 Kanikkai Raja Aseer
Kanikkai Raja Aseer Josephine M. Egan
Josephine M. Egan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 05 July 2021
Sec. Cellular Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.699661
This article is part of the Research TopicMultifaceted Cannabinoids: Regulators of Normal and Pathological Function in Metabolic and Endocrine OrgansView all 8 articles
While endocannabinoids (ECs) and cannabis were primarily studied for their nervous system effects, it is now clear that ECs are also produced in the periphery where they regulate several physiological processes, including energy storage, glucose and lipid metabolism, insulin secretion and synthesis, and hepatocyte function. Within islet of Langerhans there is an autonomous EC system (ECS). Beta (β)-cells contain all the enzymes necessary for EC synthesis and degradation; ECs are generated in response to cellular depolarization; their paracrine influence on β-cells is mostly through the cannabinoid 1 receptor (CB1R) that is present on all β-cells; they modulate basal and glucose- and incretin-induced insulin secretion, and β-cell responses to various stressors. Furthermore, there is now accumulating evidence from preclinical studies that the autonomous islet ECS is a key player in obesity-induced inflammation in islets, and β-cell damage and apoptosis from many causes can be mitigated by CB1R blockers. We will thoroughly review the literature relevant to the effects of ECs and their receptors on β-cells and the other cell types within islets. Therapeutic potential of agents targeting EC/CB1R and CB2R is highly relevant because the receptors belong to the druggable G protein-coupled receptor superfamily. Present research in the ECS must be considered preliminary, especially with regards to human islet physiology, and further research is needed in order to translate basic cellular findings into clinical practice and the use of safe, clinically approved CBR modulators with and without glucose lowering combinations presently in therapeutic use for diabetes and obesity needs to be studied.
Healthy β-cells within the pancreatic islets of Langerhans, a storehouse of insulin, are poised to produce a rapid and sensitive response to acute changes in circulating blood glucose concentrations by first rapidly secreting insulin from the stored pool, and next quickly replenishing the pool by new insulin synthesis. However, impairment of the insulin response results from chronic overnutrition and unfavorable lifestyles, that, in turn, cause β-cell damage and diabetes. The physiology and mechanism of insulin secretion from β-cells is well orchestrated and involves a series of signaling events (1–3): 1) key glucose transporters, GLUT-2 in rodents and GLUT-1 in humans, allow the entry of glucose into β-cells; 2) intracellular ATP is then generated via glycolysis; 3) an increasing ATP/ADP ratio closes the inwardly rectifying ATP-sensitive potassium (KATP) channels responsible for potassium ion efflux from the β-cells and for maintaining a resting membrane potential; 4) the resultant reduction in potassium ion efflux causes membrane depolarization and subsequent opening of L-type voltage-dependent calcium channels (VDCCs) allowing influx of calcium ions (Ca2+) into β-cells; 5) synchronous oscillations of elevated cytosolic Ca2+ and membrane excitability stimulate insulin release from β-cells into the systemic circulation. All of these events are modified by enhancing and inhibiting influences, which is where the ECS comes into play.
Much attention has been paid to the activation of membrane receptors of the G protein-coupled receptor superfamily in response to incretin secretion from the gut. This attention was warranted because it resulted in a new class of compounds, the glucagon-like peptide-1 receptor agonists (GLP-1R agonists), being now widely used to treat Type 2 diabetes (T2DM). They lower blood glucose because of increased insulin secretion and synthesis due to direct activation of adenylyl cyclase (AC) that results in increases in intracellular β-cell cAMP levels and downstream PKA activation. An added benefit is that their chronic use leads to a decrease in food intake because of a central action (4, 5). Furthermore, they have protective effects on the cardiovascular system (6–11). In contrast, there are inhibitory influences on insulin secretion due to activation of membrane G protein-coupled receptors, with somatostatin receptor activation being the most studied and clinically relevant (12, 13). More recently, it’s become clear that cannabinoid receptors (CBRs), which are also G protein-coupled receptors, are present in pancreatic islets and in β-cells specifically (14–23). We now also know that CB1R endogenous ligands and synthetic agonists contribute to inhibition of glucose-stimulated insulin secretion (GSIS) due to inhibition of AC, and they are involved in β-cell adaptation (and maladaptation) to prolonged nutrient overload and inflammation (18, 24, 25).
Endocannabinoids (ECs), anandamide (AEA) and 2-arachidonoyl glycerol (2-AG), are lipid messengers that are not pre-stored within cells but are synthesized and released on demand (26, 27). They are enzymatically produced by the essential enzymes, N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) and diacylglycerol lipase (DAGLα), respectively, from membrane phospholipid-derived precursors containing arachidonic acid (a 20-carbon membrane phospholipid) or circulating fatty acid precursors and the rate of EC release depends on their synthesis rate. ECs are degraded primarily by fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) and the degraded by-products are recycled back to cell membranes (27, 28). However, other enzymes implicated in the biosynthesis of AEA and 2-AG also exist, at least in mouse brain. Among these, 2-AG can be also produced by α,β-hydrolase-4 (ABHD4), the glycerophosphodiesterase-1 (GDE1), a soluble phospholipase A2, an unidentified phospholipase C, and phosphatases (29), whereas AEA can be produced by another isoform of DAGLβ (30). In addition, AEA can be degraded by one other enzyme, N-acylethanolamine acid amidase (31). EC synthesis rates are increased in both obese humans and rodents upon overconsumption of high caloric dietary meals rich in fats and sugars (19, 32). ECs have been found to be produced in adipose tissue, muscle (33, 34), and kidneys (35), all of which are involved in the control of energy metabolism. Moreover, ECs are also produced in gut, and they in turn at a minimum influence hepatic function and signaling through the vagal afferents (25, 36–39). Additionally, ECs are produced in stellate cells of liver (40) and islets of Langerhans: this review focuses on islets of Langerhans.
CBRs are G protein-coupled receptors, as mentioned above, that are present on cell and mitochondrial membranes (24, 41–47). CB1Rs were first described as being abundant in the pre-synaptic cells of the central nervous system (CNS) where they control retrograde synaptic signaling and regulate, amongst their many functions, appetite and reward response in the hypothalamus (44). CB2Rs are primarily considered to be associated with the immune system (42, 48). Apart from the classical CBRs, the non-classical receptors GPR55 and transient receptor potential vanilloid 1 (TRPV1) are present in high levels in the brain (49, 50) and relay signals intracellularly through Gα13 and Gq proteins (51). CBRs were subsequently also found to be present in mouse, rat, and human islets (14–21, 24, 52–56).
Islets of Langerhans are clusters of endocrine cells embedded in the pancreas that control blood glucose homeostasis. An islet is comprised of insulin-secreting β-cells, glucagon-secreting alpha (α-) cells, somatostatin-secreting delta (δ-) cells, pancreatic polypeptide-secreting PP-cells (sometimes referred to as γ-cells) and sparce ghrelin-containing ϵ-cells. β-cells are the most predominant cellular component of islets in most species – ~80% in rodents and ~50–60% in humans-the other islet endocrine cell types therefore constitute a much smaller proportion of the islet mass (57). As regards PP- and ϵ-cells there are no data with reference to the ECS.
In 1986 it was first documented that delta-9-tetrahydrocannabinol (Δ9-THC), the primary psychoactive constituent of cannabis, was involved in modulating insulin secretion (58). In the early 1990s, CB1R and CB2R were successfully cloned (59), although an exhaustive search and comprehensive knowledge of their distribution throughout the body was not delineated at the time. More than 30 years have elapsed since the first discovery of Δ9-THC’s effects on insulin secretion. With the established involvement of ECS in peripheral metabolism it seemed reasonable that the ECS might have a role in islet function; this is now being explored by several research groups (16, 21, 24, 60–65). Studies have documented the existence of a functional ECS, including EC enzymatic machinery and CBRs, in pancreatic islets (as described below). But researchers involved in studying the ECS in islets have failed to reach a consensus on exactly which endocrine islet cell types other than β-cells synthesize ECs in a regulated manner. The presence of both CB1R and CB2R in islets of mouse, rat and human has also been documented, and again, exactly which cell types have which functioning receptors is not definitive (14–21).
Starowicz et al. demonstrated for the first time that the critical biosynthetic enzymes for EC synthesis (DAGLα and NAPE-PLD) were present in islets of mouse, and that they were mostly in α-cells, whereas degrading enzymes MAGL and FAAH seemed restricted to β-cells. The authors then suggested that AEA and 2-AG participate locally as autocrine and intra islet paracrine signaling molecules in the control of secretory products from islet cells (19). However, they did not report that AEA and 2-AG were actually synthesized or secreted from α-cells. Further studies on the occurrence and cellular distribution of biosynthesizing and metabolizing enzymes inside the islets of Langerhans of mouse, rat and human have now emerged. Concerning the synthesizing enzymes in mouse islets, DAGLα is expressed in fetal α-cells, however expression levels of the enzymes (DAGLα and NAPE-PLD) were reported to be higher in β-cells in mature islets compared with α-cells (19, 24, 55). Of the degrading enzymes, MAGL and FAAH, the former was reported to be present in both α- and β-cells (mouse fetal and adult), but the latter was restricted only to β-cells (19, 24, 55). There is a poor agreement between the mouse and rat islet degrading enzymes MAGL and FAAH defined by expression of the latter enzyme mostly in α-cells, with β-cells showing a weak labeling, and the former enzyme localized to δ-cells of rat islets (20). In reviewing the literature, no data was found on the expression of synthesizing enzymes in islets of rat. However, a high glucose ‘pulse’ elevated the levels of both AEA and 2-AG in RIN-m5F rat insulinoma cells, which could be relevant to the notion of EC synthesis in β-cells as a source of ECs in rat islets (32). The distribution of synthesizing enzymes in human islets seems similar to mouse islets (24) but expression levels in the different islet cell types is unclear. DAGLα and NAPE-PLD were reported to be more abundant in β-cells than in α-cells, with one of the metabolizing enzymes FAAH reported to be confined to β-cell-rich areas (16): another report however detected FAAH mostly in α-cells (20). DAGLα and MAGL are present in both β- and α-cells (24, 55, 66).
ECs are generated within β-cells in response to cellular depolarization. When isolated mouse and human islets were subjected to depolarization by either KCl or increasing glucose concentrations, increasing amounts of ECs both 2-AG and AEA, measured by tandem mass spectrometry analysis, were generated (24). This therefore clearly shows that β-cells are generating ECs in response to depolarization. Mouse fetal α-cells produce 2-AG and α-cells in human islets are also reported to produce both ECs (55) but the stimulus or control of regulated synthesis in α-cells, if any, unlike the case for β-cells, have not been reported. EC synthesis in δ-cells has not been reported. Of note, disruption of MAGL, the enzyme key to degrade 2-AG, caused substantial increases in insulin and glucagon release from isolated human islets, demonstrating an essential role of endogenous 2-AG signaling in mediating both β-cell and α-cell function (66). Together, these reports indicate that α- and β-cells are likely sources of EC (both AEA and 2-AG) production.
Both AEA and 2-AG were reported to be overproduced in the pancreas of diet-induced hyperglycemic obese mice as compared with normal diet-fed mice (32). After 3, 8, or 14 weeks feeding of a high fat diet (HFD) to mice, increased biosynthesis of ECs and their decreased degradation within β-cells occurred, which was reflected by increased AEA or 2-AG in the whole pancreas of mice (19, 32). In fact, the HFD led to increased expression of the biosynthesizing enzymes and decreased expression of the degrading enzyme, FAAH, in β-cells, in keeping with elevated pancreatic AEA or 2-AG levels (19).
With respect to CBRs, earlier reports, based on mouse studies, suggested that CB1R is expressed exclusively on α-cells (17–19). In addition, mouse islet α- and presumably non-β-cells, whose islet cell type was not exactly defined but had the morphology of α-cells, were found to express CB2R (14, 19, 67) while later studies [including those reported by Juan-Pico et al. (14)] suggested it was also expressed in β-cells (17, 19, 21), but less so in α-cells (17). However, using laser capture of specific islet cells, CB1R was found to be specifically enriched in adult mouse β-cells and subsequent studies confirmed these results (21, 24, 55). A few studies reported that δ-cells also contain CB1R (20, 67) and CB2R (67). In addition to the classical receptors, GPR55 (a non-classical cannabinoid receptor) was mainly reported to be expressed in β-cells and in a subset of α-cells but was absent or had extremely low expression in δ-cells (53, 54). Moreover, TRPV1 was reported to be present in fetal and mature β- and α-cells in mice (55).
In rat islets, there is also lack of clarity as to which cells express CBRs. Both α- and β-cells were reported to express both CBRs (15, 17, 68). In keeping with their mouse studies, Bermudez-Silva reported that CB1R is more highly expressed relative to CB2R expression in rat α-cells compared to β-cells (17). In contrast, a more recent study reported that the abundance of both CB1R and CB2R is much higher in β-cells than non-β-cells, whose cell types were not determined (64). There is evidence that CB1R is also present in δ-cells (20). In rat, despite disparities in the degree of receptor expression, all three islet cell types (α, β and δ) express CB1R but CB2R has been so far reported to be limited to α- and β-cells (15, 17, 64, 68). The non-classical receptor GPR55 has been reported to be specifically detected in β-cells (52) and TRPV1 tended to be localized to the core portion of rat islets (and therefore presumably β-cells) (56).
In human islets, CB1R was reported to be expressed in α-cells, but in only a small proportion of β-cells (16, 17). However, another group reported that CB1R appeared confined to δ-cells and was not expressed by either α- or β-cells (20). Other reports over the last decade have documented the presence of CB1R in both α- and β-cells of human islets (24, 55). CB2R was also enriched in δ- and β-cells but not α-cells (16, 17, 24) though this is far from definitive. Whereas β-cells and a majority of α-cells express the non-classical receptor GPR55, this receptor was absent or expressed less in δ-cells (54).
Overall, an extensive body of research has demonstrated that the EC components are present within the pancreatic islets of mice, rats and humans. Among these, the majority of enzymes implicated in the synthesis and degradation of ECs have indeed been detected mostly in α- and β-cells of mouse and human but exhibit a very limited spectrum in rat islets. However, demonstration of EC enzyme localization in δ-cells of mouse, rat and human islets is lacking. CBRs appear to be localized to both β-cells in the islet core and also in the islet mantel, where α- and δ-cells predominate across the three species. The existence and function of CBRs and EC enzymes have not yet been studied in PP- and ϵ-cells. The publications and reported findings concerning cellular expression of various components of the ECS in islet cells of mouse, rat and humans are summarized in Tables 1, 2, and Figure 1. All this information is essential to have a complete picture of the importance of ECS in islets. Considering the apparent discrepancies between species, much exploration is required to further determine the detailed islet cell-type specific molecular profiling of ECS within pancreatic islets.
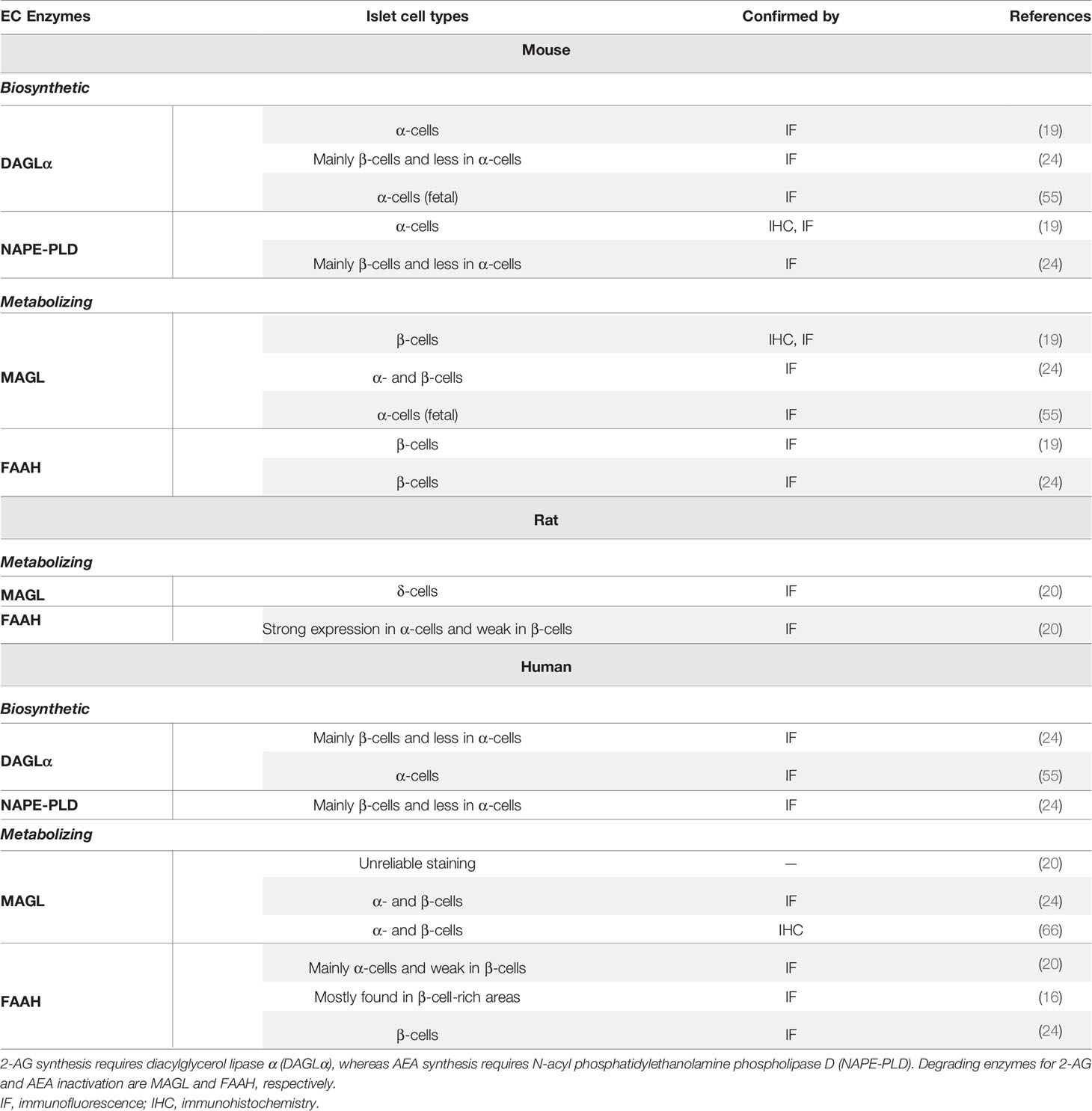
Table 1 Summary of similarities and differences of expression of biosynthetic and degrading enzymes in mouse, rat and human islets.
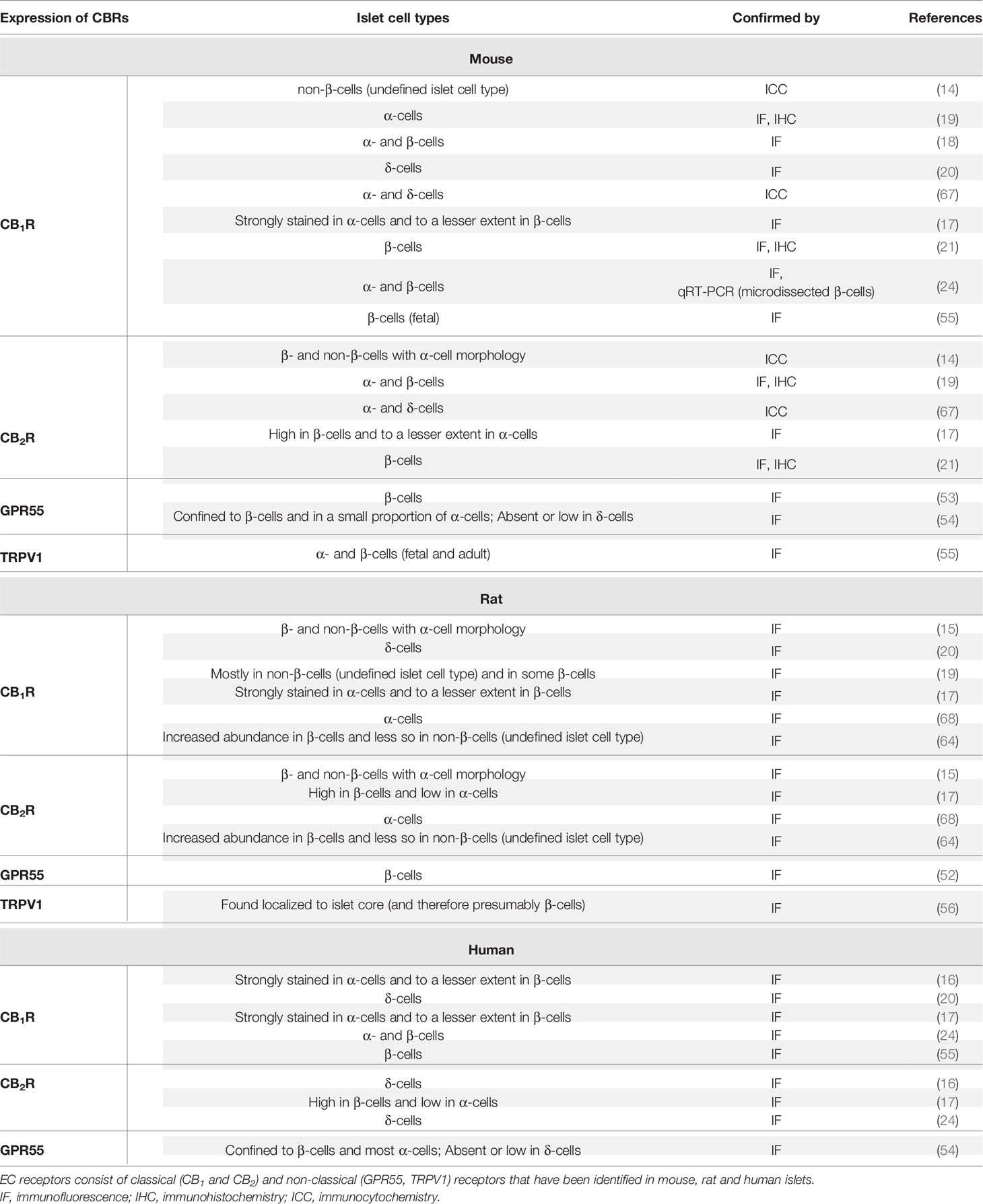
Table 2 Summary of species-specific similarities and differences in CBR expression.
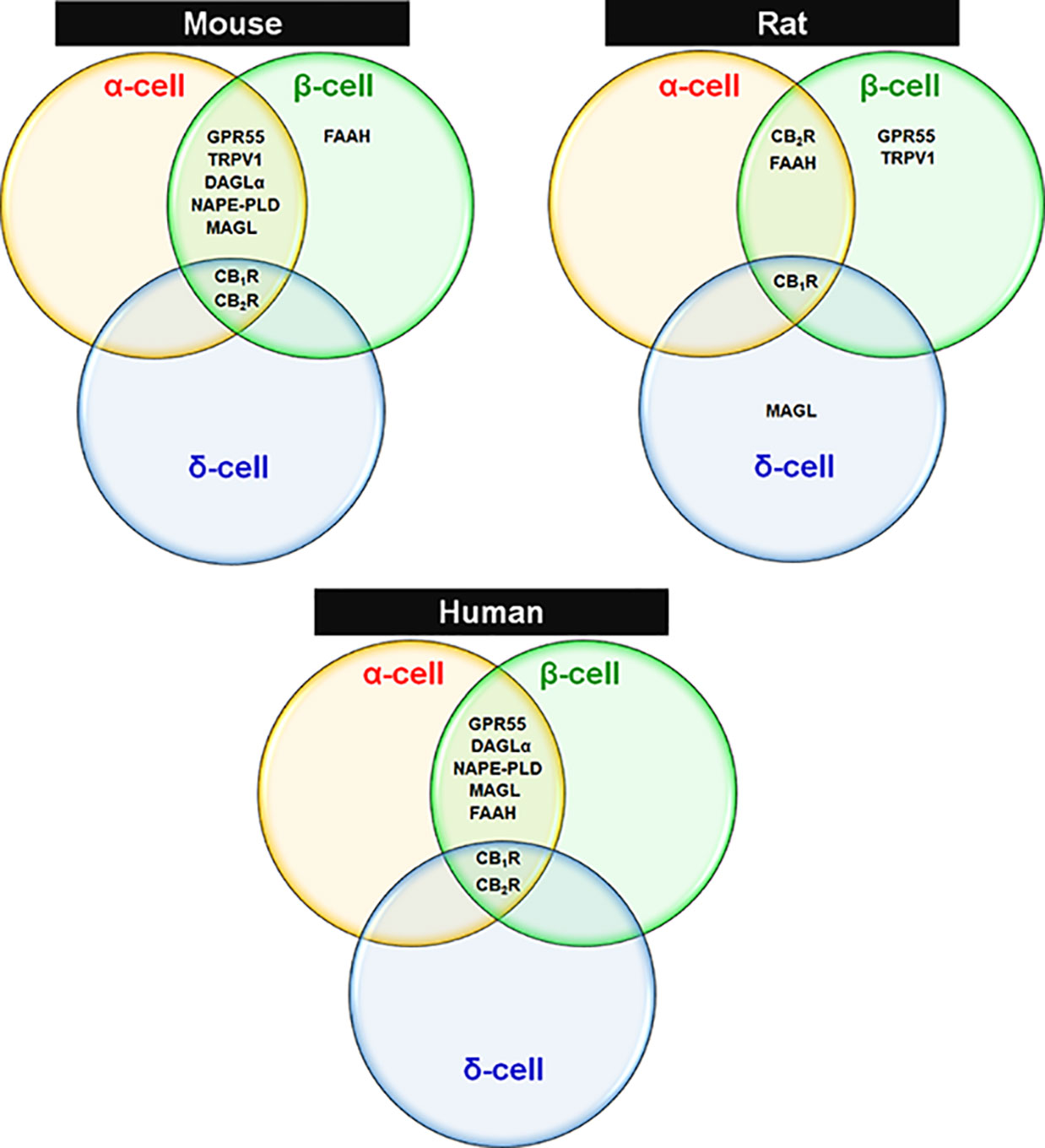
Figure 1 Expression of ECS elements in endocrine cells of mouse, rat and human islets. Venn diagrams are drawn based on reported findings and publications cited in Tables 1 and 2 regarding species-specific and islet cell type-specific ECS components.
A cardinal feature of CBR physiology is that constitutively synthesized CB1R is by no means a static component of the cellular machinery; rather approximately 70% of CB1Rs have an elimination half-life of approximately 5 hours while at least 30% of the receptors have a half-life as long as 24 hours, based on kinetic studies in neuroblastoma cells (69). CB1R expression inversely correlates with depolarization-induced increases in cytosolic Ca2+ influx, which is permissive of insulin secretion and synthesis, as mentioned above (24, 70, 71).
Since ATP synthesis, KATP and VDCC are obvious targets at the proximal and distal rate-limiting or rate-potentiating steps of the insulin secretion under the control of calcium entry into β-cells (as described above), and with the on-going type 2 diabetes epidemic there is at present a keen interest in more fully elucidating all the molecular components that affect β-cell stimulus-secretion coupling, insulin synthesis and preserve β-cell function and mass. When CB1R is activated, it signals through Gαi/o protein, thereby inhibiting adenylyl cyclase (AC) and cAMP formation with consequent reduction in protein kinase A (PKA). This slows the movement of the insulin-laden secretory granules to the cell surface. Simultaneously, KATP channels are hyperpolarized, that is, do not close so easily, and opening of Ca2+ channels (VDCC) that normally allow cytosolic Ca2+ influx is negatively impacted, resulting in further suppression of hormone secretion from β-cells (25, 62, 71). In addition, G-protein-dependent mitogen-activated protein kinases (MAPKs; ERK1/2, JNK and p38) and nitric oxide (NO) generation are impacted (72–74). Other CB1R post-receptor interactions in islet β-cells has been defined, including (1) the interactions of its Gαi subunit with the insulin receptor to form a heterotrimeric complex that directly impairs insulin signaling by inhibiting autophosphorylation of insulin receptors and thereby diminishes the trophic effects of insulin on β-cells (60); (2) EC-mediated CB1R activation tonically counters the insulin secretagogue actions of the incretins – glucagon-like peptide (GLP)-1 and glucose-dependent insulinotropic polypeptide (GIP) (25); (3) CB1R activation also alters other receptor activity besides strictly CBRs, such as TRPV1 via CB1R-dependent AC-PKA axis and/or other unclear mechanisms that in turn influence intracellular Ca2+ levels (56, 75–77). Thus, CBR effects on β-cell function are an important component of the receptor’s role in regulating calcium-evoked hormone secretion and glucose homeostasis owing to alterations in the diversity of signaling pathways and post-receptor mechanisms (Figure 2). In the next sections we will throw a spotlight on the different CB1R actions in the endocrine cells of pancreas and the involvement of this receptor in hormone secretion, β-cell mass and inflammation in the pancreatic islets.
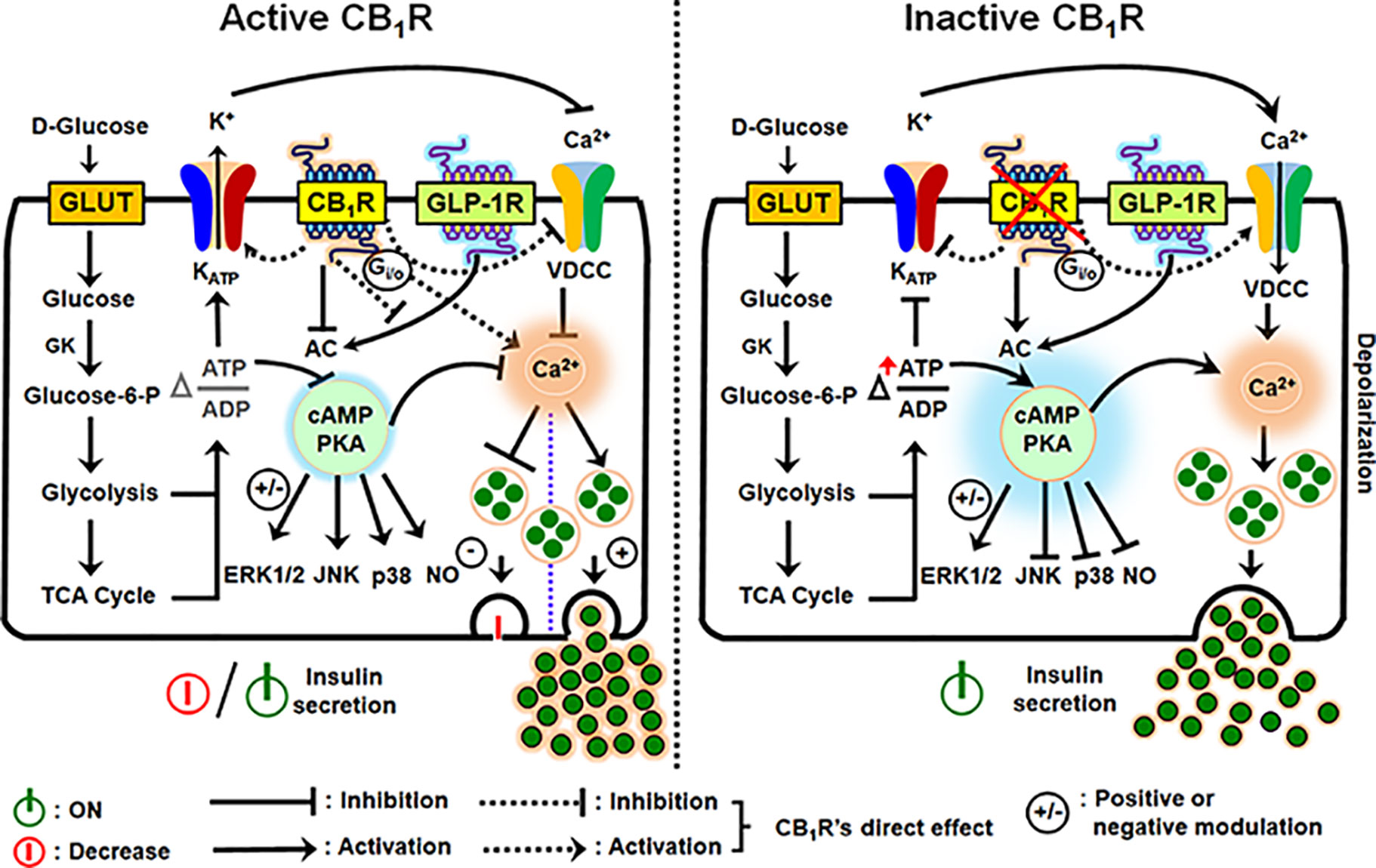
Figure 2 Schematic diagram illustrating the downstream events of CB1R activation and inhibition in the regulation of insulin secretion from β-cells. An increase in the intracellular glucose levels, which is transported by facilitated diffusion through glucose transporters, triggers the first permissive step in glucose-induced insulin secretion (GSIS). Glucose is phosphorylated by glucokinase (GK), the rate-limiting enzyme of glucose metabolism in β-cells, ATP is subsequently generated by glycolysis and the TCA cycle, causing an alteration in the intracellular ATP:ADP ratio. Increases in the [ATP/ADP] ratio closes the KATP channels and depolarizes the plasma membrane. The resultant opening of voltage-dependent calcium channels (VDCCs) and rise in cytosolic Ca2+ concentration allows for insulin exocytosis from β-cells. Although an increase in Ca2+ concentration is the primary or permissive insulin secretory event, cAMP production and protein kinase A (PKA) activity are critical in the fine tuning and potentiation of insulin secretion. The two gut-derived incretins, glucose-dependent insulinotropic peptide (GIP) and glucagon-like peptide -1 (GLP-1), are the main activators of adenylyl cyclase (AC) and subsequent cAMP formation; increases in cAMP augment GSIS by as much as 100% in non-diabetic subjects (GLP-1R is in the illustration: GIP has similar effects on AC). However, when CB1R is active, it signals through Gαi/o protein, inhibits AC, and attenuates cAMP production and protein kinase A (PKA) activity. CB1R also exerts influence on the KATP channel, decreasing potassium efflux, thereby inhibiting calcium entry through VDCCs because of hyperpolarization of the plasma membrane. In conjunction with the other forgoing events, downstream of AC-cAMP-PKA activity, CB1R activation impacts G-protein-dependent mitogen-activated protein kinases (MAPKs; ERK1/2, JNK and p38) and nitric oxide (NO) signaling, and the incretin receptors, GLP-1R and GIPR. The depicted receptor activation within the β-cell either inhibits or potentially stimulates insulin secretion under the influence of receptor-specific agonists via several mechanisms: the stimulatory role of CB1R in β-cell secretion, when reported, appears to be coupled to their capacity to increase intracellular calcium levels and reduction in cAMP production, whereas reduced intracellular cAMP formation can account for the inhibitory role of CB1R in insulin release.
Support for the evidence that CB1R is a negative regulator of insulin secretion has been demonstrated by combining small molecule studies and genetic tools in in vivo models, perfused and isolated islets, and β-cell lines: all show that activation of CB1R results in suppression of GSIS, whereas absence of CB1R enhances GSIS. CB1R antagonist treatment led to restoration of GSIS in Zucker diabetic fatty (ZDF) rats (61). Similarly, genetic and pharmacological blockade of CB1R indicated increased insulin secretion in mice (62, 63). Many CB1R antagonists (SR141716A, JD-5037, LH-21, AM251) also directly increased insulin release from mouse islets, clonal rat β-cells, mouse insulinoma βTC6 cells, and human islets (47, 53, 78) and in isolated islets from both prediabetic and diabetic mice, concomitant with improved islet function (79). However, there are discrepancies in results, going back to 2006. This will be further discussed below. We will first discuss studies which demonstrate increased insulin secretion, mostly as a result of CB1R antagonism.
The EC 2-AG blunts calcium oscillations in mouse cultured islets: calcium oscillations are permissive to pulsatile insulin release from β-cells (14). Similar observations have been obtained in isolated mouse pancreatic islets during their incubation with AEA and its non-hydrolysable analogue R(+) methanandamide, suggesting that CB1R activation promotes these effects (14). Additionally, this inhibitory effect in mouse islets have been repeated with AEA and the CB1R-selective agonist arachidonylcyclopropylamide because they both suppressed GSIS in conjunction with a decrease in Ca2+oscillations (18). The foregoing reports are congruent with the PKA/cAMP system-mediated desensitization for insulin release (80). Finally, CB1R directly modulates insulin secretion in human islets, removed from any influences from whole body. This is supported by the observation that the CB1R antagonists JD-5037 and rimonabant potentiated insulin release during perfusion of freshly isolated human islets when added to a stimulatory glucose concentration in the perfusate with more profound stimulatory effects exerted by JD-5037 than rimonabant at similar concentrations (47).
While most studies observed the inhibitory effects of CB1R activation on insulin secretion, there is data suggesting stimulatory action of CB1R on insulin secretion by direct addition of endocannabinoids AEA and 2-AG and their synthetic analogs to human and mouse islet preparations (16, 21) and rodent insulinoma cells (22, 81). GSIS is deemed to involve cytoskeletal and focal adhesion remodeling, which are key to insulin granule fusion with plasma membrane and exocytosis. CB1Rs are thought to play a role in focal adhesion kinase (FAK) phosphorylation because in INS-1E insulinoma cells FAK activation downstream of CB1Rs was found to make an essential contribution to cytoskeletal reorganization allowing for control of insulin release and potentially hypersecretion (81, 82). More precisely, the exception might be the ability of CB1R to activate FAK, which signals via Akt/PKB and ERK1/2, and the consequent FAK-mediated insulin secretion plays a role in CB1R-mediated signaling events in insulin stimulation as described for INS-1E cells (81) likely. This would also favor the physiological importance of this mechanistic link between enhanced circulating and local EC levels, and hyperinsulinemia as seen in type 2 diabetes (81). The role of CBRs in the regulation of insulin release was also reported from isolated human islets in both low and high glucose concentrations (16). This report would seem to indicate that human islets are particularly positively responsive (increased hormone secretion) to CB1R agonism with selective compounds (ACEA, for example) and ECs (AEA and 2-AG) while the effects of CB2R are inhibitory (16). CB1R-mediated cellular desynchronization indicative of opposite patterns of calcium oscillation coupled with insulin secretion and changes in gap junctions (83) associated with electrical β-cell coupling (84) could be possible pleotropic mechanisms underlying elevated insulin release. Support for potentiation of GSIS has been demonstrated in primary mouse islets and MIN6 cells during incubations with CB1R agonists 2-AG and ACEA, and, interestingly, with the CB2R agonist JWH015 – opposite to what was already published with reference to CB2R agonist in 2008 (16). Activation of both receptors elevated intracellular Ca2+ levels while inhibiting cAMP – these changes with CB1R agonist were prevented by a CB1R antagonist AM251 (21, 22). In these studies, using mouse islets and MIN6 cells, the investigators found that the insulin secretory profiles are responsive to 2-AG and ACEA in the presence of 2 and 20 mM glucose incubations reflecting the positive impact on insulin secretion primarily exerted by CB1R stimulation (21, 22). These results would reconcile that vicious cycles of overstimulation of CB1R leads to hyperstimulation of insulin secretion, hyperglycemia, lipid accumulation and consequent adipocyte hypertrophy observed in situations of obesity (21). Matias et al. (2006) had also reported enhanced insulin release due to incubation of rat insulinoma cells with HU-210, a CB1R agonist, when maintained on high glucose, and this effect was reversed by treatment with CB1R antagonist rimonabant, supporting the idea of a stimulatory role for ECs on insulin secretion (32).
From the above findings, it can be suggested that the contribution of CB1 and 2-AG to islet physiology and energy homeostasis is multifaceted, depending on the physiological or pathological conditions of the animal (16). Activation of CB1 is capable of stimulating insulin and glucagon secretion concomitant with lipogenesis in the liver and adipose tissue. As a consequence, glucose instead of being taken up by the muscle is converted to glycogen, and hence is stored instead of being oxidized (85–88). The observed rise in insulin release is also likely to facilitate the incorporation of glucose into adipocytes, thus favoring lipogenesis (16). Thus, it is possible that such events contribute to reduced energy expenditure and increased energy storage in the face of CB1R activation (16). Because of the involvement of CB1 in energy balance, Bermudez-Silva et al. (2008) has proposed that CB1 can be considered a new physiological thrifty system and that the persistent activation of this receptor could predispose individuals to obesity and type 2 diabetes upon Western diet consumption (16). Therefore, in view of the discrepancies with reports of both potentiation and suppression of insulin secretion following CB1 or CB2 receptor stimulation, several mechanisms may account for the stimulatory and inhibitory role of CB1R in β-cell secretion (Figure 2).
Another important determinant of insulin secretion are the incretins – GIP and GLP-1. They are gut hormones secreted from enteroendocrine cells into the bloodstream that greatly enhance glucose-stimulated insulin secretion in response to a meal ingestion (89). Once released into the circulation, GIP and GLP-1 activate their specific incretin receptors (GIPR and GLP-1R, respectively) on β-cells and stimulate AC activity (90, 91). Subsequently, a rise in intracellular cAMP is required for incretin-mediated insulin secretion (92). Incretin-mediated insulin secretion is inhibited by CB1R activation, and, in vivo, in addition to the enhanced early phase glucose-stimulated insulin secretion, mice deficient in CB1R have increased incretin secretion compared with control littermates (25). This suggests that CB1R, as a component of entero-insular axis, inhibits fasting in order to restrict their capacity to stimulate insulin secretion to only the fed state. Apart from directly influencing GSIS from β-cells, blockade of CB1R improves GLP-1-mediated insulin secretion in mice (25). Likewise, in the mouse insulinoma βTC6 cells and human islets, GLP-1R-mediated insulin secretion by exendin (Ex)-4 (a peptide agonist of GLP-1R) treatment was inhibited in a dose-dependent manner by both synthetic (ACEA) and endogenous (2-AG and AEA) CB1R agonists, indicative of negative EC actions in incretin-mediated insulin secretion by activated CB1R. The underlying mechanism for this likely is suppression of Ex-4-evoked increases in AC activity and reduced intracellular cAMP. Conversely, pharmacological abrogation of CB1R with either a global (AM251) or peripherally restricted (JD-5037) CB1R antagonist was reported to rescue such effects by releasing the ACEA/cannabinoid blockade of Ex-4 action in both mouse insulinoma cells and human islets (25). Similar observations have been obtained in βTC6 cells and human islets but with incubations in the presence of another synthetic CB1R agonist WIN55,212-2. Indeed, Ex-4-stimulated cAMP accumulation and insulin secretion from βTC6 cells was abolished by WIN55,212-2 in a dose-dependent manner, whereas AM251 enhanced the insulinotropic effects of Ex-4 in human islets (63). Such findings are consistent with the concept that CB1R negatively impacts incretin-mediated insulin secretion in rodents, isolated islets and β-cell lines.
In diet-induced obese mice after overnight fast, intra-peritoneal (IP) and intra-cerebroventricular administration of rimonabant caused increased glucagon secretion with concurrent increase in serum concentrations of glucose following glucose challenge (93). It is noteworthy that administration to diabetic mice of the CB1R antagonist BAR-1 led to a biphasic pattern of insulin and glucagon gene expression: their levels were reported to increase after 4 weeks of BAR-1 treatment but decrease after 8 weeks (79). In isolated human islets, CB1R agonists (2-AG and ACEA) had stimulatory effect on glucagon and somatostatin release at low glucose concentrations and ACEA-mediated glucagon release were prevented by AM251 (16). This has physiological relevance because somatostatin, as mentioned above, is an inhibitor of insulin secretion while glucagon is necessary for gluconeogenesis in order to prevent hypoglycemia (glucagon is also a stimulant to insulin secretion: all β-cells and hepatocytes express the Gs protein-coupled glucagon receptor). Separate studies reported that incubation of human islets with ACEA for approximately 2 days elevated glucagon content, thereby resulting in increased basal glucagon secretion. However, ACEA incubation for 5 days appeared to have had no effect on basal or arginine-stimulated glucagon release from human islets (94).
Islet β-cell mass, in general, is controlled by the dominant need for insulin to regulate metabolism (95) and it is regulated on the plus side by proliferation and alterations in cell size, and on the negative side by apoptosis and immune-mediated destruction. Reductions in β-cell mass has been observed in T1 and T2DM, with a more profound decline in T1DM (96, 97). Multiple lines of evidence point to CB1R as being on the negative side of β-cell mass regulation. Exposure of isolated human and mouse islets to two different CB1R antagonists SR141716A and AM251 led to increased β-cell proliferation that had first been stimulated by cytokines (98) and β-cell-specific CB1R null mice have bigger islets because of increased numbers of β-cells as do global CB1R null mice (55, 62). These observations are also consistent with the larger islets present in AM251-injected mouse (24). AM251 treatment allowed for improved β-cell recovery and mitosis of remaining β-cells after streptozotocin (STZ), a known β-cell toxin, was given to mice (60). Taken together, these studies suggest that activated CB1R negatively contributes to β-cell mass and proliferation.
Insulin is a known positive regulator of β-cell mass through IRS2/AKT downstream signaling (99). As regards mechanisms underlying CB1R effects on mass, the anti-trophic effects of CB1R activation in mice were reported to involve inhibition of insulin receptor autophosphorylation by interaction of the Gαi subunit of CB1R directly with the insulin receptor, which occurs by binding of Gαi to the activation loop of its tyrosine kinase domain (60). This is in line with the report that genetic and pharmacological disruption of CB1R enhanced insulin receptor signaling via IRS2/AKT in β-cells which led to increased β-cell mass (24). Additionally, CB1Rs regulate the expression of the anti-apoptotic protein Bcl-2 and cell cycle regulator cyclin D2 in pancreatic β-cells. Treatment of β-cell lines with WIN55,212-2 led to a decrease in the expression of Bcl-2 and cyclin D2 that in turn induced cell cycle arrest in G0/G1 phase and caspase-3-dependent apoptosis. On the contrary, genetic deletion and pharmacological blockade of CB1Rs after injury in mice led to increased levels of Bcl-2 and cyclin D2 in pancreatic β-cells (100).
There is a growing consensus that disruption of CB1R in preclinical animal models of obesity and T2DM modifies and preserves β-cell function and mass. Because ECs are synthesized during glucose stimulation, their synthesis is increased with HFDs, and high fat/sugar feeding leads to β-cell apoptosis (60), the concept of a role for β-cell-derived ECs in intra-islet inflammation was investigated. At this point, both autocrine and paracrine inflammatory actions have been attributed to CB1R activation. Consistent with an anti-inflammatory role of CB1R disruption, β-cell-specific deletion of CB1R eliminated the effects of diet-induced inflammation in β-cells via negative modulation of oxidative stress and NLRP3 inflammasome activation and led to preservation of β-cell function and survival in mice (62). Additionally, pharmacological blockade of CB1R by JD-5037 protected β-cells from inflammation and loss and allowed for normal plasma glucose levels in ZDF rats (61). To add to the complexity of action of ECs, islets have resident macrophages and both CBRs reside in those macrophages (61, 62, 101). CB1R-stimulated isolated human and rodent macrophages induced activation of NLRP3-ASC inflammasome with concomitant induction of the macrophage chemotactic protein (MCP)-1 and the release of cytokines such as IL-1β and IL-18 that resulted in apoptosis of β-cells (61). And depletion of CB1Rs in infiltrating macrophages implicated the macrophage-expressing CB1Rs as causing β-cell inflammation and cell death during obese conditions rather than cell-autonomous signaling of CB1R in β-cells as the cause. As proof, when the receptor was selectively inactivated in the macrophages of ZDF rats via IP delivery of glucan-encapsulated CB1R siRNA particles, inflammatory changes were reversed, and normoglycemia and GSIS were restored as opposed to those in rats receiving scrambled siRNA. The consequent protective actions appeared to be due to suppression of the nucleotide binding domain-like receptor protein 3-apoptosis associated speck-like protein containing caspase activation and recruitment domain (CARD) inflammasome activation and resistance to IL-1β overproduction (61). Interestingly there are intriguing results in this inflammatory space using cannabidiol. Δ9-THC protected mice from STZ-induced hyperglycemia and protected against the onset of hyperglycemia in non-obese diabetic mice (NOD; a mouse model akin to human autoimmune-mediated T1DM), indicative of protection of β-cell function and survival in the face of toxin- and autoimmune-mediated inflammation. Chronic administration of Δ9-THC appeared to prevent oxidative damage and reduce inflammation by decreasing the expression of IFN-γ, TNF-α and IL-12 in whole islets (102–104). However, it’s not evident by which mechanism or through which receptors Δ9-THC may be having its effects. It is possible that activation of CB2R and/or GPR55, or both, was the underlying protective mechanism in islets: this space is worth watching.
A growing body of evidence attests that an overactive peripheral ECS predisposes to the risk of developing diabetes and insulin resistance. Numerous studies that employ different approaches, animal models and in vitro systems have shown that an activated ECS inhibits basal insulin secretion, GSIS and incretin action. However, there are some studies that argue for stimulatory action of ECs and synthetic CB1R agonists in insulin secretion, at least with supraphysiological levels, that possibly plays a role in hyperinsulinemia as seen in obesity. Additionally, CB1R is involved in control of β-cell proliferation and islet mass, β-cell apoptosis, and toxin, inflammatory and immune-mediated β-cell destruction. There is evidence that chronic CB1R blockade (i.e., either genetic or pharmacological) ameliorates diabetes and insulin resistance and prevents β-cell loss in animal models of diabetes and obesity. Additionally, it prevented apoptosis of β-cells in isolated human islets. Clinical studies are now required to translate animal findings into treatments.
JME and KRA for conceptualization, writing, and editing. Both authors discussed the content, reviewed and approved the submitted version.
The authors are supported by the Intramural Research Program of the National Institute on Aging (NIA/NIH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Δ9-THC, delta-9-tetrahydrocannabinol; 2-AG, 2-arachidonoylglycerol; AC, adenylate cyclase; ACEA, arachidonoyl-2′-chloroethylamide; AEA, anandamide; AM251, N(piperidin-1-yl)-5-(4-iodophenyl)-1-)2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide; CB1R, Cannabinoid receptor type 1; CB2R, Cannabinoid receptor type 2; DAGLα, diacylglycerol lipase alpha; EC, endocannabinoid; ECS, endocannabinoid system; FAAH, fatty acid amide hydrolase; GLP-1R, Glucagon-like peptide 1 receptor; GPR55, G protein-coupled receptor 55; KATP channel, ATP-sensitive potassium channel; KO, Knockout; MAGL, monoacylglycerol lipase; NAPE-PLD, N-acyl-phosphatidylethanolamine phospholipase D; PKA, protein kinase A; SR141716A (rimonabant), 5-(4-Chlorophenyl)-1-(2,4dichloro-phenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3 carboxamide; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; TRPV1, transient receptor potential vanilloid 1; VDCC, Voltage-dependent calcium channel.
1. Yang SN, Shi Y, Yang G, Li Y, Yu J, Berggren PO. Ionic Mechanisms in Pancreatic β Cell Signaling. Cell Mol Life Sci (2014) 71(21):4149–77. doi: 10.1007/s00018-014-1680-6
2. Nadal A, Quesada I, Soria B. Homologous and Heterologous Asynchronicity Between Identified Alpha-, Beta- and Delta-Cells Within Intact Islets of Langerhans in the Mouse. J Physiol (1999) 517( Pt 1):85–93. doi: 10.1111/j.1469-7793.1999.0085z.x
3. Gilon P, Shepherd RM, Henquin JC. Oscillations of Secretion Driven by Oscillations of Cytoplasmic Ca2+ as Evidences in Single Pancreatic Islets. J Biol Chem (1993) 268(30):22265–8. doi: 10.1016/S0021-9258(18)41522-0
4. Hayes MR, Kanoski SE, Alhadeff AL, Grill HJ. Comparative Effects of the Long-Acting GLP-1 Receptor Ligands, Liraglutide and Exendin-4, on Food Intake and Body Weight Suppression in Rats. Obes (Silver Spring) (2011) 19(7):1342–9. doi: 10.1038/oby.2011.50
5. Dailey MJ, Moran TH. Glucagon-Like Peptide 1 and Appetite. Trends Endocrinol Metab (2013) 24(2):85–91. doi: 10.1016/j.tem.2012.11.008
6. Meneilly GS, Greig N, Tildesley H, Habener JF, Egan JM, Elahi D. Effects of 3 Months of Continuous Subcutaneous Administration of Glucagon-Like Peptide 1 in Elderly Patients With Type 2 Diabetes. Diabetes Care (2003) 26(10):2835–41. doi: 10.2337/diacare.26.10.2835
7. Egan JM, Bulotta A, Hui H, Perfetti R. GLP-1 Receptor Agonists Are Growth and Differentiation Factors for Pancreatic Islet Beta Cells. Diabetes Metab Res Rev (2003) 19(2):115–23. doi: 10.1002/dmrr.357
8. Doyle ME, Egan JM. Mechanisms of Action of Glucagon-Like Peptide 1 in the Pancreas. Pharmacol Ther (2007) 113(3):546–93. doi: 10.1016/j.pharmthera.2006.11.007
9. Martin B, Golden E, Carlson OD, Pistell P, Zhou J, Kim W, et al. Exendin-4 Improves Glycemic Control, Ameliorates Brain and Pancreatic Pathologies, and Extends Survival in a Mouse Model of Huntington’s Disease. Diabetes (2009) 58(2):318–28. doi: 10.2337/db08-0799
10. Egan JM, Chia CW. Incretin Therapy and Pancreatic Pathologies: Background Pathology Versus Drug-Induced Pathology in Rats. Diabetes (2014) 63(4):1174–8. doi: 10.2337/db13-1909
11. Kalyani RR. Glucose-Lowering Drugs to Reduce Cardiovascular Risk in Type 2 Diabetes. N Engl J Med (2021) 384(13):1248–60. doi: 10.1056/NEJMcp2000280
12. Schwetz TA, Ustione A, Piston DW. Neuropeptide Y and Somatostatin Inhibit Insulin Secretion Through Different Mechanisms. Am J Physiol Endocrinol Metab (2013) 304(2):E211–21. doi: 10.1152/ajpendo.00374.2012
13. Strowski MZ, Parmar RM, Blake AD, Schaeffer JM. Somatostatin Inhibits Insulin and Glucagon Secretion Via Two Receptors Subtypes: An In Vitro Study of Pancreatic Islets From Somatostatin Receptor 2 Knockout Mice. Endocrinology (2000) 141(1):111–7. doi: 10.1210/endo.141.1.7263
14. Juan-Pico P, Fuentes E, Bermudez-Silva FJ, Javier Diaz-Molina F, Ripoll C, Rodriguez de Fonseca F, et al. Cannabinoid Receptors Regulate Ca(2+) Signals and Insulin Secretion in Pancreatic Beta-Cell. Cell Calcium (2006) 39(2):155–62. doi: 10.1016/j.ceca.2005.10.005
15. Bermudez-Silva FJ, Sanchez-Vera I, Suarez J, Serrano A, Fuentes E, Juan-Pico P, et al. Role of Cannabinoid CB2 Receptors in Glucose Homeostasis in Rats. Eur J Pharmacol (2007) 565(1-3):207–11. doi: 10.1016/j.ejphar.2007.02.066
16. Bermudez-Silva FJ, Suarez J, Baixeras E, Cobo N, Bautista D, Cuesta-Munoz AL, et al. Presence of Functional Cannabinoid Receptors in Human Endocrine Pancreas. Diabetologia (2008) 51(3):476–87. doi: 10.1007/s00125-007-0890-y
17. Bermudez-Silva FJ, Suarez Perez J, Nadal A, Rodriguez de Fonseca F. The Role of the Pancreatic Endocannabinoid System in Glucose Metabolism. Best Pract Res Clin Endocrinol Metab (2009) 23(1):87–102. doi: 10.1016/j.beem.2008.10.012
18. Nakata M, Yada T. Cannabinoids Inhibit Insulin Secretion and Cytosolic Ca2+ Oscillation in Islet Beta-Cells Via CB1 Receptors. Regul Pept (2008) 145(1-3):49–53. doi: 10.1016/j.regpep.2007.08.009
19. Starowicz KM, Cristino L, Matias I, Capasso R, Racioppi A, Izzo AA, et al. Endocannabinoid Dysregulation in the Pancreas and Adipose Tissue of Mice Fed With a High-Fat Diet. Obes (Silver Spring) (2008) 16(3):553–65. doi: 10.1038/oby.2007.106
20. Tharp WG, Lee YH, Maple RL, Pratley RE. The Cannabinoid CB1 Receptor Is Expressed in Pancreatic Delta-Cells. Biochem Biophys Res Commun (2008) 372(4):595–600. doi: 10.1016/j.bbrc.2008.05.077
21. Li C, Bowe JE, Jones PM, Persaud SJ. Expression and Function of Cannabinoid Receptors in Mouse Islets. Islets (2010) 2(5):293–302. doi: 10.4161/isl.2.5.12729
22. Li C, Jones PM, Persaud SJ. Cannabinoid Receptors Are Coupled to Stimulation of Insulin Secretion From Mouse MIN6 Beta-Cells. Cell Physiol Biochem (2010) 26(2):187–96. doi: 10.1159/000320527
23. De Petrocellis L, Marini P, Matias I, Moriello AS, Starowicz K, Cristino L, et al. Mechanisms for the Coupling of Cannabinoid Receptors to Intracellular Calcium Mobilization in Rat Insulinoma Beta-Cells. Exp Cell Res (2007) 313(14):2993–3004. doi: 10.1016/j.yexcr.2007.05.012
24. Kim W, Doyle ME, Liu Z, Lao Q, Shin YK, Carlson OD, et al. Cannabinoids Inhibit Insulin Receptor Signaling in Pancreatic β-Cells. Diabetes (2011) 60(4):1198–209. doi: 10.2337/db10-1550
25. Gonzalez-Mariscal I, Krzysik-Walker SM, Kim W, Rouse M, Egan JM. Blockade of Cannabinoid 1 Receptor Improves GLP-1R Mediated Insulin Secretion in Mice. Mol Cell Endocrinol (2016) 423:1–10. doi: 10.1016/j.mce.2015.12.015
26. Maccarrone M, Bab I, Biro T, Cabral GA, Dey SK, Di Marzo V, et al. Endocannabinoid Signaling at the Periphery: 50 Years After THC. Trends Pharmacol Sci (2015) 36(5):277–96. doi: 10.1016/j.tips.2015.02.008
27. Lu HC, Mackie K. An Introduction to the Endogenous Cannabinoid System. Biol Psychiatry (2016) 79(7):516–25. doi: 10.1016/j.biopsych.2015.07.028
28. Gruden G, Barutta F, Kunos G, Pacher P. Role of the Endocannabinoid System in Diabetes and Diabetic Complications. Br J Pharmacol (2016) 173(7):1116–27. doi: 10.1111/bph.13226
29. Di Marzo V. Endocannabinoid Signaling in the Brain: Biosynthetic Mechanisms in the Limelight. Nat Neurosci (2011) 14(1):9–15. doi: 10.1038/nn.2720
30. Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, et al. Cloning of the First Sn1-DAG Lipases Points to the Spatial and Temporal Regulation of Endocannabinoid Signaling in the Brain. J Cell Biol (2003) 163(3):463–8. doi: 10.1083/jcb.200305129
31. Di Marzo V, Maccarrone M. FAAH and Anandamide: Is 2-AG Really the Odd One Out? Trends Pharmacol Sci (2008) 29(5):229–33. doi: 10.1016/j.tips.2008.03.001
32. Matias I, Gonthier MP, Orlando P, Martiadis V, De Petrocellis L, Cervino C, et al. Regulation, Function, and Dysregulation of Endocannabinoids in Models of Adipose and Beta-Pancreatic Cells and in Obesity and Hyperglycemia. J Clin Endocrinol Metab (2006) 91(8):3171–80. doi: 10.1210/jc.2005-2679
33. Tedesco L, Valerio A, Dossena M, Cardile A, Ragni M, Pagano C, et al. Cannabinoid Receptor Stimulation Impairs Mitochondrial Biogenesis in Mouse White Adipose Tissue, Muscle, and Liver: The Role of eNOS, P38 MAPK, and AMPK Pathways. Diabetes (2010) 59(11):2826–36. doi: 10.2337/db09-1881
34. Schonke M, Martinez-Tellez B, Rensen PC. Role of the Endocannabinoid System in the Regulation of the Skeletal Muscle Response to Exercise. Curr Opin Pharmacol (2020) 52:52–60. doi: 10.1016/j.coph.2020.05.003
35. Lecru L, Desterke C, Grassin-Delyle S, Chatziantoniou C, Vandermeersch S, Devocelle A, et al. Cannabinoid Receptor 1 Is a Major Mediator of Renal Fibrosis. Kidney Int (2015) 88(1):72–84. doi: 10.1038/ki.2015.63
36. DiPatrizio NV, Astarita G, Schwartz G, Li X, Piomelli D. Endocannabinoid Signal in the Gut Controls Dietary Fat Intake. Proc Natl Acad Sci USA (2011) 108(31):12904–8. doi: 10.1073/pnas.1104675108
37. Vianna CR, Donato J Jr, Rossi J, Scott M, Economides K, Gautron L, et al. Cannabinoid Receptor 1 in the Vagus Nerve Is Dispensable for Body Weight Homeostasis But Required for Normal Gastrointestinal Motility. J Neurosci (2012) 32(30):10331–7. doi: 10.1523/JNEUROSCI.4507-11.2012
38. Forte N, Fernandez-Rilo AC, Palomba L, Di Marzo V, Cristino L. Obesity Affects the Microbiota-Gut-Brain Axis and the Regulation Thereof by Endocannabinoids and Related Mediators. Int J Mol Sci (2020) 21(5):1554. doi: 10.3390/ijms21051554
39. Iannotti FA, Di Marzo V. The Gut Microbiome, Endocannabinoids and Metabolic Disorders. J Endocrinol (2021) 248(2):R83–97. doi: 10.1530/JOE-20-0444
40. Jeong WI, Osei-Hyiaman D, Park O, Liu J, Batkai S, Mukhopadhyay P, et al. Paracrine Activation of Hepatic CB1 Receptors by Stellate Cell-Derived Endocannabinoids Mediates Alcoholic Fatty Liver. Cell Metab (2008) 7(3):227–35. doi: 10.1016/j.cmet.2007.12.007
41. Carayon P, Marchand J, Dussossoy D, Derocq JM, Jbilo O, Bord A, et al. Modulation and Functional Involvement of CB2 Peripheral Cannabinoid Receptors During B-Cell Differentiation. Blood (1998) 92(10):3605–15. doi: 10.1182/blood.V92.10.3605
42. Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. Xxvii. Classification of Cannabinoid Receptors. Pharmacol Rev (2002) 54(2):161–202. doi: 10.1124/pr.54.2.161
43. Ligresti A, De Petrocellis L, Di Marzo V. From Phytocannabinoids to Cannabinoid Receptors and Endocannabinoids: Pleiotropic Physiological and Pathological Roles Through Complex Pharmacology. Physiol Rev (2016) 96(4):1593–659. doi: 10.1152/physrev.00002.2016
44. Sharkey KA, Wiley JW. The Role of the Endocannabinoid System in the Brain-Gut Axis. Gastroenterology (2016) 151(2):252–66. doi: 10.1053/j.gastro.2016.04.015
45. Cacciola G, Chianese R, Chioccarelli T, Ciaramella V, Fasano S, Pierantoni R, et al. Cannabinoids and Reproduction: A Lasting and Intriguing History. Pharmaceut (Basel) (2010) 3(10):3275–323. doi: 10.3390/ph3103275
46. Koch M, Varela L, Kim JG, Kim JD, Hernandez-Nuno F, Simonds SE, et al. Hypothalamic POMC Neurons Promote Cannabinoid-Induced Feeding. Nature (2015) 519(7541):45–50. doi: 10.1038/nature14260
47. Gonzalez-Mariscal I, Krzysik-Walker SM, Doyle ME, Liu QR, Cimbro R, Santa-Cruz Calvo S, et al. Human CB1 Receptor Isoforms, Present in Hepatocytes and β-Cells, Are Involved in Regulating Metabolism. Sci Rep (2016) 6:33302. doi: 10.1038/srep33302
48. Basu S, Dittel BN. Unraveling the Complexities of Cannabinoid Receptor 2 (CB2) Immune Regulation in Health and Disease. Immunol Res (2011) 51(1):26–38. doi: 10.1007/s12026-011-8210-5
49. Sawzdargo M, Nguyen T, Lee DK, Lynch KR, Cheng R, Heng HH. E Al. Identification and Cloning of Three Novel Human G Protein-Coupled Receptor Genes GPR52, PsiGPR53 and GPR55: GPR55 Is Extensively Expressed in Human Brain. Brain Res Mol Brain Res (1999) 64(2):193–8. doi: 10.1016/s0169-328x(98)00277-0
50. Muller C, Lynch DL, Hurst DP, Reggio PH. A Closer Look at Anandamide Interaction With TRPV1. Front Mol Biosci (2020) 7:144. doi: 10.3389/fmolb.2020.00144
51. Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 Is a Cannabinoid Receptor That Increases Intracellular Calcium and Inhibits M Current. Proc Natl Acad Sci USA (2008) 105(7):2699–704. doi: 10.1073/pnas.0711278105
52. Romero-Zerbo SY, Rafacho A, Diaz-Arteaga A, Suarez J, Quesada I, Imbernon M, et al. A Role for the Putative Cannabinoid Receptor GPR55 in the Islets of Langerhans. J Endocrinol (2011) 211(2):177–85. doi: 10.1530/JOE-11-0166
53. McKillop AM, Moran BM, Abdel-Wahab YH, Flatt PR. Evaluation of the Insulin Releasing and Antihyperglycaemic Activities of GPR55 Lipid Agonists Using Clonal Beta-Cells, Isolated Pancreatic Islets and Mice. Br J Pharmacol (2013) 170(5):978–90. doi: 10.1111/bph.12356
54. Liu B, Song S, Ruz-Maldonado I, Pingitore A, Huang GC, Baker D, et al. GPR55-Dependent Stimulation of Insulin Secretion From Isolated Mouse and Human Islets of Langerhans. Diabetes Obes Metab (2016) 18(12):1263–73. doi: 10.1111/dom.12780
55. Malenczyk K, Keimpema E, Piscitelli F, Calvigioni D, Bjorklund P, Mackie K, et al. Fetal Endocannabinoids Orchestrate the Organization of Pancreatic Islet Microarchitecture. Proc Natl Acad Sci USA (2015) 112(45):E6185–94. doi: 10.1073/pnas.1519040112
56. Akiba Y, Kato S, Katsube K, Nakamura M, Takeuchi K, Ishii H, et al. Transient Receptor Potential Vanilloid Subfamily 1 Expressed in Pancreatic Islet Beta Cells Modulates Insulin Secretion in Rats. Biochem Biophys Res Commun (2004) 321(1):219–25. doi: 10.1016/j.bbrc.2004.06.149
57. Dolensek J, Rupnik MS, Stozer A. Structural Similarities and Differences Between the Human and the Mouse Pancreas. Islets (2015) 7(1):e1024405. doi: 10.1080/19382014.2015.1024405
58. Laychock SG, Hoffman JM, Meisel E, Bilgin S. Pancreatic Islet Arachidonic Acid Turnover and Metabolism and Insulin Release in Response to Delta-9-Tetrahydrocannabinol. Biochem Pharmacol (1986) 35(12):2003–8. doi: 10.1016/0006-2952(86)90733-1
59. Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a Cannabinoid Receptor and Functional Expression of the Cloned Cdna. Nature (1990) 346(6284):561–4. doi: 10.1038/346561a0
60. Kim W, Lao Q, Shin YK, Carlson OD, Lee EK, Gorospe M, et al. Cannabinoids Induce Pancreatic β-Cell Death by Directly Inhibiting Insulin Receptor Activation. Sci Signal (2012) 5(216):ra23. doi: 10.1126/scisignal.2002519
61. Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, et al. Activation of the Nlrp3 Inflammasome in Infiltrating Macrophages by Endocannabinoids Mediates Beta Cell Loss in Type 2 Diabetes. Nat Med (2013) 19(9):1132–40. doi: 10.1038/nm.3265
62. Gonzalez-Mariscal I, Montoro RA, Doyle ME, Liu QR, Rouse M, O’Connell JF, et al. Absence of Cannabinoid 1 Receptor in Beta Cells Protects Against High-Fat/High-Sugar Diet-Induced Beta Cell Dysfunction and Inflammation in Murine Islets. Diabetologia (2018) 61(6):1470–83. doi: 10.1007/s00125-018-4576-4
63. Shin H, Han JH, Yoon J, Sim HJ, Park TJ, Yang S, et al. Blockade of Cannabinoid 1 Receptor Improves Glucose Responsiveness in Pancreatic Beta Cells. J Cell Mol Med (2018) 22(4):2337–45. doi: 10.1111/jcmm.13523
64. Barajas-Martinez A, Bermeo K, de la Cruz L, Martinez-Vargas M, Martinez-Tapia RJ, Garcia DE, et al. Cannabinoid Receptors Are Differentially Regulated in the Pancreatic Islets During the Early Development of Metabolic Syndrome. Islets (2020) 12(6):134–44. doi: 10.1080/19382014.2020.1849927
65. Zibolka J, Wolf A, Rieger L, Rothganger C, Jorns A, Lutz B, et al. Influence of Cannabinoid Receptor Deficiency on Parameters Involved in Blood Glucose Regulation in Mice. Int J Mol Sci (2020) 21(9):3168. doi: 10.3390/ijms21093168
66. Li C, Vilches-Flores A, Zhao M, Amiel SA, Jones PM, Persaud SJ. Expression and Function of Monoacylglycerol Lipase in Mouse β-Cells and Human Islets of Langerhans. Cell Physiol Biochem (2012) 30(2):347–58. doi: 10.1159/000339069
67. Juan-Pico P, Ropero AB, Tuduri E, Quesada I, Fuentes E, Bermudez-Silva FJ, et al. Regulation of Glucose-Induced [Ca2+]i Signals by Cannabinoid Receptors CB1 and CB2 in Pancreatic α- and δ- Cells Within Intact Islets of Langerhans. Obes Metab (2009) 5:20–8.
68. Flores LE, Alzugaray ME, Cubilla MA, Raschia MA, Del Zotto HH, Roman CL, et al. Islet Cannabinoid Receptors: Cellular Distribution and Biological Function. Pancreas (2013) 42(7):1085–92. doi: 10.1097/MPA.0b013e31828fd32d
69. McIntosh HH, Song C, Howlett AC. CB1 Cannabinoid Receptor: Cellular Regulation and Distribution in N18TG2 Neuroblastoma Cells. Brain Res Mol Brain Res (1998) 53(1-2):163–73. doi: 10.1016/s0169-328x(97)00294-5
70. Rorsman P, Braun M, Zhang Q. Regulation of Calcium in Pancreatic α- and β-Cells in Health and Disease. Cell Calcium (2012) 51(3-4):300–8. doi: 10.1016/j.ceca.2011.11.006
71. Rorsman P, Ashcroft FM. Pancreatic β-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol Rev (2018) 98(1):117–214. doi: 10.1152/physrev.00008.2017
72. Mackie K, Hille B. Cannabinoids Inhibit N-Type Calcium Channels in Neuroblastoma-Glioma Cells. Proc Natl Acad Sci USA (1992) 89(9):3825–9. doi: 10.1073/pnas.89.9.3825
73. O’Keefe L, Simcocks AC, Hryciw DH, Mathai ML, McAinch AJ. The Cannabinoid Receptor 1 and Its Role in Influencing Peripheral Metabolism. Diabetes Obes Metab (2014) 16(4):294–304. doi: 10.1111/dom.12144
74. Turu G, Hunyady L. Signal Transduction of the CB1 Cannabinoid Receptor. J Mol Endocrinol (2010) 44(2):75–85. doi: 10.1677/JME-08-0190
75. Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, et al. Vanilloid Receptors on Sensory Nerves Mediate the Vasodilator Action of Anandamide. Nature (1999) 400(6743):452–7. doi: 10.1038/22761
76. Malenczyk K, Girach F, Szodorai E, Storm P, Segerstolpe A, Tortoriello G, et al. A TRPV1-To-Secretagogin Regulatory Axis Controls Pancreatic β-Cell Survival by Modulating Protein Turnover. EMBO J (2017) 36(14):2107–25. doi: 10.15252/embj.201695347
77. Christie S, Wittert GA, Li H, Page AJ. Involvement of TRPV1 Channels in Energy Homeostasis. Front Endocrinol (Lausanne) (2018) 9:420. doi: 10.3389/fendo.2018.00420
78. Li C, Bowe JE, Huang GC, Amiel SA, Jones PM, Persaud SJ. Cannabinoid Receptor Agonists and Antagonists Stimulate Insulin Secretion From Isolated Human Islets of Langerhans. Diabetes Obes Metab (2011) 13(10):903–10. doi: 10.1111/j.1463-1326.2011.01422.x
79. Nava-Molina L, Uchida-Fuentes T, Ramos-Tovar H, Fregoso Padilla M, Rodriguez-Monroy MA, Vega AV, et al. Novel CB1 Receptor Antagonist BAR-1 Modifies Pancreatic Islet Function and Clinical Parameters in Prediabetic and Diabetic Mice. Nutr Diabetes (2020) 10(1):7. doi: 10.1038/s41387-020-0110
80. MacDonald PE, El-Kholy W, Riedel MJ, Salapatek AM, Light PE, Wheeler MB. The Multiple Actions of GLP-1 on the Process of Glucose-Stimulated Insulin Secretion. Diabetes (2002) 51(Suppl 3):S434–42. doi: 10.2337/diabetes.51.2007.s434
81. Malenczyk K, Jazurek M, Keimpema E, Silvestri C, Janikiewicz J, Mackie K, et al. CB1 Cannabinoid Receptors Couple to Focal Adhesion Kinase to Control Insulin Release. J Biol Chem (2013) 288(45):32685–99. doi: 10.1074/jbc.M113.478354
82. Rondas D, Tomas A, Soto-Ribeiro M, Wehrle-Haller B, Halban PA. Novel Mechanistic Link Between Focal Adhesion Remodeling and Glucose-Stimulated Insulin Secretion. J Biol Chem (2012) 287(4):2423–36. doi: 10.1074/jbc.M111.279885
83. Venance L, Piomelli D, Glowinski J, Giaume C. Inhibition by Anandamide of Gap Junctions and Intercellular Calcium Signalling in Striatal Astrocytes. Nature (1995) 376(6541):590–4. doi: 10.1038/376590a0
84. Ravier MA, Guldenagel M, Charollais A, Gjinovci A, Caille D, Sohl G, et al. Loss of Connexin36 Channels Alters Beta-Cell Coupling, Islet Synchronization of Glucose-Induced Ca2+ and Insulin Oscillations, and Basal Insulin Release. Diabetes (2005) 54(6):1798–807. doi: 10.2337/diabetes.54.6.1798
85. Di Marzo V, Matias I. Endocannabinoid Control of Food Intake and Energy Balance. Nat Neurosci (2005) 8(5):585–9. doi: 10.1038/nn1457
86. Cota D, Marsicano G, Tschop M, Grubler Y, Flachskamm C, Schubert M, et al. The Endogenous Cannabinoid System Affects Energy Balance Via Central Orexigenic Drive and Peripheral Lipogenesis. J Clin Invest (2003) 112(3):423–31. doi: 10.1172/JCI17725
87. Osei-Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Batkai S, et al. Endocannabinoid Activation at Hepatic CB1 Receptors Stimulates Fatty Acid Synthesis and Contributes to Diet-Induced Obesity. J Clin Invest (2005) 115(5):1298–305. doi: 10.1172/JCI23057
88. Engeli S, Bohnke J, Feldpausch M, Gorzelniak K, Janke J, Batkai S, et al. Activation of the Peripheral Endocannabinoid System in Human Obesity. Diabetes (2005) 54(10):2838–43. doi: 10.2337/diabetes.54.10.2838
89. Montrose-Rafizadeh C, Egan JM, Roth J. Incretin Hormones Regulate Glucose-Dependent Insulin Secretion in RIN 1046-38 Cells: Mechanisms of Action. Endocrinology (1994) 135(2):589–94. doi: 10.1210/endo.135.2.8033807
90. Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. Glucagon-Like Peptide I Stimulates Insulin Gene Expression and Increases Cyclic AMP Levels in a Rat Islet Cell Line. Proc Natl Acad Sci USA (1987) 84(10):3434–8. doi: 10.1073/pnas.84.10.3434
91. Thorens B. Expression Cloning of the Pancreatic Beta Cell Receptor for the Gluco-Incretin Hormone Glucagon-Like Peptide 1. Proc Natl Acad Sci USA (1992) 89(18):8641–5. doi: 10.1073/pnas.89.18.8641
92. Kim W, Egan JM. The Role of Incretins in Glucose Homeostasis and Diabetes Treatment. Pharmacol Rev (2008) 60(4):470–512. doi: 10.1124/pr.108.000604
93. Patel KN, Joharapurkar AA, Patel V, Kshirsagar SG, Bahekar R, Srivastava BK, et al. Cannabinoid Receptor 1 Antagonist Treatment Induces Glucagon Release and Shows an Additive Therapeutic Effect With GLP-1 Agonist in Diet-Induced Obese Mice. Can J Physiol Pharmacol (2014) 92(12):975–83. doi: 10.1139/cjpp-2014-0310
94. Vilches-Flores A, Franklin Z, Hauge-Evans AC, Liu B, Huang GC, Choudhary P, et al. Prolonged Activation of Human Islet Cannabinoid Receptors In Vitro Induces Adaptation But Not Dysfunction. BBA Clin (2016) 5:143–50. doi: 10.1016/j.bbacli.2016.03.009
95. Sachdeva MM, Stoffers DA. Minireview: Meeting the Demand for Insulin: Molecular Mechanisms of Adaptive Postnatal Beta-Cell Mass Expansion. Mol Endocrinol (2009) 23(6):747–58. doi: 10.1210/me.2008-0400
96. Eizirik DL, Mandrup-Poulsen T. A Choice of Death–the Signal-Transduction of Immune-Mediated Beta-Cell Apoptosis. Diabetologia (2001) 44(12):2115–33. doi: 10.1007/s001250100021
97. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-Cell Deficit and Increased Beta-Cell Apoptosis in Humans With Type 2 Diabetes. Diabetes (2003) 52(1):102–10. doi: 10.2337/diabetes.52.1.102
98. Ruz-Maldonado I, Liu B, Atanes P, Pingitore A, Huang GC, Choudhary P, et al. The Cannabinoid Ligands SR141716A and AM251 Enhance Human and Mouse Islet Function Via GPR55-Independent Signalling. Cell Mol Life Sci (2020) 77(22):4709–23. doi: 10.1007/s00018-019-03433-6
99. Elghazi L, Rachdi L, Weiss AJ, Cras-Meneur C, Bernal-Mizrachi E. Regulation of Beta-Cell Mass and Function by the Akt/protein Kinase B Signalling Pathway. Diabetes Obes Metab (2007) 9 Suppl 2:147–57. doi: 10.1111/j.1463-1326.2007.00783.x
100. Kim J, Lee KJ, Kim JS, Rho JG, Shin JJ, Song WK, et al. Cannabinoids Regulate Bcl-2 and Cyclin D2 Expression in Pancreatic β Cells. PloS One (2016) 11(3):e0150981. doi: 10.1371/journal.pone.0150981
101. Jourdan T, Szanda G, Cinar R, Godlewski G, Holovac DJ, Park JK, et al. Developmental Role of Macrophage Cannabinoid-1 Receptor Signaling in Type 2 Diabetes. Diabetes (2017) 66(4):994–1007. doi: 10.2337/db16-1199
102. Coskun ZM, Bolkent S. Oxidative Stress and Cannabinoid Receptor Expression in Type-2 Diabetic Rat Pancreas Following Treatment With Δ⁹-Thc. Cell Biochem Funct (2014) 32(7):612–9. doi: 10.1002/cbf.3058
103. Li X, Kaminski NE, Fischer LJ. Examination of the Immunosuppressive Effect of Delta9-Tetrahydrocannabinol in Streptozotocin-Induced Autoimmune Diabetes. Int Immunopharmacol (2001) 1(4):699–712. doi: 10.1016/s1567-5769(01)00003-0
Keywords: endocannabinoids, cannabinoid receptors, β-cells, islet of Langerhans, obesity, diabetes
Citation: Aseer KR and Egan JM (2021) An Autonomous Cannabinoid System in Islets of Langerhans. Front. Endocrinol. 12:699661. doi: 10.3389/fendo.2021.699661
Received: 23 April 2021; Accepted: 08 June 2021;
Published: 05 July 2021.
Edited by:
Tony Jourdan, INSERM U1231 Lipides, Nutrition, Cancer (LNC), FranceReviewed by:
Vincenzo Di Marzo, Laval University, CanadaCopyright © 2021 Aseer and Egan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kanikkai Raja Aseer, a2FuaWtrYWlyYWphLmFzZWVydmF0aGFtQG5paC5nb3Y=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.