 Claudio Giacomozzi
Claudio Giacomozzi- U.O.C. of Pediatrics, Department of Maternal and Child Health, Carlo Poma Hospital, ASST-Mantova, Mantova, Italy
Children born small for gestational age (SGA), and failing to catch-up growth in their early years, are a heterogeneous group, comprising both known and undefined congenital disorders. Care for these children must encompass specific approaches to ensure optimal growth. The use of recombinant human growth hormone (rhGH) is an established therapy, which improves adult height in a proportion of these children, but not with uniform magnitude and not in all of them. This situation is complicated as the underlying cause of growth failure is often diagnosed during or even after rhGH treatment discontinuation with unknown consequences on adult height and long-term safety. This review focuses on the current evidence supporting potential benefits from early genetic screening in short SGA children. The pivotal role that a Next Generation Sequencing panel might play in helping diagnosis and discriminating good responders to rhGH from poor responders is discussed. Information stemming from genetic screening might allow the tailoring of therapy, as well as improving specific follow-up and management of family expectations, especially for those children with increased long-term risks. Finally, the role of national registries in collecting data from the genetic screening and clinical follow-up is considered.
Introduction
Growth failure is a frequent reason for referral to a pediatric endocrinologist, and short stature in children born small for gestational age (SGA) is among the indications for the use of recombinant human growth hormone (rhGH).
Children with birth weight and/or length two standard deviations (SDs) below the mean, compared with reference standards for gestational age and gender, are defined as SGA. SGA children may or may not have a history of intra-uterine growth retardation (IUGR). SGA patients, as well as those with IUGR, have been extensively investigated and several conditions have been associated with fetal growth restrictions (1), which are classified into three main categories: maternal, placental and fetal. Maternal causes, including acquired disease (infections, chronic inflammatory disease, anemia, depression) or unhealthy lifestyle (smoking, alcohol and/or drug abuse), represent a minor percentage of causes. Maternal and placental causes are often diagnosed by ultrasound and an in-depth clinical follow-up, enabling the medical management to be tailored and accurate. In cases where gestational follow-up does not reveal any potential association, growth restriction is more likely to be related to intrinsic fetal disorders. Ultimately, if gestational follow-up is missing or inappropriate, it may be difficult to establish the cause of the growth failure.
About 90% of SGA newborns show catch-up growth in the first 2 years of life, their height normalizing by 4 years of age (2). However, about 10% of SGA children remain permanently short, and achieve an adult height less than -2 SDs (3, 4).
Recombinant Human Growth Hormone
In 2001, rhGH was approved for use in short SGA children, older than 2 years in the USA and older than 4 years in Europe, based on trials demonstrating increased height velocity and improved height gain in children born SGA compared with untreated children. The effects of rhGH administration in the short-term were evaluated by a meta-analysis including 4 independent, randomized, controlled, multicenter studies conducted over 2 years in short pre-pubertal children born SGA (5). Treated SGA children showed a dose-dependent increase in growth, with a significantly accelerating growth rate compared with the untreated group, and an improved final height (5). The first report from a long-term trial showed that a high proportion of short SGA children achieved an adult height (AH) within the target height range, without impact on the onset, progression and duration of puberty, or pubertal height gain (6).
Short SGA adolescents starting rhGH therapy at an early pubertal stage have modest and variable height gain. An AH > –2.5 SDs can be expected in only one third of patients, especially in those with a smaller height deficit at onset of rhGH treatment (7). Some authors suggest that doubling the rhGH dose, particularly during puberty (GH 2 mg/m2 per day) may improve height gain and adult height, and that the combined GH/gonadotropin-releasing hormone a treatment at the onset of puberty, may be a successful strategy to improve short stature in SGA adolescents with a height <140 cm at the onset of puberty (8, 9). However, the small number of treated subjects and the lack of further controlled randomized trials leave this issue inconclusive.
An epi-analysis of the first AH data from SGA children enrolled in four randomized trials comparing the growth-promoting efficacy of two continuous rhGH regimens (0.033 or 0.067 mg/kg per day for ~10 years, starting at ~5 years of age) (10), and an epi-analysis of the AH results published earlier than 2005 (11), confirmed that rhGH therapy is effective and safe in reducing the AH deficit in SGA patients. Contrary to previous evidence (8, 9), long-term higher rhGH doses (0.067 mg/kg per day) did not correlate with a better height gain or AH, than lower doses (0.033 mg/kg per day) (11, 12), even in adolescents (13).
In 2009, a meta-analysis of randomized, controlled trials (four RCTs including 391 children) on the impact of rhGH therapy on AH in short SGA children concluded that AH of the rhGH-treated group significantly exceeded controls by 0.9 SDs (mean height gain 1.5 SDs in treated vs 0.25 SDs in untreated SGA subjects) (14). Once again, no significant difference in AH was observed between different rhGH dose regimens. However, the response to therapy was highly variable, and additional studies were suggested to identify the good responders.
Markers for Predicting Good Responders
Despite the efforts to identify variables predictive of good response to long-term rhGH therapy and the development of prediction models (15–18), to date, no pre-treatment variables have been found to be reliable for predicting rhGH response. The ability to predict a patient’s response to rhGH would not only quantify the magnitude of height gain to be expected at the end of treatment, but, most importantly, would avoid treating non-responders, especially those who have intrinsic risk factors. The identification of good and non-responders would not only optimize the use of financial and human resources, but also prevent patient frustration and disappointment potentially after years of treatment that does not provide the expected benefit.
Clinical trials identified younger chronological and bone age at the start of treatment with rhGH as major determinants of height gain in SGA children. No other considered parameters correlated significantly with the height gain SDs from the start of rhGH treatment until AH (12). Interestingly, the AH SDs correlated positively with height SDs at the start of rhGH treatment, target height SDs, and pre-treatment height velocity SDs (12). Growth hormone secretion did not correlate with height gain in SGA children considered GH deficient by provocative tests (12). Prediction models, conversely, suggested rhGH dose was among the major predictive markers, along with weight SDs, height velocity SDs, chronological age and mid-parent height SDs (17). Conflicting data and poor applicability of predictive models to daily clinical practice have reduced the impact of these findings (18). Currently, there is no specific clinical feature that identifies a patient as one who will not benefit from rhGH therapy, unless the patient’s phenotype (and genotype) is diagnostic for a disease already known to be unresponsive to rhGH therapy.
The polymorphic deletion in exon 3 of the GH receptor (d3-GHR) has been investigated as a potential predictive marker of growth response in SGA patients, as previously shown in children with other rhGH-treated growth disorders (19). However, though the height velocity increase has been positively correlated with the presence of one or two copies of the d3-GHR allele, this association was observed only in the first and second year of treatment. No correlation was found with overall height gain and AH. In a large randomized controlled study conducted in Spain in SGA children, neither growth velocity nor height gain differed among the d3/fl-GHR genotypes over two years of rhGH therapy (20). Furthermore, the spontaneous growth rate in the untreated group did not correlate with d3/fl-GHR genotype.
Although polymorphisms in IGF1 and IGF1R genes may be potential candidate markers, only one polymorphism in the promoter region of the IGF1 gene has been associated with growth in SGA children, and its exact contribution in clinical practice remains to be elucidated (21).
Multi-allele single nucleotide polymorphisms associated with insulin sensitivity or insulin secretion have been positively associated with height velocity in the first year of rhGH therapy, but not with growth and IGF-1 responses in the long-term (22).
Overall, these data raise the question of whether the search for predictors of rhGH response among variables already linked with GH deficiency should continue, or if resources would be better spent personalizing the approach and better defining the pathogenesis underlying each individual SGA child.
Safety
The safety of rhGH therapy is a crucial issue in SGA children, as they represent an extremely heterogeneous and still poorly characterized group of patients.
A twelve-year study on rhGH therapy in SGA children has shown that rhGH therapy is well tolerated (13). Although both higher fasting and glucose-stimulated insulin levels were observed during treatment, 6 months after rhGH discontinuation, blood insulin concentrations normalized. Glycolated hemoglobin levels of SGA patients remained within normal ranges during rhGH treatment, irrespective of the dosage (0.033 versus 0.067 mg/kg/week) (23). Furthermore, rhGH therapy demonstrated a beneficial effect on serum lipid profiles, body composition, bone mineral density, and head growth.
Data on morbidity and mortality were collected during up to 25 years of follow-up in the SAGhE project (24, 25), the largest independent post-marketing surveillance study of rhGH treatment published so far. In the young adult SGA population treated with rhGH therapy during childhood, overall cancer mortality and morbidity were not increased. All-cause mortality was significantly increased when analyzed for all countries involved in the study (eight from across Europe) (SMR 1·5 [1·1–1·9]). However, when analyzed separately, the risk was significantly increased only in France (SMR 1·7 [1·2–2·4]), but not significantly in the other countries combined (SMR 1·2 [0·8–1·9]). The increased mortality risk was associated with diseases of the circulatory system. No significant association between mortality and rhGH dose, either mean daily or cumulative, was found.
Although SAGhE data are reassuring overall about mortality risk associated with rhGH therapy in SGA children, both cancer morbidity and mortality and all-cause mortality were significantly higher in the syndromic group treated with rhGH compared with the general population. These data suggest that some genetic syndromes have intrinsic risk of morbidity and mortality, which might be exacerbated by rhGH therapy. As a proportion of SGA patients harbor genetic abnormalities, the potential risk requires long-term surveillance in those treated with rhGH.
SGA Short Stature as “Neonatal Idiopathic Short Stature”
Before rhGH approval for SGA, low weight or short length at birth were well known findings in many genetic disorders, including Cornelia De Lange (1933) (26), Silver-Russell syndrome (1953-1954) (27, 28), primary IGF1 signaling anomalies (1996) (29), and more recently Gs-alpha protein-related disorders (2018) (30). Research has established the importance of distinguishing children with these conditions (who do not benefit from rhGH) from those born SGA (who might). Therefore, it seems entirely logical to expect to find more genetic defects misdiagnosed in patients who are currently labeled as SGA. Moreover, the broad individual variability of response to rhGH treatment in the SGA population is mainly due to the heterogeneous pathogenesis underlying fetal and post-natal growth restriction in these children.
Since early 2000, genetic research has resulted in the development of many sophisticated diagnostic techniques to analyze both copy number variations and punctiform mutations in an increasing number of genes simultaneously and faster. Current evidence suggests that a considerable number of SGA patients without catch-up growth carry monogenic, polygenic, or epigenetic variants, which explain their growth failure. Some short SGA patients might be considered to have a sort of neonatal idiopathic short stature (ISS), especially those without an increased risk for metabolic and cardiovascular disease in later life. SGA and ISS share the same challenges for elucidating the genetic/epigenetic cause(s) of these conditions and for finding successful treatment(s). Their similarity is supported by case studies, showing that skeletal dysplasia accounts for more than 20% of short stature in both SGA and ISS children (31). Moreover, IGF1, IGF1R, and SHOX mutations have been demonstrated in both short SGA and ISS children, although in a smaller percentage of cases (32). Both represent a group of patients categorized just on the basis of auxological parameters. In fact, the SGA condition is comprised of a large group of diseases sharing similar growth patterns but with many other different features and implications, both in children and in adults. Genome sequencing techniques have allowed the determination of defects in genes related to growth cartilage in patients with isolated short stature, who could be defined SGA only by their short birth length (33). The genes involved in endochondral ossification or cartilage extracellular matrix synthesis are responsible for very mild forms of bone dysplasia that may be clinically classified as ISS or SGA, according only to the timing of the short stature definition.
The identification of good responders is, therefore, a priority to successfully treat SGA patients, as well as ISS patients.
Advances of Genetics in SGA
The diagnostic capabilities of medical genetics are constantly evolving, and great strides forward have been made in understanding the genetic profile underlying many SGA children. Extensive research has contributed to this progress, and, in highlighting a few key papers, we acknowledge that the few cited articles are far from an exhaustive list. A representative review of what has been achieved so far, is reported by Peeters et al. (34), who demonstrated that a more advanced approach with accurate genotyping is successful in finding new potential pathways involved in the IUGR/SGA condition. This extensive genetic work-up led to identification of pathogenic variants in ELAC2, CEP57, and HNRHPH1 genes by exome sequencing, the potential involvement of chromosomal regions such as 5q.35 and 22q.11 by SNP array analysis, and different patterns of methylation in specific regions regulating the GNAS gene (already thought to potentially affect growth) by genome-wide methylation analysis.
It has been reported that about 13% of short SGA patients treated with rhGH and presenting with advanced bone age at some point of their therapy, carried an Aggrecan (ACAN) gene mutation (35). SGA patients harboring ACAN mutations are characterized by additional features such as midface hypoplasia, joint problems, and broad great toes, and seem to benefit from both rhGH and gonadotropin-releasing hormone analogue to improve their AH, although the small cohort of studied patients does not allow definitive conclusions.
An early genetic approach may help to elucidate the molecular defect underlying the SGA condition and tailor the appropriate treatment, if available. Parents might be informed in advance and more accurately on ‘what it is reasonable to expect’, and this could positively impact on patients’ and families’ compliance and, ultimately, on clinical results.
Potential Role of Genetic Screening in Short SGA Children
There is increasing evidence that genetic analysis may be used not only as a diagnostic tool but also for safety screening in SGA children.
According to the 2001 international consensus on SGA children (36), apart from Bloom syndrome and a few other chromosomal anomalies (trisomy 18 and 21, monosomy X), genetic investigations have not generally been considered in short SGA children before starting rhGH. However, many other syndromic disorders have been associated with a SGA phenotype, such as Rothmund–Thomson syndrome, Louis-Bar syndrome and Nijmegen breakage syndrome, all of which are associated with an increased risk of malignancies. Consequently, the cause of poor growth remains a ‘mystery’ in the majority of SGA children at the onset of rhGH therapy, even in a minor but extremely important percentage with potential cancer-related disorders.
Since 2001, Next Generation Sequencing (NGS) and Whole Exome Sequencing (WES) have revealed the association of an SGA phenotype with syndromic conditions not previously elucidated or whose heterogeneous phenotype-genotype correlation did not allow a proper diagnosis in a proportion of cases (37–40). It should also be considered that the percentage of diagnosis by exome sequencing for children born SGA with persistent short stature and absence of syndromic features is lower than that for syndromic children (33–38). This may suggest a wider involvement of epigenetic mechanisms in isolated short stature pathogenesis than is actually believed (37).
Potentially, genetic screening strategies (Figure 1) could increase the safety of rhGH therapy in SGA children undiagnosed at treatment onset. NGS panels, especially set up with known SGA-related genes (example shown in Table 1), may work as screening tests, with the advantage over other genetic techniques of a short response time and low cost. In the cases of syndromes with increased risk for malignancy, whose magnitude is largely unknown but might not be negligible in the SGA population, therapy with rhGH should be avoided, or, after extensive discussion with parents and in cases of severe short stature, undertaken only with specific and intensive clinical and laboratory monitoring. Although the real impact of rhGH on cancer risk in many syndromes remains unknown, the most cautious management should be pursued. In case of a ‘negative’ on genetic screening, diagnostic work-up should continue and further investigations should be arranged during treatment to get a definitive diagnosis, and to correlate the response to treatment. Indeed, the magnitude of rhGH efficacy has not been clarified in a significant portion of 'SGA-related genetic diseases’ covered in the theoretical NGS panel in Table 1. The genes included in the NGS safety screening panel should be periodically reviewed according to the latest data, to maintain safety at the highest possible level. Naturally, over time, the SGA screening NGS panel might become a diagnostic tool to investigate this population. The use of inclusion criteria for SGA genetic screening should be carefully discussed. On the one hand, selective criteria might improve the percentage of diagnoses (29), but on the other hand, might not be a sufficient safety tool, especially in patients with non-specific phenotypes (38). Among the limits of genetic screening by NGS panels would be gene methylation disorders such as Silver Russel syndrome and Temple syndrome. However, the contribution of these syndromic conditions among SGA with isolated short stature has not been demonstrated significantly so far. For these disorders, specific diagnostic pathways should be arranged before genetic screening according to phenotype, or later for those cases where features are not prognostic and genetic screening has not been helpful. Finally, safety genetic screening may prevent the premature discontinuation of rhGH due to patients’ frustration at unsatisfactory or slow results, thus preventing family disappointment, and waste of public health resources.
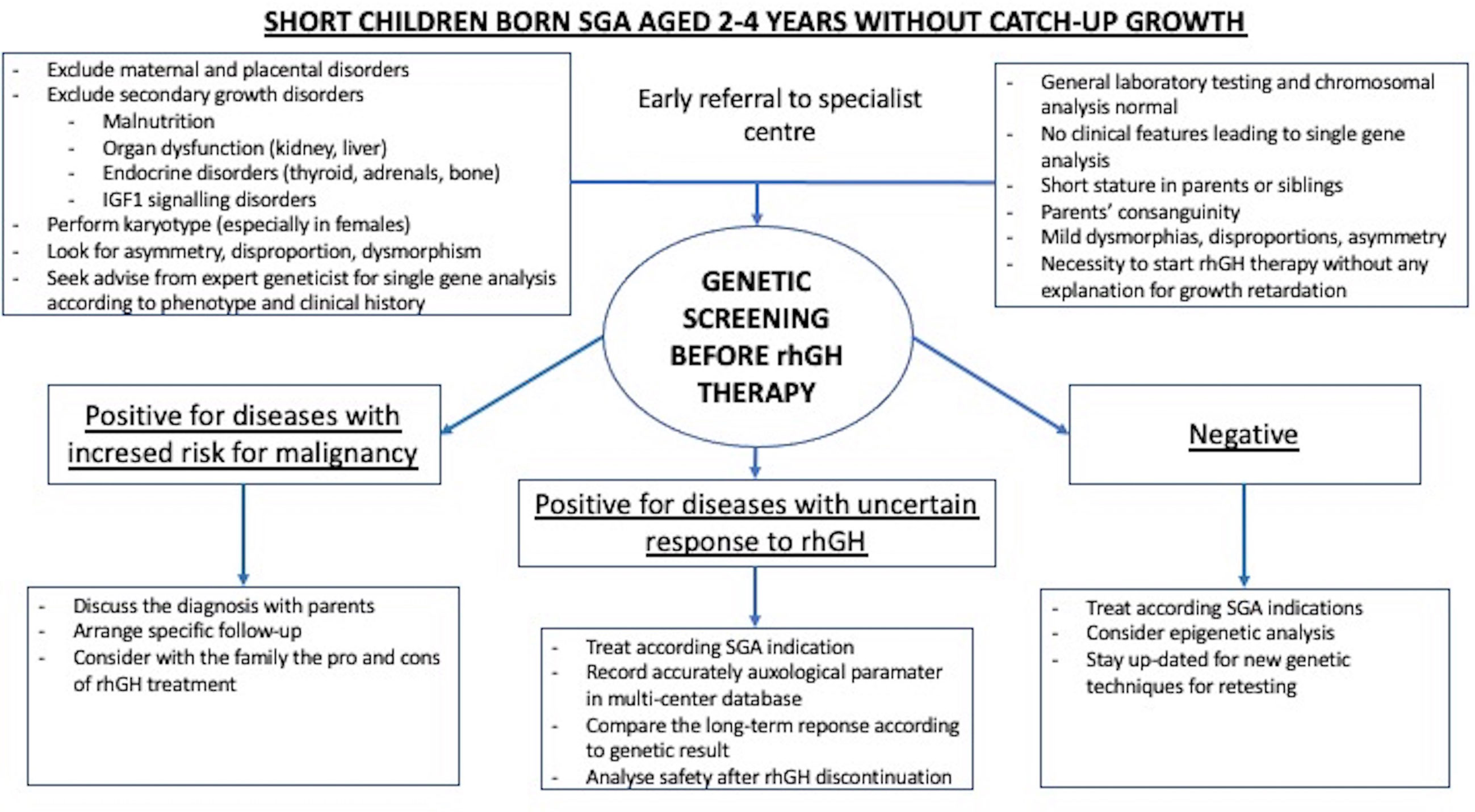
Figure 1 Example of genetic screening strategy included in the work-up and treatment of children born SGA with persistent short stature.
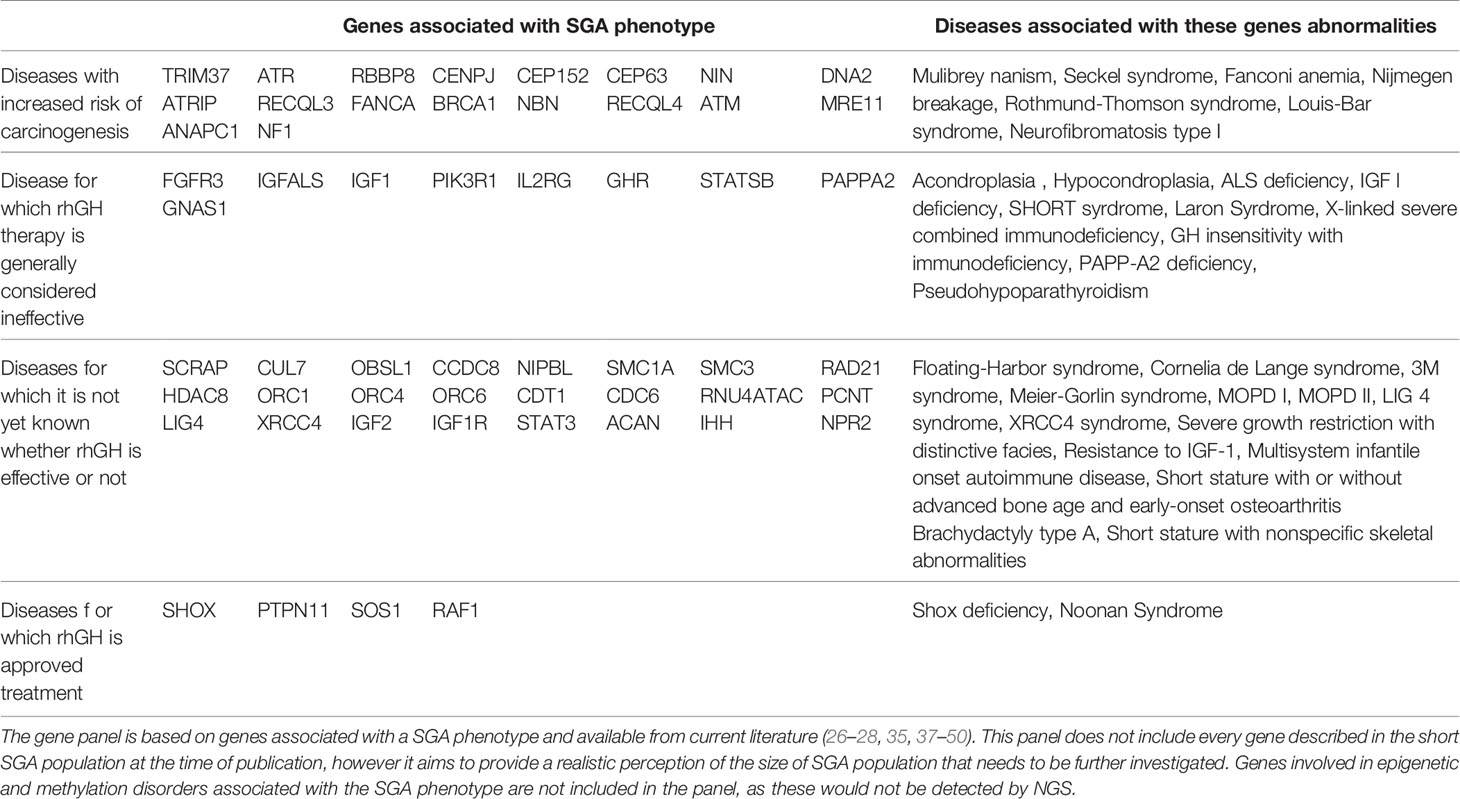
Table 1 Some genes responsible for pre-natal short stature and included in a genetic screening panel.
Potential Role of National Registries
National registries play a fundamental role in collecting data on both efficacy and safety of rhGH during active treatment and long after (51, 52). Unfortunately, the main junction where patients are lost is still the post-discontinuation follow-up, greatly limiting their use and reliability in long-term surveillance programs. Agencies regulating the registries should set up an annual recall system, based on a few but essential parameters to maintain surveillance. Modern telemedicine tools may allow long-term data collection with reduced impact on costs and human resources. Registries should also include the option to enter or adjust the underlying etiology even many years after rhGH discontinuation, to prevent bias related to misdiagnoses. A non-minor issue should be the independence of registries, as most data currently available has been collected by pharmaceutical companies. The sharing of data between different registries would be ideal to reach cohorts of sufficient size to draw conclusive results.
Conclusion
In conclusion, rhGH is currently the best available therapy to improve adult height in SGA children who do not spontaneously catch-up their growth.
Management of rhGH therapy in SGA children should go beyond prescribing the appropriate dose, IGF1 monitoring and the timing of treatment initiation - it should also include:
- Pre-arranged safety screenings, possibly standardized and internationally recognized;
- Promotion of a continuous diagnostic effort, independently dictated by patient age and duration of rhGH therapy;
- Detailed collection of efficacy and safety data by a network of interconnected registries designed to identify good and bad responders to rhGH therapy.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This article has been financially supported by Scientific Seminars International Foundation (Italy), to cover publishing cost through an educational grant received from Merck Healthcare KGaA, Darmstadt, Germany. The sponsor has had no role in data selection or in manuscript preparation.
Conflict of Interest
CG received honoraria from Novo Nordisk for consultancies as an expert board member; honoraria from Merck Serono as a medical advisor; honoraria from Ferring for article authorship. CG has no non-financial relationships (personal, political, or professional) that may potentially influence the writing of this manuscript.
Acknowledgments
I would like to thank Dr Rebecca Mant for editorial assistance and prof. Stefano Cianfarani for valuable advice.
References
1. Finken MJJ, van der Steen M, Smeets CCJ, Walenkamp MJE, de Bruin C, Hokken-Koelega ACS, et al. Children Born Small for Gestational Age: Differential Diagnosis, Molecular Genetic Evaluation, and Implications. Endocr Rev (2018) 39(6):851–94. doi: 10.1210/er.2018-00083
2. Karlberg JP, Albertsson-Wikland K, Kwan EY, Lam BC, Low LC. The Timing of Early Postnatal Catch-Up Growth in Normal, Full-Term Infants Born Short for Gestational Age. Horm Res (1997) 48(Suppl 1):17–24. doi: 10.1159/000191279.
3. Leger J, Levy-Marchal C, Bloch J, Pinet A, Chevenne D, Porquet D, et al. Reduced Final Height and Indications for Insulin Resistance in 20 Year Olds Born Small for Gestational Age: Regional Cohort Study. BMJ (1997) 315(7104):341–7. doi: 10.1136/bmj.315.7104.341
4. Jaquet D, Collin D, Lévy-Marchal C, Czernichow P. Adult Height Distribution in Subjects Born Small for Gestational Age. Horm Res (2004) 62(2):92–6. doi: 10.1159/000079709.
5. de Zegher F, Albertsson-Wikland K, Wilton P, Chatelain P, Jonsson B, Löfström A, et al. Growth Hormone Treatment of Short Children Born Small for Gestational Age: Metanalysis of Four Independent, Randomized, Controlled, Multicentre Studies. Acta Paediatr Suppl (1996) 417:27–31. doi: 10.1111/j.1651-2227.1996.tb14289.x
6. Hokken-Koelega ACS, van Pareren Y, Sas T, Arends N. Final Height Data, Body Composition and Glucose Metabolism in Growth Hormone-Treated Short Children Born Small for Gestational Age. Horm Res (2003) 60(Suppl 3):113–4. doi: 10.1159/000074511
7. Thomas M, Beckers D, Brachet C, Dotremont H, Lebrethon MC, Lysy P, et al. Adult Height After Growth Hormone Treatment At Pubertal Onset in Short Adolescents Born Small for Gestational Age: Results From a Belgian Registry-Baseds. Int J Endocrinol (2018) 2018:6421243. doi: 10.1159/000448553
8. Lem AJ, van der Kaay DC, de Ridder MA, Bakker-van Waarde WM, van der Hulst FJ, Mulder JC, et al. Adult Height in Short Children Born SGA Treated With Growth Hormone and Gonadotropin Releasing Hormone Analog: Results of a Randomized, Dose-Response GH Trial. J Clin Endocrinol Metab (2012) 97(11):4096–105. doi: 10.1210/jc.2012-1987
9. van der Steen M, Lem AJ, van der Kaay DC, Hokken-Koèelega AC. Puberty and Pubertal Growth in GH-Treated Sga Children: Effects of 2 Years of GnRHa Versus No GnRHa. J Clin Endocrinol Metab (2016) 101(5):2005–12. doi: 10.1210/jc.2016-1317
10. de Zegher F, Albertsson-Wikland K, Wollmann HA, Chatelain P, Chaussain JL, Löfström A, et al. Growth Hormone Treatment of Short Children Born Small for Gestational Age: Growth Responses With Continuous and Discontinuous Regimens Over 6 Years. J Clin Endocrinol Metab (2000) 85(8):2816–21. doi: 10.1210/jcem.85.8.6719
11. de Zegher F, Hokken-Koelega A. Growth Hormone Therapy for Children Born Small for Gestational Age: Height Gain Is Less Dose Dependent Over the Long Term Than Over the Short Term. Pediatrics (2005) 115(4):e458–62. doi: 10.1542/peds.2004-1934
12. Van Pareren Y, Mulder P, Houdijk M, Jansen M, Reeser M, Hokken-Koelega A. Adult Height After Long-Term, Continuous Growth Hormone (Gh) Treatment in Short Children Born Small for Gestational Age: Results of a Randomized, Double-Blind, Dose-Response Gh Trial. J Clin Endocrinol Metab (2003) 88(8):3584–90. doi: 10.1210/jc.2002-021172
13. Hokken-Koelega A, van Pareren Y, Arends N, Boonstra V. Efficacy and Safety of Long-Term Continuous Growth Hormone Treatment of Children Born Small for Gestational Age. Horm Res (2004) 62(Suppl 3):149–54. doi: 10.1159/000080518
14. Maiorana A, Cianfarani S. Impact of Growth Hormone Therapy on Adult Height of Children Born Small for Gestational Age. Pediatrics (2009) 124(3):e519–31. doi: 10.1542/peds.2009-0293
15. Dahlgren J, Kriström B, Niklasson A, Nierop AF, Rosberg S, Albertsson-Wikland K. Models Predicting the Growth Response to Growth Hormone Treatment in Short Children Independent of GH Status, Birth Size and Gestational Age. BMC Med Inform Decis Mak (2007) 7:40. doi: 10.1186/1472-6947-7-40
16. Kriström B, Dahlgren J, Niklasson A, Nierop AF, Albertsson-Wikland K. The First-Year Growth Response to Growth Hormone Treatment Predicts the Long-Term Prepubertal Growth Response in Children. BMC Med Inform Decis Mak (2009) 9:1. doi: 10.1186/1472-6947-9-1
17. Ranke MB, Lindberg A, KIGS International Board. Prediction Models for Short Children Born Small for Gestational Age (SGA) Covering the Total Growth Phase. Analyses Based on Data From KIGS (Pfizer International Growth Database). BMC Med Inform Decis Mak (2011) 11:38. doi: 10.1186/1472-6947-11-38
18. Wit JM, Ranke MB, Albertsson-Wikland K, Carrascosa A, Rosenfeld RG, Van Buuren S, et al. Personalized Approach to Growth Hormone Treatment: Clinical Use of Growth Prediction Models. Horm Res Paediatr (2013) 79(5):257–70. doi: 10.1159/000351025
19. Tauber M, Ester W, Auriol F, Molinas C, Fauvel J, Caliebe J, et al. Gh Responsiveness in a Large Multinational Cohort of SGA Children With Short Stature (NESTEGG) is Related to the Exon 3 GHR Polymorphism. Clin Endocrinol (Oxf) (2007) 67(3):457–61. doi: 10.1111/j.1365-2265.2007.02911.x
20. Carrascosa A, Esteban C, Espadero R, Fernández-Cancio M, Andaluz P, Clemente M, et al. The D3/Fl-Growth Hormone (Gh) Receptor Polymorphism Does Not Influence the Effect of GH Treatment (66 Microg/Kg Per Day) or the Spontaneous Growth in Short Non-GH-Deficient Small-for-Gestational-Age Children: Results From a Two-Year Controlled Prospective Study in 170 Spanish Patients. J Clin Endocrinol Metab (2006) 91(9):3281–6. doi: 10.1210/jc.2006-0685
21. Ester WA, Hokken-Koelega AC. Polymorphisms in the IGF1 and IGF1R Genes and Children Born Small for Gestational Age: Results of Large Population Studies. Best Pract Res Clin Endocrinol Metab (2008) 22(3):415–31. doi: 10.1016/j.beem.2008.03.001
22. Jensen RB, Thankamony A, Day F, Scott RA, Langenberg C, Kirk J, et al. Genetic Markers of Insulin Sensitivity and Insulin Secretion Are Associated With Spontaneous Postnatal Growth and Response to Growth Hormone Treatment in Short SGA Children: The North European SGA Study (NESGAS). J Clin Endocrinol Metab (2015) 100(3):E503–7. doi: 10.1210/jc.2014-3469
23. Tanaka T, Yokoya S, Hoshino Y, Hiro S, Ohki N. Long-Term Safety and Efficacy of Daily Recombinant Human Growth Hormone Treatment in Japanese Short Children Born Small for Gestational Age: Final Report From an Open and Multi-Center Study. Clin Pediatr Endocrinol (2018) 27(3):145–57. doi: 10.1297/cpe.27.145
24. Swerdlow AJ, Cooke R, Beckers D, Borgström B, Butler G, Carel JC, et al. Cancer Risks in Patients Treated With Growth Hormone in Childhood: The SAGhE European Cohort Study. J Clin Endocrinol Metab (2017) 102(5):1661–72. doi: 10.1210/jc.2016-2046
25. Sävendahl L, Cooke R, Tidblad A, Beckers D, Butler G, Cianfarani S, et al. Long-Term Mortality After Childhood Growth Hormone Treatment: The Saghe Cohort Study. Lancet Diabetes Endocrinol (2020) 8(8):683–92. doi: 10.1016/S2213-8587(20)30163-7
26. De Lange C. Sur Un Type Nouveau De Degenerescence (Typus Amsterlodamensis). Arch Med Enfants (1933) 36:713–9.
27. Russell A. A Syndrome of Intra-Uterine Dwarfism Recognizable at Birth With Cranio-Facial Dysostosis, Disproportionately Short Arms, and Other Anomalies (5 Examples). Proc R Soc Med (1954) 47(12):1040–4.
28. Silver HK, Kiyasu W, George J, Deamer WC. Syndrome of Congenital Hemihypertrophy, Shortness of Stature, and Elevated Urinary Gonadotropins. Pediatrics (1953) 12(4):368–76.
29. Woods KA, Camacho-Hübner C, Savage MO, Clark AJ. Intrauterine Growth Retardation and Postnatal Growth Failure Associated With Deletion of the Insulin-Like Growth Factor I Gene. N Engl J Med (1996) 335(18):1363–7. doi: 10.1056/NEJM199610313351805
30. Kawashima S, Nakamura A, Inoue T, Matsubara K, Horikawa R, Wakui K, et al. Maternal Uniparental Disomy for Chromosome 20: Physical and Endocrinological Characteristics of Five Patients. J Clin Endocrinol Metab (2018) 103(6):2083–8. doi: 10.1210/jc.2017-02780
31. Flechtner I, Lambot-Juhan K, Teissier R, Colmenares A, Baujat G, Beltrand J, et al. Unexpected High Frequency of Skeletal Dysplasia in Idiopathic Short Stature and Small for Gestational Age Patients. Eur J Endocrinol (2014) 170:677–84. doi: 10.1530/EJE-13-0864
32. Caliebe J, Broekman S, Boogaard M, Bosch CA, Ruivenkamp CA, Oostdijk W, et al. Igf1, IGF1R and SHOX Mutation Analysis in Short Children Born Small for Gestational Age and Short Children With Normal Birth Size (Idiopathic Short Stature). Horm Res Paediatr (2012) 77(4):250–60. doi: 10.1159/000338341
33. Hauer NN, Popp B, Schoeller E, Schuhmann S, Heath KE, Hisado-Oliva A, et al. Clinical Relevance of Systematic Phenotyping and Exome Sequencing in Patients With Short Stature. Genet Med (2018) 20(6):630–8. doi: 10.1038/gim.2017.159
34. Peeters S, Declerck K, Thomas M, Boudin E, Beckers D, Chivu O, et al. Dna Methylation Profiling and Genomic Analysis in 20 Children With Short Stature Who Were Born Small for Gestational Age. J Clin Endocrinol Metab (2020) 05(12):dgaa465. doi: 10.1210/clinem/dgaa465
35. van der Steen M, Pfundt R, Maas SJWH, Bakker-van Waarde WM, Odink RJ, Hokken-Koelega ACS. Acan Gene Mutations in Short Children Born SGA and Response to Growth Hormone Treatment. J Clin Endocrinol Metab (2017) 102(5):1458–67. doi: 10.1210/jc.2016-2941
36. Lee PA, Chernausek SD, Hokken-Koelega AC, Czernichow P, International Small for Gestational Age Advisory Board. International Small for Gestational Age Advisory Board Consensus Development Conference Statement: Management of Short Children Born Small for Gestational Age, April 24-October 1, 2001. Pediatrics (2003) 111(6 Pt 1):1253–61. doi: 10.1542/peds.111.6.1253
37. Stalman SE, Solanky N, Ishida M, Alemán-Charlet C, Abu-Amero S, Alders M, et al. Genetic Analyses in Small-for-Gestational-Age Newborns. J Clin Endocrinol Metab (2018) 103(3):917–25. doi: 10.1210/jc.2017-01843
38. Homma TK, Freire BL, Honjo Kawahira RS, Dauber A, Funari MFA, Lerario AM, et al. Genetic Disorders in Prenatal Onset Syndromic Short Stature Identified by Exome Sequencing. J Pediatr (2019) 215:192–8. doi: 10.1016/j.jpeds.2019.08.024
39. Freire BL, Homma TK, Funari MFA, Lerario AM, Vasques GA, Malaquias AC, et al. Multigene Sequencing Analysis of Children Born Small for Gestational Age With Isolated Short Stature. J Clin Endocrinol Metab (2019) 104(6):2023–30. doi: 10.1210/jc.2018-01971
40. Saenger P, Reiter E. Genetic Factors Associated With Small for Gestational Age Birth and the Use of Human Growth Hormone in Treating the Disorder. Int J Pediatr Endocrinol (2012) 2012(1):12. doi: 10.1186/1687-9856-2012-12
41. Murray JE, Bicknell LS, Yigit G, Duker AL, van Kogelenberg M, Haghayegh S, et al. Extreme Growth Failure Is a Common Presentation of Ligase IV Deficiency. Hum Mutat (2014) 35(1):76–85. doi: 10.1002/humu.22461
42. Janecka A, Kołodziej-Rzepa M, Biesaga B. Clinical and Molecular Features of Laron Syndrome, a Genetic Disorder Protecting From Cancer. In Vivo (2016) 30(4):375–81.
43. Fujimoto M, Hwa V, Dauber A. Novel Modulators of the Growth Hormone - Insulin-Like Growth Factor Axis: Pregnancy-Associated Plasma protein-A2 and Stanniocalcin-2. J Clin Res Pediatr Endocrinol (2017) 9(Suppl 2):1–8. doi: 10.4274/jcrpe.2017.S001
44. Ester WA, van Duyvenvoorde HA, de Wit CC, Broekman AJ, Ruivenkamp CA, Govaerts LC, et al. Two Short Children Born Small for Gestational Age With Insulin-Like Growth Factor 1 Receptor Haploinsufficiency Illustrate the Heterogeneity of its Phenotype. J Clin Endocrinol Metab (2009) 94(12):4717–27. doi: 10.1210/jc.2008-1502
45. Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, et al. Paternally Inherited Igf2 Mutation and Growth Restriction. N Engl J Med (2015) 373(4):349–56. doi: 10.1056/NEJMoa1415227
46. Gutiérrez M, Scaglia P, Keselman A, Martucci L, Karabatas L, Domené S, et al. Partial Growth Hormone Insensitivity and Dysregulatory Immune Disease Associated With De Novo Germline Activating Stat3 Mutations. Mol Cell Endocrinol (2018) 473:166–77. doi: 10.1016/j.mce.2018.01.016
47. Howell SJ, Wilton P, Lindberg A, Shalet SM. Growth Hormone Replacement and the Risk of Malignancy in Children With Neurofibromatosis. J Pediatr (1998) 133(2):201–5. doi: 10.1016/s0022-3476(98)70245-8
48. Rogvi R Á, Forman JL, Greisen G. Prematurity, Smallness-for-Gestational Age and Later Hospital Admissions: A Nation-Wide Registry Study. Early Hum Dev (2015) 91(5):299–306. doi: 10.1016/j.earlhumdev.2015.02.010
49. Ke X, Liang H, Miao H, Yang H, Wang L, Gong F, et al. Clinical Characteristics of Short Stature Patients With NPR2 Mutation and the Therapeutic Response to Rhgh. J Clin Endocrinol Metab (2020) 106(2):431–441. doi: 10.1210/clinem/dgaa842
50. Freire BL, Homma TK, Funari MFA, Lerario AM, Leal AM, Velloso EDRP, et al. Homozygous Loss of Function Brca1 Variant Causing a Fanconi-Anemia-like Phenotype, a Clinical Report and Review of Previous Patients. Eur J Med Genet (2018) 61(3):130–3. doi: 10.1016/j.ejmg.2017.11.003
51. Stochholm K, Kiess W. Long-Term Safety of Growth Hormone-a Combined Registry Analysis. Clin Endocrinol (Oxf) (2018) 88(4):515–28. doi: 10.1111/cen.13502
Keywords: small for gestational age, genetics, growth hormone, screening, short stature children, SGA
Citation: Giacomozzi C (2021) Genetic Screening for Growth Hormone Therapy in Children Small for Gestational Age: So Much to Consider, Still Much to Discover. Front. Endocrinol. 12:671361. doi: 10.3389/fendo.2021.671361
Received: 23 February 2021; Accepted: 05 May 2021;
Published: 28 May 2021.
Edited by:
Gianluca Tornese, Institute for Maternal and Child Health Burlo Garofolo (IRCCS), ItalyReviewed by:
Gabriela Vasques, Universidade de São Paulo, BrazilTiago Jeronimo Dos Santos, Instituto Hispalense de Pediatría, Spain
Copyright © 2021 Giacomozzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio Giacomozzi, ZHIuZ2lhY29tb3p6aUBnbWFpbC5jb20=