 Tania Maria Barreto Rodrigues1
Tania Maria Barreto Rodrigues1 Marlon Messias da Conceição Silva2,3Magali Maciel Freitas2Zélia Maria Costa Duarte2
Marlon Messias da Conceição Silva2,3Magali Maciel Freitas2Zélia Maria Costa Duarte2 Vitória Sousa Frutuoso2
Vitória Sousa Frutuoso2 Mariana Teixeira Rodrigues2,3
Mariana Teixeira Rodrigues2,3 Ileana Gabriela Sanchez Rubio2,3*
Ileana Gabriela Sanchez Rubio2,3*- 1Federal University of Juiz de Fora, Campus Governador Valadares, Governador Valadares, Brazil
- 2Thyroid Molecular Science Laboratory, Department of Biological Sciences, Federal University of São Paulo, Federal University of São Paulo (UNIFESP), Diadema, Brazil
- 3Structural and Functional Biology Program, Federal University of São Paulo, UNIFESP, São Paulo, Brazil
Introduction: It is rare for a euthyroid mother to carry a child with a fetal goiter. However, cases of congenital hypothyroidism (CH) caused by thyroid dyshormonogenesis have been reported. Even though gene mutations associated with fetal goiter have been reported in a few studies, the effects on intellectual development have not been investigated. This study aimed to characterize and investigate the underlying genetic mechanism of CH and neuropsychological development and growth of two siblings with CH-induced fetal goiters.
Case report: Two male siblings from a non-consanguineous marriage with CH and fetal goiter were diagnosed by ultrasonography at 32- and 26-weeks of gestation. This condition was confirmed by cordocentesis in the first pregnancy (TSH: 135 μIU/ml). The mother was euthyroid, and no intra-amniotic levothyroxine treatment was performed. Peripheral blood DNA was screened for TPO mutations. The new deletion p.Cys296Alafs*21 and the p.Arg665Trp mutation, inherited from heterozygous parents, were identified in both patients. Functional analysis showed both mutations reduced the TPO enzyme activity and impaired the membrane localization. The p.Cys296Alafs*21 mutation produces a protein product with a drastically reduced molecular weight. Additionally, a complete clinical and neuropsychological evaluation was also performed. The WISC IV test was employed to provide an overall measure of the siblings’ cognitive and intellectual abilities. No growth retardation was detected in either child. In general, both children showed normal neuropsychological development; however, they exhibited slight reduction of Processing Speed Index scores, which are sensitive to neurological and attentional factors and motor maturation activity. Notably, the younger sibling obtained significantly low scores in the Operational Memory Index, a measure of attention capacity and psychoneurological immaturity.
Conclusion: We described a new TPO compound heterozygosity that severely impaired the TPO activity and membrane localization leading to severe CH and fetal goiter. This is the first report showing the neuropsychological evaluation in patients with dyshormonogenetic fetal goiter. More studies are needed to understand the neurodevelopmental outcomes of neonates with CH-induced fetal goiters.
Introduction
Congenital hypothyroidism (CH) is the most prevalent (1:2,000–1:4,000 live births) childhood endocrine disease and the most common cause of preventable intellectual disability (1). Most CH patients (80–85%) exhibit thyroid dysgenesis, which encompasses a spectrum of abnormalities, including agenesis (a complete absence of the thyroid gland), hypoplasia (a small gland with a typical location), and ectopy (an abnormally located gland) (2). In 10–15% of the cases, CH is caused by defects in the thyroid hormone biosynthesis or dyshormonogenesis (3). Mutations in thyroid peroxidase (TPO) (4), thyroglobulin (TG) (5), dual oxidase DUOX 1, DUOX maturation factor 1 (DUOXA1) (6), dual oxidase 2 (DUOX2) (7), DUOX maturation factor 2 (DUOXA2) (8), sodium iodide symporter (NIS) (9), and pendrin (SLC26A4) (10) have been associated with dyshormonogenesis.
Secondary to dyshormonogenesis, fetal goiters can also develop. These goiters are rarely large enough to be clinically detected at birth and even more challenging during intrauterine observation. However, advances in ultrasound technology have led to identifying many cases and may suggest fetal thyroid deficiency and hypothyroidism through peripheral gland hypervascularization and paradoxical increased fetal movement (11). It is important to point out that fetal goitrous hypothyroidism can also be caused by iodine deficiency (12), massive iodide exposure (13), and maternal hyperthyroidism therapy (14).
Up to now, mutations in TG, TPO, NIS, and DUOXA2 have been identified in few dyshormonogenetic fetal goiter patients (15–28) as shown in Supplementary Table 1. These mutations are usually transmitted in an autosomal recessive fashion; however, some studies have reported monoallelic mutations (29).
The effects of thyroid hormone and its deficit on children’s psychomotor and neurological outcomes have been well-documented (30), but not in CH-induced fetal goiter patients. Before fetal thyroid hormone synthesis begins at approximately 11 weeks of gestation (WG), the fetus’s brain is protected by the maternal thyroid hormone (31). After that point, fetal thyroid hormones are necessary for healthy development. A previous study screening neonates with CH caused by a total organification defect showed that their thyroid hormone levels were about 40–60% of normal levels and likely of maternal origin (32).
This study aimed to characterize and investigate the underlying molecular genetic mechanism for CH development and neuropsychological progression and growth of two siblings with CH-induced fetal goiters.
Case Report
Clinical Presentation
The present study follows two pregnancies of an initially 32-year-old primigravida. During the first pregnancy, a routine ultrasound scan (USS) at 28 WG did not identify any polyhydramnios or other fetal abnormalities. However, the USS at 32 weeks detected a fetal goiter (Supplementary Figure 1). The mother was subsequently referred for endocrinological evaluation. The mother stated that she does not take medication and that she presented thyroid hyperplasia two years earlier. As shown in Table 1, subsequent cordocentesis confirmed fetal hypothyroidism (TSH: 135 μIU/mL; FT4: 0.57 ng/dL). At 34 WG, the fetus was delivered by cesarean without any complications due to spontaneous premature labor. The male newborn weighed 2.145 kg and had Apgar scores of ten at 1 and 5 min. The newborn presented weak suction and vomiting and remained in the intensive care unit for five days. Serum thyroid hormones were measured two days after birth, and hypothyroidism was confirmed. The neonate was subsequently treated with levothyroxine (LT4).
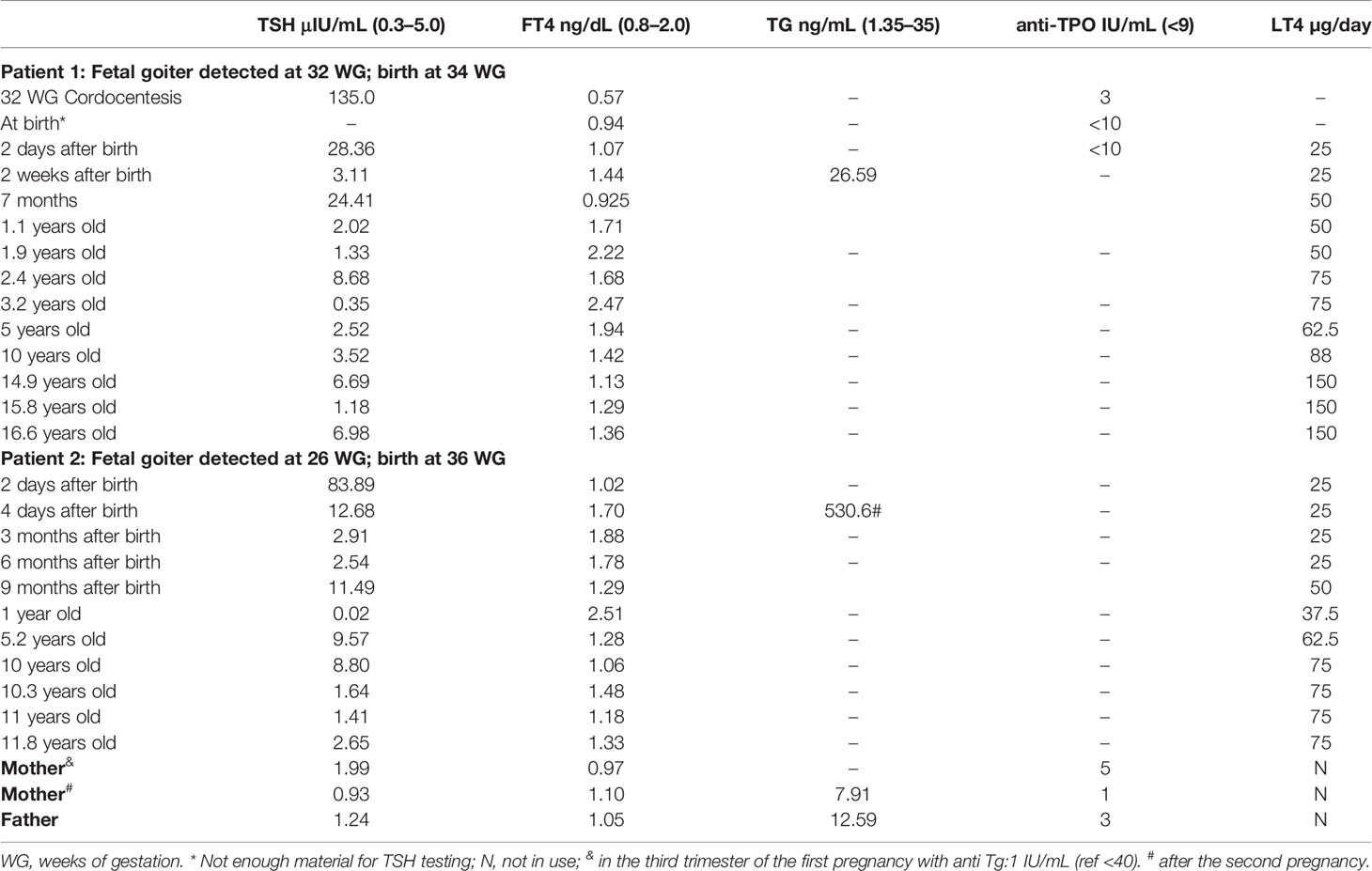
Table 1 Thyroid hormone status of Patients 1 and 2 and of their parents and LT4 dose over time.
During the second pregnancy, the USS at 22 WG revealed no abnormalities. Like the first pregnancy, four weeks later (26 weeks), a fetal goiter was detected (Supplementary Figure 1). It was decided that no intervention would be administered at the time. Due to the progressive increase of the goiter volume, delivery was induced at 35 WG. The male child was delivered without complication and weighed 2.255 kg presenting Apgar scores of nine at 1 and 5 min. Two days after birth, congenital hypothyroidism was confirmed (TSH: 83.89 μIU/mL), and LT4 treatment was initiated. Thyroid hormone status of the patients and parents and LT4 doses over time are presented in Table 1.
Both siblings and the family showed good adherence to LT4 treatment; however, the patients still have goiter detected by USS. They showed good clinical conditions and puberty stages according to Tanner (33). Considering the height and body mass indices, no growth retardation was observed according to World Health Organization (5–19 years old) (34) (Supplemental Figure 2), and there were no motor problems detected in either child during the medical follow-up. Currently, Patient 1 is 16 years old and in his second year of high school. He has a height of 175 cm and weight of 55.7 kg, Height/Age and BMI/Age z scores of 0.08 and −1.19, respectively, and bone age of 17 years. He takes 150 mcg of levothyroxine per day. Patient 2 is 11 years old. He is in elementary school. He is 148.5 cm tall and weighs 39 kg. His calculated z scores for Height/Age and BMI/Age are 0.17 and 0.16, respectively, and his bone age is 10 years. Patient 2 is also treated with levothyroxine (75 mcg/day). In May 2021, laboratory thyroid tests of the siblings’ serum were TSH 6.98 µcIU/mL and 2.65 µcIU/mL (0.27–5.00) and FT4 (1.36 ng/dL and 1.33 ng/dL (0.75–2.00) for Patients 1 and 2, respectively. In Patient 1, a slight increase in TSH level was observed, with normal levels of FT4 (Table 1). It was then decided to maintain the levothyroxine dose and to repeat the tests in two months.
Mutation Detection and Functional Assessment
The sequences analyses revealed that the genomic DNA of the two siblings contained three TPO mutations: p.Gln660Glu (C.1978C>G) and p.Arg665Trp (c.1993C>T) in exon 11 and a new deletion p.Cys296Alafs*21 (c.886delT) in exon 8. Thep.Arg665Trp mutation was present in the mother’s DNA, and the p.Gln660Glu (4) and p.Cys296Alafs*21 (35) mutations were found in the father’s DNA (Figure 1). The p.Gln660Glu mutation is on the same allele as the deletion mutation; thus, it is unlikely to contribute to the disease. Thus, the presence of both the p.Arg665Trp and the p.Cys296Alafs*21 mutations indicates compound heterozygosity in the siblings’ DNA.
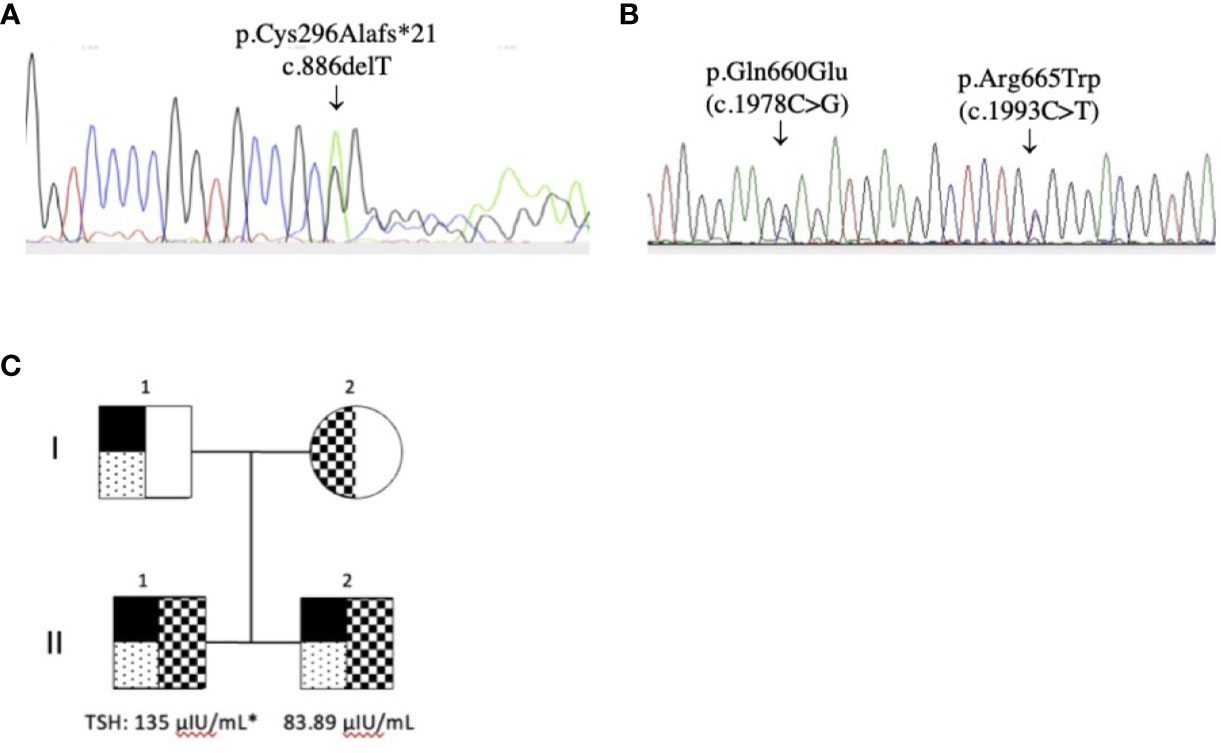
Figure 1 TPO gene mutations. (A) p.Cys296Alafs*21 (c.886delT) in exon 8 (reverse sequence) and (B) p.Gln660Glu (C.1978C>G) in exon 11 and p.Arg665Trp (c.1993C>T) in exon 11 (reverse sequence); (C) Pedigrees of the family and THS values at diagnosis (* cordocentesis). The p.Cys296Alafs*21 (c.886delT) and the p.Gln660Glu (C.1978C>G) mutation was detected in both patients (II.1 and II.2) and the father (I.1). The p.Arg665Trp (c.1993C>T) mutation was detected in both patients and the mother (I.2).
The pathogenicity of the p.Arg665Trp (Arg665Trp-TPO) and p.Cys296Alafs*21 (delT886-TPO) mutations was evaluated in HEK293 cells. The delT886-TPO mutation alters the protein reading frame and introduces a new stop codon at residue 316, resulting in a 35 kDa protein product. In comparison, the Arg665Trp-TPO missense mutant and TPO-WT migrate at a molecular weight of 103 kDa (Figure 2A). We also measured the enzymatic activities of TPO-WT, Arg665Trp-TPO, and delT886-TPO. As shown in Figure 2B, the TPO activities of Arg665Trp-TPO and delT886-TPO are significantly lower than TPO-WT levels.
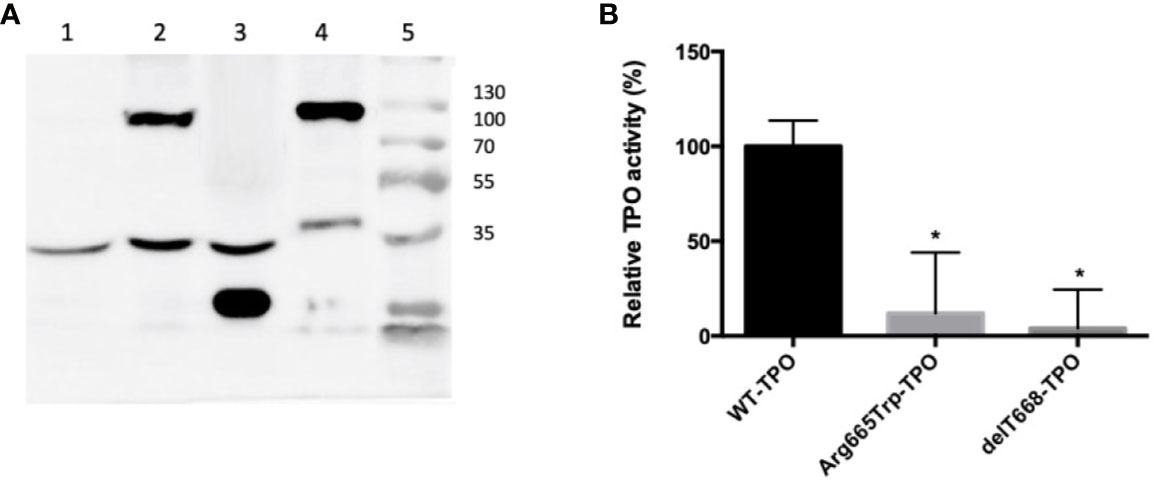
Figure 2 (A) Western blot of HEK293 extracts from cells transfected with wild-type or mutant TPOs. Lane: 1) pCDNA 3.1; Lane 2) Arg665Trp-TPO, 103 kDa; Lane 3) delT668-TPO, 35 kDa; Lane 4) TPO-WT; 103 kDa; Lane 5) molecular weight marker Thermo Scientific Page Ruler (Thermo Scientific, Carlsbad, CA). All strains expressed the endogenous control alpha-tubulin (53 kDa). (B) Extracellular enzymatic activity of HEK293 cells expressing wild-type or mutant TPOs. Enzymatic activity was assessed using the Amplex UltraRed reagent. λEXCITATION = 530 nm and λEMISSION = 560 nm. The values are expressed as a percentage of the TPO-WT transfected cells’ activity (*p < 0.05).
Next, we assessed TPO localization by immunofluorescence. Under cell permeabilizing conditions (Figures 3A–D), cytoplasmic TPO-specific immunostaining was observed in the wild-type and mutant TPO transfected cells. These results confirm TPO transfection and expression. Under non-permeabilizing conditions (Figures 3E–H), TPO-WT was localized along the cell membrane. Notably, we observed reduced membrane staining in cells transfected with delT886-TPO and Arg665Trp-TPO, indicating that these mutant proteins are not correctly inserted into the membrane (Figures 3F, G). It is also worth mentioning that no immunostaining was detected in cells transfected with empty pcDNA vector under either condition. The full methods are in the Supplementary File.
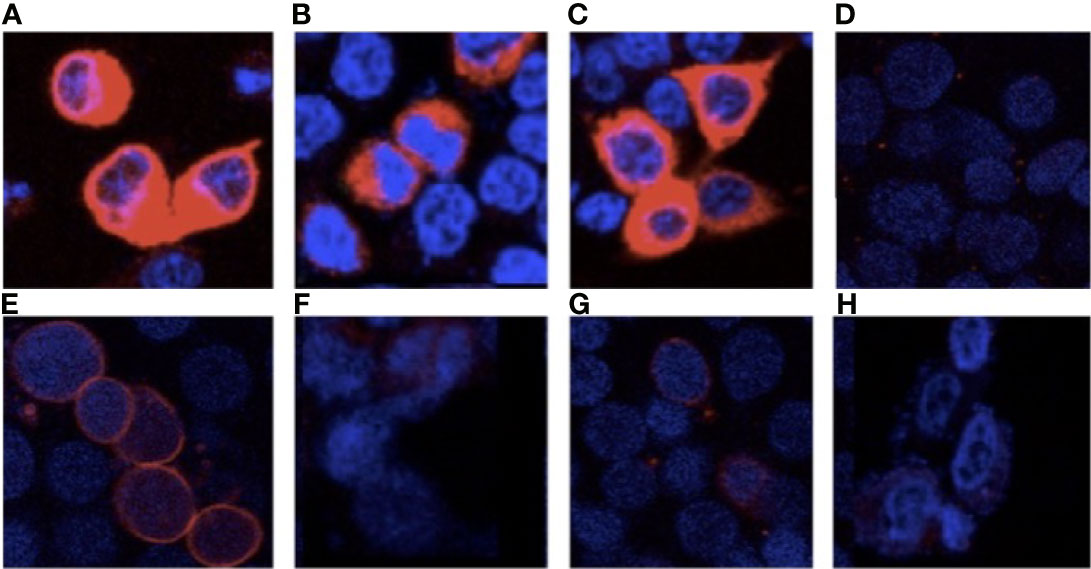
Figure 3 Cellular localization of wild-type and mutant TPOs. Immunofluorescence, using anti-TPO RPE5379- (Abcam) and AlexaFluor 594 (Invitrogen), under permeabilized conditions, Panel (A) TPO-WT, Panel (B) delT668-TPO, Panel (C) Arg665Trp-TPO, and (D) pcDNA and impermeabilized conditions Panel (E) TPO-WT, Panel (F) delT668-TPO, Panel (G) Arg665Trp-TPO, and Panel (H) pcDNA transfected HEK293 cells. Images were acquired with a Leica TCS SP8 confocal microscope at 63× magnification.
Neuropsychological Evaluation
A clinical neuropsychologist evaluated the two brothers for attention (divided, sustained, and focused), cognitive flexibility, short- and long-term memory (verbal and visual), intellectual processes (reasoning, abstract thought, and critical thinking), motor functions (movements, laterality, and others), visual functions (perception, discrimination, and visuospatial and visuoconstructive organization), praxis, general intelligence, learning ability, and language (expressive, receptive and written). The children were also evaluated for the presence of Attention Deficit Hyperactivity Disorder (ADHD). Herein, the fourth edition of the Wechsler Intelligence Scale for Children (WISC IV) test to assess the siblings’ intelligence was used. These evaluations were performed at 11 years of age for Patient 1 and 6 years for Patient 2. A summary of these results is presented in Table 2. In general, the children exhibited behavior consistent with their age, sex, and education. Moreover, the evaluations showed that both boys had adequate age-related logical reasoning, conceptual verbal formation (abstract thinking), memory and verbal fluency, semantic knowledge, intellectual curiosity, and ability to deal with abstract symbols. Quantitative analyses of the WISC-IV results and comparisons of each patient with himself using the factorial indexes, which are considered more accurate measures of intelligence, showed that both patients exhibited lower Processing Speed Index (related to the speed of mental processing and motor graph), and motor visual organization scores. Additionally, Patient 2 had a significantly lower Operational Memory Index (OMI), according to the WISC-IV. The OMI provides a measure of the child’s attention. Due to its sensitivity, it has been utilized for detecting ADHD; however, to confirm the ADHD diagnosis, new evaluations are required between 7 and 12 years of age. Although Patient 2 is currently 11 years old, the parents refused consent for revaluation.
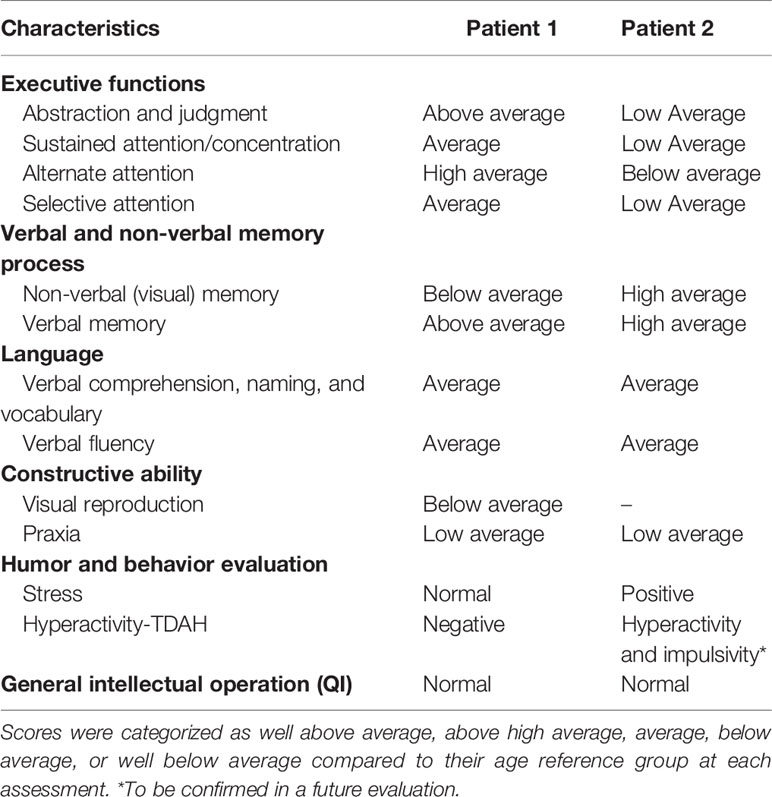
Table 2 Summary of the neuropsychological evaluation results of the dyshormonogenetic fetal goiter patients using WISC-IV Tests.
Discussion
Fetal goiter is a rare thyroid disorder with an incidence of about 1:40,000 live births. Despite being a perinatal complication with low incidence, it can cause airway obstruction and polyhydramnios and increase the fetus’ risk of other morbidities and/or mortality (18, 36). This study investigated the genetic causes of CH and fetal goiter development and their impact on neurological functions. Herein, we identified mutations in the TPO gene of two brothers from a non-consanguineous marriage who exhibited fetal hypothyroidism and are currently undergoing LT4 treatment.
The high TSH and TG levels and the patients’ fetal goiters led us to investigate TPO gene mutations by sequencing patient DNA. Herein, we identified three mutations in both patients. These mutations include p.Gln660Glu (4) and p.Cys296Alafs*21 (a new deletion) inherited from the father, and p.Arg665Trp (35) inherited from the mother.
Studies have shown that TPO protein, an essential enzyme for thyroid hormone synthesis, is localized to the apical membrane of the thyrocyte (37). TPO activity depends on the proper conformation, cellular localization and an intact catalytic site (38). The new deletion, which introduces a stop codon at amino-acid 316 and produces a 35 kDa protein, exhibits significantly reduced TPO activity (Figure 2). The deletion results in the loss of the catalytic site (exons 8–10), the transmembrane domain (exon 15), and the extracellular domain (Supplementary Figure 2). The immunofluorescence experiments demonstrate that this mutant is localized in the cytoplasm and exhibits reduced membrane localization (Figure 3). It was previously reported that the TPO mutant p.Gln660Glu displays reduced activity and membrane localization (39). It is unlikely that the p.Gln660Glu mutation contributes to the disease since it is not present in the protein as it is located downstream of the truncating mutation p.Cys296Alafs*. Thus, our results indicate compound heterozygosity involving p.Cys296Alafs*2 and p.Arg665Trp, mutations present in the paternal and maternal alleles, respectively.
It should be pointed out that the p.Arg665Trp mutation, previously identified in two patients with severe dyshormonogenesis, one with a history of neonatal goiter, was found to have reduced activity in the guaiacol oxidation assays and no membrane localization (35, 40). These results are consistent with the present study’s data, showing that Arg665Trp-TPO exhibits drastically reduced enzymatic activity and plasma membrane localization when expressed in HEK-293 cells (Figures 2 and 3). In this sense, our results provide further evidence demonstrating the severity of the p.Arg665Trp mutation.
Fetal goiter is usually diagnosed during the second or third trimester (11), which coincides with the detection at 32 and 26 WG for Patients 1 and 2 in our study. This late manifestation is because the maternal thyroid hormone supports fetal development during the first trimester, with maternal T3 regulating neuronal proliferation and the onset of neuronal cerebral cortex migration (41). During the second trimester, the fetal thyroid hormones progressively contribute more to neurogenesis, neural migration, myelination, axonal growth, dendritic arborization, and glial differentiation. Finally, in the third trimester, maternal and fetal thyroid hormones are required for central nervous system maturation (42). It has been reported that maternal hypothyroidism negatively impacts the neuropsychological development of children (43), an outcome probably due to abnormal cortical development during the first trimester (44).
On the other hand, fetal hypothyroidism appears to affect cerebral areas later in development, influencing language, associative memory, auditory processing, attention, and executive processing (45). Additional evidence associating intrauterine brain damage with fetal hypothyroidism comes from children with severe CH due to agenesia or with a thyroid gland and very low neonatal FT4. These children face a higher risk for subtle irreversible neurological deficits despite early treatment after birth (46–48) and exhibit cognitive problems with memory, attention, and visuospatial processing that can persist into early adolescence (49). Furthermore, a summary of several studies demonstrated that children diagnosed with CH by neonatal screening and who received optimal therapy presented IQ reductions of about 0.5 SD (45).
In our study, the neuropsychological assessment indicated that in general these patients with fetal goiter exhibited a normal neuropsychological development, with some high or below average scores. For example, a deficit in working memory was identified in Patient 2. This type of deficit is typically related to the management of day-to-day information difficulties. Moreover, children with this deficit can have difficulties planning, ranking, establishing priorities, distinguishing importance, and engaging in activities requiring the manipulation of temporarily stored information. They usually need more time and put forth more effort to carry out tasks. In the school context, it is common to have difficulties in mathematics operations that involve multiple steps, remembering homework instructions and simultaneously processing multiple information sources (50). A decrease in the information processing index was observed in both patients, characterized by a slow visual motor function and diminishing perceptual-motor and attention functions. This index provides a measure of the ability to decode symbols and process new information. It is sensitive to neurological factors, motor maturation, and attentional factors. Thus, children with these deficits require more time to learn the same amount of information than children of their age and are more likely to be tired since additional effort is needed to perform tasks (50).
While it is easy to speculate that the siblings’ reduced neurological skills are related to fetal hypothyroidism, it would be premature to make this conclusion. Moreover, prenatal fetal goiter treatment has been proposed to reduce goiter volume, prevent comorbidities, and provide adequate thyroid status at birth, but this therapy remains controversial. A recent study confirmed the feasibility of a conservative intrauterine LT4 treatment; however, the medications (LT4 and/or T3), recommended doses, and administration methods have not been defined. The authors state that prenatal treatment risks and benefits must be considered case by case (29). Notably, a 2020–2021 consensus updated the guidelines for the diagnosis and management of congenital hypothyroidism (CH) (51). They strongly recommended intraamniotic T4 injections in a euthyroid pregnant woman with large fetal goiter related to hydramnios and/or tracheal occlusion.
Considering the long-term medical follow-up, both siblings exhibited good clinical conditions and normal growth. They showed good adherence to LT4 treatment and adequate hormonal status along with the increase of the LT4 dose. Previous studies have also shown normal growth development of fetal goiter patients under adequate treatment (15, 21).
In conclusion, this study discovered a novel compound heterozygous mutation, including a previously unknown deletion in the TPO gene. This mutation led to severe impairment in TPO activity and membrane localization and appeared to be associated with severe intrauterine CH and fetal goiter. Under adequate treatment, the long-term follow-up showed a normal growth progression of both patients. This is also the first report investigating the neuropsychological factors in CH-induced fetal goiter patients. More studies are needed to state the neurodevelopmental outcomes of dyshormonogenetic fetal goiter.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, BankIt2432519 human MW660360.
Ethics Statement
This study was approved by the UNIFESP Ethical Committee (CAAE 59240816.2.0000.5505). Written informed consent was obtained from a parent of the patients for the publication of any potentially identifiable images or data included in this article.
Author Contributions
TR was the endocrinologist that diagnosed the fetal goiter in both patients and performed the follow-up. MS performed the mutagenesis and most of the immunofluorescence images. MF performed the DNA sequencing of the patient’s DNA. ZD performed the neuropsychological evaluation. VF performed the TPO activity evaluation. MR was responsible for the cell culture and transfection experiments. IR was the coordinator of the project. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from FAPESP 2014/24549-4, Sao Paulo State Research Foundation. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Marta Miyazawa and Dr. Rodrigo Tamura for kindly providing the HEK293 cells, Dr. Rodrigo Fortunato for supplying the pCDNA 3.1 plasmid containing the TPO-WT cDNA, Dr. Gisele Giannocco and Dr. Renato Mortara for the AlexaFluor 594 antibody and Wilson Segura, Elizabeth K. and Caroline Z. for acquiring the images in the multiuser laboratory confocal microscopy by INFAR (National Institute Pharmacology-SP) and in the confocal microscopy of the ICQAF–UNIFESP.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.671659/full#supplementary-material
References
1. Ford G, Lafranchi SH. Screening for Congenital Hypothyroidism: A Worldwide View of Strategies. Best Pract Res Clin Endocrinol Metab (2014) 28:175–87. doi: 10.1016/j.beem.2013.05.008
2. Camargo RY, Kanamura CT, Friguglietti CU, Nogueira CR, Iorcansky S, Tincani AJ, et al. Histopathological Characterization and Whole Exome Sequencing of Ectopic Thyroid: Fetal Architecture in a Functional Ectopic Gland From Adult Patient. Int J Endocrinol (2018) 2018:4682876. doi: 10.1155/2018/4682876
3. Maciel LMZ, Kimura ET, Nogueira CR, Mazeto GMFS, Magalhães PKR, Nascimento ML, et al. Congenital Hypothyroidism: Recommendations of the Thyroid Department of the Brazilian Society of Endocrinology and Metabolism. Arq Bras Endocrinol Metabol (2013) 57:184–92. doi: 10.1590/S0004-27302013000300004
4. Nascimento AC, Guedes DR, Santos CS, Knobel M, Rubio IGS, Medeiros-Neto G. Thyroperoxidase Gene Mutations in Congenital Goitrous Hypothyroidism With Total and Partial Iodide Organification Defect. Thyroid (2003) 13:1145–51. doi: 10.1089/10507250360731550
5. Rubio IGS, Medeiros-Neto G. Mutations of the Thyroglobulin Gene and its Relevance to Thyroid Disorders. Curr Opin Endocrinol Diabetes Obes (2009) 16:373–8. doi: 10.1097/MED.0b013e32832ff218
6. Hulur I, Hermanns P, Nestoris C, Heger S, Refetoff S, Pohlenz J, et al. A Single Copy of the Recently Identified Dual Oxidase Maturation Factor (DUOXA) 1 Gene Produces Only Mild Transient Hypothyroidism in a Patient With a Novel Biallelic DUOXA2 Mutation and Monoallelic DUOXA1 Deletion. J Clin Endocrinol Metab (2011) 96:841–5. doi: 10.1210/jc.2010-2321
7. Moreno JC, Klootwijk W, van Toor H, Pinto G, D’Alessandro M, Lèger A, et al. Mutations in the Iodotyrosine Deiodinase Gene and Hypothyroidism. N Engl J Med (2008) 358:1811–8. doi: 10.1056/NEJMoa0706819
8. Zamproni I, Grasberger H, Cortinovis F, Vigone MC, Chiumello G, Mora S, et al. Biallelic Inactivation of the Dual Oxidase Maturation Factor 2 (DUOXA2) Gene as a Novel Cause of Congenital Hypothyroidism. J Clin Endocrinol Metab (2008) 93:605–10. doi: 10.1210/jc.2007-2020
9. Nicola JP, Nazar M, Serrano-Nascimento C, Goulart-Silva F, Sobrero G, Testa G, et al. Iodide Transport Defect: Functional Characterization of a Novel Mutation in the Na+/I- Symporter 5′-Untranslated Region in a Patient With Congenital Hypothyroidism. J Clin Endocrinol Metab (2011) 96:1100–7. doi: 10.1210/jc.2011-0349
10. Kopp P. Mutations in the Pendred Syndrome (PDS/SLC26A) Gene: An Increasingly Complex Phenotypic Spectrum From Goiter to Thyroid Hypoplasia. J Clin Endocrinol Metab (2014) 99:67–9. doi: 10.1210/jc.2013-4319
11. Huel C, Guibourdenche J, Vuillard E, Ouahba J, Piketty M, Oury JF, et al. Use of Ultrasound to Distinguish Between Fetal Hyperthyroidism and Hypothyroidism on Discovery of a Goiter. Ultrasound Obstet Gynecol (2009) 33:412–20. doi: 10.1002/uog.6315
12. Pearce EN. Effects of Iodine Deficiency in Pregnancy. J Trace Elem Med Biol (2012) 26:131–3. doi: 10.1016/j.jtemb.2012.04.005
13. Omoto A, Kurimoto C, Minagawa M, Shozu M. A Case of Fetal Goiter That Resolved Spontaneously After Birth. J Clin Endocrinol Metab (2013) 98:3910–1. doi: 10.1210/jc.2013-1066
14. Iijima S. Current Knowledge About the In Utero and Peripartum Management of Fetal Goiter Associated With Maternal Graves’ Disease. Eur J Obstet Gynecol Reprod Biol X (2019) 3:100027. doi: 10.1016/j.eurox.2019.100027
15. Caron P, Moya CM, Malet D, Gutnisky VJ, Chabardes B, Rivolta CM, et al. Compound Heterozygous Mutations in the Thyroglobulin Gene (1143delc and 6725G→a [R2223H]) Resulting in Fetal Goitrous Hypothyroidism. J Clin Endocrinol Metab (2003) 88:3546–53. doi: 10.1210/jc.2002-021744
16. Reynolds BC, Simpson JH, Macara L, Watt AJB, Kubba H, Donaldson MDC, et al. Goitrous Congenital Hypothyroidism in a Twin Pregnancy Causing Respiratory Obstruction at Birth: Implications for Management. Acta Paediatr Int J Paediatr (2006) 95:1345–8. doi: 10.1080/08035250600711074
17. Stoppa-Vaucher S, Francoeur D, Grignon A, Alos N, Pohlenz J, Hermanns P, et al. Non-Immune Goiter and Hypothyroidism in a 19-Week Fetus: A Plea for Conservative Treatment. J Pediatr (2010) 156:1026–9. doi: 10.1016/j.jpeds.2010.01.018
18. Vasudevan P, Powell C, Nicholas AK, Scudamore I, Greening J, Park S-M, et al. Intrauterine Death Following Intraamniotic Triiodothyronine and Thyroxine Therapy for Fetal Goitrous Hypothyroidism Associated With Polyhydramnios and Caused by a Thyroglobulin Mutation. Endocrinol Diabetes Metab Case Rep (2017) 2017:17–0040. doi: 10.1530/edm-17-0040
19. Siffo S, Adrover E, Citterio CE, Miras MB, Balbi VA, Chiesa A, et al. Molecular Analysis of Thyroglobulin Mutations Found in Patients With Goiter and Hypothyroidism. Mol Cell Endocrinol (2018) 473:1–16. doi: 10.1016/j.mce.2017.12.009
20. Stern E, Kassif E, Schoenmakers N, Gruber N. A Novel Mutation in the Thyroglobulin Gene Leading to Neonatal Goiter and Congenital Hypothyroidism in an Eritrean Infant. J Clin Res Pediatr Endocrinol (2021). doi: 10.4274/jcrpe.galenos.2021.2020.0278
21. Rubio IGS, Galrao AL, Pardo V, Knobel M, Possato RF, Camargo RRY, et al. A Molecular Analysis and Long-Term Follow-Up of Two Siblings With Severe Congenital Hypothyroidism Carrying the IVS30+1G<T Intronic Thyroglobulin Mutation. Arq Bras Endocrinol Metabol (2008) 52:1337–44. doi: 10.1590/S0004-27302008000800022
22. Börgel K, Pohlenz J, Holzgreve W, Bramswig JH. Intrauterine Therapy of Goitrous Hypothyroidism in a Boy With a New Compound Heterozygous Mutation (Y453D and C800R) in the Thyroid Peroxidase Gene. A Long-Term Follow-Up. Am J Obstet Gynecol (2005) 193:857–8. doi: 10.1016/j.ajog.2005.01.060
23. Simm D, Pfarr N, Pohlenz J, Prawitt D, Dörr HG. Two Novel Mutations in the Human Thyroid Peroxidase (TPO) Gene: Genetics and Clinical Findings in Four Children. Acta Paediatr Int J Paediatr (2009) 98:1057–61. doi: 10.1111/j.1651-2227.2009.01236.x
24. Yapakç E, Kinik ST, Pohlenz J. Intrauterine Treatment of an Infant With Fetal Goitre. J Pediatr Endocrinol Metab (2010) 23:651–2. doi: 10.1515/JPEM.2010.23.7.651
25. Figueiredo CM, Falcão I, Vilaverde J, Freitas J, Oliveira MJ, Godinho C, et al. Prenatal Diagnosis and Management of a Fetal Goiter Hypothyroidism Due to Dyshormonogenesis. Case Rep Endocrinol (2018) 2018:1–4. doi: 10.1155/2018/9564737
26. Zdraveska N, Kocova M, Nicholas AK, Anastasovska V, Schoenmakers N. Genetics of Gland-in-situ or Hypoplastic Congenital Hypothyroidism in Macedonia. Front Endocrinol (Lausanne) (2020) 11:1–10. doi: 10.3389/fendo.2020.00413
27. Stoupa A, Al Hage Chehade G, Kariyawasam D, Tohier C, Bole-Feysot C, Nitschke P, et al. First Case of Fetal Goitrous Hypothyroidism Due to SLC5A5/NIS Mutations. Eur J Endocrinol (2020) 183:32805706. doi: 10.1530/EJE-20-0255
28. Tanase-Nakao K, Miyata I, Terauchi A, Saito M, Wada S, Hasegawa T, et al. Fetal Goitrous Hypothyroidism and Polyhydramnios in a Patient With Compound Heterozygous DUOXA2 Mutations. Horm Res Paediatr (2018) 90:132–7. doi: 10.1159/000491104
29. Nemescu D, Tanasa I, Stoian D, Navolan D, Vinturache A. Conservative In Utero Treatment of Fetal Dyshormonogenetic Goiter With Levothyroxine, a Systematic Literature Review. Exp Ther Med (2020) 20(3):2434–8. doi: 10.3892/etm.2020.8794
30. Prezioso G, Giannini C, Chiarelli F. Effect of Thyroid Hormones on Neurons and Neurodevelopment. Horm Res Paediatr (2018) 90:73–81. doi: 10.1159/000492129
31. Szinnai G, Lacroix L, Carré A, Guimiot F, Talbot M, Martinovic J, et al. Sodium/Iodide Symporter (NIS) Gene Expression is the Limiting Step for the Onset of Thyroid Function in the Human Fetus. J Clin Endocrinol Metab (2007) 92:70–6. doi: 10.1210/jc.2006-1450
32. Vulsma T, Gons MH, de Vijlder JJM. Maternal-Fetal Transfer of Thyroxine in Congenital Hypothyroidism Due to a Total Organification Defect or Thyroid Agenesis. N Engl J Med (1989) 321:13–6. doi: 10.1056/NEJM198907063210103
33. Tanner J. Psysical Growth and Development. In: Forjar J, Amell C, editors. Textbook of Paediatrics. Cambridge: Harvard University Press. (1978) p. 249–303.
34. WHO | World Health. AnthroPlus Manual for Personal Computers Manual: Software for Assessing Growth of the Word’s Children and Adolescents. Geneva: WHO (2009).
35. Umeki K, Kotani T, Kawano JI, Suganuma T, Yamamoto I, Aratake Y, et al. Two Novel Missense Mutations in the Thyroid Peroxidase Gene, R665W and G771R, Result in a Localization Defect and Cause Congenital Hypothyroidism. Eur J Endocrinol (2002) 146:491–8. doi: 10.1530/eje.0.1460491
36. Blumenfeld YJ, Davis A, Milan K, Chueh J, Hudgins L, Barth RA, et al. Conservatively Managed Fetal Goiter: An Alternative to In Utero Therapy. Fetal Diagn Ther (2013) 34:184–7. doi: 10.1159/000353387
37. Rousset B, Dupuy C, Miot F, Dumont J. Thyroid Hormone Synthesis And Secretion. South Dartmouth (MA): Endotext (2000–2015).
38. Libert F, Ruel J, Ludgate M, Swillens S, Alexander N, Vassart G, et al. Thyroperoxidase, an Auto-Antigen With a Mosaic Structure Made of Nuclear and Mitochondrial Gene Modules. EMBO J (1987) 6:4193–6. doi: 10.1002/j.1460-2075.1987.tb02766.x
39. Deladoëy J, Pfarr N, Vuissoz JM, Parma J, Vassart G, Biesterfeld S, et al. Pseudodominant Inheritance of Goitrous Congenital Hypothyroidism Caused by TPO Mutations: Molecular and in Silico Studies. J Clin Endocrinol Metab (2008) 93:627–33. doi: 10.1210/jc.2007-2276
40. Zhao D, Li Y, Shan Z, Teng W, Li J, Teng X. Functional Analysis of Thyroid Peroxidase Gene Mutations Resulting in Congenital Hypothyroidism. Clin Endocrinol (Oxf) (2020) 93:499–507. doi: 10.1111/cen.14253
41. Kester MHA, Martinez De Mena R, Obregon MJ, Marinkovic D, Howatson A, Visser TJ, et al. Iodothyronine Levels in the Human Developing Brain: Major Regulatory Roles of Iodothyronine Deiodinases in Different Areas. J Clin Endocrinol Metab (2004) 89:3117–28. doi: 10.1210/jc.2003-031832
42. Williams GR. Neurodevelopmental and Neurophysiological Actions of Thyroid Hormone. J Neuroendocrinol (2008) 20:784–94. doi: 10.1111/j.1365-2826.2008.01733.x
43. Haddow JE, Palomaki GE, Allan WC, Williams JR, Knight GJ, Gagnon J, et al. Maternal Thyroid Deficiency During Pregnancy and Subsequent Neuropsychological Development of the Child. N Engl J Med (1999) 341:2015–7. doi: 10.1056/NEJM199908193410801
44. Rami A, Patel AJ, Rabié A. Thyroid Hormone and Development of the Rat Hippocampus: Morphological Alterations in Granule and Pyramidal Cells. Neuroscience (1986) 19:1217–26. doi: 10.1016/0306-4522(86)90135-1
45. Rovet JF. The Role of Thyroid Hormones for Brain Development and Cognitive Function. Endocr Dev (2014) 26:26–43. doi: 10.1159/000363153
46. Rovet JF, Ehrlich RM, Sorbara DL. Neurodevelopment in Infants and Preschool Children With Congenital Hypothyroidism: Etiological and Treatment Factors Affecting Outcome. J Pediatr Psychol (1992) 17:187–213. doi: 10.1093/jpepsy/17.2.187
47. Kooistra L, Laane C, Vulsma T, Schellekens JMH, van der Meere JJ, Kalverboer AF. Motor and Cognitive Development in Children With Congenital Hypothyroidism: A Long-Term Evaluation of the Effects of Neonatal Treatment. J Pediatr (1994) 124:903–9. doi: 10.1016/S0022-3476(05)83178-6
48. Glorieux J, Dussault J, Van Vliet G. Intellectual Development at Age 12 Years of Children With Congenital Hypothyroidism Diagnosed by Neonatal Screening. J Pediatr (1992) 121:581–4. doi: 10.1016/S0022-3476(05)81150-3
49. Rovet JF, Ehrlich R. Psychoeducational Outcome in Children With Early-Treated Congenital Hypothyroidism. Pediatrics (2000) 105:515–22. doi: 10.1542/peds.105.3.515
50. Weiss L, Saklofske D, Prifitera A, Holdnack J. Wisc-Iv Advanced Clinical Interpretation. Cambridge: Academic Press (2006).
51. Van Trotsenburg P, Stoupa A, Léger J, Rohrer T, Peters C, Fugazzola L, et al. Congenital Hypothyroidism: A 2020-2021 Consensus Guidelines Update - An Endo-European Reference Network Initiative Endorsed by the European Society for Pediatric Endocrinology and the European Society for Endocrinology. Thyroid (2021) 31:387–419. doi: 10.1089/thy.2020.0333
52. Perry RJ, Hollman AS, Wood AM, Donaldson MDC. Ultrasound of the Thyroid Gland in the Newborn: Normative Data. Arch Dis Child Fetal Neonatal Ed (2002) 87:209–11. doi: 10.1136/fn.87.3.F209
53. Fortunato RS, De Souza ECL, Ameziane-el Hassani R, Boufraqech M, Weyemi U, Talbot M, et al. Functional Consequences of Dual Oxidase-Thyroperoxidase Interaction at the Plasma Membrane. J Clin Endocrinol Metab (2010) 95:5403–11. doi: 10.1210/jc.2010-1085
Keywords: fetal goiter, congenital hypothyroidism, thyroid peroxidase, mutations, neuropsychological evaluation, growth
Citation: Rodrigues TMB, Silva MMdC, Freitas MM, Duarte ZMC, Frutuoso VS, Rodrigues MT and Rubio IGS (2021) Case Report: Functional Analysis and Neuropsychological Evaluation of Dyshormonogenetic Fetal Goiter in Siblings Caused by Novel Compound Hyterozygous TPO Gene Mutations. Front. Endocrinol. 12:671659. doi: 10.3389/fendo.2021.671659
Received: 15 March 2021; Accepted: 21 May 2021;
Published: 18 June 2021.
Edited by:
Ronald Cohen, University of Chicago, United StatesReviewed by:
Alexandra Dumitrescu, University of Chicago Medicine, United StatesTania M. Ortiga-Carvalho, Federal University of Rio de Janeiro, Brazil
Copyright © 2021 Rodrigues, Silva, Freitas, Duarte, Frutuoso, Rodrigues and Rubio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ileana Gabriela Sanchez Rubio, aWxlYW5hLnJ1YmlvQHVuaWZlc3AuYnI=