 Sandra Heller
Sandra Heller Michael Karl Melzer1,2
Michael Karl Melzer1,2 Alexander Kleger
Alexander Kleger- 1Department of Internal Medicine I, Ulm University Hospital, Ulm, Germany
- 2Department of Urology, Ulm University Hospital, Ulm, Germany
- 3Université de Paris, Institut Cochin, INSERM U1016, CNRS UMR-8104, Paris, France
Diabetes, as one of the major diseases in industrial countries, affects over 350 million people worldwide. Type 1 (T1D) and type 2 diabetes (T2D) are the most common forms with both types having invariable genetic influence. It is accepted that a subset of all diabetes patients, generally estimated to account for 1–2% of all diabetic cases, is attributed to mutations in single genes. As only a subset of these genes has been identified and fully characterized, there is a dramatic need to understand the pathophysiological impact of genetic determinants on β-cell function and pancreatic development but also on cell replacement therapies. Pluripotent stem cells differentiated along the pancreatic lineage provide a valuable research platform to study such genes. This review summarizes current perspectives in applying this platform to study monogenic diabetes variants.
Introduction
Diabetes, as one of the major diseases in industrial countries, affects over 350 million people worldwide. Type 1 (T1D) and type 2 diabetes (T2D) are the most common forms. T2D accounts for most diabetes cases and is a multifactorial metabolic disease where insulin deficiency is caused by insulin resistance in target organs and pancreatic β-cell failure. The current diabetes classifications are insufficient to explain the large clinical and biological variability of diabetes, suggesting an unrecognized level of heterogeneity (1). T1D is described as a chronic autoimmune disease against insulin-producing β-cells leading to hyperglycemia. T1D results from the combination of multiple factors, including environment, genes, and a prominent role of the immune system. Genetic studies have long recognized that mutations of the human leukocyte antigens (HLAs) within the Major Histocompatibility Complex (MHC) represent major genetic risk factors in T1D (2, 3). More recently, genome-wide association studies (GWAS) and candidate gene approaches have identified more than 50 other loci contributing to T1D risk, including INS, PTPN22, CTLA4, and GLIS3 genes (4, 5). In addition, mutations in several genes, such as AIRE, FOXP3, and STAT (6, 7), may cause rare monogenic forms of autoimmune diabetes.
Similarly, genetic studies of T2D identified many single nucleotide polymorphisms (SNPs) associated with T2D risk, which are located near several functionally relevant genes such as PPARG (8), WFS1 (9), KCNJ11 (10), KLF14 (9), ANK1 (11), INS (12), HNF1A (9), HNF1B (13), and GLIS3 (14). In addition to genetic predisposition, environmental factors and epigenetic changes are influencing the pathophysiology of T2D, which may contribute to the additional variance in susceptibility.
Overall, genetic studies of T1D and T2D resulted in the identification of many disease-associated variants, most of which, with the exception of the HLA locus for T1D, contribute to a small increase in disease risk (5, 15, 16). These studies have provided valuable information on putative genes and the mechanisms involved in diabetes. For example, many genes identified by T2D GWAS are expressed in human islets (17) and may regulate β-cell mass and function (18). While a large proportion of T1D susceptibility genes are surprisingly not related to the immune system (19), studies from D. Eizirik’s group have also shown that >60% of these genes are expressed in β-cells, and their expression is affected upon exposure to cytokines, viruses, and double-stranded RNA, a by-product of viral infection, in human and rodent β-cells (19–21). Altogether, these studies suggest an essential role of mechanisms acting at the level of β-cells in the etiology of both T1D and T2D (19–21).
Interestingly, several of these genes are involved in both monogenic diabetes (rare variants) and multifactorial diabetes (frequent variants). This is the case of the insulin gene (INS, monogenic neonatal diabetes, Maturity Onset of Diabetes in the Young (MODY) and multifactorial T1D), as well as KCNJ11, WFS1, HNF1A, HNF1B (neonatal, syndromic, or MODY monogenic diabetes, and multifactorial T2D) and GLIS3 (neonatal monogenic diabetes and multifactorial T1D and T2D). This plethora of genes involved in common multifactorial and rare monogenic forms of diabetes suggests that some disease mechanisms and biological pathways may be shared between different forms of diabetes. The identification and detailed study of genes responsible for monogenic diabetes are therefore extremely valuable to investigate important genes and pathways involved in both monogenic diabetes and common forms of diabetes. Noteworthy, it has become evident most recently that a subset of all diabetes patients, generally estimated to account for 1–5% of all diabetic cases, is attributed to mutations in single genes (22, 23).
As only a subset of these genes has been identified and fully characterized, there is a dramatic need to understand the pathophysiological impact of genetic determinants on β-cell function and pancreatic development but also on cell replacement therapies. Although islet transplantation can lead to insulin-independency of diabetic patients for 5 years or longer, this therapeutic option is only accessible for a rare number of patients due to the limited number of cadaveric human islets and complex handling (24). On the other hand, the use of human pluripotent stem cells [hPSCs, induced pluripotent (iPSC) and embryonic stem cells (hESC)] may bypass this need by generating mature β-cells in vitro upon improving the current protocols of β-cell generation.
PSCs have been used as a relevant model system to elucidate pathophysiological mechanisms in diseases such as diabetes, blood disorders, defined neurological disorders, and genetic liver disease (25–27). Induced pluripotent stem cells (iPSCs) allow dissecting monogenic human disease mechanisms (28) as well as mechanisms of genetically complex human disorders such as schizophrenia (29). This opens promising perspectives in both regenerative medicine but also in drug development to screen for innovative, “druggable” targets (30) and to develop ex vivo gene-targeting therapies (28). Given the still high intra- and interpatient variability of patient-derived iPSCs, controls are the key for a precise analysis (31). Recent advances in the development of genomic editing tools such as the Zinc-finger or clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 technology have further revolutionized this research field. Now researchers can precisely modify a human pluripotent stem cell genome with (i) high efficiency, (ii) on a single-base resolution, (iii) without altering the pluripotent capacity, and (iv) with negligible off-target effects to provide isogenic controls and to facilitate data interpretation. In turn, these recent tools represent novel state-of-the-art disease-in-a-dish models and will pioneer research fields aiming to understand also the mechanisms underlying monogenic diseases (32–35). Human pancreatic disease modeling is highly dependent on reliable and efficient differentiation protocols for human PSCs. We and others have recently challenged the currently existing protocols (36), first, to optimize the step toward pancreatic progenitor cells (37), second, to drive maturation in a 3D environment (38–40), and third, to increase yields of true monohormonal β-cells (41–44). In turn, optimized differentiation platforms now allow for appropriately modeling complex pancreatic diseases such as diabetes (45). A schematic overview of currently available disease modeling tools for diabetes employing hPSCs is presented in Figure 1.
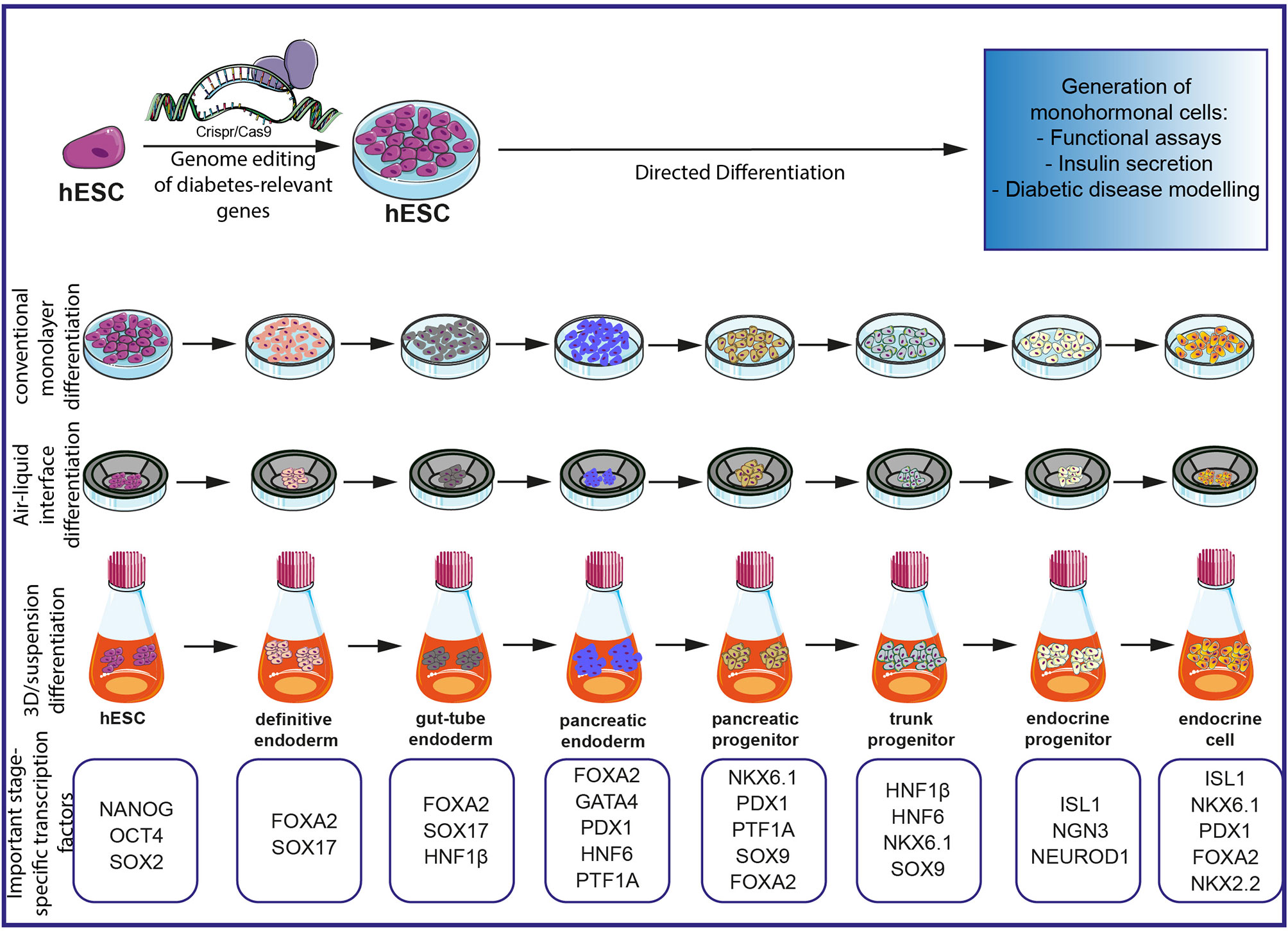
Figure 1 Schematic overview of currently existing protocols for the investigation of diabetes on an hPSC-based platform. Gene editing with CRISPR/Cas9 allows precise editing of diabetes-relevant genes and generation of hESC for further differentiation experiments. Different differentiation protocols allow the generation of monohormonal cells by passing through different milestones during embryonic development. Important stage-specific transcription factors are indicated below the schematics. Subsequent analysis of monohormonal β-cells, including insulin secretions assays, can be performed and generate hypotheses about the influence of specific genetic variants. The figure was modified from Smart Servier Medical Art (https://smart.servier.com/) under a Creative Common Attribution 3.0 Generic License.
Maturity-Onset Diabetes of the Young
So far, 14 subtypes of maturity-onset diabetes of the young (MODY) have been described to be caused by mainly heterozygous dominant mutations in genes for pancreas-specific transcription factors as well as enzymes, hormones, and ion channels (46, 47). These mutations impair endocrine function at various levels ranging from alterations in development, glucose sensing, synthesis, and storage of insulin to inappropriate secretion of insulin in β-cells. The most frequently identified mutations are located in the HNF4A gene (MODY1) (48) with a frequency of 4–10% (49–51), in the GCK gene (MODY2) (52) with 30–60% (49–51, 53) and in the HNF1A gene (MODY3) (54) with 30–50% (49–51) depending on the study population. MODY1 patients are particularly characterized by defective glucose-stimulated insulin secretion possibly caused by disrupted gene expression playing a role in glucose transport and glycolysis (48, 55, 56). MODY2 due to glucokinase deficiency often results in mild hyperglycemia during early life (57). These patients have a defect in glucose-stimulated insulin secretion caused by impaired glucose sensitivity in β-cells. MODY3 patients develop β-cell dysfunction and hyperglycemia caused by impairment of glucose-dependent insulin secretion (58).
In addition, rare MODY cases [accounting for up to 6% of all MODY forms (49)] have been diagnosed with mutations in PDX1 (59), HNF1B (60), NEUROD1 (61, 62), KLF11 (63), CEL (64), PAX4 (65), INS (66), BLK (67), ABCC8 (68), KCNJ11 (69), and APPL1 (70) (known as MODY4-14).
mpaired functions of proteins caused by pathogenic variants can vary depending on the nature of the mutation, therefore causing a spectrum of clinical manifestations. Patients harboring heterozygous HNF1B mutations suffer from MODY, but may also feature pancreas exocrine dysfunction as well as kidney and liver abnormalities (71) and vaginal and uterine malformation (72). Few specified cases of NEUROD1 mutations are characterized mainly by early onset diabetes (61, 62, 73), but patients with neurological defects such as pituitary gland hypoplasia, growth hormone deficiency, epilepsy, and intellectual disability have also been described (74, 75). CEL mutations cause early onset diabetes associated with exocrine pancreatic dysfunction and chronic pancreatitis (76, 77). In addition to the diabetic phenotype in patients with PAX4 mutations, diabetic complications such as retinopathy and nephropathy have been observed (65, 78). MODY-causing INS mutations have been associated with early onset diabetes as well as ketoacidosis in some cases (79), whereas rare cases with BLK mutations have also been associated with overweight (67).
No other clear clinical manifestation besides diabetes has been described for patients with heterozygous mutations in KLF11 (MODY7), ABCC8 (MODY12), KCNJ11 (MODY13), and APPL1 (MODY14). A summary of confirmed MODY-causing mutations as well as of other prominent clinical features is presented in Table 1.
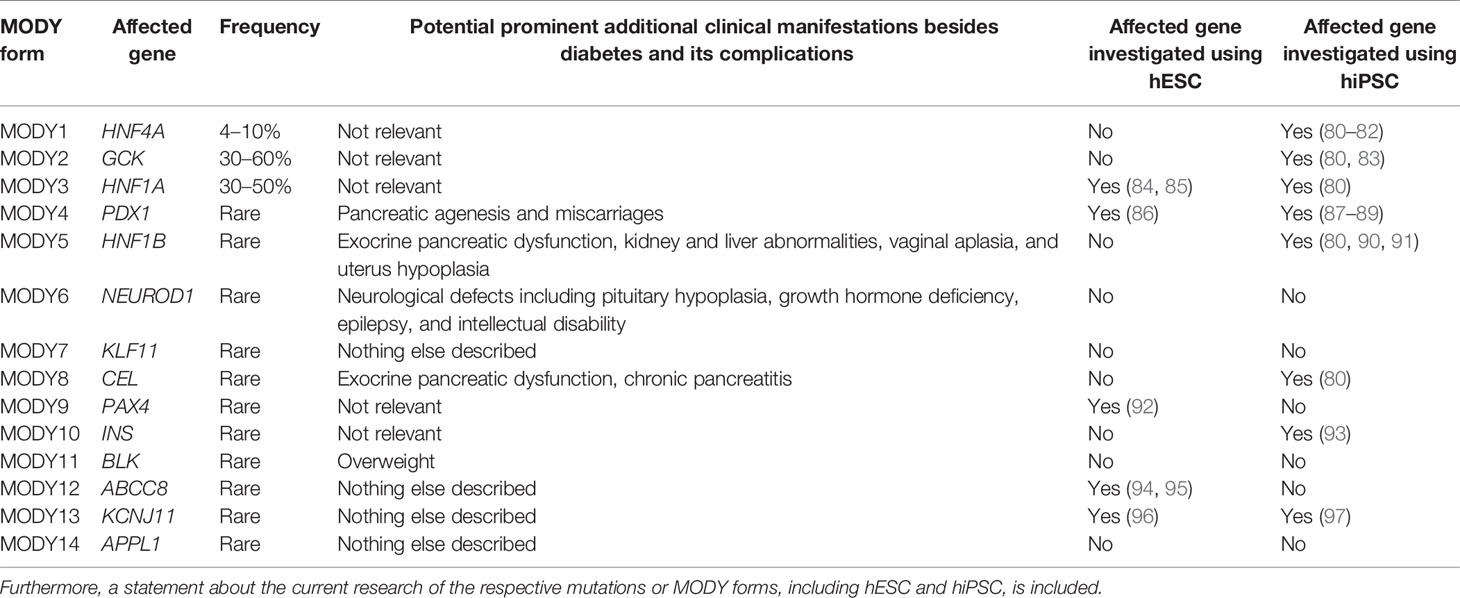
Table 1 Different MODY forms, including their frequencies, affected genes, and potential other prominent clinical manifestations are presented.
Noteworthy, the homozygous status for mutations in several MODY genes has been found to lead to extreme clinical presentations, contrasting with the less severe early onset diabetes observed in heterozygous carriers. For example, homozygous mutations in the PDX1 gene result in early onset diabetes associated with pancreatic agenesis and maternal miscarriages (59, 98).
A subset of patients with MODY-like phenotype doesn’t carry any mutation in the known MODY genes, suggesting the involvement of additional genes. The identification of these additional genes responsible for rare MODY forms is now facilitated by the availability of large databases of diabetic cases and control cohorts that enable increased efficiency to detect novel genes with rare contributing variants (including MODY-like effects) (99), compared to earlier studies with smaller sample size (100). In addition, the availability of large databases of control subjects (e.g., gnomAD, TOPMED) provides now the possibility to estimate the frequency of rare coding variants in candidate genes, hence allowing for efficient association and burden-testing for rare monogenic contributions, such as MODY. Consequently, recent studies identified RFX6 as a novel MODY gene (101) and WFS1, PPARG, and GLIS3 have recently been proposed as potential candidates for these rare MODY forms (101–103).
Taken into consideration the overlap in genes involved in common multifactorial and rare monogenic forms of diabetes, the specific analysis of monogenic pathogenic variants can therefore reveal novel interaction partners and gene targets that might be helpful to better understand the mechanisms involved in the onset of T1D and T2D.
Permanent Neonatal Diabetes Mellitus
Permanent neonatal diabetes mellitus (PNDM) is the second form of monogenic diabetes. It is characterized by hyperglycemia and partial or complete insulin deficiency in patients in the first 6 months postnatal (104, 105). Moreover, patients with PNDM may suffer from intrauterine growth retardation, glycosuria, ketoacidosis, failure to thrive as well as various clinical features depending on the gene. Mutations in more than 20 genes with monogenic contribution important for β-cell development have been identified to cause PNDM (46). Treatment of PNDM includes oral sulfonylureas or insulin therapy and may require pancreatic enzyme replacement for infants with pancreatic aplasia or hypoplasia.
Some genes, including ABCC8 (106), GCK (107), INS (108), KCNJ11 (109), and PDX1 (110) may alternatively cause PNMD or MODY, with various severity and clinical features depending on the gene, nature of the mutation, and genotype (homozygous or heterozygous). Common variants in these genes may also be associated with multifactorial T1D or T2D.
In addition to diabetes, KCNJ11-PNMD patients may also have neurological features such as developmental delay and epilepsy (DEND syndrome) (111). Similarly, pathogenic homozygous PDX1-PNDM patients have pancreatic agenesis and pancreatic hypoplasia leading to exocrine pancreatic insufficiency (110, 112). Pancreatic agenesis is furthermore caused by homozygous mutations in another pancreatic transcription factor, PTF1A (113). Here, PNDM patients additionally suffer from severe intrauterine growth retardation, cerebellar agenesis, and neurological dysfunction.
In addition, PNDM may manifest in the context of specific syndromes. Homozygous mutations in EIF2AK3 cause Wolcott–Rallison syndrome, characterized by PNDM, exocrine pancreas dysfunction, and abnormalities such as liver failure, developmental delay, and epiphyseal dysplasia (114). Inactivating GATA4 variants can induce pancreatic agenesis or hypoplasia, causing PNDM but also lead to extrapancreatic symptoms such as cardiac and neurodevelopmental abnormalities (115). Similarly, GATA6 mutations cause pancreatic agenesis leading to PNDM, together with abnormalities of the heart, biliary tract, and gut development (116). Homozygous mutations in GLIS3 cause PNDM together with congenital hypothyroidism associated with congenital glaucoma, hepatic fibrosis, and polycystic kidneys (117). In addition to neonatal diabetes, NEUROD1 mutations cause cerebellar hypoplasia, sensorineural deafness, and visual impairment (118), whereas NEUROG3 mutations affect intestinal development leading to congenital malabsorptive diarrhea (119–121). Mutations in PAX6, encoding a transcription factor involved in β-cell development as well as eye and brain development, cause neonatal diabetes combined with abnormalities of the central nervous system and visual system (e.g. microencephaly, optic nerve defects, microphthalmia) (122). In addition to PNDM, mutations in the transcription factor RFX6 cause pancreatic hypoplasia, intestinal atresia, and gall bladder hypoplasia (123, 124). Patients with Wolfram syndrome caused by mutations in WFS1 suffer from early onset diabetes as well as optic atrophy, deafness, ataxia, and dementia (125). Other neonatal diabetes syndromes have been described for mutations in SLC19A2 (associated with thiamin-responsive megaloblastic anemia, neurological disorders, cardiac abnormalities, and deafness) (126, 127), MNX1 (associated with growth retardation, delayed central nervous system development, hypoplastic lungs, renal maldevelopment, skeletal dysplasia) (128), NKX2.2 (further leading to growth retardation, delayed central nervous system development, constipation) (128), and IER3IP1 (additional microcephaly, CNS maldevelopment) (129). Furthermore, some mutations in the glucose transporter SLC2A2 can cause neonatal diabetes prior to the Fanconi–Bickel syndrome associated with glycosuria, galactosemia, aminoaciduria, proteinuria, hepatomegaly, as well as glucose and galactose intolerance (130, 131). Franco et al. recently showed that mutations in YIPF5 cause neonatal diabetes associated with microcephaly and epilepsy (132).
Interestingly, an overlapping phenotype between PNDM and autoimmune T1D was observed for a patient with an activating mutation in the STAT3 gene (133). Although the autoimmune-mediated destruction of β-cells was prominent Saarimäki-Vire et al. revealed an additional mechanism (PNDM) due to the observed pancreatic hypoplasia (134).
An overview of all described genes leading to the development of PNDM if affected by mutations is presented in Table 2.
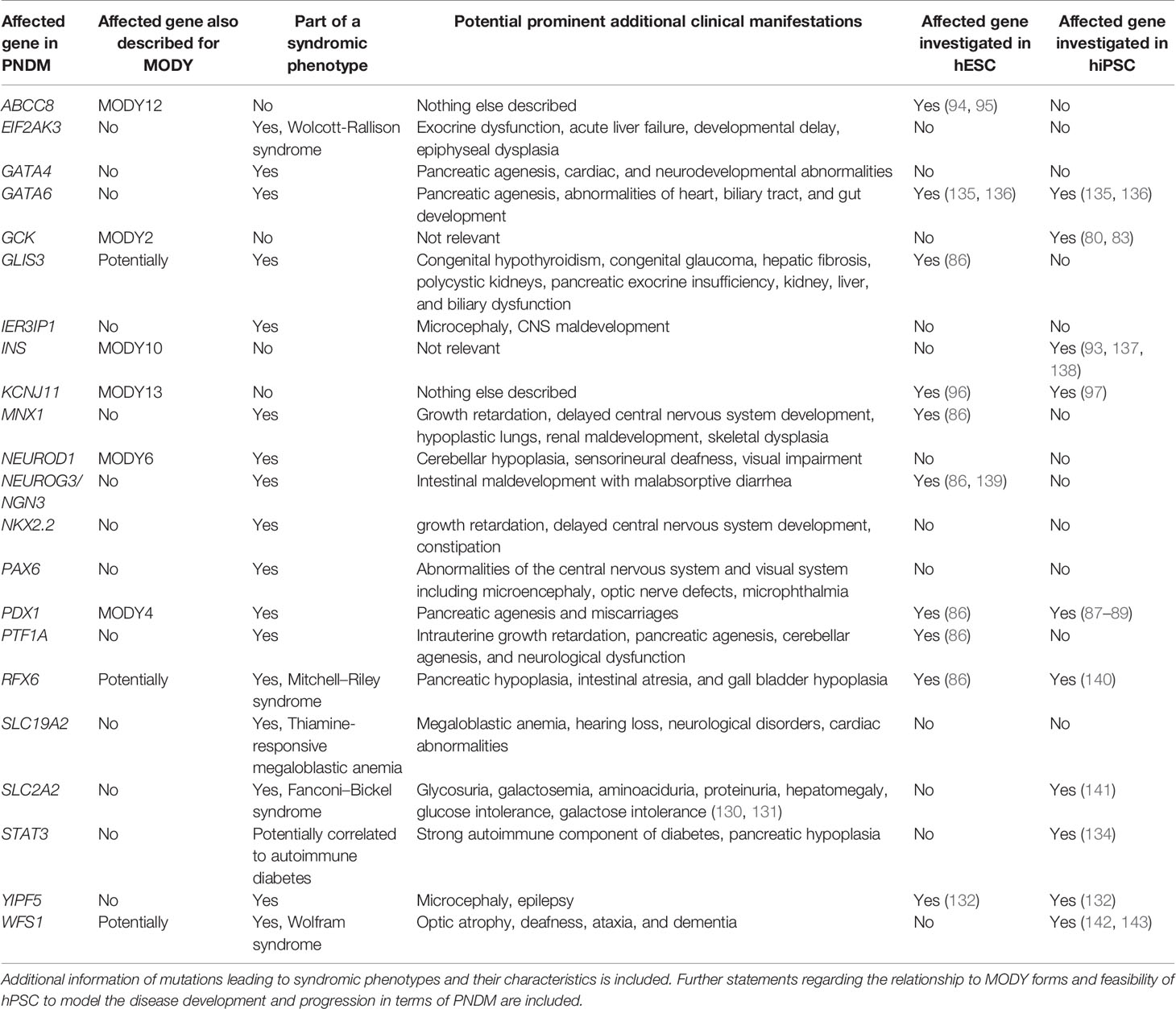
Table 2 Overview of mutations in genes that can lead to PNDM.
Modeling Pancreatic Endocrine Development
The detailed pathomechanisms of monogenic diabetes are not yet fully understood since mouse models do not completely recapitulate the human disease phenotype (121, 144, 145), and patient samples such as β-cells have very limited availability. Moreover, animal models with a specific knockout of MODY genes show species-specific differences that do not entirely recapitulate the patient phenotype (146–150). Therefore, even more suitable disease models are crucial to develop an adequate therapy.
In the recent years, human pluripotent stem cells have been deployed as a suitable human model system. On the one hand, human embryonic stem cells (hESCs) can be subjected to a directed differentiation protocol to investigate different mechanisms during differentiation of mature pancreatic β-cells. Additionally, patient-specific induced pluripotent stem cells (iPSCs) can be generated from materials such as fibroblasts and keratinocytes, allowing to address various genetic backgrounds of patients. Of note, gene-mutated iPSCs show high heterogeneity in terms of differentiation efficiency and are best controlled with isogenic, repaired lines. Furthermore, patient-specific iPSCs are a useful tool for biobanking because of their unlimited expansion capacity. Subsequently, these cells are differentiated in vitro into disease-relevant cell types such as pancreatic endocrine cells or β-cells.
To better understand the importance of certain genes in the maturation of β-cells, genetic engineering may be performed with hESCs and iPSCs. Here, state-of-the-art gene-editing tools such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the more recent clustered regularly interspaced short palindromic repeats (CRISPRs)/Cas allow the generation of specific point mutations or gene knockouts (KOs). A potential option of genetic engineering might involve gene correction in iPSCs. This allows the generation of autologous β-cells for transplantation that may circumvent immune reaction and donor scarcity.
The generation of pancreatic endocrine cells is achieved by different differentiation protocols. Established protocols try to mimic signaling pathways of in vivo embryonic developmental stages by involving different/various combinations of growth factors, cytokines, and small molecules known/reported to guide the stem cells through stages of definitive endoderm, gut-tube endoderm, pancreatic endoderm, pancreatic progenitors, and endocrine progenitors to finally yield mature mono-hormonal endocrine cells (Figure 1).
Overall differentiation models of hESCs and hiPSCs provide a versatile tool to study the influence of genetic disorders on β-cell development in the human pancreas as well as the embryonic development of the human pancreas itself. Different published protocols whose concepts are described below allow the investigation of multiple facets of β-cell maturation during different steps of embryonic development. However, the procedure itself may have a huge influence on the phenotype of differentiated cells. This might lead to the bias that a “good” differentiation protocol can overcome the inherent genotypic features, which would rather underestimate the phenotypic features of a certain genotype. This is, for example, the case for a GATA6-mutant iPSC cell line with a severe phenotypic loss of endoderm and β-cell differentiation capacity in a differentiation condition involving low levels of retinoic acid. On the other hand, high levels of retinoic acid mask this phenotype (136). The same principle might apply to a “poor” differentiation protocol that may overestimate the phenotypic properties of a certain genotype.
Generation of Mature Pancreatic β-Cells Requires Complex and Sophisticated Differentiation Protocols
During the last decade, differentiation protocols have been adapted to achieve a more mature state of in vitro differentiated β-cells. Since earlier pancreatic endocrine differentiation protocols in monolayers yield mainly an immature or heterogeneous population of polyhormonal cells lacking robust insulin secretion in response to glucose stimulation, a prerequisite of β-cells (36, 151), novel in vitro approaches including different culture conditions have been established. Recent protocols include a transition to 3D culture using a suspension culture system with spinner flasks and orbital shaker generating endocrine spheres (41, 42). Alternatively, a switch from the initial culture in monolayer to an air–liquid interphase culture stage promoting basal and apical cell polarity generates even more functionally mature β-cells (152) (Figure 1).
Air–liquid interphase culture systems require spotting the cells from the pancreatic endoderm stage on filters. Upon formation of small cell clusters, the differentiation is further improved as measured by NGN3 and insulin expression (43). This transition might help to mimic the natural 3D environment and cell orientation within the developing tissue, thus promoting in vitro differentiation (153).
Further progress in β-cell maturation was also achieved by reaggregating immature cells into enriched β-cell clusters using an insulin-driven fluorescence reporter (154). Veres et al. combined cellular reaggregation and β-cell purification using CD49a to enrich endocrine cells and promote functional maturation of β-cells able to maintain their identity for several weeks in culture (155).
An alternative approach is to enrich precursor cells in the differentiation process. A recent study demonstrated that enrichment of anterior definitive endoderm with CD177 results in a more homogenous pancreatic progenitor population and subsequent better functional maturation (156).
Moreover, besides optimizing technical conditions for differentiation, the modulation of signaling pathways and cytoskeleton is a promising mean to increase the β-cell yield. Inhibition of certain pathways such as ROCKII and WNT using specific inhibitors promoted maturation (157, 158). Hogrebe et al. investigated the role of the actin cytoskeleton in promoting the expression of pancreas-specific transcription factors such as NEUROG3 during differentiation (159). The manipulation of actin polymerization during early developmental stages influences the expression of transcription factors important for the specification of lineage fate in pancreatic progenitors. Depolymerization of the cytoskeleton during endocrine induction further improved the functionality of derived β-cells, also allowing for a planar differentiation protocol.
Another strategy to improve differentiation efficacy is to modulate the basic content of cell culture media. Two studies explored metabolic changes during β-cell maturation (160, 161). Adaptation of nutrient-sensing via mTORC1 signaling during the transition from fetal to adult pancreatic islets can be recapitulated by reduced amino acid content in differentiation media, further advancing cellular insulin content and glucose-stimulated insulin secretion (160). Similarly, epigenomic characterization of primary and in vitro differentiated pancreatic cells revealed that entrainment to cycles of fasting and feeding leads to circadian control of genes important for energy and insulin metabolism, further improving β-cell function (161).
Protocol improvements resulted in more mature β-cells and faster reversal of diabetes after transplantation in mice. However, manifold successful approaches show that regulation of human pancreas development is still not fully understood, and various adaptations to endocrine differentiation protocols are difficult to compare because they use different cell lines and culture methods as well as slightly different functional assays. Therefore, more research is necessary to determine the appropriate combination of culture methods, cytokine and small molecule cocktails, purification markers, and metabolic modifications, generating a protocol that robustly produces the desired pancreatic cell types.
For potential clinical use, several questions/issues regarding the composition of transplanted cells containing only β-cells or more than one endocrine cell type, the best transplantation site, and whether transplanted cells benefit from co-transplantation with other cell types such as mesenchymal stem cells need to be answered. Moreover, long-term survival and functionality of transplanted cells exceeding the life span of mice have to be addressed. Additionally, the possibility of teratoma formation from the remaining progenitor cells even after prolonged time has to be eliminated. Encapsulation of cells in suitable biomaterials such as alginates or synthetic polymer hydrogels might not only reduce the risk of tumor formation but also provide protection from the immune rejection of the host, removing the need for lifelong immunosuppression.
Although the latest research significantly improved our knowledge about transcriptional regulation, signaling pathways as well as metabolic adaptation during in vitro differentiation and maturation of β-cells and paved the way for future clinical use, more research is necessary until in vitro-generated pancreatic endocrine cells can be used as potential diabetes therapy.
Pluripotent Stem Cell Models to Understand Monogenic Diabetes
Although iPSC could be successfully generated from T1D and T2D patients, complex autoimmune reactions, environmental influence, as well as multifactorial genetic factors hampered the intimate recapitulation of pathogenesis (162–164). Despite the complexity of T1D and T2D pathogenesis, some recent approaches have been performed to model T1D using pluripotent stem cells. Co-culture studies of iPSCs derived from T1D patients together with immune cells are one such way to model the mechanisms of T1D in vitro (165). Yet, it has to be kept in mind that this kind of model requires additional prerequisites such as environmental factors and complex composition of different immune cell types as recently reviewed by Joshi et al. (166). Modeling T2D in vitro is far more complex as many more different pathogenic mechanisms can cause or even interact to promote T2D development, including multiple genetic and environmental factors. This fact makes it even harder to investigate T2D by pluripotent stem cell-based approaches solely in vitro.
Whereas modeling T1D and T2D using PSCs remains challenging due to their complex nature, monogenic alterations leading to a MODY or PNMD diabetes phenotype are ideal to be investigated by PSC-based approaches. The role of specific variants of the respective genes has already been investigated using pancreatic differentiation of pluripotent stem cells. Compared to various genetic and environmental aspects contributing to other diabetes types, single mutations present in monogenic diabetes allow tighter control of the observed phenotypes. Deciphered mechanisms for the development of MODY, which were uncovered using hPSC-based systems, are presented in Figure 2 (MODY1, 2, 3, 4, 5, 10, 13).
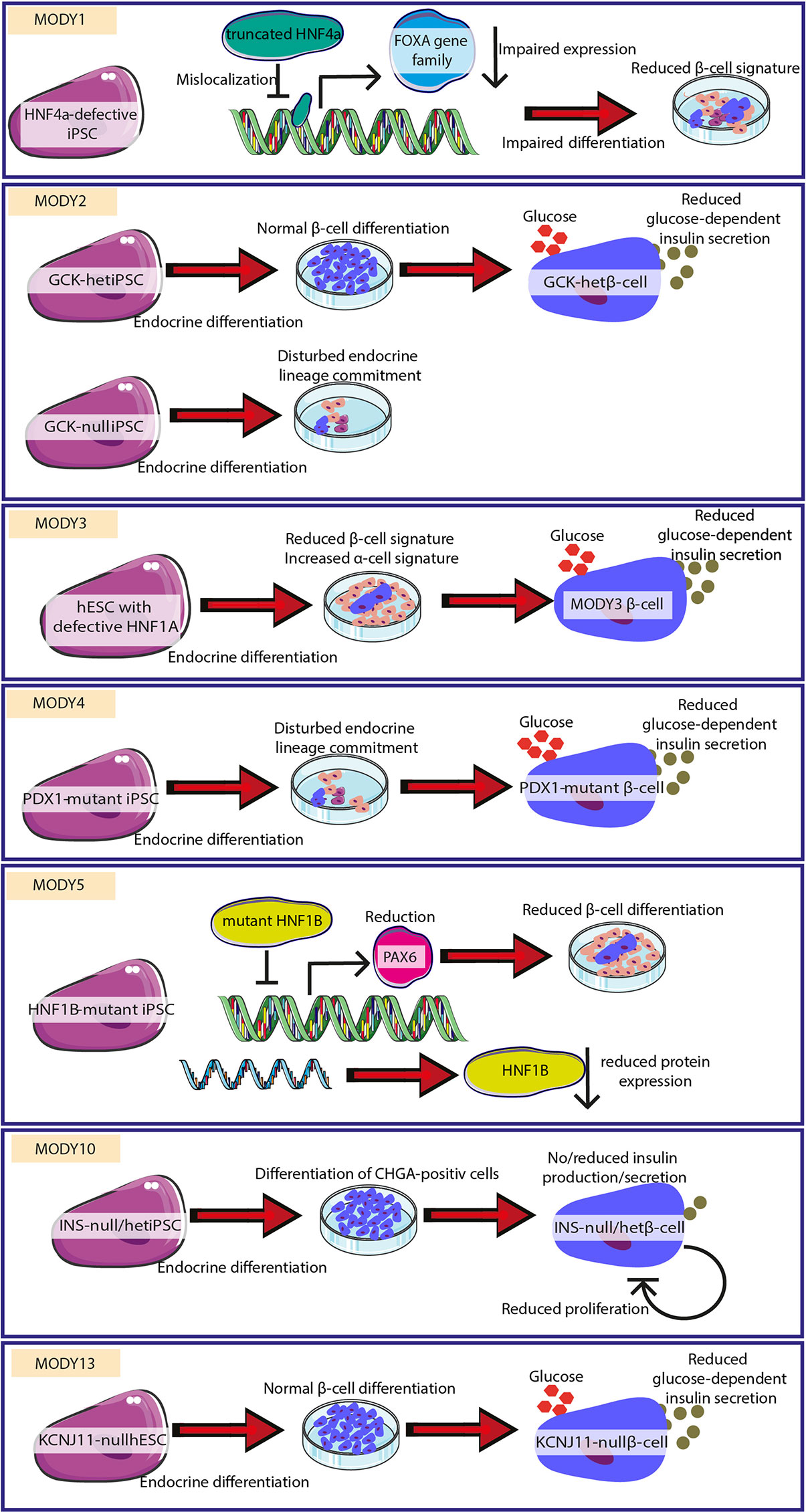
Figure 2 Successful uncovering of pathomechanisms for different MODY forms. Different MODY forms were modeled by employing hESC/hiPSC with respective mutations. Mechanisms leading to monogenic diabetes could be delineated/characterized. In MODY1, mutated HNF4a leads to reduced FOXA gene family expression and impaired β-cell signature. MODY2 is characterized by reduced differentiation or reduced glucose-dependent insulin secretion. MODY3 is caused by reduced β-cell differentiation and insulin secretion. MODY4 shows reduced endocrine lineage entrance and impaired insulin secretion. MODY5 is caused by diminished β-cell differentiation. MODY10 is highlighted by lacking production and secretion of insulin. MODY13 is characterized by impaired glucose-dependent insulin secretion. The figure was modified from Smart Servier Medical Art (https://smart.servier.com/) under a Creative Common Attribution 3.0 Generic License.
Teo et al. showed that karyotypically normal iPSC expressing pluripotency markers and able to differentiate in all three germ layers could be derived from different MODY patients (MODY1, MODY2, MODY3, MODY5, and MODY8) (80) and can serve as a tool to study the role of the respective genes in pancreatic development. A more detailed study of these MODY1 iPSCs with premature HNF4A protein truncation revealed impaired foregut and hepatopancreatic progenitor development. These events were associated with HNF4A mislocalization and reduced expression of target genes such as the FOXA gene family, HNF1B, PDX1, GATA4, and RFX6 (81). In turn, impaired activation of target genes disturbs β-cell gene signatures. Prior to that study, iPSCs from MODY1 patients with a nonsense mutation were characterized (82). Here, the patient phenotype was caused by a reduction in levels of functional HNF4A accompanied by increased expression of pancreatic transcription factors and pancreatic hormones as a compensatory mechanism (82).
Understanding the role of transcription factor HNF1A in MODY3, human ESC lacking one or both alleles have been differentiated to study endocrine development (84). HNF1A deficiency increased expression of markers for α-cells but reduced expression of β-cell markers suggesting a role in endocrine hormone expression. In addition, HNF1A is required for insulin secretion, in line with hyperglycemia observed in patients. Moreover, mutated cells show metabolic defects in glycolysis and mitochondrial respiration, also typical for T2D. This observation, together with the finding that frequent HNF1A variants are associated with T2D, suggests a link between mechanisms identified in MODY and in common T2D. Furthermore, iPSCs from a patient harboring heterozygous HNF1A mutation generated by non-integrative viral transduction, show a normal karyotype, and express pluripotency factors (85). These cells can be used for further functional analysis of this specific mutation.
Relevant defects in the GATA6 gene disrupt the endoderm differentiation by decreased cell survival. Later, the pancreas specification and β-cell function were identified using iPSC and genome-edited ESC (135, 136). By circumventing the developmental block at the endoderm stage, cell lines with GATA6 mutations were differentiated with low dose retinoic acid to mimic severe patient phenotype. These cells failed to show normal insulin secretion after glucose stimulation and harbor defective insulin processing (136).
Fibroblasts from patients with heterozygous point mutations in the PDX1 transactivation domain were successfully reprogrammed to iPSCs and can be used to study diabetes-associated pathomechanisms (87–89). Further pancreatic differentiation reveals that mutations in the PDX1 transactivation domain disturb the pancreatic endocrine lineage development and result in impairment of the glucose-responsive function of β-cells (88).
The analysis of patient-derived iPSCs with HNF1B mutations (MODY5) suffering from early onset diabetes and pancreatic hypoplasia revealed a compensatory increase in markers of definitive endoderm and pancreatic transcription factor expression such as PDX1 (90). Additionally, downregulation of transcription factor PAX6, important for islet development (167), may result in the observed patient phenotype (90). Furthermore, iPSCs were generated from a Japanese MODY5 patient with a truncated HNF1B variant in order to account for differences in insulin sensitivity and insulin response depending on the genetic background (91). Yabe et al. compared iPSCs derived from healthy and patient skin fibroblasts and detected degradation of mutant mRNA by the nonsense-mediated decay pathway in differentiated patient-derived iPSCs (91).
A more systematic analysis of pancreatic transcription factors PDX1, RFX6, PTF1A, GLIS3, MNX1, NGN3, HES1, and ARX, partly identified in monogenic variants of MODY and PNDM, characterized the transcriptional control and corresponding defects at several developmental stages (86). This study highlights especially the role of RFX6 in controlling pancreatic progenitor numbers and differences of NEUROG3 requirement in humans and mice. Mutations in WFS1, causing Wolfram syndrome, lead to chronic endoplasmic reticulum stress activating the unfolded protein response, which impairs survival of β-cells and neurons (168–170). This could be recapitulated using iPSC with WFS1 variants (142). A recent publication from Maxwell et al. also characterized iPSC from a Wolfram syndrome patient (143). Patient-specific iPSCs harboring a pathogenic variant of WFS1 were corrected using CRISPR/Cas9 technology. This study used a differentiation protocol with cytoskeletal modification, which significantly improved differentiation efficiency compared to previously tested suspension culture in these cell lines (159). Corrected cells showed higher WFS1 expression and robust insulin secretion, probably benefiting from reduced ER stress and improved mitochondrial respiratory capacity in endocrine cells (143). In turn, further maturation of in vitro generated β-cells allows better identification of effects also in later stages, additionally providing potential use for β-cell replacement therapy.
Homozygous mutations in the insulin gene (INS) are known to lead to PNDM. Pancreatic differentiation of patient iPSC results in CHGA-positive endocrine cells expressing β-cell markers NKX6.1, PDX1, and MAFA but lacking insulin expression (93). Gene correction rescued the phenotype and prevented diabetes in a streptozotocin mouse model, providing a future tool for patient cell therapy. Another study involved iPSCs generated from patients with neonatal diabetes and heterozygous insulin mutations disturbing proper proinsulin folding (137). Patient-derived iPSCs show normal pancreatic differentiation comparable to corrected isogenic iPSC but have reduced insulin expression. Moreover, INS mutation increases ER stress and hampers proliferation of β-cells but without increased apoptosis promoting diabetes development in patients. In addition, fibroblasts from a PNDM patient harboring an intronic INS mutation have been efficiently generated and may serve as a diabetes model to characterize the expected aberrant splicing (138).
Another study characterized iPSCs from MODY2 patients with a heterozygous GCK mutation (83). Similar to control cells, these iPSCs differentiated into insulin-producing β-cells but showed reduced insulin secretion in response to glucose stimulation. In addition, iPSCs with two inactive GCK alleles also showed reduced differentiation efficiency recapitulating the functional impairment observed in patients and mouse models.
After the identification of YIPF5 mutations causing a novel PNDM syndrome, Franco et al. characterized in vitro differentiated patient-derived iPSCs harboring a homozygous YIPF5 mutation as well as genome-edited ESC in addition to a β-cell line (132). Functional impairment of YIPF5, responsible for trafficking between the endoplasmic reticulum and the Golgi apparatus, caused proinsulin retention at the ER resulting in ER stress-induced apoptosis and β-cell failure and, thus, diabetes.
In order to understand the role of activating mutations in STAT3 during pancreatic development, iPSCs derived from patient fibroblasts were subjected to pancreatic differentiation and revealed a premature endocrine differentiation later preferentially forming α-cells which is in line with the observed phenotype of the patient (134). This defect results from enhanced nuclear localization of the mutant protein and NEUROG3 activation and could be rescued by correction of the STAT3 mutation.
Inactivating mutations in ABCC8 resulting in excess insulin secretion have been successfully employed for modeling congenital hyperinsulinism using ABCC8-deficient ESC (94, 95). In contrast, activating mutations in ABCC8 have been described in diabetes (171). Thus, using the hPSC-systems, a better characterization of the components of the β-cell ATP-sensitive potassium channel may be obtained to understand the function of β-cells and associated pathomechanisms such as diabetes (caused by activating mutations) or the vice-versa effect of congenital hyperinsulinism (caused by inactivating mutations).
In the context of T2D susceptibility genes identified in GWAS, Zeng et al. generated ESC with null alleles for KCNJ11, also associated with MODY13 (96). Although the loss of KCNJ11 does not affect in vitro differentiation towards β-like cells and insulin production, these cells show impaired glucose-stimulated insulin secretion (96). In addition to hESCs with KCNJ11 mutations, iPSCs have been generated from peripheral blood mononuclear cells with a heterozygous activating mutation in KCNJ11 and are available for mechanistic studies as well as drug testing in differentiated pancreatic cells (97).
Skin fibroblasts from patients with Michell–Riley syndrome were used to generate iPSCs harboring a homozygous nonsense mutation in RFX6 (140). Pancreatic differentiation revealed an impaired formation of pancreatic endoderm and thus, supports the impaired formation of endocrine cells in the pancreas in line with the patient phenotype.
In order to better understand the impact of different pathogenic NEUROG3 variants, Zhang et al. expressed NEUROG3 mutant proteins at physiological levels in NEUROG3 knockout ESC during pancreatic and intestinal differentiation determining the ability to rescue the generation of endocrine cells (139). Depending on the variant, expression resulted either in the decreased or abolished formation of pancreatic endocrine cells recapitulating the respective patient phenotype. Moreover, these effects could be retraced to be caused by impairment of NEUROG3 protein stability, DNA-binding affinity, and protein dimerization. Those features can differ in various tissues, a fact that emphasizes the importance of considering the relationship between protein structure and function.
Adenoviral PAX4 overexpression during pancreatic differentiation of ESCs results in decreased glucagon-positive cells promoting the formation of monohormonal insulin-positive cells supporting its role in cell fate specification (92). This suggests a crucial role of intact PAX4 in the development of healthy monohormonal insulin-positive cells.
A recent study reported the generation of iPSCs with a homozygous mutation in the SLC2A2 gene (141). Peripheral mononuclear blood cells from a patient suffering from Fanconi–Bickel syndrome accompanied by early onset diabetes were reprogrammed using a non-integrating Sendai virus vector. These cells can be used to study the pathogenesis associated with defects in the GLUT2 glucose transporter in pancreatic β-cells.
In addition to KO mouse models, genome-engineered and patient-specific hPSCs have helped to get more insight into developmental and mechanistic processes as well as transcriptional networks (81). Not only do they provide additional information but sometimes even highlight the species-specific differences that make them even more crucial for a better understanding of monogenic diabetes.
A conditional Hnf4a KO in mice did not result in a diabetic phenotype but revealed that expression of the potassium channel subunit Kir6.2 regulating insulin secretion is promoted by Hnf4a (148). Patient-specific iPSCs inform about changes in transcriptional network (81). Similarly, Hnf1a mouse models with heterozygous KO present without developing diabetes, whereas a homozygous KO impairs β-cell function by reducing the insulin secretion. Stem cell-based models provided further details characterizing the developmental, transcriptional, and metabolic role of HNF1a (84). Furthermore, loss of Wfs1 in mice showed impairment of β-cell development and function with a mild diabetic phenotype (149). Patient-specific iPSCs provided deeper insight into endoplasmatic reticulum stress and were used to test a possible therapeutic approach (142). Since GATA6 haploinsufficiency resulting in pancreatic agenesis in patients cannot be recapitulated in mice (172, 173), effects of GATA6 gene dosage on pancreatic differentiation in vitro helped in understanding the clinical presentation of different patients (135). Furthermore, hPSC models facilitate the characterization of disease-specific variants. For example, a Hnf1b heterozygous KO in mice is not associated with a diabetic phenotype (150), but patient-specific iPSC carrying a heterozygous variant of HNF1B helped to explain the MODY5 phenotype (90). In mice, Neurog3 is essential for the development of the endocrine pancreas (144), but the disease phenotype slightly differs in humans (121). Expression of different disease-associated NEUROG3 variants during in vitro differentiation helped in explaining various phenotypes in patients (139).
Taken together, a comparison of human and mouse model systems can provide further insight into the role of specific genes but also highlights the species-specific differences concerning, for example, transcription factor activity. That explains why human PSC-based models are crucially needed to compensate for those specific differences.
Therefore, genome editing in PSC or even patient-specific hiPSC provide a versatile approach to study developmental and functional effects of selected diabetes genes and variants and complement or even contradict data obtained from mouse models.
So far, many stem-cell-based models exist that characterize monogenic mutations resulting in early-onset diabetes. These models nicely elucidate the diabetic patient phenotype and help in understanding the common pathways in β-cell development and function. Altogether, improved screening for pathogenic variants in combination with thorough functional analysis will be the first step to precision medicine in diabetes therapy.
Conclusion/Outlook
In vitro pancreatic differentiation of pluripotent stem cells is a powerful tool to better understand pancreatic development and the specific role of the involved transcription factors. Identification and characterization of specific variants in monogenic diabetes help in characterizing the complex transcriptional network and in overcoming phenotypic differences between patients and corresponding mouse models. In addition, these model systems provide the basis for drug development and testing that could benefit both patients of monogenic and multifactorial diabetes. Ideally, iPSCs of genetically disordered persons could be repaired and serve as a major source for tissue engineering and regeneration, e.g. β-cells in the case of monogenic diabetes.
Yet, some major roadblocks need to be kept in mind before translating genetically repaired iPSCs into clinics. First, epigenetic modifications of iPSCs which are derived from their originating tissue, might reduce their differentiation capacity and subsequent function as well as immune tolerance after autologous transplantation (174). Furthermore, genetic aberrations after reprogramming might bear tumorigenic potential and thus provoke carcinogenesis in the transplanted iPSC-derived tissues (174). Additionally, iPSC-derived tissues need to be manufactured according to SOPs and GMP guidelines which need lots of effort to implement those prerequisites into standard clinical care (175).
Altogether, these studies further urge the involvement of pluripotent stem cells in deciphering the underlying pathomechanisms as well as the affected genes, particularly when monogenic diabetes displays discrete clinical phenotypes and needs specific treatment depending on the subtype.
Author Contributions
All authors wrote the paper. MM constructed figures. All authors contributed to the article and approved the submitted version.
Funding
ANR-DFG collaborative research project (ANR-18-CE92-0031, DFG KL 2544/5-1) to CJ and AK; DFG funding with the identifiers KL 2544/6-1 (“Heisenberg-Programm”), KL 2544/7-1 (“Sachbeihilfe”) and KL 2544/1-2 (HEIST RTG) to AK; Agence Nationale pour la Recherche (ANR-09-GENO-021), the European Foundation for the Study of Diabetes/JDRF/Novo Nordisk, the Assistance Publique-Hôpitaux de Paris Programme Hospitalier de Recherche Clinique, and France Génomique (project DIAPED) to CJ. Novo Nordisk was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. MM received funding from Ulm University in the Clinician Scientist Program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The figures were modified from Smart Servier Medical Art (https://smart.servier.com/) under a Creative Common Attribution 3.0 Generic License.
Abbreviations
CRISPR, Clustered regularly interspaced short palindromic repeats; ER, Endoplasmatic reticulum; GWAS, Genome-wide association studies; hESC, Human embryonic stem cell; HLA, Human leucocyte antigen; hPSC, Human pluripotent stem cell; iPSC, Induced pluripotent stem cell; JOD, Juvenile-onset diabetes; KO, Knockout; MHC, Major histocompatibility complex; MODY, Maturity Onset of Diabetes in the Young; PNDM, Permanent neonatal diabetes mellitus; SNP, Single nucleotide polymorphism; T1D, Type 1 diabetes; T2D, Type 2 diabetes.
References
1. Merger SR, Leslie RD, Boehm BO. The Broad Clinical Phenotype of Type 1 Diabetes At Presentation. Diabetes Med (2013) 30:170–8. doi: 10.1111/dme.12048
2. Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE, et al. Localization of Type 1 Diabetes Susceptibility to the MHC Class I Genes HLA-B and HLA-A. Nature (2007) 450:887–92. doi: 10.1038/nature06406
3. Nerup J, Platz P, Andersen OO, Christy M, Lyngsøe J, Poulsen J, et al. HL-a Antigens and Diabetes Mellitus. Lancet (1974) 304:864–6. doi: 10.1016/S0140-6736(74)91201-X
4. Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine Mapping of Type 1 Diabetes Susceptibility Loci and Evidence for Colocalization of Causal Variants With Lymphoid Gene Enhancers. Nat Genet (2015) 47:381–6. doi: 10.1038/ng.3245
5. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-Wide Association Study and Meta-Analysis Find That Over 40 Loci Affect Risk of Type 1 Diabetes. Nat Genet (2009) 41:703–7. doi: 10.1038/ng.381
6. Fabbri M, Frixou M, Degano M, Fousteri G. Type 1 Diabetes in STAT Protein Family Mutations: Regulating the Th17/Treg Equilibrium and Beyond. Diabetes (2019) 68:258–65. doi: 10.2337/db18-0627
7. Hattersley AT, Patel KA. Precision Diabetes: Learning From Monogenic Diabetes. Diabetologia (2017) 60:769–77. doi: 10.1007/s00125-017-4226-2
8. Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, et al. Genome-Wide Trans-Ancestry Meta-Analysis Provides Insight Into the Genetic Architecture of Type 2 Diabetes Susceptibility. Nat Genet (2014) 46:234–44. doi: 10.1038/ng.2897
9. Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve Type 2 Diabetes Susceptibility Loci Identified Through Large-Scale Association Analysis. Nat Genet (2010) 42:579. doi: 10.1038/ng.609
10. Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, et al. Replication of Genome-Wide Association Signals in UK Samples Reveals Risk Loci for Type 2 Diabetes. Science (2007) 316:1336–41. doi: 10.1126/science.1142364
11. Imamura M, Maeda S, Yamauchi T, Hara K, Yasuda K, Morizono T, et al. A Single-Nucleotide Polymorphism in ANK1 is Associated With Susceptibility to Type 2 Diabetes in Japanese Populations. Hum Mol Genet (2012) 21:3042–9. doi: 10.1093/hmg/dds113
12. Ng MC, Shriner D, Chen BH, Li J, Chen W-M, Guo X, et al. Meta-Analysis of Genome-Wide Association Studies in African Americans Provides Insights Into the Genetic Architecture of Type 2 Diabetes. PloS Genet (2014) 10:e1004517. doi: 10.1371/journal.pgen.1004517
13. Li H, Gan W, Lu L, Dong X, Han X, Hu C, et al. A Genome-Wide Association Study Identifies GRK5 and RASGRP1 as Type 2 Diabetes Loci in Chinese Hans. Diabetes (2013) 62:291–8. doi: 10.2337/db12-0454
14. Cho YS, Chen C-H, Hu C, Long J, Ong RTH, Sim X, et al. Meta-Analysis of Genome-Wide Association Studies Identifies Eight New Loci for Type 2 Diabetes in East Asians. Nat Genet (2012) 44:67. doi: 10.1038/ng.1019
15. Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, et al. New Genetic Loci Implicated in Fasting Glucose Homeostasis and Their Impact on Type 2 Diabetes Risk. Nat Genet (2010) 42:105–16. doi: 10.1038/ng.520
16. Morris JP4, Cano DA, Sekine S, Wang SC, Hebrok M. Beta-Catenin Blocks Kras-Dependent Reprogramming of Acini Into Pancreatic Cancer Precursor Lesions in Mice. J Clin Invest (2010) 120:508–20. doi: 10.1172/JCI40045
17. Cnop M, Abdulkarim B, Bottu G, Cunha DA, Igoillo-Esteve M, Masini M, et al. RNA Sequencing Identifies Dysregulation of the Human Pancreatic Islet Transcriptome by the Saturated Fatty Acid Palmitate. Diabetes (2014) 63:1978–93. doi: 10.2337/db13-1383
18. Florez JC. Newly Identified Loci Highlight Beta Cell Dysfunction as a Key Cause of Type 2 Diabetes: Where are the Insulin Resistance Genes? Diabetologia (2008) 51:1100–10. doi: 10.1007/s00125-008-1025-9
19. Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, et al. Genetics of Type 1 Diabetes: What’s Next? Diabetes (2010) 59:1561–71. doi: 10.2337/db10-0076
20. Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, et al. The Human Pancreatic Islet Transcriptome: Expression of Candidate Genes for Type 1 Diabetes and the Impact of Pro-Inflammatory Cytokines. PloS Genet (2012) 8:e1002552. doi: 10.1371/journal.pgen.1002552
21. Santin I, Moore F, Colli ML, Gurzov EN, Marselli L, Marchetti P, et al. PTPN2, a Candidate Gene for Type 1 Diabetes, Modulates Pancreatic Beta-Cell Apoptosis Via Regulation of the BH3-only Protein Bim. Diabetes (2011) 60:3279–88. doi: 10.2337/db11-0758
22. Schwitzgebel VM. Many Faces of Monogenic Diabetes. J Diabetes Invest (2014) 5:121–33. doi: 10.1111/jdi.12197
23. Shepherd M, Shields B, Hammersley S, Hudson M, McDonald TJ, Colclough K, et al. Systematic Population Screening, Using Biomarkers and Genetic Testing, Identifies 2.5% of the U.K. Pediatric Diabetes Population With Monogenic Diabetes. Diabetes Care (2016) 39:1879–88. doi: 10.2337/dc16-0645
24. Bellin MD, Barton FB, Heitman A, Harmon JV, Kandaswamy R, Balamurugan AN, et al. Potent Induction Immunotherapy Promotes Long-Term Insulin Independence After Islet Transplantation in Type 1 Diabetes. Am J Transplant (2012) 12:1576–83. doi: 10.1111/j.1600-6143.2011.03977.x
25. Maehr R, Chen S, Snitow M, Ludwig T, Yagasaki L, Goland R, et al. Generation of Pluripotent Stem Cells From Patients With Type 1 Diabetes. Proc Natl Acad Sci USA (2009) 106:15768–73. doi: 10.1073/pnas.0906894106
26. Raya A, Rodriguez-Piza I, Guenechea G, Vassena R, Navarro S, Barrero MJ, et al. Disease-Corrected Haematopoietic Progenitors From Fanconi Anaemia Induced Pluripotent Stem Cells. Nature (2009) 460:53–9. doi: 10.1038/nature08129
27. Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, et al. Modeling Inherited Metabolic Disorders of the Liver Using Human Induced Pluripotent Stem Cells. J Clin Invest (2010) 120:3127–36. doi: 10.1172/JCI43122
28. Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, et al. Recapitulation of Premature Ageing With iPSCs From Hutchinson-Gilford Progeria Syndrome. Nature (2011) 472:221–5. doi: 10.1038/nature09879
29. Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, et al. Modelling Schizophrenia Using Human Induced Pluripotent Stem Cells. Nature (2011) 473(7346):221–5. doi: 10.1038/nature09915
30. Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, et al. Modelling the Long QT Syndrome With Induced Pluripotent Stem Cells. Nature (2011) 471:225–9. doi: 10.1038/nature09747
31. Thatava T, Kudva YC, Edukulla R, Squillace K, De Lamo JG, Khan YK, et al. Intrapatient Variations in Type 1 Diabetes-Specific iPS Cell Differentiation Into Insulin-Producing Cells. Mol Ther J Am Soc Gene Ther (2013) 21:228–39. doi: 10.1038/mt.2012.245
32. Maetzel D, Sarkar S, Wang H, Abi-Mosleh L, Xu P, Cheng AW, et al. Genetic and Chemical Correction of Cholesterol Accumulation and Impaired Autophagy in Hepatic and Neural Cells Derived From Niemann-Pick Type C Patient-Specific iPS Cells. Stem Cell Rep (2014) 2:866–80. doi: 10.1016/j.stemcr.2014.03.014
33. Sarkar S, Carroll B, Buganim Y, Maetzel D, Ng AH, Cassady JP, et al. Impaired Autophagy in the Lipid-Storage Disorder Niemann-Pick Type C1 Disease. Cell Rep (2013) 5:1302–15. doi: 10.1016/j.celrep.2013.10.042
34. Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, et al. Isogenic Human iPSC Parkinson’s Model Shows Nitrosative Stress-Induced Dysfunction in MEF2-PGC1alpha Transcription. Cell (2013) 155:1351–64. doi: 10.1016/j.cell.2013.11.009
35. Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L, Glatza M, et al. Genetic Correction of a LRRK2 Mutation in Human iPSCs Links Parkinsonian Neurodegeneration to ERK-dependent Changes in Gene Expression. Cell Stem Cell (2013) 12:354–67. doi: 10.1016/j.stem.2013.01.008
36. D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, et al. Production of Pancreatic Hormone–Expressing Endocrine Cells From Human Embryonic Stem Cells. Nat Biotechnol (2006) 24:1392–401. doi: 10.1038/nbt1259
37. Nostro MC, Sarangi F, Yang C, Holland A, Elefanty AG, Stanley EG, et al. Efficient Generation of NKX6-1+ Pancreatic Progenitors From Multiple Human Pluripotent Stem Cell Lines. Stem Cell Rep (2015) 4:591–604. doi: 10.1016/j.stemcr.2015.02.017
38. Hohwieler M, Illing A, Hermann PC, Mayer T, Stockmann M, Perkhofer L, et al. Human Pluripotent Stem Cell-Derived Acinar/Ductal Organoids Generate Human Pancreas Upon Orthotopic Transplantation and Allow Disease Modelling. Gut (2017) 66:473–86. doi: 10.1136/gutjnl-2016-312423
39. Hohwieler M, Perkhofer L, Liebau S, Seufferlein T, Muller M, Illing A, et al. Stem Cell-Derived Organoids to Model Gastrointestinal Facets of Cystic Fibrosis. U Eur Gastroenterol J (2017) 5:609–24. doi: 10.1177/2050640616670565
40. Hohwieler M, Renz S, Liebau S, Lin Q, Lechel A, Klaus J, et al. “Miniguts” From Plucked Human Hair Meet Crohn’s Disease. Z Gastroenterol (2016) 54:748–59. doi: 10.1055/s-0042-105520
41. Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, et al. Controlled Induction of Human Pancreatic Progenitors Produces Functional Beta-Like Cells In Vitro. EMBO J (2015) 34:1759–72. doi: 10.15252/embj.201591058
42. Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of Functional Human Pancreatic β Cells In Vitro. Cell (2014) 159:428–39. doi: 10.1016/j.cell.2014.09.040
43. Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, et al. Reversal of Diabetes With Insulin-Producing Cells Derived In Vitro From Human Pluripotent Stem Cells. Nat Biotechnol (2014) 32:1121–33. doi: 10.1038/nbt.3033
44. Cheng X, Ying L, Lu L, Galvao AM, Mills JA, Lin HC, et al. Self-Renewing Endodermal Progenitor Lines Generated From Human Pluripotent Stem Cells. Cell Stem Cell (2012) 10:371–84. doi: 10.1016/j.stem.2012.02.024
45. Hua H, Shang L, Martinez H, Freeby M, Gallagher MP, Ludwig T, et al. iPSC-derived Beta Cells Model Diabetes Due to Glucokinase Deficiency. J Clin Invest (2013) 123:3146–53. doi: 10.1172/JCI67638
46. Carmody D, Støy J, Greeley SAW, Bell GI, Philipson LH. A Clinical Guide to Monogenic Diabetes. Genetic Diagnosis of Endocrine Disorders: Elsevier (2016) p. 21–30. doi: 10.1016/B978-0-12-800892-8.00002-6
47. Bishay RH, Greenfield JR. A Review of Maturity Onset Diabetes of the Young (MODY) and Challenges in the Management of Glucokinase-MODY. Med J Aust (2016) 205:480–5. doi: 10.5694/mja16.00458
48. Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, et al. Mutations in the Hepatocyte Nuclear Factor-4α Gene in Maturity-Onset Diabetes of the Young (MODY1). Nature (1996) 384:458–60. doi: 10.1038/384458a0
49. Shields B, Hicks S, Shepherd M, Colclough K, Hattersley AT, Ellard S. Maturity-Onset Diabetes of the Young (MODY): How Many Cases are We Missing? Diabetologia (2010) 53:2504–8. doi: 10.1007/s00125-010-1799-4
50. Søvika O, Irgens HU, Molnes J, Sagena JV, Bjørkhaug L, Ræder H, et al. Monogenic diabetes mellitus in Norway. Norsk Epidemiol (2013) 23. doi: 10.5324/nje.v23i1.1603
51. Schober E, Rami B, Grabert M, Thon A, Kapellen T, Reinehr T, et al. Phenotypical Aspects of Maturity-Onset Diabetes of the Young (MODY Diabetes) in Comparison With Type 2 Diabetes Mellitus (T2DM) in Children and Adolescents: Experience From a Large Multicentre Database. Diabetic Med (2009) 26:466–73. doi: 10.1111/j.1464-5491.2009.02720.x
52. Sakura H, Eto K, Kadowaki H, Simokawa K, Ueno H, Koda N, et al. Structure of the Human Glucokinase Gene and Identification of a Missense Mutation in a Japanese Patient With Early-Onset non-Insulin-Dependent Diabetes Mellitus. J Clin Endocrinol Metab (1992) 75:1571–3. doi: 10.1210/jcem.75.6.1464666
53. Froguel P, Zouali H, Vionnet N, Velho G, Vaxillaire M, Sun F, et al. Familial Hyperglycemia Due to Mutations in Glucokinase–Definition of a Subtype of Diabetes Mellitus. New Engl J Med (1993) 328:697–702. doi: 10.1056/NEJM199303113281005
54. Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, et al. Mutations in the Hepatocyte Nuclear factor-1alpha Gene in Maturity-Onset Diabetes of the Young (MODY3). Nature (1996) 384:455–8. doi: 10.1038/384455a0
55. Stoffel M, Duncan SA. The Maturity-Onset Diabetes of the Young (MODY1) Transcription Factor HNF4α Regulates Expression of Genes Required for Glucose Transport and Metabolism. Proc Natl Acad Sci (1997) 94:13209–14. doi: 10.1073/pnas.94.24.13209
56. Byrne MM, Sturis J, Fajans SS, Ortiz FJ, Stoltz A, Stoffel M, et al. Altered Insulin Secretory Responses to Glucose in Subjects With a Mutation in the MODY1 Gene on Chromosome 20. Diabetes (1995) 44:699–704. doi: 10.2337/diabetes.44.6.699
57. Velho G, Blanche H, Vaxillaire M, Bellanne-Chantelot C, Pardini V, Timsit J, et al. Identification of 14 New Glucokinase Mutations and Description of the Clinical Profile of 42 MODY-2 Families. Diabetologia (1997) 40:217–24. doi: 10.1007/s001250050666
58. Byrne MM, Sturis J, Menzel S, Yamagata K, Fajans SS, Dronsfield MJ, et al. Altered Insulin Secretory Responses to Glucose in Diabetic and Nondiabetic Subjects With Mutations in the Diabetes Susceptibility Gene MODY3 on Chromosome 12. Diabetes (1996) 45:1503–10. doi: 10.2337/diab.45.11.1503
59. Staffers DA, Ferrer J, Clarke WL, Habener JF. Early-Onset Type-Ll Diabetes Mellitus (MODY4) Linked to IPF1. Nat Genet (1997) 17:138–9. doi: 10.1038/ng1097-138
60. Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in Hepatocyte Nuclear Factor–1β Gene (TCF2) Associated With MODY. Nat Genet (1997) 17:384–5. doi: 10.1038/ng1297-384
61. Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, et al. Mutations in NEUROD1 are Associated With the Development of Type 2 Diabetes Mellitus. Nat Genet (1999) 23:323–8. doi: 10.1038/15500
62. Szopa M, Ludwig-Galezowska AH, Radkowski P, Skupien J, Machlowska J, Klupa T, et al. A Family With the Arg103Pro Mutation in the NEUROD1 Gene Detected by Next-Generation Sequencing–Clinical Characteristics of Mutation Carriers. Eur J Med Genet (2016) 59:75–9. doi: 10.1016/j.ejmg.2016.01.002
63. Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, Dina C, Hamid YH, Joly E, et al. Role of Transcription Factor KLF11 and its Diabetes-Associated Gene Variants in Pancreatic Beta Cell Function. Proc Natl Acad Sci (2005) 102:4807–12. doi: 10.1073/pnas.0409177102
64. Torsvik J, Johansson S, Johansen A, Ek J, Minton J, Ræder H, et al. Mutations in the VNTR of the Carboxyl-Ester Lipase Gene (CEL) are a Rare Cause of Monogenic Diabetes. Hum Genet (2010) 127:55. doi: 10.1007/s00439-009-0740-8
65. Plengvidhya N, Kooptiwut S, Songtawee N, Doi A, Furuta H, Nishi M, et al. PAX4 Mutations in Thais With Maturity Onset Diabetes of the Young. J Clin Endocrinol Metab (2007) 92:2821–6. doi: 10.1210/jc.2006-1927
66. Haneda M, Chan SJ, Kwok S, Rubenstein AH, Steiner DF. Studies on Mutant Human Insulin Genes: Identification and Sequence Analysis of a Gene Encoding [SerB24] Insulin. Proc Natl Acad Sci (1983) 80:6366–70. doi: 10.1073/pnas.80.20.6366
67. Borowiec M, Liew CW, Thompson R, Boonyasrisawat W, Hu J, Mlynarski WM, et al. Mutations At the BLK Locus Linked to Maturity Onset Diabetes of the Young and β-Cell Dysfunction. Proc Natl Acad Sci (2009) 106:14460–5. doi: 10.1073/pnas.0906474106
68. Bowman P, Flanagan S, Edghill E, Damhuis A, Shepherd M, Paisey R, et al. Heterozygous ABCC8 Mutations are a Cause of MODY. Diabetologia (2012) 55:123–7. doi: 10.1007/s00125-011-2319-x
69. Bonnefond A, Philippe J, Durand E, Dechaume A, Huyvaert M, Montagne L, et al. Whole-Exome Sequencing and High Throughput Genotyping Identified KCNJ11 as the Thirteenth MODY Gene. PloS One (2012) 7:e37423. doi: 10.1371/journal.pone.0037423
70. Prudente S, Jungtrakoon P, Marucci A, Ludovico O, Buranasupkajorn P, Mazza T, et al. Loss-of-Function Mutations in APPL1 in Familial Diabetes Mellitus. Am J Hum Genet (2015) 97:177–85. doi: 10.1016/j.ajhg.2015.05.011
71. Dubois-Laforgue D, Cornu E, Saint-Martin C, Coste J, Bellanné-Chantelot C, Timsit J. Diabetes, Associated Clinical Spectrum, Long-Term Prognosis, and Genotype/Phenotype Correlations in 201 Adult Patients With Hepatocyte Nuclear Factor 1B (HNF1B) Molecular Defects. Diabetes Care (2017) 40:1436–43. doi: 10.2337/dc16-2462
72. Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A Novel Syndrome of Diabetes Mellitus, Renal Dysfunction and Genital Malformation Associated With a Partial Deletion of the pseudo-POU Domain of Hepatocyte Nuclear Factor-1beta. Hum Mol Genet (1999) 8:2001–8. doi: 10.1093/hmg/8.11.2001
73. Abreu G, Tarantino RM, Cabello PH, Zembrzuski VM, da Fonseca ACP, Rodacki M, et al. The First Case of NEUROD1-MODY Reported in Latin America. Mol Genet Genomic Med (2019) 7:e989. doi: 10.1002/mgg3.989
74. Ağladıoğlu SY, Aycan Z, Çetinkaya S, Baş VN, Önder A, Kendirci HNP, et al. Maturity Onset Diabetes of Youth (MODY) in Turkish Children: Sequence Analysis of 11 Causative Genes by Next Generation Sequencing. J Pediatr Endocrinol Metab (2016) 29:487–96. doi: 10.1515/jpem-2015-0039
75. Horikawa Y, Enya M, Mabe H, Fukushima K, Takubo N, Ohashi M, et al. NEUROD1-Deficient Diabetes (MODY6): Identification of the First Cases in Japanese and the Clinical Features. Pediatr Diabetes (2018) 19:236–42. doi: 10.1111/pedi.12553
76. Fjeld K, Weiss FU, Lasher D, Rosendahl J, Chen J-M, Johansson BB, et al. A Recombined Allele of the Lipase Gene CEL and its Pseudogene CELP Confers Susceptibility to Chronic Pancreatitis. Nat Genet (2015) 47:518–22. doi: 10.1038/ng.3249
77. Ræder H, Johansson S, Holm PI, Haldorsen IS, Mas E, Sbarra V, et al. Mutations in the CEL VNTR Cause a Syndrome of Diabetes and Pancreatic Exocrine Dysfunction. Nat Genet (2006) 38:54–62. doi: 10.1038/ng1708
78. Jo W, Endo M, Ishizu K, Nakamura A, Tajima T. A Novel PAX4 Mutation in a Japanese Patient With Maturity-Onset Diabetes of the Young. Tohoku J Exp Med (2011) 223:113–8. doi: 10.1620/tjem.223.113
79. Ushijima K, Fukami M, Ayabe T, Narumi S, Okuno M, Nakamura A, et al. Comprehensive Screening for Monogenic Diabetes in 89 Japanese Children With Insulin-Requiring Antibody-Negative Type 1 Diabetes. Pediatr Diabetes (2018) 19:243–50. doi: 10.1111/pedi.12544
80. Teo AK, Windmueller R, Johansson BB, Dirice E, Njolstad PR, Tjora E, et al. Derivation of Human Induced Pluripotent Stem Cells From Patients With Maturity Onset Diabetes of the Young. J Biol Chem (2013) 288:5353–6. doi: 10.1074/jbc.C112.428979
81. Ng NHJ, Jasmen JB, Lim CS, Lau HH, Krishnan VG, Kadiwala J, et al. HNF4A Haploinsufficiency in MODY1 Abrogates Liver and Pancreas Differentiation From Patient-Derived Induced Pluripotent Stem Cells. Iscience (2019) 16:192–205. doi: 10.1016/j.isci.2019.05.032
82. Braverman-Gross C, Nudel N, Ronen D, Beer NL, McCarthy MI, Benvenisty N. Derivation and Molecular Characterization of Pancreatic Differentiated MODY1-Ipscs. Stem Cell Res (2018) 31:16–26. doi: 10.1016/j.scr.2018.06.013
83. Hua H, Shang L, Martinez H, Freeby M, Gallagher MP, Ludwig T, et al. iPSC-derived Beta Cells Model Diabetes Due to Glucokinase Deficiency. J Clin Invest (2017) 127:1115. doi: 10.1172/JCI92775
84. Cardenas-Diaz FL, Osorio-Quintero C, Diaz-Miranda MA, Kishore S, Leavens K, Jobaliya C, et al. Modeling Monogenic Diabetes Using Human ESCs Reveals Developmental and Metabolic Deficiencies Caused by Mutations in HNF1A. Cell Stem Cell (2019) 25:273–89.e5. doi: 10.1016/j.stem.2019.07.007
85. Griscelli F, Ezanno H, Soubeyrand M, Feraud O, Oudrhiri N, Bonnefond A, et al. Generation of an Induced Pluripotent Stem Cell (iPSC) Line From a Patient With Maturity-Onset Diabetes of the Young Type 3 (MODY3) Carrying a Hepatocyte Nuclear Factor 1-Alpha (HNF1A) Mutation. Stem Cell Res (2018) 29:56–9. doi: 10.1016/j.scr.2018.02.017
86. Zhu Z, Li QV, Lee K, Rosen BP, González F, Soh C-L, et al. Genome Editing of Lineage Determinants in Human Pluripotent Stem Cells Reveals Mechanisms of Pancreatic Development and Diabetes. Cell Stem Cell (2016) 18:755–68. doi: 10.1016/j.stem.2016.03.015
87. Wang X, Chen S, Burtscher I, Sterr M, Hieronimus A, Machicao F, et al. Generation of a Human Induced Pluripotent Stem Cell (iPSC) Line From a Patient Carrying a P33T Mutation in the PDX1 Gene. Stem Cell Res (2016) 17:273–6. doi: 10.1016/j.scr.2016.08.004
88. Wang X, Sterr M, Burtscher I, Böttcher A, Beckenbauer J, Siehler J, et al. Point Mutations in the PDX1 Transactivation Domain Impair Human β-Cell Development and Function. Mol Metab (2019) 24:80–97. doi: 10.1016/j.molmet.2019.03.006
89. Wang X, Chen S, Burtscher I, Sterr M, Hieronimus A, Machicao F, et al. Generation of a Human Induced Pluripotent Stem Cell (iPSC) Line From a Patient With Family History of Diabetes Carrying a C18R Mutation in the PDX1 Gene. Stem Cell Res (2016) 17:292–5. doi: 10.1016/j.scr.2016.08.005
90. Teo AKK, Lau HH, Valdez IA, Dirice E, Tjora E, Raeder H, et al. Early Developmental Perturbations in a Human Stem Cell Model of MODY5/HNF1B Pancreatic Hypoplasia. Stem Cell Rep (2016) 6:357–67. doi: 10.1016/j.stemcr.2016.01.007
91. Yabe SG, Iwasaki N, Yasuda K, Hamazaki TS, Konno M, Fukuda S, et al. Establishment of Maturity-Onset Diabetes of the Young-Induced Pluripotent Stem Cells From a Japanese Patient. J Diabetes Invest (2015) 6:543–7. doi: 10.1111/jdi.12334
92. Gage BK, Baker RK, Kieffer TJ. Overexpression of PAX4 Reduces Glucagon Expression in Differentiating Hescs. Islets (2014) 6:e29236. doi: 10.4161/isl.29236
93. Ma S, Viola R, Sui L, Cherubini V, Barbetti F, Egli D. β Cell Replacement After Gene Editing of a Neonatal Diabetes-Causing Mutation At the Insulin Locus. Stem Cell Rep (2018) 11:1407–15. doi: 10.1016/j.stemcr.2018.11.006
94. Guo D, Liu H, Gao G, Ruzi A, Wang K, Wu H, et al. Generation of an Abcc8 Heterozygous Mutation Human Embryonic Stem Cell Line Using CRISPR/Cas9. Stem Cell Res (2016) 17:670–2. doi: 10.1016/j.scr.2016.11.014
95. Guo D, Liu H, Ruzi A, Gao G, Nasir A, Liu Y, et al. Modeling Congenital Hyperinsulinism With ABCC8-deficient Human Embryonic Stem Cells Generated by CRISPR/Cas9. Sci Rep (2017) 7:1–8. doi: 10.1038/s41598-017-03349-w
96. Zeng H, Guo M, Zhou T, Tan L, Chong CN, Zhang T, et al. An Isogenic Human ESC Platform for Functional Evaluation of Genome-Wide-Association-Study-Identified Diabetes Genes and Drug Discovery. Cell Stem Cell (2016) 19:326–40. doi: 10.1016/j.stem.2016.07.002
97. Griscelli F, Feraud O, Ernault T, Oudrihri N, Turhan AG, Bonnefond A, et al. Generation of an Induced Pluripotent Stem Cell (iPSC) Line From a Patient With Maturity-Onset Diabetes of the Young Type 13 (MODY13) With a the Potassium Inwardly-Rectifying Channel, Subfamily J, Member 11 (KCNJ11) Mutation. Stem Cell Res (2017) 23:178–81. doi: 10.1016/j.scr.2017.07.023
98. Gragnoli C, Stanojevic V, Gorini A, Von Preussenthal GM, Thomas MK, Habener JF. Ipf-1/MODY4 Gene Missense Mutation in an Italian Family With Type 2 and Gestational Diabetes. Metabolism (2005) 54:983–8. doi: 10.1016/j.metabol.2005.01.037
99. Mahajan A, Wessel J, Willems SM, Zhao W, Robertson NR, Chu AY, et al. Refining the Accuracy of Validated Target Identification Through Coding Variant Fine-Mapping in Type 2 Diabetes. Nat Genet (2018) 50:559–71. doi: 10.1038/s41588-018-0084-1
100. Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, et al. The Genetic Architecture of Type 2 Diabetes. Nature (2016) 536:41–7. doi: 10.1038/nature18642
101. Patel KA, Kettunen J, Laakso M, Stančáková A, Laver TW, Colclough K, et al. Heterozygous RFX6 Protein Truncating Variants are Associated With MODY With Reduced Penetrance. Nat Commun (2017) 8:1–8. doi: 10.1038/s41467-017-00895-9
102. Shim YJ, Kim JE, Hwang S-K, Choi BS, Choi BH, Cho E-M, et al. Identification of Candidate Gene Variants in Korean MODY Families by Whole-Exome Sequencing. Hormone Res Paediatr (2015) 83:242–51. doi: 10.1159/000368657
103. Johansson S, Irgens H, Chudasama KK, Molnes J, Aerts J, Roque FS, et al. Exome Sequencing and Genetic Testing for MODY. PloS One (2012) 7:e38050. doi: 10.1371/journal.pone.0038050
104. Fösel S. Transient and Permanent Neonatal Diabetes. Eur J Pediatr (1995) 154:944–8. doi: 10.1007/BF01958635
105. Hattersley A, Bruining J, Shield J, Njolstad P, Donaghue KC. The Diagnosis and Management of Monogenic Diabetes in Children and Adolescents. Pediatr Diabetes (2009) 10:33–42. doi: 10.1111/j.1399-5448.2009.00571.x
106. Babenko AP, Polak M, Cavé H, Busiah K, Czernichow P, Scharfmann R, et al. Activating Mutations in the ABCC8 Gene in Neonatal Diabetes Mellitus. New Engl J Med (2006) 355:456–66. doi: 10.1056/NEJMoa055068
107. Njølstad PR, Søvik O, Cuesta-Muñoz A, Bjørkhaug L, Massa O, Barbetti F, et al. Neonatal Diabetes Mellitus Due to Complete Glucokinase Deficiency. New Engl J Med (2001) 344:1588–92. doi: 10.1056/NEJM200105243442104
108. Støy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, et al. Insulin Gene Mutations as a Cause of Permanent Neonatal Diabetes. Proc Natl Acad Sci (2007) 104:15040–4. doi: 10.1073/pnas.0707291104
109. Flanagan S, Edghill E, Gloyn A, Ellard S, Hattersley A. Mutations in KCNJ11, Which Encodes Kir6. 2, are a Common Cause of Diabetes Diagnosed in the First 6 Months of Life, With the Phenotype Determined by Genotype. Diabetologia (2006) 49:1190–7. doi: 10.1007/s00125-006-0246-z
110. Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic Agenesis Attributable to a Single Nucleotide Deletion in the Human IPF1 Gene Coding Sequence. Nat Genet (1997) 15:106–10. doi: 10.1038/ng0197-106
111. Gloyn AL, Diatloff-Zito C, Edghill EL, Bellanné-Chantelot C, Nivot S, Coutant R, et al. KCNJ11 Activating Mutations are Associated With Developmental Delay, Epilepsy and Neonatal Diabetes Syndrome and Other Neurological Features. Eur J Hum Genet (2006) 14:824–30. doi: 10.1038/sj.ejhg.5201629
112. Schwitzgebel VM, Mamin A, Brun T, Ritz-Laser B, Zaiko M, Maret A, et al. Agenesis of Human Pancreas Due to Decreased Half-Life of Insulin Promoter Factor 1. J Clin Endocrinol Metab (2003) 88:4398–406. doi: 10.1210/jc.2003-030046
113. Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, Coleman RJ, Garrett C, et al. Mutations in PTF1A Cause Pancreatic and Cerebellar Agenesis. Nat Genet (2004) 36:1301–5. doi: 10.1038/ng1475
114. Delépine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, Encoding Translation Initiation Factor 2-α Kinase 3, is Mutated in Patients With Wolcott-Rallison Syndrome. Nat Genet (2000) 25:406–9. doi: 10.1038/78085
115. D’Amato E, Giacopelli F, Giannattasio A, d’Annunzio G, Bocciardi R, Musso M, et al. Genetic Investigation in an Italian Child With an Unusual Association of Atrial Septal Defect, Attributable to a New Familial GATA4 Gene Mutation, and Neonatal Diabetes Due to Pancreatic Agenesis. Diabetic Med (2010) 27:1195–200. doi: 10.1111/j.1464-5491.2010.03046.x
116. Allen HL, Flanagan SE, Shaw-Smith C, De Franco E, Akerman I, Caswell R, et al. GATA6 Haploinsufficiency Causes Pancreatic Agenesis in Humans. Nat Genet (2012) 44:20–2. doi: 10.1038/ng.1035
117. Senée V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec J-C, et al. Mutations in GLIS3 are Responsible for a Rare Syndrome With Neonatal Diabetes Mellitus and Congenital Hypothyroidism. Nat Genet (2006) 38:682–7. doi: 10.1038/ng1802
118. Rubio-Cabezas O, Minton JA, Kantor I, Williams D, Ellard S, Hattersley AT. Homozygous Mutations in NEUROD1 are Responsible for a Novel Syndrome of Permanent Neonatal Diabetes and Neurological Abnormalities. Diabetes (2010) 59:2326–31. doi: 10.2337/db10-0011
119. Pinney SE, Oliver-Krasinski J, Ernst L, Hughes N, Patel P, Stoffers DA, et al. Neonatal Diabetes and Congenital Malabsorptive Diarrhea Attributable to a Novel Mutation in the Human Neurogenin-3 Gene Coding Sequence. J Clin Endocrinol Metab (2011) 96:1960–5. doi: 10.1210/jc.2011-0029
120. Rubio-Cabezas O, Jensen JN, Hodgson MI, Codner E, Ellard S, Serup P, et al. Permanent Neonatal Diabetes and Enteric Anendocrinosis Associated With Biallelic Mutations in NEUROG3. Diabetes (2011) 60:1349–53. doi: 10.2337/db10-1008
121. Wang J, Cortina G, Wu SV, Tran R, Cho J-H, Tsai M-J, et al. Mutant Neurogenin-3 in Congenital Malabsorptive Diarrhea. New Engl J Med (2006) 355:270–80. doi: 10.1056/NEJMoa054288
122. Solomon BD, Pineda-Alvarez DE, Balog JZ, Hadley D, Gropman AL, Nandagopal R, et al. Compound Heterozygosity for Mutations in PAX6 in a Patient With Complex Brain Anomaly, Neonatal Diabetes Mellitus, and Microophthalmia. Am J Med Genet Part A (2009) 149:2543–6. doi: 10.1002/ajmg.a.33081
123. Spiegel R, Dobbie A, Hartman C, de Vries L, Ellard S, Shalev SA. Clinical Characterization of a Newly Described Neonatal Diabetes Syndrome Caused by RFX6 Mutations. Am J Med Genet Part A (2011) 155:2821–5. doi: 10.1002/ajmg.a.34251
124. Smith SB, Qu H-Q, Taleb N, Kishimoto NY, Scheel DW, Lu Y, et al. Rfx6 Directs Islet Formation and Insulin Production in Mice and Humans. Nature (2010) 463:775–80. doi: 10.1038/nature08748
125. Bonnycastle LL, Chines PS, Hara T, Huyghe JR, Swift AJ, Heikinheimo P, et al. Autosomal Dominant Diabetes Arising From a Wolfram Syndrome 1 Mutation. Diabetes (2013) 62:3943–50. doi: 10.2337/db13-0571
126. Olsen BS, Hahnemann JM, Schwartz M, Østergaard E. Thiamine-Responsive Megaloblastic Anaemia: A Cause of Syndromic Diabetes in Childhood. Pediatr Diabetes (2007) 8:239–41. doi: 10.1111/j.1399-5448.2007.00251.x
127. Shaw-Smith C, Flanagan SE, Patch AM, Grulich-Henn J, Habeb AM, Hussain K, et al. Recessive SLC19A2 Mutations are a Cause of Neonatal Diabetes Mellitus in Thiamine-Responsive Megaloblastic Anaemia. Pediatr Diabetes (2012) 13:314–21. doi: 10.1111/j.1399-5448.2012.00855.x
128. Flanagan SE, De Franco E, Allen HL, Zerah M, Abdul-Rasoul MM, Edge JA, et al. Analysis of Transcription Factors Key for Mouse Pancreatic Development Establishes NKX2-2 and MNX1 Mutations as Causes of Neonatal Diabetes in Man. Cell Metab (2014) 19:146–54. doi: 10.1016/j.cmet.2013.11.021
129. Poulton CJ, Schot R, Kia SK, Jones M, Verheijen FW, Venselaar H, et al. Microcephaly With Simplified Gyration, Epilepsy, and Infantile Diabetes Linked to Inappropriate Apoptosis of Neural Progenitors. Am J Hum Genet (2011) 89:265–76. doi: 10.1016/j.ajhg.2011.07.006
130. Sansbury F, Flanagan S, Houghton J, Shen FS, Al-Senani A, Habeb A, et al. SLC2A2 Mutations can Cause Neonatal Diabetes, Suggesting GLUT2 may Have a Role in Human Insulin Secretion. Diabetologia (2012) 55:2381–5. doi: 10.1007/s00125-012-2595-0
131. Santer R, Schneppenheim R, Dombrowski A, Götze H, Steinmann B, Schaub J. Mutations in GLUT2, the Gene for the Liver-Type Glucose Transporter, in Patients With Fanconi-Bickel Syndrome. Nat Genet (1997) 17:324–6. doi: 10.1038/ng1197-324
132. De Franco E, Lytrivi M, Ibrahim H, Montaser H, Wakeling MN, Fantuzzi F, et al. YIPF5 Mutations Cause Neonatal Diabetes and Microcephaly Through Endoplasmic Reticulum Stress. J Clin Invest (2020) 130(12):6338–53. doi: 10.1172/JCI141455
133. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating Germline Mutations in STAT3 Cause Early-Onset Multi-Organ Autoimmune Disease. Nat Genet (2014) 46:812–4. doi: 10.1038/ng.3040
134. Saarimäki-Vire J, Balboa D, Russell MA, Saarikettu J, Kinnunen M, Keskitalo S, et al. An Activating STAT3 Mutation Causes Neonatal Diabetes Through Premature Induction of Pancreatic Differentiation. Cell Rep (2017) 19:281–94. doi: 10.1016/j.celrep.2017.03.055
135. Shi Z-D, Lee K, Yang D, Amin S, Verma N, Li QV, et al. Genome Editing in hPSCs Reveals GATA6 Haploinsufficiency and a Genetic Interaction With GATA4 in Human Pancreatic Development. Cell Stem Cell (2017) 20:675–88.e6. doi: 10.1016/j.stem.2017.01.001
136. Tiyaboonchai A, Cardenas-Diaz FL, Ying L, Maguire JA, Sim X, Jobaliya C, et al. GATA6 Plays an Important Role in the Induction of Human Definitive Endoderm, Development of the Pancreas, and Functionality of Pancreatic β Cells. Stem Cell Rep (2017) 8:589–604. doi: 10.1016/j.stemcr.2016.12.026
137. Balboa D, Saarimäki-Vire J, Borshagovski D, Survila M, Lindholm P, Galli E, et al. Insulin Mutations Impair Beta-Cell Development in a Patient-Derived iPSC Model of Neonatal Diabetes. Elife (2018) 7:e38519. doi: 10.7554/eLife.38519
138. Panova AV, Klementieva NV, Sycheva AV, Goliusova DV, Khokhlov NV, Zubkova NA, et al. Generation of an Induced Pluripotent Stem Cell Line MNDINSi001-A From a Patient With Neonatal Diabetes Caused by a Heterozygous INS Mutation. Stem Cell Res (2020) 47:101929. doi: 10.1016/j.scr.2020.101929
139. Zhang X, McGrath PS, Salomone J, Rahal M, McCauley HA, Schweitzer J, et al. A Comprehensive Structure-Function Study of Neurogenin3 Disease-Causing Alleles During Human Pancreas and Intestinal Organoid Development. Dev Cell (2019) 50:367–80.e7. doi: 10.1016/j.devcel.2019.05.017
140. Trott J, Alpagu Y, Tan EK, Shboul M, Dawood Y, Elsy M, et al. Mitchell-Riley Syndrome iPSCs Exhibit Reduced Pancreatic Endoderm Differentiation Due to a Mutation in RFX6. Development (2020) 147(21):dev194878. doi: 10.1242/dev.194878
141. Elsayed AK, Aghadi M, Al-Khawaga S, Hussain K, Abdelalim EM. Derivation of a Human Induced Pluripotent Stem Cell Line (Qbrii007-A) From a Patient Carrying a Homozygous Intronic Mutation (C. 613-7T> G) in the SLC2A2 Gene. Stem Cell Res (2020) 44:101736. doi: 10.1016/j.scr.2020.101736
142. Shang L, Hua H, Foo K, Martinez H, Watanabe K, Zimmer M, et al. β-Cell Dysfunction Due to Increased ER Stress in a Stem Cell Model of Wolfram Syndrome. Diabetes (2014) 63:923–33. doi: 10.2337/db13-0717
143. Maxwell KG, Augsornworawat P, Velazco-Cruz L, Kim MH, Asada R, Hogrebe NJ, et al. Gene-Edited Human Stem Cell–Derived β Cells From a Patient With Monogenic Diabetes Reverse Preexisting Diabetes in Mice. Sci Trans Med (2020) 12(540):eaax9106. doi: 10.1126/scitranslmed.aax9106
144. Gradwohl G, Dierich A, LeMeur M, Guillemot F. Neurogenin3 is Required for the Development of the Four Endocrine Cell Lineages of the Pancreas. Proc Natl Acad Sci (2000) 97:1607–11. doi: 10.1073/pnas.97.4.1607
145. Rodríguez-Seguí S, Akerman I, Ferrer J. GATA Believe it: New Essential Regulators of Pancreas Development. J Clin Invest (2012) 122:3469–71. doi: 10.1172/JCI65751
146. Garcia-Gonzalez MA, Carette C, Bagattin A, Chiral M, Makinistoglu MP, Garbay S, et al. A Suppressor Locus for MODY3-Diabetes. Sci Rep (2016) 6:33087. doi: 10.1038/srep35697
147. Pontoglio M, Sreenan S, Roe M, Pugh W, Ostrega D, Doyen A, et al. Defective Insulin Secretion in Hepatocyte Nuclear Factor 1alpha-Deficient Mice. J Clin Invest (1998) 101:2215–22. doi: 10.1172/JCI2548
148. Gupta RK, Vatamaniuk MZ, Lee CS, Flaschen RC, Fulmer JT, Matschinsky FM, et al. The MODY1 Gene HNF-4α Regulates Selected Genes Involved in Insulin Secretion. J Clin Invest (2005) 115:1006–15. doi: 10.1172/JCI200522365
149. Riggs A, Bernal-Mizrachi E, Ohsugi M, Wasson J, Fatrai S, Welling C, et al. Mice Conditionally Lacking the Wolfram Gene in Pancreatic Islet Beta Cells Exhibit Diabetes as a Result of Enhanced Endoplasmic Reticulum Stress and Apoptosis. Diabetologia (2005) 48:2313–21. doi: 10.1007/s00125-005-1947-4
150. Haumaitre C, Barbacci E, Jenny M, Ott M, Gradwohl G, Cereghini S. Lack of TCF2/vHNF1 in Mice Leads to Pancreas Agenesis. Proc Natl Acad Sci (2005) 102:1490–5. doi: 10.1073/pnas.0405776102
151. Nostro MC, Sarangi F, Ogawa S, Holtzinger A, Corneo B, Li X, et al. Stage-Specific Signaling Through Tgfβ Family Members and WNT Regulates Patterning and Pancreatic Specification of Human Pluripotent Stem Cells. Development (2011) 138:861–71. doi: 10.1242/dev.055236
152. Rezania A, Bruin JE, Riedel MJ, Mojibian M, Asadi A, Xu J, et al. Maturation of Human Embryonic Stem Cell–Derived Pancreatic Progenitors Into Functional Islets Capable of Treating Pre-Existing Diabetes in Mice. Diabetes (2012) 61:2016–29. doi: 10.2337/db11-1711
153. Cortijo C, Gouzi M, Tissir F, Grapin-Botton A. Planar Cell Polarity Controls Pancreatic Beta Cell Differentiation and Glucose Homeostasis. Cell Rep (2012) 2:1593–606. doi: 10.1016/j.celrep.2012.10.016
154. Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, et al. Recapitulating Endocrine Cell Clustering in Culture Promotes Maturation of Human Stem-Cell-Derived β Cells. Nat Cell Biol (2019) 21:263–74. doi: 10.1038/s41556-018-0271-4
155. Veres A, Faust AL, Bushnell HL, Engquist EN, Kenty JH-R, Harb G, et al. Charting Cellular Identity During Human In Vitro β-Cell Differentiation. Nature (2019) 569:368–73. doi: 10.1038/s41586-019-1168-5
156. Mahaddalkar PU, Scheibner K, Pfluger S, Sterr M, Beckenbauer J, Irmler M, et al. Generation of Pancreatic β Cells From CD177+ Anterior Definitive Endoderm. Nat Biotechnol (2020) 38:1061–72. doi: 10.1038/s41587-020-0492-5
157. Ghazizadeh Z, Kao D-I, Amin S, Cook B, Rao S, Zhou T, et al. ROCKII Inhibition Promotes the Maturation of Human Pancreatic Beta-Like Cells. Nat Commun (2017) 8:1–12. doi: 10.1038/s41467-017-00129-y
158. Vethe H, Ghila L, Berle M, Hoareau L, Haaland ØA, Scholz H, et al. The Effect of Wnt Pathway Modulators on Human iPSC-derived Pancreatic Beta Cell Maturation. Front Endocrinol (2019) 10:293. doi: 10.3389/fendo.2019.00293
159. Hogrebe NJ, Augsornworawat P, Maxwell KG, Velazco-Cruz L, Millman JR. Targeting the Cytoskeleton to Direct Pancreatic Differentiation of Human Pluripotent Stem Cells. Nat Biotechnol (2020) 38:460–70. doi: 10.1038/s41587-020-0430-6
160. Helman A, Cangelosi AL, Davis JC, Pham Q, Rothman A, Faust AL, et al. A Nutrient-Sensing Transition At Birth Triggers Glucose-Responsive Insulin Secretion. Cell Metab (2020) 31:1004–16.e5. doi: 10.1016/j.cmet.2020.04.004
161. Alvarez-Dominguez JR, Donaghey J, Rasouli N, Kenty JH, Helman A, Charlton J, et al. Circadian Entrainment Triggers Maturation of Human In Vitro Islets. Cell Stem Cell (2020) 26:108–22.e10. doi: 10.1016/j.stem.2019.11.011
162. Kim MJ, Lee EY, You Y-H, Yang HK, Yoon K-H, Kim J-W. Generation of iPSC-derived Insulin-Producing Cells From Patients With Type 1 and Type 2 Diabetes Compared With Healthy Control. Stem Cell Res (2020) 48:101958. doi: 10.1016/j.scr.2020.101958
163. Millman JR, Xie C, Van Dervort A, Gürtler M, Pagliuca FW, Melton DA. Generation of Stem Cell-Derived β-Cells From Patients With Type 1 Diabetes. Nat Commun (2016) 7:1–9. doi: 10.1038/ncomms11463
164. Hosokawa Y, Toyoda T, Fukui K, Baden MY, Funato M, Kondo Y, et al. Insulin-Producing Cells Derived From ‘Induced Pluripotent Stem Cells’ of Patients With Fulminant Type 1 Diabetes: Vulnerability to Cytokine Insults and Increased Expression of Apoptosis-Related Genes. J Diabetes Invest (2018) 9:481–93. doi: 10.1111/jdi.12727
165. Leite NC, Sintov E, Meissner TB, Brehm MA, Greiner DL, Harlan DM, et al. Modeling Type 1 Diabetes In Vitro Using Human Pluripotent Stem Cells. Cell Rep (2020) 32:107894. doi: 10.1016/j.celrep.2020.107894
166. Joshi K, Cameron F, Tiwari S, Mannering SI, Elefanty AG, Stanley EG. Modeling Type 1 Diabetes Using Pluripotent Stem Cell Technology. Front Endocrinol (2021) 12:260. doi: 10.3389/fendo.2021.635662
167. Sander M, Neubüser A, Kalamaras J, Ee HC, Martin GR, German MS. Genetic Analysis Reveals That PAX6 is Required for Normal Transcription of Pancreatic Hormone Genes and Islet Development. Genes Dev (1997) 11:1662–73. doi: 10.1101/gad.11.13.1662
168. Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, Bernal-Mizrachi E, et al. A Gene Encoding a Transmembrane Protein is Mutated in Patients With Diabetes Mellitus and Optic Atrophy (Wolfram Syndrome). Nat Genet (1998) 20:143–8. doi: 10.1038/2441
169. Fonseca SG, Fukuma M, Lipson KL, Nguyen LX, Allen JR, Oka Y, et al. WFS1 is a Novel Component of the Unfolded Protein Response and Maintains Homeostasis of the Endoplasmic Reticulum in Pancreatic β-Cells. J Biol Chem (2005) 280:39609–15. doi: 10.1074/jbc.M507426200
170. Yamada T, Ishihara H, Tamura A, Takahashi R, Yamaguchi S, Takei D, et al. WFS1-Deficiency Increases Endoplasmic Reticulum Stress, Impairs Cell Cycle Progression and Triggers the Apoptotic Pathway Specifically in Pancreatic β-Cells. Hum Mol Genet (2006) 15:1600–9. doi: 10.1093/hmg/ddl081
171. Flanagan SE, Clauin S, Bellanné-Chantelot C, de Lonlay P, Harries LW, Gloyn AL, et al. Update of Mutations in the Genes Encoding the Pancreatic Beta-Cell KATP Channel Subunits Kir6. 2 (KCNJ11) and Sulfonylurea Receptor 1 (ABCC8) in Diabetes Mellitus and Hyperinsulinism. Hum Mutat (2009) 30:170–80. doi: 10.1002/humu.20838
172. Carrasco M, Delgado I, Soria B, Martín F, Rojas A. GATA4 and GATA6 Control Mouse Pancreas Organogenesis. J Clin Invest (2012) 122:3504–15. doi: 10.1172/JCI63240
173. Xuan S, Borok MJ, Decker KJ, Battle MA, Duncan SA, Hale MA, et al. Pancreas-Specific Deletion of Mouse Gata4 and Gata6 Causes Pancreatic Agenesis. J Clin Invest (2012) 122:3516–28. doi: 10.1172/JCI63352
174. Garreta E, Sanchez S, Lajara J, Montserrat N, Belmonte JCI. Roadblocks in the Path of iPSC to the Clinic. Curr Transplant Rep (2018) 5:14–8. doi: 10.1007/s40472-018-0177-x
Keywords: pluripotent stem cells, diabetes, monogenic variants, Maturity Onset of Diabetes in the Young, type 2 diabetes, type 1 diabetes
Citation: Heller S, Melzer MK, Azoitei N, Julier C and Kleger A (2021) Human Pluripotent Stem Cells Go Diabetic: A Glimpse on Monogenic Variants. Front. Endocrinol. 12:648284. doi: 10.3389/fendo.2021.648284
Received: 12 January 2021; Accepted: 13 April 2021;
Published: 17 May 2021.
Edited by:
Shuibing Chen, Cornell University, United StatesReviewed by:
Holger Andreas Russ, University of Colorado Anschutz Medical Campus, United StatesJeffery Sivert Tessem, Brigham Young University, United States
Copyright © 2021 Heller, Melzer, Azoitei, Julier and Kleger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Heller, c2FuZHJhLmhlbGxlckB1bmktdWxtLmRl
Cécile Julier, Y2VjaWxlLmp1bGllckBpbnNlcm0uZnI=
Alexander Kleger, YWxleGFuZGVyLmtsZWdlckB1bmktdWxtLmRl