 Vinay Singh Tanwar
Vinay Singh Tanwar Marpadga A. Reddy
Marpadga A. Reddy Rama Natarajan
Rama Natarajan- Department of Diabetes Complications and Metabolism, Arthur Riggs Diabetes and Metabolism Research Institute, Beckman Research Institute of City of Hope, Duarte, CA, United States
Chronic metabolic disorders such as obesity and diabetes are associated with accelerated rates of macrovascular and microvascular complications, which are leading causes of morbidity and mortality worldwide. Further understanding of the underlying molecular mechanisms can aid in the development of novel drug targets and therapies to manage these disorders more effectively. Long non-coding RNAs (lncRNAs) that do not have protein-coding potential are expressed in a tissue- and species-specific manner and regulate diverse biological processes. LncRNAs regulate gene expression in cis or in trans through various mechanisms, including interaction with chromatin-modifying proteins and other regulatory proteins and via posttranscriptional mechanisms, including acting as microRNA sponges or as host genes of microRNAs. Emerging evidence suggests that major pathological factors associated with diabetes such as high glucose, free fatty acids, proinflammatory cytokines, and growth factors can dysregulate lncRNAs in inflammatory, cardiac, vascular, and renal cells leading to altered expression of key inflammatory genes and fibrotic genes associated with diabetic vascular complications. Here we review recent reports on lncRNA characterization, functions, and mechanisms of action in diabetic vascular complications and translational approaches to target them. These advances can provide new insights into the lncRNA-dependent actions and mechanisms underlying diabetic vascular complications and uncover novel lncRNA-based biomarkers and therapies to reduce disease burden and mortality.
Introduction
Diabetes is a global epidemic affecting nearly 463 million people worldwide and is projected to increase by 700 million in 2045 (1). The increasing prevalence of diabetes significantly augments the risk for multiple associated complications, including cardiovascular diseases (such as atherosclerosis and hypertension), diabetic nephropathy (DN; also known as diabetic kidney disease), diabetic cardiomyopathy (DCM), and diabetic retinopathy (DR) (2–4). The morbidity and mortality associated with these complications significantly increase the global economic burden. Therapeutic interventions and lifestyle modifications are helpful to decrease the incidence of diabetes but are not always effective in preventing the progression of vascular complications. Therefore, identification of new mechanisms and related novel therapies to mitigate the impact of diabetes and associated complications is urgently warranted. Chronic high glucose (HG) and increased levels of other pathological factors like advanced glycation end products (AGE)s, saturated free fatty acids (FFAs), and low density lipoprotein (LDL) cholesterol can dysregulate the functions of numerous cell types, including inflammatory and immune cells, endothelial cells (ECs), vascular smooth muscle cells (VSMCs), cardiac myocytes, and renal cells in target tissues, leading to various complications of diabetes (2). Multiple underlying mechanisms such as activation of various signaling pathways and downstream transcription factors that promote expression and production of inflammatory cytokines, fibrotic factors, oxidative stress, and other related changes have been described (2, 5–8). Despite such knowledge of traditional regulatory mechanisms driving cellular dysfunction in diabetic complications, the continued increase in disease incidence underscores the need to explore other mediators and mechanisms. Clinical studies with diabetic patients have revealed that memory of prior episodes of hyperglycemia can have long-term deleterious effects with continued risk and progression of complications despite subsequent glycemic control, a phenomenon known as “metabolic memory” or “legacy effect” (5, 9). In addition, accumulating evidence suggests key roles for epigenetic changes and non-coding RNAs in regulating genes, cellular functions, and phenotypes associated with diabetes complications and metabolic memory. Here we review recent reports on the characterization, functions, and mechanisms of action of long non-coding RNAs (lncRNAs) in diabetic vascular complications and translational approaches to target them.
Epigenetics and Long Non-Coding RNAs
Genome-wide association studies (GWAS) have implicated multiple risk loci for diabetes complications such as DKD, but the functional significance of many of these genomic regions is not yet fully understood (10). Because many human disease risk loci are often located in non-coding regions and enhancers, they likely influence the expression of associated genes via epigenetic mechanisms. Epigenetics plays an important role in the integration of cellular responses to diverse genetic, metabolic, and environmental factors (11, 12) and refers to changes in gene expression and function without alteration in the DNA sequence leading to altered phenotypes (11). Epigenetic modifications, such as chromatin histone post-translational modifications (PTMs) and DNA methylation (DNAme), regulate chromatin structure and function, which are further modulated by non-coding RNAs, such as microRNAs (miRNAs) and lncRNAs. Epigenetic modifications associated with cis-elements, such as proximal promoters and distal enhancers, fine-tune transcriptional activity via regulation of chromatin structure and access to transcription factors (TFs). In general, histone PTMs such as histone H3/H4 lysine acetylation (H3/H4K9/14ac) and H3K4methylation (H3K4me1/2/3) regulate transcriptional activation, whereas H3K9me2/3, H3K27me3, and promoter DNAme are associated with gene repression (13). Epigenetic mechanisms are tightly regulated by the balance between the actions of histone or DNA modifying enzymes (“writers”) and actions of enzymes that remove these modifications (“erasers”), and the “readers” that interact with these modifications to regulate chromatin structure (14, 15). LncRNAs can fine-tune these epigenetic processes via direct interaction of lncRNAs with chromatin, chromatin modifiers, or other nuclear proteins (16, 17). For instance, the interaction of lncRNA Xist with polycomb repressor complex 2 (PRC2), which regulates histone H3K27me3, plays a key role in X chromosome inactivation (17). Whereas interaction of lncRNA Kcnq1ot1 with the histone H3K9-methyltransferase G9a (EHMT2) increases repressive modification H3K9me2 and inhibits target genes (17). Increasing evidence also implicates the dysregulation of such epigenetic mechanisms and lncRNAs in diabetes and complications (9, 18–21). Further in-depth investigations into the precise mechanisms by which lncRNAs and associated epigenetic mechanisms regulate genes and processes associated with diabetic complications can lead to the development of more effective therapies and clinical management.
LncRNA-Dependent Mechanisms of Gene Regulation
Advances in the next generation RNA sequencing revealed the complexity and diversity of the transcription process and led to the discovery of thousands of lncRNAs in the mammalian genome (22–24). LncRNAs are transcripts that are >200 nucleotides long and have many features similar to protein-coding genes, including splicing of the transcripts and RNA polymerase II-mediated transcription. However, lncRNAs do not code for any proteins, with the exception of some that are reported to encode small peptides (25). Therefore, while characterizing novel lncRNA transcripts, it is important to use rigorous bioinformatics tools and experimental approaches to verify that they truly lack coding potential (26–29). LncRNAs have also been identified by integrating RNA-seq data with chromatin immunoprecipitation linked to sequencing (ChIP-seq) data, which can reveal enrichment of H3K4me3 marks at transcriptional start sites and H3K36me3 at gene bodies and can distinguish genomic regions from which lncRNAs can be transcribed (30, 31). LncRNAs can be classified as intergenic transcripts, divergent transcripts adjacent to coding transcripts on the opposite strand, anti-sense transcripts of protein-coding genes, and enhancer RNAs (eRNAs) that are transcribed near enhancers (Figure 1) (32). In addition, alternative splicing generates multiple isoforms of the same lncRNA, which may affect its subcellular localization and functions. In general, lncRNAs show tissue-specific expression patterns and are expressed in low abundance compared to protein-coding genes, except for some highly expressed lncRNAs such as MALAT1 (Metastasis-associated lung carcinoma transcript 1) (22, 33).
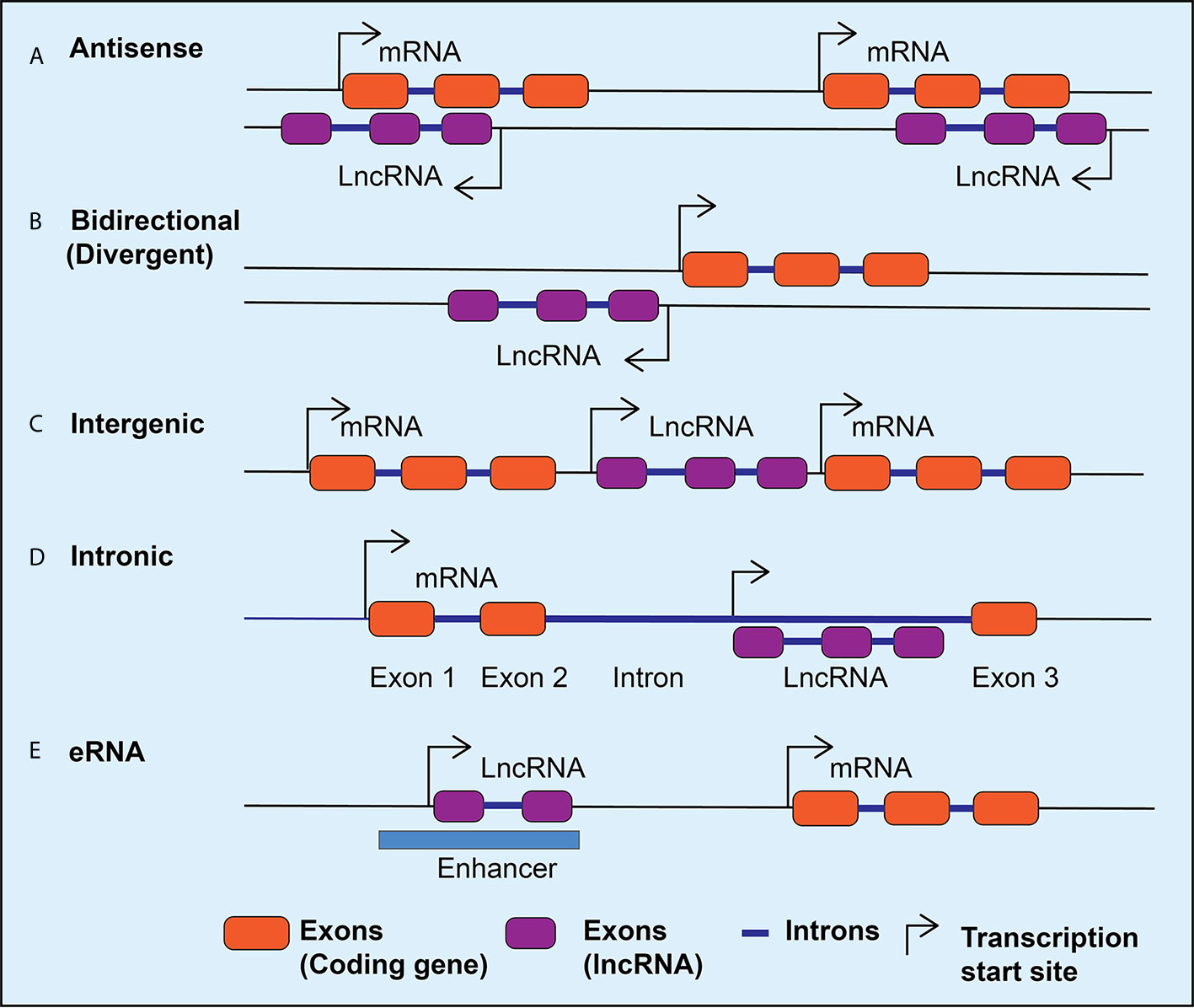
Figure 1 LncRNA classification. Schematic diagram depicting different classes of lncRNAs expressed in mammalian cells. The lncRNAs can be expressed as anti-sense lncRNAs from the anti-sense strand overlapping coding genes (A), as bidirectional or divergent transcripts from a shared promoter with coding genes (B), as intergenic (C), or intronic (D), and as enhancer RNAs (eRNAs) from the enhancers (E). Arrows indicate the direction of transcription.
The functions and mechanisms of actions of lncRNAs generally depend on their subcellular localization. Biochemical and epigenomic approaches have revealed that lncRNAs regulate genes via transcriptional and post-transcriptional mechanisms (32, 34–36). Nuclear lncRNAs can regulate epigenetic mechanisms involved in gene transcription and function of enhancers via interactions with chromatin, chromatin regulatory proteins, other RNA binding proteins, and TFs (Figure 2) (17, 32). On the other hand, cytoplasmic lncRNAs can alter mRNA stability or translation, regulate the expression and function of miRNAs and their targets by acting as sponges for miRNAs, and influence the actions of signaling proteins (Figure 2) (32, 35). It is now well established that lncRNAs are involved in many biological processes like cell proliferation, differentiation, inflammatory and immune responses (37). Emerging evidence shows that dysregulated expression and functions of lncRNAs can modulate pathologic gene expression associated with diabetes and its complications, including cardiovascular diseases (CVDs), DN, DCM, and DR (18, 21).
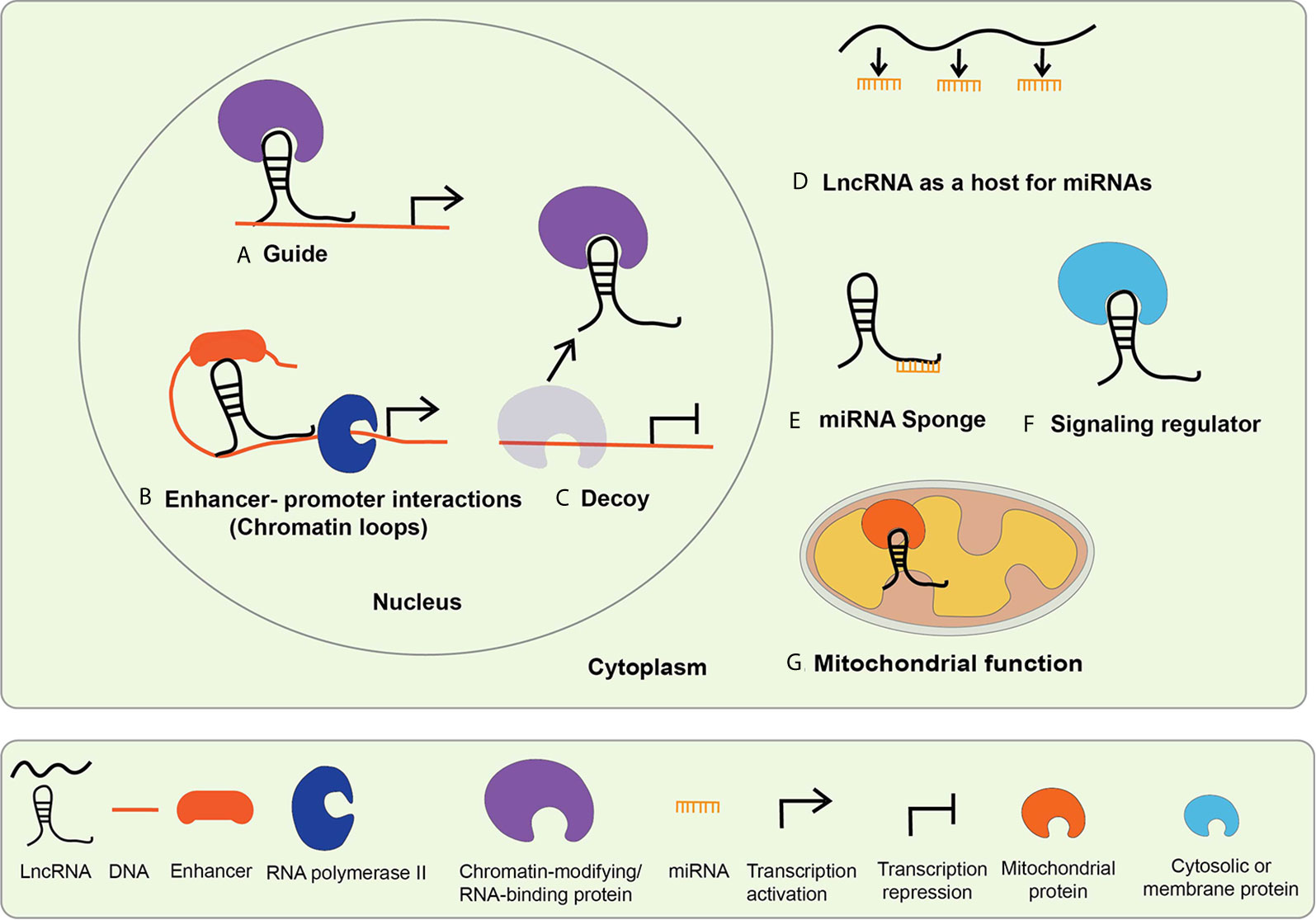
Figure 2 Mechanisms of lncRNA mediated gene regulation. The mechanisms of lncRNA actions are dependent upon their subcellular localization. LncRNAs localized in the nucleus mediate transcriptional regulation, whereas those located in the cytoplasm regulate via post-transcriptional mechanisms. Nuclear lncRNAs regulate transcription by (A) acting as guide lncRNAs that interact with chromatin-modifying and RNA-binding proteins to facilitate their recruitment on regulatory DNA sequences to activate or repress gene expression; (B) mediating enhancer promoter interactions-LncRNAs promote long-range enhancer-promoter interactions via chromatin looping; (C) acting as decoys that interact with the regulatory proteins to sequester them and prevent their normal functions. Cytosolic lncRNA mechanism of action includes: (D) serving as host genes for miRNAs, i.e. they harbor miRNAs within their exonic or intronic sequences; (E) acting as miRNA Sponges or competitive endogenous RNA (ceRNA)s, compete with mRNAs for shared complementary miRNA binding sites; (F) interacting with cytosolic or membrane proteins to regulate their functions and signaling processes; (G) serving as mitochondrial protein activators-lncRNAs interact with key mitochondrial proteins to modulate mitochondrial functions such as fatty acid oxidation.
Role of LncRNAs in Inflammatory Processes and Diabetic Cardiovascular Complications
Chronic inflammation is associated with both diabetes as well as most of the associated complications, including CVDs (2, 7). Monocytes and macrophages are innate immune cells that regulate inflammatory responses and host defense against invading microbial pathogens (38, 39). After acute exposure to environmental and metabolic cues like HG, FFAs, viruses, or endotoxin, these cells respond by inducing inflammatory cytokines via activation of nuclear factor kappa B (NF-κB) TF mediated pathways. This inflammatory phase is normally followed by an inflammation resolution phase (40, 41). However, recurrent exposure to these insults can weaken the inflammation resolution pathways leading to chronic inflammation associated with multiple disease conditions, including diabetes and its complications (41–43). Different stages of inflammation and inflammation resolution are tightly regulated by distinct gene expression patterns and phenotypic modulation of monocytes and macrophages via complex interactions between cis-regulatory elements on chromatin and transcription factors/regulators (44–46). Alterations in these transcription programs and dysregulated expression of inflammatory genes contribute to uncontrolled inflammation and diabetic vascular complications.
In diabetes, increases in levels of deleterious lipids and FFAs in the circulation along with HG and growth factors like Angiotensin II (AngII) accelerate the risk for CVDs, including hypertension and atherosclerosis (47, 48). Hyperglycemia and these atherogenic factors promote inflammation in monocytes/macrophages, vascular smooth muscles, and endothelial cells, endothelial dysfunction, trans-endothelial migration of monocytes, foam cell formation, and VSMC migration and proliferation, key events in hypertension, restenosis, and atherosclerotic lesion formation (47). In advanced stages of atherosclerosis, macrophage apoptosis, defective efferocytosis, and dysregulated extracellular matrix (ECM) deposition in VSMCs play important roles in plaque stability (47). In diabetes, all these processes are significantly enhanced and result in accelerated atherosclerosis and unstable plaques, which are prone to rupture and thrombosis (48). Accumulating bodies of evidence show that a number lncRNAs are involved in the dysregulated functions of monocytes, macrophages, endothelial cells, and VSMCs associated with inflammation and the pathogenesis of atherosclerosis under diabetic or non-diabetic states (Table 1 and Figure 3) (21, 86–88), suggesting their potential use as biomarkers and therapeutic targets. The following sub-sections highlight recent advances in the roles played by key candidate lncRNAs expressed in these inflammatory and vascular cells in processes related to diabetic CVDs.
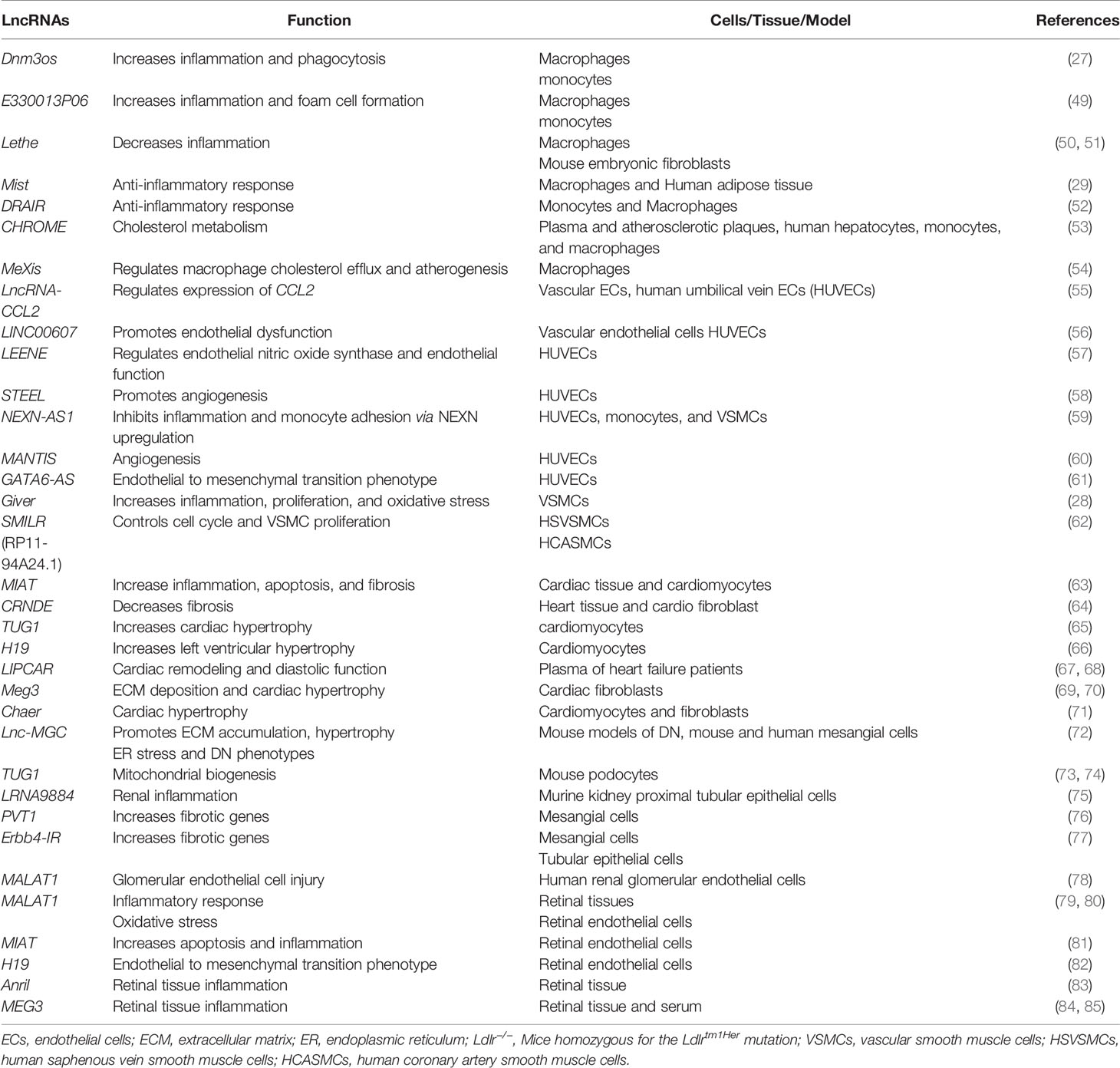
Table 1 Long non-coding RNAs and their functions in inflammation and diabetes complications.
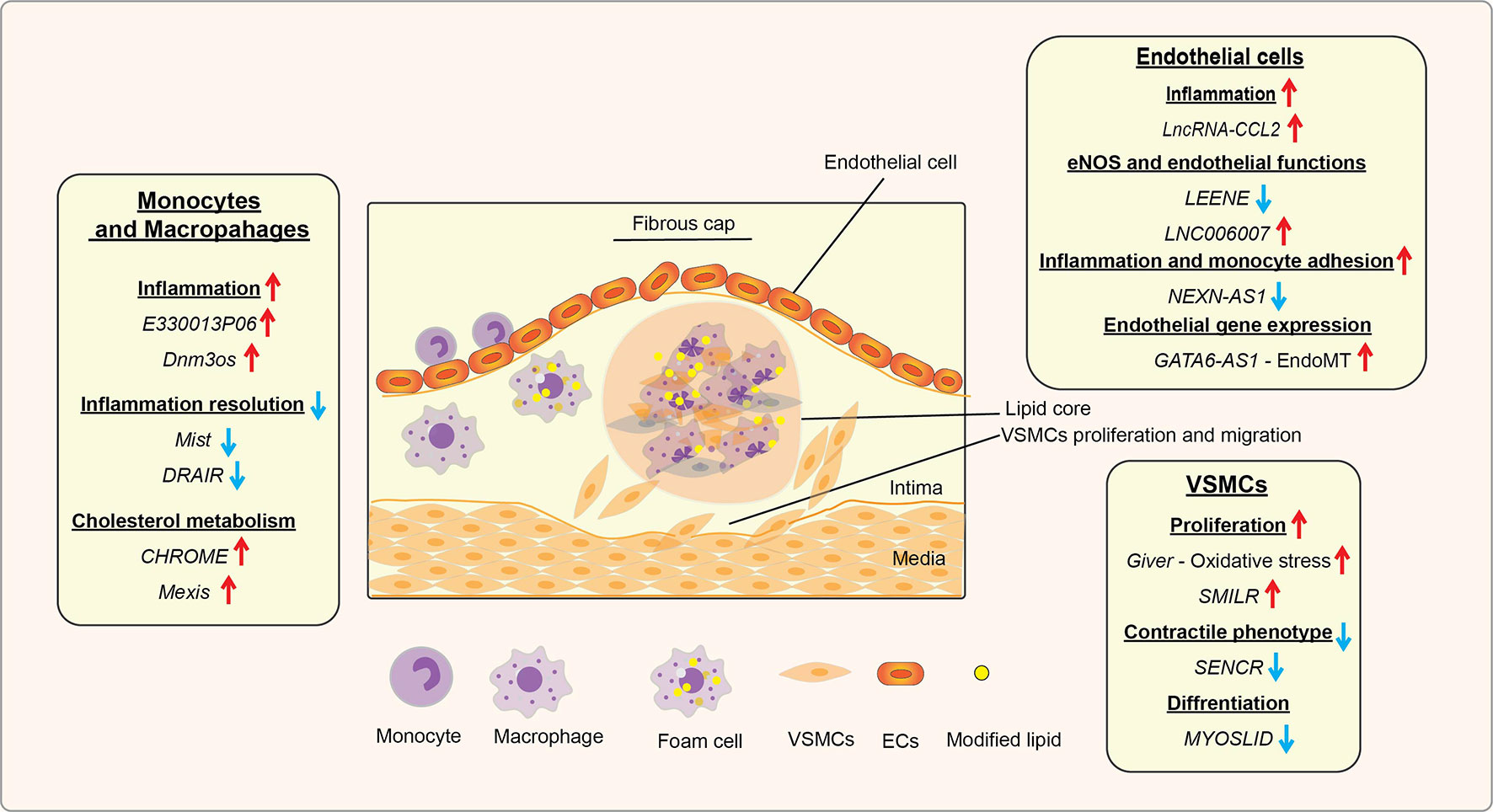
Figure 3 Role of lncRNAs in inflammatory processes and diabetic cardiovascular complications. A schematic showing the roles of candidate lncRNAs with functions in endothelial cells (ECs), monocytes and macrophages, and vascular smooth muscle cells (VSMCs) during the initiation and progression of inflammatory CVDs, such as atherosclerosis, that are enhanced in diabetes. The reported roles of lncRNAs in processes related to atherosclerosis and related CVDs are indicated under relevant cell types (monocytes/macrophages, ECs, and VSMC). Arrows indicate the direction of changes in the expression or function. CVDs, Cardiovascular diseases; EndoMT, Endothelial to mesenchymal transition.
LncRNA Functions in Monocytes and Macrophages
Several studies have shown that lncRNA mediated mechanisms regulate several key monocyte/macrophage functions associated with differentiation, inflammation, and innate immunity (89, 90). Since increased infiltration of inflammatory monocytes/macrophages into target organs and enhanced inflammation are key features of most diabetic vascular complications, lncRNAs that affect the expression of inflammatory genes in these cells are likely to be involved in diabetes, obesity, and diabetic CVDs. RNA-sequencing identified several dysregulated lncRNAs in bone marrow-derived macrophages (BMDM) from type 2 diabetic (T2D) db/db mice relative to control db/+ mice, and the function of one of these lncRNAs E330013P06 (E33) was further characterized (49). Expression of the lncRNA E33 was upregulated in macrophages from db/db mice, and high-fat diet (HFD) plus streptozotocin (STZ) induced T2D mice but not in STZ-induced type 1 diabetic (T1D) mice, suggesting a T2D disease-specific regulation of lncRNA E33 (49). Furthermore, diabetogenic factors, like HG and palmitic acid (PA), upregulated lncRNA E33 expression in vitro in mouse macrophages. Over-expression of the lncRNA E33 increased the expression of inflammatory genes Il6, Tnf, Ptgs2, and Ccl2 and promoted foam-cell formation (proatherogenic phenotype) in mouse macrophages. In contrast, siRNA-mediated knockdown of lncRNA E33 inhibited HG and PA-induced upregulation of these inflammatory genes in primary mouse BMDM. Interestingly, the human ortholog of lncRNA E33 was also upregulated in CD14+ monocytes from T2D patients. These results suggest that lncRNA E33 is an important regulator of inflammation and foam cell formation in diabetes and diabetic vascular complications, and its functions are conserved in humans and mice.
Another lncRNA, Dynamin3 opposite strand (Dnm3os), was also induced in macrophages from T2D mice, HFD-induced insulin-resistant mice, T1D mice, and diabetic Apoe−/− mice (a mouse model of accelerated atherosclerosis), indicating that Dnm3os plays a key role in diabetes and accelerated atherosclerosis. Dnm3os was upregulated by PA via activation of NF-κB, supporting its pro-inflammatory properties (27). Stable overexpression of Dnm3os increased basal and PA-induced Il6 and Tnf genes in mouse RAW macrophages. Furthermore, Dnm3os overexpression upregulated gene networks associated with inflammation, immune response, chemotaxis, phagocytosis, and wound healing, key processes associated with CVDs and diabetic vascular complications. Conversely, Dnm3os knockdown with siRNAs in macrophages from diabetic db/db mice ameliorated the enhanced expression of inflammatory genes (Il6, Tnf, Itgax, and Nfkbiz) and phagocytosis of E.coli bioparticles, clearly supporting its role in diabetes-induced inflammation. Further, mechanistic studies showed that Dnm3os overexpression increased global levels of chromatin marks such as histone H3K9ac and H3K27ac associated with active transcription, suggesting it may act via epigenetic mechanisms.
Additionally, RNA-pulldown assays and RNA-fluorescence in-situ hybridization (RNA-FISH) revealed that Dnm3os interacts with the multifunctional nucleolar protein nucleolin, which is known to have anti-inflammatory and protective roles in macrophages. Nucleolin protein levels were also reduced in macrophages treated with HG and PA and in macrophages of T2D db/db mice. Further studies with nucleolin knockdown and ChIP assays suggested that, under normal conditions, the interaction with nucleolin restrains the actions of Dnm3os and maintains compact chromatin near inflammatory gene promoters. However, under diabetic conditions, the altered levels of Dnm3os and nucleolin disrupt this homeostasis, and augments the interaction of Dnm3os with histone acetyltransferases, leading to H3K9ac enrichment, chromatin relaxation, and increased inflammatory gene expression. Further highlighting its relevance, human DNM3OS levels were increased in CD14+ monocytes from T2D patients and in vitro in PA-treated macrophages derived from CD14+ monocytes from non-diabetic humans. Moreover, GapmeR-mediated knockdown of human DNM3OS in human THP1 monocytes inhibited inflammatory genes (IL6, TNF, and ITGAX) and phagocytosis similar to actions of the mouse ortholog, suggesting DNM3OS upregulation in humans likely contributes to enhanced inflammation in diabetes. However, Dnm3os levels have not been examined in endothelial cells (ECs) and VSMCs in diabetes.
Resolution of the inflammatory response is required for normal homeostasis, tissue repair, and wound healing (91, 92). Apart from increases in pro-inflammatory lncRNAs, reductions of “protective” lncRNAs that restrain, or mediate inflammation resolution could also contribute to enhanced inflammation and impaired wound healing in diabetes. LncRNA Lethe (named after the mythological river of forgetfulness for its role in negative feedback) was induced by pro-inflammatory cytokines (TNF-α and IL-1β) and anti-inflammatory dexamethasone in mouse embryonic fibroblasts (50). Lethe, a chromatin-associated lncRNA, suppressed inflammatory gene expression by inhibiting the DNA binding activity of RelA (p65), the active subunit of NF-κB (50). Interestingly, Lethe expression was downregulated while the oxidative stress gene NADPH oxidase 2 (Nox2) was upregulated by HG in mouse macrophages. Overexpression of Lethe attenuated HG-induced Nox2 and reactive oxygen species (ROS) production as well as NF-κB activation in macrophages. Furthermore, Lethe expression was downregulated in vivo in peritoneal macrophages, BMDM and in wounds from T2D db/db mice, suggesting a potential protective role for Lethe against impaired wound healing in diabetes (51).
Similarly, an anti-inflammatory function was demonstrated for a novel lncRNA Macrophage Inflammation-Suppressing Transcript (Mist) in macrophages during obesity. Mist was one of the lncRNAs differentially expressed in peritoneal macrophages (PMs) from HFD-induced obese insulin-resistant mice relative to control-diet fed mice (29). LncRNA Mist and its adjacent coding gene Fabp5 were both downregulated in PMs and adipose tissue macrophages from HFD-fed obese mice. Mist was found to inhibit the expression of inflammatory genes Tnf, Il1b Nos2, and Il6, and foam cell formation in mouse macrophages. Mechanistic studies, including RNA pull-down coupled with mass spectrometry, showed that Mist interacts with Poly (ADP)-ribose polymerase-1 (PARP1) protein. Disruption of this interaction by Gapmer mediated knockdown of Mist led to increased enrichment of PARP1 and chromatin PARylation, a permissive epigenetic modification, at the Il6 and Tnf promoters and their upregulation. Expression of the human ortholog MIST was also downregulated by pro-inflammatory stimuli, and its expression in human adipose stromal vascular fractions (enriched in macrophages) inversely correlated with obesity, insulin resistance, and metabolic dysfunction (29). Together these results supported the notion that MIST has anti-inflammatory properties and its downregulation in obesity contributes to augmented inflammation.
Furthermore, lncRNAs can also modulate monocyte activation associated with chronic inflammation in human T2D. Recently, RNA-seq analysis of CD14+ monocytes from T2D patients versus healthy controls revealed dysregulated expression of several lncRNAs along with reduced levels of protective genes in T2D, which could be associated with enhanced monocytosis and inflammatory phenotype of monocytes. Interestingly, a novel lncRNA DRAIR (Diabetes Regulated anti-inflammatory RNA) was found to be downregulated in T2D monocytes. The lncRNA DRAIR is divergently expressed adjacent to Cytoplasmic polyadenylation element-binding protein 2 gene (CPEB2), which belongs to CPEB family of proteins with anti-inflammatory functions (93), suggesting a potential role for DRAIR in inflammation. Furthermore, HG and PA downregulated DRAIR in CD14+monocytes from healthy normal controls. On the other hand, DRAIR was upregulated by anti-inflammatory cytokines IL-4 and IL-13 via KLF4, a key transcription factor that promotes anti-inflammatory phenotype in macrophages. Gain- and loss-of-function experiments showed that DRAIR increases anti-inflammatory genes like IL1RN and macrophage differentiation markers like CD36, whereas DRAIR inhibits pro-inflammatory TNF and IL1B, and EC-monocyte binding and phagocytosis, supporting anti-inflammatory functions for DRAIR. Mouse orthologous Drair was also downregulated in T2D db/db mice and its knockdown with locked nucleic acid (LNA) modified GapmeRs in macrophages in vitro and in vivo in non-diabetic mice enhanced Tnf, Il1b, and Il6 genes. Further studies with Chromatin Isolation by RNA Purification assays (ChIRP-seq) and ChIRP-Mass spectrometry revealed that DRAIR interacts with chromatin and chromatin-modifying proteins, including G9a, which mediates H3K9me2. Moreover, G9a was upregulated in monocytes from T2D subjects and in human monocytes treated with HG+PA, and its knockdown with siRNAs increased anti-inflammatory genes. Furthermore, DRAIR overexpression increased anti-inflammatory genes via inhibition of G9a recruitment and enrichment of repressive H3K9me2 at their promoters. These studies suggest that inhibition of DRAIR mediated epigenetic mechanisms via its downregulation and increased G9a could contribute to monocyte dysfunction and inflammation in diabetes (52).
Hyperlipidemia, characterized by high cholesterol, LDL, and triglyceride levels, is a strong driver of atherosclerosis in animal models and humans and is associated with high cholesterol diet and diabetes. Macrophages from hyperlipidemic Ldlr−/− mice and Apoe−/− mice fail to effectively clear LDL from the circulation leading to the development of spontaneous atherosclerotic lesions. Nuclear receptors such as the liver X receptor (LXR) family play key roles in the regulation of genes involved in cholesterol homeostasis (94). LncRNAs regulated by LXR such as CHROME (Cholesterol Homeostasis Regulator of MiRNA Expression) and MeXis (Macrophage-expressed LXR-induced sequence) were shown to regulate cholesterol metabolism genes in macrophages (Table 1 and Figure 3) (53, 54). MeXis was identified as an amplifier of LXR-dependent transcription of Abca1, which plays a critical role in regulating cholesterol efflux. Additionally, CHROME was shown to promote cholesterol efflux and HDL biogenesis by restraining key functionally related miRNAs that downregulate important genes (including Abca1) related to cholesterol homeostasis and atherosclerosis (53, 54). However, further studies are needed to extend these findings to diabetic CVDs.
Together these reports show that lncRNAs in monocytes/macrophages can regulate inflammatory processes and lipid metabolism via interactions with chromatin factors and signaling proteins to modulate the epigenetic states at the promoters of key genes associated with CVDs in diabetes. Further characterization of such lncRNAs in humans could lead to their utility as novel biomarkers for early detection and/or therapeutic targets to reduce inflammation, promote cholesterol homeostasis, or inhibit the accelerated progression of CVDs, like atherosclerosis and hypertension in diabetes.
LncRNA Functions in Endothelial Cells
The endothelium makes up the innermost layer of the vasculature, and disruption of endothelial homeostasis leads to endothelial dysfunction in diabetes and several diabetic vascular complications, including CVDs (2, 47). In addition, many lncRNAs implicated in EC homeostasis are regulated by local mechanical, hypoxic, and inflammatory conditions associated with CVD (86).
Atherosclerotic lesions are more localized in the aortic arch where disturbed blood flow patterns promote atherogenic genes and EC apoptosis. By examining the regulatory role of blood flow-dependent lncRNAs expressed in EC can provide new insights into EC functions, and several lncRNAs are regulated by changes in mechanical (hemodynamic) forces (57, 58). Nitric oxide (NO) produced by endothelial nitric oxide synthase (eNOS) has vital vasodilatory functions and is critical for EC homeostasis. Pathological flow patterns and inflammatory conditions involved in CVDs downregulate eNOS (NOS3) via epigenetic mechanisms (95). ECs subjected to physiological flow increased not only eNOS, but also a lncRNA LEENE (lncRNA that enhances eNOS expression) an eRNA expressed from a nearby enhancer, in KLF2 and KLF4 dependent manner (57). LEENE upregulated eNOS expression via epigenetic mechanisms such as promoting enhancer-promoter interactions and recruitment of RNA pol II at the eNOS promoter (57). Moreover, LEENE knockdown increased monocyte-EC adhesion further supporting its anti-inflammatory functions. Similar functions and regulation were also exhibited by a mouse ortholog BY707159.1 suggesting conserved functions across species. Another EC enriched nuclear lncRNA, Spliced-transcript endothelial-enriched lncRNA (STEEL), was shown to regulate several genes involved in EC functions, including angiogenesis (58). STEEL also induced the expression of KLF2 and its target eNOS via increasing eNOS promoter recruitment of PARP1, which is implicated in EC dysfunction (58). The authors suggested that STEEL plays an important role in transcriptional regulation of EC identity. Furthermore, a search for epigenetically regulated lncRNAs by shear stress in ECs led to the discovery of lncRNA MANTIS, which was regulated by a histone demethylase JARID1B (60). MANTIS, a nuclear lncRNA, was shown to regulate the expression of genes associated with angiogenesis via interaction with BRG1, the catalytic subunit of the SWI/SNF complex, promoting chromatin remodeling and recruitment of RNA pol II. MANTIS expression was downregulated in idiopathic pulmonary hypertension in humans and rats and increased during atherosclerosis regression in primates, suggesting it might have protective functions in ECs (60).
In addition to hemodynamic forces, hypoxia was also shown to regulate lncRNAs such as lncRNA GATA6-AS in ECs (61). GATA6-AS could promote endothelial-mesenchymal transition and regulate hypoxia as well as angiogenesis-related genes, including PTGS2 and POSTN implicated in the development of CVDs (61). This study suggested that GATA6-AS regulates angiogenesis-related genes and endothelial function via epigenetic mechanisms, at least in part, by binding to a fraction of nuclear Lysyl oxidase-like 2 (LOXL2) in vitro and in vivo experiments. LOXL2 was identified as a transcription co-repressor that reduces the permissive histone modification H3K4me3 via deamination of the trimethylated lysine 4 (96). Interaction with GATA6-AS was shown to inhibit LOXL2 function and increase H3K4me3 levels at angiogenesis-related target gene promoters.
lncRNA-CCL2 and LINC00607 are involved in regulating inflammation in human umbilical vein ECs (HUVECs) (55, 56). LncRNA-CCL2, a divergent transcript expressed adjacent to CCL2, was induced by the pro-inflammatory cytokine IL-1β in primary ECs, immortalized HUVECs and dermal microvascular ECs. In addition, LncRNA-CCL2 upregulated CCL2, which codes for chemokine CCL2 involved in inflammatory cell recruitment via post-transcriptional mechanisms involving interactions with RNA binding proteins (55). Moreover, increased lncRNA-CCL2 levels correlated with increased CCL2 levels in primary HUVECs treated with IL-1β and unstable symptomatic human atherosclerotic plaques.
Emerging studies show that chromatin-associated lncRNAs can also have functional roles in ECs. An integrative Omics study characterized HG plus TNF-α induced changes in ECs using a combination of single-cell RNA-seq, Hi-C (to identify DNA-DNA interactions), and in situ mapping of RNA–genome interactome (iMARGI) (to identify RNA-chromatin interactions genome-wide). HG + TNF-α treatment induced inter-chromosomal RNA-chromatin interactions that were particularly clustered around super enhancers from many chromosomes (56). One of the key chromatin-associated lncRNAs induced by HG and TNF-α, LINC00607, regulated SERPINE1, a known pro-inflammatory and pro-fibrotic gene. Furthermore, bioinformatics analysis of the co-expression gene networks in arterial ECs obtained from diabetic versus control human donors could closely substantiate HG + TNF-α induced RNA-chromatin interactions in vitro. This report highlights the putative role of chromatin-associated RNAs in regulating genes associated with EC dysfunction under diabetic conditions.
Another nuclear lncRNA NEXN-AS1 and nearby gene NEXN (Nexilin F-actin-binding protein) were downregulated in atherosclerotic plaques from humans (59). NEXN-AS1 was shown to upregulate NEXN, an atheroprotective gene, via interacting with chromatin upstream of NEXN promoter and inhibiting the transcription repressor BAZ1A (also known as ATP-utilizing chromatin assembly and remodeling factor 1 [ACF1]) to promote open chromatin formation. NEXN and NEXN-AS1 knockdown enhanced pro-inflammatory genes such as CCL2, TNF, and IL6 in ECs. Furthermore, heterozygous NEXN+/− Apoe−/− mice on Western diet showed significantly more atherosclerosis relative to NEXN+/+ Apoe−/− mice, suggesting that NEXN-AS1 mediates atheroprotective effects via NEXN regulation (59).
Overall, these reports clearly show that lncRNAs are important epigenetic regulators of endothelial function and could therefore be valuable therapeutic targets for CVDs. It is yet to be determined if dysregulation of many of these lncRNAs is associated with accelerated vascular inflammation, EC differentiation and dysfunction in diabetes/metabolic disease.
LncRNA Functions in VSMCs
Evidence shows that lncRNAs can also modulate various VSMC functions, including phenotypic switching, proliferation, oxidative stress, and inflammation-key processes associated with accelerated CVDs and other vascular complications of diabetes. Several known and novel lncRNAs that are regulated by growth factors, inflammatory cytokines and in CVDs have been identified using sequencing approaches in rat and human VSMCs (28, 88, 97). VSMC lncRNAs were identified for the first time by integration of datasets from RNA-seq and ChIP-seq (H3K4me3/H3K36me3) from rat VSMCs treated with vehicle or Angiotensin II (AngII), a potent VSMC growth factor that promotes hypertension and atherosclerosis (30). AngII-induced differential expression of 491 lncRNAs, including Lnc-Ang362, which is co-transcribed with miRNAs miR-221 and miR-222 that are implicated in VSMC proliferation (98). Accordingly, Lnc-Ang362 knockdown with siRNAs downregulated miR-222 and miR-221 and inhibited VSMC proliferation, demonstrating that non-coding RNAs can play functional roles in AngII-induced VSMC dysfunction related to CVDs.
A novel lncRNA Giver (growth factor- and proinflammatory cytokine-induced vascular cell-expressed RNA) located near the nuclear receptor Nr4a3 was also markedly induced by AngII, growth factors, HG, and inflammatory cytokines (28). Further studies revealed that Giver is a nuclear lncRNA, regulated by the nuclear receptor Nr4A3 and promoted oxidative stress, inflammatory gene expression, and proliferation in VSMC. Giver-induced gene expression was mediated, at least in part, by its interaction with several nuclear proteins, such as NONO (Non-POU Domain Containing Octamer Binding) protein and regulation of epigenetic histone PTMs at target genes. Interestingly, human GIVER was upregulated in arteries from hypertensive patients but significantly attenuated in hypertensive patients taking ACE (angiotensin-converting enzyme) inhibitors and angiotensin receptor blockers, supporting a role for GIVER in AngII signaling and hypertension.
Another VSMC-specific lncRNA, Smooth Muscle Enriched Long Non-coding RNA (SMILR), was identified using RNA-seq of human VSMCs treated with platelet derived growth factor (PDGF) and inflammatory cytokine (IL-1β) (62). Knockdown of SMILR decreased VSMC proliferation and conversely, its overexpression increased VSMC proliferation. SMILR, a cytoplasmic enriched lncRNA, was found to regulate the late phase of the mitotic cycle via interaction with mRNA of a key mitotic centromere protein and RNA binding protein Staufen1. Moreover, SMILR was upregulated in unstable human atherosclerotic plaques and positively correlated with elevated levels of inflammatory C-reactive protein, suggesting that targeting SMILR may confer protection from atherosclerosis. VSMC de-differentiation and phenotypic switching play key roles in VSMC dysfunction and CVDs like hypertension and atherosclerosis. Smooth muscle and endothelial cell-enriched migration/differentiation-associated long non-coding RNA (SENCR) was one of the first lncRNAs shown to be expressed in both human ECs and VSMCs. SENCR could stabilize VSMC contractile phenotype and inhibit migration, suggesting it has protective functions in the vasculature (97). More recently, another lncRNA MYOcardin-induced Smooth muscle LncRNA, Inducer of Differentiation (MYOSLID) was reported to be the first VSMC-selective lncRNA that acts as a novel amplifier of the VSMC differentiation program via distinct mechanisms. Furthermore, MYOSLID expression was reduced in CVD, supporting a vasculo-protective role (99).
Together these studies show lncRNAs act via novel (including epigenetic) mechanisms in VSMCs and have important functional roles related CVDs that could be exploited for therapy (Table 1 and Figure 3).
Role of LncRNAs in Diabetic Cardiomyopathy (DCM)
Diabetic cardiomyopathy (DCM) is also a common and severe complication of diabetes characterized by myocardium dysfunction, cardiac fibrosis, and microvascular abnormalities. DCM occurs even in the absence of traditional risk factors like hypertension and coronary artery disease (100). Factors associated with diabetes, including HG, AGEs and lipids, increase myocardial fibrosis, cardiomyocyte hypertrophy and stiffness, and inflammation, which contribute to DCM (100). Accumulating evidence suggests that lncRNAs play a key role in DCM (Table 1 and Figure 4) (101).
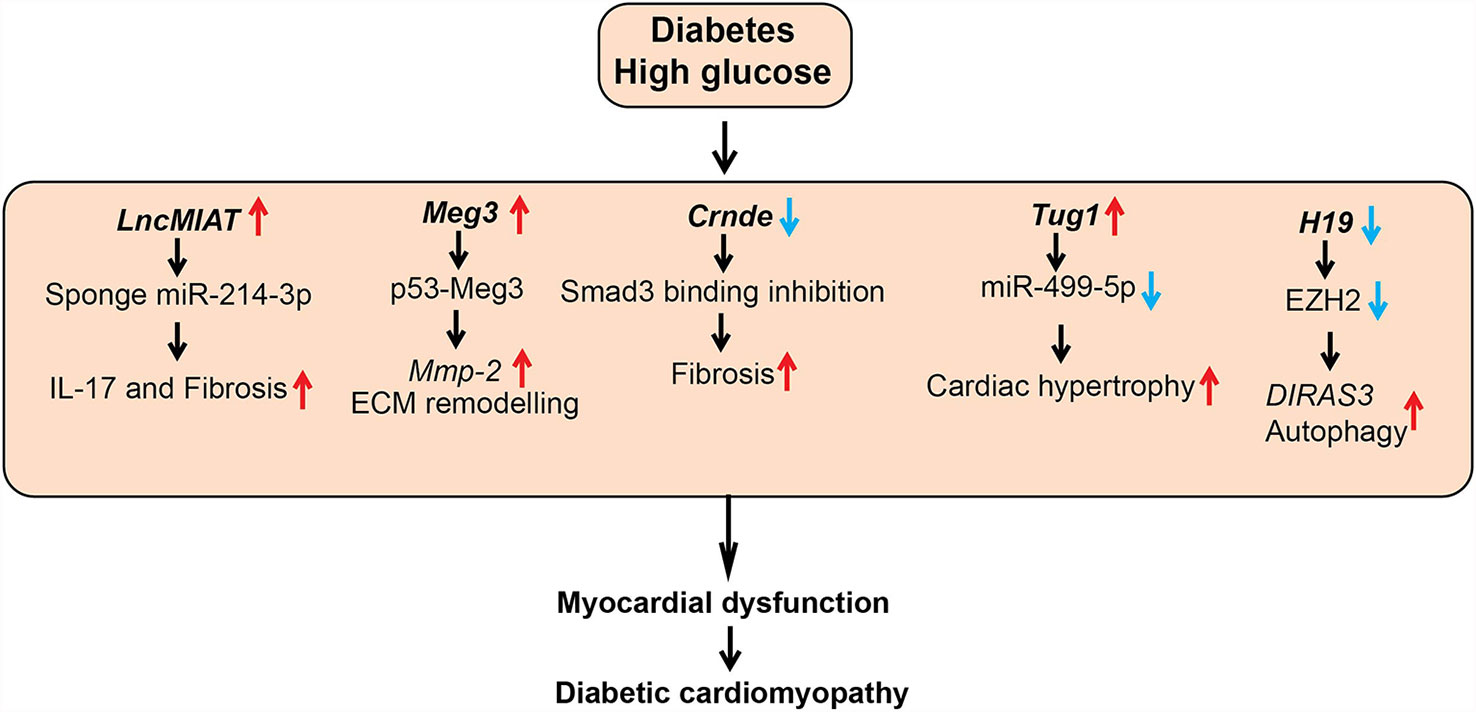
Figure 4 Role of lncRNAs in diabetic cardiomyopathy (DCM). A schematic showing dysregulated function of lncRNAs in the pathogenesis of DCM, such as cardiac hypertrophy, fibrosis, and inflammation. Arrows indicate the direction of changes in the expression or function. Mmp-2, Matrix Metalloproteinase-2; ECM, Extracellular matrix; Smad3, SMAD family member 3; EZH2, Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit; DIRAS3, DIRAS Family GTPase 3.
A number of studies investigated the role of the lncRNA Myocardial Infarction Associated Transcript (MIAT) in DCM. MIAT was identified as a susceptible locus for myocardial infarction on human chromosome 22q12 (102). MIAT levels were upregulated in cardiac tissues and serum from patients with DCM and in animal models of DCM along with several inflammatory cytokines, including IL-17, IL-1β, IL-6, and TNF-α. HG upregulated MIAT and these inflammatory cytokines and promoted apoptosis in cardiomyocytes. HG-effects were inhibited by siRNA-mediated knockdown of MIAT, but they were augmented by MIAT overexpression. Mechanistic studies showed that MIAT acts as a competing endogenous RNA to inhibit functions of miRNAs, such as miR-214-3p and miR-22-3p, that target inflammatory and apoptosis genes. Furthermore, lentiviral-based knockdown of MIAT in diabetic animals improved cardiac ejection fraction, decreased inflammatory cytokines (IL-17, IL-6, IL-1β, and TNF- α) and fibrotic markers (collagen I and III) in heart tissue (left ventricle), inhibited apoptosis, and ameliorated DCM (63, 103).
Expression of another lncRNA, colorectal neoplasia differentially expressed (CRNDE), was also upregulated and negatively correlated with the myocardial fibrosis markers, including COL1A1 in human heart tissue. Crnde expression was upregulated in cardiac fibroblasts treated with Transforming growth factor-beta1 (TGF-β1) and angiotensin II, and in heart tissues from a mice model of myocardial infarction. Adeno-Associated Virus (AAV)-mediated overexpression of Crnde in vivo in a mouse model of DCM decreased the deposition of fibrosis marker collagen in the hearts of the mice with DCM compared to control. Furthermore, Crnde overexpression in cardiac fibroblasts in vitro reduced TGF-β1 induced expression of myofibroblast markers (Col1a1, Col3a1, and Acta2). Crnde was found to inhibit the binding of SMAD family member 3 (Smad3) to Smad binding element and downregulate the expression of key myofibroblast marker genes such as Acta2. However, further studies are needed to determine if human CRNDE has function and mechanism of action similar to the murine ortholog (64).
Additionally, LncRNA TUG1 (taurine upregulated gene1) was upregulated in cardiomyocytes of diabetic mice. Lentiviral-mediated TUG1 knockdown (si-TUG1) improved diastolic dysfunction in a mice model of DCM but had no effect on hyperglycemia and dyslipidemia. TUG1 inhibition also decreased the expression of key hypertrophic markers as well as fibrotic area in db/db mice. Furthermore, this study reported that TUG1 mediates cardiac hypertrophy in DCM via inhibition of miR-499-5p (65). Dysregulated autophagy also promotes cardiac dysfunction in DCM, and the role of lncRNAs such as H19 in autophagy has been reported. H19 was downregulated in the myocardium of a rat model of DCM and lentiviral-mediated overexpression of H19 decreased autophagy and improved left ventricular dysfunction. Furthermore, overexpression of H19 decreased levels of diabetes-induced autophagy markers, such as LC3-II, BECN1, and ATG7, in cardiomyocytes. Mechanistic investigations demonstrated that H19 represses autophagy inducer DIRAS3 via recruiting the repressive histone H3K27me3 methyltransferase EZH2 and increasing promoter H3K27me3. Thus, H19 downregulation in diabetes can de-repress epigenetic mechanisms to upregulate DIRAS3 and activate autophagy. Results from this study suggest that overexpression of H19 could be an approach to alleviate progression of DCM (66).
LncRNA Meg3 was reported to be enriched in cardiac fibroblasts (CFs) and dysregulated during pressure overload–induced cardiac remodeling. Meg3 knockdown with GapmeRs led to dysregulation of several matrix metalloprotease (MMP)s in CFs. Meg3, a chromatin-associated lncRNA interacted with p53 and this interaction is required for the transcription of Mmp2 promoter in CFs. Accordingly, Meg3 knockdown reduced basal and TGF-β1-induced expression of Mmp2 in CFs. Furthermore, GapmeR mediated knockdown of Meg3 in vivo decreased ECM deposition, cardiomyocyte hypertrophy and improved impaired diastolic function in a mice model of transverse aortic constriction-induced cardiac hypertrophy/heart failure (69). This study did not investigate the role of lncRNA Meg3 in DCM. However, another study reported that HG upregulates MEG3 expression in human cardiomyocytes, and MEG3 knockdown attenuated HG-induced apoptosis. Their mechanistic studies revealed that MEG3 binds to miR-145 and upregulates the miR-145 target PDCD4, which promotes apoptosis in cardiomyocytes. Overall these studies suggest that targeting MEG3 may ameliorate DCM (70).
Another lncRNA Chaer (cardiac-hypertrophy-associated epigenetic regulator) was also found to be essential for the development of cardiac hypertrophy. Chaer induced cardiac hypertrophy by interacting with Ezh2 (component of the repressive PRC2 complex) via a 66-mer motif. This interaction inhibits PRC2 function and reduces deposition of repressive epigenetic mark H3K27me3 at the promoter regions of genes mediating hypertrophy. Inhibition of Chaer before the onset of pressure overload attenuated cardiac hypertrophy and dysfunction in mice. However, the role of lncRNA Chaer in DCM was not investigated (71).
Interestingly, circulating levels of the mitochondrial lncRNA LIPCAR (long intergenic non-coding RNA predicting cardiac remodeling) expression were downregulated early in patients with myocardial infarction but increased in later stages. LIPCAR expression was associated with chronic heart failure and its expression inversely correlated with diastolic function in T2D patients (67, 68). Furthermore, changes in circulating lncRNAs MIAT and SENCR were also associated with cardiac remodeling (i.e. changes in the size, shape, structure, and function of the heart) in patients with T2D (68). Overall, these reports support a role for lncRNAs in the regulation of fibrosis, inflammation, and hypertrophy in cardiac cells related to cardiac function. Further studies are needed to clarify utility of these lncRNAs as biomarkers and therapeutic targets in DCM.
The Role of LncRNAs in Diabetic Nephropathy
Diabetic nephropathy (DN), a leading cause of renal failure, is characterized by micro- and macro-albuminuria and fibrosis in most renal cells and progressive renal dysfunction. Diabetes-induced HG and TGF-β1 regulate lncRNAs involved in DN. Often HG mediates its effect via TGF-β1, a potent profibrotic factor augmented in the diabetic kidney, and a major player in DN pathology. TGF-β1 induced activation of endoplasmic reticulum (ER) stress, PI3K/AKT signaling, and Smad TFs play key roles in expression of genes associated with DN (18). In recent years, multiple HG- and/or TGF-β1-induced lncRNAs have been shown to regulate genes associated with renal function, fibrosis, apoptosis, autophagy, inflammation, and ER stress in key affected renal cells like mesangial cells, tubular epithelial cells, endothelial cells, and podocytes, and therefore implicated in DN (18) (Table 1 and Figure 5).
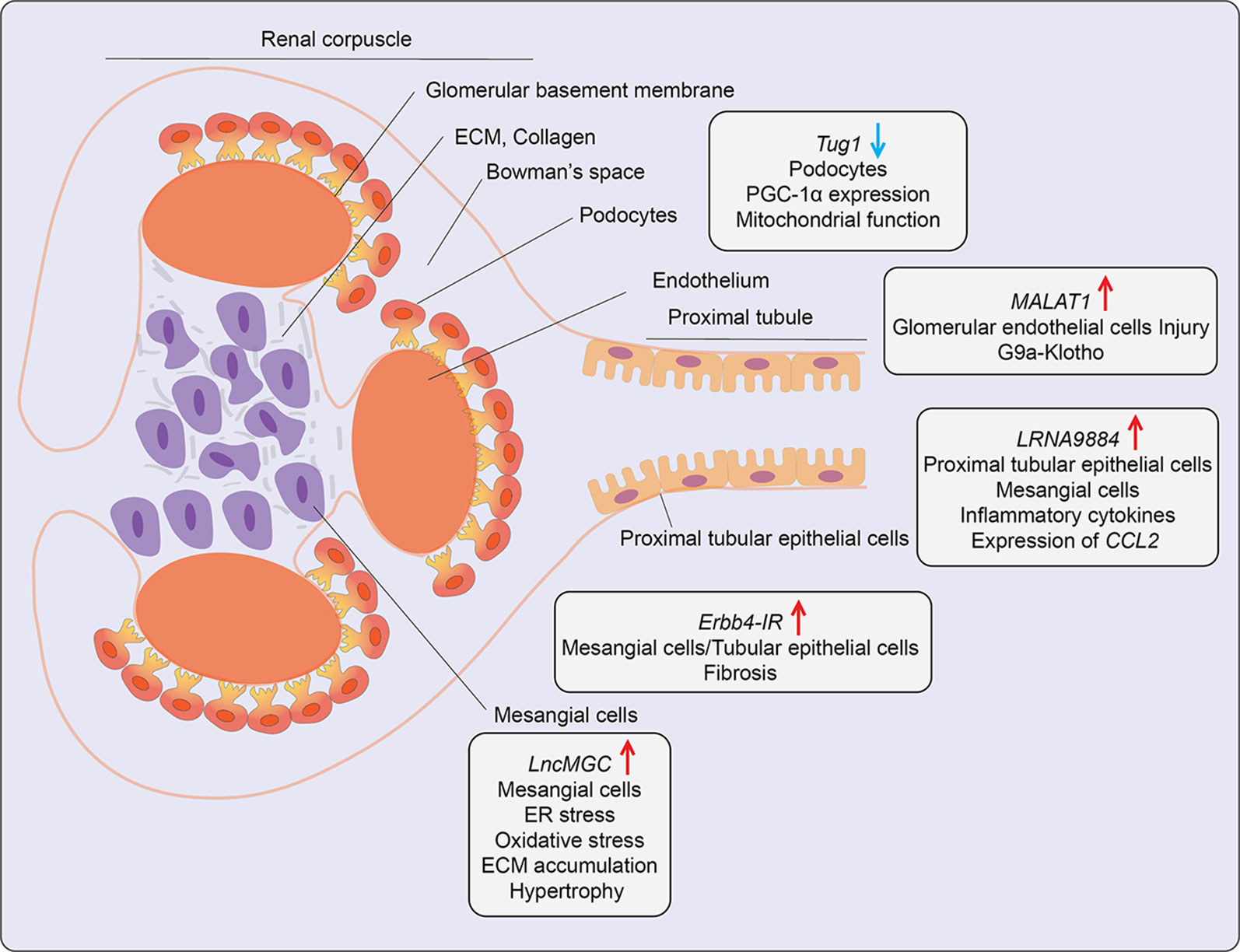
Figure 5 Role of LncRNAs in diabetic nephropathy (DN). A schematic showing the roles of candidate lncRNAs with functions in the indicated kidney cell types during development of diabetic nephropathy. Arrows indicate the direction of changes in the expression or function. ER, Endoplasmic Reticulum; CCL2, C-C Motif Chemokine Ligand 2; ECM, Extracellular matrix; G9a, Histone H3-lysine 9-dimethyl-transferase; PGC-1α, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha.
A lncRNA known as lncRNA megacluster (lnc-MGC) was found to play a key role in the pathogenesis of early DN (72). Lnc-MGC is a host gene for miR-379 cluster, i. e., harbors miRNAs in its exonic and intronic sequences and these non-coding RNAs are present within one of the largest miRNA clusters in the genome. The expression of lnc-MGC and key component mature cluster miRNAs located within lnc-MGC were upregulated in the renal glomeruli of T1D and T2D mice (72). Both HG and TGF-β1 increased the expression of lnc-MGC and several cluster miRNAs located within it, such as miR-379, miR-494, miR-495, and miR-377 in mouse renal mesangial cells via the ER stress- related TF CHOP in collaboration with Smad2/3 TFs. The cluster miRNAs derived from lnc-MGC, including miR379 were observed to regulate target genes involved in various functions related to DN, such as protein synthesis, ER stress, RNA binding, and protein translation. Notably, targeting lnc-MGC using LNA-modified GapmeR antisense oligos in vivo in diabetic mice downregulated lnc-MGC and key cluster miRNAs, including miR-379, but upregulated miR-379 targets, such as ER Degradation Enhancer, Mannosidase Alpha-Like 3 (Edem3), a regulator of ER stress. In parallel, this GapmeR also ameliorated diabetes-induced expression of profibrotic genes Tgfβ1, Col1a2, Col4a1, and Ctgf in renal glomeruli and reduced key features of early DN in mice (72). Moreover, miR-379 knockout mice created with CRISPR-Cas9 editing were also protected from DN as shown by decreases in renal fibrosis, hypertrophy, albuminuria, and podocyte dysfunction (104). Since a GapmeR targeting human ortholog of lnc-MGC was effective in reducing fibrosis in human renal cells, these data highlight the translational potential of targeting pathological renal ncRNAs like lnc-MGC or miR-379 for the treatment of DN. In other studies, GWAS identified the association of lncRNA PVT1(plasmacytoma variant translocation1) locus with end-stage renal disease attributed to T1D and T2D (105, 106). Experimental studies demonstrated that lncRNA PVT1 plays a key role in HG-induced fibrotic genes in mesangial cells, possibly via acting as host gene for miRNAs such as miR-1207-5p (76), further supporting the utility of targeting miRNAs hosted within lncRNAs for DN treatment.
Podocyte apoptosis and effacement are key pathological events associated with glomerular dysfunction and proteinuria in DN. The role of lncRNAs in podocyte dysfunction has been evaluated. One report demonstrated that lncRNA Tug1 expression was downregulated in glomerular podocytes from kidneys of T2D db/db mice relative to control mice and in glomeruli from human subjects with DN (73). Further studies demonstrated that HG suppresses Tug1 expression via recruitment of TF Carbohydrate response element-binding protein (ChREBP) and multiple co-repressors to the Tug1 promoter (74). Tug1 was mostly localized in the nucleus and upregulated peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α), a master transcriptional regulator of mitochondrial biogenesis. Tug1 was shown to regulate Ppargc1a expression through interaction with chromatin at Tug1 binding sites in the upstream region (73). Tug1 overexpression improved mitochondrial function in podocytes of diabetic mice. Thus, Tug1 downregulation in diabetes appears to inhibit the expression of PGC-1α and its target genes involved in mitochondrial biogenesis, resulting in podocyte apoptosis and glomerular dysfunction. Another report showed lncRNA9884 is upregulated in kidneys of diabetic db/db mice, and its expression was positively correlated with DN progression (75). This lncRNA was primarily expressed in glomerular mesangial and tubular epithelial cells and induced in the diabetic milieu via Smad3 binding to its promoter. Downregulation of kidney-specific lncRNA9884 in db/db mice reduced glomerular dysfunction, fibrosis, and proteinuria independent of body weight and blood glucose levels. Mechanistically, nuclear-enriched lncRNA9884 directly interacts with chromatin at the promoters of inflammatory genes such as Ccl2 and increases their expression (75). The detailed mechanisms regulating this lncRNA in DN, as well as its human relevance, are not clear.
The role of AGEs and their actions via their receptor (RAGE) signaling drives DN pathogenesis. AGEs induced a novel lncRNA Erbb4-IR in a Smad3-dependent manner in mouse mesangial cells and tubular epithelial cells. Erbb4-IR induced fibrotic genes like collagen via sponging the miR-29b (which targets collagens) in mesangial cells and tubular epithelial cells. Expression of Erbb4-IR was also increased in db/db mice, and its knockdown in vivo ameliorated features of DN, including microalbuminuria, creatinine, and fibrosis (77). LncRNA MALAT1 was also upregulated (78). Interestingly, nuclear lncRNA MALAT1 expression was also upregulated in kidneys from DN patients and glomeruli from DN mice. It correlated with reduced levels of Klotho, a component of the endocrine fibroblast growth factor (FGF) receptor complex. Reduced levels of Klotho are implicated in renal disease (107). A similar inverse relationship between MALAT1 and Klotho was observed in HG-treated human renal glomerular endothelial cells (HRGECs), human renal tubular epithelial cells (HK2), and podocytes (78). Furthermore, MALAT1 overexpression in HRGECs promoted glucose-induced endothelial cell injury by inhibiting Klotho expression via enrichment of G9a and corresponding repressive histone modifications at its promoter.
It is thus becoming increasingly evident that many lncRNAs have critical functions in multiple renal cells, and their dysregulation in diabetes can modulate DN progression. Targeting critical renal lncRNAs with various RNA targeting modalities can confer protection against DN in mice, but more work is needed to extend these approaches for human DN treatment.
Role of LncRNAs in Diabetic Retinopathy
Diabetic retinopathy (DR) is a major microvascular complication affecting a significant proportion of diabetic individuals and is a major cause of blindness. Dysfunction of retinal ECs and microvascular tissue homeostasis induced by HG, hypoxia, and growth factors are key processes in diabetes-induced retinal damage and retinopathies (108). Several lncRNAs associated with microvascular endothelial dysfunction have been described (Table 1 and Figure 6). Expression of lncRNA MIAT was upregulated in retinal tissues from diabetic humans, rat, and mice and in retinal ECs treated with HG (81). Moreover, knockdown of MIAT decreased apoptosis, inflammation, and phosphorylation of AKT kinase. Studies in cultured ECs showed that MIAT acts as a miR-150-5p sponge and increases the expression of miR-150-5p target, vascular endothelial growth factor (VEGF), a potent angiogenic factor that promotes retinopathy. Further, intraocular injection of siRNAs targeting MIAT ameliorated DR in diabetic rats, possibly through MIAT knockdown in ECs as well as non-ECs. Since intraocular injections with anti-VEGF antibodies are already in clinical use, in the future, combination therapy with lncRNA-based oligonucleotides could be evaluated for potential superior beneficial effects, especially for patients refractory to anti-VEGF antibodies.
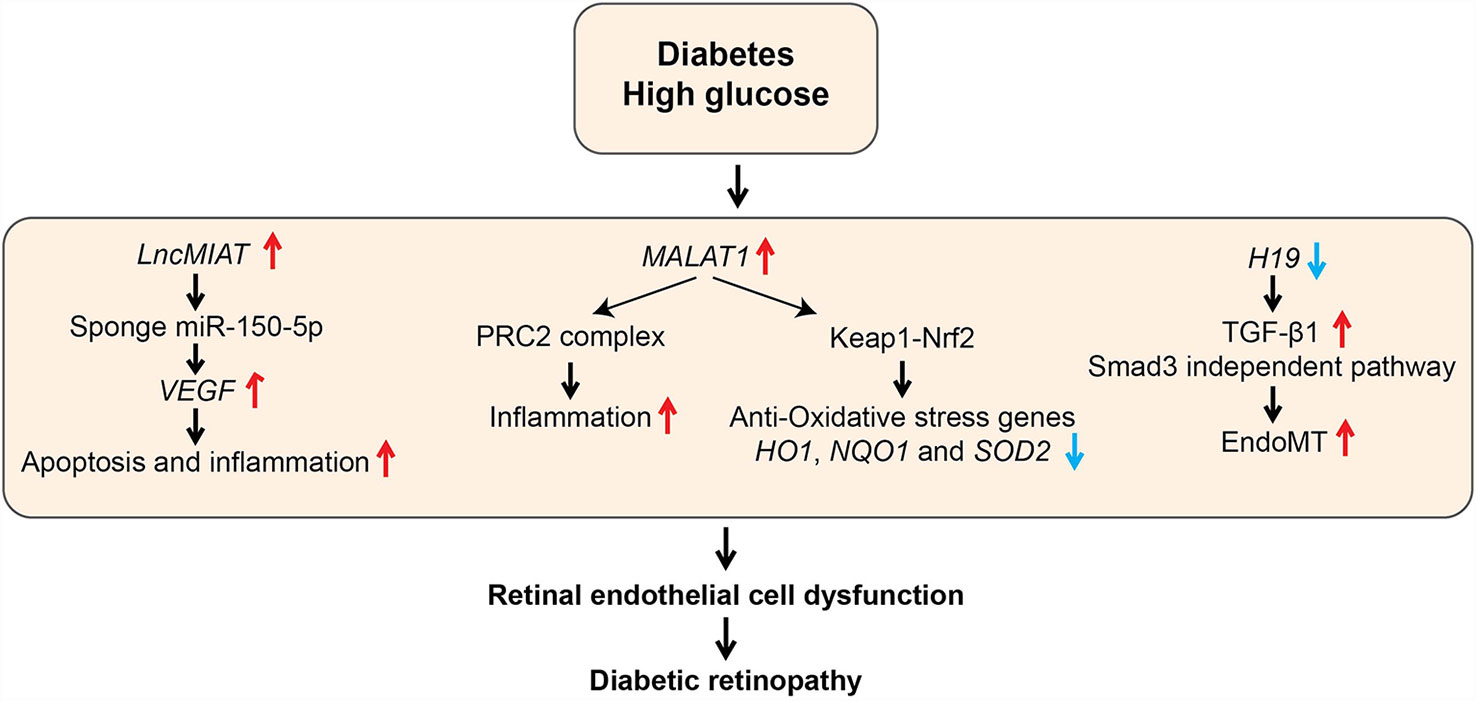
Figure 6 Role of LncRNAs in diabetic retinopathy (DR). The schematic diagram shows the processes involved in the regulation of genes mediated by candidate lncRNAs during various pathological processes associated with DR. EndoMT, Endothelial to mesenchymal transition; HO1, Heme oxygenase-1; Keap1, Kelch Like ECH Associated Protein 1; NQO1, NAD(P)H Quinone Dehydrogenase 1; Nrf2, Nuclear factor erythroid 2-related factor2; PRC2, Polycomb Repressive Complex 2; SOD2, Superoxide dismutase 2; VEGF, Vascular Endothelial Growth Factor.
LncRNA MALAT1 was also demonstrated to have a role in inflammation and retinal EC dysfunction associated with DR. Malat1 was upregulated in retinas of rats with DR, and its knockdown with short hairpin RNA ameliorated retinal inflammation and retinal vessel impairment in these rats (109). Furthermore, MALAT1 was also upregulated by HG along with inflammatory cytokines in human retinal ECs (HRECs), which was abolished by MALAT1 knockdown with GapmeRs (79). In this study, the role of complex epigenetic mechanisms, including MALAT1 interaction with EZH2 and dysregulation of DNA methyltransferases such as DNMT1 have been implicated in MALAT1-dependent and -independent production of HG-induced inflammatory genes in HRECs (79). However, further studies are needed to clarify the precise role of these epigenetic mechanisms in the pathogenesis of human DR. Recent studies showed that MALAT1 also plays a central role in inhibiting the anti-oxidant defense pathway to promote inflammation by regulating nuclear factor erythroid 2-related factor2 (NRF2), a master regulator of the anti-oxidant defense pathway that protects retinas from oxidative stress. NRF2 function is negatively regulated by Kelch-like ECH-associated protein 1 (KEAP1). But, KEAP1 levels are increased in DR, which enhances oxidative stress and retinal cell death (110). HG upregulated MALAT1 via transcriptional activation by Sp1 in HRECs. In parallel, HG also increased KEAP1 and inhibited nuclear translocation of Nrf2 as well as transcription of anti-oxidant genes HO1 and SOD2. MALAT1 knockdown with siRNAs abrogated all these HG-induced effects in HRECs (80). Furthermore, similar changes in MALAT1, KEAP1, and anti-oxidant genes were also demonstrated in retinal microvessels from diabetic rats and humans with DR (80). These studies collectively support the notion that MALAT1 promotes inflammation and oxidative stress in DR, and MALAT1 could be a viable target for DR treatment.
Endothelial to mesenchymal transition (EndoMT) also plays an essential role in EC dysfunction and development of DR. EndoMT can be induced by TGF-β signaling, oxidative stress, and hypoxia, and leads to loss of EC phenotype and acquisition of mesenchymal cell features, including increased inflammatory cytokines, proliferation, and migration. Accumulating evidence suggests a role of lncRNAs like H19 in EndoMT in the pathogenesis of DR. H19 was downregulated in retinas of humans with DR and H19 knockout could exacerbate DR in mice. Mechanistic studies in retinal ECs showed that downregulation of H19 by HG promotes TGF-β1 -induced EndoMT. In contrast, H19 overexpression inhibited EndoMT via downregulation of TGF-β1 expression (82). These results suggest protective functions for H19 in DR. Dysregulation of lncRNAs MEG3 (Maternally Expressed Gene 3) and ANRIL (Antisense Non-coding RNA in the INK4 Locus) in retinal cells also contributed to increased inflammation in DR. However, detailed mechanisms of action of these lncRNAs were not examined (83–85). These reports, taken together, illustrate the involvement of lncRNAs in controlling retinal microvascular EC homeostasis and pathology in DN.
Approaches to Target LncRNAs In Vivo
As detailed in the earlier sections, it is clear that dysregulated expression and functions of multiple lncRNAs play pivotal roles in the pathogenesis of various diabetic vascular complications. Thus, developing therapies targeting lncRNAs and their key effector molecules can have tremendous translational potential. Recently, RNA-based therapies have been gaining a lot of momentum in general, but relatively much less progress has been made in the field of diabetes complications. Unlike miRNAs that can be targeted quite easily in vitro and in vivo, targeting lncRNAs can be more challenging, especially in vivo, due to their relatively large size, complex structure, low expression, tissue/cell specificity, and low conservation across species. The expression of lncRNAs can be modulated using RNA interference (RNAi) with siRNAs, small antisense oligos (ASOs), or CRISPR-Cas9 based genome editing of lncRNA loci. Endogenous lncRNA expression can be enhanced using small activating RNAs (saRNAs) or by guiding transcriptional activators to lncRNA gene promoters using CRISPR-Cas9 genome editing (111–113).
RNAi approaches utilize siRNAs, which are 20–25 nucleotides long double-stranded ribonucleotides that, after delivery into the cytoplasm, are loaded into the RNA-induced silencing complex to induce target RNA degradation. ASO approaches mostly use LNA-modified GapmeRs, 16-nucleotide long single-stranded oligos that are DNA-RNA hybrids containing a central DNA sequence (Gap) flanked by LNA nucleotides (72, 114, 115). The central DNA hybridizes with the complementary sequence in the target lncRNA and initiates the cleavage of target lncRNA by RNase H-dependent mechanisms (114). LNA modifications increase affinity and base pairing specificity of the GapmeRs, while phosphorothioate backbones confer resistance to degradation by endogenous nucleases (114). In contrast, saRNAs are used to overexpress endogenous lncRNAs or to restore normal levels when they are downregulated under different disease states. saRNAs are 21-nucleotide long double-stranded RNAs with two nucleotide overhangs and contain complementary sequences to the promoter regions. After delivery into cells, they are loaded into the saRNA-AGO2 complex, which enters the nucleus and binds to the target promoter and activates transcription (113). Each of these methods has its own merits and pitfalls, and lncRNA knockdown efficiency is also influenced by subcellular localization. siRNAs are, in general, very efficient, and design algorithms have been well developed, but they may not be as efficient as GapmeRs to knockdown nuclear lncRNAs. On the other hand, LNA GapmeRs working via RNase H mechanisms can efficiently target both nuclear and cytoplasmic lncRNAs. In addition, dual localized lncRNAs may require both siRNAs and GapmeRs for efficient knockdown (115). However, GapmeR design algorithms are less well developed compared to siRNA design; therefore, they require testing of several GapmeRs before an optimal one can be selected. In addition, a recent study showed that targeting lncRNAs with GapmeRs at the 5’ sequence could lead to immature transcriptional termination (116). In addition, dual localized lncRNAs may require both siRNAs and GapmeRs for efficient knockdown. Recently, siRNA-based therapies have reached clinical development; the chemically modified siRNA targeting PCSK9 (Inclisiran) for CVD showed an efficient reduction in LDL levels in Phase II clinical trials (117). As described earlier in this review, GapmeRs targeting lncRNAs have been used in animal models of diabetes and in human cells to ameliorate features of diabetic complications (72, 118, 119), thus, providing proof of principle for future evaluation in human clinical trials. However, extensive studies are needed to determine safe and efficient delivery vehicles and organ-, tissue-, or cell-specific accumulation of GapmeRs to reduce off-target effects before they are evaluated in clinical trials.
CRISPR-Cas9 editing is increasingly emerging as a viable approach to address some concerns with RNA therapies while also providing potentially long-term gene editing at specific loci. CRISPR-Cas9 editing can be used for a partial or complete deletion of the lncRNA locus. Locus-specific CRISPR activation (CRISPRa) or interference (CRISPRi) methods are also widely used to overexpress or inhibit lncRNA expression, respectively. In these approaches, catalytically dead Cas9 nuclease is fused with transcriptional activator or repressor proteins, which are directed to specific lncRNA loci by guide RNA (gRNA)s (111, 120). The CRISPRa or CRISPRi modifies the chromatin environment at the promoters, leading to activation or repression of lncRNAs transcription. However, CRISPR-Cas9 editing also has its own limitations (121), including off-target effects, but recent technical advances have significantly improved specificity (121). Other challenges include difficulty in achieving cell-type-specific targeting and the possibility of unanticipated side effects due to long-term alteration of lncRNA expression. Furthermore, unlike coding genes, deleting the first exon of a lncRNA gene may not silence specific lncRNA completely, and alternate transcripts with similar activity may still be expressed. Since lncRNAs with multiple exons may occupy large genomic regions, and deletion of the entire locus may elicit unintended deleterious effects. Additionally, targeting bidirectional and lncRNAs overlapping with the promoter of a neighboring coding gene via CRISPR-Cas9 editing may inadvertently deregulate neighboring genes leading to undesirable effects. For such lncRNAs, RNAi or ASO-mediated knockdown approach would be more appropriate (122). However, there has been significant recent progress in the direct clinical application of CRISPR-Cas9 editing (123). These reports offer great promise for CRISPR-based therapy to correct defective coding or ncRNAs for human diseases, including diabetic complications.
Of note, modulation of lncRNA expression can also be achieved via transfecting lncRNA transcripts into cells or via injecting viral vectors into the mice. Recombinant adeno-associated virus (rAAV)-based gene therapy is now gaining interest and in various stages of clinical trials for the treatment of multiple diseases (124). The viral vector transgenes up to ~5 kb flanked by AAV inverted terminal repeats are inserted into rAAV genomes (125). The hepatic delivery of lncRNA Lexis in mice models of hypercholesterolemia and atherosclerosis to overexpress Lexis led to a significant reduction of hypercholesterolemia and ameliorated atherosclerosis burden versus control vector treated mice (126). These reports demonstrate the feasibility of lncRNA mimetic therapeutic strategy for diabetic CVDs, such as atherosclerosis and possibly other diabetes complications. One advantage of rAAVs is that, unlike adenoviral vectors, they are less immunogenic and widely used in in vivo settings (124). Most of these methods have been applied for targeting the mRNA or DNA sequence of a single gene implicated in monogenic diseases. However, extending them for the treatment of polygenic, multifactorial diseases and complex disorders, like diabetes and its complications, requires further investigation of precise cell-specific targeting approaches, optimal design for enhanced bioavailability, reduced toxicity, and efficient delivery into target tissues.
Summary and Perspectives
LncRNAs are emerging as not only important regulatory RNAs that function in many cellular processes during normal states but also as critical modulators of disease states. They can play significant roles in the pathology of diabetic complications by regulating genes that affect inflammation, fibrosis, ER stress, oxidative stress, and mitochondrial dysfunction. With rapid advances in sequencing technologies, transcriptomics, and epigenomics, as well as bioinformatic tools, we will undoubtedly continue to identify more lncRNAs involved in the development of diabetic complications. Advances in single-cell sequencing will be invaluable to identify lncRNAs expressed in distinct cell types in diabetes, which, in turn, would enhance the development of targeted therapies. Interestingly, data analysis of 10 transcriptomes from granulocytes from different individuals revealed that lncRNAs expression varies amongst individuals at levels that are greater than protein-coding genes (127). It will be useful to understand whether expression of a specific lncRNA is associated with an individual’s susceptibility to diabetic complications. Additionally, valuable catalogs of lncRNAs expressed in normal cells and tissues are available (128). These data sets can be compared to datasets from transcriptomic analyses of cells and tissues from diabetic patients to uncover dysregulated lncRNAs to gain more insights into their function in diabetic vascular complications.
In addition to dysregulated lncRNAs, genetic variation within lncRNA regions can modulate function of lncRNAs and drive disease pathogenesis. Most of the genetic variants associated with cardiometabolic traits and diabetes are present in non-coding regions of the genome. Several hundreds of lncRNA regions harbor disease-associated single nucleotide polymorphisms (SNPs), suggesting the importance of these lncRNA regions in disease traits (129). A recent study mapped regulatory variants that modulate DNA binding of the coronary artery disease-associated TF TCF21 (130). Integration of epigenome-wide association studies (EWAS) and GWAS identified several genetic variations that correlated with epigenetic mechanisms associated with TCF21 binding in human coronary artery VSMC (130, 131). Because, TCF21 function is also dysregulated in diabetes complications, these and other reports suggest a link between genetic and epigenetic variations in modulating vascular complications (131, 132). Similar studies with SNPs in lncRNA loci and cis-regulatory elements regulated by them can further advance our understanding of the regulation and functional roles of lncRNAs in diabetes complications. Studies from our group showed that enhancers, super-enhancers, and eRNAs are dysregulated in VSMCs treated with AngII (133). These enhancers harbored SNPs associated with CVDs. Since lncRNAs can regulate the functions of these enhancers and super-enhancers, any genetic variations affecting enhancer activity can alter lncRNA functions. Such chromatin-lncRNA cross-talk and epigenetic mechanisms could be involved in vascular cell dysfunction associated with diabetic CVD. Moreover, although eRNAs are implicated as important regulators of gene expression, their function in diabetes complications is not fully clear. Recent studies showed that enhancer associated lncRNAs such as CARMN and LEENE regulate cardiac cell differentiation and EC function respectively (57, 134). It likely that additional eRNAs will be characterized under diabetic conditions in the upcoming years.
One impediment in extending lncRNA-based studies to the clinic is that lncRNAs are either not expressed in all species or exhibit low sequence conservation across species. For example, lncADAL and CHROME, which regulate adipocyte differentiation and cholesterol homeostasis, respectively, are primarily expressed only in humans. Whereas lnc-MGC is expressed in both mice and humans and have similar functional roles related to DN, but their nucleotide sequences are not highly conserved (53, 72, 135). However, evidence shows that, even in the absence of nucleotide sequence conservation, secondary structures and lncRNA functions can be conserved across species (136). Interestingly, evidence shows non-conserved lncRNAs harbor a significant number of SNPs associated with cardiometabolic traits than conserved lncRNAs, which can enhance our understanding of the roles of key SNPs and lncRNAs in driving disease pathogenesis (129). Moreover, lncRNAs expressed from syntenic gene loci often showed limited sequence conservation but had functional similarities in mice and humans (27, 29, 49, 52). These studies demonstrate the importance of prioritizing not only conserved but also non-conserved lncRNAs for future studies. Notably, a recent study demonstrated that the function of non-conserved human lncRNAs in metabolism can be studied in vivo using a humanized liver mouse model (137). Therefore, investigations are likely to also focus on lncRNA functions and functional lncRNA domains conserved across species through prediction of secondary structure using bioinformatics tools and experimental approaches, such as humanized mice and domain-specific chromatin isolation by RNA purification (dChIRP) (136–138). Identification of lncRNAs harboring genetic variations associated with diabetes complications could provide important insights into the cross-talk between transcriptomics, genetics, and epigenetics underlying diabetic complications that could inform the development of much needed novel therapeutic strategies. Additionally, lncRNA expression profiles in circulating biofluids can yield valuable non-invasive biomarkers for early detection of diabetic complications, a major unmet need in clinical management. With the rapid advances in genomic sequencing, single-cell sequencing, spatial transcriptomics, and RNA targeting approaches, lncRNA research is clearly expected to grow exponentially in the upcoming years.
Author Contributions
VT, MR, and RN conceived and designed the study. VT, MR, and RN wrote and critically revised the manuscript for important intellectual content. VT prepared the figures. All authors contributed to the article and approved the submitted version.
Funding
We gratefully acknowledge funding from the National Institutes of Health (NIDDK and NHLBI) R01 DK065073, R01 HL106089, R01 DK081705, DP3 DK106917, the Schaeffer Foundation, and the Wanek Family Project for the Cure of Type 1 Diabetes at City of Hope.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. International Diabetes Federation. Idf Diabetes Atlas. 9th. Brussels, Belgium: International Diabetes Federation (2019).
2. Forbes JM, Cooper ME. Mechanisms of Diabetic Complications. Physiol Rev (2013) 93:137–88. doi: 10.1152/physrev.00045.2011
3. Nathan DM. Long-Term Complications of Diabetes Mellitus. N Engl J Med (1993) 328:1676–85. doi: 10.1056/NEJM199306103282306
4. Rask-Madsen C, King GL. Vascular Complications of Diabetes: Mechanisms of Injury and Protective Factors. Cell Metab (2013) 17:20–33. doi: 10.1016/j.cmet.2012.11.012
5. Giacco F, Brownlee M. Oxidative Stress and Diabetic Complications. Circ Res (2010) 107:1058–70. doi: 10.1161/CIRCRESAHA.110.223545
6. Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A Glimpse of Various Pathogenetic Mechanisms of Diabetic Nephropathy. Annu Rev Pathol (2011) 6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150
7. Yan SF, Ramasamy R, Schmidt AM. Mechanisms of Disease: Advanced Glycation End-Products and Their Receptor in Inflammation and Diabetes Complications. Nat Clin Pract Endocrinol Metab (2008) 4:285–93. doi: 10.1038/ncpendmet0786
8. Schmidt AM. Diabetes Mellitus and Cardiovascular Disease. Arterioscler Thromb Vasc Biol (2019) 39:558–68. doi: 10.1161/ATVBAHA.119.310961
9. Reddy MA, Zhang E, Natarajan R. Epigenetic Mechanisms in Diabetic Complications and Metabolic Memory. Diabetologia (2015) 58:443–55. doi: 10.1007/s00125-014-3462-y
10. Sullivan KM, Susztak K. Unravelling the Complex Genetics of Common Kidney Diseases: From Variants to Mechanisms. Nat Rev Nephrol (2020) 16:628–40. doi: 10.1038/s41581-020-0298-1
11. Russo VE, Martienssen RA, Riggs AD. Epigenetic Mechanisms of Gene Regulation. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press (1996).
12. Feil R, Fraga MF. Epigenetics and the Environment: Emerging Patterns and Implications. Nat Rev Genet (2012) 13:97–109. doi: 10.1038/nrg3142
13. Allis CD, Jenuwein T. The Molecular Hallmarks of Epigenetic Control. Nat Rev Genet (2016) 17:487–500. doi: 10.1038/nrg.2016.59
14. Zhou VW, Goren A, Bernstein BE. Charting Histone Modifications and the Functional Organization of Mammalian Genomes. Nat Rev Genet (2011) 12:7–18. doi: 10.1038/nrg2905
15. Chen ZX, Riggs AD. DNA Methylation and Demethylation in Mammals. J Biol Chem (2011) 286:18347–53. doi: 10.1074/jbc.R110.205286
16. Morris KV, Mattick JS. The Rise of Regulatory RNA. Nat Rev Genet (2014) 15:423–37. doi: 10.1038/nrg3722
17. Long Y, Wang X, Youmans DT, Cech TR. How do lncRNAs Regulate Transcription? Sci Adv (2017) 3:eaao2110. doi: 10.1126/sciadv.aao2110
18. Kato M, Natarajan R. Epigenetics and Epigenomics in Diabetic Kidney Disease and Metabolic Memory. Nat Rev Nephrol (2019) 15:327–45. doi: 10.1038/s41581-019-0135-6
19. Nilsson E, Ling C. DNA Methylation Links Genetics, Fetal Environment, and an Unhealthy Lifestyle to the Development of Type 2 Diabetes. Clin Epigenet (2017) 9:105. doi: 10.1186/s13148-017-0399-2
20. Rodriguez H, El-Osta A. Epigenetic Contribution to the Development and Progression of Vascular Diabetic Complications. Antioxid Redox Signal (2018) 29:1074–91. doi: 10.1089/ars.2017.7347
21. Das S, Reddy MA, Natarajan R. Role of Epigenetic Mechanisms Regulated by Enhancers and Long Noncoding RNAs in Cardiovascular Disease. Curr Opin Cardiol (2020) 35:234–41. doi: 10.1097/HCO.0000000000000728
22. Mercer TR, Mattick JS. Structure and Function of Long Noncoding RNAs in Epigenetic Regulation. Nat Struct Mol Biol (2013) 20:300–7. doi: 10.1038/nsmb.2480
23. Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative Annotation of Human Large Intergenic Noncoding RNAs Reveals Global Properties and Specific Subclasses. Genes Dev (2011) 25:1915–27. doi: 10.1101/gad.17446611
24. Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, et al. Ab Initio Reconstruction of Cell Type-Specific Transcriptomes in Mouse Reveals the Conserved Multi-Exonic Structure of Lincrnas. Nat Biotechnol (2010) 28:503–10. doi: 10.1038/nbt.1633
25. Hartford CCR, Lal A. When Long Noncoding Becomes Protein Coding. Mol Cell Biol (2020) 40:e00528–19. doi: 10.1128/MCB.00528-19
26. Kang YJ, Yang DC, Kong L, Hou M, Meng YQ, Wei L, et al. CPC2: A Fast and Accurate Coding Potential Calculator Based on Sequence Intrinsic Features. Nucleic Acids Res (2017) 45:W12–6. doi: 110.1093/nar/gkx428
27. Das S, Reddy MA, Senapati P, Stapleton K, Lanting L, Wang M, et al. Diabetes Mellitus-Induced Long Noncoding Rna Dnm3os Regulates Macrophage Functions and Inflammation Via Nuclear Mechanisms. Arterioscler Thromb Vasc Biol (2018) 38:1806–20. doi: 10.1161/ATVBAHA.117.310663
28. Das S, Zhang E, Senapati P, Amaram V, Reddy MA, Stapleton K, et al. A Novel Angiotensin II-Induced Long Noncoding Rna Giver Regulates Oxidative Stress, Inflammation, and Proliferation in Vascular Smooth Muscle Cells. Circ Res (2018) 123:1298–312. doi: 10.1161/CIRCRESAHA.118.313207
29. Stapleton K, Das S, Reddy MA, Leung A, Amaram V, Lanting L, et al. Novel Long Noncoding RNA, Macrophage Inflammation-Suppressing Transcript (Mist), Regulates Macrophage Activation During Obesity. Arterioscler Thromb Vasc Biol (2020) 40:914–28. doi: 10.1161/ATVBAHA.119.313359
30. Leung A, Trac C, Jin W, Lanting L, Akbany A, Saetrom P, et al. Novel Long Noncoding RNAs are Regulated by Angiotensin II in Vascular Smooth Muscle Cells. Circ Res (2013) 113:266–78. doi: 10.1161/CIRCRESAHA.112.300849
31. Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin Signature Reveals Over a Thousand Highly Conserved Large non-Coding RNAs in Mammals. Nature (2009) 458:223–7. doi: 10.1038/nature07672
32. Kung JT, Colognori D, Lee JT. Long Noncoding RNAs: Past, Present, and Future. Genetics (2013) 193:651–69. doi: 10.1534/genetics.112.146704
33. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE V7 Catalog of Human Long Noncoding RNAs: Analysis of Their Gene Structure, Evolution, and Expression. Genome Res (2012) 22:1775–89. doi: 10.1101/gr.132159.111
34. Kopp F, Mendell JT. Functional Classification and Experimental Dissection of Long Noncoding Rnas. Cell (2018) 172:393–407. doi: 10.1016/j.cell.2018.01.011
35. Noh JH, Kim KM, McClusky WG, Abdelmohsen K, Gorospe M. Cytoplasmic Functions of Long Noncoding Rnas. Wiley Interdiscip Rev RNA (2018) 9:e1471. doi: 10.1002/wrna.1471
36. Engreitz JM, Ollikainen N, Guttman M. Long non-Coding RNAs: Spatial Amplifiers That Control Nuclear Structure and Gene Expression. Nat Rev Mol Cell Biol (2016) 17:756–70. doi: 10.1038/nrm.2016.126
37. Statello L, Guo CJ, Chen LL, Huarte M. Gene Regulation by Long non-Coding RNAs and its Biological Functions. Nat Rev Mol Cell Biol (2021) 22:96–118. doi: 10.1038/s41580-020-00315-9
38. Murray PJ, Wynn TA. Protective and Pathogenic Functions of Macrophage Subsets. Nat Rev Immunol (2011) 11:723–37. doi: 10.1038/nri3073
39. Serhan CN, Levy BD. Resolvins in Inflammation: Emergence of the Pro-Resolving Superfamily of Mediators. J Clin Invest (2018) 128:2657–69. doi: 10.1172/JCI97943
40. Serhan CN, Savill J. Resolution of Inflammation: The Beginning Programs the End. Nat Immunol (2005) 6:1191–7. doi: 10.1038/ni1276
41. Fullerton JN, Gilroy DW. Resolution of Inflammation: A New Therapeutic Frontier. Nat Rev Drug Discov (2016) 15:551–67. doi: 10.1038/nrd.2016.39
42. Das S, Shah R, Dimmeler S, Freedman JE, Holley C, Lee JM, et al. Noncoding RNAs in Cardiovascular Disease: Current Knowledge, Tools and Technologies for Investigation, and Future Directions: A Scientific Statement From the American Heart Association. Circ Genom Precis Med (2020) 13:e000062. doi: 10.1161/HCG.0000000000000062
43. Huarte M. The Emerging Role of lncRNAs in Cancer. Nat Med (2015) 21:1253–61. doi: 10.1038/nm.3981
44. Kuznetsova T, Prange KHM, Glass CK, de Winther MPJ. Transcriptional and Epigenetic Regulation of Macrophages in Atherosclerosis. Nat Rev Cardiol (2020) 17:216–28. doi: 10.1038/s41569-019-0265-3
45. Murray PJ. On Macrophage Diversity and Inflammatory Metabolic Timers. Nat Rev Immunol (2020) 20:89–90. doi: 10.1038/s41577-019-0260-2
46. Glass CK, Natoli G. Molecular Control of Activation and Priming in Macrophages. Nat Immunol (2016) 17:26–33. doi: 10.1038/ni.3306
47. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers (2019) 5:56. doi: 10.1038/s41572-019-0106-z
48. Sugiyama T, Yamamoto E, Bryniarski K, Xing L, Fracassi F, Lee H, et al. Coronary Plaque Characteristics in Patients With Diabetes Mellitus Who Presented With Acute Coronary Syndromes. J Am Heart Assoc (2018) 7:e009245. doi: 10.1161/JAHA.118.009245
49. Reddy MA, Chen Z, Park JT, Wang M, Lanting L, Zhang Q, et al. Regulation of Inflammatory Phenotype in Macrophages by a Diabetes-Induced Long Noncoding RNA. Diabetes (2014) 63:4249–61. doi: 10.2337/db14-0298
50. Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge RM, Chang HY. A Mammalian Pseudogene lncRNA at the Interface of Inflammation and Anti-Inflammatory Therapeutics. Elife (2013) 2:e00762. doi: 10.7554/eLife.00762
51. Zgheib C, Hodges MM, Hu J, Liechty KW, Xu J. Long non-Coding RNA Lethe Regulates Hyperglycemia-Induced Reactive Oxygen Species Production in Macrophages. PloS One (2017) 12:e0177453. doi: 10.1371/journal.pone.0177453
52. Reddy MA, Amaram V, Das S, Tanwar VS, Ganguly R, Wang M, et al. Lncrna DRAIR is Downregulated in Diabetic Monocytes and Modulates Inflammatory Phenotype Via Epigenetic Mechanisms. JCI Insight (2021) 6:e143289. doi: 10.1172/jci.insight.143289
53. Hennessy EJ, van Solingen C, Scacalossi KR, Ouimet M, Afonso MS, Prins J, et al. The Long Noncoding RNA CHROME Regulates Cholesterol Homeostasis in Primate. Nat Metab (2019) 1:98–110. doi: 10.1038/s42255-018-0004-9
54. Sallam T, Jones M, Thomas BJ, Wu X, Gilliland T, Qian K, et al. Transcriptional Regulation of Macrophage Cholesterol Efflux and Atherogenesis by a Long Noncoding RNA. Nat Med (2018) 24:304–12. doi: 10.1038/nm.4479
55. Khyzha N, Khor M, DiStefano PV, Wang L, Matic L, Hedin U, et al. Regulation of CCL2 Expression in Human Vascular Endothelial Cells by a Neighboring Divergently Transcribed Long Noncoding RNA. Proc Natl Acad Sci U.S.A. (2019) 116:16410–9. doi: 10.1073/pnas.1904108116
56. Calandrelli R, Xu L, Luo Y, Wu W, Fan X, Nguyen T, et al. Stress-Induced RNA-chromatin Interactions Promote Endothelial Dysfunction. Nat Commun (2020) 11:5211. doi: 10.1038/s41467-020-18957-w
57. Miao Y, Ajami NE, Huang TS, Lin FM, Lou CH, Wang YT, et al. Enhancer-Associated Long non-Coding RNA LEENE Regulates Endothelial Nitric Oxide Synthase and Endothelial Function. Nat Commun (2018) 9:292. doi: 10.1038/s41467-017-02113-y
58. Man HSJ, Sukumar AN, Lam GC, Turgeon PJ, Yan MS, Ku KH, et al. Angiogenic Patterning by STEEL, an Endothelial-Enriched Long Noncoding RNA. Proc Natl Acad Sci U S A (2018) 115:2401–6. doi: 10.1073/pnas.1715182115
59. Hu YW, Guo FX, Xu YJ, Li P, Lu ZF, McVey DG, et al. Long Noncoding RNA Nexn-AS1 Mitigates Atherosclerosis by Regulating the Actin-Binding Protein NEXN. J Clin Invest (2019) 129:1115–28. doi: 10.1172/JCI98230
60. Leisegang MS, Fork C, Josipovic I, Richter FM, Preussner J, Hu J, et al. Long Noncoding Rna MANTIS Facilitates Endothelial Angiogenic Function. Circulation (2017) 136:65–79. doi: 10.1161/CIRCULATIONAHA.116.026991
61. Neumann P, Jae N, Knau A, Glaser SF, Fouani Y, Rossbach O, et al. The Lncrna GATA6-AS Epigenetically Regulates Endothelial Gene Expression Via Interaction With LOXL2. Nat Commun (2018) 9:237. doi: 10.1038/s41467-017-02431-1
62. Ballantyne MD, Pinel K, Dakin R, Vesey AT, Diver L, Mackenzie R, et al. Smooth Muscle Enriched Long Noncoding RNA (Smilr) Regulates Cell Proliferation. Circulation (2016) 133:2050–65. doi: 10.1161/CIRCULATIONAHA.115.021019
63. Zhou X, Zhang W, Jin M, Chen J, Xu W, Kong X. Lncrna MIAT Functions as a Competing Endogenous RNA to Upregulate DAPK2 by Sponging miR-22-3p in Diabetic Cardiomyopathy. Cell Death Dis (2017) 8:e2929. doi: 10.1038/cddis.2017.321
64. Zheng D, Zhang Y, Hu Y, Guan J, Xu L, Xiao W, et al. Long Noncoding RNA Crnde Attenuates Cardiac Fibrosis Via Smad3-Crnde Negative Feedback in Diabetic Cardiomyopathy. FEBS J (2019) 286:1645–55. doi: 10.1111/febs.14780
65. Zhao L, Li W, Zhao H. Inhibition of Long non-Coding RNA TUG1 Protects Against Diabetic Cardiomyopathy Induced Diastolic Dysfunction by Regulating Mir-499-5p. Am J Transl Res (2020) 12:718–30.
66. Zhuo C, Jiang R, Lin X, Shao M. Lncrna H19 Inhibits Autophagy by Epigenetically Silencing of DIRAS3 in Diabetic Cardiomyopathy. Oncotarget (2017) 8:1429–37. doi: 10.18632/oncotarget.13637
67. Kumarswamy R, Bauters C, Volkmann I, Maury F, Fetisch J, Holzmann A, et al. Circulating Long Noncoding RNA, LIPCAR, Predicts Survival in Patients With Heart Failure. Circ Res (2014) 114:1569–75. doi: 10.1161/CIRCRESAHA.114.303915
68. de Gonzalo-Calvo D, Kenneweg F, Bang C, Toro R, van der Meer RW, Rijzewijk LJ, et al. Circulating Long-non Coding RNAs as Biomarkers of Left Ventricular Diastolic Function and Remodelling in Patients With Well-Controlled Type 2 Diabetes. Sci Rep (2016) 6:37354. doi: 10.1038/srep37354
69. Piccoli MT, Gupta SK, Viereck J, Foinquinos A, Samolovac S, Kramer FL, et al. Inhibition of the Cardiac Fibroblast-Enriched Lncrna Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ Res (2017) 121:575–83. doi: 10.1161/CIRCRESAHA.117.310624
70. Chen Y, Zhang Z, Zhu D, Zhao W, Li F. Long non-Coding RNA MEG3 Serves as a ceRNA for microRNA-145 to Induce Apoptosis of AC16 Cardiomyocytes Under High Glucose Condition. Biosci Rep (2019) 39:BSR20190444. doi: 10.1042/BSR20190444
71. Wang Z, Zhang XJ, Ji YX, Zhang P, Deng KQ, Gong J, et al. The Long Noncoding RNA Chaer Defines an Epigenetic Checkpoint in Cardiac Hypertrophy. Nat Med (2016) 22:1131–9. doi: 10.1038/nm.4179
72. Kato M, Wang M, Chen Z, Bhatt K, Oh HJ, Lanting L, et al. An Endoplasmic Reticulum Stress-Regulated lncRNA Hosting a microRNA Megacluster Induces Early Features of Diabetic Nephropathy. Nat Commun (2016) 7:12864. doi: 10.1038/ncomms12864
73. Long J, Badal SS, Ye Z, Wang Y, Ayanga BA, Galvan DL, et al. Long Noncoding RNA Tug1 Regulates Mitochondrial Bioenergetics in Diabetic Nephropathy. J Clin Invest (2016) 126:4205–18. doi: 10.1172/JCI87927
74. Long J, Galvan DL, Mise K, Kanwar YS, Li L, Poungvarin N, et al. Role for Carbohydrate Response Element-Binding Protein (ChREBP) in High Glucose-Mediated Repression of Long Noncoding RNA Tug1. J Biol Chem (2020) 295:15840–52. doi: 10.1074/jbc.RA120.013228
75. Zhang YY, Tang PM, Tang PC, Xiao J, Huang XR, Yu C, et al. LRNA9884, a Novel Smad3-DependentPromotes Diabetic Kidney Injury in Db/Db Mice Via Enhancing Mcp-1-Dependent Renal Inflammation. Diabetes (2019) 68:1485–98. doi: 10.2337/db18-1075
76. Alvarez ML, DiStefano JK. Functional Characterization of the Plasmacytoma Variant Translocation 1 Gene (PVT1) in Diabetic Nephropathy. PloS One (2011) 6:e18671. doi: 10.1371/journal.pone.0018671
77. Sun SF, Tang PMK, Feng M, Xiao J, Huang XR, Li P, et al. Novel Lncrna Erbb4-IR Promotes Diabetic Kidney Injury in Db/Db Mice by Targeting Mir-29b. Diabetes (2018) 67:731–44. doi: 10.2337/db17-0816
78. Li Y, Ren D, Xu G. Long Noncoding RNA MALAT1 Mediates High Glucose-Induced Glomerular Endothelial Cell Injury by Epigenetically Inhibiting Klotho Via Methyltransferase G9a. IUBMB Life (2019) 71:873–81. doi: 10.1002/iub.2009
79. Biswas S, Thomas AA, Chen S, Aref-Eshghi E, Feng B, Gonder J, et al. Malat1: An Epigenetic Regulator of Inflammation in Diabetic Retinopathy. Sci Rep (2018) 8:6526. doi: 10.1038/s41598-018-24907-w
80. Radhakrishnan R, Kowluru RA. Long Noncoding Rna MALAT1 and Regulation of the Antioxidant Defense System in Diabetic Retinopathy. Diabetes (2021) 70:227–39. doi: 10.2337/db20-0375
81. Yan B, Yao J, Liu JY, Li XM, Wang XQ, Li YJ, et al. lncRNA-MIAT Regulates Microvascular Dysfunction by Functioning as a Competing Endogenous RNA. Circ Res (2015) 116:1143–56. doi: 10.1161/CIRCRESAHA.116.305510
82. Thomas AA, Biswas S, Feng B, Chen S, Gonder J, Chakrabarti S. Lncrna H19 Prevents Endothelial-Mesenchymal Transition in Diabetic Retinopathy. Diabetologia (2019) 62:517–30. doi: 10.1007/s00125-018-4797-6
83. Wei JC, Shi YL, Wang Q. Lncrna ANRIL Knockdown Ameliorates Retinopathy in Diabetic Rats by Inhibiting the NF-kappaB Pathway. Eur Rev Med Pharmacol Sci (2019) 23:7732–9. doi: 10.26355/eurrev_201909_18982
84. Zhao Y, Chen X, Tong XL. Effect of Lncrna MEG3 on Retinopathy in Diabetic Rats Through Regulating Fox01 Expression. Eur Rev Med Pharmacol Sci (2019) 23:9163–70. doi: 10.26355/eurrev_201911_19406
85. Zhang D, Qin H, Leng Y, Li X, Zhang L, Bai D, et al. Lncrna MEG3 Overexpression Inhibits the Development of Diabetic Retinopathy by Regulating TGF-beta1 and VEGF. Exp Ther Med (2018) 16:2337–42. doi: 10.3892/etm.2018.6451
86. Jae N, Heumuller AW, Fouani Y, Dimmeler S. Long non-Coding RNAs in Vascular Biology and Disease. Vascul Pharmacol (2019) 114:13–22. doi: 10.1016/j.vph.2018.03.003
87. Fasolo F, Di Gregoli K, Maegdefessel L, Johnson JL. Non-Coding RNAs in Cardiovascular Cell Biology and Atherosclerosis. Cardiovasc Res (2019) 115:1732–56. doi: 10.1093/cvr/cvz203
88. Leung A, Amaram V, Natarajan R. Linking Diabetic Vascular Complications With Lncrnas. Vasc Pharmacol (2019) 114:139–44. doi: 10.1016/j.vph.2018.01.007
89. Chen YG, Satpathy AT, Chang HY. Gene Regulation in the Immune System by Long Noncoding Rnas. Nat Immunol (2017) 18:962–72. doi: 10.1038/ni.3771
90. Mowel WK, Kotzin JJ, McCright SJ, Neal VD, Henao-Mejia J. Control of Immune Cell Homeostasis and Function by Lncrnas. Trends Immunol (2018) 39:55–69. doi: 10.1016/j.it.2017.08.009
91. den Dekker A, Davis FM, Kunkel SL, Gallagher KA. Targeting Epigenetic Mechanisms in Diabetic Wound Healing. Transl Res (2019) 204:39–50. doi: 10.1016/j.trsl.2018.10.001
92. Boniakowski AE, Kimball AS, Jacobs BN, Kunkel SL, Gallagher KA. Macrophage-Mediated Inflammation in Normal and Diabetic Wound Healing. J Immunol (2017) 199:17–24. doi: 10.4049/jimmunol.1700223
93. Ivshina M, Alexandrov IM, Vertii A, Doxsey S, Richter JD. CPEB Regulation of TAK1 Synthesis Mediates Cytokine Production and the Inflammatory Immune Response. Mol Cell Biol (2015) 35:610–8. doi: 10.1128/MCB.00800-14
94. Lee SD, Tontonoz P. Liver X Receptors at the Intersection of Lipid Metabolism and Atherogenesis. Atherosclerosis (2015) 242:29–36. doi: 10.1016/j.atherosclerosis.2015.06.042
95. Ku KH, Dubinsky MK, Sukumar AN, Subramaniam N, Feasson MYM, Nair R, et al. In Vivo Function of Flow-Responsive Cis-Dna Elements of the Endothelial Nitric Oxide Synthase Gene: A Role for Chromatin-Based Mechanisms. Circulation (2021). doi: 10.1161/CIRCULATIONAHA.120.051078
96. Millanes-Romero A, Herranz N, Perrera V, Iturbide A, Loubat-Casanovas J, Gil J, et al. Regulation of Heterochromatin Transcription by Snail1/LOXL2 During Epithelial-to-Mesenchymal Transition. Mol Cell (2013) 52:746–57. doi: 10.1016/j.molcel.2013.10.015
97. Bell RD, Long X, Lin M, Bergmann JH, Nanda V, Cowan SL, et al. Identification and Initial Functional Characterization of a Human Vascular Cell-Enriched Long Noncoding RNA. Arterioscler Thromb Vasc Biol (2014) 34:1249–59. doi: 10.1161/ATVBAHA.114.303240
98. Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A Necessary Role of miR-221 and miR-222 in Vascular Smooth Muscle Cell Proliferation and Neointimal Hyperplasia. Circ Res (2009) 104:476–87. doi: 10.1161/CIRCRESAHA.108.185363
99. Zhao J, Zhang W, Lin M, Wu W, Jiang P, Tou E, et al. Myoslid Is a Novel Serum Response Factor-Dependent Long Noncoding RNA That Amplifies the Vascular Smooth Muscle Differentiation Program. Arterioscler Thromb Vasc Biol (2016) 36:2088–99. doi: 10.1161/ATVBAHA.116.307879
100. Jia G, Hill MA, Sowers JR. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ Res (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
101. Raut SK, Khullar M. The Big Entity of New Rna World: Long Non-Coding Rnas in Microvascular Complications of Diabetes. Front Endocrinol (Lausanne) (2018) 9:300. doi: 10.3389/fendo.2018.00300
102. Ishii N, Ozaki K, Sato H, Mizuno H, Susumu S, Takahashi A, et al. Identification of a Novel non-Coding RNA, MIAT, That Confers Risk of Myocardial Infarction. J Hum Genet (2006) 51:1087–99. doi: 10.1007/s10038-006-0070-9
103. Qi Y, Wu H, Mai C, Lin H, Shen J, Zhang X, et al. LncRNA-MIAT-Mediated Mir-214-3p Silencing is Responsible for IL-17 Production and Cardiac Fibrosis in Diabetic Cardiomyopathy. Front Cell Dev Biol (2020) 8:243. doi: 10.3389/fcell.2020.00243
104. Kato M, Abdollahi M, Tunduguru R, Tsark W, Chen Z, Wu X, et al. miR-379 Deletion Ameliorates Features of Diabetic Kidney Disease by Enhancing Adaptive Mitophagy Via FIS1. Commun Biol (2021) 4:30. doi: 10.1038/s42003-020-01516-w
105. Millis MP, Bowen D, Kingsley C, Watanabe RM, Wolford JK. Variants in the Plasmacytoma Variant Translocation Gene (PVT1) are Associated With End-Stage Renal Disease Attributed to Type 1 Diabetes. Diabetes (2007) 56:3027–32. doi: 10.2337/db07-0675
106. Hanson RL, Craig DW, Millis MP, Yeatts KA, Kobes S, Pearson JV, et al. Identification of PVT1 as a Candidate Gene for End-Stage Renal Disease in Type 2 Diabetes Using a Pooling-Based Genome-Wide Single Nucleotide Polymorphism Association Study. Diabetes (2007) 56:975–83. doi: 10.2337/db06-1072
107. Lin Y, Kuro-o M, Sun Z. Genetic Deficiency of Anti-Aging Gene Klotho Exacerbates Early Nephropathy in STZ-induced Diabetes in Male Mice. Endocrinology (2013) 154:3855–63. doi: 10.1210/en.2013-1053
108. Wong TY, Cheung CM, Larsen M, Sharma S, Simo R. Diabetic Retinopathy. Nat Rev Dis Primers (2016) 2:16012. doi: 10.1038/nrdp.2016.12
109. Liu JY, Yao J, Li XM, Song YC, Wang XQ, Li YJ, et al. Pathogenic Role of lncRNA-MALAT1 in Endothelial Cell Dysfunction in Diabetes Mellitus. Cell Death Dis (2014) 5:e1506. doi: 10.1038/cddis.2014.466
110. Kowluru RA, Mishra M. Epigenetic Regulation of Redox Signaling in Diabetic Retinopathy: Role of Nrf2. Free Radic Biol Med (2017) 103:155–64. doi: 10.1016/j.freeradbiomed.2016.12.030
111. Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, et al. RNA-Guided Gene Activation by CRISPR-Cas9-based Transcription Factors. Nat Methods (2013) 10:973–6. doi: 10.1038/nmeth.2600
112. Liu SJ, Lim DA. Modulating the Expression of Long non-Coding RNAs for Functional Studies. EMBO Rep (2018) 19:e46955. doi: 10.15252/embr.201846955
113. Kwok A, Raulf N, Habib N. Developing Small Activating RNA as a Therapeutic: Current Challenges and Promises. Ther Deliv (2019) 10:151–64. doi: 10.4155/tde-2018-0061
114. Lim KRQ, Yokota T. Invention and Early History of Gapmers. Methods Mol Biol (2020) 2176:3–19. doi: 10.1007/978-1-0716-0771-8_1
115. Lennox KA, Behlke MA. Cellular Localization of Long non-Coding RNAs Affects Silencing by RNAi More Than by Antisense Oligonucleotides. Nucleic Acids Res (2016) 44:863–77. doi: 10.1093/nar/gkv1206
116. Lee JS, Mendell JT. Antisense-Mediated Transcript Knockdown Triggers Premature Transcription Termination. Mol Cell (2020) 77:1044–.e3. doi: 10.1016/j.molcel.2019.12.011
117. Raal FJ, Kallend D, Ray KK, Turner T, Koenig W, Wright RS, et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N Engl J Med (2020) 382:1520–30. doi: 10.1056/NEJMoa1913805
118. Simion V, Zhou H, Haemmig S, Pierce JB, Mendes S, Tesmenitsky Y, et al. A Macrophage-Specific lncRNA Regulates Apoptosis and Atherosclerosis by Tethering HuR in the Nucleus. Nat Commun (2020) 11:6135. doi: 10.1038/s41467-020-19664-2
119. Haemmig S, Yang D, Sun X, Das D, Ghaffari S, Molinaro R, et al. Long Noncoding RNA SNHG12 Integrates a DNA-PK-mediated DNA Damage Response and Vascular Senescence. Sci Transl Med (2020) 12:eaaw1868. doi: 10.1126/scitranslmed.aaw1868
120. Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided Platform for Sequence-Specific Control of Gene Expression. Cell (2013) 152:1173–83. doi: 10.1016/j.cell.2013.02.022
121. Doudna JA. The Promise and Challenge of Therapeutic Genome Editing. Nature (2020) 578:229–36. doi: 10.1038/s41586-020-1978-5
122. Goyal A, Myacheva K, Gross M, Klingenberg M, Duran Arque B, Diederichs S. Challenges of CRISPR/Cas9 Applications for Long non-Coding RNA Genes. Nucleic Acids Res (2017) 45:e12. doi: 10.1093/nar/gkw883
123. Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, et al. Crispr-Cas9 Gene Editing for Sickle Cell Disease and Beta-Thalassemia. N Engl J Med (2021) 384:252–60. doi: 10.1056/NEJMoa2031054
124. Naso MF, Tomkowicz B, Perry WL 3rd, Strohl WR. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs (2017) 31:317–34. doi: 10.1007/s40259-017-0234-5
125. Berry GE, Asokan A. Cellular Transduction Mechanisms of Adeno-Associated Viral Vectors. Curr Opin Virol (2016) 21:54–60. doi: 10.1016/j.coviro.2016.08.001
126. Tontonoz P, Wu X, Jones M, Zhang Z, Salisbury D, Sallam T. Long Noncoding Rna Facilitated Gene Therapy Reduces Atherosclerosis in a Murine Model of Familial Hypercholesterolemia. Circulation (2017) 136:776–8. doi: 10.1161/CIRCULATIONAHA.117.029002
127. Kornienko AE, Dotter CP, Guenzl PM, Gisslinger H, Gisslinger B, Cleary C, et al. Long non-Coding RNAs Display Higher Natural Expression Variation Than Protein-Coding Genes in Healthy Humans. Genome Biol (2016) 17:14. doi: 10.1186/s13059-016-0873-8
128. Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, et al. An Atlas of Human Long non-Coding RNAs With Accurate 5’ Ends. Nature (2017) 543:199–204. doi: 10.1038/nature21374
129. Foulkes AS, Selvaggi C, Cao T, O’Reilly ME, Cynn E, Ma P, et al. Nonconserved Long Intergenic Noncoding Rnas Associate With Complex Cardiometabolic Disease Traits. Arterioscler Thromb Vasc Biol (2021) 41:501–11. doi: 10.1161/ATVBAHA.120.315045
130. Zhao Q, Dacre M, Nguyen T, Pjanic M, Liu B, Iyer D, et al. Molecular Mechanisms of Coronary Disease Revealed Using Quantitative Trait Loci for TCF21 Binding, Chromatin Accessibility, and Chromosomal Looping. Genome Biol (2020) 21:135. doi: 10.1186/s13059-020-02049-5
131. Nagao M, Lyu Q, Zhao Q, Wirka RC, Bagga J, Nguyen T, et al. Coronary Disease-Associated Gene TCF21 Inhibits Smooth Muscle Cell Differentiation by Blocking the Myocardin-Serum Response Factor Pathway. Circ Res (2020) 126:517–29. doi: 10.1161/CIRCRESAHA.119.315968
132. Maezawa Y, Onay T, Scott RP, Keir LS, Dimke H, Li C, et al. Loss of the Podocyte-Expressed Transcription Factor Tcf21/Pod1 Results in Podocyte Differentiation Defects and FSGS. J Am Soc Nephrol (2014) 25:2459–70. doi: 10.1681/ASN.2013121307
133. Das S, Senapati P, Chen Z, Reddy MA, Ganguly R, Lanting L, et al. Regulation of Angiotensin II Actions by Enhancers and Super-Enhancers in Vascular Smooth Muscle Cells. Nat Commun (2017) 8:1467. doi: 10.1038/s41467-017-01629-7
134. Ounzain S, Micheletti R, Arnan C, Plaisance I, Cecchi D, Schroen B, et al. CARMEN, a Human Super Enhancer-Associated Long Noncoding RNA Controlling Cardiac Specification, Differentiation and Homeostasis. J Mol Cell Cardiol (2015) 89:98–112. doi: 10.1016/j.yjmcc.2015.09.016
135. Zhang X, Xue C, Lin J, Ferguson JF, Weiner A, Liu W, et al. Interrogation of Nonconserved Human Adipose lincRNAs Identifies a Regulatory Role of linc-ADAL in Adipocyte Metabolism. Sci Transl Med (2018) 10:eaar5987. doi: 10.1126/scitranslmed.aar5987
136. Johnsson P, Lipovich L, Grander D, Morris KV. Evolutionary Conservation of Long non-Coding RNAs; Sequence, Structure, Function. Biochim Biophys Acta (2014) 1840:1063–71. doi: 10.1016/j.bbagen.2013.10.035
137. Ruan X, Li P, Chen Y, Shi Y, Pirooznia M, Seifuddin F, et al. In Vivo Functional Analysis of non-Conserved Human lncRNAs Associated With Cardiometabolic Traits. Nat Commun (2020) 11:45. doi: 10.1038/s41467-019-13688-z
Keywords: diabetes, diabetes complications, long non-coding RNAs, inflammation, fibrosis, epigenetics
Citation: Tanwar VS, Reddy MA and Natarajan R (2021) Emerging Role of Long Non-Coding RNAs in Diabetic Vascular Complications. Front. Endocrinol. 12:665811. doi: 10.3389/fendo.2021.665811
Received: 09 February 2021; Accepted: 01 June 2021;
Published: 21 June 2021.
Edited by:
Sangeeta Dhawan, City of Hope National Medical Center, United StatesReviewed by:
Saumik Biswas, Western University, CanadaSatish K. Raut, Dr. D. Y. Patil Biotechnology & Bioinformatics Institute, India
Copyright © 2021 Tanwar, Reddy and Natarajan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rama Natarajan, Uk5hdGFyYWphbkBjb2gub3Jn
†These authors have contributed equally to this work