 Marc Tatar
Marc Tatar- Department of Ecology and Evolutionary Biology, Brown University, Providence, RI, United States
Mutations of the insulin-like receptor in Drosophila extend lifespan. New research suggests this receptor operates in two modes. The first extends lifespan while slowing reproduction and reducing growth. The second strongly extends lifespan without impairing growth or reproduction; it confers longevity assurance. The mutation that confers longevity assurance resides in the kinase insert domain, which contains a potential SH2 binding site for substrate proteins. We apply a recent model for the function of receptor tyrosine kinases to propose how insulin receptor structure can modulate aging. This concept hypothesizes that strong insulin-like ligands promote phosphorylation of high threshold substrate binding sites to robustly induce reproduction, which impairs survival as a consequence of trade-offs. Lower levels of receptor stimulation provide less kinase dimer stability, which reduces reproduction and extends lifespan by avoiding reproductive costs. Environmental conditions that favor diapause alter the expression of insulin ligands to further repress the stability of the interacting kinase domains, block phosphorylation of low threshold substrates and thus induce a unique molecular program that confers longevity assurance. Mutations of the insulin receptor that block low-phosphorylation site interactions, such as within the kinase insert domain, can extend lifespan while maintaining overall dimer stability. These flies are long-lived while maintaining reproduction and growth. The kinase insert domain of Drosophila provides a novel avenue from which to seek signaling of the insulin/insulin-like growth factor system of humans that modulate aging without impacting reproduction and growth, or incurring insulin resistance pathology.
Introduction
Mutations of the insulin/IGF tyrosine kinase receptor slow aging in Drosophila and C. elegans, and perhaps as well in humans (1–3). These invertebrates have single insulin/IGF-like receptors, InR in Drosophila and DAF-2 in C. elegans. Besides aging, these receptors regulate traits including development, growth, metabolism, reproduction, sleep, behavior, and Dauer/diapause (4–10). In mammals, a family of insulin, IGF, relaxin, and insulin-like peptides modulate many functions including metabolism, cell cycle, development, reproduction, cognition, and vascular physiology (11–13), where adult insulin and IGF1 signals via three dimeric receptors [IR, IGF1-R, IR/IGF1R hybrid (14)]. In contrast, the single invertebrate insulin-like receptors respond to a number of unique insulin-like ligands, seven in Drosophila and as many as 40 in C. elegans (15, 16). Despite their centrality, little is understood about how these invertebrate insulin-like ligands control such an array of distinct phenotypes. Here we explore a potential solution. We integrate new observations derived from single amino acid substitutions of Drosophila InR (17) with the receptor tyrosine kinase (RTK) threshold model of Zinkle and Mohammadi (18). We will propose that the level of insulin-stimulated dimer stability determines which substrate binding sites are activated to impact specific traits. Mutations of InR may slow aging because they reduce overall receptor dimer stability or because they directly modify binding sites. This model suggests how insulin-like receptors might slow aging without insulin-resistance and how diverse Drosophila insulin-like ligands control unique sets of traits. The model provides a framework to understand where and how modified insulin/IGF signaling can affect human aging.
The Threshold Model of Receptor Tyrosine Kinase Signaling
Receptor tyrosine kinases (RTK) are single-pass transmembrane proteins that transduce extracellular ligand binding into kinase activity. Strongly bound ligands are thought to induce sustained kinase activity to promote outputs distinct from those of weak ligands, which produce transient or low kinase activity; the intensity and duration of intracellular signaling pathways determines the cellular response (19). As reviewed in Zinkle and Mohammadi (18), this process was first proposed for rat PC12 cells where the duration of MAPK activation differentially promotes neurite outgrowth versus cell proliferation, independent of ligand or receptor identity (19). In a second example, isoforms of fetal growth factor (FGF) ligand FGF8a and FGF8b differentially induce the midbrain to differentiate or expand. This specificity, however, is based on the relative abundance of each isoform and the associated magnitude of Ras/MAPK induction, not upon the ligand identity (20).
RTK also phosphorylate binding sites within their juxtamembrane (JM), C-terminal tail, and kinase domains. These sites recruit adapter proteins including those with Src homology 2 (SH2), phosphotyrosine-binding (PTB), and SH3 domain-binding sites. The identity of recruited substrate specifies which transduction pathways the receptor activates (21–23). Thus, mutation of one docking site can alter one particular outcome without affecting others, for instance when mutation of the Grb2-recruitment site on the canine kidney cell MET receptor blocks tubulogenesis without disrupting cell dissociation (24). In this view, the quality of the receptor-protein interaction determines the cellular response.
Zinkle and Mohammadi (18) integrate how the intensity of activation and the quality of interactions determine RTK function. Ligand binding causes receptor tyrosine kinase protomers to dimerize or in the case of IR preformed dimers cause the intracellular domains to structurally reorient (25). Repositioning of IR intracellular domains is induced when insulin binds multiple ectodomain sites upon both protomers to affect hinge motions that bring each internal kinase domains into proximity, permitting them to asymmetrically transphosphorylate A-loop tyrosine residues (26–28). This transactivation stimulates subsequent kinase activity to phosphorylate endodomain tyrosine residues and substrate binding proteins. Central to the model (18), the level of stability between the repositioned intracellular domains determines which endodomain tyrosine residues are phosphorylated, where adaptor binding sites have unique phosphorylation thresholds. High affinity insulin ligands will have fast on-rates and slow off-rates at receptor binding sites and thus continuously stabilize the dimer to phosphorylate both low- and high-threshold sites (Figure 1A). Relatively weak or transient ligands will have slower on-rates and faster off-rates and consequently induce weak dimer stability that only activates binding sites with low phosphorylation thresholds. As a general point for the model relevant for any RTK, although thresholds are ordered, cellular responses need not be nested because signals from a high threshold site can inhibit the output from lower threshold sites (Figure 1B).
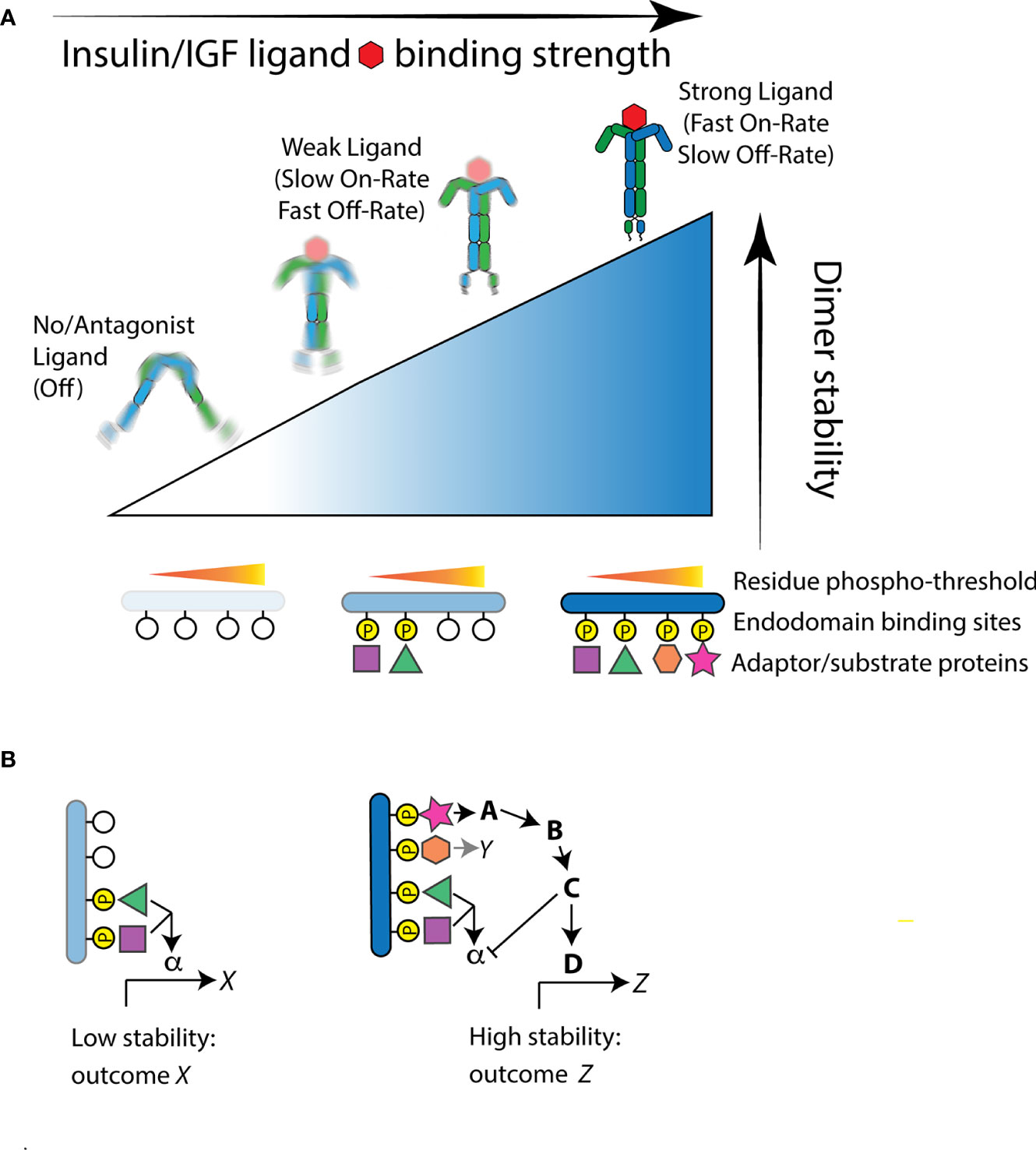
Figure 1 The Receptor Tyrosine Kinase threshold model of Zinkle and Mohammadi applied to the insulin-like receptor. (A) Structural reorientation of insulin-like protomers is induced by ligand binding. Strong binding ligands have fast-on/slow-off rates and produce highly stable interactions of the internal kinase domains. This permits phosphorylation of endodomain substrate binding sites that have high as well as low thresholds, thus recruiting a full complement of available substrate proteins. Weaker ligands produce moderate kinase domain interaction stability and thus only induce residue phosphorylation at sites with a relatively low stability threshold. In the absence of ligand, or when the receptor is bound by an antagonist ligand, protomers fail to reorient or are highly unstable. In this state, the kinase domains do not phosphorylate substrate residue sites and few if any substrate binding protein are engaged. (B) Signal feedback among hierarchical thresholds can produce unnested signaling outcomes. As an example: In weak stability activation of the receptor, low threshold binding protein interactions activate a signal pathway through the substrate protein α to induce a transcriptional program X. This program is not necessarily activated, however, when the receptor gains greater stability, even though the substrate protein α is recruited. A high threshold substrate interaction that activates binding protein A may simultaneously propagate signaling to induce the transcriptional program Z and repress signaling otherwise propagated by α.
Overall, Zinkle and Mohammadi synthesize both perspectives of RTK operation: the intensity and duration of dimer stability regulates which binding proteins are activated, and these substrates specify the cellular outcome of the stimulated receptor. Here we develop how this threshold model helps explain control of aging by insulin-like receptors. First we describe longevity-extending mutations of Drosophila InR and C. elegans daf-2, and introduce known adaptor proteins of InR.
The Drosophila and C. elegans Insulin-Like Receptors
Gems, Patel, and colleagues classified multiple mutations of the C. elegans insulin-like receptor daf-2 (29, 30). “Class 1” mutants include substitutions in the extracellular CR, L2, and FnIII domains. These induce dauer, an alternative quiescent developmental stage, and promote adult longevity. “Class 2” substitutions reside in the L1 ligand pocket, the CR ectodomain, and the intracellular tyrosine kinase domain. These alleles induce dauer and extend lifespan, but also variously affect feeding, reproduction, movement, and growth (29). Class 1 and Class 2 alleles stimulate unique transcriptional profiles (30). To explain these differences, Patel (30) suggested Class 1 mutants reduce DAF-2 abundance and thus activate the transcription factor DAF-16/FOXO. Class 2 alleles were thought to increase receptor perdurance and thereby reduce interaction with Ras-associated substrates while retaining signal induction of PI3K/Akt. From extensive phenotypic analyses, these authors suggest the DAF-2 receptor has two distinct functional outputs.
We recently studied how mutations in Drosophila InR affect aging (17). InR is generated from three alternative 5’UTRs (31, 32) to produce isoforms differing by a 368-amino acid C-terminal tail (33–35). Based on our analysis of codon substitutions, InR appears to modulate aging through distinct modes (Table 1). As transheterozygotes, Mode 1 alleles increase survival, decrease egg production, reduce body size, and repress insulin-stimulated Akt phosphorylation (17). Among genotypes from these alleles, lifespan negatively correlates with egg production (Figure 2A), consistent with theory for how aging arises when selection optimizes fitness (47). These pro-longevity mutations produce amino acid substitutions in the extracellular FnIII domain (extracellular V810D), and in conserved residues of the kinase A-loop and the kinase C-lobe (Figure 2B). As a group, these substitutions are likely to destabilize protomer endodomain interaction or directly inhibit kinase catalytic function (27, 28).

Table 1 Phenotypes of Drosophila insulin/IGF receptor and substrate protein mutations.
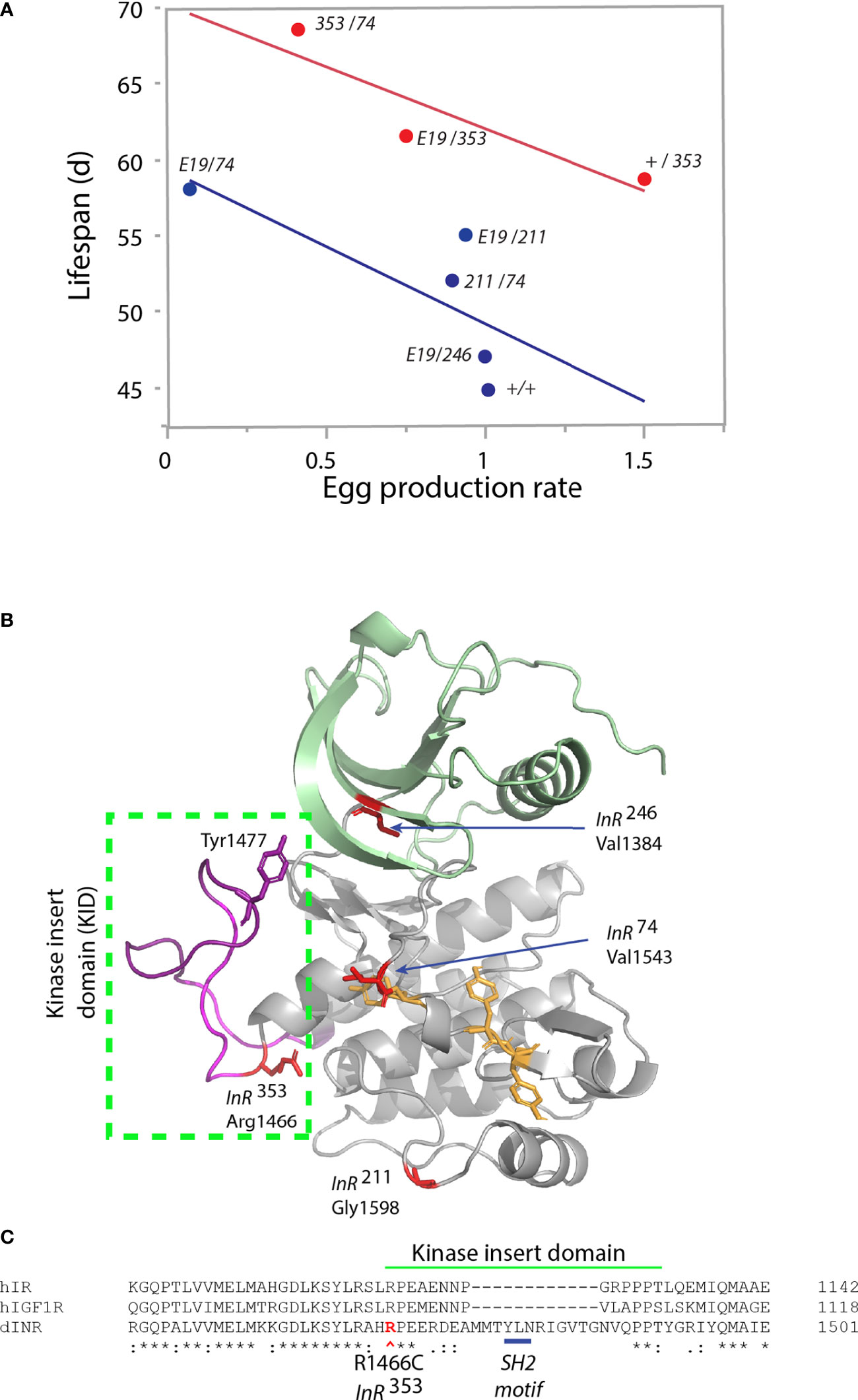
Figure 2 Characteristics of single amino acid substitution of the Drosophila insulin-like receptor. (A) Relationship of genotype average median lifespan relative to rate of egg production, from Yamamoto (17). Egg production rate is number of eggs produced daily scaled by the ovary size (number of ovarioles: ovary subunits). Lifespan is the average of median survival among replicate trials and independent genetic accessions of each genotype. Blue regression: genotypes lacking the InR353 allele (“353” in figure). Red regression: genotypes that include one InR353 allele. “E19” is the allele InRE19, a V810D substitution in the extracellular FnIII domain (17). Other alleles (InR74, InR211, InR246; in figure “74,” “211,” “246”) are substitutions within the kinase domain. The +/+ genotype is wildtype coisogenic will all mutant alleles, co-derived by homologous recombination; details in Yamamoto (17). (B) Model of kinase domain structure for the Drosophila insulin-like receptor. Subdomains: N-lobe (green), C-lobe (gray), A-loop tyrosines (yellow), Kinase Insert Domain (magenta) within box. Substitution residues for kinase domain mutations in red; Tyrosine1477 of the proposed SH2 binding site within the KID. (C) Partial amino acid alignment of the C-lobe to include region of the KID in human IR, human IGF1R and Drosophila InR [alignments and nomenclature from (17)]. Site of the InR353 substitution in red (R1466C), site of the proposed SH2 motif underlined in blue.
Mode 2 is represented by the dominant allele InR353 (17). Adult heterozygotes (wildtype/InR353) have robustly increased lifespan but remarkably so without decreasing reproduction or growth (Table 1). Unlike Mode 1 flies, tissue from the InR353 heterozygotes strongly induces pAkt in response to insulin—they are not insulin resistant. When InR353 is combined with Mode 1 alleles, adults lay fewer eggs and lifespan is increased by the combined effects of reduced survival costs of reproduction added to the longevity assured by InR353 (Figure 2A).
The InR353 substitution Arg1466Cys lies within the kinase insert domain (KID) (Figure 2C), an unstructured peptide segment that interrupts the kinase domain of many RTKs (48). Arg1466 of Drosophila is homologous to Arg1092 of the human insulin receptor (49). In humans, the insulin receptor mutation Arg1092Glu produces Donohue syndrome where heterozygotes are largely normal while homozygotes are strongly insulin resistant, small, and inviable (50). Overall, the function of kinase insert domains is poorly understood but whereas the human IR and IGFR domains are short, the longer Drosophila KID contains a potential SH2 binding motif (Tyr1477-Leu-Asn; Figure 2C). This site may recruit an adaptor protein, potentially Grb2 as seen in the KID of mammalian PDGFR, CSF1R, and Kit (48). We hypothesize the InR353 substitution disrupts this receptor-protein interaction to induce longevity assurance—a homeostatic program that increases somatic survival independent of reproductive trade-offs (51).
Adaptor and Substrate Proteins of Drosophila InR
A number of receptor-adaptor protein interactions are documented for the Drosophila insulin receptor. The C-terminal tail of InR recruits Chico (homolog of IRS1-4), although apparently without phosphorylating this substrate (52, 53). The tail likewise contains YXXM motifs to recruit the p85/p60 subunit of PI3-kinase (34), and PXXP sequences for the SH2/SH3 adapter Dock (homolog of mammalian Nck) (54). Dock modulates photoreceptor axon guidance but does not affect growth. No data address if protein interactions with the C-terminal tail affect aging.
The juxtamembrane domain (JM) of InR also recruits Chico, using NPXY residues conserved in the human insulin receptor (42, 53). Interaction between InR and Chico is mediated by the SH2B1 adaptor protein Lnk (44, 45, 55). In mammals, SH2B1 is recruited to insulin receptor A-loop phosphotyrosines (56, 57). In Drosophila, Lnk colocalizes InR and Chico to promote phosphorylation of Akt (55). Genetic loss of Lnk extends longevity, reduces body size, and represses fecundity (Table 1).
Mutation of chico itself slows aging (Table 1). Appropriate for the centennial of insulin discovery, chico is debated to harken back to 1919, potentially as an allele of the mutation flipper identified by Bridges and Mohr (see https://flybase.org/reports/FBgn0000675). Modern chico mutant alleles are transposon insertions initially characterized to elevate lipids, and impair cell size and number (42, 58). Homozygotes of the mutant chico1 are small, long-lived, and sterile; wildtype/chico1 heterozygotes are also long-lived and similar to wildtype/InR353 these adults have normal growth and fertility (36–38) (Table 1).
Chico is a substrate adapter protein. It recruits SH2/SH3 domain-containing proteins including the p85/p60 subunit of PI3K and the Grb2 homolog Drk (Downstream of receptor kinase) (42, 53, 59). Oldham expressed chico-transgenes in chico1 homozygotes (59). Wildtype chico-transgenes rescued body size and fertility. Transgenes that only restored Grb2/Drk binding did not rescue these traits while those that restored p60/PI3K restored growth and reproduction. Slack (40) used this design to study aging. The exceptional longevity of chico1 heterozygotes reverted to normal by addition of a wildtype chico transgene but not when the chico transgene contained only functional p60/PI3K sites or only functional Grb2/Drk sites. Overall, Chico controls p60/PI3K/Akt to modulate growth, metabolism, and longevity, but its effects through Grb2/Drk appear to be limited to aging.
As in mammals, activated InR phosphorylates Akt to repress Drosophila Foxo, the homolog of mammalian FOXO1-4 and C. elegans DAF-16. As seen for daf-16, foxo is required for insulin receptor mutations to extend Drosophila lifespan (39, 46). Gene targets of these transcription factors in both invertebrates reveal many distal mechanisms to slow aging (60–62). Parallel to Akt-Foxo, Drosophila Grb2/Drk regulates Ras to control signaling through Erk (63). Slack (40) demonstrated Chico acts through Ras-Erk to regulate the E-twenty-six transcription factor Anterior Open (Aop). Aop is required for chico mutations to extend lifespan, however no data yet shows if this interaction is downstream of InR rather than other potential IRS-regulatory receptors (64).
These observations provide three touchpoints. First, mutations of InR may affect aging through altered kinase activity while another may act by altering adapter protein interaction. Second, InR353 and chico1 are dominant alleles that produce long-lived adults that are unexpectedly large and fecund. Third, Chico appears to signal through SH2-Grb2/Drk-Ras to modulate aging without affecting growth or reproduction, while we suggest the InR kinase insert domain contains an unrecognized SH2 binding motif. The Arg1466Cys substitution of InR353 within the KID may destabilize Grb2/Drk direct signaling to slow aging. These observations can be integrated with the RTK threshold model to hypothesize how InR regulates aging.
Hypothesis: Stability Thresholds to Regulate Aging
Zinkle and Mohammadi (18) propose stimulated RTK have varied levels of dimer stability that progressively phosphorylate adaptor binding sites, each with a characteristic threshold. Activated binding sites interact with specific adaptor proteins to stimulate unique cellular outcomes. We envision this model operates within insulin-like receptors (Figure 3A). In Drosophila InR, sites with high thresholds may include those that recruit Lnk and Chico while sites with a relatively Low phosphorylation threshold might recruit Grb2/Drk. In conditions favoring full reproduction, abundant, strong insulin ligands interact with InR to stabilize protomer kinase domain interaction. Strong transphosphorylation and extensive kinase activity phosphorylate both low (Grb2/Drk) and high threshold receptor binding sites (Chico, Lnk), and efficiently phosphorylate substrate proteins. The activated substrates transduce signals through Akt, TOR, Ras, AMPK, and GSK to promote growth and reproduction. These conditions are permissive for aging because lifetime reproductive success is optimized through the balance of egg production with associated survival costs.
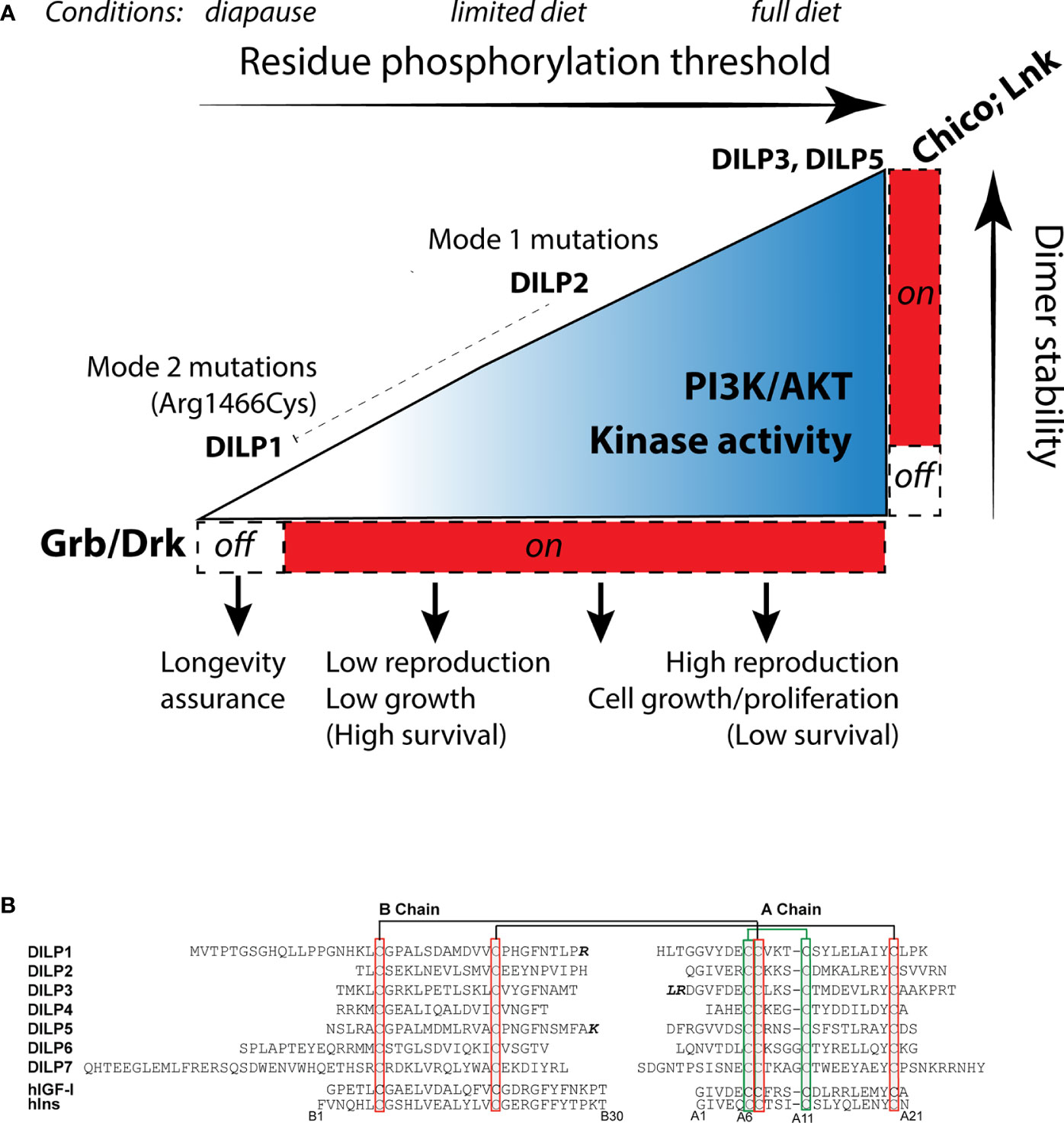
Figure 3 How insulin-like receptors and ligands may modulate aging relative to reproduction, growth and metabolism through the Receptor Tyrosine Kinase threshold model. (A) The environment determines the level of insulin-like receptor dimer stability through control of Drosophila insulin like peptides (DILP). Peptides with high binding activity (DILP5, perhaps DILP3) promote stable protomer kinase interaction, leading to strong kinase catalytic activity and phosphorylation of high threshold substrate binding sites, activation of the adaptor proteins LNK, Chico and Grb, and signal transduction through AKT and Ras. This stimulates reproduction and growth. Limited diet (or moderating environments) reduce DILP5 and DILP3 but retain DILP2 expression (65). DILP2 has reduced kinase dimer stability, potentially sufficient to activate AKT and RAS through PI3K and Grb/Drk recruitment but with less kinase catalytic activity. This state sustains less reproduction. DILP2 represses expression of dilp1. In extreme environments, flies enter diapause and express dilp1. We propose DILP1 is a competitive receptor antagonist. Kinase dimer stability is minimized. High/moderate threshold sites required to activate Akt are not phosphorylated, ceasing reproduction. Low threshold sites required to activate Grb/Drk are not activated. This impairs Ras signaling, which induces systems to support somatic survival (longevity assurance). This state produces reproductive diapause. The InR353 mutation (Arg1466Cys), we propose, inhibits phosphorylation of Grb/Drk by the KID but does not (as observed) affect the ability of the receptor to induce phosphorylation of Akt. The mutation unleashes the longevity assurance program of diapause while bypassing the loss of dimer stability that would otherwise inhibit reproduction. (B) Sequences of the Drosophila insulin-like peptides DILP1-7, B- and A-chains, using cleavage sites predicted by Gronke (66), with potential alternatives where the additional residues are noted in bold. Chains are aligned across bridge cysteines. Human insulin and insulin growth factor 1 for comparison, numbered from insulin.
In restricted conditions such as limited diet, adults secrete fewer or different DILPs (65). We propose this moderately reduces dimer stability to a level that dampens kinase activity while Akt is still phosphorylated. The receptor propagates less intense signaling, which reduces reproduction and correspondingly increases survival. In extreme conditions, such a season that induces diapause, we propose the endocrine state minimizes InR dimer stability so that low threshold residues become dephosphorylated. The SH2 motif of the kinase insert domain may represent such a site. It may be activated in normal conditions by insulin ligands to induce Grb2/Drk-Ras/Erk signaling. However, in diapause conditions key insulin ligands are repressed (67). We hypothesize this will destabilize InR dimers to dephosphorylate the SH2/Grb/Drk site of the KID, and thereby blunt Erk signaling to release somatic maintenance programs that retards somatic aging. Because of the hierarchy within the threshold model, high-phosphorylation threshold sites of InR will not be activated in this state of low dimer stability; Akt will not transduce pro-reproductive signaling. This mechanism models InR regulation of reproductive diapause; it simultaneously stalls reproduction and assures somatic survival until favorable environmental conditions return (68, 69).
This model may explain how some insulin receptor mutations slow aging without affecting reproduction or insulin sensitivity (Mode 2). We hypothesize the Arg1466Cyr substitution disrupts how Grb/Drk is recruited to the SH2 binding motif of the KID. This mutation, however, does not destabilize the dimer and heterozygous receptors therefore phosphorylate Akt and retain kinase activity that propagate reproduction and growth. Although, balancing this hypothesis, the drug Trametinib, a selective MEK1 and MEK2 inhibitor, extends fly lifespan while reducing fecundity (70). In contrast, Mode 1 mutations have reduced kinase activity and are therefore insulin resistant (17). We propose these mutations somewhat increase dimer instability, but not to an extent that dephosphorylates Try1477. Fecundity and growth are reduced with moderate loss of dimer stability, and longevity is increased by mitigating survival costs of reproduction.
Drosophila Insulin-Like Ligands
In this threshold model, receptor dimer stability will be modulated by the quantity, quality, and bioavailability of insulin-like ligands. Drosophila has seven insulin-like loci, dilp1-7 (49, 66, 71). Based on dilp sequence from 12 Drosophila species, Gronke (66) concluded these ligand peptides contain conserved cysteine disulfide bridges, bioactive A and B chains, and functional signal peptides (Figure 3B). DILP1, DILP6, and DILP7 are notable for their extended B-chain N termini. DILP6 has a short C-peptide sequence and may thus more resemble mammalian IGF. An alternative insulin-like peptide was subsequently identified, dilp8, which encodes a relaxin-like ligand that stimulates G protein-coupled signaling (72, 73). The insulin-like peptide genes are expressed in varied tissues from embryo to adult, and early work showed mutants of these loci affect growth and metabolism (49, 74, 75). In normal adults, dilp2-3, and 5 are primarily produced in median neurosecretory cells (MNC) where they are released into the brain, into secondary endocrine organs, and into circulation (74). In contrast, adult dilp1 is only expressed in MNC during reproductive diapause (76). The MNCs derive from anterior neuroectoderm of the fly embryo, orthologous to vertebrate adenohypophyseal placoid that is the developmental source of mammalian islet-like endocrine cells (77). As well, dilp6 is expressed in the fat body, a tissue with liver- and adipose-related function (78, 79).
Synthetic and recombinant peptides have been used to reveal the function of individual DILPs. Dimeric recombinant DILP5 binds human insulin receptors in a manner consistent with negative cooperativity (80), and when injected into rats and Drosophila the recombinant hormone transiently lowers circulating sugar. DILP5 also interacts with the insect-binding protein Imp-L2 (80, 81), likely to antagonize circulating insulin (82, 83). Notably, elevated Imp-L2 is associated with extended lifespan, even in conditions where dilp2, dilp3, and dilp5 mRNA are elevated (84–86). It is not known which insulins aside from DILP5 bind to Imp-L2.
A synthetic DILP2 was compared to DILP5 when these peptides stimulated Drosophila S2 cells in culture (87). These peptides induced broadly similar signaling elements (Akt, Erk, S6K) and transcriptional profiles, but they also revealed unique outputs. DILP5 produced high, continuous phosphorylation of Akt whereas DILP2 only induced a transient response. In a phosphoproteomic scan, DILP2 equally increased and decreased the number of total phosphorylation sites while DILP5 overwhelmingly increased total phosphorylation. Several specific proteins were differentially phosphorylated by these peptides. Notably, glycogen phosphorylase did not respond to DILP5 but the enzyme was dephosphorylated and inactivated in cells stimulated by DILP2, a response typical for human insulin. Conversely, elevated glycogen phosphorylase activity was found in dilp2 mutant flies, which are long lived, while transgenic expression of GlyP was sufficient to extend lifespan. These data demonstrate measurable differences among specific DILPs acting through a common receptor. And they remind us that the action of insulin-like peptides in aging can involve non-genomic, cellular metabolic regulation independent of canonical FOXO transcription factors.
Understanding DILP function in vivo is complicated because mutation of one dilp changes the expression of others (66). Nonetheless, abundant data shows longevity is extended when dilp2 is reduced alone or with other insulins (66, 79, 88). dilp2 expression in adults is greatest on diet of low protein and high sugar (65). In contrast, adult dilp1 is absent under normal conditions soon after eclosion, but is elevated 14-fold in dilp2 mutants and 4-fold during diapause (76, 89). Post (89) demonstrated that dilp1 is required for loss of dilp2 to extend lifespan, but dilp1 is not required for the loss of dilp2 to induce dilp3 and dilp5 or stimulate phosphorylation of Akt. In contrast, loss of dilp2 represses pErk in a dilp1 dependent manner. DILP1 and DILP2 appear to have countervailing functions associated with diapause, longevity, and Erk signaling.
These observations suggest how Drosophila insulin-like peptides might regulate the outcomes of InR. We tentatively propose DILP5 (and perhaps DILP3) strongly stabilizes InR dimers; DILP2 transiently stabilizes the dimer; DILP1 inhibits InR stability and competitively blocks other insulin-like ligands. In good environments, DILP2, DILP3, and DILP5 promote dimer stability and kinase activity. This activates pAKT and pERK signal transduction to promote growth and reproduction. In this state DILP2 simultaneously represses dilp1. Conditions of limited diet repress dilp3 and dilp5 but not dilp2 (65); dimer stability is moderately reduced. This state still phosphorylates Akt but diminishes kinase signaling, which down-regulates reproduction and improves survival. At the extreme, in diapause, dilp1 is transcribed. Abundant DILP1 inhibits the binding of other insulin ligands to the receptor, minimizes dimer stability, prevents Akt phosphorylation to retard reproduction, and extinguishes Grb2/Drk-Erk signaling to induce systems of longevity assurance.
This sketch is speculative and incomplete. No work yet reveals how DILP1 or DILP2 interact with InR, or how any DILP affects dimer stability or substrate protein interaction. We have not considered DILP6, perhaps the most IGF-like fly ligand, which non-autonomous affects aging through its action in the fat body (78, 79). Little functional data are available for DILP3 despite its abundance in adults. There is much work ahead.
The Paradox Of Insulin Resistance and Longevity
How could altered insulin-like signaling support healthy human aging as found in C. elegans and Drosophila? One solution argues the domain-defined functions of the invertebrate insulin-like receptor are distributed across the mammalian IR and IGFR receptors. The Arg1466Cys substitution of the Drosophila kinase insert domain promotes longevity without impairing growth and reproduction, or incurring loss of kinase activity (stimulated pAkt). Similar outcomes arise in chico heterozygotes and when the SH2/Grb site of Chico is blocked. None of these genotypes are particularly hyperglycemic or insulin resistant (17, 40). Instead, insulin resistance occurs in InR genotypes where we predict the mutations reduce stability of activated protomers. These outcomes suggest we identify where the longevity assurance function of the Drosophila KID translates to human IR or IGFR. While the human kinase insert domains share the KID sequence Arg-Pro-Glu where Arg1466Cys is substituted in Drosophila InR353, the human KID are small and lack the SH2 motif proposed for Drosophila. It is possible in the evolution of insulin-like receptors that some ancestral KID functions were integrated into the four insulin receptor substrates of mammals, as may also be the case of the Drosophila IRS-like C-terminal tail. In particular, IRS2 contains SH2 binding motifs that recruit Grb2, and mice mutant for IRS2 are long-lived (90). This property of IRS2 could involve interactions with IGF1R (91, 92). Notably, human polymorphisms in IGF1R are associated with survival to extreme age (3), and mice heterozygous for IGF1R are long-lived in some genetic backgrounds (93). It would be interesting to explore how these IGF1R genotypes affect specific phospho-sites of IRS2, and whether they alter Grb/Ras/Erk signaling.
If human aging can be modulated by IGFR-IRS2, insulin resistance is not required to slow aging (94), which is otherwise a paradox attributable to invertebrate models. Insulin resistance and slow aging indeed covary in Drosophila but the traits are decoupled in a mutation of the KID that potentially avoids loss of high dimer-stability signaling. Parallel benefits in humans might occur through elements of IGFR-mediated signaling rather than through reduced insulin sensitivity.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the author, without undue reservation.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
MT has been supported to study the role of insulin signaling in aging by the National Institutes of Health through awards NIH R01AG16632 and R37AG024360.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks the extended work and careful experimentation provided by Rochele Yamamoto, Aleksandra Norton, and the whole team of the Tatar lab. The author is grateful for the years of mentoring and discussion on insulin receptor biology provided by P. de Meyts, R. Kohanski, B. Garofalo, B. Forbes, Y. Suh, X. Bai, M. Mohammadi, and S. Takahashi. The kinase domain structure model of Figure 2 was generously provided by Rebecca Page (University of Connecticut Health Sciences Center).
References
1. Albani D, Batelli S, Polito L, Vittori A, Pesaresi M, Gajo GB, et al. A polymorphic variant of the insulin-like growth factor 1 (IGF-1) receptor correlates with male longevity in the Italian population: a genetic study and evaluation of circulating IGF-1 from the “Treviso Longeva (TRELONG)” study. BMC Geriatr (2009) 9:19. doi: 10.1186/1471-2318-9-19
2. Altintas O, Park S, Lee SJ. The role of insulin/IGF-1 signaling in the longevity of model invertebrates, C. elegans and D. melanogaster. BMB Rep (2016) 49:81–92. doi: 10.5483/BMBRep.2016.49.2.261
3. Suh Y, Atzmon G, Cho M-O, Hwang D, Liu B, Leahy DJ, et al. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci (2008) 105:3438–3442%R. doi: 10.1073/pnas.0705467105
4. Texada MJ, Koyama T, Rewitz K. Regulation of Body Size and Growth Control. Genetics (2020) 216:269–313. doi: 10.1534/genetics.120.303095
5. Geminard C, Arquier N, Layalle S, Bourouis M, Slaidina M, Delanoue R, et al. Control of Metabolism and Growth Through Insulin-Like Peptides in Drosophila. Diabetes (2006) 55:S5–S8%R. doi: 10.2337/db06-S001
6. Kuo TH, Fedina TY, Hansen I, Dreisewerd K, Dierick HA, Yew JY, et al. Insulin signaling mediates sexual attractiveness in Drosophila. PloS Genet (2012) 8:e1002684. doi: 10.1371/journal.pgen.1002684
7. Nelson DW, Padgett RW. Insulin worms its way into the spotlight. Genes Dev (2003) 17:813–8. doi: 10.1101/gad.1090203
8. Metaxakis A, Tain LS, Gronke S, Hendrich O, Hinze Y, Birras U. and L. Partridge, Lowered insulin signalling ameliorates age-related sleep fragmentation in Drosophila. PloS Biol (2014) 12:e1001824. doi: 10.1371/journal.pbio.1001824
9. Honda Y, Tanaka M, Honda S. Modulation of longevity and diapause by redox regulation mechanisms under the insulin-like signaling control in Caenorhabditis elegans. Exp Gerontol (2008) 43:520. doi: 10.1016/j.exger.2008.02.009
10. Murphy CT, Hu PJ. Insulin/insulin-like growth factor signaling in C. elegans. WormBook. ed. The C. elegans Research Community (2013). doi: 10.1895/wormbook.1.164.1
11. Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev (2018) 98:2133–223. doi: 10.1152/physrev.00063.2017
12. LeRoith D, Roberts CT Jr. Insulin-like growth factors and their receptors in normal physiology and pathological states. J Pediatr Endocrinol (1993) 6:251–5. doi: 10.1515/JPEM.1993.6.3-4.251
13. Park JI, Chang CL, Hsu SY. New Insights into biological roles of relaxin and relaxin-related peptides. Rev Endocr Metab Disord (2005) 6:291–6. doi: 10.1007/s11154-005-6187-x
14. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev (2009) 30:586–623. doi: 10.1210/er.2008-0047
15. Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science (2003) 299:1346–51. doi: 10.1126/science.1081447
16. Fernandes de Abreu DA, Caballero A, Fardel P, Stroustrup N, Chen Z, Lee K, et al. An insulin-to-insulin regulatory network orchestrates phenotypic specificity in development and physiology. PloS Genet (2014) 10:e1004225. doi: 10.1371/journal.pgen.1004225
17. Yamamoto R, Palmer MJ, Koski H, Curtis-Joseph N, Tatar M. Aging modulated by the Drosophila insulin receptor through distinct structure-defined mechanisms. Genetics (2021) 217(2):iyaa037. doi: 10.1093/genetics/iyaa037
18. Zinkle A, Mohammadi M. A threshold model for receptor tyrosine kinase signaling specificity and cell fate determination. F1000Research (2018) 7(F1000 Faculty Rev):872. doi: 10.12688/f1000research.14143.1
19. Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell (1995) 80:179–85. doi: 10.1016/0092-8674(95)90401-8
20. Sato T, Araki I, Nakamura H. Inductive signal and tissue responsiveness defining the tectum and the cerebellum. Development (2001) 128:2461–9.
21. Vasudevan HN, Mazot P, He F, Soriano P. Receptor tyrosine kinases modulate distinct transcriptional programs by differential usage of intracellular pathways. eLife (2015) 4:e07186. doi: 10.7554/eLife.07186
22. Boller S, Joblin BA, Xu L, Item F, Trub T, Boschetti N, et al. From signal transduction to signal interpretation: an alternative model for the molecular function of insulin receptor substrates. Arch Physiol Biochem (2012) 118:148–55. doi: 10.3109/13813455.2012.671333
23. Liu BA, Jablonowski K, Shah EE, Engelmann BW, Jones RB, Nash PD. SH2 domains recognize contextual peptide sequence information to determine selectivity. Mol Cell Proteomics: MCP (2010) 9:2391–404. doi: 10.1074/mcp.M110.001586
24. Royal I, Fournier TM, Park M. Differential requirement of Grb2 and PI3-kinase in HGF/SF-induced cell motility and tubulogenesis. J Cell Physiol (1997) 173:196–201. doi: 10.1002/(SICI)1097-4652(199711)173:2<196::AID-JCP20>3.0.CO;2-D
25. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell (2010) 141:1117–34. doi: 10.1016/j.cell.2010.06.011
26. Gutmann T, Kim KH, Grzybek M, Walz T, Coskun U. Visualization of ligand-induced transmembrane signaling in the full-length human insulin receptor. J Cell Biol (2018) 217:1643–9. doi: 10.1083/jcb.201711047
27. Uchikawa E, Choi E, Shang G, Yu H, Bai XC. Activation mechanism of the insulin receptor revealed by cryo-EM structure of the fully liganded receptor-ligand complex. eLife (2019) 8:e48630. doi: 10.7554/eLife.48630
28. Chen L, Marsiglia WM, Chen H, Katigbak J, Erdjument-Bromage H, Kemble DJ, et al. Molecular basis for receptor tyrosine kinase A-loop tyrosine transphosphorylation. Nat Chem Biol (2020) 16:267–77. doi: 10.1038/s41589-019-0455-7
29. Gems D, Sutton ML, Sundermeyer PS, Albert KV, King ML, Edgley PL, et al. Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics (1998) 150:129–55.
30. Patel DS, Garza-Garcia A, Nanji M, McElwee JJ, Ackerman D, Driscoll PC, et al. Clustering of genetically defined allele classes in the Caenorhabditis elegans DAF-2 insulin/IGF-1 receptor. Genetics (2008) 178:931–46. doi: 10.1534/genetics.107.070813
31. Fernandez R, Tabarini D, Azpiazu N, Frasch M, Schlessinger J. The Drosophila insulin receptor homolog: a gene essential for embryonic development encodes two receptor isoforms with different signaling potential. EMBO J (1995) 14:3373–84. doi: 10.1002/j.1460-2075.1995.tb07343.x
32. Marr MT 2nd, D’Alessio JA, Puig O, Tjian R. IRES-mediated functional coupling of transcription and translation amplifies insulin receptor feedback. Genes Dev (2007) 21:175–83. doi: 10.1101/gad.1506407
33. Marin-Hincapie M, Garofalo RS. The carboxly terminal extension of the Drosophila insulin receptor homologue binds IRS-1 and influences cell survival. J Biol Chem (1999) 247:24987–94. doi: 10.1074/jbc.274.35.24987
34. Yenush L, Fernandez R, Myers J, Grammer M.G, T.C., Sun XJ, Blenis J, et al. The Drosophila insulin receptor activates multiple signaling pathways but requires insulin receptor substrate proteins for DNA synthesis. Mol Cell Biol (1996) 16:2509–17. doi: 10.1128/MCB.16.5.2509
35. Ruan Y, Chen C, Cao Y, Garofalo RS. The Drosophila insulin receptor contains a novel carboxyl-terminal extension likely to play an important role in signal transduction. J Biol Chem (1995) 270:4236–43. doi: 10.1074/jbc.270.9.4236
36. Bai H, Post S, Kang P, Tatar M. Drosophila Longevity Assurance Conferred by Reduced Insulin Receptor Substrate Chico Partially Requires d4eBP. PloS One (2015) 10:e0134415. doi: 10.1371/journal.pone.0134415
37. Tu MP, Epstein D, Tatar M. The demography of slow aging in male and female Drosophila mutant for the insulin-receptor substrate homologue chico. Aging Cell (2002) 1:75–80. doi: 10.1046/j.1474-9728.2002.00010.x
38. Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science (2001) 292:104–6. doi: 10.1126/science.1057991
39. Yamamoto R, Tatar M. Insulin receptor substrate chico acts with the transcription factor FOXO to extend Drosophila lifespan. Aging Cell (2011) 10:729–32. doi: 10.1111/j.1474-9726.2011.00716.x
40. Slack C, Alic N, Foley A, Cabecinha M, Hoddinott MP. and L. Partridge, The Ras-Erk-ETS-Signaling Pathway Is a Drug Target for Longevity. Cell (2015) 162:72–83. doi: 10.1016/j.cell.2015.06.023
41. Libert S, Chao Y, Zwiener J, Pletcher SD. Realized immune response is enhanced in long-lived puc and chico mutants but is unaffected by dietary restriction. Mol Immunol (2008) 45:810–7. doi: 10.1016/j.molimm.2007.06.353
42. Bohni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, et al. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell (1999) 97:865–75. doi: 10.1016/S0092-8674(00)80799-0
43. Slack C, Werz C, Wieser D, Alic N, Foley A, Stocker H, et al. and L. Partridge, Regulation of lifespan, metabolism, and stress responses by the Drosophila SH2B protein, Lnk. PloS Genet (2010) 6:e1000881. doi: 10.1371/journal.pgen.1000881
44. Song W, Ren D, Li W, Jiang L, Cho KW, Huang P, et al. SHB2 regulation of growth, metabolism, and longevity in both insects and mammals. Cell Metab (2010) 11:427–37. doi: 10.1016/j.cmet.2010.04.002
45. Werz C, Kohler K, Hafen E, Stocker H. The Drosophila SH2B family adaptor Lnk acts in parallel to chico in the insulin signaling pathway. PloS Genet (2009) 5:e1000596. doi: 10.1371/journal.pgen.1000596
46. Slack C, Giannakou ME, Foley A, Goss M. and L. Partridge, dFOXO-independent effects of reduced insulin-like signaling in Drosophila. Aging Cell (2011) 10:735–48. doi: 10.1111/j.1474-9726.2011.00707.x
47. Flatt T. Survival costs of reproduction in Drosophila. Exp Gerontol (2011) 46:369–75. doi: 10.1016/j.exger.2010.10.008
48. Locascio LE, Donoghue DJ. KIDs rule: regulatory phosphorylation of RTKs. Trends Biochem Sci (2013) 38:75–84. doi: 10.1016/j.tibs.2012.12.001
49. Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr Biol (2001) 11:213–21. doi: 10.1016/S0960-9822(01)00068-9
50. Takahashi Y, Kadowaki H, Momomura K, Fukushima Y, Orban T, Okai T, et al. A homozygous kinase-defective mutation in the insulin receptor gene in a patient with leprechaunism. Diabetologia (1997) 40:412–20. doi: 10.1007/s001250050695
51. Dillin A, Gottschling DE, Nystrom T. The good and the bad of being connected: the integrons of aging. Curr Opin Cell Biol (2014) 26:107–12. doi: 10.1016/j.ceb.2013.12.003
52. Li CR, Guo D, Pick L. Independent signaling by Drosophila insulin receptor for axon guidance and growth. Front Physiol (2014) 4:385. doi: 10.3389/fphys.2013.00385
53. Poltilove RM, Jacobs AR, Haft CR, Xu P, Taylor SI. Characterization of Drosophila insulin receptor substrate. J Biol Chem (2000) 275:23346–54. doi: 10.1074/jbc.M003579200
54. Song J, Wu L, Chen Z, Kohanski RA, Pick L. Axons guided by insulin receptor in Drosophila visual system. Science (2003) 300:502–5. doi: 10.1126/science.1081203
55. Almudi I, Poernbacher I, Hafen E, Stocker H. The Lnk/SH2B adaptor provides a fail-safe mechanism to establish the Insulin receptor-Chico interaction. Cell Commun Signal (2013) 11:26. doi: 10.1186/1478-811X-11-26
56. Rui L. SH2B1 regulation of energy balance, body weight, and glucose metabolism. World J Diabetes (2014) 5:511–26. doi: 10.4239/wjd.v5.i4.511
57. Morris DL, Cho KW, Zhou Y, Rui L. SH2B1 enhances insulin sensitivity by both stimulating the insulin receptor and inhibiting tyrosine dephosphorylation of insulin receptor substrate proteins. Diabetes (2009) 58:2039–47. doi: 10.2337/db08-1388
58. Berg CA, Spradling AC. Studies on the rate and site-specificity of P element transposition. Genetics (1991) 127:515–24. doi: 10.1093/genetics/127.3.515
59. Oldham S, Stocker H, Laffargue M, Wittwer F, Wymann M, Hafen E. The Drosophila insulin/IGF receptor controls growth and size by modulating PtdInsP(3) levels. Development (2002) 129:4103–9.
60. Murphy CT. The search for DAF-16/FOXO transcriptional targets: approaches and discoveries. Exp Gerontol (2006) 41:910–21. doi: 10.1016/j.exger.2006.06.040
61. Alic N, Giannakou ME, Papatheodorou I, Hoddinott MP, Andrews TD, Bolukbasi E. and L. Partridge, Interplay of dFOXO and two ETS-family transcription factors determines lifespan in Drosophila melanogaster. PloS Genet (2014) 10:e1004619. doi: 10.1371/journal.pgen.1004619
62. Bai H, Kang P, Hernandez AM, Tatar M. Activin signaling targeted by insulin/dFOXO regulates aging and muscle proteostasis in Drosophila. PloS Genet (2013) 9:e1003941. doi: 10.1371/journal.pgen.1003941
63. Raabe T, Olivier JP, Dickson B, Liu X, Gish GD, Pawson T, et al. Biochemical and genetic analysis of the Drk SH2/SH3 adaptor protein of Drosophila. EMBO J (1995) 14:2509–18. doi: 10.1002/j.1460-2075.1995.tb07248.x
64. Shaw LM. The insulin receptor substrate (IRS) proteins: at the intersection of metabolism and cancer. Cell Cycle (2011) 10:1750–6. doi: 10.4161/cc.10.11.15824
65. Post S, Tatar M. Nutritional Geometric Profiles of Insulin/IGF Expression in Drosophila melanogaster. PloS One (2016) 11:e0155628. doi: 10.1371/journal.pone.0155628
66. Gronke S, Clarke DF, Broughton S, Andrews TD. and L. Partridge, Molecular evolution and functional characterization of Drosophila insulin-like peptides. PloS Genet (2010) 6:e1000857. doi: 10.1371/journal.pgen.1000857
67. Nassel DR, Kubrak OI, Liu Y, Luo J, Lushchak OV. Factors that regulate insulin producing cells and their output in. Front Physiol (2013) 4:252. doi: 10.3389/fphys.2013.00252
68. Kubrak OI, Kucerova L, Theopold U, Nassel DR. The sleeping beauty: how reproductive diapause affects hormone signaling, metabolism, immune response and somatic maintenance in Drosophila melanogaster. PloS One (2014) 9:e113051. doi: 10.1371/journal.pone.0113051
69. Arpagaus M. Vertebrate insulin induces diapause termination in Pieris brassicae pupae. Roux’s Archieve Dev Biol (1987) 196:527–30. doi: 10.1007/BF00399877
70. Castillo-Quan JI, Tain LS, Kinghorn KJ, Li L, Grönke S, Hinze Y, et al. and L. Partridge, A triple drug combination targeting components of the nutrient-sensing network maximizes longevity. Proc Natl Acad Sci (2019) 116:20817–9. doi: 10.1073/pnas.1913212116
71. Wu Q, Brown MR. Signaling and function of insulin-like peptides in insects. Annu Rev Entomol (2006) 51:1–24. doi: 10.1146/annurev.ento.51.110104.151011
72. Garelli A, Gontijo AM, Miguela V, Caparros E, Dominguez M. Imaginal discs secrete insulin-like peptide 8 to mediate plasticity of growth and maturation. Science (2012) 336:579–82. doi: 10.1126/science.1216735
73. Vallejo DM, Juarez-Carreno S, Bolivar J, Morante J, Dominguez M. A brain circuit that synchronizes growth and maturation revealed through Dilp8 binding to Lgr3. Science (2015) 350:aac6767. doi: 10.1126/science.aac6767
74. Ikeya T, Galic M, Belawat P, Nairz K, Hafen E. Nutrient-dependent expression of insulin-like peptides from neuroendocrine cells in the CNS contributes to growth regulation in Drosophila. Curr Biol (2002) 12:1293–300. doi: 10.1016/S0960-9822(02)01043-6
75. Rulifson EJ, Kim SK, Nusse R. Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science (2002) 296:1118–20. doi: 10.1126/science.1070058
76. Liu Y, Liao S, Veenstra JA, Nassel DR. Drosophila insulin-like peptide 1 (DILP1) is transiently expressed during non-feeding stages and reproductive dormancy. Sci Rep (2016) 6:26620. doi: 10.1038/srep26620
77. Wang S, Tulina N, Carlin DL, Rulifson EJ. The origin of islet-like cells in Drosophila identifies parallels to the vertebrate endocrine axis. Proc Natl Acad Sci U.S.A. (2007) 104:19873–8. doi: 10.1073/pnas.0707465104
78. Slaidina M, Delanoue R, Gronke S, Partridge L, Leopold P. A Drosophila insulin-like peptide promotes growth during nonfeeding states. Dev Cell (2009) 17:874–84. doi: 10.1016/j.devcel.2009.10.009
79. Bai H, Kang P, Tatar M. Drosophila insulin-like peptide-6 (dilp6) expression from fat body extends lifespan and represses secretion of Drosophila insulin-like peptide-2 from the brain. Aging Cell (2012) 11:978–85. doi: 10.1111/acel.12000
80. Sajid W, Kulahin N, Schluckebier G, Ribel U, Henderson HR, Tatar M, et al. Structural and biological properties of the Drosophila insulin-like peptide 5 show evolutionary conservation. J Biol Chem (2011) 286:661–73. doi: 10.1074/jbc.M110.156018
81. Roed NK, Viola CM, Kristensen O, Schluckebier G, Norrman M, Sajid W, et al. Structures of insect Imp-L2 suggest an alternative strategy for regulating the bioavailability of insulin-like hormones. Nat Commun (2018) 9:3860. doi: 10.1038/s41467-018-06192-3
82. Honegger B, Galic M, Kohler K, Wittwer F, Brogiolo W, Hafen E, et al. Imp-L2, a putative homolog of vertebrate IGF-binding protein 7, counteracts insulin signaling in Drosophila and is essential for starvation resistance. J Biol (2008) 7:10. doi: 10.1186/jbiol72
83. Arquier N, Geminard C, Bourouis M, Jarretou G, Honegger B, Paix A, et al. Drosophila ALS regulates growth and metabolism through functional interaction with insulin-like peptides. Cell Metab (2008) 7:333–8. doi: 10.1016/j.cmet.2008.02.003
84. Flatt T, Min KJ, D’Alterio C, Villa-Cuesta E, Cumbers J, Lehmann R, et al. Drosophila germ-line modulation of insulin signaling and lifespan. Proc Natl Acad Sci U.S.A. (2008) 105:6368–73. doi: 10.1073/pnas.0709128105
85. Alic N, Hoddinott MP, Vinti G. and L. Partridge, Lifespan extension by increased expression of the Drosophila homologue of the IGFBP7 tumour suppressor. Aging Cell (2011) 10:137–47. doi: 10.1111/j.1474-9726.2010.00653.x
86. Paik D, Jang YG, Lee YE, Lee YN, Yamamoto R, Gee HY, et al. Misexpression screen delineates novel genes controlling Drosophila lifespan. Mech Ageing Dev (2012) 133:234–45. doi: 10.1016/j.mad.2012.02.001
87. Post S, Karashchuk G, Wade JD, Sajid W, De Meyts P, Tatar M. Drosophila Insulin-Like Peptides DILP2 and DILP5 Differentially Stimulate Cell Signaling and Glycogen Phosphorylase to Regulate Longevity. Front Endocrinol (2018) 9:245. doi: 10.3389/fendo.2018.00245
88. Hwangbo DS, Gershman B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature (2004) 429:562–6. doi: 10.1038/nature02549
89. Post S, Liao S, Yamamoto R, Veenstra JA, Nassel DR, Tatar M. Drosophila insulin-like peptide dilp1 increases lifespan and glucagon-like Akh expression epistatic to dilp2. Aging Cell (2019) 18:e12863. doi: 10.1111/acel.12863
90. Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science (2007) 317:369–72. doi: 10.1126/science.1142179
91. White MF. IRS2 integrates insulin/IGF1 signalling with metabolism, neurodegeneration and longevity. Diabetes Obes Metab (2014) 16 Suppl 1:4–15. doi: 10.1111/dom.12347
92. He W, Craparo A, Zhu Y, O’Neill TJ, Wang LM, Pierce JH, et al. Interaction of insulin receptor substrate-2 (IRS-2) with the insulin and insulin-like growth factor I receptors. Evidence for two distinct phosphotyrosine-dependent interaction domains within IRS-2. J Biol Chem (1996) 271:11641–5. doi: 10.1074/jbc.271.20.11641
93. Xu J, Gontier G, Chaker Z, Lacube P, Dupont J, Holzenberger M. Longevity effect of IGF-1R(+/-) mutation depends on genetic background-specific receptor activation. Aging Cell (2014) 13:19–28. doi: 10.1111/acel.12145
Keywords: aging, Drosophila, insulin receptor, insulin, IGF, reproduction, longevity, insulin resistance
Citation: Tatar M (2021) Aging Regulated Through a Stability Model of Insulin/Insulin Growth Factor Receptor Function. Front. Endocrinol. 12:649880. doi: 10.3389/fendo.2021.649880
Received: 05 January 2021; Accepted: 08 February 2021;
Published: 11 March 2021.
Edited by:
Jeff M. P. Holly, University of Bristol, United KingdomReviewed by:
Andrzej Bartke, Southern Illinois University School of Medicine, United StatesCunming Duan, University of Michigan, United States
Sebastian Grönke, Max-Planck-Gesellschaft (MPG), Germany
Copyright © 2021 Tatar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc Tatar, TWFyY19UYXRhckBCcm93bi5lZHU=