 Georgia Pitsava
Georgia Pitsava Nikolaos Settas2
Nikolaos Settas2 Fabio R. Faucz
Fabio R. Faucz Constantine A. Stratakis
Constantine A. Stratakis- 1Division of Intramural Population Health Research, Eunice Kennedy Shriver National Institutes of Child Health and Human Development, National Institutes of Health, Bethesda, MD, United States
- 2Section on Endocrinology and Genetics, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, United States
Succinate dehydrogenase (SDH) is a key respiratory enzyme that links Krebs cycle and electron transport chain and is comprised of four subunits SDHA, SDHB, SDHC and SDHD. All SDH-deficient tumors are caused by or secondary to loss of SDH activity. As many as half of the familial cases of paragangliomas (PGLs) and pheochromocytomas (PHEOs) are due to mutations of the SDHx subunits. Gastrointestinal stromal tumors (GISTs) associated with SDH deficiency are negative for KIT/PDGFRA mutations and present with distinctive clinical features such as early onset (usually childhood or adolescence) and almost exclusively gastric location. SDH-deficient GISTs may be part of distinct clinical syndromes, Carney-Stratakis syndrome (CSS) or dyad and Carney triad (CT). CSS is also known as the dyad of GIST and PGL; it affects both genders equally and is inherited in an autosomal dominant manner with incomplete penetrance. CT is a very rare disease; PGL, GIST and pulmonary chondromas constitute CT which shows female predilection and may be a mosaic disorder. Even though there is some overlap between CT and CSS, as both are due to SDH deficiency, CSS is caused by inactivating germline mutations in genes encoding for the SDH subunits, while CT is mostly caused by a specific pattern of methylation of the SDHC gene and may be due to germline mosaicism of the responsible genetic defect.
Introduction
Succinate dehydrogenase (SDH - also known as mitochondrial complex II or succinate-ubiquinone oxydoreductase) is the only enzyme that is concurrently both a functional member of both the Krebs cycle (or citric acid or tricarboxylic acid cycle) and the electron transport chain (ETC), where it provides electrons for oxidative phosphorylation (1). It is comprised of four mitochondrial subunit proteins: SDHA, SDHB, SDHC, SDHD encoded by nuclear genes, mapped to 5p15.22, 1p36.13, 1q23.3 and 11q23.1, respectively (Figure 1). SDHA is a flavoprotein and SDHB is an iron-sulfur protein; together they make up the main catalytic component of the complex. The other two subunits, SDHC and SDHD are two integral membrane proteins that anchor the complex to the inner mitochondrial membrane (3, 4). Additionally, the succinate dehydrogenase assembly factor 2 (SDHAF2) is required for the flavination and thus normal function of SDHA (4).
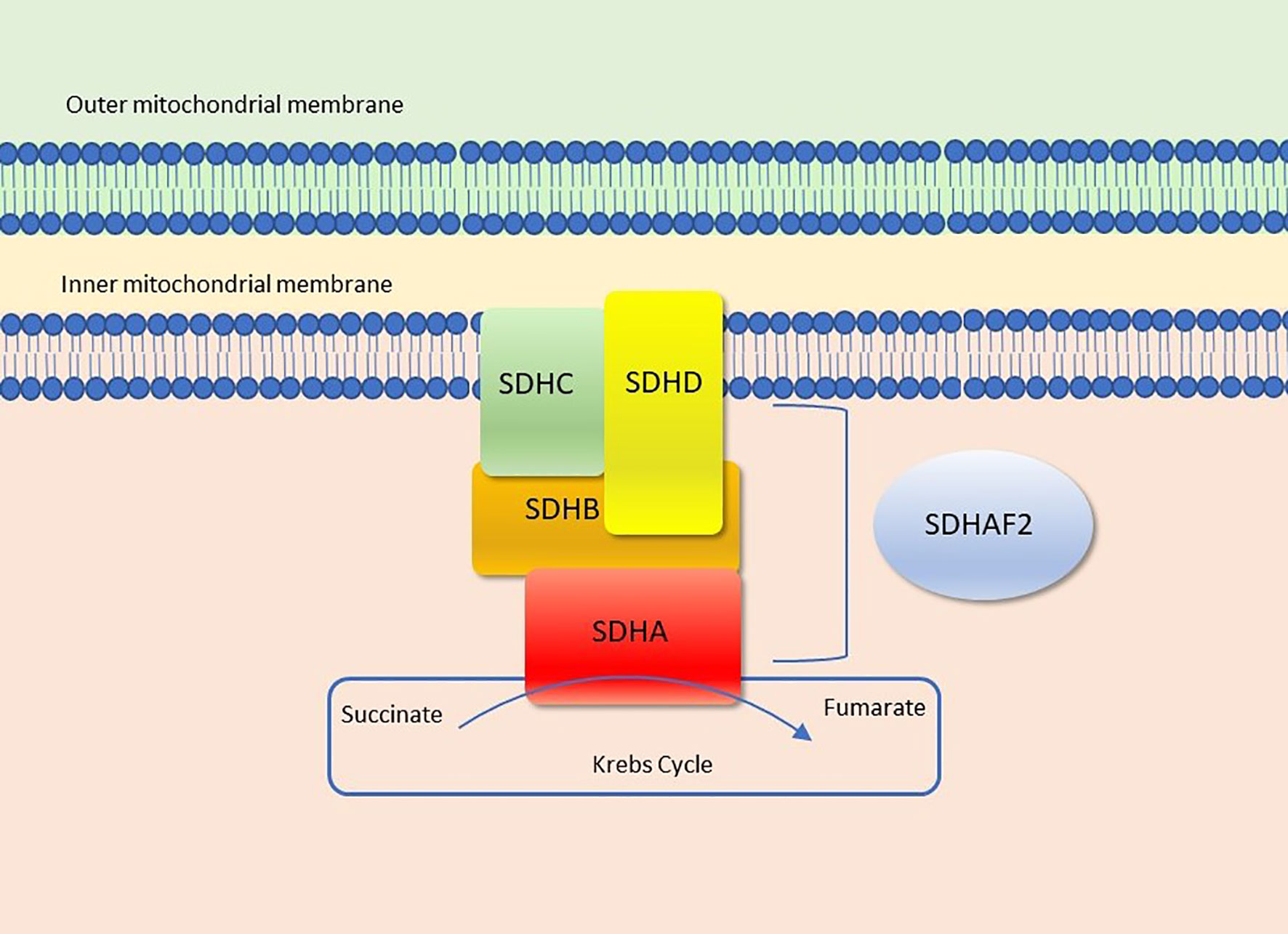
Figure 1 Succinate dehydrogenase (Complex II). Figure modified from Settas et al. (2).
Genetic alterations in any of the four SDHx genes (SDHA, SDHB, SDHC, SDHD) or SDHAF2 lead to SDH complex dysfunction and loss of SDHB expression (5). This loss of SDHB can be detected rapidly by immunohistochemistry (IHC) and thus, loss of immunohistochemical staining for SDHB is used as the hallmark of SDH-deficient tumors (6–9).
How Do SDHx Mutations Lead to Tumorigenesis?
It is not completely clear how the dysfunction of SDH leads to neoplasia; several mechanisms have been proposed. One of them is the activation of pseudohypoxia pathway (10). This mechanism implies that due to SDH deficiency, succinate is accumulated; this inhibits propyl hydroxylases (PHDs) resulting in induction of the hypoxic response despite normoxic conditions (pseudohypoxia) (11, 12). At the cellular level, the three α subunits of the hypoxia inducible factor-1 (HIF-1α, HIF2α, HIF3α), are hydroxylated by PHDs 1, 2 and 3 (also known as Egln2, Egln1 and Egln3), which are oxygen-dependent enzymes. The hydroxylated HIFαs are then targeted by von Hippel-Lindau (VHL) protein for degradation in the proteasome. In order for the HIFs to be recognized by the VHL, hydroxylation of two proline residues on HIFα is required by PHDs. In the case that the SHDx genes are mutated, propyl hydroxylases are inhibited by the accumulated succinate, hydroxylation of HIF-1αs is decreased and therefore they escape degradation. As a result, they translocate to the nucleus, they dimerize with HIFβ and create a complex that activates genes that induce angiogenesis, cell proliferation and glycolysis (12, 13). This mechanism was further supported by additional studies (12, 14–17). Additionally, besides succinate, the accumulation of reactive oxygen species (ROS) in mitochondria, leading to loss of function of the SDH enzyme, has also been implicated in tumor pathogenesis. ROS are mainly produced in complex I (NADH-ubiquinone oxidoreductase) and complex III (ubiquinone-cytochrome c oxidoreductase) in ETC (18). Recently, Xiao et al. demonstrate that SDHx knockdown increases intracellular levels of succinate; subsequently, this acts as an alpha-ketoglutarate competitor, inhibiting a-KG-dependent dioxygenases, JIp1, which is involved in sulfur metabolism and Jhd1 which belongs to the JmjC-domain-containing histone demethylase (JHDM) enzymes. That could lead to tumor formation by causing epigenetic changes (11, 19). The corresponding human JHDM, JMJD2D, was shown to be inhibited by accumulation of succinate as well (20).
Mutations in the SDHx subunits have been implicated in familial paragangliomas (PGLs) and pheochromocytomas (PHEOs), gastric stromal tumors (GISTs), Carney-Stratakis syndrome (CSS), and rarely in Carney triad (CT) and a few other tumors (21–30). This review focuses on SDH-deficient tumors, the two relevant genetic conditions, CSS and CT, and an association (3PAS), and their clinical, pathological and molecular characteristics.
SDH-Deficient Paragangliomas and Pheochromocytomas
Pheochromocytomas and paragangliomas are rare neuroendocrine neoplasms derived from chromaffin cells (31). Tumors arising from the adrenal medulla, which is the largest paraganglion in the body, are termed PHEOs, while those derived from the sympathetic and parasympathetic paraganglia are known as PGLs (31). Extraadrenal locations most commonly include the head and neck, mainly the carotid body, jugular foramen, middle ear, but can also occur in the thorax, abdomen and pelvis (32). PGLs/PHEOs can be either sporadic or hereditary. As many as 35% of them are due to genetic predisposition (33). To date, more than 20 susceptibility genes have been identified (34). Germline mutations of SDHB, SDHC, and SDHD genes are responsible for approximately 50% of hereditary paragangliomas (4, 24, 25, 35–38) and pheochromocytomas (24, 36, 39). Recently, mutations in SDHA (21) and SDHAF2 were also identified in hereditary PHEOs and PGLs (40). In addition, multiple reports have shown that these tumors have high incidence in patients with cyanotic congenital heart disease (41–43).
Depending on the SDHx subunit that is mutated, PGL syndromes have different characteristics (Table 1): SDHD (PGL1) (OMIM#168000)-mutated PGLs are more common in the head and neck and appear to have very high lifetime penetrance as 75% of carriers will have manifestations by 40 years old (44). Mutations in SDHB gene as the susceptibility gene for PGL4 (OMIM#115310) are more likely to be in the abdomen and show very high metastatic risk, but lower penetrance compared to PGL1 (~40% of carriers manifest the disease by age 40) (45). On the other hand, in SDHC (PGL3) (OMIM#605373) gene mutations, much rarer than the previous two, tumors are more commonly located in the carotid body (35, 46, 47) and have a low malignant potential (45). Mutations in SDHA and SDHAF2 are associated with PGL5 (OMIM#614165) and PGL2 (OMIM#601650) respectively and are very rare. A patient with any type of PGL will present in any of the following contexts: a) because of signs and/or symptoms of excess catecholamine secretion (e.g. hypertension, headache, palpitations, hyperhidrosis, tremor); b) because of an incidental finding on an imaging study; c) because of signs and/or symptoms due to a local mass (various signs and/or symptoms depending on the location); and d) after a genetic testing was performed in the case of familial disease. Histologically, SDH-deficient PHEOs/PGLs have a nested architecture with round cells and prominent vasculature (4).
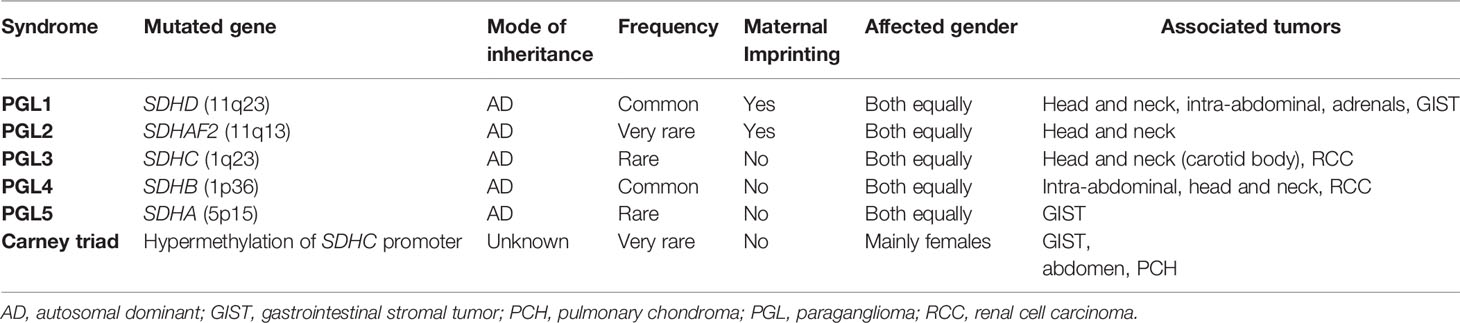
Table 1 Characteristics of SDH-deficient pheochromocytoma and paraganglioma.
PHEOs can occur as part of PGL1 and PGL4 and about 3% of them are attributed to SDH deficiency (6). The rest of them are either sporadic or they are associated with other familial syndromes such as VHL, MEN2 and NF. What could differentiate SDH-deficient PHEOs is the negative SDHB IHC and the secretion solely of noradrenaline (and/or dopamine) in contrast to the others that secret both adrenaline and noradrenaline (47). In addition, PHEOs caused by SDHB mutations show higher malignancy risk (47).
Family history is not always helpful in predicting hereditary PHEOs/PGLs because of phenotypic heterogeneity, incomplete penetrance and in the case of PGL1 and PGL2, maternal imprinting (5, 25, 48). It is interesting that in PHEOs/PGLs that appear to be sporadic based on family history, germline mutations were found in up to 25% of cases (49–51). Therefore, all patients with PHEOs/PGLs (sporadic and hereditary cases) should undergo genetic testing and counseling after IHC is performed (5, 6).
SDH-Deficient GISTs
GISTs are the most common neoplasms of the gastrointestinal tract of mesenchymal origin and more than 5000 cases are diagnosed each year in the US alone (52). They originate from the interstitial cells of Cajal (53), the pacemaker cells that regulate peristalsis in the digestive tract (54). Most GISTs occurring in adults are driven by activating mutations in KIT proto-oncogene receptor tyrosine kinase (KIT) (75-80% of cases) or platelet-derived growth factor receptor A (PDGFRA) (5-15%) genes (55–58). The rest (10-15%), that lack KIT and PDGFRA gene mutations, are described as ‘wild type GISTs’ (WT GISTs) and comprise most of pediatric GISTs (59, 60). SDH-deficient GISTs are the majority of WT GISTs (50% of these tumors are associated with hypermethylation of the SDHC promoter locus (CT), 30% with germline SDHA mutations (4), while 20% is associated with mutations in SDHB, SDHC, SDHD (Table 2) (61). The rest harbor mutations in NF-1, BRAF, ARID1A, ARID1B, CBL, NRAS, HRAS, KRAS, EGFR1, MAX, MEN1, PIK3CA and ETV6-NTRK3 fusion genes; these patients are usually older (same as KIT/PDGFRA + tumors) and they have more aggressive disease (62–72) (Figure 2). It is important to identify these mutations as it can be useful in the treatment plan.
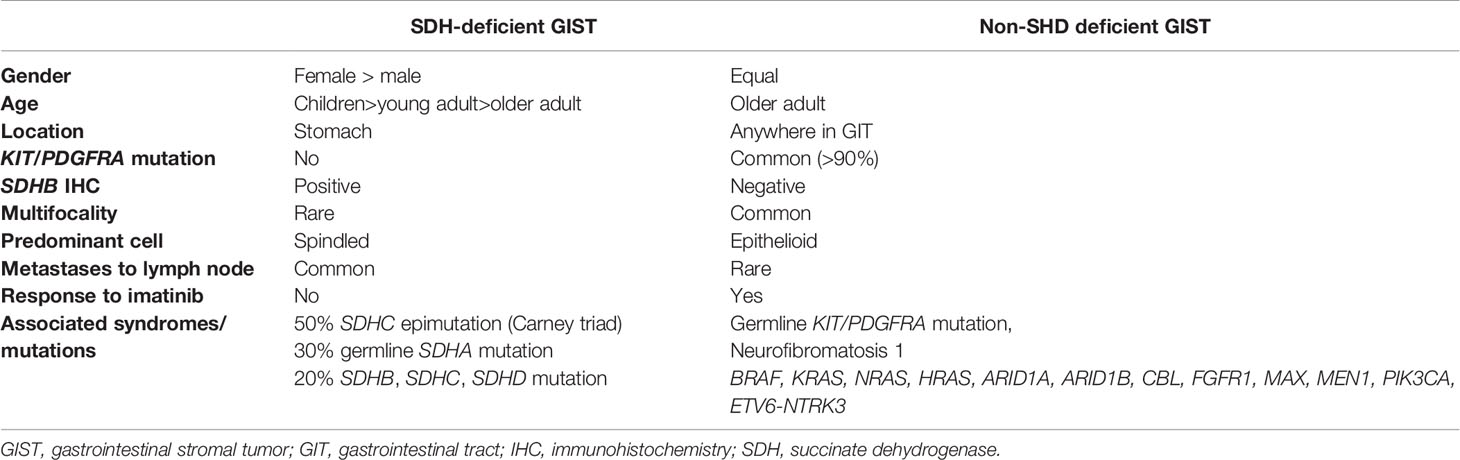
Table 2 Comparison of SDH-deficient GISTs and SDH-competent GISTs.
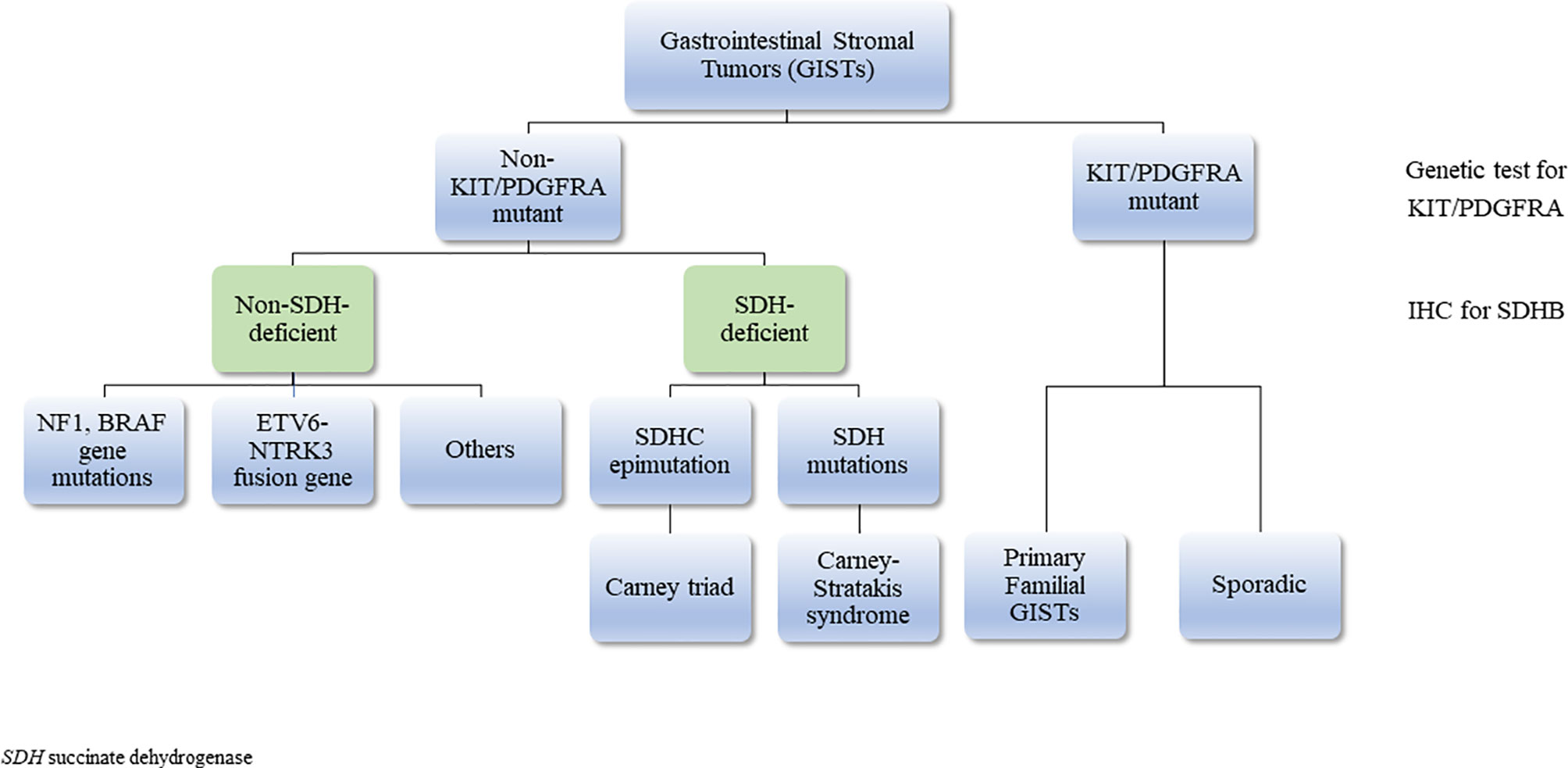
Figure 2 Classification of gastrointestinal stromal tumors (GISTs).
SDH- deficient GISTs exhibit unique features which are summarized in Table 2. Briefly, they manifest predominantly in females, at a young age. They arise almost exclusively in the stomach (61, 73–79) and they frequently have early lymphovascular invasion and consequent involvement of the lymph nodes (76), and less frequently of the liver (61), and do not frequently respond to imatinib (80). However, even in the setting of metastatic disease, they have an indolent clinical course. Histologically, these tumors exhibit multinodular growth pattern with epithelioid cells and they are multifocal. In addition, it was found that SDH-deficient GISTs overexpress insulin-like growth factor receptor (IGF1R) (81), and that this upregulation is highly specific of SDH-deficient GISTs (61, 78, 82, 83). The underlying molecular mechanism is unknown, but it could possibly be due to genetic amplification (61). Stratakis and his group also showed that immunohistochemistry that is negative for SDHB can be used to identify SDH-deficient GISTs caused by SDHB, SDHC or SDHD mutations (75). SDH-deficient GISTs can be sporadic or may present as part of two syndromes, CT (84) and CSS (26, 85).
Carney Triad (CT)
Going back, in 1977 Dr. J. Aidan Carney described the association of three uncommon tumors- GISTs, PGLs and pulmonary chondroma (PCH) (86). Among other characteristics, the young age (median 18 years old), the female predilection, the multifocality and the concurrence of rare tumors suggested a genetic etiology (87). This association was later referred to as CT (OMIM #604287). Afterwards, adrenocortical adenoma and esophageal leiomyoma were added as components of the triad (88). The etiology of CT is not yet clear but recent data have implicated SDHC. In a cohort of 37 patients, comparative genomic hybridization demonstrated no mutations of any of the SDHx subunits. Instead, it revealed the most frequent and largest genomic change to be the deletion of 1q12-q21, a region where SDHC gene resides (84). Later, Haller et al. demonstrated that aberrant DNA hypermethylation is present at specific sequences of the SDHC gene (in the promoter and first exon) in patients with CT; this methylation leads to reduced SDHC mRNA expression (89). A genome-wide DNA study confirmed the SDHC gene promoter hypermethylation in both CT and WT-GISTs (90). Today, SDHC-specific methylation is considered the molecular signature of CT and is used as simple diagnostic test to identify lesions that may be part of CT in patients that are suspected to be affected by the condition.
Carney-Stratakis Syndrome (CSS)
In 2002, Dr Carney and Dr Stratakis, described a new condition, that is today known as Carney-Stratakis syndrome (CSS) (OMIM #606864) (also reported as the “paraganglioma and gastric stromal sarcoma syndrome” or Carney-Stratakis dyad) (91). This newly described genetic disorder included only two types of tumors, PGLs/PHEOs and GISTs and is inherited in an autosomal dominant manner with incomplete penetrance. It affects both males and females during childhood and adolescence. Later, in 2007, Dr. Stratakis and his group identified inactivating mutations in the SDHB, SDHC and SDHD subunits as responsible for CSS (26, 92), with subunits B and D being mutated in higher frequency. Pasini et al. studied patients with CSS who developed GIST and they identified germline mutations in SDHB, SDHC and SDHD (26). Hemizygosity/homozygosity for the mutant allele was found in the GISTs of the affected individuals which is consistent with the tumor suppressor activity of SDHx genes (26). SDHA loss-of-function mutations have also been identified in patients with CSS (74). Surprisingly, patients harboring SDHA mutations demonstrated impressively long survival (93).
3PAS
Over the years, the co-existence of PHEOs/PGLs and pituitary adenomas (PAs) was thought to be a coincidence due to the rarity of those endocrine tumors (23). However, in some cases, they may have a common pathogenic mechanism. The first case of a patient with PHEO and acromegaly was described in 1952 (94). Since then, more than 80 such cases have been published (95). In 2012, Xekouki et al. described an individual within a family history with multiple PGLs and PHEOs caused by a germline SDHD mutation; in addition, the individual had an aggressive growth hormone (GH)-secreting PA, and loss of heterozygosity at the SDHD locus in the pituitary tumor along with increased levels of HIF-1α (96). Since then, the co-existence of those tumors, not recognized as a distinct entity before, has been known as 3PAs. More cases of PAs in patients with SDH mutations have been described, supporting the evidence that SDH deficiency plays a role in pituitary tumors (97–99).
SDH-deficient PAs that are part of 3PAs are more commonly macroadenomas and they frequently exhibit different phenotypes within the same family, such as prolactinomas, somatotropinomas and non-functional adenomas (95). Most of the time they respond poorly to somatostatin analogues and they require multiple treatments (95). In addition, PHEOs/PGLs in patients with 3PAs are often bilateral and/or multiple and tend to recur (95). In a cohort study of 19 patients with PHEO/PGL and PA, 9 of them had SDHx mutations. In PAs caused by mutations in any of the SDHx subunits intracytoplasmic vacuoles were present, a histological characteristic specific to those kinds of tumors (100). One could speculate that those vacuoles could possibly be autophagic bodies, as it is known that activation of autophagy is related to hypoxia-related pathways (101, 102); moreover, autophagy has been found to contribute to chemo- and radio-therapy resistance (103, 104).
SDH-Deficient Renal Cell Carcinoma
SDH-deficient renal carcinoma was first recognized in 2004 (22) and later was accepted as a distinct type of renal cell carcinoma (RCC) (4, 105). It is rare, as it is estimated to account for 0.05-0.2% of all renal carcinomas (106). The mean age is 38 to 40 years (107) and there is a slight male predisposition (106, 108). In most of them, SDHB (83%) germline mutation is present (80), but few cases with SDHC and SDHD mutations have been reported as well (106–109). SDHA mutation in RCC was reported for the first time recently by Yakirevich et al. (110), followed by other reports (111, 112). In a cohort study, 36 SDH-deficient RCCs from 27 patients were studied; all of them were negative for SDHB and positive for SDHA by IHC. In addition, genetic testing was performed in 17 of these patients and they all harbored a germline SDHx mutation (16 SDHB, 1 SDHC) (106). In another study, 37 tumors exhibiting morphologic features of SDH-deficient RCC were evaluated; of them 11 showed immunohistochemical loss of SDHB and 1 out of 11 cases loss of SDHA (in this case no SDHB gene mutation was detected by sequencing and SDHA gene was not evaluated) (108).
Morphologically, SHD-deficient RCCs exhibit distinctive features, being made of cuboidal cells with variable cysts and ‘bubbly’ eosinophilic cytoplasm with flocculent inclusions. They also exhibit a solid, nested or tubular growth pattern (80, 106–108, 110, 113, 114). The hallmark of these tumors is loss of SDH immunohistochemical expression. Therefore, in renal tumors with morphology suggestive of SDH-deficient RCC or syndromic disease (younger age, family history of RCC, personal or family history of other SDH-deficient tumors) IHC for SDHB should be performed (106, 112). It is possible that SDHA-deficient RCCs may exhibit slightly different morphologic features such as papillary, cribiform-like architecture, higher nuclear grade and areas of solid growth pattern (110–112). However, very few cases have been reported so far and it is difficult to make any definitive associations.
In addition, this distinct type of RCCs is negative for c-kit, cytokeratin 7 (CK7), carbonic anhydrase IX (CAIX), CD117 and vimentin, while it is immunoreactive for PAX8 and kidney-specific cadherin. These markers can be useful in the case that IHC is unavailable (106–108).
Although most SHD-deficient RCCs have a good prognosis, and the risk of metastasis is estimated to be 11%, some of them- those with high-grade nuclear atypia, tumor necrosis or sarcomatoid differentiation- may behave aggressively reaching metastatic rates as high as 70% (106, 108, 115).
Other SDH-Deficient Tumors
Apart from PGLs/PHEOs, GISTs, PAs and renal cell carcinomas discussed above, there is not much evidence that SDHx deficiency contributes significantly to other neoplasms. Thyroid carcinoma associated with either SDHB or SDHD has been reported in a few individuals (46, 51). Patients with PTEN-negative Cowden and Cowden-like syndromes, have also been reported in association with either SDHB or SDHD variants (27). Neuroblastoma (28) and bilateral adrenal medullary hyperplasia (29) have been linked to SDHB mutations. Moreover, a case of testicular seminoma has been reported in association with SDHD mutation (30). While a variety of tumors has been reported in association with SDH mutations we cannot say for sure if there is a causal relationship between them due to the very limited number of cases.
Loss of SDHB Immunohistochemistry as an Important Tool of Validating SDH Mutations
SDHx genes act as tumor suppressor genes (116). SDHx germline heterozygous inactivating mutations affect the protein function and predispose to hereditary neoplasms; subsequently, loss of heterozygosity (LOH) in the tumor level results in complete loss of SDH activity (14). Loss of immunohistochemical staining for SDHB has been proved to be a robust and reliable marker for syndromic disease resulting from germline mutation of SDHA, SDHB, SDHC or SDHD (6–9). In addition, in the case of double-hit inactivation of SDHA, IHC for SDHA becomes negative as well (9, 21). Thus, tumors associated with bi-allelic inactivation of SDHA stain negative for SDHB and SDHA, while tumors caused by inactivating mutations in SDHB, SDHC or SDHD show negative staining only for SDHB. In every case, caution should be taken when interpreting the results and further clinical and genetic assessment should ensue.
SDH-Deficient Tumors: Clinical Considerations and Genetic Counseling
Clinical features of the tumors discussed above should be taken into careful consideration. It is very important, in the case of PGLs/PHEOs, to be aware of any catecholamine excess symptoms (such as hypertension, hyperhidrosis, palpitations, headache) as well as signs and/or symptoms of a local mass. Depending on the tumor location they may vary. Tumors located in the carotid body may present with voice hoarseness, neck fullness, cough, dysphagia or clinically palpable mass in the lateral upper neck. When located in the middle ear (glomus tympanicum) patients may present with palsies of the cranial nerves VII, IX, X, XI and/or XII (117). It is recommended that these patients undergo imaging in order to detect metastatic disease or new tumors (118–120). In the case of SDHA, SDHC and SDHD mutations, because of the slow-growing tumors, MRI screening is suggested every three to five years (120). In individuals with SDHB mutations, due to the rapidly growing nature of these tumors, it should be performed every two years (120). A recent study demonstrated that the most optimal diagnostic imaging included MRI/CT and 111In-octreotide scintigraphy (121). Other studies showed higher sensitivity and more detailed imaging (regardless of genetic mutation and familial or sporadic cases) using 68Ga-DOTA-peptides PET/CT, which targets the abundantly expressed somatostatin receptors in those tumors, compared to conventional CT or MRI (122–128); in addition, more lesions were identified in the case of head and neck paragangliomas (HNPGLs) using that compared to all other imaging techniques (126) (including [18F]-fluorohydroyphenylalanine ([18F]-FDOPA) PET/CT, currently the gold standard for head and neck paragangliomas) (119, 129, 130). Patients with PA should be carefully examined for any symptoms of prolactin (PRL) or GH hypersecretion or visual disturbances, as most PAs that occur in the context of 3PAs are PRL- or GH- secreting macroadenomas or non-functional PAs. Complete pituitary hormone evaluation should also be performed to rule out other pituitary tumors. Hormonal testing should also be performed in the case of concurrent PGLs/PHEOs either in the index case or any family member. If there are no abnormal findings, based on the most recent recommendations, biochemical tests, including testing for PGLs/PHEOs, should be performed annually (118, 119). Pituitary MRI is indicated in the case of abnormal biochemistry or clinical findings. SDH-deficient PAs are treated the same as sporadic (131–134). In the case of renal cancer, patients may complain about flank pain and/or hematuria, whereas in GISTs abdominal pain or fullness may be the main issue.
Genetic Counseling and Genetic Testing
It could be suggested that in the presence of SDH deficiency a careful and detailed medical and family history should be obtained even in patients with apparently ‘sporadic’ PGLs/PHEOs, GISTs or PAs due to the variable expression and decreased penetrance of those conditions. Patients and family members should be referred for genetic counseling. Genetic testing for SDHx mutations in any of the above patients, particularly if there are other family members with any of those tumors (do not only include first-degree relatives) should be performed. Doctors should be aware of CT or CSS especially in the case of a KIT or PDFGRA negative GIST. In the case that genetic testing is unavailable or cannot be performed, SDHB IHC could be performed.
Summary
SDH-deficient tumors are often an indicator of a genetic, tumor-predisposition syndrome, associated with germline mutations in any of the SDHx subunits: SDHA, SDHB, SDHC, SDHD or rarely SDHAF2. In the case of CT, epimutation of SDHC promoter locus is the cause. Identifying the genetic basis of SDH-deficient tumors has helped in identifying individuals in high risk and introduce screening to them and their families. Thus, better clinical care can be provided as early detection and treatment have become more feasible.
Author Contributions
GP contributed to writing the draft of the paper, to writing the final version of the paper, and critically revised it. FF and NS contributed to writing the final version of the paper and critically revised it. CS conceived the study and contributed to writing the final version of the paper and critically revised it. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Intramural Research Program, Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), Bethesda, Md 20892, USA.
Conflict of Interest
CAS holds patent on the PRKAR1A, PDE11A, and GPR101 genes and/or their function and his laboratory has received research funding from Pfizer Inc. FRF holds patent on the GPR101 gene and/or its function. CAS is receiving compensation by ELPEN, Inc. Neither Pfizer, Inc nor ELPEN, Inc had any role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, et al. Crystal Structure of Mitochondrial Respiratory Membrane Protein Complex Ii. Cell (2005) 121(7):1043–57. doi: 10.1016/j.cell.2005.05.025
2. Settas N, Faucz FR, Stratakis CA. Succinate dehydrogenase (SDH) deficiency, Carney triad and the epigenome. Mol Cell Endocrinol (2017) 469:107–11. doi: 10.1016/j.mce.2017.07.018
3. Huang S, Millar AH. Succinate Dehydrogenase: The Complex Roles of a Simple Enzyme. Curr Opin Plant Biol (2013) 16(3):344–9. doi: 10.1016/j.pbi.2013.02.007
4. Gill AJ. Succinate Dehydrogenase (Sdh)-Deficient Neoplasia. Histopathology (2018) 72(1):106–16. doi: 10.1111/his.13277
5. Gill AJ. Succinate Dehydrogenase (SDH) and Mitochondrial Driven Neoplasia. Pathology (2012) 44(4):285–92. doi: 10.1097/PAT.0b013e3283539932
6. Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, Meyer-Rochow GY, et al. Immunohistochemistry for SDHB Triages Genetic Testing of SDHB, SDHC, and SDHD in Paraganglioma-Pheochromocytoma Syndromes. Hum Pathol (2010) 41(6):805–14. doi: 10.1016/j.humpath.2009.12.005
7. van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM, et al. An Immunohistochemical Procedure to Detect Patients With Paraganglioma and Phaeochromocytoma With Germline Sdhb, SDHC, or SDHD Gene Mutations: A Retrospective and Prospective Analysis. Lancet Oncol (2009) 10(8):764–71. doi: 10.1016/S1470-2045(09)70164-0
8. Papathomas TG, Oudijk L, Persu A, Gill AJ, van Nederveen F, Tischler AS, et al. Sdhb/Sdha Immunohistochemistry in Pheochromocytomas and Paragangliomas: A Multicenter Interobserver Variation Analysis Using Virtual Microscopy: A Multinational Study of the European Network for the Study of Adrenal Tumors (EnsT). Mod Pathol (2015) 28(6):807–21. doi: 10.1038/modpathol.2015.41
9. Korpershoek E, Favier J, Gaal J, Burnichon N, van Gessel B, Oudijk L, et al. Sdha Immunohistochemistry Detects Germline Sdha Gene Mutations in Apparently Sporadic Paragangliomas and Pheochromocytomas. J Clin Endocrinol Metab (2011) 96(9):E1472–6. doi: 10.1210/jc.2011-1043
10. Gottlieb E, Tomlinson IP. Mitochondrial Tumour Suppressors: A Genetic and Biochemical Update. Nat Rev Cancer (2005) 5(11):857–66. doi: 10.1038/nrc1737
11. Bardella C, Pollard PJ, Tomlinson I. Sdh Mutations in Cancer. Biochim Biophys Acta (2011) 1807(11):1432–43. doi: 10.1016/j.bbabio.2011.07.003
12. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate Links Tca Cycle Dysfunction to Oncogenesis by Inhibiting HIF-Alpha Prolyl Hydroxylase. Cancer Cell (2005) 7(1):77–85. doi: 10.1016/j.ccr.2004.11.022
13. Raimundo N, Baysal BE, Shadel GS. Revisiting the TCA Cycle: Signaling to Tumor Formation. Trends Mol Med (2011) 17(11):641–9. doi: 10.1016/j.molmed.2011.06.001
14. Gimenez-Roqueplo AP, Favier J, Rustin P, Mourad JJ, Plouin PF, Corvol P, et al. The R22X Mutation of the SDHD Gene in Hereditary Paraganglioma Abolishes the Enzymatic Activity of Complex II in the Mitochondrial Respiratory Chain and Activates the Hypoxia Pathway. Am J Hum Genet (2001) 69(6):1186–97. doi: 10.1086/324413
15. Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Kerlan V, Plouin PF, et al. Functional Consequences of a SDHB Gene Mutation in an Apparently Sporadic Pheochromocytoma. J Clin Endocrinol Metab (2002) 87(10):4771–4. doi: 10.1210/jc.2002-020525
16. Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, et al. Accumulation of Krebs Cycle Intermediates and Over-Expression of HIF1alpha in Tumours Which Result From Germline FH and SDH Mutations. Hum Mol Genet (2005) 14(15):2231–9. doi: 10.1093/hmg/ddi227
17. Lopez-Jimenez E, Gomez-Lopez G, Leandro-Garcia LJ, Munoz I, Schiavi F, Montero-Conde C, et al. Research Resource: Transcriptional Profiling Reveals Different Pseudohypoxic Signatures in SDHB and VHL-related Pheochromocytomas. Mol Endocrinol (2010) 24(12):2382–91. doi: 10.1210/me.2010-0256
18. Raha S, McEachern GE, Myint AT, Robinson BH. Superoxides From Mitochondrial Complex Iii: The Role of Manganese Superoxide Dismutase. Free Radic Biol Med (2000) 29(2):170–80. doi: 10.1016/S0891-5849(00)00338-5
19. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of Alpha-KG-Dependent Histone and DNA Demethylases by Fumarate and Succinate That are Accumulated in Mutations of FH and SDH Tumor Suppressors. Genes Dev (2012) 26(12):1326–38. doi: 10.1101/gad.191056.112
20. Smith EH, Janknecht R, Maher LJ,3. Succinate Inhibition of Alpha-Ketoglutarate-Dependent Enzymes in a Yeast Model of Paraganglioma. Hum Mol Genet (2007) 16(24):3136–48. doi: 10.1093/hmg/ddm275
21. Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, et al. SDHA is a Tumor Suppressor Gene Causing Paraganglioma. Hum Mol Genet (2010) 19(15):3011–20. doi: 10.1093/hmg/ddq206
22. Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, et al. Early-Onset Renal Cell Carcinoma as a Novel Extraparaganglial Component of SDHB-associated Heritable Paraganglioma. Am J Hum Genet (2004) 74(1):153–9. doi: 10.1086/381054
23. Xekouki P, Stratakis CA. Succinate Dehydrogenase (Sdhx) Mutations in Pituitary Tumors: Could This be a New Role for Mitochondrial Complex II and/or Krebs Cycle Defects? Endocr Relat Cancer (2012) 19(6):C33–40. doi: 10.1530/ERC-12-0118
24. Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, et al. Gene Mutations in the Succinate Dehydrogenase Subunit Sdhb Cause Susceptibility to Familial Pheochromocytoma and to Familial Paraganglioma. Am J Hum Genet (2001) 69(1):49–54. doi: 10.1086/321282
25. Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a Mitochondrial Complex II Gene, in Hereditary Paraganglioma. Science (2000) 287(5454):848–51. doi: 10.1126/science.287.5454.848
26. Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, et al. Clinical and Molecular Genetics of Patients With the Carney-Stratakis Syndrome and Germline Mutations of the Genes Coding for the Succinate Dehydrogenase Subunits Sdhb, SDHC, and SDHD. Eur J Hum Genet (2008) 16(1):79–88. doi: 10.1038/sj.ejhg.5201904
27. Ni Y, Zbuk KM, Sadler T, Patocs A, Lobo G, Edelman E, et al. Germline Mutations and Variants in the Succinate Dehydrogenase Genes in Cowden and Cowden-like Syndromes. Am J Hum Genet (2008) 83(2):261–8. doi: 10.1016/j.ajhg.2008.07.011
28. Schimke RN, Collins DL, Stolle CA. Paraganglioma, Neuroblastoma, and a SDHB Mutation: Resolution of a 30-Year-Old Mystery. Am J Med Genet A (2010) 152A(6):1531–5. doi: 10.1002/ajmg.a.33384
29. Grogan RH, Pacak K, Pasche L, Huynh TT, Greco RS. Bilateral Adrenal Medullary Hyperplasia Associated With an SDHB Mutation. J Clin Oncol (2011) 29(8):e200–2. doi: 10.1200/JCO.2010.32.2156
30. Galera-Ruiz H, Gonzalez-Campora R, Rey-Barrera M, Rollon-Mayordomo A, Garcia-Escudero A, Fernandez-Santos JM, et al. W43x SDHD Mutation in Sporadic Head and Neck Paraganglioma. Anal Quant Cytol Histol (2008) 30(2):119–23.
31. Rindi G, Klimstra DS, Abedi-Ardekani B, Asa SL, Bosman FT, Brambilla E, et al. A Common Classification Framework for Neuroendocrine Neoplasms: An International Agency for Research on Cancer (IARC) and World Health Organization (Who) Expert Consensus Proposal. Mod Pathol (2018) 31(12):1770–86. doi: 10.1038/s41379-018-0110-y
32. Lee KY, Oh YW, Noh HJ, Lee YJ, Yong HS, Kang EY, et al. Extraadrenal Paragangliomas of the Body: Imaging Features. AJR Am J Roentgenol (2006) 187(2):492–504. doi: 10.2214/AJR.05.0370
33. Raygada M, Pasini B, Stratakis CA. Hereditary Paragangliomas. Adv Otorhinolaryngol (2011) 70:99–106. doi: 10.1159/000322484
34. Vicha A, Musil Z, Pacak K. Genetics of Pheochromocytoma and Paraganglioma Syndromes: New Advances and Future Treatment Options. Curr Opin Endocrinol Diabetes Obes (2013) 20(3):186–91. doi: 10.1097/MED.0b013e32835fcc45
35. Niemann S, Muller U. Mutations in SDHC Cause Autosomal Dominant Paraganglioma, Type 3. Nat Genet (2000) 26(3):268–70. doi: 10.1038/81551
36. Astuti D, Douglas F, Lennard TW, Aligianis IA, Woodward ER, Evans DG, et al. Germline SDHD Mutation in Familial Phaeochromocytoma. Lancet (2001) 357(9263):1181–2. doi: 10.1016/S0140-6736(00)04378-6
37. Gimm O, Armanios M, Dziema H, Neumann HP, Eng C. Somatic and Occult Germ-Line Mutations in SDHD, a Mitochondrial Complex II Gene, in Nonfamilial Pheochromocytoma. Cancer Res (2000) 60(24):6822–5.
38. Baysal BE, Willett-Brozick JE, Lawrence EC, Drovdlic CM, Savul SA, McLeod DR, et al. Prevalence of SDHB, SDHC, and SDHD Germline Mutations in Clinic Patients With Head and Neck Paragangliomas. J Med Genet (2002) 39(3):178–83. doi: 10.1136/jmg.39.3.178
39. van Nederveen FH, Korpershoek E, Lenders JW, de Krijger RR, Dinjens WN. Somatic SDHB Mutation in an Extraadrenal Pheochromocytoma. N Engl J Med (2007) 357(3):306–8. doi: 10.1056/NEJMc070010
40. Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, et al. SDH5, a Gene Required for Flavination of Succinate Dehydrogenase, is Mutated in Paraganglioma. Science (2009) 325(5944):1139–42. doi: 10.1126/science.1175689
41. Opotowsky AR, Moko LE, Ginns J, Rosenbaum M, Greutmann M, Aboulhosn J, et al. Pheochromocytoma and Paraganglioma in Cyanotic Congenital Heart Disease. J Clin Endocrinol Metab (2015) 100(4):1325–34. doi: 10.1210/jc.2014-3863
42. Song MK, Kim GB, Bae EJ, Lee YA, Kim HY, Min SK, et al. Pheochromocytoma and Paraganglioma in Fontan Patients: Common More Than Expected. Congenit Heart Dis (2018) 13(4):608–16. doi: 10.1111/chd.12625
43. Agarwal S, Jindal I, Balazs A, Paul D. Catecholamine-Secreting Tumors in Pediatric Patients With Cyanotic Congenital Heart Disease. J Endocr Soc (2019) 3(11):2135–50. doi: 10.1210/js.2019-00226
44. Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, et al. Clinical Presentation and Penetrance of Pheochromocytoma/Paraganglioma Syndromes. J Clin Endocrinol Metab (2006) 91(3):827–36. doi: 10.1210/jc.2005-1862
45. Schiavi F, Boedeker CC, Bausch B, Peczkowska M, Gomez CF, Strassburg T, et al. Predictors and Prevalence of Paraganglioma Syndrome Associated With Mutations of the SDHC Gene. JAMA (2005) 294(16):2057–63. doi: 10.1001/jama.294.16.2057
46. Ricketts CJ, Forman JR, Rattenberry E, Bradshaw N, Lalloo F, Izatt L, et al. Tumor Risks and Genotype-Phenotype-Proteotype Analysis in 358 Patients With Germline Mutations in SDHB and SDHD. Hum Mutat (2010) 31: (1):41–51. doi: 10.1002/humu.21136
47. Benn DE, Robinson BG, Clifton-Bligh RJ. 15 YEARS of PARAGANGLIOMA: Clinical Manifestations of Paraganglioma Syndromes Types 1-5. Endocr Relat Cancer (2015) 22(4):T91–103. doi: 10.1530/ERC-15-0268
48. van der Mey AG, Maaswinkel-Mooy PD, Cornelisse CJ, Schmidt PH, van de Kamp JJ. Genomic Imprinting in Hereditary Glomus Tumours: Evidence for New Genetic Theory. Lancet (1989) 2(8675):1291–4. doi: 10.1016/S0140-6736(89)91908-9
49. Korpershoek E, Van Nederveen FH, Dannenberg H, Petri BJ, Komminoth P, Perren A, et al. Genetic Analyses of Apparently Sporadic Pheochromocytomas: The Rotterdam Experience. Ann N Y Acad Sci (2006) 1073:138–48. doi: 10.1196/annals.1353.014
50. Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, et al. Germ-Line Mutations in Nonsyndromic Pheochromocytoma. N Engl J Med (2002) 346(19):1459–66. doi: 10.1056/NEJMoa020152
51. Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, et al. Distinct Clinical Features of Paraganglioma Syndromes Associated With SDHB and SDHD Gene Mutations. JAMA (2004) 292(8):943–51. doi: 10.1001/jama.292.8.943
52. Barnett CM, Corless CL, Heinrich MC. Gastrointestinal Stromal Tumors: Molecular Markers and Genetic Subtypes. Hematol Oncol Clin North Am (2013) 27(5):871–88. doi: 10.1016/j.hoc.2013.07.003
53. Perez-Atayde AR, Shamberger RC, Kozakewich HW. Neuroectodermal Differentiation of the Gastrointestinal Tumors in the Carney Triad. An Ultrastructural and Immunohistochemical Study. Am J Surg Pathol (1993) 17(7):706–14. doi: 10.1097/00000478-199307000-00008
54. Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal Pacemaker Cell Tumor (Gipact): Gastrointestinal Stromal Tumors Show Phenotypic Characteristics of the Interstitial Cells of Cajal. Am J Pathol (1998) 152(5):1259–69.
55. Schaefer IM, Marino-Enriquez A, Fletcher JA. What is New in Gastrointestinal Stromal Tumor? Adv Anat Pathol (2017) 24(5):259–67. doi: 10.1097/PAP.0000000000000158
56. Huss S, Kunstlinger H, Wardelmann E, Kleine MA, Binot E, Merkelbach-Bruse S, et al. A Subset of Gastrointestinal Stromal Tumors Previously Regarded as Wild-Type Tumors Carries Somatic Activating Mutations in KIT Exon 8 (P.D419del). Mod Pathol (2013) 26(7):1004–12. doi: 10.1038/modpathol.2013.47
57. Rossi S, Gasparotto D, Miceli R, Toffolatti L, Gallina G, Scaramel E, et al. Kit, PDGFRA, and BRAF Mutational Spectrum Impacts on the Natural History of Imatinib-Naive Localized Gist: A Population-Based Study. Am J Surg Pathol (2015) 39(7):922–30. doi: 10.1097/PAS.0000000000000418
58. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-Function Mutations of C-Kit in Human Gastrointestinal Stromal Tumors. Science (1998) 279(5350):577–80. doi: 10.1126/science.279.5350.577
59. Mei L, Smith SC, Faber AC, Trent J, Grossman SR, Stratakis CA, et al. Gastrointestinal Stromal Tumors: The GIST of Precision Medicine. Trends Cancer (2018) 4(1):74–91. doi: 10.1016/j.trecan.2017.11.006
60. Boikos SA, Pappo AS, Killian JK, LaQuaglia MP, Weldon CB, George S, et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol (2016) 2(7):922–8. doi: 10.1001/jamaoncol.2016.0256
61. Miettinen M, Lasota J. Succinate Dehydrogenase Deficient Gastrointestinal Stromal Tumors (Gists) - a Review. Int J Biochem Cell Biol (2014) 53:514–9. doi: 10.1016/j.biocel.2014.05.033
62. Agaram NP, Wong GC, Guo T, Maki RG, Singer S, Dematteo RP, et al. Novel V600E Braf Mutations in Imatinib-Naive and Imatinib-Resistant Gastrointestinal Stromal Tumors. Genes Chromosomes Cancer (2008) 47(10):853–9. doi: 10.1002/gcc.20589
63. Brenca M, Rossi S, Polano M, Gasparotto D, Zanatta L, Racanelli D, et al. Transcriptome Sequencing Identifies ETV6-NTRK3 as a Gene Fusion Involved in GIST. J Pathol (2016) 238(4):543–9. doi: 10.1002/path.4677
64. Hostein I, Faur N, Primois C, Boury F, Denard J, Emile JF, et al. Braf Mutation Status in Gastrointestinal Stromal Tumors. Am J Clin Pathol (2010) 133(1):141–8. doi: 10.1309/AJCPPCKGA2QGBJ1R
65. Lasota J, Felisiak-Golabek A, Wasag B, Kowalik A, Zieba S, Chlopek M, et al. Frequency and Clinicopathologic Profile of PIK3CA Mutant GISTs: Molecular Genetic Study of 529 Cases. Mod Pathol (2016) 29(3):275–82. doi: 10.1038/modpathol.2015.160
66. Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal Stromal Tumors in Patients With Neurofibromatosis 1: A Clinicopathologic and Molecular Genetic Study of 45 Cases. Am J Surg Pathol (2006) 30(1):90–6. doi: 10.1097/01.pas.0000176433.81079.bd
67. Miranda C, Nucifora M, Molinari F, Conca E, Anania MC, Bordoni A, et al. KRAS and BRAF Mutations Predict Primary Resistance to Imatinib in Gastrointestinal Stromal Tumors. Clin Cancer Res (2012) 18(6):1769–76. doi: 10.1158/1078-0432.CCR-11-2230
68. Patil DT, Rubin BP. Genetics of Gastrointestinal Stromal Tumors: A Heterogeneous Family of Tumors? Surg Pathol Clin (2015) 8(3):515–24. doi: 10.1016/j.path.2015.05.006
69. Weldon CB, Madenci AL, Boikos SA, Janeway KA, George S, von Mehren M, et al. Surgical Management of Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Pediatric and Wildtype Gist Clinic. J Clin Oncol (2017) 35(5):523–8. doi: 10.1200/JCO.2016.68.6733
70. Shi E, Chmielecki J, Tang CM, Wang K, Heinrich MC, Kang G, et al. FGFR1 and NTRK3 Actionable Alterations in “Wild-Type” Gastrointestinal Stromal Tumors. J Transl Med (2016) 14(1):339. doi: 10.1186/s12967-016-1075-6
71. Pantaleo MA, Urbini M, Indio V, Ravegnini G, Nannini M, De Luca M, et al. Genome-Wide Analysis Identifies MEN1 and MAX Mutations and a Neuroendocrine-Like Molecular Heterogeneity in Quadruple Wt Gist. Mol Cancer Res (2017) 15(5):553–62. doi: 10.1158/1541-7786.MCR-16-0376
72. Nannini M, Urbini M, Astolfi A, Biasco G, Pantaleo MA. The Progressive Fragmentation of the KIT/PDGFRA Wild-Type (Wt) Gastrointestinal Stromal Tumors (Gist). J Transl Med (2017) 15(1):113. doi: 10.1186/s12967-017-1212-x
73. Elston MS, Sehgal S, Dray M, Phillips E, Conaglen JV, Clifton-Bligh RJ, et al. A Duodenal Sdh-Deficient Gastrointestinal Stromal Tumor in a Patient With a Germline Sdhb Mutation. J Clin Endocrinol Metab (2017) 102(5):1447–50. doi: 10.1210/jc.2017-00165
74. Dwight T, Benn DE, Clarkson A, Vilain R, Lipton L, Robinson BG, et al. Loss of SDHA Expression Identifies Sdha Mutations in Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumors. Am J Surg Pathol (2013) 37(2):226–33. doi: 10.1097/PAS.0b013e3182671155
75. Gaal J, Stratakis CA, Carney JA, Ball ER, Korpershoek E, Lodish MB, et al. Sdhb Immunohistochemistry: A Useful Tool in the Diagnosis of Carney-Stratakis and Carney Triad Gastrointestinal Stromal Tumors. Mod Pathol (2011) 24(1):147–51. doi: 10.1038/modpathol.2010.185
76. Miettinen M, Wang ZF, Sarlomo-Rikala M, Osuch C, Rutkowski P, Lasota J. Succinate Dehydrogenase-Deficient Gists: A Clinicopathologic, Immunohistochemical, and Molecular Genetic Study of 66 Gastric Gists With Predilection to Young Age. Am J Surg Pathol (2011) 35(11):1712–21. doi: 10.1097/PAS.0b013e3182260752
77. Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, et al. Defects in Succinate Dehydrogenase in Gastrointestinal Stromal Tumors Lacking KIT and PDGFRA Mutations. Proc Natl Acad Sci USA (2011) 108(1):314–8. doi: 10.1073/pnas.1009199108
78. Chou A, Chen J, Clarkson A, Samra JS, Clifton-Bligh RJ, Hugh TJ, et al. Succinate Dehydrogenase-Deficient Gists are Characterized by IGF1R Overexpression. Mod Pathol (2012) 25(9):1307–13. doi: 10.1038/modpathol.2012.77
79. Wagner AJ, Remillard SP, Zhang YX, Doyle LA, George S, Hornick JL. Loss of Expression of SDHA Predicts SDHA Mutations in Gastrointestinal Stromal Tumors. Mod Pathol (2013) 26(2):289–94. doi: 10.1038/modpathol.2012.153
80. Evenepoel L, Papathomas TG, Krol N, Korpershoek E, de Krijger RR, Persu A, et al. Toward an Improved Definition of the Genetic and Tumor Spectrum Associated With SDH Germ-Line Mutations. Genet Med (2015) 17(8):610–20. doi: 10.1038/gim.2014.162
81. Janeway KA, Zhu MJ, Barretina J, Perez-Atayde A, Demetri GD, Fletcher JA. Strong Expression of IGF1R in Pediatric Gastrointestinal Stromal Tumors Without Igf1r Genomic Amplification. Int J Cancer (2010) 127(11):2718–22. doi: 10.1002/ijc.25247
82. Lasota J, Wang Z, Kim SY, Helman L, Miettinen M. Expression of the Receptor for Type I Insulin-Like Growth Factor (IGF1R) in Gastrointestinal Stromal Tumors: An Immunohistochemical Study of 1078 Cases With Diagnostic and Therapeutic Implications. Am J Surg Pathol (2013) 37(1):114–9. doi: 10.1097/PAS.0b013e3182613c86
83. Wang YM, Gu ML, Ji F. Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumors. World J Gastroenterol (2015) 21(8):2303–14. doi: 10.3748/wjg.v21.i8.2303
84. Matyakhina L, Bei TA, McWhinney SR, Pasini B, Cameron S, Gunawan B, et al. Genetics of Carney Triad: Recurrent Losses At Chromosome 1 But Lack of Germline Mutations in Genes Associated With Paragangliomas and Gastrointestinal Stromal Tumors. J Clin Endocrinol Metab (2007) 92(8):2938–43. doi: 10.1210/jc.2007-0797
85. Stratakis CA, Carney JA. The Triad of Paragangliomas, Gastric Stromal Tumours and Pulmonary Chondromas (Carney Triad), and the Dyad of Paragangliomas and Gastric Stromal Sarcomas (Carney-Stratakis Syndrome): Molecular Genetics and Clinical Implications. J Intern Med (2009) 266(1):43–52. doi: 10.1111/j.1365-2796.2009.02110.x
86. Carney JA, Sheps SG, Go VL, Gordon H. The Triad of Gastric Leiomyosarcoma, Functioning Extra-Adrenal Paraganglioma and Pulmonary Chondroma. N Engl J Med (1977) 296(26):1517–8. doi: 10.1056/NEJM197706302962609
87. Zhang L, Smyrk TC, Young WF Jr, Stratakis CA, Carney JA. Gastric Stromal Tumors in Carney Triad are Different Clinically, Pathologically, and Behaviorally From Sporadic Gastric Gastrointestinal Stromal Tumors: Findings in 104 Cases. Am J Surg Pathol (2010) 34(1):53–64. doi: 10.1097/PAS.0b013e3181c20f4f
88. Carney JA. Gastric Stromal Sarcoma, Pulmonary Chondroma, and Extra-Adrenal Paraganglioma (Carney Triad): Natural History, Adrenocortical Component, and Possible Familial Occurrence. Mayo Clin Proc (1999) 74(6):543–52. doi: 10.4065/74.6.543
89. Haller F, Moskalev EA, Faucz FR, Barthelmess S, Wiemann S, Bieg M, et al. Aberrant DNA Hypermethylation of SDHC: A Novel Mechanism of Tumor Development in Carney Triad. Endocr Relat Cancer (2014) 21(4):567–77. doi: 10.1530/ERC-14-0254
90. Killian JK, Miettinen M, Walker RL, Wang Y, Zhu YJ, Waterfall JJ, et al. Recurrent Epimutation of SDHC in Gastrointestinal Stromal Tumors. Sci Transl Med (2014) 6(268):268ra177. doi: 10.1126/scitranslmed.3009961
91. Carney JA, Stratakis CA. Familial Paraganglioma and Gastric Stromal Sarcoma: A New Syndrome Distinct From the Carney Triad. Am J Med Genet (2002) 108(2):132–9. doi: 10.1002/ajmg.10235
92. McWhinney SR, Pasini B, Stratakis CA, International Carney T. Carney-Stratakis Syndrome C. Familial Gastrointestinal Stromal Tumors and Germ-Line Mutations. N Engl J Med (2007) 357(10):1054–6. doi: 10.1056/NEJMc071191
93. Pantaleo MA, Lolli C, Nannini M, Astolfi A, Indio V, Saponara M, et al. Good Survival Outcome of Metastatic SDH-Deficient Gastrointestinal Stromal Tumors Harboring SDHA Mutations. Genet Med (2015) 17(5):391–5. doi: 10.1038/gim.2014.115
94. Iversen K. Acromegaly Associated With Phaeochromocytoma. Acta Med Scand (1952) 142(1):1–5. doi: 10.1111/j.0954-6820.1952.tb13837.x
95. Xekouki P, Brennand A, Whitelaw B, Pacak K, Stratakis CA. The 3pas: An Update on the Association of Pheochromocytomas, Paragangliomas, and Pituitary Tumors. Horm Metab Res (2019) 51(7):419–36. doi: 10.1055/a-0661-0341
96. Xekouki P, Pacak K, Almeida M, Wassif CA, Rustin P, Nesterova M, et al. Succinate Dehydrogenase (Sdh) D Subunit (Sdhd) Inactivation in a Growth-Hormone-Producing Pituitary Tumor: A New Association for SDH? J Clin Endocrinol Metab (2012) 97(3):E357–66. doi: 10.1210/jc.2011-1179
97. Dwight T, Mann K, Benn DE, Robinson BG, McKelvie P, Gill AJ, et al. Familial SDHA Mutation Associated With Pituitary Adenoma and Pheochromocytoma/Paraganglioma. J Clin Endocrinol Metab (2013) 98(6):E1103–8. doi: 10.1210/jc.2013-1400
98. Papathomas TG, Gaal J, Corssmit EP, Oudijk L, Korpershoek E, Heimdal K, et al. Non-Pheochromocytoma (Pcc)/Paraganglioma (Pgl) Tumors in Patients With Succinate Dehydrogenase-Related PCC-PGL Syndromes: A Clinicopathological and Molecular Analysis. Eur J Endocrinol (2014) 170(1):1–12. doi: 10.1530/EJE-13-0623
99. Gill AJ, Toon CW, Clarkson A, Sioson L, Chou A, Winship I, et al. Succinate Dehydrogenase Deficiency is Rare in Pituitary Adenomas. Am J Surg Pathol (2014) 38(4):560–6. doi: 10.1097/PAS.0000000000000149
100. Denes J, Swords F, Rattenberry E, Stals K, Owens M, Cranston T, et al. Heterogeneous Genetic Background of the Association of Pheochromocytoma/Paraganglioma and Pituitary Adenoma: Results From a Large Patient Cohort. J Clin Endocrinol Metab (2015) 100(3):E531–41. doi: 10.1210/jc.2014-3399
101. Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial Autophagy is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J Biol Chem (2008) 283(16):10892–903. doi: 10.1074/jbc.M800102200
102. Zakeri Z, Lockshin RA. Cell Death: History and Future. Adv Exp Med Biol (2008) 615:1–11. doi: 10.1007/978-1-4020-6554-5_1
103. Lee JG, Shin JH, Shim HS, Lee CY, Kim DJ, Kim YS, et al. Autophagy Contributes to the Chemo-Resistance of non-Small Cell Lung Cancer in Hypoxic Conditions. Respir Res (2015) 16:138. doi: 10.1186/s12931-015-0285-4
104. Chaachouay H, Fehrenbacher B, Toulany M, Schaller M, Multhoff G, Rodemann HP. Ampk-Independent Autophagy Promotes Radioresistance of Human Tumor Cells Under Clinical Relevant Hypoxia In Vitro. Radiother Oncol (2015) 116(3):409–16. doi: 10.1016/j.radonc.2015.08.012
105. Srigley JR, Delahunt B, Eble JN, Egevad L, Epstein JI, Grignon D, et al. The International Society of Urological Pathology (Isup) Vancouver Classification of Renal Neoplasia. Am J Surg Pathol (2013) 37(10):1469–89. doi: 10.1097/PAS.0b013e318299f2d1
106. Gill AJ, Hes O, Papathomas T, Sedivcova M, Tan PH, Agaimy A, et al. Succinate Dehydrogenase (Sdh)-Deficient Renal Carcinoma: A Morphologically Distinct Entity: A Clinicopathologic Series of 36 Tumors From 27 Patients. Am J Surg Pathol (2014) 38(12):1588–602. doi: 10.1097/PAS.0000000000000292
107. Wang G, Rao P. Succinate Dehydrogenase-Deficient Renal Cell Carcinoma: A Short Review. Arch Pathol Lab Med (2018) 142(10):1284–8. doi: 10.5858/arpa.2017-0199-RS
108. Williamson SR, Eble JN, Amin MB, Gupta NS, Smith SC, Sholl LM, et al. Succinate Dehydrogenase-Deficient Renal Cell Carcinoma: Detailed Characterization of 11 Tumors Defining a Unique Subtype of Renal Cell Carcinoma. Mod Pathol (2015) 28(1):80–94. doi: 10.1038/modpathol.2014.86
109. Gill AJ, Lipton L, Taylor J, Benn DE, Richardson AL, Frydenberg M, et al. Germline SDHC Mutation Presenting as Recurrent Sdh Deficient GIST and Renal Carcinoma. Pathology (2013) 45(7):689–91. doi: 10.1097/PAT.0000000000000018
110. Yakirevich E, Ali SM, Mega A, McMahon C, Brodsky AS, Ross JS, et al. A Novel Sdha-Deficient Renal Cell Carcinoma Revealed by Comprehensive Genomic Profiling. Am J Surg Pathol (2015) 39(6):858–63. doi: 10.1097/PAS.0000000000000403
111. Jiang Q, Zhang Y, Zhou YH, Hou YY, Wang JY, Li JL, et al. A Novel Germline Mutation in SDHA Identified in a Rare Case of Gastrointestinal Stromal Tumor Complicated With Renal Cell Carcinoma. Int J Clin Exp Pathol (2015) 8(10):12188–97.
112. Ozluk Y, Taheri D, Matoso A, Sanli O, Berker NK, Yakirevich E, et al. Renal Carcinoma Associated With a Novel Succinate Dehydrogenase A Mutation: A Case Report and Review of Literature of a Rare Subtype of Renal Carcinoma. Hum Pathol (2015) 46(12):1951–5. doi: 10.1016/j.humpath.2015.07.027
113. Miettinen M, Sarlomo-Rikala M, McCue P, Czapiewski P, Langfort R, Waloszczyk P, et al. Mapping of Succinate Dehydrogenase Losses in 2258 Epithelial Neoplasms. Appl Immunohistochem Mol Morphol (2014) 22(1):31–6. doi: 10.1097/PAI.0b013e31828bfdd3
114. Gill AJ, Pachter NS, Chou A, Young B, Clarkson A, Tucker KM, et al. Renal Tumors Associated With Germline SDHB Mutation Show Distinctive Morphology. Am J Surg Pathol (2011) 35(10):1578–85. doi: 10.1097/PAS.0b013e318227e7f4
115. Ricketts CJ, Shuch B, Vocke CD, Metwalli AR, Bratslavsky G, Middelton L, et al. Succinate Dehydrogenase Kidney Cancer: An Aggressive Example of the Warburg Effect in Cancer. J Urol (2012) 188(6):2063–71. doi: 10.1016/j.juro.2012.08.030
116. Hensen EF, Bayley JP. Recent Advances in the Genetics of SDH-related Paraganglioma and Pheochromocytoma. Fam Cancer (2011) 10(2):355–63. doi: 10.1007/s10689-010-9402-1
117. Suarez C, Rodrigo JP, Bodeker CC, Llorente JL, Silver CE, Jansen JC, et al. Jugular and Vagal Paragangliomas: Systematic Study of Management With Surgery and Radiotherapy. Head Neck (2013) 35(8):1195–204. doi: 10.1002/hed.22976
118. Plouin PF, Amar L, Dekkers OM, Fassnacht M, Gimenez-Roqueplo AP, Lenders JW, et al. European Society of Endocrinology Clinical Practice Guideline for Long-Term Follow-Up of Patients Operated on for a Phaeochromocytoma or a Paraganglioma. Eur J Endocrinol (2016) 174(5):G1–G10. doi: 10.1530/EJE-16-0033
119. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2014) 99(6):1915–42. doi: 10.1210/jc.2014-1498
120. CI NSW. Risk Management for SDH-related Paraganglioma-Phaeochromocytoma Predisposition Syndromes. Available at: https://www.eviq.org.au/cancer-genetics/risk-management/3558-risk-management-for-sdh-related-paraganglioma#cancer-and-tumour-risk-management-guidelines.
121. Gimenez-Roqueplo AP, Caumont-Prim A, Houzard C, Hignette C, Hernigou A, Halimi P, et al. Imaging Work-Up for Screening of Paraganglioma and Pheochromocytoma in SDHx Mutation Carriers: A Multicenter Prospective Study From the PGL.EVA Investigators. J Clin Endocrinol Metab (2013) 98(1):E162–73. doi: 10.1210/jc.2012-2975
122. Han S, Suh CH, Woo S, Kim YJ, Lee JJ. Performance of (68)Ga-DOTA-Conjugated Somatostatin Receptor-Targeting Peptide PET in Detection of Pheochromocytoma and Paraganglioma: A Systematic Review and Metaanalysis. J Nucl Med (2019) 60(3):369–76. doi: 10.2967/jnumed.118.211706
123. Janssen I, Chen CC, Millo CM, Ling A, Taieb D, Lin FI, et al. Pet/Ct Comparing (68)Ga-DOTATATE and Other Radiopharmaceuticals and in Comparison With CT/MRI for the Localization of Sporadic Metastatic Pheochromocytoma and Paraganglioma. Eur J Nucl Med Mol Imaging (2016) 43(10):1784–91. doi: 10.1007/s00259-016-3357-x
124. Chang CA, Pattison DA, Tothill RW, Kong G, Akhurst TJ, Hicks RJ, et al. (68)Ga-DOTATATE and (18)F-FDG PET/CT in Paraganglioma and Pheochromocytoma: Utility, Patterns and Heterogeneity. Cancer Imaging (2016) 16(1):22. doi: 10.1186/s40644-016-0084-2
125. Castinetti F, Kroiss A, Kumar R, Pacak K, Taieb D. 15 YEARS of PARAGANGLIOMA: Imaging and Imaging-Based Treatment of Pheochromocytoma and Paraganglioma. Endocr Relat Cancer (2015) 22(4):T135–45. doi: 10.1530/ERC-15-0175
126. Janssen I, Chen CC, Taieb D, Patronas NJ, Millo CM, Adams KT, et al. 68Ga-DOTATATE PET/CT in the Localization of Head and Neck Paragangliomas Compared With Other Functional Imaging Modalities and CT/MRI. J Nucl Med (2016) 57(2):186–91. doi: 10.2967/jnumed.115.161018
127. Janssen I, Blanchet EM, Adams K, Chen CC, Millo CM, Herscovitch P, et al. Superiority of [68Ga]-DOTATATE PET/CT to Other Functional Imaging Modalities in the Localization of SDHB-Associated Metastatic Pheochromocytoma and Paraganglioma. Clin Cancer Res (2015) 21(17):3888–95. doi: 10.1158/1078-0432.CCR-14-2751
128. Archier A, Varoquaux A, Garrigue P, Montava M, Guerin C, Gabriel S, et al. Prospective Comparison of (68)Ga-DOTATATE and (18)F-FDOPA PET/CT in Patients With Various Pheochromocytomas and Paragangliomas With Emphasis on Sporadic Cases. Eur J Nucl Med Mol Imaging (2016) 43(7):1248–57. doi: 10.1007/s00259-015-3268-2
129. Taieb D, Timmers HJ, Hindie E, Guillet BA, Neumann HP, Walz MK, et al. Eanm 2012 Guidelines for Radionuclide Imaging of Phaeochromocytoma and Paraganglioma. Eur J Nucl Med Mol Imaging (2012) 39(12):1977–95. doi: 10.1007/s00259-012-2215-8
130. King KS, Chen CC, Alexopoulos DK, Whatley MA, Reynolds JC, Patronas N, et al. Functional Imaging of SDHx-related Head and Neck Paragangliomas: Comparison of 18F-Fluorodihydroxyphenylalanine, 18f-Fluorodopamine, 18f-Fluoro-2-Deoxy-D-Glucose PET, 123i-Metaiodobenzylguanidine Scintigraphy, and 111In-Pentetreotide Scintigraphy. J Clin Endocrinol Metab (2011) 96(9):2779–85. doi: 10.1210/jc.2011-0333
131. Aghi MK, Chen CC, Fleseriu M, Newman SA, Lucas JW, Kuo JS, et al. Congress of Neurological Surgeons Systematic Review and Evidence-Based Guidelines on the Management of Patients With Nonfunctioning Pituitary Adenomas: Executive Summary. Neurosurgery (2016) 79(4):521–3. doi: 10.1227/NEU.0000000000001386
132. Katznelson L, Laws ER Jr, Melmed S, Molitch ME, Murad MH, Utz A, et al. Acromegaly: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2014) 99(11):3933–51. doi: 10.1210/jc.2014-2700
133. Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, et al. Diagnosis and Treatment of Hyperprolactinemia: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2011) 96(2):273–88. doi: 10.1210/jc.2010-1692
Keywords: Succinate dehydrogenase (SDH), GIST, paraganglioma, Carney triad, Carney-Stratakis syndrome, SDHB
Citation: Pitsava G, Settas N, Faucz FR and Stratakis CA (2021) Carney Triad, Carney-Stratakis Syndrome, 3PAS and Other Tumors Due to SDH Deficiency. Front. Endocrinol. 12:680609. doi: 10.3389/fendo.2021.680609
Received: 15 March 2021; Accepted: 12 April 2021;
Published: 03 May 2021.
Edited by:
Enzo Lalli, UMR7275 Institut de pharmacologie moléculaire et cellulaire (IPMC), FranceReviewed by:
Attila Patocs, Semmelweis University, HungaryAndre Lacroix, Université de Montréal, Canada
Copyright © 2021 Pitsava, Settas, Faucz and Stratakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabio R. Faucz, ZmFiaW8uZmF1Y3pAbmloLmdvdg==