 Qian Li1
Qian Li1 Bichao Lu2
Bichao Lu2 Jia Yang2Chao Li2Yanchun Li3
Jia Yang2Chao Li2Yanchun Li3 Hui Chen2Naishi Li1Lian Duan1
Hui Chen2Naishi Li1Lian Duan1 Feng Gu1*
Feng Gu1* Jianmin Zhang2*
Jianmin Zhang2* Weibo Xia1*
Weibo Xia1*- 1Department of Endocrinology, Key Laboratory of Endocrinology, NHC, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Beijing, China
- 2Department of Immunology, Research Center on Pediatric Development and Diseases, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and School of Basic Medicine, Peking Union Medical College, State Key Laboratory of Medical Molecular Biology, Beijing, China
- 3Department of Radiation Oncology, Stanford University, School of Medicine, Stanford, CA, United States
The aquaporin 2 (AQP2) plays a critical role in water reabsorption to maintain water homeostasis. AQP2 mutation leads to nephrogenic diabetes insipidus (NDI), characterized by polyuria, polydipsia, and hypernatremia. We previously reported that a novel AQP2 mutation (G215S) caused NDI in a boy. In this study, we aimed to elucidate the cell biological consequences of this mutation on AQP2 function and clarify the molecular pathogenic mechanism for NDI in this patient. First, we analyzed AQP2 expression in Madin-Darby canine kidney (MDCK) cells by AQP2-G215S or AQP2-WT plasmid transfection and found significantly decreased AQP2-G215S expression in cytoplasmic membrane compared with AQP2-WT, independent of forskolin treatment. Further, we found co-localization of endoplasmic reticulum (ER) marker (Calnexin) with AQP2-G215S rather than AQP2-WT in MDCK cells by immunocytochemistry. The functional analysis showed that MDCK cells transfected with AQP2-G215S displayed reduced water permeability compared with AQP2-WT. Visualization of AQP2 structure implied that AQP2-G215S mutation might interrupt the folding of the sixth transmembrane α-helix and/or the packing of α-helices, resulting in the misfolding of monomer and further impaired formation of tetramer. Taken together, these findings suggested that AQP2-G215S was misfolded and retained in the ER and could not be translocated to the apical membrane to function as a water channel, which revealed the molecular pathogenic mechanism of AQP2-G215S mutation and explained for the phenotype of NDI in this patient.
Introduction
Nephrogenic diabetes insipidus (NDI) is characterized by impaired arginine vasopressin (AVP)-induced water reabsorption in the kidney, leading to polyuria, polydipsia, and hypernatremia. The most severe outcomes include impaired mental development, dilation of the urinary tract, and death (1–3). NDI can be secondary to other clinical conditions, such as drugs (e.g., lithium and cisplatin therapy) and electrolyte abnormalities, or caused by mutations in the vasopressin V2 receptor (AVPR2, OMIM#304800) or AQP2 (OMIM#125800, 107777) (4–7). AVPR2 accounts for X-linked cases of NDI. Current therapeutic options for congenital NDI focus on ameliorating symptoms rather than curing, which are limited and only partially effective (3).
AQP2 is a key factor for maintaining normal body water homeostasis. When the plasma osmolality increases, antidiuretic hormone (AVP) is released from the pituitary gland and binds to AVPR2 in principal cells of the kidney collecting duct, resulting in the accumulation of AQP2 in the apical plasma membrane, which is responsible for water reabsorption (8, 9). AQP2 forms a homotetramer in the plasma membrane, and each monomer is composed of 271 amino acids, containing six transmembrane spanning regions with the intracellular COOH terminus, which is essential for correct routing of AQP2. AVP increases phosphorylation of AQP2 at ser256 and ser269, which is important for the accumulation of AQP2 (10–12). K63-linked ubiquitylation of lys270 is critical for the internalization and degradation of AQP2 from the plasma membrane (13). Up to now, there were 65 mutations of AQP2 reported to cause NDI, and missense/nonsense mutation is the most common mutation type (14). More than 90% of mutations are inherited in autosome recessive mode, which can be categorized into three types, depending on the structural analysis: (i) the pore features (e.g. A70D), (ii) the tetramer assembly (e.g. T126M), and (iii) the monomer folding (e.g. A47V). The remaining 10% of mutations are inherited in a dominant trait, involving the C-terminal tail for AQP2 routing (e.g. R254L) (15–19). Most autosomal recessive cases had severe phenotypes in contrast to autosomal dominant NDI (20).
We previously reported a homozygous missense mutation AQP2-G215S (substitution of Gly215 with Serine) caused NDI in a boy for the first time (21). The initial symptom occurred at 4 months, and the male patient had a total urine volume greater than 4 L in 24 h when diagnosed. Sequence alignment of AQP2 proteins indicated Gly215 showed a 100% conservation among six different species. In the study, we aimed to elucidate the cell biological consequences of this mutation on AQP2 function and clarify the molecular pathogenic mechanism for NDI in this patient.
Materials And Methods
Cell Culture
MDCK cell line was obtained from Cell Resource Center, Basic Medicine Institute, Chinese Academy of Medical Sciences. MDCK cells were cultured in Eagle’s Minimum Essential Medium with 10% fetal bovine serum. All experiments were performed with the approval of the institutional review board and ethics committee of PUMCH, and written informed consents were obtained from the patients.
Plasmid Construction
Coding sequence of human AQP2 was cloned into a vector containing the pCMV6 promoter. Mutant AQP2-G215S plasmid was constructed by site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kits, Agilent) and confirmed by sequence analysis.
Electroporation
Cells were resuspended in 100 μl buffer, and 2 μg plasmid was added. Cells were transferred to a sterile 0.2-cm cuvette (Cell Line Nucleofector™ Kit L, Lonza) and electroporated using Lonza® Nucleofector® II electroporation system according to the manual and protocol. After transfection, cells were gently resuspended in pre-warmed medium. Indomethacin (5 × 10−5 M, Selleck) was added to culture medium 24 h after electroporation and incubated overnight. The medium was replaced with fresh medium containing indomethacin (5 × 10−5 M, Selleck) plus forskolin (5 × 10−5 M, Selleck) or not and incubated for 2 h. Then, the cells were harvested for protein extraction or fixed for immunocytochemistry.
Western Blot
Total cellular proteins were extracted using RIPA buffer containing protease and phosphatase inhibitors. Total membrane and cytoplasmic membrane protein were extracted by a kit (Minute™ Plasma Membrane Protein Isolation and Cell Fractionation Kit, invent BIOTECHNOLOGIES) according to the protocol. All procedures were performed on ice, and protease and phosphatase inhibitors were added into buffer A before use. 20 to 50 × 106 cells were prepared for plasma membrane protein isolation. Equal amounts of protein were loaded on a 12% SDS-polyacrylamide gel and transferred to nitrocellulose membranes. Membranes were blocked with 5% non-fat dry milk for 2 h, followed by incubation with primary antibodies (AQP2 polyclonal antibody, 1:1000, cell signaling technology, #3487; Cadherin antibody, 1:1000, Cell Signaling Technology, ab16505; Calnexin, 1:1000, Thermo Fisher Scientific, MA3-027) over night. Membranes were washed and incubated with HRP-conjugated goat anti-rabbit IgG. Bands were visualized by enhanced chemiluminescence (Pierce™ ECL Western Blotting Substrate kit, Thermo Fisher Scientific).
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde in PBS (pH 7.4) and blocked with 5% normal goat serum and 0.2% Triton X-100. Primary antibodies, including Calnexin (Thermo Fisher Scientific), were diluted in 5% normal goat serum and incubated overnight. Alexa Fluor 594- and Alexa Fluor 488-labeled secondary antibodies were used. Samples were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and mounted on glass slides using the ProLong antifade kit (Thermo Fisher Scientific).
Transcellular Osmotic Water Permeability Measurements
Cells derived from 0.33 cm2 of confluent monolayers were seeded onto 0.33-cm2 polycarbonate filters (Costar, Cambridge, U.S.A.). On the second day after seeding, the medium was aspirated and replaced by fresh medium in the presence of indomethacin (5 × 10−5 M, Selleck) to reduce basal intracellular cyclic adenosine monophosphate (cAMP) levels. Three days after seeding, osmotic water transport was assayed in the presence of indomethacin with or without adding forskolin (5 × 10−5 M, Selleck), by incubation of the apical compartment with 150 µl of 0.5× KHB (1× KHB contains 1.2 mM MgSO4, 128 mM NaCl, 5 mM KCl, 2 mM NaHPO4, 10 mM NaAc, 20 mM HEPES, 1 mM CaCl2, 1 mM L-alanine, 4 mM L-Lactate; pH=7.4), containing 30 mg/L phenol-red and the addition of 800 µl KHB to the basal compartment. After incubation for 2 h at 37°C, the content of the apical compartment was mixed with a pipette, and two aliquots of 50 µl per insert were put into Eppendorf tubes and diluted to 600 µl with Tris-buffered saline (TBS: 20 mM Tris, 73 mM NaCl; pH=7.6) containing 1% (w/v) extrane (Merck, Darmstadt, Germany). After mixing and centrifugation, absorbance at 479 nm was measured. The osmotic water transport (Pf) was calculated from the acquired absorbances as described (22, 23).
Visualization of AQP2 Structure
To visualize the potential impact of the G215S mutation, we built the crystal structure of G215S tetramer using homology modelling with Prime (24). The template structure was human Aquaporin 2 (PDB ID: 4NEF) with 99% sequence identity (17). The snapshots were prepared using VMD 1.9.3 (25).
Data Analysis and Statistics
All results represented at least three independent replications. All data were represented as mean ± SEM. Statistical analysis was performed using GraphPad Prism software (San Diego, California USA, www.graphpad.com). Differences between groups were analyzed as appropriate using t test or one-way ANOVA and post-hoc Tukey’s multiple comparison tests. P < 0.05 (two tailed) was considered to be statistically significant.
Results
Expression of AQP2-G215S Was Decreased in the Cell Membrane Compared With AQP2-WT
We expressed AQP2-WT and AQP2-G215S in MDCK cells by electroporation with plasmid, a cell line demonstrated to be a typical model for AQP2 function (26). Since AQP2 is a membrane protein and functions as a water channel in the apical membrane of principle cell, we further isolated the cytoplasmic membrane and total membrane and detect AQP2 expression by WB (as shown in Figure 1). The results showed that the expression of AQP2 was similar in the total membrane of AQP2-WT-transfected cells and AQP2-G215S-transfected cells (p=0.44). However, AQP2 was decreased in the cell membrane of AQP2-G215S–transfected cells compared with AQP2-WT-transfected cells (p<0.01), even after treated with forskolin (p<0.001), suggesting that mutant AQP2-G215S may not be transported to the cell membrane.
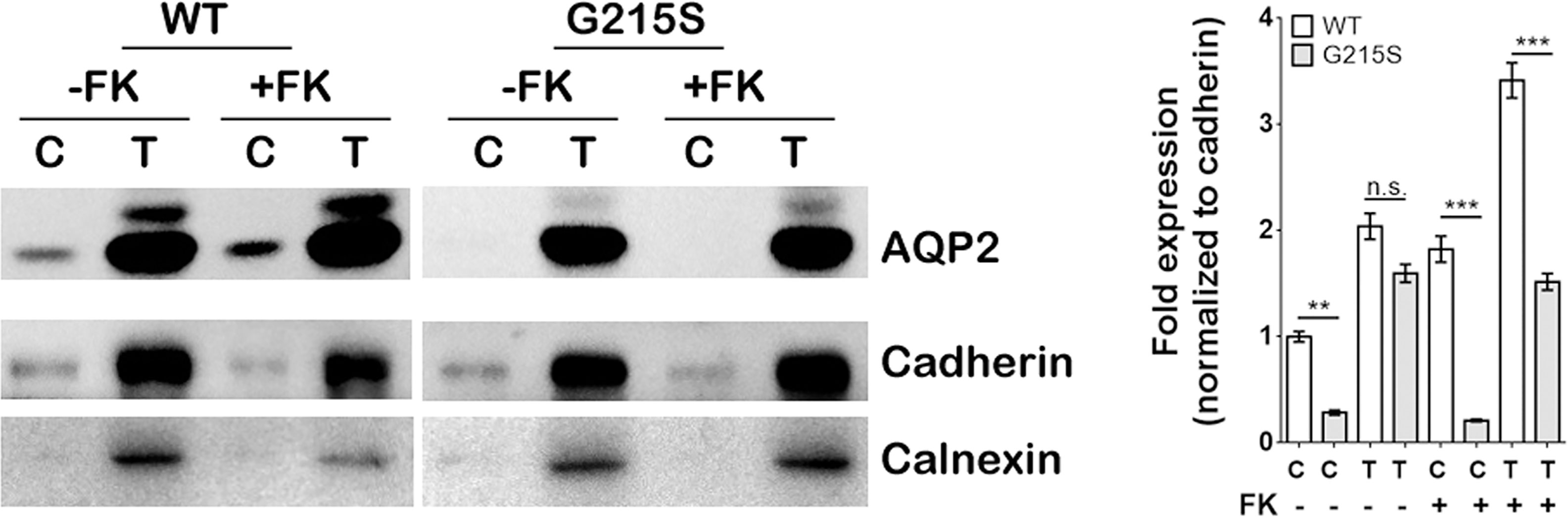
Figure 1 Expression of AQP2-G215S was decreased in the cell membrane compared with AQP2-WT. MDCK cells were transfected with AQP2-WT and AQP2-G215S plasmids. Total membrane proteins (labeled as T) and cytoplasmic membrane proteins (labeled as C) were harvested for AQP2, Pan-cadherin, and calnexin immunoblotting. Pan-Cadherin is the membrane marker, and calnexin is the endoplasmic marker. Protein fold expression normalized to cadherin is shown. WT, wild type; FK, forskolin. Data were shown as mean ± SEM. n = 3, **p < 0.01, ***p < 0.001, n.s.=no statistically significant difference.
AQP2-G215S Is Retained in Endoplasmic Reticulum
We further analyzed the subcellular localization of AQP2-G215S in MDCK cells by immunocytochemistry. MDCK cells were electroporated with AQP2-WT or AQP2-G215S plasmid, then fixed and stained to detect the expression of AQP2 and ER marker (Calnexin). Our results showed co-localization of AQP2 with Calnexin in AQP2-G215S-transfected cells compared with AQP2-WT-transfected cells with or without the stimulation of Forskolin (Figure 2), which suggested that AQP2-G215S was retained in endoplasmic reticulum, in contrast to AQP2-WT.
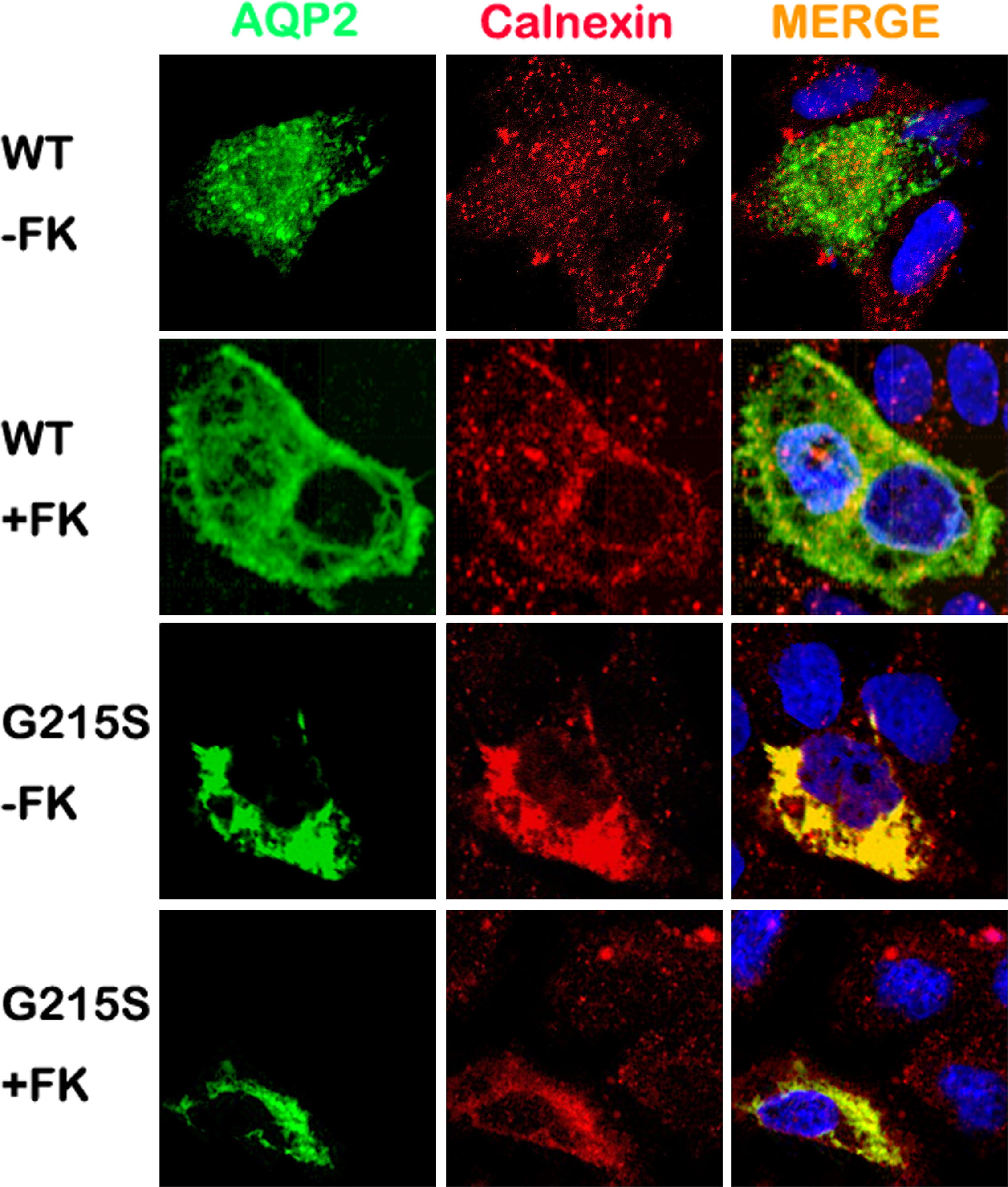
Figure 2 AQP2-G215S was retained in endoplasmic reticulum. MDCK cells were electroporated with AQP2-WT and AQP2-G215S plasmids and stained for AQP2 and endoplasmic reticulum marker (Calnexin). WT, wild type; FK, forskolin.
AQP2-G215S Displays Impaired Transcellular Osmotic Water Permeability
We analyzed the transcellular osmotic water permeability of AQP2-G215S- and AQP2-WT-transfected MDCK cells. The osmotic water transport (Pf) of AQP2-WT-transfected cells was higher than AQP2-WT–transfected cells (24 ± 2.2 vs 5.5 ± 1.0 μm/s, p<0.01) (Figure 3). With the stimulation of forskolin, the Pf of AQP2-WT-transfected cells was increased (57.8 ± 5.1 μm/s, p<0.001), whereas the Pf of AQP2-G215S did not change apparently and remained a much lower level than AQP2-WT (p<0.001). These results indicated that the permeability of MDCK cells transfected with AQP2-G215S was impaired, which further supported retention of AQP2-G215S mutant in endoplasmic reticulum.
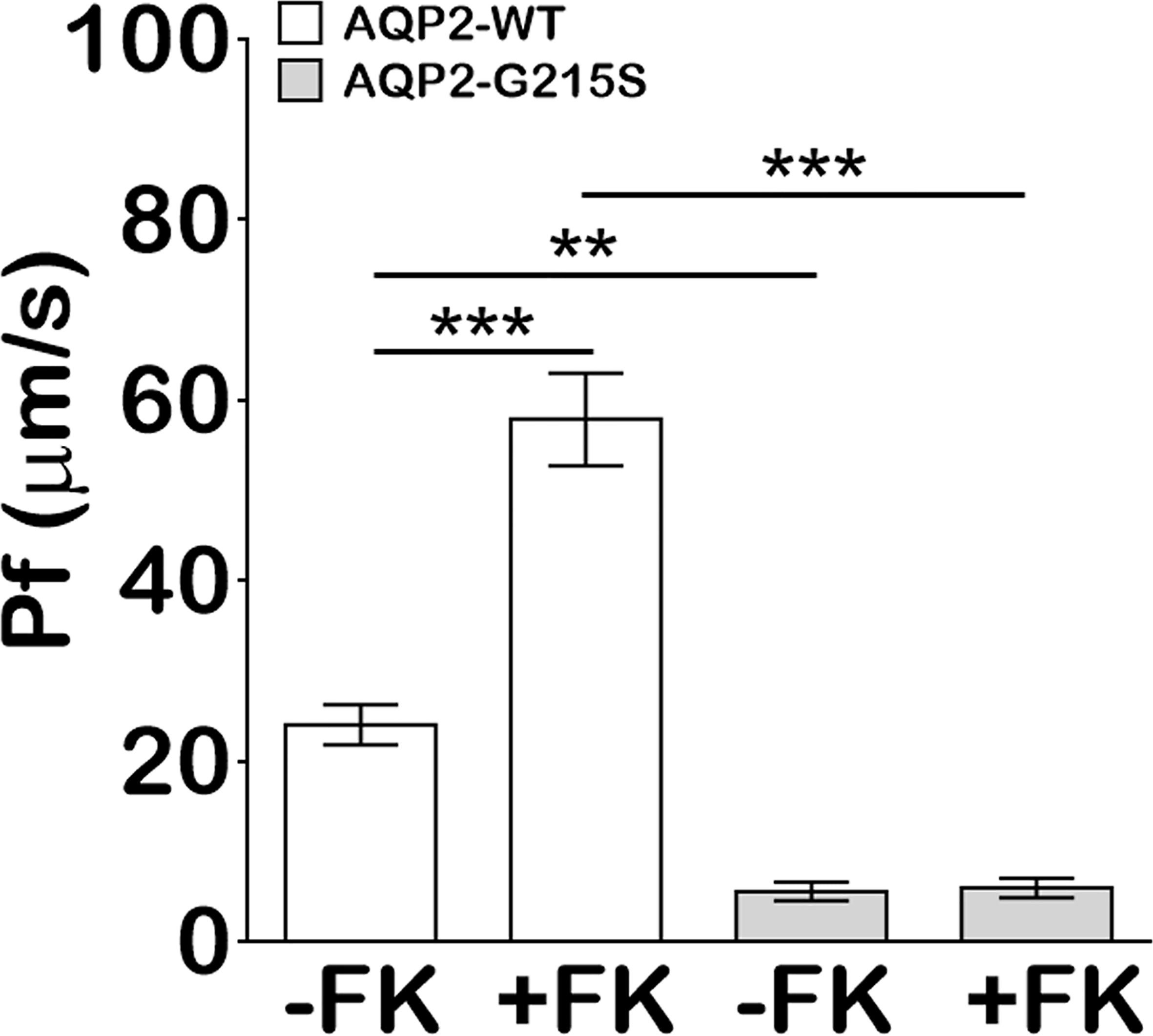
Figure 3 MDCK cells transfected with AQP2-G215S displayed impaired transcellular osmotic water permeability. Cells were seeded onto 0.33 cm2 polycarbonate filters, and replaced with fresh medium in the presence of 5 × 10−5 M indomethacin. Osmotic water transport was assayed with or without 5 × 10−5 M forskolin. The osmotic water transport (Pf) of AQP2-G215S-transfected and AQP2-WT-transfected cells was calculated as described in Material and Methods. WT, wild type; FK, Froskolin. Data were shown as mean ± SEM. n = 3, **p < 0.01, ***p < 0.001.
Structural Visualization of AQP2−G215S
Gly215 was located at alpha-helix in the sixth transmembrane spanning of AQP2 monomer as shown in Figures 4A, B. Amino acids surrounding S215 were on the same monomer, including V142, L143, and L139, suggesting that G215S mutation mostly destabilized AQP2 monomer by changing interactions within intra-monomer helices (Figure 4C). Further, G215S, introducing a polar side chain of S215 into a hydrophobic environment, was thermodynamically unfavorable (Figure 4D). Based on these information, the serine substitution at Gly215 probably interrupted folding of the sixth transmembrane α-helix and/or the packing of α-helices, resulting in the misfolding of monomer, which may further influence the formation of functional AQP2 tetramer.
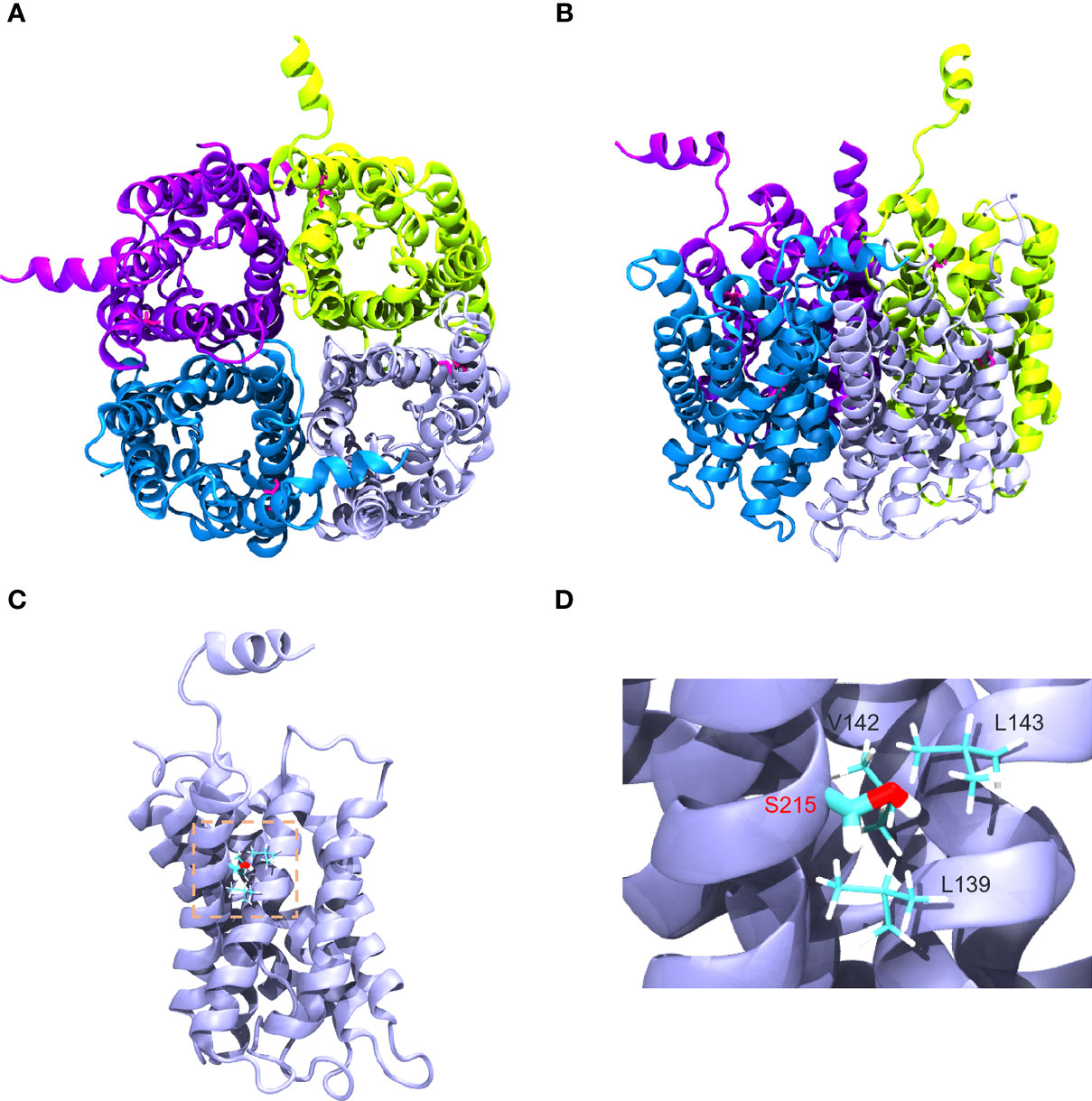
Figure 4 Structural visualization of AQP2−G215S. (A, B) The structure of AQP2 (PDB ID is 4NEF) was visualized as a tetramer. (C, D) Gly215 was indicated as red in the sixth membrane spanning alpha-helix of the AQP2 monomer as shown.
Discussion
In this study, we aimed to elucidate cell biological consequences of a G215S mutation in AQP2 that was discovered in a boy with severe NDI (21). The AQP2 mutation was inherited from his parents, who had a heterozygous genotype and normal phenotypes, which supported an autosomal recessive inheritance model. There was a variety of cell lines and animal models applied to clarify molecular action of AQP2 and the pathogenic mechanism of NDI. MDCK, a kidney epithelia cell line, has been widely used as a model to study NDI, because AQP2 transfection reconstituted vasopressin-regulated transcellular osmotic water transport in principle cells of human renal collect duct (26–28). Here, we first constructed AQP2-G215S and AQP2-WT plasmids and analyzed the expression of AQP2-G215S and AQP2-WT by Western blot. Results showed that there was no difference of AQP2 in total membrane. AQP2 forms a homotetramer in the plasma membrane for water reabsorption, so we further isolated cell membrane of MDCK cells and found there was significantly decreased AQP2 expression in cell membrane of AQP2-G215S-transfected cells compared with AQP2-WT-transfected cells, which suggested that AQP2-G215S could not be translocated to cell membrane.
AQP2 monomer is a glycosylated membrane protein with six-pass-transmembrane domain, which is folded and assembled in the ER. During the processing, high-mannose sugar moieties are attached to Asn123 of AQP2, which is part of a canonical N-glycosylation consensus site (N123-X-T125), and the high-mannose sugar groups are removed in the Golgi complex en route to the plasma membrane (29, 30). To investigate the subcellular localization of AQP2-G215S and AQP2-WT in MDCK cells, immunocytochemistry was performed, and results showed co-localization of ER marker (Calnexin) with AQP2-G215S rather than AQP2-WT in MDCK cells with or without the stimulation of forskolin. These results suggested that the AQP2-G215S was retained in the ER, which was also reported in a previous study. For example, David et al. demonstrated that AQP2-F204V mutant was retained in the ER in a renal cell line and in vivo (31). Nannette also reported that most AQP2 missense mutants in recessive NDI are retained in the ER (32). There were other studies showing misrouting of AQP2 to Golgi complex (33) or late endosomes or lysosome (34). Here, we demonstrated mutant protein, AQP2-G215S, was retained in the ER for the first time. Functional analysis further demonstrated reduced water permeability in AQP2-G215S-transfected cells compared with AQP2-WT–transfected cells, which further supported that AQP2-G215S mutation was retained in the ER. We further performed structure analysis of AQP2-G215S mutant, and results showed that serine substitution at Gly215 probably interrupted the folding of the sixth transmembrane α-helix and/or the packing of α-helices, resulting in the misfolding of monomer, which may further influence the formation of functional AQP2 tetramer. There were also other studies aiming to elucidate the structural basis for mutations of human aquaporins. For instance, Luisa Calvanese built a 3D molecular model for AQP mutants and explored the effect of mutations on structural feature, which provided a rationale for interpreting mutations. Their results suggested that S216P caused impaired monomer folding, similar to G215S. Taken together, these results showed that AQP2-G215S mutant may be misfolded and retained in the ER and could not be translocated to the apical membrane to function as a water channel. There were limitations in our study, for example, cell biological consequences of this mutation were only investigated in MDCK cell line. Besides, novel therapy is further expected to improve the prognosis of NDI. Up to now, some chemical chaperones were shown to correct the trafficking and folding defects of AQP2 mutants and show efficacy in NDI treatment, and gene editing may correct such mutation and cure diseases in the future (35)
In conclusion, we investigated cell biological consequences of a novel mutation (AQP2-G215S) discovered in a boy with NDI. Results showed that AQP2-G215S mutant may be misfolded and retained in the ER and could not be translocated to the apical membrane to function as water channel. This knowledge elucidated the potential molecular mechanism for NDI in this patient.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics Statement
Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
Study design: FG, JZ, and WX. Study conduct: QL, BL, JY, and CL. Data collection: QL, BL, JY, and CL. Data analysis: QL, BL, JY, CL, YL, HC, NL, LD, FG, JZ, and WX. Data interpretation: QL, BL, JY, CL, YL, HC, NL, LD, FG, JZ, and WX. Drafting manuscript: QL. Revising manuscript content: QL, BL, JY, CL, HC, NL, LD, FG, JZ, and WX. Approving final version of manuscript: QL, FG, JZ, and WX. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Fund (No. 81670814), National Key R&D Program of China (2018YFA 0800801), and National Natural Science Fund (No. 81970757).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We appreciate our patients and their families for their participating in this study.
References
1. Hillman DA, Neyzi O, Porter P, Cushman A, Talbot NB. Renal (Vasopressin-Resistant) Diabetes Insipidus; Definition of the Effects of a Homeostatic Limitation in Capacity to Conserve Water on the Physical, Intellectual and Emotional Development of a Child. Pediatrics (1958) 21:430–5 doi: 10.1016/S0022-3476(58)80276-0
2. THea Y. Congenital Nephrogenic Diabetes Insipidus Presented With Bilateral Hydronephrosis: Genetic Analysis of V2R Gene Mutations. Yonsei Med J (2006) 47:126–30. doi: 10.3349/ymj.2006.47.1.126
3. Bockenhauer D, Bichet DG. Pathophysiology, Diagnosis and Management of Nephrogenic Diabetes Insipidus. Nat Rev Nephrol (2015) 11(10):576–88. doi: 10.1038/nrneph.2015.89
4. Bockenhauer D, Hoff WVT, Dattani M, Lehnhardt A, Subtirelu M, Hildebrandt F, Bichet DG. Secondary Nephrogenic Diabetes Insipidus as a Complication of Inherited Renal Diseases. Nephron Physiol (2010) 116: 23–9. doi: 10.1159/000320117
5. Deen P. Requirement of Human Renal Water Channel Aquaporin-2 for Vasopressindependent Concentration of Urine. Science (1994) 264:92–5. doi: 10.1126/science.8140421
6. Lolait SJEA. Cloning and Characterization of a Vasopressin V2 Receptor and Possible Link to Nephrogenic Diabetes Insipidus. Nature (1992) 357:336–9. doi: 10.1038/357336a0
7. Thompson MD HG, Percy ME, Bichet DG, Cole DE. G Protein-Coupled Receptor Mutations and Human Genetic Disease. Methods Mol Biol (2014) 1175:153–87. doi: 10.1007/978-1-4939-0956-8_8
8. Jung HJ KT. Molecular Mechanisms Regulating Aquaporin-2 in Kidney Collecting Duct. Am J Physiol Renal Physiol (2016) 311(6):F1318–F28. doi: 10.1152/ajprenal.00485.2016
9. CW B. Central Mechanisms of Osmosensation and Systemic Osmoregulation. Nat Rev Neurosci (2008) 9(7):519–31. doi: 10.1038/nrn2400
10. Fushimi K SS. Marumo F Phosphorylation of Serine 256 is Required for cAMP-dependent Regulatory Exocytosis of the Aquaporin-2 Water Channel. J Biol Chem (1997) 272(23):14800–4. doi: 10.1074/jbc.272.23.14800
11. al HJE. Vasopressin-Stimulated Increase in Phosphorylation at Ser269 Potentiates Plasma Membrane Retention of Aquaporin-2. J Biol Chem (2008) 283(36):24617–27. doi: 10.1074/jbc.M803074200
12. al H. Quantitative Phosphoproteomics of Vasopressin-Sensitive Renal Cells: Regulation of Aquaporin-2 Phosphorylation at Two Sites. Proc Natl Acad Sci USA (2006) 103(18):7159–64. doi: 10.1073/pnas.0600895103
13. al K. Short-Chain Ubiquitination Mediates the Regulated Endocytosis of the Aquaporin-2 Water Channel. Proc Natl Acad Sci USA (2006) 103(48):18344–9. doi: 10.1073/pnas.0604073103
14. Milano S, Carmosino M, Gerbino A, Svelto M, Procino G. Hereditary Nephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. Update. Int J Mol Sci (2017) 18(11):2385. doi: 10.3390/ijms18112385
15. Moeller HB, Rittig S, Fenton RA. Nephrogenic Diabetes Insipidus: Essential Insights Into the Molecular Background and Potential Therapies for Treatment. Endocr Rev (2013) 34(2):278–301. doi: 10.1210/er.2012-1044
16. Calvanese L, D’Auria G, Vangone A, Falcigno L, Oliva R. Structural Basis for Mutations of Human Aquaporins Associated to Genetic Diseases. Int J Mol Sci (2018) 19(6):1577. doi: 10.3390/ijms19061577
17. Frick A EU, de Mattia F, Oberg F, Hedfalk K, Neutze R, de Grip WJ, et al. X-Ray Structure of Human Aquaporin 2 and Its Implications for Nephrogenic Diabetes Insipidus and Trafficking. Proc Natl Acad Sci USA (2014) 111(17):6305–10. doi: 10.1073/pnas.1321406111
18. El Tarazi A, Lussier Y, Da Cal S, Bissonnette P, Bichet DG. Functional Recovery of AQP2 Recessive Mutations Through Hetero-Oligomerization With Wild-Type Counterpart. Sci Rep (2016) 6:33298. doi: 10.1038/srep33298
19. de Mattia F, Savelkoul PJ, Kamsteeg EJ, Konings IB, van der Sluijs P, Mallmann R, et al. Lack of Arginine Vasopressin-Induced Phosphorylation of Aquaporin-2 Mutant AQP2-R254L Explains Dominant Nephrogenic Diabetes Insipidus. J Am Soc Nephrol (2005) 16(10):2872–80. doi: 10.1681/ASN.2005010104
20. Babey M KP, Robertson GL. Familial Forms of Diabetes Insipidus: Clinical and Molecular Characteristics. Nat Rev Endocrinol (2011) 7:701–14. doi: 10.1038/nrendo.2011.100
21. Jing Cen MN, Duan L, Gu F. Novel Autosomal Recessive Gene Mutations in Aquaporin-2 in Two Chinese Congenital Nephrogenic Diabetes Insipidus Pedigrees. Int J Clin Exp Med (2015) 8(3):3629–39. doi: 10.1016/S0022-3476(58)80276-0
22. Peter MT, Deen SN, René JM, van Os CH. Apical and Basolateral Expression of Aquaporin-1 in Transfected MDCK and LLC-PK Cells and Functional Evaluation of Their Transcellular Osmotic Water Permeabilities. Pflügers Arch- Eur J Physiol (1997) 1997(433):780–7. doi: 10.1007/s004240050345
23. Jovov NKW B, Lewis SA. A Spectroscopic Method for Assessing Confluence of Epithelial Cell Cultures. Am J Physiol (1991) 261:C1196–203. doi: 10.1152/ajpcell.1991.261.6.C1196
24. Jacobson MPP, D. L., Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins (2004) 55:351–67. doi: 10.1002/prot.10613
25. Humphrey W, Dalke A, Schulten K. Vmd - Visual Molecular Dynamics. J Molec Graphics (1996) 14:33–8. doi: 10.1016/0263-7855(96)00018-5
26. Deen PM RJ, Mulders SM, Errington RJ, van Baal J, van Os CH. Aquaporin-2 Transfection of Madin-Darby Canine Kidney Cells Reconstitutes Vasopressin-Regulated Transcellular Osmotic Water Transport. J Am Soc Nephrol (1997) 8:1493–501. doi: 10.1681/ASN.V8101493
27. Richardson JC SV, Simmons NL. Identification of Two Strains of MDCK Cells Which Resemble Separate Nephron Tubule Segments. Biochim Biophys Acta (1981) 673:26–36. doi: 10.1016/0304-4165(81)90307-X
28. William L, Rice WL, Mamuya F, McKee M, Păunescu TG, Jenny Lu HA. Polarized Trafficking of AQP2 Revealed in Three Dimensional Epithelial Culture. PloS One (2015) 10(7):e0131719. doi: 10.1371/journal.pone.0131719
29. Ineke Braakman DNH. Protein Folding in the Endoplasmic Reticulum. Cold Spring Harb Perspect Biol (2013) 5(5):a013201. doi: 10.1101/cshperspect.a013201
30. Baumgarten R1 VDPM, Wetzels JF, Van Os CH, Deen PM. Glycosylation is Not Essential for Vasopressin-Dependent Routing of Aquaporin-2 in Transfected Madin-Darby Canine Kidney Cells. J Am Soc Nephrol (1998) 9(9):1553–9. doi: 10.1681/ASN.V991553
31. David J Lloyd FWH, Tarantino LM. Nicholas Gekakis. Diabetes Insipidus in Mice With a Mutation in Aquaporin-2. PloS Genet (2005) 1(2):e20. doi: 10.1371/journal.pgen.0010020
32. Marr N, Bichet DG, Hoefs S, Savelkoul PJ, Konings IB, De Mattia F, et al. Cell-Biologic and Functional Analyses of Five New Aquaporin-2 Missense Mutations That Cause Recessive Nephrogenic Diabetes Insipidus. J Am Soc Nephrol (2002) 13(9):2267–77. doi: 10.1097/01.ASN.0000027355.41663.14
33. Mulders SM1 KN, Van Lieburg AF, Monnens LA, Leumann E, Wühl E, Schober E, et al. An Aquaporin-2 Water Channel Mutant Which Causes Autosomal Dominant Nephrogenic Diabetes Insipidus is Retained in the Golgi Complex. J Clin Invest (1998) 102(1):57–66. doi: 10.1172/JCI2605
34. al MNE. Heteroligomerization of an Aquaporin-2 Mutant With Wild-Type Aquaporin-2 and Their Misrouting to Late Endosomes/Lysosomes Explains Dominant Nephrogenic Diabetes Insipidus. Hum Mol Genet (2002) 11(7):779–89. doi: 10.1093/hmg/11.7.779
Keywords: aquaporin 2, nephrogenic diabetes insipidus, water reabsorption, polydipsia, hypernatremia
Citation: Li Q, Lu B, Yang J, Li C, Li Y, Chen H, Li N, Duan L, Gu F, Zhang J and Xia W (2021) Molecular Characterization of an Aquaporin−2 Mutation Causing Nephrogenic Diabetes Insipidus. Front. Endocrinol. 12:665145. doi: 10.3389/fendo.2021.665145
Received: 07 February 2021; Accepted: 06 July 2021;
Published: 27 August 2021.
Edited by:
Ralf Jockers, Université de Paris, FranceReviewed by:
Miles Douglas Thompson, University of California, San Diego, United StatesEmma Olesen, University of Copenhagen, Denmark
Copyright © 2021 Li, Lu, Yang, Li, Li, Chen, Li, Duan, Gu, Zhang and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianmin Zhang, anpoYW5nNDJAMTYzLmNvbQ==; Weibo Xia, eGlhd2VpYm84MzAxQDE2My5jb20=; Feng Gu, Z3VmZW5nQGd1c21lZHNjaS5jbg==