 Juyi Li
Juyi Li Meng Shu2†
Meng Shu2† Aiping Deng
Aiping Deng Si Jin
Si Jin Hongmei Zhang
Hongmei Zhang- 1Department of Pharmacy, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Endocrinology, Institute of Geriatric Medicine, Liyuan Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Department of Pain, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 4Department of Traditional Chinese Medicine, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 5Department of Endocrinology, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Objective: To determine the pathogenic gene and explore the clinical characteristics of maturity-onset diabetes of the young type 2 (MODY2) pedigree caused by a mutation in the glucokinase (GCK) gene.
Methods: Using whole-exome sequencing (WES), the pathogenic gene was detected in the proband—a 20-year-old young man who was accidentally found with hyperglycemia, no ketosis tendency, and a family history of diabetes. The family members of the proband were examined. In addition, relevant clinical data were obtained and genomic DNA from peripheral blood was obtained. Pathologic variants of the candidate were verified by Sanger sequencing technology, and cosegregation tests were conducted among other family members and non-related healthy controls. After adjusting the treatment plan based on the results of genetic testing, changes in biochemical parameters, such as blood glucose levels and HAblc levels were determined.
Results: In the GCK gene (NM_000162) in exon 9, a heterozygous missense mutation c.1160C > T (p.Ala387Val) was found in the proband, his father, uncle, and grandmother. Thus mutation, which was found to co-segregate with diabetes, was the first discovery of such a mutation in the Asian population. After stopping hypoglycemic drug treatment, good glycemic control was achieved with diet and exercise therapy.
Conclusion: GCK gene mutation c.1160C > T (p.Ala387Val) is the pathogenic gene in the GCK-MODY pedigree. Formulating an optimized and personalized treatment strategy can reduce unnecessary excessive medical treatment and adverse drug reactions, and maintain a good HbA1c compliance rate
Introduction
Maturity-onset diabetes of the young (MODY) is a genetically heterogeneous type of monogenic diabetes mellitus that is characterized by autosomal dominant inheritance, an early age at onset, and pancreatic β-cell dysfunction (1). MODY is a diabetes subtype that is distinct from type 1 diabetes (T1D) and type 2 diabetes (T2D) (2). Identification of the MODY subtype is critical due to important therapeutic implications (3). It shows a different clinical heterogeneity due to its different mutation genes, therefore, disease judgment, prognosis, and treatment are also different (4). So far, 14 MODY-related genes have been discovered, including KCNJ11, ABCC8, INS, GCK, IPF1, PTF1A, GLIS3, FOX3, EIF2AK3, GLUT2, HNF1A, HNF1B, HNF4A, and PAX4 (5–9). Among these genes, heterozygous mutations in HNF1A, HNF4A, and GCK have been identified as the root cause of more than 90% of MODY cases (10). Moreover, mutations in GCK, HNF1A, and HNF4A HNF1B are the most common causes of MODY, accounting for 32%, 52%, 10% and 6% of all infected patients in the UK, respectively (3). The frequency of this causality, however, may differ between the Asian and Caucasian population. A genetic screening study among the Chinese population included 74 clinically highly suspected MODY patients from 59 unrelated families with a detection rate in HNF1A and HNF4A of 13.6% and 1.7%, respectively (11). In another MODY genetic screening study of 76 unrelated families, GCK, HNF1A and HNF4A accounted for 18.42%, 15.79% and 2.63% of all MODY cases, respectively (12). In the Chinese population, few cases of MODY have been identified, and investigation of large sample population is lacking. These data suggest that there may be unidentified genetic variants that play a role in MODY cases in the Asian population. Early identification and accurate classification of MODY cases is an important factor that is critical to guide precise treatment and determine patient prognosis.
The clinical manifestations of MODY depend on the molecular genetic basis. Patients with MODY resulting from mutations in genes that encode transcription factors present with more overt diabetes, often with a striking family history and frequent diabetic complications (13, 14). In contrast, patients with mutations in the GCK gene present with mild, stable hyperglycemia from birth, are mostly treated by diet alone and rarely suffer from diabetic complications (15, 16). In general, patients with GCK-MODY do not need pharmacological therapy (17). During illness or routine screening, genetic testing of family members can prevent the anxiety that is associated with hyperglycemia. Therefore, a correct genetically-based diagnosis of MODY is crucial to confirm the diagnosis and establish a treatment plan. Incorrect classification of MODY subtypes may lead to unnecessary treatment and increase personal healthcare expenditures.
Next generation sequencing is characterized by high throughput and low cost, which is conducive to clear research objectives and accuracy as well as for improving timeliness, to obtain required and high-quality results from the limited DNA information (18, 19). Application of this technique in the diagnosis of MODY diabetes improves the accuracy of clinical diagnosis.
Whole exome sequencing (WES) can improve molecular diagnostics of monogenic diabetes, thus genetic testing has important implications for MODY (20). In our study, WES was used to conduct genetic and functional studies on a family of suspected monogenic diabetes patients in China to identify disease-causing genes in a family and to explore the clinical characteristics of MODY patients caused by gene mutations. This may provide guidance for precise treatment of MODY and targeted individualized preventive measures for all carriers bearing a genetic mutation.
Materials and Methods
Participants
All subjects completed questionnaires regarding medical and family history, and information was supplemented by information from medical records. Diagnosis and the classification of diabetes complied with the latest guidelines presented by the American Diabetes Association (ADA) (21). This study was approved by the Ethics Committee of Wuhan Central Hospital, and participants signed informed consent for participation in the study.
DNA Extraction and Whole Exome Sequencing
Clinical data were obtained from all patients suspected of MODY, and 5ml of fasting blood was collected using EDTA for anticoagulation purposes. According to the manufacturer’s instructions, a DNA Extraction Kit (Tiangen Biotech, Beijing, China) was used to isolate genomic DNA of peripheral blood obtained from the MODY family. For high-throughput sequencing, the SureSelect Human All Exon V5 kit (Agilent technologies Inc, USA) was used for sample preparation and pretreatment, and samples underwent WES using the Illumina HiSeq 2500 sequencing platform.
Molecular Genetic Analyses
The sequence was aligned with the human genome reference sequence (UCSC hg19, GRCh37), compared, and the SNPs (Single Nucleotide Polymorphism) and InDels (Insertion-Deletion) variants were obtained after alignment with publicly available databases (1000 Genomes, ExAC (Exome Aggregation Consortium), dbSNP, ESP (NHLBI Exome Sequencing Project), and gnomAD (Genome Aggregation Database)). For mutations that were not detected in the database, further analysis of the impact that mutation on changes in the genetic code is required to distinguish between synonymous mutations and missense mutations. Next, several silico prediction tools (SIFT, PolyPhen2, Mutation Taster, Mutation Assessor, FATHMM, GERP-plus, PhyloP100, PhastCons100) were used to predict the influence of identified nonsynonymous mutations on protein structure and function.
Sanger Sequencing
It is known that high-throughput sequencing has a certain rate of mismeasurement. Therefore, Sanger sequencing should be performed in the proband and his family to verify variants identified by next generation sequencing. Polymerase chain reaction (PCR) amplification of target gene fragments: Primer design: Based on the location of the mutation in the target gene, primers were designed at the genome-wide level. Primer synthesis: completed by Beijing Kinco Biotechnology Co., Ltd. Target gene amplification: the target gene was amplified using Golden Mix (green) Golden Star T6 Super PCR Mix (1.1×) from Beijing Kinco Biotechnology Co., Ltd. (Beijing, China). Subsequently, the amplified product was subjected to agarose gel electrophoresis, and the target band as cut out of the gel and recovered using a DNA gel imager. The PCR product fragments were sequenced as follows: After the PCR product was tapped, recovered, and purified, the product was sequenced using an ABI3730XL Automatic Sequencer (Applied Biosystems, Foster City, CA). The sequencing results were compared and analyzed with genome sequences that were downloaded from the Genebank database (using AutoAssembler 2.0 software).
Structure Modeling
The Swiss-Prot repository (http://web.expasy.org/docs/swiss-prot_guideline.html) was used to simulate the three-dimensional (3D) structure of the protein sequence of the GCK gene bearing a missense mutation.
Medication Plan Adjustment and Follow Up the Effect of Drug Treatment
Based on the genetic testing results and related literature reports, the patient’s medication plan was adjusted in a timely manner. Glycemic control was assessed via monitoring blood glucose levels of each subject (at least one blood glucose measurement per day) and by rechecking HbA1c levels.
Results
Characteristics of Study Participants
Baseline characteristics of the study subjects are displayed in Table 1. The pedigree of this family is displayed in Figure 1. Physical and laboratory analyses showed that all subjects were negative for conventionally-assessed islet autoantibodies and urine sugar. Oral hypoglycemic drugs are ineffective for diabetic patients, and patients with diabetes show impaired beta-cell islet function. In this family, patients with diabetes were previously diagnosed as T2D. In addition, the proband’s grandmother (I-2) suffered from cataracts and previous suffered a stroke, but there were no sequelae. The proband’s father (II-2) had high blood pressure.
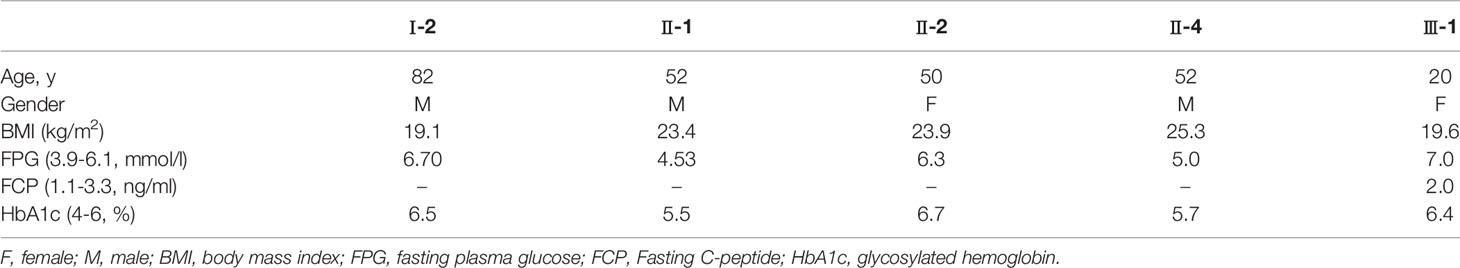
Table 1 Physical and laboratory examination.
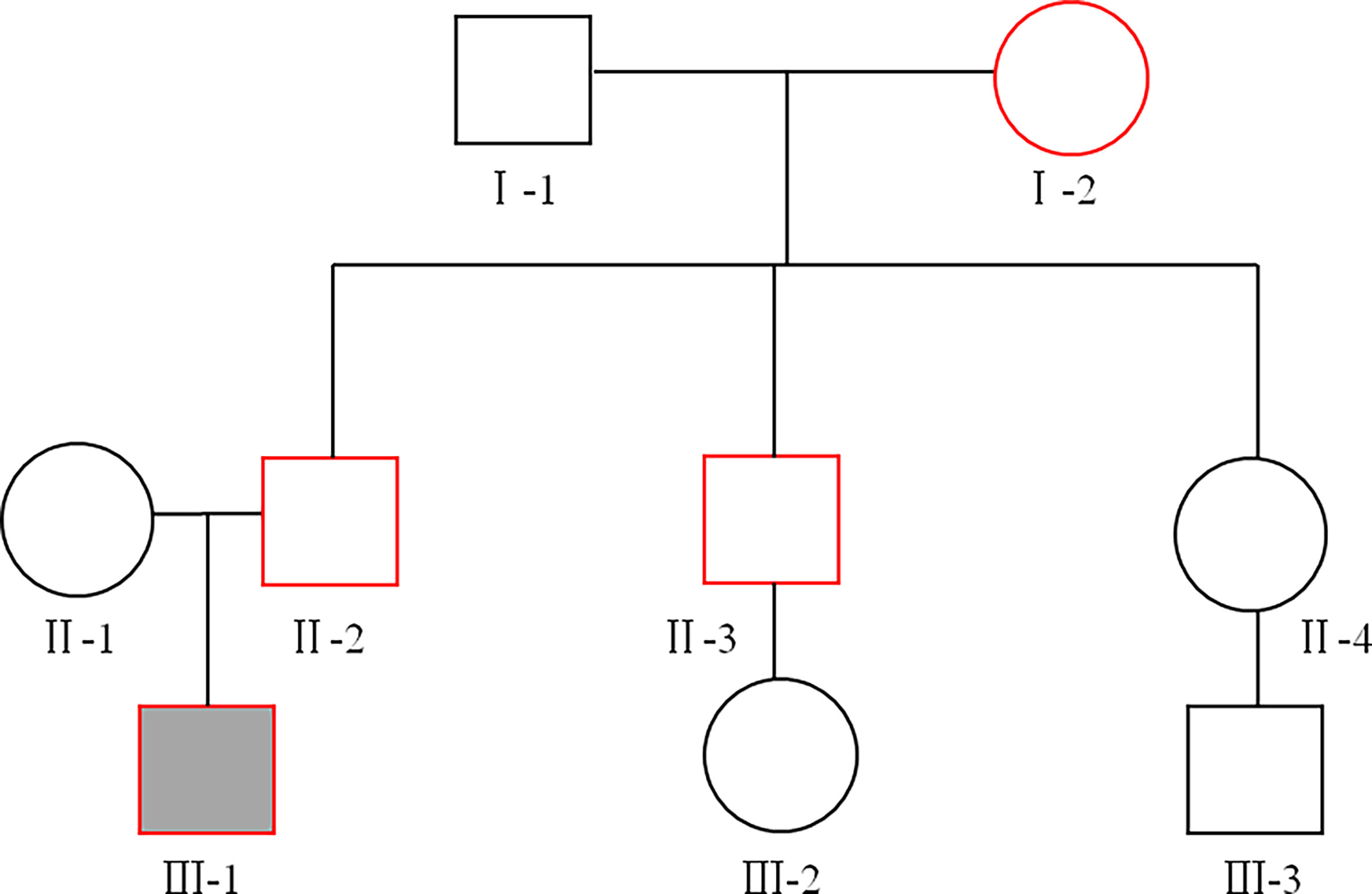
Figure 1 The family pedigree. Squares represent males, circles represent females. The proband is represented by a solid black box, and the red line indicates patients with diabetes. The proband (III-1), his father (II-2), his uncle (II-3), and his grandmother (I-2) were diagnosed with type 2 diabetes (T2D) at the age of 18, 42, 43, and 60, respectively.
Variant Detection
Exome Capture Statistics are shown in Table 2. In brief, a total of 130076 mutations were identified by WES, including 118031 SNPs and 12045 InDels. Among them, the numbers of non-synonymous SNVs (Single nucleotide variant), synonymous SNVs, frameshift deletions, frameshift insertions, non-frameshift deletions, and non-frameshift insertions were 56218, 58449, 2708, 2091, 3165, and 2389, respectively.
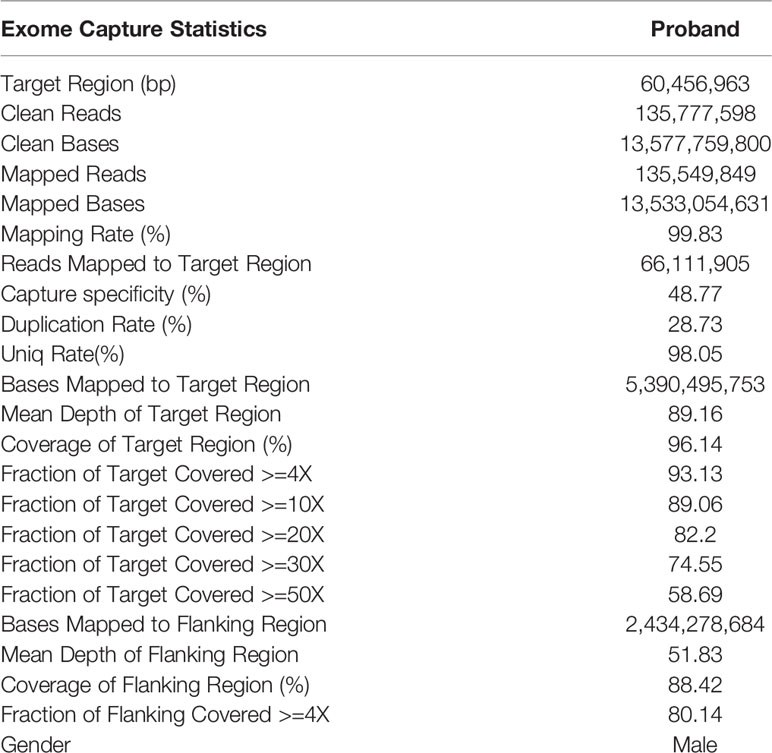
Table 2 Details of whole exome sequencing (WES).
Genetic and Bioinformatics Analyses
To identify key biological functions in the family of the proband, variants were filtered according to their genome site location, allele frequency, and functional consequences, excluding intronic and nonsense variants. Mutation sites are presented in Table 3. Functional consequences of the coding variant were estimated by several prediction programs including SIFT, PolyPhen2, Mutation Taster, Mutation Assessor, FATHMM, GERP-plus, PhyloP100, and PhastCons100 (Table 4), and the results suggest that the alteration predicted was probably pathogenic, had a deleterious effect on protein performance, and was closely related to diabetes. Sequencing in other relatives of this family and 200 non-related healthy controls revealed that affected relatives (I-2, II-2, II-3) were heterozygous for the same mutation, which was not found in non-related healthy controls (Tables 5, 6, and Figure 2). Taken together, the data demonstrate that only heterozygous missense mutations with predicted destructive effects were found in diabetic patients, thereby indicating that the variant in the GCK gene co-segregated with diabetes. The mutation was located on chromosome 7, position 44185189, c.1160C>T, NM_000162, p.Ala387Val, namely rs193921338. The p.Ala387Val mutation was analyzed for evolutionary conservation, which showed that the mutation was located in a highly conserved region among multiple animal species (Table 7). In the ExAC database (http://exac.broadinstitute.org/), the variant was categorized as a rare change with a mutation frequency of 0.00001145. In addition, in this database, the mutation was classified as likely pathogenic. Therefore, based on the findings described above and the clinical condition of this family, this rare missense mutation was considered a disease-causing mutation.

Table 3 Information of candidate pathogenic gene loci.

Table 4 Pathogenicity results of candidate gene mutation sites predicted by bioinformatics analysis.
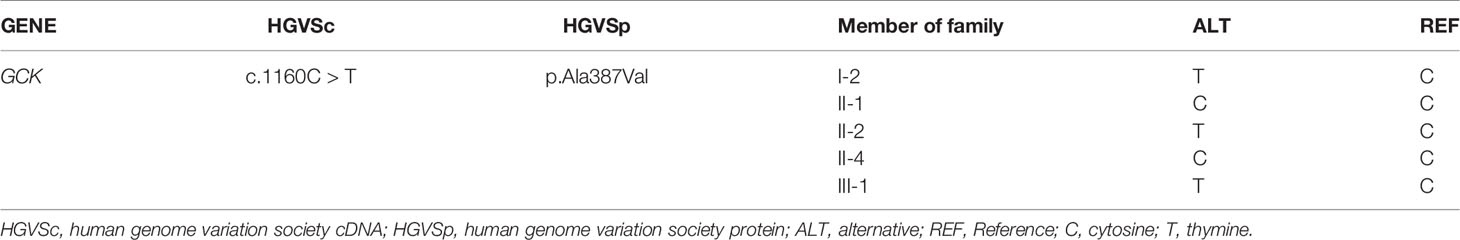
Table 5 Sanger sequencing results of candidate gene loci in family members of the proband.

Table 6 Sanger sequencing results of the c.1160C > T mutation in the GCK gene in 200 non-related healthy controls.
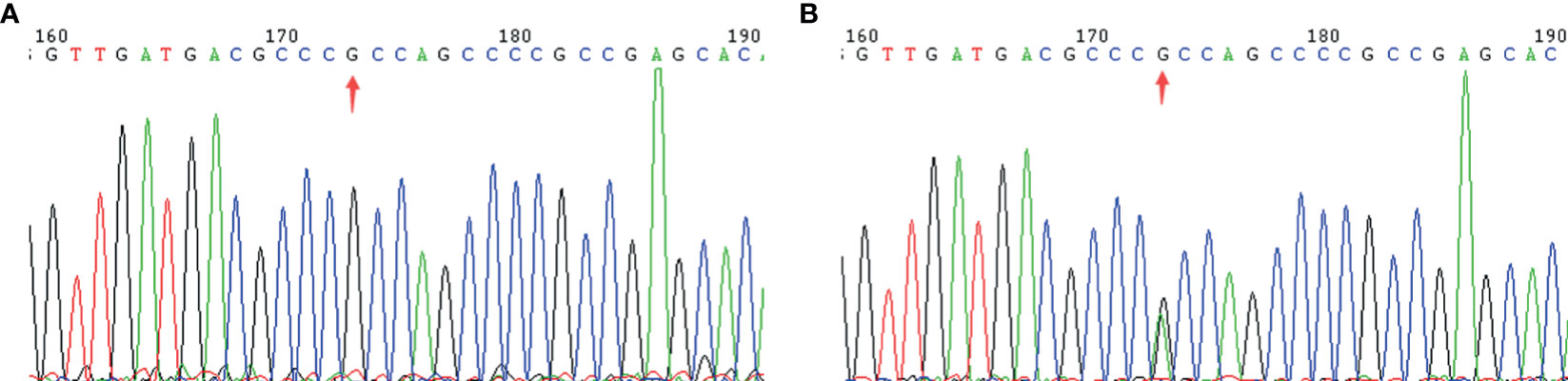
Figure 2 A mutation in the GCK gene causes MODY2. (A): GCK wild type; (B): GCK heterozygote.
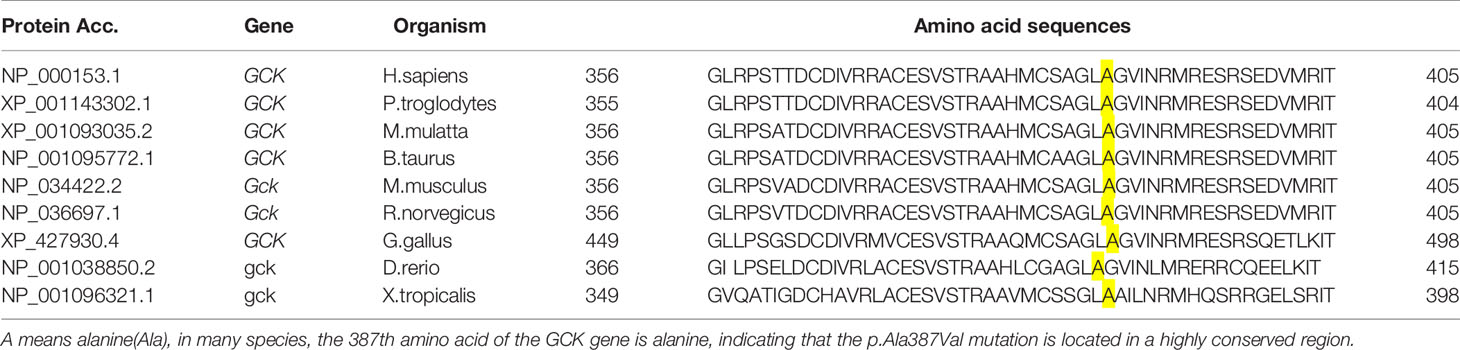
Table 7 Evolutionary conservation analysis of the p.Ala387Val mutation in the GCK gene.
Prediction of Protein Structure
Homology models of the tertiary protein structure of wild-type and p.Ala387Val GCK proteins were predicted by Swiss-Prot (Figure 3), and the mutated protein structure was compared with the normal (non-mutated) protein structure. A change in amino acids results from a nonsynonymous substitution. A close-up of amino acid change analysis shows that the number of chemical bonds and hydrophilicity of amino acid side chains have changed, which may cause structural disorder and change protein function.
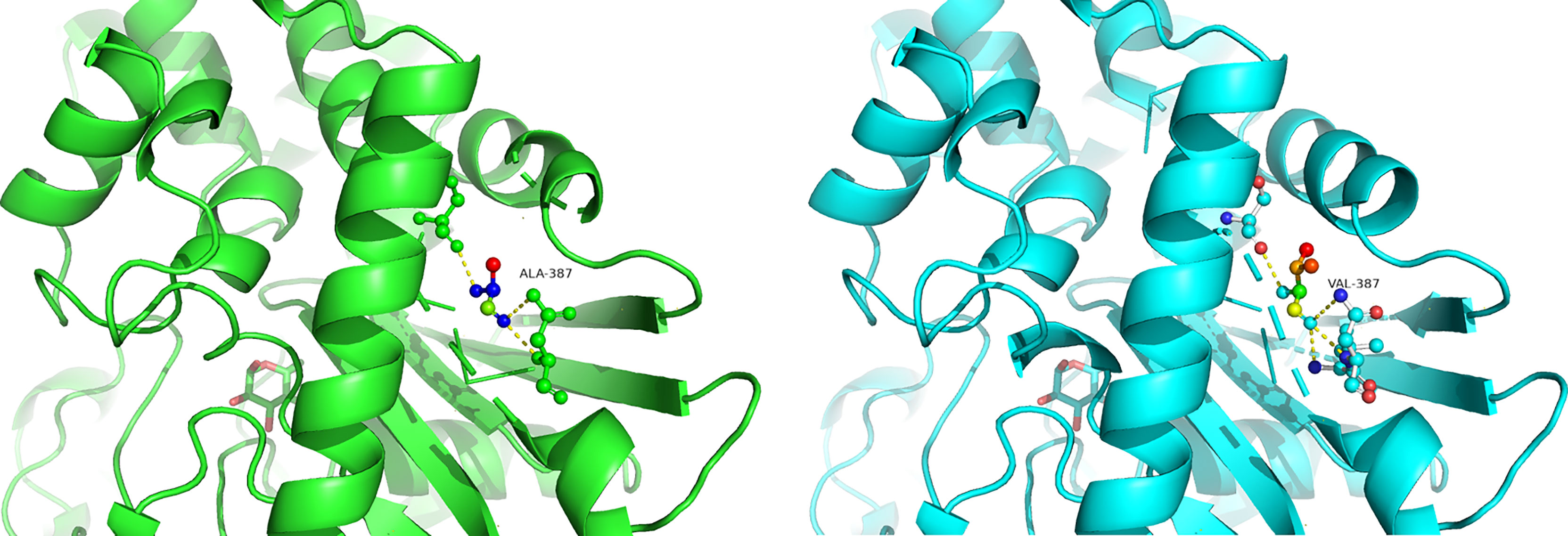
Figure 3 Predicted computational tertiary structure. The wild genotype is presented on the left; the mutant genotype is presented on the right.
Clinical Follow-Up After Drug Withdrawal
To validate the type of diabetes caused by the mutation in the GCK gene, for which no additional hypoglycemic drugs are needed, a follow-up observation was performed after drug withdrawal (Table 8). The results of blood glucose monitoring showed that after discontinuation of an oral hypoglycemic drug (metformin), the proband’s blood glucose level was still maintained at a stable level and there was no need for excessive drug therapy to control his blood glucose level.

Table 8 Fluctuations of self-monitoring blood glucose (SMBG) and HbA1c during follow-up analysis.
Discussion
MODY is a type of monogenic diabetes that is inherited in an autosomal dominant pattern with different prevalence rates in different ethnic groups (22, 23). In this study, we used WES to study the sequence variations of all genes in a suspected MODY family of early-onset diabetes. In this family, there were 5 cases of abnormal glucose metabolism, among which the proband was diagnosed with diabetes when he was younger than 25 years of age. Three generations of his immediate family members were diagnosed with diabetes, which conforms to the characteristics of autosomal dominant inheritance and meets the diagnostic criteria for MODY (24). GCK-MODY is caused by a mutation in the GCK gene, which leads to decreased activity of this enzyme, and is one of the most common characteristics of MODY (25). In previous studies, it has been shown that the prevalence of MODY caused by the mutation in the GCK gene in the Chinese population is 0.21%, and the prevalence of MODY in Chinese diabetic population is 1.3% (26).
Patients with GCK-MODY present with a mild clinical phenotype, which does not lead to serious complications of diabetes (27–29). In general, blood glucose levels can be controlled by adjustments of diet and exercise. Insulin or oral hypoglycemic drugs are not effective in lowering the blood glucose levels or HbA1c levels in GCK-MODY patients, so there is no need to over-treat the patient (other than during pregnancy) (30). In the general population, in patients with GCK-MODY who are not pregnant, ensuring a lifestyle modification involving diet and exercise is often the only necessary intervention (31, 32). This can significantly help improve the patient’s quality of life (33). In our study, we found that adherence to a healthy diet and exercise can help patients with GCK-MODY achieve good control of their blood glucose level.
So far, 620 types of mutations have been identified in the GCK gene in a total of 1441 families (34, 35), and there are still many gene variants to be discovered. This study was the first to report a GCK-MODY family in the Chinese population bearing by a heterozygous missense mutation in the GCK gene c.1160C > T (p.Ala387Val). Data analysis showed that the mutation involved a rare change with a mutation frequency of 0.00001145 of which the variant was predicted to be likely pathogenic. Recent evidence has suggested that the coding region in which the mutation is located is a highly conserved region in many species. The mutation can cause changes in protein structure and affect protein stability, thereby affecting functional changes. Therefore, we have reason to believe that the GCK gene is the disease-causing gene in this diabetes family, and the pathogenic mutation site is rs193921338, which was first discovered in the Chinese population. For GCK-MODY, correct molecular genetic diagnosis is critical (36). Young patients, especially children, suffer from diabetes, and there is no typical clinical evidence of T1DM or T2DM. Therefore, GCK-MODY should be highly suspected, and further investigation and screening of the GCK gene mutation should be conducted in the future (37). These findings will help predict the clinical course of the disease and affect a patient’s diagnosis and treatment plan. In a cross-sectional and longitudinal study involving 799 patients with a heterozygous mutation in the GCK gene in the UK, patients with a GCK gene mutation showed no significant changes in blood glucose levels and HbA1c levels after drug treatment (38). In this study, follow-up blood glucose monitoring after drug withdrawal of the proband showed that blood glucose control was sufficient and drug therapy was not required. In addition, several GCK-MODY patients in this family developed ocular complications (cataracts) or cardiovascular disease (hypertension, stroke) later in life. Interestingly, these complications are relatively mild and did not significantly increase their mortality.
In conclusion, for patients with a high clinical suspicion of MODY, a definite diagnosis should be made through genetic testing. Correct diagnosis helps patients to choose a personal treatment plan and avoid overtreatment. In our study, we reported a GCK-MODY family with a mutation in exon 9 of the GCK gene c.1160C > T (p.Ala387Val), which, according to our knowledge, was the first time to be detected in the Chinese population. For this GCK-MODY family, exercise and diet were sufficient as a therapeutic approach. The underlying molecular mechanism of action of GCK gene c.1160C > T (p.Ala387Val) participation in diabetes remains to be elucidated and future prospective investigations are warranted.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI SRA: PRJNA744432.
Ethics Statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
Conceived and designed the experiments: JL and HZ. Performed the experiments: JL, XW, AD, CW, JW, SJ, and HZ. Analyzed the data: JL and MS. Wrote the paper: MS and JL. JL and MS contributed equally. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Grants from the National Natural Science Foundation of China (81900719, 81800704), Grants from the Health and Family Planning Commission of Wuhan City (Grant Number WX18M02).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation with the authors at time of review.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank all the volunteers that participated in this study.
References
1. Gloyn AL. Glucokinase (GCK) Mutations in Hyper- and Hypoglycemia: Maturity-onset Diabetes of the Young, Permanent Neonatal Diabetes, and Hyperinsulinemia of Infancy. Hum Mutat (2003) 22(5):353–62. doi: 10.1002/humu.10277
2. Todd JN, Srinivasan S, Pollin TI. Advances in the Genetics of Youth-Onset Type 2 Diabetes. Curr Diabetes Rep (2018) 18(8):57. doi: 10.1007/s11892-018-1025-1
3. Urakami T. Maturity-Onset Diabetes of the Young (MODY): Current Perspectives on Diagnosis and Treatment. Diabetes Metab Syndrome Obes: Targets Ther (2019) 12:1047–56. doi: 10.2147/DMSO.S179793
4. Bishay RH, Greenfield JR. A Review of Maturity Onset Diabetes of the Young (MODY) and Challenges in the Management of Glucokinase-MODY. Med J Aust (2016) 205(10):480–5. doi: 10.5694/mja16.00458
5. Anuradha S, Radha V, Mohan V. Association of Novel Variants in the Hepatocyte Nuclear Factor 4A Gene With Maturity Onset Diabetes of the Young and Early Onset Type 2 Diabetes. Clin Genet (2011) 80(6):541–9. doi: 10.1111/j.1399-0004.2010.01577.x
6. Kooptiwut S, Plengvidhya N, Chukijrungroat T, Sujjitjoon J, Semprasert N, Furuta H, et al. Defective PAX4 R192H Transcriptional Repressor Activities Associated With Maturity Onset Diabetes of the Young and Early Onset-Age of Type 2 Diabetes. J Diabetes Its Complications (2012) 26(4):343–7. doi: 10.1016/j.jdiacomp.2012.03.025
7. Zhuang L, Zhao Y, Zhao W, Li M, Yu M, Lu M, et al. The E23K and A190A Variations of the KCNJ11 Gene are Associated With Early-Onset Type 2 Diabetes and Blood Pressure in the Chinese Population. Mol Cell Biochem (2015) 404(1-2):133–41. doi: 10.1007/s11010-015-2373-7
8. Gragnoli C, Stanojevic V, Gorini A, Von Preussenthal GM, Thomas MK, Habener JF. Ipf-1/MODY4 Gene Missense Mutation in an Italian Family With Type 2 and Gestational Diabetes. Metabolism (2005) 54(8):983–8. doi: 10.1016/j.metabol.2005.01.037
9. Ivanoshchuk DE, Shakhtshneider EV, Rymar OD, Ovsyannikova AK, Mikhailova SV, Fishman VS, et al. The Mutation Spectrum of Maturity Onset Diabetes of the Young (Mody)-Associated Genes Among Western Siberia Patients. J Pers Med (2021) 11(1):57. doi: 10.3390/jpm11010057
10. Naylor RN, John PM, Winn AN, Carmody D, Greeley SAW, Philipson LH, et al. Cost-Effectiveness of MODY Genetic Testing: Translating Genomic Advances Into Practical Health Applications. Diabetes Care (2014) 37(1):202–9. doi: 10.2337/dc13-0410
11. Wang X, Wang T, Yu M, Zhang H, Ping F, Zhang Q, et al. Screening of HNF1A and HNF4A Mutation and Clinical Phenotype Analysis in a Large Cohort of Chinese Patients With Maturity-Onset Diabetes of the Young. Acta Diabetol (2019) 56(3):281–8. doi: 10.1007/s00592-018-1232-x
12. Liang H, Zhang Y, Li M, Yan J, Yang D, Luo S, et al. Recognition of Maturity-Onset Diabetes of the Young in China. J Diabetes Investig (2021) 12(4):501–9. doi: 10.1111/jdi.13378
13. Weinreich SS, Bosma A, Henneman L, Rigter T, Spruijt CT, Grimbergen AJ, et al. A Decade of Molecular Genetic Testing for MODY: A Retrospective Study of Utilization in The Netherlands. Eur J Hum Genet (2015) 23(1):29–33. doi: 10.1038/ejhg.2014.59
14. Fu J, Wang T, Liu J, Wang X, Zhang Q, Li M, et al. Using Clinical Indices to Distinguish Mody2 (Gck Mutation) and MODY3 (Hnf1a Mutation) From Type 1 Diabetes in a Young Chinese Population. Diabetes Ther (2019) 10(4):1381–90. doi: 10.1007/s13300-019-0647-x
15. Shields BM, McDonald TJ, Ellard S, Campbell MJ, Hyde C, Hattersley AT. The Development and Validation of a Clinical Prediction Model to Determine the Probability of MODY in Patients With Young-Onset Diabetes. Diabetologia (2012) 55(5):1265–72. doi: 10.1007/s00125-011-2418-8
16. Thomson KL, Gloyn AL, Colclough K, Batten M, Allen LIS, Beards F, et al. Identification of 21 Novel Glucokinase (GCK) Mutations in UK and European Caucasians With Maturity-Onset Diabetes of the Young (MODY). Hum Mutat (2003) 22(5):417–. doi: 10.1002/humu.9186
17. Carmody D, Naylor RN, Bell CD, Berry S, Montgomery JT, Tadie EC, et al. Gck-MODY in the US National Monogenic Diabetes Registry: Frequently Misdiagnosed and Unnecessarily Treated. Acta Diabetol (2016) 53(5):703–8. doi: 10.1007/s00592-016-0859-8
18. Flannick J, Mercader JM, Fuchsberger C, Udler MS, Mahajan A, Wessel J, et al. Exome Sequencing of 20,791 Cases of Type 2 Diabetes and 24,440 Controls. Nature (2019) 570(7759):71–6. doi: 10.1038/s41586-019-1231-2
19. Aksyonov SA, Bittner M, Bloom LB, Reha-Krantz LJ, Gould IR, Hayes MA, et al. Multiplexed DNA Sequencing-by-Synthesis. Analytical Biochem (2006) 348(1):127–38. doi: 10.1016/j.ab.2005.10.001
20. Johansson S, Irgens H, Chudasama KK, Molnes J, Aerts J, Roque FS, et al. Exome Sequencing and Genetic Testing for MODY. PloS One (2012) 7(5):e38050. doi: 10.1371/journal.pone.0038050
21. American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2019. Diabetes Care (2018) 42(Supplement 1):S13–28. doi: 10.2337/dc19-S002
22. Hattersley AT, Patel KA. Precision Diabetes: Learning From Monogenic Diabetes. Diabetologia (2017) 60(5):769–77. doi: 10.1007/s00125-017-4226-2
23. Harron KL, Feltbower RG, McKinney PA, Bodansky HJ, Campbell FM, Parslow RC. Rising Rates of All Types of Diabetes in South Asian and Non-South Asian Children and Young People Aged 0-29 Years in West Yorkshire, U.K., 1991-2006. Diabetes Care (2011) 34(3):652–4. doi: 10.2337/dc10-1512
24. Thanabalasingham G, Owen KR. Diagnosis and Management of Maturity Onset Diabetes of the Young (MODY). Bmj (2011) 343(oct19 3):d6044–d. doi: 10.1136/bmj.d6044
25. Gardner DS, Tai ES. Clinical Features and Treatment of Maturity Onset Diabetes of the Young (MODY). Diabetes Metab Syndr Obes (2012) 5:101–8. doi: 10.2147/DMSO.S23353
26. Ma Y, Han X, Zhou X, Li Y, Gong S, Zhang S, et al. A New Clinical Screening Strategy and Prevalence Estimation for Glucokinase Variant-Induced Diabetes in an Adult Chinese Population. Genet Med (2018) 21(4):939–47. doi: 10.1038/s41436-018-0282-3
27. Colom C, Corcoy R. Maturity Onset Diabetes of the Young and Pregnancy. Best Pract Res Clin Endocrinol Metab (2010) 24(4):605–15. doi: 10.1016/j.beem.2010.05.008
28. Froguel P, Zouali H, Vionnet N, Velho G, Vaxillaire M, Sun F, et al. Familial Hyperglycemia Due to Mutations in Glucokinase. Definition of a Subtype of Diabetes Mellitus. New Engl J Med (1993) 328(10):697–702. doi: 10.1056/NEJM199303113281005
29. Pearson ER, Velho G, Clark P, Stride A, Shepherd M, Frayling TM, et al. Beta-Cell Genes and Diabetes: Quantitative and Qualitative Differences in the Pathophysiology of Hepatic Nuclear factor-1alpha and Glucokinase Mutations. Diabetes (2001) 50 Suppl 1:S101–S7. doi: 10.2337/diabetes.50.2007.S101
30. Chakera AJ, Steele AM, Gloyn AL, Shepherd MH, Shields B, Ellard S, et al. Recognition and Management of Individuals With Hyperglycemia Because of a Heterozygous Glucokinase Mutation. Diabetes Care (2015) 38(7):1383–92. doi: 10.2337/dc14-2769
31. Hosokawa Y, Higuchi S, Kawakita R, Hata I, Urakami T, Isojima T, et al. Pregnancy Outcome of Japanese Patients With Glucokinase-Maturity-Onset Diabetes of the Young. J Diabetes Investig (2019) 10(6):1586–9. doi: 10.1111/jdi.13046
32. Pinés Corrales PJ, López Garrido MP, Louhibi Rubio L, Aznar Rodríguez S, López Jiménez LM, Lamas Oliveira C, et al. Importance of Clinical Variables in the Diagnosis of MODY2 and MODY3. Endocrinol Nutr (2011) 58(7):341–6. doi: 10.1016/j.endonu.2011.05.002
33. Bonfig W, Hermanns S, Warncke K, Eder G, Engelsberger I, Burdach S, et al. Gck-MODY (Mody 2) Caused by a Novel p.Phe330Ser Mutation. ISRN Pediatr (2011) 2011:676549. doi: 10.5402/2011/676549
34. Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanné-Chantelot C, Ellard S, et al. Update on Mutations in Glucokinase (GCK), Which Cause Maturity-Onset Diabetes of the Young, Permanent Neonatal Diabetes, and Hyperinsulinemic Hypoglycemia. Hum Mutat (2009) 30(11):1512–26. doi: 10.1002/humu.21110
35. Ellard S, Beards F, Allen LI, Shepherd M, Ballantyne E, Harvey R, et al. A High Prevalence of Glucokinase Mutations in Gestational Diabetic Subjects Selected by Clinical Criteria. Diabetologia (2000) 43(2):250–3. doi: 10.1007/s001250050038
36. Ma ML, Ping F, Chang YS, Li YX. A Three-Year Follow-Up Observation of a Pedigree of Maturity Onset Diabetes of the Young Caused by a Novel Mutation of Glucokinase and Literature Review. Zhonghua Nei Ke Za Zhi (2020) 59(5):366–71. doi: 10.3760/cma.j.cn112138-20191118-00759
37. Kleinberger JW, Pollin TI. Undiagnosed MODY: Time for Action. Curr Diab Rep (2015) 15(12):110. doi: 10.1007/s11892-015-0681-7
Keywords: glucokinase, monogenic diabetes, mutation, MODY2, pedigree
Citation: Li J, Shu M, Wang X, Deng A, Wen C, Wang J, Jin S and Zhang H (2021) Precision Therapy for a Chinese Family With Maturity-Onset Diabetes of the Young. Front. Endocrinol. 12:700342. doi: 10.3389/fendo.2021.700342
Received: 26 April 2021; Accepted: 08 July 2021;
Published: 05 August 2021.
Edited by:
Juan Zheng, Huazhong University of Science and Technology, Wuhan, ChinaReviewed by:
Changhan Ouyang, Hubei University of Science and Technology, ChinaGuidong Dai, Kaili University, China
Copyright © 2021 Li, Shu, Wang, Deng, Wen, Wang, Jin and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Si Jin, amluc2lAaHVzdC5lZHUuY24=; Hongmei Zhang, emhtNzAwMUAxNjMuY29t
†These authors have contributed equally to this work