 Lise Coderre
Lise Coderre Lyna Debieche1,2
Lyna Debieche1,2 Joëlle Plourde
Joëlle Plourde Sylvie Lesage
Sylvie Lesage- 1Immunology-Oncology Section, Maisonneuve-Rosemont Hospital Research Center, Montréal, QC, Canada
- 2Département de médecine, Université de Montréal, Montréal, QC, Canada
- 3Division of Cardiovascular and Metabolic Diseases, Institut de recherche clinique de Montréal, Montréal, QC, Canada
- 4Département de nutrition, Université de Montréal, Montréal, QC, Canada
- 5Cystic Fibrosis Clinic, Centre Hospitalier de l’Université de Montréal (CHUM), Montréal, QC, Canada
- 6Département de microbiologie, infectiologie et immunologie, Université de Montréal, Montréal, QC, Canada
Cystic fibrosis (CF) is a genetic disease caused by mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR). Cystic fibrosis-related diabetes (CFRD) is the most common comorbidity, affecting more than 50% of adult CF patients. Despite this high prevalence, the etiology of CFRD remains incompletely understood. Studies in young CF children show pancreatic islet disorganization, abnormal glucose tolerance, and delayed first-phase insulin secretion suggesting that islet dysfunction is an early feature of CF. Since insulin-producing pancreatic β-cells express very low levels of CFTR, CFRD likely results from β-cell extrinsic factors. In the vicinity of β-cells, CFTR is expressed in both the exocrine pancreas and the immune system. In the exocrine pancreas, CFTR mutations lead to the obstruction of the pancreatic ductal canal, inflammation, and immune cell infiltration, ultimately causing the destruction of the exocrine pancreas and remodeling of islets. Both inflammation and ductal cells have a direct effect on insulin secretion and could participate in CFRD development. CFTR mutations are also associated with inflammatory responses and excessive cytokine production by various immune cells, which infiltrate the pancreas and exert a negative impact on insulin secretion, causing dysregulation of glucose homeostasis in CF adults. In addition, the function of macrophages in shaping pancreatic islet development may be impaired by CFTR mutations, further contributing to the pancreatic islet structural defects as well as impaired first-phase insulin secretion observed in very young children. This review discusses the different factors that may contribute to CFRD.
Introduction
Cystic fibrosis (CF; OMIM 219700) is the most frequent autosomal recessive disease in individuals of European ancestry with approximately 1 in 3000 births (1). This disease, for which there is no cure, is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, a cAMP-regulated anion channel responsible for chloride and bicarbonate transport (1–3). To date, more than 2000 CFTR variants have been identified with more than 300 variants known to directly cause CF (https://cftr2.org/mutations_history). CFTR mutations are grouped into six different classes; mutations in class I-III are considered severe while those in classes IV-VI are mild, in line with the severity of the pathology resulting from these mutations (1, 3, 4).
Mutations in CFTR cause dysregulation of ion and water transport across cellular membranes, which leads to the accumulation of dehydrated mucus and thickened secretions in organs such as the lungs and the exocrine pancreas (3, 4). Consequently, CF patients show a deterioration in pulmonary function associated with recurrent pulmonary infections, a major cause of morbidity and mortality. In the pancreas, a similar phenomenon leads to pancreatic exocrine insufficiency in the vast majority of patients, resulting in nutritional deficiency characterized by poor growth or weight gain in spite normal or increased food intake. To mitigate this nutritional deficiency, CF patients must frequently take pancreatic enzymes and vitamins. In addition to the lungs and pancreas, CFTR mutations can affect other tissues leading, for example, to male infertility.
With the advent of new treatments, the life-expectancy of CF patients has increased considerably from 10 years of age to now more than 50 years in some countries (5, 6). The prolonged life expectancy has led to the emergence of additional complications such as CF-related diabetes (CFRD). CFRD is the most common comorbidity of CF and its development has been associated with a decrease in lung function and survival (7, 8). This review will discuss the etiology of CFRD and highlight the emerging potential role of inflammation in the progression to CFRD.
Comparison of CFRD to Other Forms of Diabetes
Diabetes is a general term encompassing metabolic diseases characterized by dysregulation of blood glucose levels. The pancreas is the primary organ responsible for regulating glucose homeostasis. It is composed of exocrine and endocrine tissues. While the exocrine pancreas produces proteolytic enzymes to facilitate digestion, the endocrine tissue, named islets of Langerhans, is dispersed as cell clusters throughout the exocrine tissue (9). Each islet contains a cluster of α, β, δ, and PP cells, producing various hormones involved in glucose homeostasis (9, 10).
Diabetes is characterized by hyperglycemia. Chronic hyperglycemia contributes directly to an increased risk of vascular diseases, renal failure, blindness, amputation, and neuropathy as well as decreased life expectancy while severe hyperglycemia can lead to coma and death (11). While the most common forms of diabetes are type 1 (T1D) and type 2 diabetes (T2D), diabetes of the exocrine pancreas (DEP) is also getting recognition as an important cause of diabetes [reviewed in (12, 13)]. In T1D, representing less than 10% of overall cases, the immune system attacks and destroys insulin-producing pancreatic β-cells (14); patients living with T1D are dependent on exogenous insulin injections to regulate glucose levels (14). T2D, representing close to 90% of overall cases, is characterized by insulin resistance, whereby tissues such as the liver, muscle, and brain do not respond efficiently to insulin resulting in impaired glucose homeostasis. In addition, T2D patients exhibit quantitative and qualitative defects in insulin production. The combined effects of insulin resistance and abnormal insulin secretion together with other important mechanisms such as dysregulated secretion and/or action of incretin hormones and kidney glucose reabsorption lead to increased blood glucose levels (15, 16). Early treatment of T2D targets three different pathways, alone or in combination by: 1) improving insulin sensitivity via either changes in lifestyle or pharmacologic approaches; 2) increasing insulin secretion; and 3) reducing kidney glucose reabsorption (17). Over time, the ability of pancreatic β-cells to produce insulin is reduced in most T2D patients and exogenous insulin may be required to maintain appropriate glucose levels (18). A major complication of insulin therapy is hypoglycemia, which is associated with physical (e.g. falls) and/or psychological complications (e.g. fear) and, in its most severe form, can lead to coma or death (19). DEP, also named type 3c diabetes or pancreatogenic diabetes, refers to patients that develop diabetes secondary to exocrine pancreatic diseases such as acute and chronic pancreatitis, pancreatic cancer, alcohol-induced pancreatitis and CFRD. DEP is often underdiagnosed and may represent between 1-9% of diabetes cases (20, 21). Alcohol consumption is a leading cause of DEP (22), and alcohol-induced pancreatitis is associated with the accumulation of mucus in small ductal cells and destruction of the exocrine pancreas (22, 23). Interestingly, alcohol consumption is also associated with a reduction of the expression and membrane localization of CFTR in human pancreas (24, 25), and CFTR variants are important risk factors for DEP (26, 27). These observations highlight the important role of CFTR in the maintenance of pancreatic functions beyond CF. Still, because of the heterogeneous nature of the underlying mechanism driving the lesions in the exocrine pancreas for DEP, these diseases tend to exhibit very different pathophysiological features (18). One common feature of various forms of DEP is the reduction of insulin secretion. Consequently, DEP patients resort to the administration of exogenous insulin to regulate their blood glucose levels.
CFRD is a heterogeneous disease that shares some characteristics of T1D, T2D and alcohol-induced diabetes, such as insulin insufficiency. Similar to other forms of DEP, CFRD is associated with pancreatic inflammation, fibrosis and fatty infiltration leading to a reduction of the number of islets and impaired insulin secretion. The development of CFRD in CF patients is not associated with β-cell autoimmunity or HLA haplotypes that are linked to T1D susceptibility (28, 29). This suggests that for a majority of CF patients, the development of diabetes is not due to an autoimmune destruction of the β-cells. Nevertheless, a small subset of CF patients present evidence of an autoimmune response directed towards insulin-producing pancreatic β-cells. Specifically, autoantibodies to β-cell antigens, such as glutamic acid decarboxylase, insulin, or protein tyrosine phosphatase, which can be present in T1D patients (30, 31), have been detected in 0.8%-8.5% of CF patients (28, 29, 32). In this subset of CF patients, diabetes onset occurs earlier and is associated with a higher risk for acute complications, such as severe hypoglycemia and ketoacidosis (32). Thus for these autoantibody-positive CF patients, the autoimmune response to β-cell antigens is associated with the progression and severity of CFRD. Whether HLA haplotypes linked to T1D susceptibility or whether autoreactive T cells are more prevalent in autoantibody-positive CF patients relative to autoantibody-negative patients remains to be determined. From a clinical standpoint, CFRD and T1D share common features: onset is mostly in young patients, the diagnosis is typically not associated with obesity and insulin therapy is very frequently the therapeutic option. Of interest, a CFTR variant has recently been linked to T1D susceptibility suggesting a further association between CFRD and T1D pathologies (33).
CFRD also shares some features of T2D, such as insulin resistance and pancreatic amyloid deposition (34–37). Though some older studies failed to identify insulin resistance as a component of dysglycemia in CF, most recent studies show that CFRD patients exhibit both peripheral and hepatic insulin resistance similar to T2D (37–41). The hepatic insulin resistance coupled with a higher rate of gluconeogenesis result in a net increase in hepatic glucose production, which contributes to the hyperglycemia in CF patients (38, 39). Prospective observational cohort studies show that in a context of significantly reduced insulin secretion, variations of insulin sensitivity are associated with variations of glucose tolerance in adult patients with CF (41). Pancreatic amyloid deposition is also characteristic of CFRD patients. It is observed in 69% of CFRD patients, whereas it is absent in CF patients without diabetes (35). Interestingly, amyloid deposits are also observed in diabetics suffering from chronic pancreatitis and pancreatic cancer (42). As for T2D, this amyloid deposition in CFRD is progressive; it is generally not observed in children with CF (36, 43, 44). Because these deposits are detected late in the disease process, amyloidosis is probably a consequence rather than a cause of diabetes. Moreover, a recent study has found elevated levels of inflammatory markers in T2D and CFRD patients compared to control subjects, and these markers were associated with diabetic complications in both groups of patients (45). Of note, systemic inflammation is usually associated with insulin resistance. From a clinical standpoint, the onset of diabetes in both CFRD and T2D is preceded by a long phase of glucose intolerance usually characterized by postprandial glucose excursions (46).
A most interesting commonality between T2D and CFRD is the view that lung function may contribute to glucose dysregulation. In a T2D cross sectional study, lower forced expiratory volume in one second (FEV1), a measure of lung function, was associated with higher levels of plasma glucose in both control and T2D subjects (47). Similarly, in CF children, there is an inverse association between lower arterial oxygen saturation at night and glucose excursion during the OGTT (48). Prospective studies have also reported that reduced lung function increases the risk of T2D (49, 50). It would be interesting to examine whether reduced lung function in CF accelerates the progression to CFRD.
Altogether, results from these studies suggest that CFRD is a heterogeneous disease and that progression to CFRD is regulated by a multitude of factors. A key specific point of abnormal glucose homeostasis in patients living with CF is an association with an increased risk of accelerated weight loss, lung function reduction, and accordingly, a marked increased risk of early death (51, 52). Historically, it was reported that such risks start in the pre-diabetic glucose intolerance phase (53). Recently published data, however, indicate that improvement of overall CF management could modify some of the previously reported associations. For example, CFRD onset is no longer preceded by a reduction in lung function and/or accelerated weight loss in the majority of patients (53).
Risk Factors Associated With the Development of CFRD
Age, genotype and the presence of exocrine pancreatic deficiency are defined risk factors for CFRD. Notably, similar to insulin resistance (7, 37, 53, 54), the prevalence of CFRD increases with age; around 2% of children, 19% of adolescents, and up to 50% of adult CF patients are diabetic (7). In addition, patients carrying severe mutations such as class I and II mutations have a higher risk of diabetes and pancreatic insufficiency compared to those with milder CFTR mutations (54). Pancreatic insufficiency is closely associated with dysglycemia as well as low body mass index (BMI) and FEV1 (53). However, not all patients bearing identical CFTR mutations will develop CFRD and variation in diabetes onset as well as disease severity among CF patients with the same mutation suggest that additional factors contribute to CFRD. Genome wide association studies (GWAS) indicate that while there is only weak genetic association between T1D and CFRD (55), CFRD shares some genetic etiology with T2D, including loci associated with β-cell function (55, 56). In fact, a family history of T2D increases by 3-fold the risk of developing CFRD (56). GWAS have also highlighted the contribution of gene modifiers to CFRD. For example, variants of SLC26A9, a chloride channel, are associated with CFRD onset (55, 56) and patients that express higher levels of this transporter develop diabetes later (57). While SLC26A9 variants are specific for CFRD, other variants such as TCF7L2 are associated with both CFRD and T2D (55).
In addition to age and genotype, gender also contributes to CFRD susceptibility, but its role is more complex. Indeed, CFRD is more prevalent in women with CF in spite of higher insulin secretion compared to men with CF (58). One possible explanation for this counterintuitive observation is the fact that bacterial infections occur earlier and more frequently in women than men with CF, often leading to a systemic inflammatory response (59). Both acute and low grade systemic infections as well as corticosteroid use are known to contribute to insulin resistance and diabetes (60–63). Interestingly, Harding et al. showed that TNF-α levels, a marker of systemic inflammation, correlated with worse clinical status in CF subjects with impaired glucose tolerance (64). Altogether, this suggests that heightened systemic inflammation present in women relative to men with CF exacerbates insulin resistance and contributes to CFRD development.
CFTR mutations also affect the hepatobiliary system and liver diseases are risk factors for CFRD (65). Of note, liver biopsies from CF patients revealed fatty degeneration and liver fibrosis, which can progress to liver cirrhosis (66). Studies have shown that there is a strong association between liver steatosis and insulin resistance [reviewed in (67)]. In addition, crude markers of hepatic damage are associated with higher probability of dysglycemia and surrogate markers of insulin resistance and this effect was predominantly observed in men with CF (68). As mentioned above, CF patients present with hepatic insulin resistance and increased gluconeogenesis contributing to hyperglycemia (38, 39). The mechanism leading to CF liver diseases is not completely understood. Still, because CFTR is expressed in bile ducts and not in hepatocytes, the prevalent hypothesis is that, similar to exocrine pancreas, inspissated bile will obstruct small ducts causing liver injury and inflammation, resulting in liver fibrosis (69).
Overall, multiple factors are associated with increased risk or accelerated progression to CFRD, such as age, gender, CFTR genotype, genetic modifiers, the degree of pancreatic exocrine deficiency, lung function, liver disease, inflammation, the presence of β-cell specific autoantibodies, etc. Defining how the combination of these factors drives the progression to CFRD in each individual would increase our understanding of the heterogeneity of disease severity and onset.
Glucose Homeostasis in CF Patients
In diabetic patients, hyperglycemia can be diagnosed by quantifying fasting, post-glucose charge, or random plasma glucose levels, as well as glycosylated hemoglobin (A1c), a biomarker of mean plasma glucose over 3 months (70). Diabetes is usually preceded by a phase of glucose intolerance, which can affect fasting and/or postprandial glucose values. In CF patients, abnormal glucose tolerance (AGT) is diagnosed in more than 50% of adult CF patients.
Studies of CF children suggest that the incidence of AGT do not follow a linear progression across age group. Notably, the prevalence of AGT is higher in 2-4 year old children than in those over 5 years of age, suggesting that glucose abnormalities occur very early in life and that the severity of the dysregulation of glucose homeostasis may be age-dependent. A transient increase in the prevalence of AGT has also been observed in an animal model of CF, namely, in CFTR-null ferrets (71). Indeed, in young CFTR-null ferrets pancreatic inflammation and hyperglycemia peak in two month old kits and resolve thereafter (72, 73). These changes in glucose homeostasis in CFTR-null ferrets correlate with the destruction of the exocrine pancreas by proteolytic enzymes and remodeling of the endocrine pancreatic tissue, including the insulin-producing β-cells (73). Based on the observations in CFTR-null ferrets, it is tempting to suggest that AGT in children may result from deleterious exocrine and endocrine pancreatic tissue remodeling that increases the risk of developing CFRD later in life.
AGT increases the risk of developing diabetes in CF patients, and the detection of AGT in children aged 6-10 years old is associated with early onset CFRD (74). Inversely, cross-sectional analysis of adult CF subjects showing normal glucose tolerance [i.e. with peak blood glucose levels below 8 mmol/L during an oral glucose tolerance test (OGTT)] did not develop diabetes over the next decade (75). However, it should be noted that the number of CF patients with defects in glucose homeostasis is likely to be dramatically underestimated. Studies using continuous glucose monitoring show that 75% of young children (2-6 years old) and most adults with CF exhibit some glucose abnormality, despite showing normal variations in glucose levels when subjected to an OGTT (76–78). Still, questions persist about which parameters and glucose thresholds derived from continuous glucose monitoring should be used to diagnose CFRD and also to predict the risk of adverse clinical outcome especially rapid BMI and/or FEV1 decline.
The widespread glucose abnormalities observed in CF subjects are primarily due to altered insulin secretion, especially a reduction in first-phase insulin secretion (79–81). AGT is detectable in very young infants, in line with the fact that CF patients exhibit early endocrine pancreatic abnormalities, such as β-cell dysfunction and/or reduced β-cell mass. Indeed, while there are no significant changes in pancreatic islet size and distribution, histopathological analysis of pancreas from neonates and young children reveal a reduced number of insulin-positive β-cells per islets. Among insulin-positive β-cells, there is also a decrease in β-cell proliferation and neogenesis (36). Pancreatic β-cells are thus considerably reduced in young CF patients and are not being replaced. These changes correlate with the reported abnormalities in insulin secretion in both pancreatic sufficient and insufficient CF children (82, 83). Still, the decrease in insulin production in CF patients is not reliably predictive of CFRD onset. Interestingly, the decrease in β-cells is concomitant with an increase in the proportion of glucagon- and somatostatin- positive α- and δ-cells, respectively (36). Furthermore, secretion of glucagon, somatostatin and pancreatic polypeptide is dysregulated in CF patients and may participate in abnormal glucose regulation of CFRD patients (84–86). Altogether, these results suggest that, in CF patients, the endocrine dysfunctions are not limited to β-cells; rather the function of the whole endocrine pancreas is severely affected.
Indirect EffectS of CFTR Mutation on β-Cell Function
Earlier studies of CF were mostly from histological sections of pancreas autopsies from both CF and control subjects. They were very detailed and generated extensive descriptions of pancreatic injury. However, because they were mostly from autopsies, they tended to represent later stages of the disease or more severe mutations. More recent immunochemistry studies provide a detailed description of islet cellular composition, as well as markers of cell proliferation and β-cell neogenesis. In addition, isolation of human pancreatic islets from CF patients has allowed for more dynamic studies such as cellular electrophysiology as well as insulin secretion furthering our comprehension of CFRD. To determine whether these observations are generalizable to all CFRD patients, these studies would benefit from gaining access to larger patient cohorts, allowing, for instance, to establish the impact of mutation class, age, gender and other variables. Regardless, these studies have revealed important aspects of CFRD pathology, and we discuss them in greater detail.
In the human pancreas, the CFTR protein is detected in endocrine α-cells as well as in the exocrine pancreas (Figure 1). However, its presence in β-cells remains controversial, with most studies reporting very low levels of CFTR expression in human β-cells (87–91). In human fetal tissues, CFTR mRNA expression is found in the ducts of exocrine pancreas but not directly in the pancreatic islets (89, 92). More recently, in situ analysis showed that CFTR mRNA is detectable in only 0.45% of insulin-positive β-cells, in accordance with other studies which detected low levels of CFTR mRNA in islets (88, 90). However, CFTR protein and activity are not detected in human β-cells (90). Together, these findings suggest that only a small fraction of human β-cells potentially express CFTR. As such, the global alteration in β-cell insulin secretion in CF patients is most likely due to the impact of CFTR mutations in non β-cells.
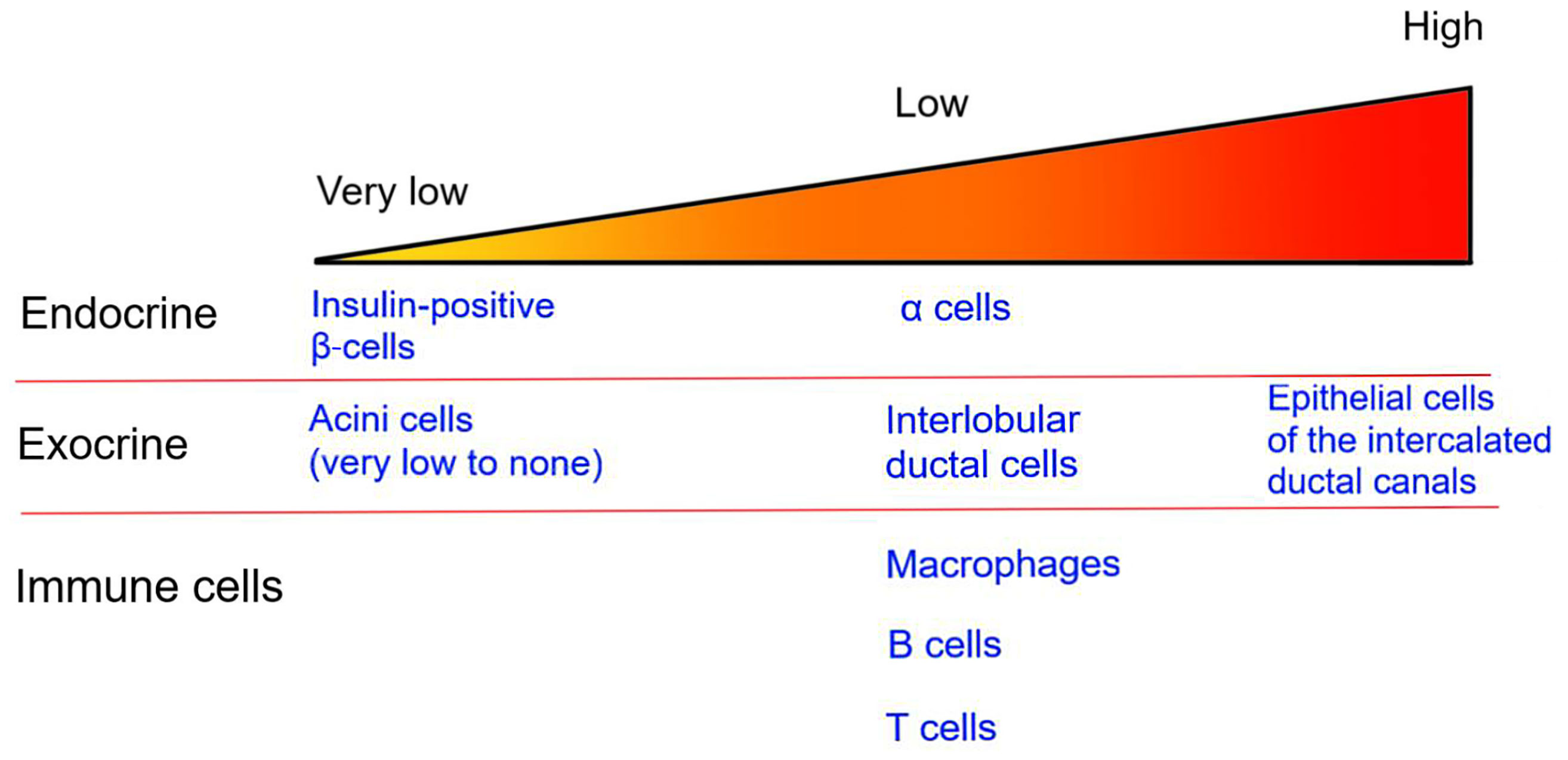
Figure 1 CFTR expression in the pancreas. The image represents the relative level of CFTR expression in pancreatic endocrine and exocrine tissues as well as in immune cells. Specific cell types are listed.
The islets, which represent only 1-2% of the pancreatic mass, are surrounded by the exocrine pancreas. Within the exocrine pancreas, CFTR has not been detected in the acini cells and is expressed at low levels in the interlobular duct, but at high levels in the epithelial cells of the intercalated duct canal (Figure 1). Notably, CFTR expression in the ductal system is detected early during embryonic development, starting at week 12 of gestation (92–94). Appropriate CFTR function is necessary for the development of the exocrine pancreas, as mutations in CFTR result in profound structural abnormalities in this tissue (95). With disease progression, the pancreatic ductal system of CF subjects shows high levels of protein concentration, such as trypsin, leading to protein precipitation and calcification. All these changes favor tubule dilation eventually provoking acini disruption and the release of pancreatic proteolytic enzymes within the pancreas (96, 97). This process, which starts in utero, continues postnatally (98). Notably, pancreatic structural abnormalities are detected at birth in CF infants (97, 99). In most CF subjects, the release of proteolytic enzymes will cause the destruction of the exocrine pancreas, followed by the activation of tissue repair mechanisms, fibrosis and the eventual replacement of the exocrine pancreas tissue by fat (Figure 2). This process leads to remodeling of the pancreas with islets often found clustering together (100). The destruction of the exocrine pancreas results in pancreatic insufficiency in 85% of CF patients while up to 20% of pancreatic sufficient CF patients will develop pancreatitis.
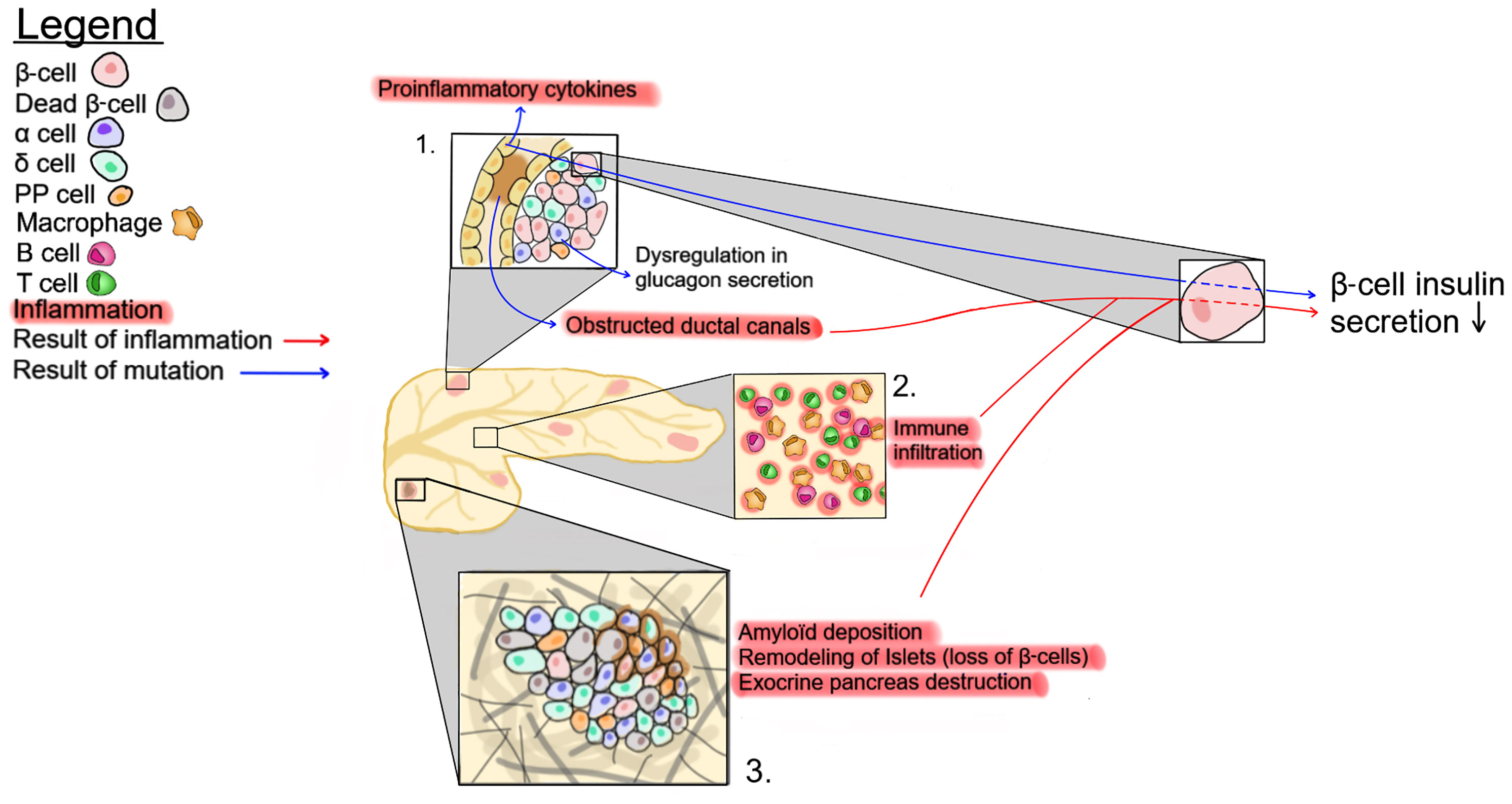
Figure 2 Possible mechanisms affecting insulin secretion in CF patients. 1- CFTR mutations eventually lead to ductal obstruction, which causes an increase in proinflammatory cytokines. 2- The accrued immune infiltration in the pancreas of CF patients. 3- Changes in pancreatic tissue structure, such as amyloid deposition, exocrine tissue destruction and β-cell loss. These three pathways impact insulin production by β-cells.
There is a close physical connection between the ductal system and pancreatic islets, as well as a strong association between markers of exocrine pancreatic damage, such as circulating immunoreactive trypsinogen, and the risk of developing CFRD (101). Recently, this association has been strengthened by the identification of genetic variants, including PRSS1 and SLC26A9, coding for cationic trypsinogen and a chloride channel, respectively, as predictors of CFRD (102). This suggests that the destruction of the exocrine pancreas and the resulting inflammation and fibrosis may contribute to disorganization, dysfunction, and destruction of the endocrine tissue (Figure 2). Accordingly, early histological examinations of CF pancreas by Andersen and others, described the presence of fibrosis as well as of cysts in infant pancreatic tissues at autopsy (96, 99). However, a recent study by Bogdani et al. show some fibrosis but very little fatty infiltration in the pancreas of children under 4 years of age (36) while CF adults show islet sclerosis and reduced β-cell areas (36, 100). It should be noted that results from older CF patients with severe pancreatic disease seem to separate into two broad phenotypes, a more fibrotic phenotype or one with a lipoatrophic pattern i.e. fat tissue enveloping pancreatic islets (36, 100). At present, it is unclear whether CFTR genotype, modifier genes or other factors play a role in the generation of these different patterns but they seem to be independent of CFRD (36, 100). It is also possible that, similar to the CFTR-null ferret, fibrosis will ultimately be followed by fatty infiltration (73). Altogether, these observations suggest that mutations in CFTR lead to alterations in the exocrine pancreas resulting in inflammation and tissue repair, which consequently modulate the pancreatic islet structure.
The sum of these correlative studies is not sufficient to determine if defective CFTR function in pancreatic ductal cells can directly influence insulin secretion by pancreatic β-cells. To address this, Shik Mun et al. designed a two-chamber in vitro system, wherein they cultured pancreatic ductal epithelial cells and islets in two interconnected chambers. Interestingly, chemical inhibition of CFTR function had no direct effect on isolated islet cells. However, it specifically affected the function of ductal cells, which resulted in a 50% decrease in insulin secretion by pancreatic islet cells (103). This study demonstrates that inhibition of CFTR function in ductal cells impedes insulin secretion likely through soluble mediators.
Findings from the latter study exploiting a two-chamber in vitro system suggest that improving CFTR function in ductal cells may ameliorate glucose homeostasis in CF patients (103). This has been addressed somewhat clinically through the administration of novel drugs, namely lumacaftor, a corrector that binds to and improves CFTR folding and expression and ivacaftor, a potentiator, which enhances CFTR function by facilitating channel opening. Early studies with ivacaftor alone suggested that this treatment may have a beneficial effect on glucose tolerance and the development of CFRD (104, 105). However, in more recent studies, CF patients treated with both lumacaftor and ivacaftor did not improve glucose tolerance or insulin secretion (106–108). It could be that administration of the drugs occurred too late in the disease process since most of the pancreatic CF manifestations are already present at birth. Future studies should investigate the effect of lumacaftor/ivacaftor in very young children and/or over a longer time-period.
In addition to their role in bicarbonate production and pH maintenance, pancreatic ductal epithelial cells have the capacity to secrete various growth factors and cytokines/chemokines, which may be affected by CFTR mutations (Figure 2). Ductal cells constitutively express CD40, and in vitro activation of purified pancreatic ductal epithelial cells with CD40L increases the secretion of multiple inflammatory cytokines and chemokines, including MIP-1β, IL-6, IL-8, TNF-α, IL-1β, IFN-γ, and granulocyte-macrophage colony-stimulating factor (GM-CSF). In addition, similar to the morphological changes of the pancreas (fibrosis; fat infiltration), the composition of the leukocytic infiltration was found to change over time, being composed primarily of macrophages and T lymphocytes in young patients, and of T lymphocytes with very few, if any, macrophages in adult CF patients at a time when the destruction of the exocrine pancreas is almost complete and remodeling of the endocrine pancreas has occurred (36, 43). This is consistent with the role of macrophages and lymphocytes in inflammation, tissue remodeling and fibrosis which is predominant in young children pancreas (109). These observations suggest that the inflammatory response in CF patients is a dynamic process that may contribute to the pathology.
Possible Contribution of Immune Cells in Progression to CFRD in CF Patients
Although CFTR is expressed at low levels in immune cells (Figure 1), CFTR mutations are associated with alterations of both innate and adaptive immune responses in CF subjects (110–112). Thus, in addition to the inflammation generated by the destruction/damage of the exocrine pancreas, dysregulation of the immune system may contribute to impaired insulin secretion and the development of CFRD (Figure 2).
Macrophages are one of the immune cell types that express CFTR (113–115). Indeed, mutations in CFTR have an impact on macrophage function, arguably promoting a proinflammatory phenotype (112, 113, 116, 117). In healthy humans and in mice, macrophages are detected early in the embryonic pancreas, and they contribute to pancreatic growth as well as to the establishment of β-cells in perinatal life (118, 119). Mice deficient in macrophages show abnormal islet morphogenesis, smaller islets, and a reduction in insulin mass (118, 120). Macrophages also play an important role in angiogenesis and tissue repair mechanisms, and can increase β-cell proliferation to maintain islet mass following pancreatic damage (121, 122). As mentioned above, intra-islet infiltration of monocytes/macrophages are increased in young CF patients, but are absent in both adult CF patients and control subjects (36, 43). This suggests that the presence of macrophages in the pancreatic islet tissue is a transitory phenomenon in CF. Alterations in macrophage function as a consequence of CFTR mutations in young CF patients may impact pancreatic development and thus contribute to enhanced susceptibility to CFRD. Of note, inflammatory macrophages are also found in the pancreatic islets of T1D and T2D patients, as well as in chronic pancreatitis (123–125). In chronic pancreatitis, activated macrophages participate in lymphocyte recruitment through the secretion of chemokines as well as in the progression of fibrosis in concert with pancreatic stellate cells (126, 127).
Macrophages also regulate insulin secretion, in part through soluble mediators such as IL-1β (128). Immunoreactivity for IL-1β was observed in pancreatic islets of CFRD patients as well as in young children with CF (43). In contrast to T2D, CF islets do not express the IL-1β antagonist, IL-1Rα, suggesting that the bio-activity of IL-1β is even higher in CFRD than in T2D (43). Macrophages may thus play a general role in all forms of diabetes and pancreatic inflammatory conditions, including CFRD. Additional studies are required to assess the phenotype and function of macrophages within the pancreatic tissue of CF patients, and to define whether altered macrophage function in CF patients contributes to the development of CFRD.
In addition to macrophages, there is growing evidence that T cell responses are altered in CF patients. T cells play an important role in both T1D and T2D, suggesting that they may also contribute to CFRD. Specifically, autoreactive T cells destroy β-cells directly contributing to T1D pathology, whereas in T2D, T cells contribute to systemic inflammation (129–131). T cells are also recruited in the pancreas during acute and chronic pancreatitis [reviewed in (132)]. T cell function in CF patients has been studied primarily in the lungs, often in the context of pulmonary infections. These studies revealed a reduction in regulatory T cells, as well as an increase in cytokine and chemokine production compatible with a bias towards a Th2 and/or a Th17 phenotype (133–137). T cell production of IL-17 is notably increased in CF patients (138). This is of interest to CFRD as the abundance of Th17 cells, which produce IL-17, is elevated in the blood of both T1D and T2D patients (131, 139). The presence of Th17 cells in the blood may thus contribute to diabetic pathologies. In addition to these systemic phenotypes, CF patients have lymphocytic infiltrates in pancreatic islets (36, 90). The T cells infiltrating the pancreas in CF patients secrete proinflammatory cytokines, known to inhibit insulin secretion [reviewed in (140)] Proinflammatory T cells in CF may thus contribute to CFRD by producing cytokines that increase the level of systemic inflammation, by infiltrating islets, and by destroying pancreatic β-cells, as well as by inhibiting insulin production by producing cytokines in the vicinity of β-cells.
B lymphocytes also express CFTR, and are likely to contribute to CFRD, at least in patients presenting with autoantibodies to islet antigens. Indeed, as mentioned previously, β-cell autoantibody-positive CF patients tend to develop CFRD earlier than autoantibody-negative individuals (32). Studies in Cftr-/- mice suggest that CFTR mutations directly affect B lymphocyte function. Uninfected Cftr-/- mice have higher levels of the B cell survival factor, B cell-activating factor (BAFF), a member of TNF cytokine family, as well as an increased number of lung lymphoid follicles compared to control mice (141). Increased levels of BAFF and lymphoid follicles were also observed in CF patients (141). Moreover, the pancreas of newborn CFTR-/- pigs present with a higher proportion of activated B lymphocytes, likely producing antibodies. These results suggest that CFTR alters B lymphocyte homeostasis, promoting the accumulation of B cells, which may contribute to CFRD. Interestingly, in addition to β-cell autoantibodies, some CF patients also exhibit autoantibodies to actin, and to double-stranded DNA (142). Overall, these data suggest that autoimmunity is a relatively frequent occurrence in CF patients. However, whether this effect is intrinsic to B lymphocytes or a consequence of recurrent lung infections remains to be demonstrated.
Discussion
The etiology of CFRD is complex with reduced insulin secretion playing a dominant role. However, despite numerous studies, the causal factor(s) implicated in disease onset and progression remains to be identified. Severe CFTR mutations cause alterations in the exocrine pancreas, which attract immune cells to this tissue leading to inflammation, islet disorganization, loss of β-cell mass, and reduced insulin production. The onset of exocrine pancreatic destruction in utero may explain, at least partially, why glucose abnormalities and altered insulin secretion are detected in very young infants. However, despite these changes, CFRD is rare in infants and not all CF patients progress to CFRD in adulthood, suggesting that other mechanisms contribute to disease onset. Immune pathways contribute to the normal development of the pancreas, but also to the pathogenesis of T1D and T2D and could play a similar role in CFRD. In particular, macrophages are important for normal islet homeostasis and CFTR mutations in macrophages may affect their function contributing to islet dysfunction and impaired insulin secretion. CF patients also show other alterations in immune function such as enhanced cytokine secretion by T cells and a bias toward Th2 and Th17 responses in the lung, as well as accumulation of B cells and the production of autoantibodies. These changes could contribute to the development of CFRD either directly or through their crosstalk with macrophages. Thus, despite intense research, numerous questions remain on the causes of CFRD onset in CF patients that should be the focus of future studies.
Author Contributions
LC supervised LD and wrote the final version of the manuscript. LD wrote the first draft of the manuscript with LC. JP prepared the figures and edited the manuscript. RR-L wrote parts of the manuscript and edited the manuscript. SL supervised the study and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
LD: holds a Diabete-Quebec studentship. RR-L: J-A DeSève diabetes research chair and CF Canada operating grant. SL: Research Scholar Emeritus awardee from the FRQS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Dr Mary Stevenson for a critical review of the manuscript.
References
1. O’Sullivan BP, Freedman SD. Cystic Fibrosis. Lancet (2009) 373(9678):1891–904. doi: 10.1016/S0140-6736(09)60327-5
2. Riordan JR. CFTR Function and Prospects for Therapy. Annu Rev Biochem (2008) 77:701–26. doi: 10.1146/annurev.biochem.75.103004.142532
3. Rowe SM, Miller S, Sorscher EJ. Cystic Fibrosis. N Engl J Med (2005) 352(19):1992–2001. doi: 10.1056/NEJMra043184
5. Stephenson AL, Sykes J, Stanojevic S, Quon BS, Marshall BC, Petren K, et al. Survival Comparison of Patients With Cystic Fibrosis in Canada and the United States: A Population-Based Cohort Study. Ann Intern Med (2017) 166(8):537–46. doi: 10.7326/M16-0858
6. Stephenson AL, Tom M, Berthiaume Y, Singer LG, Aaron SD, Whitmore GA, et al. A Contemporary Survival Analysis of Individuals With Cystic Fibrosis: A Cohort Study. Eur Respir J (2015) 45(3):670–9. doi: 10.1183/09031936.00119714
7. Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic Fibrosis-Related Diabetes: Current Trends in Prevalence, Incidence, and Mortality. Diabetes Care (2009) 32(9):1626–31. doi: 10.2337/dc09-0586
8. Moran A, Becker D, Casella SJ, Gottlieb PA, Kirkman MS, Marshall BC, et al. Epidemiology, Pathophysiology, and Prognostic Implications of Cystic Fibrosis-Related Diabetes: A Technical Review. Diabetes Care (2010) 33(12):2677–83. doi: 10.2337/dc10-1279
9. Rickels MR, Norris AW, Hull RL. A Tale of Two Pancreases: Exocrine Pathology and Endocrine Dysfunction. Diabetologia (2020) 63(10):2030–9. doi: 10.1007/s00125-020-05210-8
10. Brereton MF, Vergari E, Zhang Q, Clark A. Alpha-, Delta- and PP-Cells: Are They the Architectural Cornerstones of Islet Structure and Co-Ordination? J Histochem Cytochem (2015) 63(8):575–91. doi: 10.1369/0022155415583535
11. Petersmann A, Muller-Wieland D, Muller UA, Landgraf R, Nauck M, Freckmann G, et al. Definition, Classification and Diagnosis of Diabetes Mellitus. Exp Clin Endocrinol Diabetes (2019) 127(S 01):S1–7. doi: 10.1055/a-1018-9078
12. Wynne K, Devereaux B, Dornhorst A. Diabetes of the Exocrine Pancreas. J Gastroenterol Hepatol (2019) 34(2):346–54. doi: 10.1111/jgh.14451
13. Petrov MS, Basina M. Diagnosis of Endocrine Disease: Diagnosing and Classifying Diabetes in Diseases of the Exocrine Pancreas. Eur J Endocrinol (2021) 184(4):R151–R63. doi: 10.1530/EJE-20-0974
14. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 Diabetes. Lancet (2014) 383(9911):69–82. doi: 10.1016/S0140-6736(13)60591-7
15. Goldstein BJ. Insulin Resistance as the Core Defect in Type 2 Diabetes Mellitus. Am J Cardiol (2002) 90(5A):3G–10G. doi: 10.1016/s0002-9149(02)02553-5
16. Roden M, Shulman GI. The Integrative Biology of Type 2 Diabetes. Nature (2019) 576(7785):51–60. doi: 10.1038/s41586-019-1797-8
17. Diabetes Canada Clinical Practice Guidelines Expert C, Lipscombe L, Butalia S, Dasgupta K, Eurich DT, MacCallum L, et al. Pharmacologic Glycemic Management of Type 2 Diabetes in Adults: 2020 Update. Can J Diabetes (2020) 44(7):575–91. doi: 10.1016/j.jcjd.2020.08.001
18. Yu CG, Fu Y, Fang Y, Zhang N, Sun RX, Zhao D, et al. Fighting Type-2 Diabetes: Present and Future Perspectives. Curr Med Chem (2019) 26(10):1891–907. doi: 10.2174/0929867324666171009115356
19. Diabetes Canada Clinical Practice Guidelines Expert C, Yale JF, Paty B, Senior PA. Hypoglycemia. Can J Diabetes (2018) 42 Suppl 1:S104–8. doi: 10.1016/j.jcjd.2017.10.010
20. Hardt PD, Brendel MD, Kloer HU, Bretzel RG. Is Pancreatic Diabetes (Type 3c Diabetes) Underdiagnosed and Misdiagnosed? Diabetes Care (2008) 31 Suppl 2:S165–9. doi: 10.2337/dc08-s244
21. Hart PA, Bellin MD, Andersen DK, Bradley D, Cruz-Monserrate Z, Forsmark CE, et al. Type 3c (Pancreatogenic) Diabetes Mellitus Secondary to Chronic Pancreatitis and Pancreatic Cancer. Lancet Gastroenterol Hepatol (2016) 1(3):226–37. doi: 10.1016/S2468-1253(16)30106-6
22. Klochkov A, Kudaravalli P, Lim Y, Sun Y. Alcoholic Pancreatitis. Treasure Island (FL: StatPearls (2021).
23. Sarles H, Sarles JC, Camatte R, Muratore R, Gaini M, Guien C, et al. Observations on 205 Confirmed Cases of Acute Pancreatitis, Recurring Pancreatitis, and Chronic Pancreatitis. Gut (1965) 6(6):545–59. doi: 10.1136/gut.6.6.545
24. Maleth J, Balazs A, Pallagi P, Balla Z, Kui B, Katona M, et al. Alcohol Disrupts Levels and Function of the Cystic Fibrosis Transmembrane Conductance Regulator to Promote Development of Pancreatitis. Gastroenterology (2015) 148(2):427–39.e16. doi: 10.1053/j.gastro.2014.11.002
25. Patman G. Pancreatitis: Mislocalization of CFTR by Alcohol Promotes Pancreatitis. Nat Rev Gastroenterol Hepatol (2015) 12(1):2. doi: 10.1038/nrgastro.2014.204
26. Kondo S, Fujiki K, Ko SB, Yamamoto A, Nakakuki M, Ito Y, et al. Functional Characteristics of L1156F-CFTR Associated With Alcoholic Chronic Pancreatitis in Japanese. Am J Physiol Gastrointest Liver Physiol (2015) 309(4):G260–9. doi: 10.1152/ajpgi.00015.2014
27. Aghdassi AA, Weiss FU, Mayerle J, Lerch MM, Simon P. Genetic Susceptibility Factors for Alcohol-Induced Chronic Pancreatitis. Pancreatology (2015) 15(4 Suppl):S23–31. doi: 10.1016/j.pan.2015.05.476
28. Gottlieb PA, Yu L, Babu S, Wenzlau J, Bellin M, Frohnert BI, et al. No Relation Between Cystic Fibrosis-Related Diabetes and Type 1 Diabetes Autoimmunity. Diabetes Care (2012) 35(8):e57. doi: 10.2337/dc11-2327
29. Lanng S, Thorsteinsson B, Pociot F, Marshall MO, Madsen HO, Schwartz M, et al. Diabetes Mellitus in Cystic Fibrosis: Genetic and Immunological Markers. Acta Paediatr (1993) 82(2):150–4. doi: 10.1111/j.1651-2227.1993.tb12628.x
30. Knip M, Siljander H, Ilonen J, Simell O, Veijola R. Role of Humoral Beta-Cell Autoimmunity in Type 1 Diabetes. Pediatr Diabetes (2016) 17 Suppl 22:17–24. doi: 10.1111/pedi.12386
31. Pihoker C, Gilliam LK, Hampe CS, Lernmark A. Autoantibodies in Diabetes. Diabetes (2005) 54 Suppl 2:S52–61. doi: 10.2337/diabetes.54.suppl_2.s52
32. Konrad K, Kapellen T, Lilienthal E, Prinz N, Bauer M, Thon A, et al. Does Beta-Cell Autoimmunity Play a Role in Cystic Fibrosis-Related Diabetes? Analysis Based on the German/Austrian Diabetes Patienten Verlaufsdokumentation Registry. Diabetes Care (2016) 39(8):1338–44. doi: 10.2337/dc16-0020
33. Chiou J, Geusz RJ, Okino ML, Han JY, Miller M, Melton R, et al. Interpreting Type 1 Diabetes Risk With Genetics and Single-Cell Epigenomics. Nature (2021) 594(7863):398–402. doi: 10.1038/s41586-021-03552-w
34. Marzban L, Park K, Verchere CB. Islet Amyloid Polypeptide and Type 2 Diabetes. Exp Gerontol (2003) 38(4):347–51. doi: 10.1016/s0531-5565(03)00004-4
35. Couce M, O’Brien TD, Moran A, Roche PC, Butler PC. Diabetes Mellitus in Cystic Fibrosis Is Characterized by Islet Amyloidosis. J Clin Endocrinol Metab (1996) 81(3):1267–72. doi: 10.1210/jcem.81.3.8772610
36. Bogdani M, Blackman SM, Ridaura C, Bellocq JP, Powers AC, Aguilar-Bryan L. Structural Abnormalities in Islets From Very Young Children With Cystic Fibrosis May Contribute to Cystic Fibrosis-Related Diabetes. Sci Rep (2017) 7(1):17231. doi: 10.1038/s41598-017-17404-z
37. Colomba J, Boudreau V, Lehoux-Dubois C, Desjardins K, Coriati A, Tremblay F, et al. The Main Mechanism Associated With Progression of Glucose Intolerance in Older Patients With Cystic Fibrosis Is Insulin Resistance and Not Reduced Insulin Secretion Capacity. J Cyst Fibros (2019) 18(4):551–6. doi: 10.1016/j.jcf.2019.01.009
38. Hardin DS, LeBlanc A, Para L, Seilheimer DK. Hepatic Insulin Resistance and Defects in Substrate Utilization in Cystic Fibrosis. Diabetes (1999) 48(5):1082–7. doi: 10.2337/diabetes.48.5.1082
39. Hardin DS, Ahn C, Rice J, Rice M, Rosenblatt R. Elevated Gluconeogenesis and Lack of Suppression by Insulin Contribute to Cystic Fibrosis-Related Diabetes. J Investig Med (2008) 56(3):567–73. doi: 10.2310/JIM.0b013e3181671788
40. Austin A, Kalhan SC, Orenstein D, Nixon P, Arslanian S. Roles of Insulin Resistance and Beta-Cell Dysfunction in the Pathogenesis of Glucose Intolerance in Cystic Fibrosis. J Clin Endocrinol Metab (1994) 79(1):80–5. doi: 10.1210/jcem.79.1.8027259
41. Boudreau V, Coriati A, Hammana I, Ziai S, Desjardins K, Berthiaume Y, et al. Variation of Glucose Tolerance in Adult Patients With Cystic Fibrosis: What Is the Potential Contribution of Insulin Sensitivity? J Cyst Fibros (2016) 15(6):839–45. doi: 10.1016/j.jcf.2016.04.004
42. Ueberberg S, Nauck MA, Uhl W, Montemurro C, Tannapfel A, Clark A, et al. Islet Amyloid in Patients With Diabetes Due to Exocrine Pancreatic Disorders, Type 2 Diabetes, and Nondiabetic Patients. J Clin Endocrinol Metab (2020) 105(8):2595–605. doi: 10.1210/clinem/dgaa176
43. Hull RL, Gibson RL, McNamara S, Deutsch GH, Fligner CL, Frevert CW, et al. Islet Interleukin-1beta Immunoreactivity Is an Early Feature of Cystic Fibrosis That May Contribute to Beta-Cell Failure. Diabetes Care (2018) 41(4):823–30. doi: 10.2337/dc17-1387
44. Maloy AL, Longnecker DS, Greenberg ER. The Relation of Islet Amyloid to the Clinical Type of Diabetes. Hum Pathol (1981) 12(10):917–22. doi: 10.1016/s0046-8177(81)80197-9
45. Aulich J, Cho YH, Januszewski AS, Craig ME, Selvadurai H, Wiegand S, et al. Associations Between Circulating Inflammatory Markers, Diabetes Type and Complications in Youth. Pediatr Diabetes (2019) 20(8):1118–27. doi: 10.1111/pedi.12913
46. Costa M, Potvin S, Berthiaume Y, Gauthier L, Jeanneret A, Lavoie A, et al. Diabetes: A Major Co-Morbidity of Cystic Fibrosis. Diabetes Metab (2005) 31(3 Pt 1):221–32. doi: 10.1016/s1262-3636(07)70189-1
47. Lange P, Groth S, Kastrup J, Mortensen J, Appleyard M, Nyboe J, et al. Diabetes Mellitus, Plasma Glucose and Lung Function in a Cross-Sectional Population Study. Eur Respir J (1989) 2(1):14–9.
48. Suratwala D, Chan JS, Kelly A, Meltzer LJ, Gallagher PR, Traylor J, et al. Nocturnal Saturation and Glucose Tolerance in Children With Cystic Fibrosis. Thorax (2011) 66(7):574–8. doi: 10.1136/thx.2010.142141
49. Engstrom G, Hedblad B, Nilsson P, Wollmer P, Berglund G, Janzon L. Lung Function, Insulin Resistance and Incidence of Cardiovascular Disease: A Longitudinal Cohort Study. J Intern Med (2003) 253(5):574–81. doi: 10.1046/j.1365-2796.2003.01138.x
50. Ford ES, Mannino DM, National H. Nutrition Examination Survey Epidemiologic Follow-Up S. Prospective Association Between Lung Function and the Incidence of Diabetes: Findings From the National Health and Nutrition Examination Survey Epidemiologic Follow-Up Study. Diabetes Care (2004) 27(12):2966–70. doi: 10.2337/diacare.27.12.2966
51. Milla CE, Warwick WJ, Moran A. Trends in Pulmonary Function in Patients With Cystic Fibrosis Correlate With the Degree of Glucose Intolerance at Baseline. Am J Respir Crit Care Med (2000) 162(3 Pt 1):891–5. doi: 10.1164/ajrccm.162.3.9904075
52. Lanng S, Thorsteinsson B, Nerup J, Koch C. Influence of the Development of Diabetes Mellitus on Clinical Status in Patients With Cystic Fibrosis. Eur J Pediatr (1992) 151(9):684–7. doi: 10.1007/BF01957574
53. Potter KJ, Boudreau V, Shohoudi A, Mailhot M, Tremblay F, Lavoie A, et al. Influence of Pre-Diabetic and Pancreatic Exocrine States on Pulmonary and Nutritional Status in Adults With Cystic Fibrosis. J Cyst Fibros (2020). doi: 10.1016/j.jcf.2020.11.022
54. Lewis C, Blackman SM, Nelson A, Oberdorfer E, Wells D, Dunitz J, et al. Diabetes-Related Mortality in Adults With Cystic Fibrosis. Role of Genotype and Sex. Am J Respir Crit Care Med (2015) 191(2):194–200. doi: 10.1164/rccm.201403-0576OC
55. Aksit MA, Pace RG, Vecchio-Pagan B, Ling H, Rommens JM, Boelle PY, et al. Genetic Modifiers of Cystic Fibrosis-Related Diabetes Have Extensive Overlap With Type 2 Diabetes and Related Traits. J Clin Endocrinol Metab (2020) 105(5):1401–15. doi: 10.1210/clinem/dgz102
56. Blackman SM, Commander CW, Watson C, Arcara KM, Strug LJ, Stonebraker JR, et al. Genetic Modifiers of Cystic Fibrosis-Related Diabetes. Diabetes (2013) 62(10):3627–35. doi: 10.2337/db13-0510
57. Lam AN, Aksit MA, Vecchio-Pagan B, Shelton CA, Osorio DL, Anzmann AF, et al. Increased Expression of Anion Transporter SLC26A9 Delays Diabetes Onset in Cystic Fibrosis. J Clin Invest (2020) 130(1):272–86. doi: 10.1172/JCI129833
58. Coriati A, Belson L, Ziai S, Haberer E, Gauthier MS, Mailhot G, et al. Impact of Sex on Insulin Secretion in Cystic Fibrosis. J Clin Endocrinol Metab (2014) 99(5):1767–73. doi: 10.1210/jc.2013-2756
59. Chmiel JF, Berger M, Konstan MW. The Role of Inflammation in the Pathophysiology of CF Lung Disease. Clin Rev Allergy Immunol (2002) 23(1):5–27. doi: 10.1385/CRIAI:23:1:005
60. Granados A, Chan CL, Ode KL, Moheet A, Moran A, Holl R. Cystic Fibrosis Related Diabetes: Pathophysiology, Screening and Diagnosis. J Cyst Fibros (2019) 18 Suppl 2:S3–9. doi: 10.1016/j.jcf.2019.08.016
61. Olesen HV, Drevinek P, Gulmans VA, Hatziagorou E, Jung A, Mei-Zahav M, et al. Cystic Fibrosis Related Diabetes in Europe: Prevalence, Risk Factors and Outcome; Olesen et al. J Cyst Fibros (2020) 19(2):321–7. doi: 10.1016/j.jcf.2019.10.009
62. Marshall BC, Butler SM, Stoddard M, Moran AM, Liou TG, Morgan WJ. Epidemiology of Cystic Fibrosis-Related Diabetes. J Pediatr (2005) 146(5):681–7. doi: 10.1016/j.jpeds.2004.12.039
63. Yki-Jarvinen H, Sammalkorpi K, Koivisto VA, Nikkila EA. Severity, Duration, and Mechanisms of Insulin Resistance During Acute Infections. J Clin Endocrinol Metab (1989) 69(2):317–23. doi: 10.1210/jcem-69-2-317
64. Hardin DS, Leblanc A, Marshall G, Seilheimer DK. Mechanisms of Insulin Resistance in Cystic Fibrosis. Am J Physiol Endocrinol Metab (2001) 281(5):E1022–8. doi: 10.1152/ajpendo.2001.281.5.E1022
65. Minicucci L, Lorini R, Giannattasio A, Colombo C, Iapichino L, Reali MF, et al. Liver Disease as Risk Factor for Cystic Fibrosis-Related Diabetes Development. Acta Paediatr (2007) 96(5):736–9. doi: 10.1111/j.1651-2227.2007.00250.x
66. Parisi GF, Di Dio G, Franzonello C, Gennaro A, Rotolo N, Lionetti E, et al. Liver Disease in Cystic Fibrosis: An Update. Hepat Mon (2013) 13(8):e11215. doi: 10.5812/hepatmon.11215
67. Bugianesi E, Moscatiello S, Ciaravella MF, Marchesini G. Insulin Resistance in Nonalcoholic Fatty Liver Disease. Curr Pharm Des (2010) 16(17):1941–51. doi: 10.2174/138161210791208875
68. Colomba J, Netedu SR, Lehoux-Dubois C, Coriati A, Boudreau V, Tremblay F, et al. Hepatic Enzyme ALT as a Marker of Glucose Abnormality in Men With Cystic Fibrosis. PloS One (2019) 14(7):e0219855. doi: 10.1371/journal.pone.0219855
69. Fiorotto R, Strazzabosco M. Pathophysiology of Cystic Fibrosis Liver Disease: A Channelopathy Leading to Alterations in Innate Immunity and in Microbiota. Cell Mol Gastroenterol Hepatol (2019) 8(2):197–207. doi: 10.1016/j.jcmgh.2019.04.013
70. Diabetes Canada Clinical Practice Guidelines Expert C, Punthakee Z, Goldenberg R, Katz P. Definition, Classification and Diagnosis of Diabetes, Prediabetes and Metabolic Syndrome. Can J Diabetes (2018) 42 Suppl 1:S10–S5. doi: 10.1016/j.jcjd.2017.10.003
71. Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ, et al. Disease Phenotype of a Ferret CFTR-Knockout Model of Cystic Fibrosis. J Clin Invest (2010) 120(9):3149–60. doi: 10.1172/JCI43052
72. Olivier AK, Yi Y, Sun X, Sui H, Liang B, Hu S, et al. Abnormal Endocrine Pancreas Function at Birth in Cystic Fibrosis Ferrets. J Clin Invest (2012) 122(10):3755–68. doi: 10.1172/JCI60610
73. Rotti PG, Xie W, Poudel A, Yi Y, Sun X, Tyler SR, et al. Pancreatic and Islet Remodeling in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Knockout Ferrets. Am J Pathol (2018) 188(4):876–90. doi: 10.1016/j.ajpath.2017.12.015
74. Ode KL, Frohnert B, Laguna T, Phillips J, Holme B, Regelmann W, et al. Oral Glucose Tolerance Testing in Children With Cystic Fibrosis. Pediatr Diabetes (2010) 11(7):487–92. doi: 10.1111/j.1399-5448.2009.00632.x
75. Bonhoure A, Potter KJ, Colomba J, Boudreau V, Bergeron C, Desjardins K, et al. Peak Glucose During an Oral Glucose Tolerance Test Is Associated With Future Diabetes Risk in Adults With Cystic Fibrosis. Diabetologia (2021) 64:1332–41. doi: 10.1007/s00125-021-05423-5
76. Nguyen CQT, Denis MH, Chagnon M, Rabasa-Lhoret R, Mailhot G. Abnormal Glucose Tolerance in a Pediatric Cystic Fibrosis Cohort: Trends in Clinical Outcomes and Associated Factors in the Preceding Years. Nutr Metab Cardiovasc Dis (2021) 31(1):277–85. doi: 10.1016/j.numecd.2020.07.044
77. Schiaffini R, Brufani C, Russo B, Fintini D, Migliaccio A, Pecorelli L, et al. Abnormal Glucose Tolerance in Children With Cystic Fibrosis: The Predictive Role of Continuous Glucose Monitoring System. Eur J Endocrinol (2010) 162(4):705–10. doi: 10.1530/EJE-09-1020
78. Moreau F, Weiller MA, Rosner V, Weiss L, Hasselmann M, Pinget M, et al. Continuous Glucose Monitoring in Cystic Fibrosis Patients According to the Glucose Tolerance. Horm Metab Res (2008) 40(7):502–6. doi: 10.1055/s-2008-1062723
79. Cucinotta D, De Luca F, Arrigo T, Di Benedetto A, Sferlazzas C, Gigante A, et al. First-Phase Insulin Response to Intravenous Glucose in Cystic Fibrosis Patients With Different Degrees of Glucose Tolerance. J Pediatr Endocrinol (1994) 7(1):13–7. doi: 10.1515/jpem.1994.7.1.13
80. Anzeneder L, Kircher F, Feghelm N, Fischer R, Seissler J. Kinetics of Insulin Secretion and Glucose Intolerance in Adult Patients With Cystic Fibrosis. Horm Metab Res (2011) 43(5):355–60. doi: 10.1055/s-0031-1275270
81. Hammana I, Potvin S, Tardif A, Berthiaume Y, Coderre L, Rabasa-Lhoret R. Validation of Insulin Secretion Indices in Cystic Fibrosis Patients. J Cyst Fibros (2009) 8(6):378–81. doi: 10.1016/j.jcf.2009.08.007
82. Yi Y, Norris AW, Wang K, Sun X, Uc A, Moran A, et al. Abnormal Glucose Tolerance in Infants and Young Children With Cystic Fibrosis. Am J Respir Crit Care Med (2016) 194(8):974–80. doi: 10.1164/rccm.201512-2518OC
83. Wooldridge JL, Szczesniak RD, Fenchel MC, Elder DA. Insulin Secretion Abnormalities in Exocrine Pancreatic Sufficient Cystic Fibrosis Patients. J Cyst Fibros (2015) 14(6):792–7. doi: 10.1016/j.jcf.2015.02.009
84. Moran A, Diem P, Klein DJ, Levitt MD, Robertson RP. Pancreatic Endocrine Function in Cystic Fibrosis. J Pediatr (1991) 118(5):715–23. doi: 10.1016/s0022-3476(05)80032-0
85. Meacham LR, Caplan DB, McKean LP, Buchanan CN, Parks JS, Culler FL. Preservation of Somatostatin Secretion in Cystic Fibrosis Patients With Diabetes. Arch Dis Child (1993) 68(1):123–5. doi: 10.1136/adc.68.1.123
86. Lanng S, Thorsteinsson B, Roder ME, Orskov C, Holst JJ, Nerup J, et al. Pancreas and Gut Hormone Responses to Oral Glucose and Intravenous Glucagon in Cystic Fibrosis Patients With Normal, Impaired, and Diabetic Glucose Tolerance. Acta Endocrinol (Copenh) (1993) 128(3):207–14. doi: 10.1530/acta.0.1280207
87. Edlund A, Esguerra JL, Wendt A, Flodstrom-Tullberg M, Eliasson L. CFTR and Anoctamin 1 (ANO1) Contribute to cAMP Amplified Exocytosis and Insulin Secretion in Human and Murine Pancreatic Beta-Cells. BMC Med (2014) 12:87. doi: 10.1186/1741-7015-12-87
88. Di Fulvio M, Bogdani M, Velasco M, McMillen TS, Ridaura C, Kelly L, et al. Heterogeneous Expression of CFTR in Insulin-Secreting Beta-Cells of the Normal Human Islet. PloS One (2020) 15(12):e0242749. doi: 10.1371/journal.pone.0242749
89. White MG, Maheshwari RR, Anderson SJ, Berlinguer-Palmini R, Jones C, Richardson SJ, et al. In Situ Analysis Reveals That CFTR Is Expressed in Only a Small Minority of Beta-Cells in Normal Adult Human Pancreas. J Clin Endocrinol Metab (2020) 105(5):1366–74. doi: 10.1210/clinem/dgz209
90. Hart NJ, Aramandla R, Poffenberger G, Fayolle C, Thames AH, Bautista A, et al. Cystic Fibrosis-Related Diabetes Is Caused by Islet Loss and Inflammation. JCI Insight (2018) 3(8):e98240. doi: 10.1172/jci.insight.98240
91. Strong TV, Boehm K, Collins FS. Localization of Cystic Fibrosis Transmembrane Conductance Regulator mRNA in the Human Gastrointestinal Tract by in Situ Hybridization. J Clin Invest (1994) 93(1):347–54. doi: 10.1172/JCI116966
92. Tizzano EF, Chitayat D, Buchwald M. Cell-Specific Localization of CFTR mRNA Shows Developmentally Regulated Expression in Human Fetal Tissues. Hum Mol Genet (1993) 2(3):219–24. doi: 10.1093/hmg/2.3.219
93. Trezise AE, Chambers JA, Wardle CJ, Gould S, Harris A. Expression of the Cystic Fibrosis Gene in Human Foetal Tissues. Hum Mol Genet (1993) 2(3):213–8. doi: 10.1093/hmg/2.3.213
94. Hyde K, Reid CJ, Tebbutt SJ, Weide L, Hollingsworth MA, Harris A. The Cystic Fibrosis Transmembrane Conductance Regulator as a Marker of Human Pancreatic Duct Development. Gastroenterology (1997) 113(3):914–9. doi: 10.1016/s0016-5085(97)70187-2
95. Wilschanski M, Novak I. The Cystic Fibrosis of Exocrine Pancreas. Cold Spring Harb Perspect Med (2013) 3(5):a009746. doi: 10.1101/cshperspect.a009746
96. Porta EA, Stein AA, Patterson P. Ultrastructural Changes of the Pancreas and Liver in Cystic Fibrosis. Am J Clin Pathol (1964) 42:451–65. doi: 10.1093/ajcp/42.5.451
97. Andersen DH. Cystic Fibrosis of the Pancreas. J Chronic Dis (1958) 7(1):58–90. doi: 10.1016/0021-9681(58)90185-1
98. Wilschanski M, Durie PR. Patterns of GI Disease in Adulthood Associated With Mutations in the CFTR Gene. Gut (2007) 56(8):1153–63. doi: 10.1136/gut.2004.062786
99. Di Sant’Agnese PE, Andersen DH. Cystic Fibrosis of the Pancreas. Prog Pediat Study (1948) 1 Vol.):160–76. doi: 10.1016/0002-9343(56)90040-7
100. Lohr M, Goertchen P, Nizze H, Gould NS, Gould VE, Oberholzer M, et al. Cystic Fibrosis Associated Islet Changes may Provide a Basis for Diabetes. An Immunocytochemical and Morphometrical Study. Virchows Arch A Pathol Anat Histopathol (1989) 414(2):179–85. doi: 10.1007/BF00718598
101. Soave D, Miller MR, Keenan K, Li W, Gong J, Ip W, et al. Evidence for a Causal Relationship Between Early Exocrine Pancreatic Disease and Cystic Fibrosis-Related Diabetes: A Mendelian Randomization Study. Diabetes (2014) 63(6):2114–9. doi: 10.2337/db13-1464
102. Lin YC, Keenan K, Gong J, Panjwani N, Avolio J, Lin F, et al. Cystic Fibrosis-Related Diabetes Onset Can Be Predicted Using Biomarkers Measured at Birth. Genet Med (2021) 23:927–33. doi: 10.1038/s41436-020-01073-x
103. Shik Mun K, Arora K, Huang Y, Yang F, Yarlagadda S, Ramananda Y, et al. Patient-Derived Pancreas-on-a-Chip to Model Cystic Fibrosis-Related Disorders. Nat Commun (2019) 10(1):3124. doi: 10.1038/s41467-019-11178-w
104. Volkova N, Moy K, Evans J, Campbell D, Tian S, Simard C, et al. Disease Progression in Patients With Cystic Fibrosis Treated With Ivacaftor: Data From National US and UK Registries. J Cyst Fibros (2020) 19(1):68–79. doi: 10.1016/j.jcf.2019.05.015
105. Bellin MD, Laguna T, Leschyshyn J, Regelmann W, Dunitz J, Billings J, et al. Insulin Secretion Improves in Cystic Fibrosis Following Ivacaftor Correction of CFTR: A Small Pilot Study. Pediatr Diabetes (2013) 14(6):417–21. doi: 10.1111/pedi.12026
106. Colombo C, Foppiani A, Bisogno A, Gambazza S, Dacco V, Nazzari E, et al. Lumacaftor/ivacaftor in Cystic Fibrosis: Effects on Glucose Metabolism and Insulin Secretion. J Endocrinol Invest (2021). doi: 10.1007/s40618-021-01525-4
107. Thomassen JC, Mueller MI, Alejandre Alcazar MA, Rietschel E, van Koningsbruggen-Rietschel S. Effect of Lumacaftor/Ivacaftor on Glucose Metabolism and Insulin Secretion in Phe508del Homozygous Cystic Fibrosis Patients. J Cyst Fibros (2018) 17(2):271–5. doi: 10.1016/j.jcf.2017.11.016
108. Li A, Vigers T, Pyle L, Zemanick E, Nadeau K, Sagel SD, et al. Continuous Glucose Monitoring in Youth With Cystic Fibrosis Treated With Lumacaftor-Ivacaftor. J Cyst Fibros (2019) 18(1):144–9. doi: 10.1016/j.jcf.2018.07.010
109. Mack M. Inflammation and Fibrosis. Matrix Biol (2018) 68-69:106–21. doi: 10.1016/j.matbio.2017.11.010
110. Painter RG, Valentine VG, Lanson NA Jr, Leidal K, Zhang Q, Lombard G, et al. CFTR Expression in Human Neutrophils and the Phagolysosomal Chlorination Defect in Cystic Fibrosis. Biochemistry (2006) 45(34):10260–9. doi: 10.1021/bi060490t
111. Lara-Reyna S, Holbrook J, Jarosz-Griffiths HH, Peckham D, McDermott MF. Dysregulated Signalling Pathways in Innate Immune Cells With Cystic Fibrosis Mutations. Cell Mol Life Sci (2020) 77(22):4485–503. doi: 10.1007/s00018-020-03540-9
112. Turton KB, Ingram RJ, Valvano MA. Macrophage Dysfunction in Cystic Fibrosis: Nature or Nurture? J Leukoc Biol (2021) 109(3):573–82. doi: 10.1002/JLB.4RU0620-245R
113. Bruscia EM, Zhang PX, Ferreira E, Caputo C, Emerson JW, Tuck D, et al. Macrophages Directly Contribute to the Exaggerated Inflammatory Response in Cystic Fibrosis Transmembrane Conductance Regulator-/- Mice. Am J Respir Cell Mol Biol (2009) 40(3):295–304. doi: 10.1165/rcmb.2008-0170OC
114. Yoshimura K, Nakamura H, Trapnell BC, Chu CS, Dalemans W, Pavirani A, et al. Expression of the Cystic Fibrosis Transmembrane Conductance Regulator Gene in Cells of Non-Epithelial Origin. Nucleic Acids Res (1991) 19(19):5417–23. doi: 10.1093/nar/19.19.5417
115. Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, et al. CFTR Regulates Phagosome Acidification in Macrophages and Alters Bactericidal Activity. Nat Cell Biol (2006) 8(9):933–44. doi: 10.1038/ncb1456
116. Kopp BT, Abdulrahman BA, Khweek AA, Kumar SB, Akhter A, Montione R, et al. Exaggerated Inflammatory Responses Mediated by Burkholderia Cenocepacia in Human Macrophages Derived From Cystic Fibrosis Patients. Biochem Biophys Res Commun (2012) 424(2):221–7. doi: 10.1016/j.bbrc.2012.06.066
117. Lara-Reyna S, Scambler T, Holbrook J, Wong C, Jarosz-Griffiths HH, Martinon F, et al. Metabolic Reprograming of Cystic Fibrosis Macrophages via the IRE1alpha Arm of the Unfolded Protein Response Results in Exacerbated Inflammation. Front Immunol (2019) 10:1789. doi: 10.3389/fimmu.2019.01789
118. Banaei-Bouchareb L, Gouon-Evans V, Samara-Boustani D, Castellotti MC, Czernichow P, Pollard JW, et al. Insulin Cell Mass Is Altered in Csf1op/Csf1op Macrophage-Deficient Mice. J Leukoc Biol (2004) 76(2):359–67. doi: 10.1189/jlb.1103591
119. Mussar K, Pardike S, Hohl TM, Hardiman G, Cirulli V, Crisa L. A CCR2+ Myeloid Cell Niche Required for Pancreatic Beta Cell Growth. JCI Insight (2017) 2(15):e93834. doi: 10.1172/jci.insight.93834
120. Calderon B, Carrero JA, Ferris ST, Sojka DK, Moore L, Epelman S, et al. The Pancreas Anatomy Conditions the Origin and Properties of Resident Macrophages. J Exp Med (2015) 212(10):1497–512. doi: 10.1084/jem.20150496
121. Tessem JS, Jensen JN, Pelli H, Dai XM, Zong XH, Stanley ER, et al. Critical Roles for Macrophages in Islet Angiogenesis and Maintenance During Pancreatic Degeneration. Diabetes (2008) 57(6):1605–17. doi: 10.2337/db07-1577
122. Van Gassen N, Van Overmeire E, Leuckx G, Heremans Y, De Groef S, Cai Y, et al. Macrophage Dynamics Are Regulated by Local Macrophage Proliferation and Monocyte Recruitment in Injured Pancreas. Eur J Immunol (2015) 45(5):1482–93. doi: 10.1002/eji.201445013
123. Korpos E, Kadri N, Kappelhoff R, Wegner J, Overall CM, Weber E, et al. The Peri-Islet Basement Membrane, a Barrier to Infiltrating Leukocytes in Type 1 Diabetes in Mouse and Human. Diabetes (2013) 62(2):531–42. doi: 10.2337/db12-0432
124. Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. Islet-Associated Macrophages in Type 2 Diabetes. Diabetologia (2009) 52(8):1686–8. doi: 10.1007/s00125-009-1410-z
125. Yadav D, Notahara K, Smyrk TC, Clain JE, Pearson RK, Farnell MB, et al. Idiopathic Tumefactive Chronic Pancreatitis: Clinical Profile, Histology, and Natural History After Resection. Clin Gastroenterol Hepatol (2003) 1(2):129–35. doi: 10.1053/cgh.2003.50016
126. Xue J, Sharma V, Hsieh MH, Chawla A, Murali R, Pandol SJ, et al. Alternatively Activated Macrophages Promote Pancreatic Fibrosis in Chronic Pancreatitis. Nat Commun (2015) 6:7158. doi: 10.1038/ncomms8158
127. Wynn TA, Barron L. Macrophages: Master Regulators of Inflammation and Fibrosis. Semin Liver Dis (2010) 30(3):245–57. doi: 10.1055/s-0030-1255354
128. Dalmas E. Innate Immune Priming of Insulin Secretion. Curr Opin Immunol (2019) 56:44–9. doi: 10.1016/j.coi.2018.10.005
129. Burrack AL, Martinov T, Fife BT. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front Endocrinol (Lausanne) (2017) 8:343. doi: 10.3389/fendo.2017.00343
130. Xia C, Rao X, Zhong J. Role of T Lymphocytes in Type 2 Diabetes and Diabetes-Associated Inflammation. J Diabetes Res (2017) 2017:6494795. doi: 10.1155/2017/6494795
131. Jagannathan-Bogdan M, McDonnell ME, Shin H, Rehman Q, Hasturk H, Apovian CM, et al. Elevated Proinflammatory Cytokine Production by a Skewed T Cell Compartment Requires Monocytes and Promotes Inflammation in Type 2 Diabetes. J Immunol (2011) 186(2):1162–72. doi: 10.4049/jimmunol.1002615
132. Bhatia R, Thompson C, Ganguly K, Singh S, Batra SK, Kumar S. Alcohol and Smoking Mediated Modulations in Adaptive Immunity in Pancreatitis. Cells (2020) 9(8):1880. doi: 10.3390/cells9081880
133. Hartl D, Griese M, Kappler M, Zissel G, Reinhardt D, Rebhan C, et al. Pulmonary T(H)2 Response in Pseudomonas Aeruginosa-Infected Patients With Cystic Fibrosis. J Allergy Clin Immunol (2006) 117(1):204–11. doi: 10.1016/j.jaci.2005.09.023
134. Tiringer K, Treis A, Fucik P, Gona M, Gruber S, Renner S, et al. A Th17- and Th2-Skewed Cytokine Profile in Cystic Fibrosis Lungs Represents a Potential Risk Factor for Pseudomonas Aeruginosa Infection. Am J Respir Crit Care Med (2013) 187(6):621–9. doi: 10.1164/rccm.201206-1150OC
135. Hector A, Schafer H, Poschel S, Fischer A, Fritzsching B, Ralhan A, et al. Regulatory T-Cell Impairment in Cystic Fibrosis Patients With Chronic Pseudomonas Infection. Am J Respir Crit Care Med (2015) 191(8):914–23. doi: 10.1164/rccm.201407-1381OC
136. Kreindler JL, Steele C, Nguyen N, Chan YR, Pilewski JM, Alcorn JF, et al. Vitamin D3 Attenuates Th2 Responses to Aspergillus Fumigatus Mounted by CD4+ T Cells From Cystic Fibrosis Patients With Allergic Bronchopulmonary Aspergillosis. J Clin Invest (2010) 120(9):3242–54. doi: 10.1172/JCI42388
137. Chan YR, Chen K, Duncan SR, Lathrop KL, Latoche JD, Logar AJ, et al. Patients With Cystic Fibrosis Have Inducible IL-17+IL-22+ Memory Cells in Lung Draining Lymph Nodes. J Allergy Clin Immunol (2013) 131(4):1117–29, 29 e1-5. doi: 10.1016/j.jaci.2012.05.036
138. Tan HL, Regamey N, Brown S, Bush A, Lloyd CM, Davies JC. The Th17 Pathway in Cystic Fibrosis Lung Disease. Am J Respir Crit Care Med (2011) 184(2):252–8. doi: 10.1164/rccm.201102-0236OC
139. Honkanen J, Nieminen JK, Gao R, Luopajarvi K, Salo HM, Ilonen J, et al. IL-17 Immunity in Human Type 1 Diabetes. J Immunol (2010) 185(3):1959–67. doi: 10.4049/jimmunol.1000788
140. Wang C, Guan Y, Yang J. Cytokines in the Progression of Pancreatic Beta-Cell Dysfunction. Int J Endocrinol (2010) 2010:515136. doi: 10.1155/2010/515136
141. Polverino F, Lu B, Quintero JR, Vargas SO, Patel AS, Owen CA, et al. CFTR Regulates B Cell Activation and Lymphoid Follicle Development. Respir Res (2019) 20(1):133. doi: 10.1186/s12931-019-1103-1
Keywords: diabetes, cystic fibrosis, risk factors, immune function, CFTR
Citation: Coderre L, Debieche L, Plourde J, Rabasa-Lhoret R and Lesage S (2021) The Potential Causes of Cystic Fibrosis-Related Diabetes. Front. Endocrinol. 12:702823. doi: 10.3389/fendo.2021.702823
Received: 30 April 2021; Accepted: 06 July 2021;
Published: 30 July 2021.
Edited by:
Åke Sjöholm, Gävle Hospital, SwedenReviewed by:
Helen Thomas, University of Melbourne, AustraliaEtienne Larger, Université de Paris, France
Copyright © 2021 Coderre, Debieche, Plourde, Rabasa-Lhoret and Lesage. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvie Lesage, c3lsdmllLmxlc2FnZUB1bW9udHJlYWwuY2E=