. Endocr J (2018) 65:1019–28. doi: 10.1507/endocrj.EJ18-0156
34. de Filippis T, Gelmini G, Paraboschi E, Vigone MC, Di Frenna M, Marelli F, et al. A Frequent Oligogenic Involvement in Congenital Hypothyroidism. Hum Mol Genet (2017) 26:2507–14. doi: 10.1093/hmg/ddx145
35. Persani L, Rurale G, de Filippis T, Galazzi E, Muzza M, Fugazzola L. Genetics and Management of Congenital Hypothyroidism. Best Pract Res Clin Endocrinol Metab (2018) 32:387–96. doi: 10.1016/j.beem.2018.05.002
36. Zheng X, Ma SG, Qiu YL, Guo ML, Shao XJ. A Novel C.554+5C>T Mutation in the DUOXA2 Gene Combined With P.R885Q Mutation in the DUOX2 Gene Causing Congenital Hypothyroidism. J Clin Res Pediatr Endocrinol (2016) 8:224–7. doi: 10.4274/jcrpe.2380
37. Al Taji E, Biebermann H, Límanová Z, Hníková O, Zikmund J, Dame C, et al. Screening for Mutations in Transcription Factors in a Czech Cohort of 170 Patients With Congenital and Early-Onset Hypothyroidism: Identification of a Novel PAX8 Mutation in Dominantly Inherited Early-Onset Non-Autoimmune Hypothyroidism. Eur J Endocrinol (2007) 156:521–9. doi: 10.1530/EJE-06-0709
38. Stoupa A, Al Hage Chehade G, Chaabane R, Kariyawasam D, Szinnai G, Hanein S, et al. High Diagnostic Yield of Targeted Next-Generation Sequencing in a Cohort of Patients With Congenital Hypothyroidism Due to Dyshormonogenesis. Front Endocrinol (Lausanne) (2020) 11:545339. doi: 10.3389/fendo.2020.545339
39. Luca P, Tiziana de F, Carla C, Davide G. Genetics in Endocrinology: Genetic Diagnosis of Endocrine Diseases by NGS: Novel Scenarios and Unpredictable Results and Risks. Eur J Endocrinol (2018) 179:R111–23. doi: 10.1530/EJE-18-0379
40. Wassner AJ. Unraveling the Genetics of Congenital Hypothyroidism: Challenges and Opportunities. J Clin Endocrinol Metab (2020) 105:e3464–5. doi: 10.1210/clinem/dgaa454
Keywords: hypothyroidism, whole-exome sequencing, biallelic variant, oligogenic variant, characteristic
Citation: Long W, Guo F, Yao R, Wang Y, Wang H, Yu B and Xue P (2021) Genetic and Phenotypic Characteristics of Congenital Hypothyroidism in a Chinese Cohort. Front. Endocrinol. 12:705773. doi: 10.3389/fendo.2021.705773
Received: 06 May 2021; Accepted: 16 August 2021;
Published: 03 September 2021.
Edited by:
Noriyuki Koibuchi, Gunma University, JapanReviewed by:
Hector Manuel Targovnik, University of Buenos Aires, ArgentinaKeisuke Nagasaki, Niigata University, Japan
Copyright © 2021 Long, Guo, Yao, Wang, Wang, Yu and Xue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Yu, YmlueXVAbmptdS5lZHUuY24=; Peng Xue, eHVlcGVuZ0BjemVkdS5jbg==