 Laura Costanza De Angelis1,2*
Laura Costanza De Angelis1,2* Giorgia Brigati1
Giorgia Brigati1 Giulia Polleri1
Giulia Polleri1 Mariya Malova1,2
Mariya Malova1,2 Alessandro Parodi1,2Diego Minghetti1Andrea Rossi3,4Paolo Massirio1,2Cristina Traggiai1Mohamad Maghnie2,5
Alessandro Parodi1,2Diego Minghetti1Andrea Rossi3,4Paolo Massirio1,2Cristina Traggiai1Mohamad Maghnie2,5 Luca Antonio Ramenghi1,2
Luca Antonio Ramenghi1,2- 1Neonatal Intensive Care Unit, Department Mother and Child, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 2Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (DINOGMI), University of Genoa, Genoa, Italy
- 3Department of Health Sciences (DISSAL), University of Genoa, Genoa, Italy
- 4Neuroradiology Unit, Istituti di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Giannina Gaslini, Genoa, Italy
- 5Department of Pediatrics, IRCCS Istituto Giannina Gaslini, Genoa, Italy
Neonatal hypoglycemia is a common condition. A transient reduction in blood glucose values is part of a transitional metabolic adaptation following birth, which resolves within the first 48 to 72 h of life. In addition, several factors may interfere with glucose homeostasis, especially in case of limited metabolic stores or increased energy expenditure. Although the effect of mild transient asymptomatic hypoglycemia on brain development remains unclear, a correlation between severe and prolonged hypoglycemia and cerebral damage has been proven. A selective vulnerability of some brain regions to hypoglycemia including the second and the third superficial layers of the cerebral cortex, the dentate gyrus, the subiculum, the CA1 regions in the hippocampus, and the caudate-putamen nuclei has been observed. Several mechanisms contribute to neuronal damage during hypoglycemia. Neuronal depolarization induced by hypoglycemia leads to an elevated release of glutamate and aspartate, thus promoting excitotoxicity, and to an increased release of zinc to the extracellular space, causing the extensive activation of poly ADP-ribose polymerase-1 which promotes neuronal death. In this review we discuss the cerebral glucose homeostasis, the mechanisms of brain injury following neonatal hypoglycemia and the possible treatment strategies to reduce its occurrence.
Introduction
Hypoglycemia is a common condition in the neonatal population (1). It has a significant prevalence in at risk infants, with that of 47% in large-for-gestational age (LGA) infants, 52% in small-for-gestational age (SGA) infants, 48% in neonates of diabetic mothers, and 54% in late preterm infants (2). In infants born before 33 weeks, the prevalence of hypoglycemia is nearly 34% (3).
A transient reduction in blood glucose values immediately after birth is part of a transitional metabolic adaptation that generally resolves within the first hours of life as glucose levels gradually increase to reach adult values (blood glucose > 70 mg/dl) within the first 72 to 96 h (4, 5). However, a minority of neonates experience a prolonged and severe hypoglycemia, usually associated with specific risk factors (6).
Although adverse effects on the neurodevelopmental outcome have been proven for severe and persistently low glucose levels (7–10), the long-term significance of transient asymptomatic neonatal hypoglycemia remains unclear. Additionally, while a “neurologically safe” blood glucose numerical threshold has not yet been identified, guidelines for the optimization of detection and prompt treatment of hypoglycemic neonates have been empirically established to avoid neurologic sequelae (11). A summary of the main expert opinions on the definition of neonatal hypoglycemia is presented in Table 1. In spite of the inhomogeneous definitions, no evidence of the superiority of hypoglycemia management according to different blood glucose thresholds has currently been established.
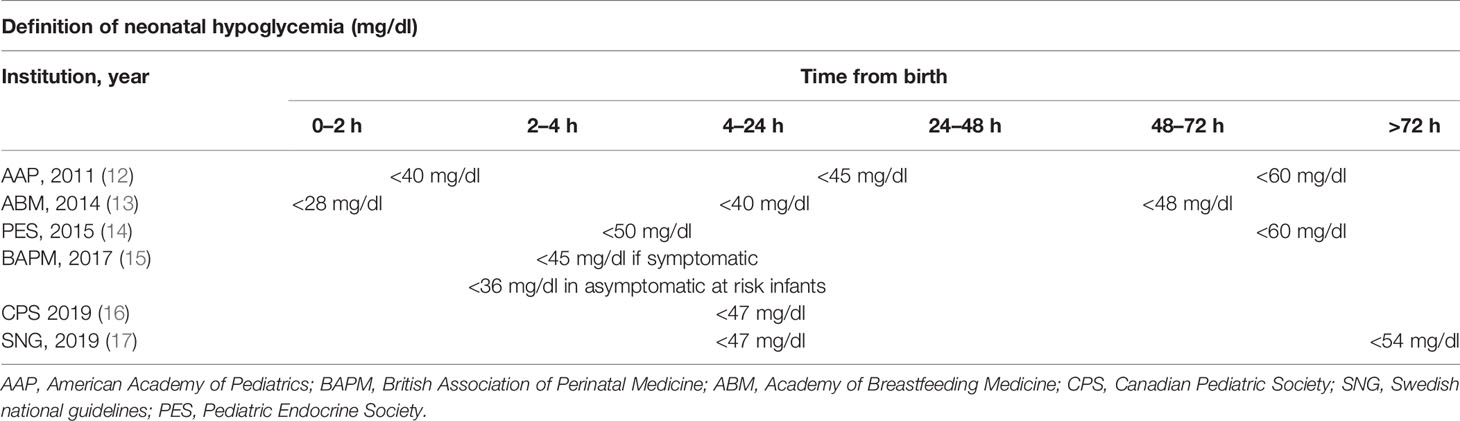
Table 1 Definition of neonatal hypoglycemia in the first 72 h from birth, according to the more widespread recommendations.
The aim of this review is to discuss the cerebral glucose homeostasis, the mechanisms of brain injury following neonatal hypoglycemia and the possible treatment strategies to reduce its occurrence.
Transition of Glucose Homeostasis
Glucose is the main energy source for fetal development. During pregnancy, its homeostasis entirely depends on continuous glucose supply from the maternal circulation (18). Glucose transportation across the placenta is mediated by facilitate diffusion, dependent on the maternal-fetal concentration gradient (19, 20). About the 70% of the maternal glucose is allocated to the fetus while the 30% is consumed by the placenta (21). Fetal glucose metabolism is regulated by fetal insulin production, which increases with pregnancy progression and enhances glucose utilization by insulin-sensitive tissues, including skeletal muscle, liver, heart, and adipose tissue (22).
At birth, the interruption of glucose transfer causes a prompt feedback of neonatal glucose control leading to a consistent increase in plasma catecholamines, glucagon (and glucagon receptors) and cortisol, and a decrease in plasma insulin levels (23). These endocrine changes are essential for inducing hepatic glycogenolysis and gluconeogenesis thus maintaining glucose homeostasis. In addition, lipolysis, fatty acid oxidation, and proteolysis actively concur to sustain blood glucose levels in the neonatal period (24). This metabolic pattern allows the prompt activation of catabolic processes and the utilization of endogenous energy stores (25).
Hepatic glycogenolysis is the fastest mechanism that allows an increase of blood glucose levels after birth. The high rate of glycogenolysis leads to hastened depletion of hepatic glycogen stores, especially in preterm infants in which liver glycogen deposits are limited. Gluconeogenesis is not immediately effective after birth due to the initial low enzymatic activity of this metabolic pathway. Gluconeogenesis slowly starts after some hours from birth and reaches its maturation after 12 h (26).
Glucose oxidation supports about 70% of the energy requirement of the brain. Ketone bodies and lactate are important alternative fuels to reduce the glucose requirement. Hepatic ketogenesis markedly increases during the first hours from birth, to provide alternative fuels for brain metabolism in term infants. However this metabolic pattern is severely limited in preterm infants due to a lack of fat stores in adipose tissue, which eventually results in failure of lipolysis (27).
During the first days of life interferences of neonatal glucose homeostasis may happen, especially in case of limited metabolic stores or increased energy expenditure (28). Most studies have shown that premature infants are more vulnerable than full-term infants to remain normoglycemic in the first week of life. This depends on several mechanism such as insulin resistance, deficient proinsulin processing by pancreatic beta cells (29), abnormal glucagon response (30), and lack of suppression of hepatic glucose release following intravenous glucose infusion that have been suggested to contribute to hyperglycemia. Predisposition to hypoglycemia is due to low glucose-6-phosphatase activity (31), the existence of an incompletely coordinated counter-regulatory system (32), increased basal metabolism of glucose, a lower capacity for production of alternative energy sources from their already insufficient stores (27), and presence of clinical conditions associated with hypoglycemia, such as perinatal asphyxia, hypoxia, sepsis, and hypothermia. As a consequence, it is essential to provide exogenous glucose at birth, especially in preterm infants (33).
Brain Energetics and Glucose Sensing Neurons
The largest proportion of brain energy is consumed for neuronal computation, information processing, and maintaining ion gradients across neuronal membranes. Glucose metabolism provides several precursors as well as the energy for the biosynthesis of neurotransmitters (34). Brain function impairments caused by hypoglycemia seems to begin prior to any detectable drop in overall brain ATP. Neuronal activity may be suppressed as an energy conserving strategy. The release of ADP, induced by hypoglycemia, suppresses excitatory postsynaptic potential, and ATP produced by glycolysis (during euglycemia) has a special role in maintaining glutamatergic neurotransmission (35).
Production of glutamate vesicles is a glucose-dependent process. The membrane of glutamate vesicles contains enzymes responsible for several ATP generating steps of glycolysis, so the rate of glutamate refilling these vesicles is reduced in the absence of glucose resulting in the inhibition of all neuronal glutamate activity. With severe hypoglycemia, ATP levels decline simultaneous to glycogen store depletion.
Glucose homeostasis is based on an accurate and prompt detection of blood glucose variations. Glucose sensing neurons are a specialized group of neurons which modify their electrical activity according to extracellular glucose levels (36). Among them, glucose-excited neurons increase their activity in response to an increase of interstitial glucose levels while glucose-inhibited neurons reduce their firing rate. As glucose concentrations decrease, GI neurons are excited, whereas GE neurons decrease their activity (37). Glucose sensing neurons are mainly situated in the hypothalamic nuclei, the prefrontal cortex, the hippocampus, the paraventricular thalamus, and the amygdala (38–40).
The glucose level in the brain is about 30% of the blood level, as the presence of the blood-brain-barrier (BBB) prevents its free diffusion. The glucose supply to the brain is ensured by the high affinity of the glucose transporter type 1 (GLUT1) of brain endothelial cells (41). Glucose subsequently enter the neurons through GLUT1 and the glucose transporter type 3 (GLUT3) (42).
Some studies demonstrated that hypothalamic glucose levels do not fluctuate significantly following meals. Conversely, glucose levels in the arcuate nucleus were observed to slightly increase after fasting due to an increased permeability of the BBB. Thus, only profound alterations of blood glucose levels induce changes in brain glucose levels (43). Nevertheless, the presence of specialized neurons able to detect changes above 2.5 mM suggests that, at least in confined areas (such as those around fenestrated capillaries in the BBB), glucose levels may be increased to levels closer to that of the blood. The modification in cerebral fluid glucose caused by a change in blood sugar was observed to be delayed and dampened. BBB permeability to blood glucose changes depend on various pathophysiological conditions, including hyperglycemia and uncontrolled diabetes mellitus (44).
As the maintenance of a constant and adequate supply of sugar for the brain is a priority, it is obvious that the ability to detect changes in blood glucose levels by sensitive neurons situated behind the BBB is not useful to glucose homeostasis because it only foresees changes in brain glucose. It is likely that this ability serves more as a “fail safe system”. These neurons can detect basic conditions to compare information and create anticipatory responses (43).
Besides neurons, astrocytes have also been reported to be involved in glucose sensing, and due to their role in blood flow regulation, they may regulate it according to the neuronal metabolic state (45).
Hypoglycemic Brain Damage
Many factors have been implicated in neuronal death induced by severe hypoglycemia. This event is not only a consequence of energy failure but also as a result of a sequence of events induced by hypoglycemia. Interestingly, correction of plasma glucose concentration alone does not interrupt this process. The main events induced by hypoglycemia are now discussed.
Activation of Neuronal Glutamate Receptors
Hypoglycemia causes neuronal depolarization, resulting in a significant elevations in brain extracellular glutamate concentrations (46). It has been demonstrated that the ablation of presynaptic glutamatergic terminals prevents the rise of glutamate in the extracellular space and hypoglycemic neuronal death (47). In addition, impaired glutamate astrocyte reuptake causes an increased glutamate level leading to an accumulation of aspartate in brain tissues (48). As a consequence, increased levels of aspartate may activate some glutamate receptors subtypes, contributing to excitotoxicity. The sustained glutamate receptor activation is the first step of the process leading to neuronal cell death.
Oxidative Stress
Oxidative DNA damage plays a critical role in neuronal cell death. Mitochondria have been implicated as a source of reactive oxygen species (ROS) in many disorders, and mitochondria taken from a hypoglycemic brain exhibit an increased capacity to generate ROS in response to glutamate excitotoxicity. However, ROS can also be generated by several other sources. NADPH oxidase is an enzyme studied in neutrophils which synthesizes superoxides. It has been observed that superoxide production in neuron cells affected by hypoglycemia occurs mainly during glucose reperfusion.
Moreover, the rate of superoxide production is influenced by blood glucose concentrations achieved in the immediate post-hypoglycemic period (49). The selective vulnerability of brain regions to hypoglycemia has been demonstrated. The II and III superficial layers of the cerebral cortex, the dentate gyrus, the subiculum, the CA1 regions in the hippocampus, and the caudate –putamen, emerged as the main susceptible areas to hypoglycemic insult (50, 51). Although the physiopathology underlying this different susceptibility still remains unclear, oxidative stress seems to play a central role (52).
Neuronal Zinc Release
Zinc (as Zn2+) is a neuromodulator stored within vesicles in presynaptic terminals, and it is massively released from into the extracellular space during pathological conditions such as ischemia, seizure, brain trauma, and hypoglycemia (49, 53). Zinc significantly affects the activity of many receptors including NMDA, GABA-A, ATP, and glycine receptors as well as voltage-gated Na+ and Ca2+ channels and it has been associated with the promotion of neuronal death (53, 54). It induces profound mitochondrial dysfunction triggering mitochondrial depolarization and leading to PARP-1 activation, probably as a result of increased production of reactive oxygen species (ROS) (55, 56). In addition, it has been observed that zinc contributes to neuronal energy failure by glycolisis inhibition at GAPDH level (57).
Activation of Poly-ADP-Ribose Polymerase–1 (PARP1)
PARP-1 has a catalytic domain that shows structural homologies to other ADP-ribosyl-transferase enzymes. It uses the ADP ribose group of NAD+ to form branched ADP ribose polymers on specific acceptor proteins near DNA strand breaks or kinks. These polymers facilitate the DNA repair process and prevent chromatid exchange. During oxidative stress, reactive species damage all cellular components, including nucleic acids. PARP-1 activation takes place in order to facilitate the DNA repair machinery. While adequate PARP activation facilitates DNA repair, extensive PARP activation, caused by extensive damage from a sustained action of glutamate, induces mitochondrial permeability transition and mitochondrial damage that culminates in cell death. Moreover, activation of PARP-1 consumes cytosolic NAD+ required for glycolytic process; therefore, capacitating neurons to utilize glucose because of PARP-1 activation, even when glucose availability is restored. Pyruvate and other non-glucose substrates can be metabolized without cytosolic NAD+ and can rescue cells from PARP-1 induced cell death (35).
Blood Glucose Variability and Brain Damage
Cerebral blood flow (CBF) is regulated by neurons and astrocytes. Under resting conditions, local CBF is highest in brain regions with the highest local glucose metabolism. During functional activation, the increase in local CBF usually correlates with an increased cerebral metabolic rate of glucose (34). Neurotransmitter-mediated signaling, particularly by glutamate, plays a key role in regulating CBF, and much of this control is mediated by astrocytes. Traditionally, it was thought that active neurons generate a metabolic signal (hypoxemia, hypoglycemia, or hypercapnia), which triggers an increase in blood flow.
To sustain neuronal function, the brain has evolved “neurovascular coupling” mechanisms to increase the flow of blood to regions where neurons are independently active of glucose metabolism-induced signaling. However, it seems that changes in the lactate levels and its metabolic products may be at least partially responsible for vasodilatation during neuronal activation. During acute hypoglycemia, resting CBF only increases significantly when blood and brain glucose are dramatically reduced. Neither hyperglycemia nor mild/moderate hypoglycemia significantly changes the blood flow responses to functional activation. Giordani et al. demonstrated that acute hyperglycemia induced a reduction in cerebral vasomotor reactivity, both in normal control subjects and in patients with diabetes mellitus (58). To date, there are still no conclusive studies on the clinical impact of neonatal glycemic variability on cerebral flow and neurological development.
Magnetic Resonance Imaging and Topography of The Lesions
Another controversial topic in the field of neonatal hypoglycemia is the correlation between hypoglycemic insult and brain damage as visualized by magnetic resonance imaging (MRI), and further correlation of brain damage with the outcome. Occipital lobe damage is thought to predominate, with case series describing patterns of posterior injury evident already in the acute phase on diffusion-weighted imaging (DWI), which correlated with significant structural injury later on (Figure 1). Moreover, the predilection for occipital and, to a minor extent, parietal zones in the hypoglycemic brain has been demonstrated in various studies by computed tomography, MRI, and single-photon emission computed tomography blood flow scans (59–64). In a study including 45 term infants with hypoglycemia, diffusion restriction in the mesial occipital poles was observed in half of the cohort with early imaging (65).
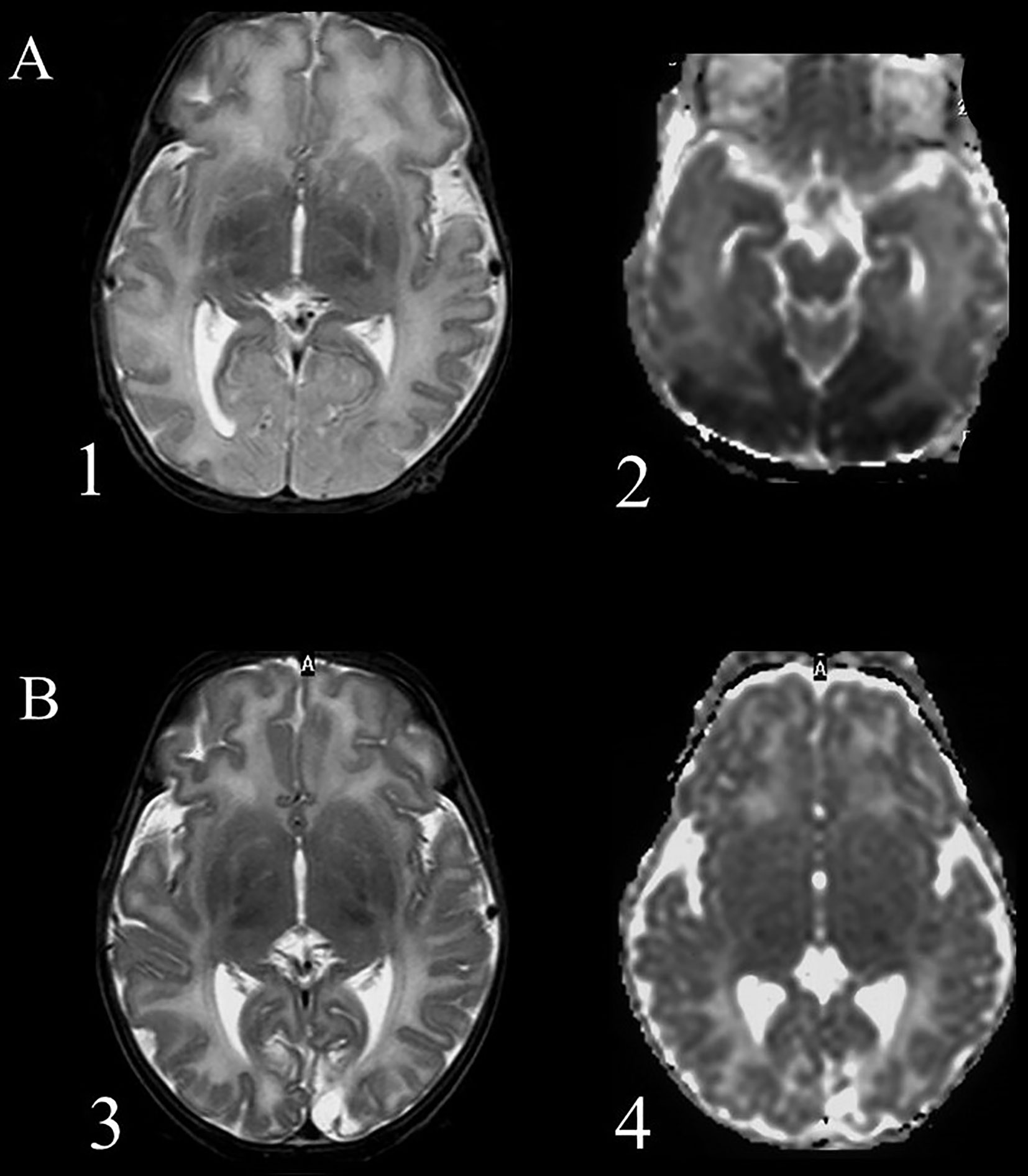
Figure 1 Magnetic resonance imaging of a term newborn with apnea and seizures associated with severe hypoglycemia. (A) scans performed after 5 days from the event (acute phase), 1 = axial T2 weighted imaging showing an extensive cortical and subcortical damage, manly in the occipital lobes, with loss of cortical differentiation; 2 = Diffusion weighted imaging shows a large posterior subcortical area of reduction of the apparent diffusion coefficient. (B) scans performed after 5 weeks (chronic phase): 3 = Presence of an occipital atrophic area, more extended within the left occipital lobe; 4 = Diffusion weighted imaging shows an increased water diffusion in the affected areas and a smaller cavitation.
Reduction of regional cerebral glucose use, or a local expression deficit of the glucose membrane transporter proteins, have been named as possible contributors to the localization of damage (64). Another factor in play could be active synaptogenesis and axonal migration in the occipital lobe, as well as elevated levels of aspartate stimulating newly developed receptors for excitatory amino acids, causing selective death of the postsynaptic neurons (66). A MRI study in patients with symptomatic hypoglycemia associated with metabolic disease showed a correlation between pattern of damage and age at clinical presentation, suggesting a correlation with brain maturation processes: parieto-occipital white matter lesions were observed in infants with hypoglycemia occurring from the neonatal period to 6 months of age, while the basal ganglia damage and parieto-temporal cortex involvement were present in older infants (67).
On the other hand, several reports suggest that the brain damage connected with neonatal hypoglycemia could be more heterogeneous than previously thought. In a study of 35 term symptomatic infants, white matter abnormalities were not limited to the posterior regions. Basal ganglia/thalamic abnormalities, hemorrhages, and middle cerebral artery infarctions were seen, and cortical involvement was present in half of the cases. In the cohort of that study, 65% of infants presented impairments related to the white matter damage involvement at 18 months of age (68). Another study investigating the effect of hypoglycemia in a cohort of 94 term neonates at risk for neonatal encephalopathy has observed that the only type of brain lesion significantly associated with hypoglycemia was injury in the corticospinal tract (odds ratio 3.72, 95% CI 1.02 – 13.57, P=0.047) (8).
Neurodevelopmental Outcomes in Infants With Hypoglycemia
Neonatal hypoglycemia has been associated with various forms of neurological impairment, including developmental delay, seizures, visual processing problems, and cognitive difficulties (Table 2), but many questions regarding which patients are most at risk, imaging-outcome correlation, or follow-up options remain unanswered (68, 69).
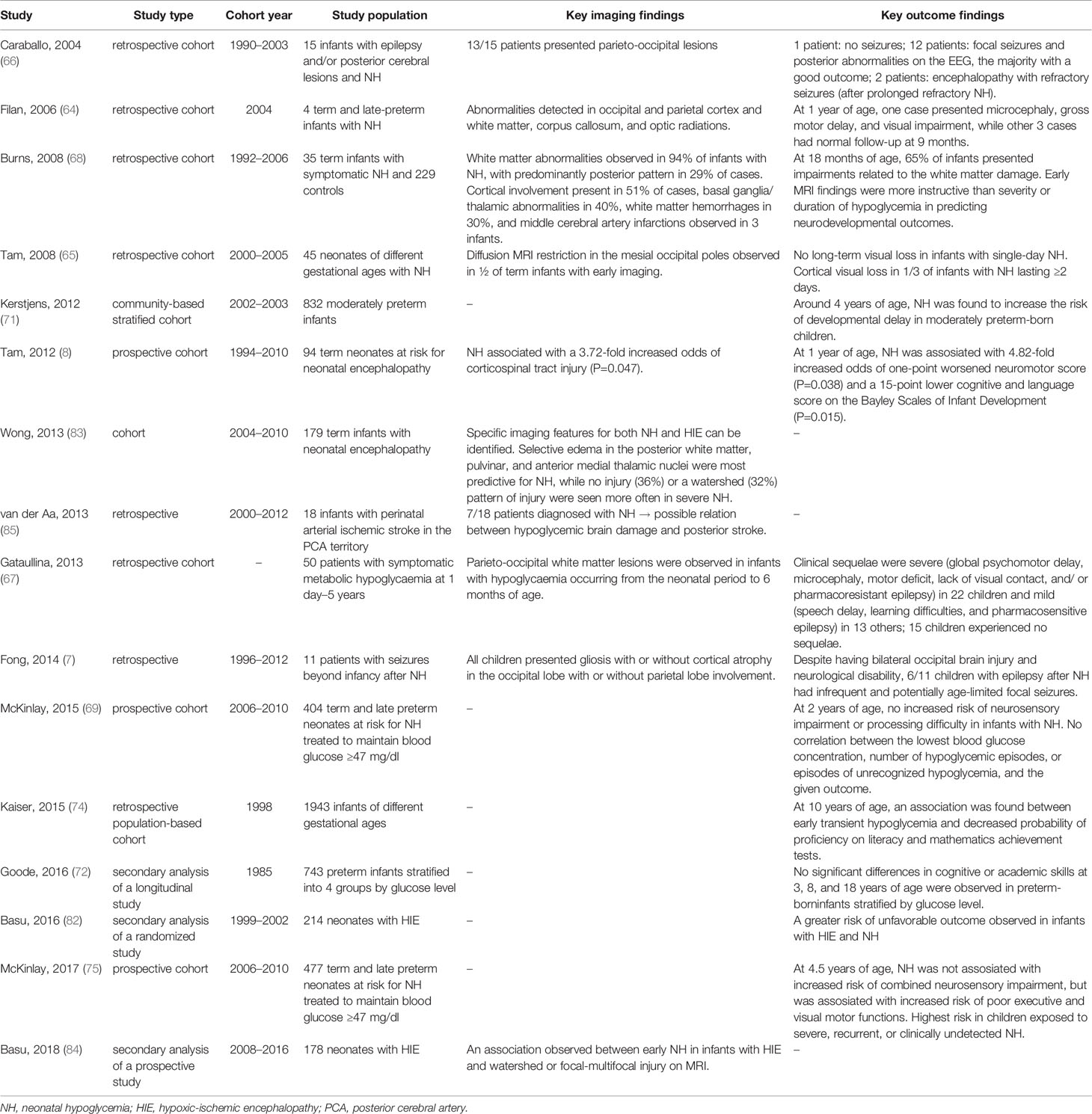
Table 2 A summary of cited studies regarding neurodevelopmental outcomes and other brain injuries in infants with neonatal hypoglycemia.
A recent meta-analysis considering neurodevelopmental outcomes after neonatal hypoglycemia underlined the need for additional studies to determine the best strategies for improving long-term outcomes in neonates at risk: the authors found a correlation between hypoglycemic events and an increased risk of visual motor impairment and executive dysfunction in early childhood and an increased risk of literacy and numeracy problems in later childhood, but the quality of evidence was low, and data on long-term outcomes were not available (70).
One of the reasons for disagreeing evidence in the field could be the difficulty associated with defining neonatal hypoglycemia, starting from debates on the thresholds for intervention, including a need to take into consideration person-to-person variability in glucose homeostasis and reaction to hypoglycemia (68). For example, in a large cohort study involving 832 moderately preterm children, different clinical parameters were evaluated in association with the developmental delay at 4 years of age, and only NH was found to increase the risk of developmental delay (71). Another cohort study that included 745 preterm infants, had patients stratified into 4 groups by glucose level, and cognitive, academic, and behavioral outcomes were assessed at 3, 8, and 18 years of age. In this study, the authors did not observe any significant differences in cognitive or academic skills between the groups at any age (72). In term infants, a small study evaluated neurodevelopmental outcomes at 18 months of age in 35 patients with symptomatic neonatal hypoglycemia. No relationship was found between the severity or duration of hypoglycemia and outcomes (68).
Moreover, while many authors agree that severe, persistent hypoglycemia can cause seizures and brain injury, the prognostic meaning of transient hypoglycemia remains controversial (73, 74).
A big retrospective population-based cohort study involving 1943 infants of different gestational ages found a clear association between early transient hypoglycemia and decreased probability of proficiency on literacy and mathematics achievement tests at age 10 years, leading to considerable conversation regarding the possible usefulness of universal newborn glucose screening (74). At the same time, one of the biggest prospective cohort studies on the topic (CHYLD Study), which included 404 late preterm and term neonates at risk for hypoglycemia, studied infants who were treated to maintain a blood glucose concentration of at least 47 mg/dl. Neurodevelopmental follow-up was assessed first at 2 years, and then at 4.5 years of age (69, 75). At two years, those with treated hypoglycemia were observed to not exhibit an increased risk of neurosensory impairment or processing difficulty. No correlation was found between the lowest blood glucose concentration, number of hypoglycemic episodes, or episodes of unrecognized hypoglycemia, and the given outcome (69). At 4 and a half years, hypoglycemia was not associated with an increased risk of combined neurosensory impairment, but was associated with an increased risk of low executive function and visual motor function, with the highest risk in children exposed to severe, recurrent, or clinically undetected hypoglycemia.
Another interesting result of that study was the association of cognitive delay with higher glucose concentration and less glucose stability after treatment, observed as a longer time outside the range of 54-72 mg/dl in the first two days after birth. Authors suggest that glucose reperfusion injury in the case of overly rapid or overly intense correction of hypoglycemia may exacerbate oxidative stress (76), even if data on biological effect of glycemic variability in neonates is almost absent in literature (77).
Visual Deficits
Visual outcomes connected with neonatal hypoglycemia seem to depend on the severity and length of hypoglycemic insult, even if various comorbidities, difficulties in testing vision, and frequent absence of school-age follow-up makes the interpretation of the present data difficult (78). MRI that was performed promptly after insult could be of help in predicting visual outcomes. In a study involving 25 neonates of different gestational ages, diffusion restriction in the mesial occipital poles was associated with cortical visual deficits. It is interesting to note that restricted diffusion was observed in term but not in preterm infants. In the same study, visual outcomes seemed to correlate with the duration of the hypoglycemic insult: cortical visual loss was documented for one-third of infants with hypoglycemia measured over 2 days, while no long-term visual loss was observed in infants with neonatal hypoglycemia documented on a single day only (65).
Epilepsy
Epilepsy with seizures protracting beyond infancy is one of most threatening consequences of NH, with the majority of cases described as being of occipital origin (79, 80). The presence of epilepsy is rarely the only symptom of previous neonatal hypoglycemia, with patients often presenting with other neurological problems, such as visual disturbances, developmental delay, or pervasive developmental disorders. As for other outcomes, electroclinical features and an evolution of the disease seem to depend on the severity of the initial insult and the eventual presence of other comorbidities (66). Even if cases of epileptic encephalopathy with refractory seizures has been described, the literature suggests that in a lot of cases, epilepsy can be mild, focal, and age-limited with favorable outcomes in adolescence (7).
Hypoglycemia and Hypoxic-Ischemic Encephalopathy
Negative influence of hypoglycemic insult on future neurodevelopment is exacerbated by the presence of other comorbidities most significantly by hypoxic-ischemic encephalopathy (HIE). Hypoglycemia tend to have both an additive and potentiating role in producing brain injury. The number of ATP molecules that can be produced from one molecule of glucose goes down drastically with the transition from aerobic to anaerobic conditions. Thus, hypoxemia and ischemia increase cerebral demand for glucose, exposing brain cells to a significantly increased risk of damage in the case of low blood glucose levels (66). An interesting study by Basu et al. compared 60 infants with HIE to 60 controls, exploring the relationship between plasma glucose level and the neurological status of the patients. Authors have observed lower mean plasma glucose level in the asphyxiated group when compared to the controls. Furthermore, a negative linear correlation between the glucose level and different stages of HIE was observed, which led to the conclusion that the severity of encephalopathy varies with the severity of hypoglycemia (81). Moreover, the CoolCap Study of therapeutic hypothermia for neonates with HIE observed a greater risk of negative outcome in infants who presented with hypoglycemia (82).
Even if HIE and hypoglycemia can have a combined negative effect on the brain, it seems possible to diversify MRI patterns of the two types of injury. Indeed, in a study including 179 term infants, authors were able to predict hypoglycemia from only the MRI result (selective edema in the posterior white matter, pulvinar, and anterior medial thalamic nuclei) with a positive predictive value of 82% and negative predictive value of 78% (83). Another study including 178 neonates with HIE showed a distinct association between early glycemic profile in infants with HIE and specific patterns of brain injury on MRI, with predominant watershed or focal-multifocal injury being more frequent in infants with hypoglycemia (84).
Hypoglycemia as a Risk Factor for Other Brain Lesions
Another question that needs to be addressed is a possible role of alterations in glucose metabolism as a risk factor of other brain lesions. An interesting result comes from work by van der Aa et al., which described a cohort of infants with perinatal arterial ischemic stroke in the territory of the posterior cerebral artery. Seven patients out of 18 included in the study were diagnosed with neonatal hypoglycemia, leading to doubts about the possible relation between hypoglycemic brain damage and posterior stroke. A prevalent pattern of injury observed in the study was a unilateral occipital damage with a sharp demarcation line, different from what is habitually described for the hypoglycemic brain. The authors suggested that hypoglycemia may have acted as a risk factor for stroke development, underlining that the exact role of hypoglycemia in the pathogenesis of posterior stroke requires further investigation (85).
In preterm infants, the risk of germinal matrix hemorrhage - intraventricular hemorrhage is reported to be increased in case of hyperglycemia and higher glycemic variability, while no association seems to be present with hypoglycemia (86, 87). On the other hand, when intraventricular hemorrhage occurs atypically in term infants, hypoglycemia could be an underlying risk factor, as in 3 out of 35 term symptomatic infants with in one MRI study (68).
It has been suggested that less mature white matter could be more vulnerable to the negative effect of hypoglycemia, although no direct evidence of this possibility seem to exist at the moment. It is interesting to note how multiple punctate white matter lesions compatible with hemorrhage in the periventricular white matter were described both in term infants suffering from hypoglycemia and in extremely preterm infants (68, 88). Nevertheless, a role of hypoglycemia as a risk factor for punctate white matter lesions in preterm infants has not been yet investigated.
Prevention of Neonatal Brain Damage Hypoglycemia
Although the actual risk of neurologic injuries and abnormal outcomes, prevention of neonatal hypoglycemia still remains challenging. Neonatal hypoglycemia is frequently asymptomatic or associated with aspecific symptoms, and a precise glycemic threshold for brain injury development is not clearly established. The screening programs available to date displays substantial issues, including overtreatment of the physiologic glycemic reduction after birth and interference with breastfeeding, excessive blood sampling in at-risk euglycemic infants and missed early diagnosis of hypoglycemia in infants not included in the at-risk categories (89). Screening strategies in asymptomatic infants are suggested in the presence of several maternal or neonatal conditions possibly affecting the metabolic adaptation after birth (Table 3) (14, 90–92).
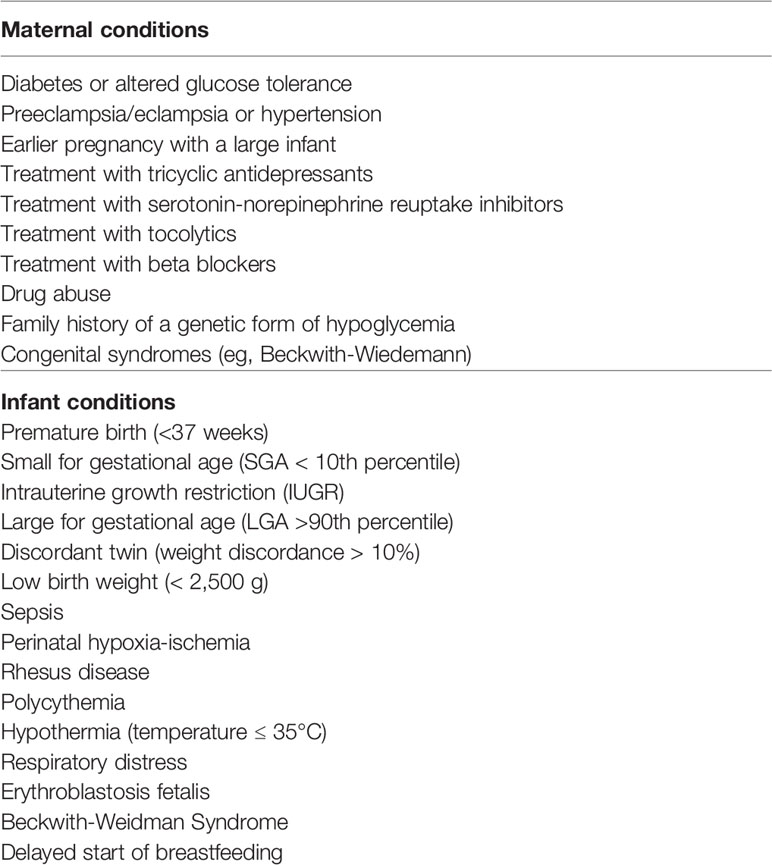
Table 3 Risk factors associated with hypoglycemia.
Routine measurements of blood glucose concentrations are recommended for infants with a risk factor for compromised metabolic adaptation or infants showing signs and symptoms suggestive of hypoglycemia. Many guidelines considered universal glucose monitoring for hypoglycemia in healthy, asymptomatic, term infants born after uncomplicated pregnancy and delivery to be an inappropriate procedure. Therefore, glucose screening should be reserved only for infants at risk. Glucose monitoring should be initiated within 3 h of life, or at any time infants showed symptoms of hypoglycemia. The decision to discontinue blood glucose monitoring depends on the glycemic trend and the treatment needed (2).
Early and exclusive breastfeeding meets the nutritional and metabolic needs of healthy, term newborn infants. A prompt initiation of breastfeeding immediately after birth reduces the risk of hypoglycemia. In addition, skin to skin practice help maintaining an adequate body temperature thus reducing energy expenditure and glucose consumption, while stimulating milk production (93, 94). Infants at risk for hypoglycemia must be fed within the first hour of life, before performing the first plasma glucose detection, and supplementary feeding with mother’s own breast milk or infant formula is recommended for all infants at risk for hypoglycemia. On the contrary, feeding with dextrose solution in the postnatal period may cause negative metabolic effects including increased insulin secretion, decreased glucagon secretion, delay of gluconeogenesis and ketogenic homeostasis. Prevention with dextrose gel has been recently proposed as an alternative to early supplementary feedings, but its efficacy on neonatal hypoglycemia prevention is still unclear (95).
It is commonly accepted that symptomatic infants should be promptly treated, in order to avoid severe and prolonged hypoglycemia which may result in neurologic injury (68). On the other hand, the management of hypoglycemia in asymptomatic infant poses more problems.
Frequent milk feedings associated with repeated glucose measurements represent the current standard treatment for asymptomatic hypoglycemia in high risk neonates (96). In infants with very low blood glucose or persistent hypoglycemia in spite of feedings, it is commonly accepted to start intravenous glucose administration. However, as improvement in long term outcomes following a rapid correction of asymptomatic hypoglycemia still remains unclear, and may expose to the risk of brain damage associated with a sudden increase in blood glucose levels and high glucose variability, we stress the importance of oral correction over the intravenous route in asymptomatic hypoglycemia.
Several screening and treatment programs have been proposed, with slight differences on operative thresholds and treatment strategies, aiming at the prevention of neonatal hypoglycemia through the following interventions:
- early feeding and blood glucose monitoring in infants considered at risk;
- use feeding supplementation as the first step to treat mild and asymptomatic hypoglycemia;
- promptly start intravenous dextrose infusion in case of severe, prolonged, and symptomatic hypoglycemia.
Though this approach seems the most reasonable, we also suggest to:
- Carefully evaluate maternal and pregnancy history and perinatal comorbidities for a prompt identification of at-risk infants;
- Perform a complete physical evaluation to exclude even mild symptoms of hypoglycemia;
- Carefully calculate the dextrose infusion rate to avoid dangerous glycemic oscillations;
- Intensively support breastfeeding through frequent and personalized breastfeeding interventions;
- Closely monitor glucose levels to titrate dextrose dose in order to avoid hyperglicemia, significant glycemic oscillations or recurrent episodes of hypoglycemia, which are associated with an increased risk of neurologic damage (71).
Discussion
Despite its significant prevalence, neonatal hypoglycemia remains a challenging condition. The transition of glucose homeostasis at birth leads to a temporary reduction of blood glucose levels in the first hours of life which is rarely symptomatic in healthy infants. However, the absence of clear blood glucose cut-off values in the first hours of life and the lack of universally accepted guidelines render its management largely uncertain. At the same time, though a causative role of hypoglycemia in brain damage has been demonstrated, a precise damage topography and the relationship between the entity and length of hypoglycemia and the extent of brain injury have not been clarified yet. Interpersonal variability in glucose homeostasis and the confounding effects of comorbidities especially in preterm infants may largely explain the uncertain results of the studies. In addition, the effect of glycemic oscillations often associated with hypoglycemia treatment could play an important role in increasing the risk of brain damage, possibly through enhancing oxidative stress responses.
Although few evidences are available to precisely establish operative guidelines to reduce hypoglycemia-related brain damage, we suggest to carefully assess the possible risk factors for hypoglycemia, to intensively support breastfeeding and to precisely titrate the dextrose dose.
Collaborative studies involving specific neonatal subpopulations together with comparable animal models may help discovery new insights in brain damage after neonatal hypoglycemia.
Author Contributions
LR, LD, and GB contributed conception of the study. GP, GB, AP, CT, PM, and MaM performed a literature search and wrote sections of the manuscript. LD, MoM, and DM critically revised the manuscript and wrote section of the manuscript. AR provided the brain magnetic resonance imaging and wrote a section of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ADP, adenosine-di-phosphate; ATP, adenosine-tri-phosphate; BBB, blood-brain-barrier; CBF, cerebral blood flow; DWI, diffusion-weighted imaging; GABA, gamma-aminobutyric acid; GE, glucose-excited; GI, glucose-inhibited; GLUT-1, glucose transporter-1; GLUT-3, glucose transporter-3; HIE, hypoxic-ischemic encephalopathy; LGA, large-for-gestational age; MRI, magnetic resonance imaging; NADPH, nicotinamide adenine dinucleotide phosphate; NAD+, nicotinamide adenine dinucleotide; NH, neonatal hypoglycemia; NMDA, N-methyl-D-aspartate; PARP-1, poly-ADP-ribose polymerase-1; ROS, reactive oxygen species; SGA, small-for-gestational age.
References
1. Puchalski ML, Russell TL, Karlsen KA. Neonatal Hypoglycemia: Is There a Sweet Spot? Crit Care Nurs Clin North Am (2018) 30:467–80. doi: 10.1016/j.cnc.2018.07.004
2. Harris DL, Weston PJ, Harding JE. Incidence of neonatal hypoglycemia in babies identified as at risk. J Pediatr (2012) 16:787–91. doi: 10.1016/j.jpeds.2012.05.022
3. Mitchell NA, Grimbly C, Rosolowsky ET, O’Reilly M, Yaskina M, Cheung PY, et al. Incidence and Risk Factors for Hypoglycemia During Fetal-to-Neonatal Transition in Premature Infants. Front Pediatr (2020) 8:34. doi: 10.3389/fped.2020.00034
4. Thompson-Branch A, Havranek T. Neonatal Hypoglycemia. Pediatr Rev (2017) 38:147–57. doi: 10.1542/pir.2016-0063
5. Güemes M, Rahman SA, Hussain K. What is a normal blood glucose? Arch Dis Child (2016) 101:569–74. doi: 10.1136/archdischild-2015-308336
6. Lord K, De León DD. Hyperinsulinism in the Neonate. Clin Perinatol (2018) 45:61–74. doi: 10.1016/j.clp.2017.10.007
7. Fong CY, Harvey AS. Variable outcome for epilepsy after neonatal hypoglycaemia. Dev Med Child Neurol (2014) 56:1093–9. doi: 10.1111/dmcn.12496
8. Tam EW, Haeusslein LA, Bonifacio SL, Glass HC, Rogers EE, Jeremy RJ, et al. Hypoglycemia is associated with increased risk for brain injury and adverse neurodevelopmental outcome in neonates at risk for encephalopathy. J Pediatr (2012) 161:88–93. doi: 10.1016/j.jpeds.2011.12.047
9. Kim JH, Koh SB. Extensive white matter injury in hypoglycemic coma. Neurology (2007) 68:1074. doi: 10.1212/01.wnl.0000258546.83251.36
10. Yang G, Zou LP, Wang J, Shi X, Tian S, Yang X, et al. Neonatal hypoglycemic brain injury is a cause of infantile spasms. Exp Ther Med (2016) 11:2066–70. doi: 10.3892/etm.2016.3107
11. Cornblath M, Hawdon JM, Williams AF, Aynsley-Green A, Ward-Platt MP, Schwartz R, et al. Controversies regarding definition of neonatal hypoglycemia: suggested operational thresholds. Pediatrics (2000) 105:1141–5. doi: 10.1542/peds.105.5.1141
12. Committee on Fetus and Newborn, Adamkin DH. Postnatal glucose homeostasis in late-preterm and term infants. Pediatrics (2011) 127:575–9. doi: 10.1542/peds.2010-3851
13. Wight N, Marinelli KA. Academy of Breastfeeding Medicine. ABM clinical protocol #1: guidelines for blood glucose monitoring and treatment of hypoglycemia in term and late-preterm neonates, revised 2014. Breastfeed Med (2014) 9:173–9. doi: 10.1089/bfm.2014.9986
14. Thornton PS, Stanley CA, De Leon DD, Harris D, Haymond MW, Hussain K, et al. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J Pediatr (2015) 167:238–45. doi: 10.1016/j.jpeds.2015.03.057
15. Hawdon JM. Identification and Management of Neonatal Hypoglycemia in the Full-Term Infant. British Association of Perinatal Medicine Framework for Practice, 2017. J Hum Lact (2019) 35:521–3. doi: 10.1177/0890334419846128
16. Narvey MR, Marks SD. The screening and management of newborns at risk for low blood glucose. Paediatr Child Health (2019) 24:536–54. doi: 10.1093/pch/pxz134
17. Wackernagel D, Gustafsson A, Edstedt Bonamy AK, Reims A, Ahlsson F, Elfving M, et al. Swedish national guideline for prevention and treatment of neonatal hypoglycaemia in newborn infants with gestational age ≥35 weeks. Acta Paediatr (2020) 109:31–44. doi: 10.1111/apa.14955
18. Kalhan S, Parimi P. Gluconeogenesis in the fetus and neonate. Semin Perinatol (2000) 24:94–106. doi: 10.1053/sp.2000.6360
19. Holme AM, Roland MC, Lorentzen B, Michelsen TM, Henriksen T. Placental glucose transfer: a human in vivo study. PLoS One (2015) 10:e0117084 e0117084. doi: 10.1371/journal.pone.0117084
20. Illsley NP. Glucose transporters in the human placenta. Placenta (2000) 21:14–22. doi: 10.1053/plac.1999.0448
21. Michelsen TM, Holme AM, Holm MB, Roland MC, Haugen G, Powell TL, et al. Uteroplacental Glucose Uptake and Fetal Glucose Consumption: A Quantitative Study in Human Pregnancies. J Clin Endocrinol Metab (2019) 104:873–82. doi: 10.1210/jc.2018-01154
22. Hay WW Jr. Energy and substrate requirements of the placenta and fetus. Proc Nutr Soc (1991) 50:321–36. doi: 10.1079/PNS19910042
24. Hume R, Burchell A, Williams FL, Koh DK. Glucose homeostasis in the newborn. Early Hum Dev (2005) 81:95–101. doi: 10.1016/j.earlhumdev.2004.10.005
26. Girard J. Gluconeogenesis in late fetal and early neonatal life. Biol Neonate (1986) 50:237–58. doi: 10.1159/000242605
27. Mitanchez D. Glucose regulation in preterm newborn infants. Horm Res (2007) 68:265–71. doi: 10.1159/000104174
28. Hoseth E, Joergensen A, Ebbesen F, Moeller M. Blood glucose levels in a population of healthy, breast fed, term infants of appropriate size for gestational age. Arch Dis Child Fetal Neonatal Ed (2000) 83:F117–9. doi: 10.1136/fn.83.2.f117
29. Mitanchez-Mokhtari D, Lahlou N, Kieffer F, Magny JF, Roger M, Voyer M. Both relative insulin resistance and defective islet beta-cell processing of proinsulin are responsible for transient hyperglycemia in extremely preterm infants. Pediatrics (2004) 113:537–41. doi: 10.1542/peds.113.3.537
30. Jackson L, Burchell A, McGeechan A, Hume R. An inadequate glycaemic response to glucagon is linked to insulin resistance in preterm infants? Arch Dis Child Fetal Neonatal Ed (2003) 88:F62–6. doi: 10.1136/fn.88.1.f62
31. Hume R, Burchell A. Abnormal expression of glucose-6-phosphatase in preterm infants. Arch Dis Child (1993) 68:202–4. doi: 10.1136/adc.68.2.202
32. Jackson L, Williams FL, Burchell A, Coughtrie MW, Hume R. Plasma catecholamines and the counterregulatory responses to hypoglycemia in infants: a critical role for epinephrine and cortisol. J Clin Endocrinol Metab (2004) 89:6251–6. doi: 10.1210/jc.2004-0550
33. Mesotten D, Joosten K, van Kempen A, Verbruggen S. ESPGHAN/ESPEN/ESPR/CSPEN working group on pediatric parenteral nutrition. ESPGHAN/ESPEN/ESPR/CSPEN guidelines on pediatric parenteral nutrition: Carbohydrates. Clin Nutr (2018) 37:2337–43. doi: 10.1016/j.clnu.2018.06.947
34. Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci (2013) 36:587–97. doi: 10.1016/j.tins.2013.07.001
35. Suh SW, Hamby AM, Swanson RA. Hypoglycemia, brain energetics, and hypoglycemic neuronal death. Glia (2007) 55:1280–6. doi: 10.1002/glia.20440
36. Bentsen MA, Mirzadeh Z, Schwartz MW. Revisiting How the Brain Senses Glucose-And Why. Cell Metab (2019) 29:11–7. doi: 10.1016/j.cmet.2018.11.001
37. Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes (2002) 51:2056–65. doi: 10.2337/diabetes.51.7.2056
38. Routh VH. Glucose-sensing neurons: are they physiologically relevant? Physiol Behav (2002) 76:403–13. doi: 10.1016/s0031-9384(02)00761-8
39. Alvarsson A, Stanley SA. Remote control of glucose-sensing neurons to analyze glucose metabolism. Am J Physiol Endocrinol Metab (2018) 315:E327–39. doi: 10.1152/ajpendo.00469.2017
40. McNay EC, McCarty RC, Gold PE. Fluctuations in brain glucose concentration during behavioral testing: dissociations between brain areas and between brain and blood. Neurobiol Learn Mem (2001) 75:325–37. doi: 10.1006/nlme.2000.3976
41. Devraj K, Klinger ME, Myers RL, Mokashi A, Hawkins RA, Simpson IA. GLUT-1 glucose transporters in the blood-brain barrier: differential phosphorylation. J Neurosci Res (2011) 89:1913–25. doi: 10.1002/jnr.22738
42. Mueckler M. Facilitative glucose transporters. Eur J Biochem (1994) 219:713–25. doi: 10.1111/j.1432-1033.1994.tb18550.x
43. Fioramonti X, Chrétien C, Leloup C, Pénicaud L. Recent Advances in the Cellular and Molecular Mechanisms of Hypothalamic Neuronal Glucose Detection. Front Physiol (2017) 8:875. doi: 10.3389/fphys.2017.00875
44. Hwang JJ, Jiang L, Hamza M, Sanchez Rangel E, Dai F, Belfort-DeAguiar R, et al. Blunted rise in brain glucose levels during hyperglycemia in adults with obesity and T2DM. JCI Insight (2017) 2:e95913. doi: 10.1172/jci.insight.95913
45. Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature (2008) 456:745–9. doi: 10.1038/nature07525
46. Engelsen B. Neurotransmitter glutamate: its clinical importance. Acta Neurol Scand (1986) 74:337–55. doi: 10.1111/j.1600-0404.1986.tb03524.x
47. Wieloch T, Engelsen B, Westerberg E, Auer R. Lesions of the glutamatergic cortico-striatal projections in the rat ameliorate hypoglycemic brain damage in the striatum. Neurosci Lett (1985) 58:25–30. doi: 10.1016/0304-3940(85)90323-4
48. Aral YZ, Gücüyener K, Atalay Y, Hasanoğlu A, Türkyilmaz C, Sayal A, et al. Role of excitatory aminoacids in neonatal hypoglycemia. Acta Paediatr Jpn (1998) 40:303–6. doi: 10.1111/j.1442-200X.1998.tb01936.x
49. Suh SW, Gum ET, Hamby AM, Chan PH, Swanson RA. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J Clin Invest (2007) 117:910–8. doi: 10.1172/JCI30077
50. Burke SP, Nadler JV. Effects of glucose deficiency on glutamate/aspartate release and excitatory synaptic responses in the hippocampal CA1 area in vitro. Brain Res (1989) 500:333–42. doi: 10.1016/0006-8993(89)90329-6
51. Auer RN. Progress review: hypoglycemic brain damage. Stroke (1986) 17:699–708. doi: 10.1161/01.str.17.4.699
52. Haces ML, Montiel T, Massieu L. Selective vulnerability of brain regions to oxidative stress in a non-coma model of insulin-induced hypoglycemia. Neuroscience (2010) 165:28–38. doi: 10.1016/j.neuroscience.2009.10.003
53. Suh SW, Garnier P, Aoyama K, Chen Y, Swanson RA. Zinc release contributes to hypoglycemia-induced neuronal death. Neurobiol Dis (2004) 16:538–45. doi: 10.1016/j.nbd.2004.04.017
54. Sensi SL, Paoletti P, Koh JY, Aizenman E, Bush AI, Hershfinkel M. The neurophysiology and pathology of brain zinc. J Neurosci (2011) 31:16076–85. doi: 10.1523/JNEUROSCI.3454-11.2011
55. Kim YH, Koh JY. The role of NADPH oxidase and neuronal nitric oxide synthase in zinc-induced poly(ADP-ribose) polymerase activation and cell death in cortical culture. Exp Neurol (2002) 177:407–18. doi: 10.1006/exnr.2002.7990
56. Weiss JH, Sensi SL, Koh JY. Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci (2000) 21:395–401. doi: 10.1016/S0165-6147(00)01541-8
57. Sheline CT, Behrens MM, Choi DW. Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J Neurosci (2000) 20:3139–46. doi: 10.1523/JNEUROSCI.20-09-03139.2000
58. Giordani I, Di Flaviani A, Picconi F, Malandrucco I, Ylli D, Palazzo P, et al. Acute hyperglycemia reduces cerebrovascular reactivity: the role of glycemic variability. J Clin Endocrinol Metab (2014) 99:2854–60. doi: 10.1210/jc.2014-1087
59. Spar JA, Lewine JD, Orrison WW Jr. Neonatal hypoglycemia: CT and MR findings. AJNR Am J Neuroradiol (1994) 15:1477–8.
60. Barkovich AJ, Ali FA, Rowley HA, Bass N. Imaging patterns of neonatal hypoglycemia. AJNR Am J Neuroradiol (1998) 19:523–8.
61. Traill Z, Squier M, Anslow P. Brain imaging in neonatal hypoglycaemia. Arch Dis Child Fetal Neonatal Ed (1998) 79:F145–7. doi: 10.1136/fn.79.2.f145
62. Chiu NT, Huang CC, Chang YC, Lin CH, Yao WJ, Yu CY. Technetium-99m-HMPAO brain SPECT in neonates with hypoglycemic encephalopathy. J Nucl Med (1998) 39:1711–3.
63. Kinnala A, Rikalainen H, Lapinleimu H, Parkkola R, Kormano M, Kero P. Cerebral magnetic resonance imaging and ultrasonography findings after neonatal hypoglycemia. Pediatrics (1999) 103:724–9. doi: 10.1542/peds.103.4.724
64. Filan PM, Inder TE, Cameron FJ, Kean MJ, Hunt RW. Neonatal hypoglycemia and occipital cerebral injury. J Pediatr (2006) 148:552–5. doi: 10.1016/j.jpeds.2005.11.015
65. Tam EW, Widjaja E, Blaser SI, Macgregor DL, Satodia P, Moore AM. Occipital lobe injury and cortical visual outcomes after neonatal hypoglycemia. Pediatrics (2008) 122:507–12. doi: 10.1542/peds.2007-2002
66. Caraballo RH, Sakr D, Mozzi M, Guerrero A, Adi JN, Cersósimo RO, et al. Symptomatic occipital lobe epilepsy following neonatal hypoglycemia. Pediatr Neurol (2004) 31:24–9. doi: 10.1016/j.pediatrneurol.2003.12.008
67. Gataullina S, De Lonlay P, Dellatolas G, Valayannapoulos V, Napuri S, Damaj L, et al. Topography of brain damage in metabolic hypoglycaemia is determined by age at which hypoglycaemia occurred. Dev Med Child Neurol (2013) 55:162–6. doi: 10.1111/dmcn.12045
68. Burns CM, Rutherford MA, Boardman JP, Cowan FM. Patterns of cerebral injury and neurodevelopmental outcomes after symptomatic neonatal hypoglycemia. Pediatrics (2008) 122:65–74. doi: 10.1542/peds.2007-2822
69. McKinlay CJ, Alsweiler JM, Ansell JM, Anstice NS, Chase JG, Gamble GD, et al. CHYLD Study Group. Neonatal glycemia and neurodevelopmental outcomes at 2 years. N Engl J Med (2015) 373:1507–18. doi: 10.1056/NEJMoa1504909
70. Shah R, Harding J, Brown J, McKinlay C. Neonatal Glycaemia and Neurodevelopmental Outcomes: A Systematic Review and Meta-Analysis. Neonatology (2019) 115:116–26. doi: 10.1159/000492859
71. Kerstjens JM, Bocca-Tjeertes IF, de Winter AF, Reijneveld SA, Bos AF. Neonatal morbidities and developmental delay in moderately preterm-born children. Pediatrics (2012) 130:e265–72. doi: 10.1542/peds.2012-0079
72. Goode RH, Rettiganti M, Li J, Lyle RE, Whiteside-Mansell L, Barrett KW, et al. Developmental Outcomes of Preterm Infants With Neonatal Hypoglycemia. Pediatrics (2016) 138:e20161424. doi: 10.1542/peds.2016-1424
73. Harding JE, Harris DL, Hegarty JE, Alsweiler JM, McKinlay CJ. An emerging evidence base for the management of neonatal hypoglycaemia [published correction appears in Early Hum Dev (2017) 108:61]. Early Hum Dev (2017) 104:51–6. doi: 10.1016/j.earlhumdev.2016.12.009
74. Kaiser JR, Bai S, Gibson N, Holland G, Lin TM, Swearingen CJ, et al. Association Between Transient Newborn Hypoglycemia and Fourth-Grade Achievement Test Proficiency: A Population-Based Study. JAMA Pediatr (2015) 169:913–21. doi: 10.1001/jamapediatrics.2015.1631
75. McKinlay CJD, Alsweiler JM, Anstice NS, Burakevych N, Chakraborty A, Chase JG, et al. Children With Hypoglycemia and Their Later Development (CHYLD) Study Team. Association of Neonatal Glycemia With Neurodevelopmental Outcomes at 4.5 Years. JAMA Pediatr (2017) 171:972–83. doi: 10.1001/jamapediatrics.2017.1579
76. Ennis K, Dotterman H, Stein A, Rao R. Hyperglycemia accentuates and ketonemia attenuates hypoglycemia-induced neuronal injury in the developing rat brain. Pediatr Res (2015) 77:84–90. doi: 10.1038/pr.2014.146
77. Fendler W, Walenciak J, Mlynarski W, Piotrowski A. Higher glycemic variability in very low birth weight newborns is associated with greater early neonatal mortality. J Matern Fetal Neonatal Med (2012) 25:1122–6. doi: 10.3109/14767058.2011.624220
78. Paudel N, Chakraborty A, Anstice N, Jacobs RJ, Hegarty JE, Harding JE, et al. CHYLD Study Group. Neonatal Hypoglycaemia and Visual Development: A Review. Neonatology (2017) 112:47–52. doi: 10.1159/000456705
79. Alkalay AL, Flores-Sarnat L, Sarnat HB, Moser FG, Simmons CF. Brain imaging findings in neonatal hypoglycemia: case report and review of 23 cases. Clin Pediatr (2005) 44:783–90. doi: 10.1177/000992280504400906
80. Yalnizoglu D, Haliloglu G, Turanli G, Cila A, Topcu M. Neurologic outcome in patients with MRI pattern of damage typical for neonatal hypoglycemia. Brain Dev (2007) 29:285–92. doi: 10.1016/j.braindev.2006.09.011
81. Basu P, Som S, Choudhuri N, Das H. Contribution of the blood glucose level in perinatal asphyxia. Eur J Pediatr (2009) 168:833–8. doi: 10.1007/s00431-008-0844-5
82. Basu SK, Kaiser JR, Guffey D, Minard CG, Guillet R, Gunn AJ, et al. Hypoglycaemia and hyperglycaemia are associated with unfavourable outcome in infants with hypoxic ischaemic encephalopathy: a post hoc analysis of the CoolCap Study. Arch Dis Child Fetal Neonatal Ed (2016) 101:F149–55. doi: 10.1136/archdischild-2015-308733
83. Wong DS, Poskitt KJ, Chau V, Miller SP, Roland E, Hill A, et al. Brain injury patterns in hypoglycemia in neonatal encephalopathy. AJNR Am J Neuroradiol (2013) 34:1456–61. doi: 10.3174/ajnr.A3423
84. Basu SK, Ottolini K, Govindan V, Mashat S, Vezina G, Wang Y, et al. Early Glycemic Profile Is Associated with Brain Injury Patterns on Magnetic Resonance Imaging in Hypoxic Ischemic Encephalopathy. J Pediatr (2018) 203:137–43. doi: 10.1016/j.jpeds.2018.07.041
85. van der Aa NE, Dudink J, Benders MJ, Govaert P, van Straaten HL, Porro GL, et al. Neonatal posterior cerebral artery stroke: clinical presentation, MRI findings, and outcome. Dev Med Child Neurol (2013) 55:283–90. doi: 10.1111/dmcn.12055
86. Auerbach A, Eventov-Friedman S, Arad I, Peleg O, Bdolah-Abram T, Bar-Oz B, et al. Long duration of hyperglycemia in the first 96 hours of life is associated with severe intraventricular hemorrhage in preterm infants. J Pediatr (2013) 163:388–93. doi: 10.1016/j.jpeds.2013.01.051
87. Jagła M, Szymońska I, Starzec K, Kwinta P. Impact of early glycemic variability on mortality and neurologic outcome of very low birth weight infants: Data from a continuous glucose monitoring system. Dev Period Med (2019) 23:7–14.
88. Niwa T, de Vries LS, Benders MJ, Takahara T, Nikkels PG, Groenendaal F. Punctate white matter lesions in infants: new insights using susceptibility-weighted imaging. Neuroradiology (2011) 53:669–79. doi: 10.1007/s00234-011-0872-0
89. Mukhopadhyay S, Wade KC, Dhudasia MB, Skerritt L, Chou JH, Dukhovny D, et al. Clinical impact of neonatal hypoglycemia screening in the well-baby care. J Perinatol (2020) 40:1331–8. doi: 10.1038/s41372-020-0641-1
90. Rozance PJ, Hay WW. Hypoglycemia in newborn infants: features associated with adverse outcomes. Biol Neonate (2006) 90:74–86. doi: 10.1159/000091948
91. Lubchenco LO, Bard H. Incidence of hypoglycemia in newborn infants classified by birth weight and gestational age. Pediatrics (1971) 47:831–8.
92. Stenninger E, Schollin J, Aman J. Early postnatal hypoglycemia in newborn infants of diabetic mothers. Acta Paediatr (1997) 86:1374–6. doi: 10.1111/j.1651-2227.1997.tb14916.x
93. Breastfeeding and the use of human milk. American Academy of Pediatrics. Work Group on Breastfeeding. Pediatrics (1997) 100:1035–9. doi: 10.1542/peds.100.6.1035
94. Wyllie J, Bruinenberg J, Roehr CC, Rüdiger M, Trevisanuto D, Urlesberger B. European resuscitation council guidelines for resuscitation 2015. Resuscitation (2015) 95:249–63. doi: 10.1016/j.resuscitation.2015.07.029
95. Hegarty JE, Harding JE, Gamble GD, Crowther CA, Edlin R, Alsweiler JM. Prophylactic oral dextrose gel for newborn babies at risk of neonatal hypoglycaemia: a randomised controlled dosefinding trial (the Pre-hPOD Study). PLoS Med (2016) 13:e1002155. doi: 10.1371/journal.pmed.1002155
Keywords: neonatal hypoglycemia, brain damage, glucose homeostasis, glucose sensing neurons, brain energetics
Citation: De Angelis LC, Brigati G, Polleri G, Malova M, Parodi A, Minghetti D, Rossi A, Massirio P, Traggiai C, Maghnie M and Ramenghi LA (2021) Neonatal Hypoglycemia and Brain Vulnerability. Front. Endocrinol. 12:634305. doi: 10.3389/fendo.2021.634305
Received: 27 November 2020; Accepted: 15 February 2021;
Published: 16 March 2021.
Edited by:
Ester Vitacolonna, University of Studies G. d’Annunzio Chieti and Pescara, ItalyReviewed by:
Dario Iafusco, University of Campania Luigi Vanvitelli, ItalySara Parrettini, University of Perugia, Italy
Copyright © 2021 De Angelis, Brigati, Polleri, Malova, Parodi, Minghetti, Rossi, Massirio, Traggiai, Maghnie and Ramenghi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Costanza De Angelis, bGFsbGFkZUBnbWFpbC5jb20=