 Hélène Duez
Hélène Duez Benoit Pourcet
Benoit Pourcet
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 25 February 2021
Sec. Molecular and Structural Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.630536
This article is part of the Research Topic Nuclear Receptors and Coregulators in Metabolism and Immunity View all 14 articles
The innate immune system is the first line of defense specialized in the clearing of invaders whether foreign elements like microbes or self-elements that accumulate abnormally including cellular debris. Inflammasomes are master regulators of the innate immune system, especially in macrophages, and are key sensors involved in maintaining cellular health in response to cytolytic pathogens or stress signals. Inflammasomes are cytoplasmic complexes typically composed of a sensor molecule such as NOD-Like Receptors (NLRs), an adaptor protein including ASC and an effector protein such as caspase 1. Upon stimulation, inflammasome complex components associate to promote the cleavage of the pro-caspase 1 into active caspase-1 and the subsequent activation of pro-inflammatory cytokines including IL-18 and IL-1β. Deficiency or overactivation of such important sensors leads to critical diseases including Alzheimer diseases, chronic inflammatory diseases, cancers, acute liver diseases, and cardiometabolic diseases. Inflammasomes are tightly controlled by a two-step activation regulatory process consisting in a priming step, which activates the transcription of inflammasome components, and an activation step which leads to the inflammasome complex formation and the subsequent cleavage of pro-IL1 cytokines. Apart from the NF-κB pathway, nuclear receptors have recently been proposed as additional regulators of this pathway. This review will discuss the role of nuclear receptors in the control of the NLRP3 inflammasome and the putative beneficial effect of new modulators of inflammasomes in the treatment of inflammatory diseases including colitis, fulminant hepatitis, cardiac ischemia–reperfusion and brain diseases.
Any living organism has to adapt to a specific environment and share common resources with others. To this purpose, organisms may collaborate in a reciprocal relationship from which each one of them benefits for its own survival. On the other hand, organisms may also be subject to threats from pathogenic offenders or from the environment itself, against which they have to defend themselves. The immune system is fundamental to anticipate and to preserve organisms from these threats. For that purpose, a specific system has been developed to allow the detection of two major classes of molecular signals, the pathogen-associated-molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) (1). PAMP and DAMP classification appears to be based on their biological sources rather than their chemical structures (1). PAMPs derive from pathogens including microbes and their products, while DAMPs originate from environmental disturbances such as the abnormal accumulation of endogenous compounds and cellular or subcellular damage. A common sensor system, defined as pattern-recognition receptors (PRRs), is able to detect both PAMPs and DAMPs. PRRs which are encoded by innate immune cells such as resident macrophages thus serve as sentinels of environmental changes including the presence of microbes and sterile tissue injury. In addition to PRRs, DAMPs are also detected by non-PRR receptors including receptor for advanced glycation end products (RAGEs), triggering receptors expressed in myeloid cells (TREMs), G-protein-coupled receptors (GPCRs), and ion channel (2). This allows the innate immune system to integrate various deleterious environmental changes to deliver the appropriate response according to nature of the threat (1).
PRRs can be distinguished based on their cellular location and the chemical nature of their ligand. Five main classes have been described: membrane-bound Toll-Like Receptors (TLRs) and C-Lectin Receptors (CLRs), cytoplasmic NOD-like receptors (NLR) and Retinoid acid-inducible gene I (RIG-1)-like receptors (RLRs), and multiple intracellular DNA sensors (CDSs) including cyclic GMP-AMP synthase (cGAS) and absent in melanoma 2 (AIM2) (2). Although TLRs were known to be activated by bacterial wall components such as LPS or proteoglycans, DAMPs including nucleic acids released from damaged cells are able to activate TLR3, TLR7, and TLR9 for instance, while intracellular proteins and extracellular matrix components released after tissue damage are able to induce a TLR2 or a TLR4-dependent signaling cascade (2). In addition, CLRs, usually known to be activated by fungi, are also able to detect lectin-derived compounds such as dendritic cell natural killer lectin group receptor 1 (DNGR1), macrophage-inducible C-type lectin (MINCLE), and Dectin-1 (2). RLRs are able to detect non-self RNA from microbial origin but also inappropriately masked self 5′ppp-RNA such as RNA generated during the unfolded protein response (2). CDSs are able to detect cytoplasmic (cGAS and AIM2) and damaged DNA in the nucleus (AIM2 only) (2). Finally, NLRs, which recognize bacterial compounds such as flagellin, are also able to detect crystals, ATP, amyloid fibers, glucose, or mitochondrial DNA. Therefore, PRRs and non-PPRs are able to sense extracellular and intracellular DAMPs, thus allowing a thorough surveillance of potential threats. Importantly, extracellular signals are considered as low-threat and resolvable problems, while cytosolic signals represent high-threat encounters that may induce pyroptosis, known as an interleukin (IL)-1β and IL-18-triggered cell death program induced by cytosolic PRRs only, mainly inflammasomes (1). When activated by DAMPs, PRRs and non-PRRs then trigger a so-called sterile inflammation, i.e. not induced by microbes. Therefore, a sustained activation of these receptors leads to inflammatory diseases including ischemia–reperfusion injury, colitis, systemic lupus erythematous, gout, neurodegenerative diseases, diabetes, atherosclerosis, hepatitis, rheumatoid arthritis, cancer, lung diseases, and gut diseases (2).
Inflammation is characterized by the production of a plethora of secreted immunomodulatory signaling molecules such as histamine, cytokines, chemokines, and lipid derivatives (1). The IL-1 cytokine family is a major cytokine family that includes IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β and IL-36γ. Except for IL-1α, IL-1 cytokines are produced as inactive pro-cytokines and require maturation to biologically active forms by enzymatic cleavage. For instance, pro-IL-1β and pro-IL-18, the most studied IL-1 family members, are processed by the proteolytic activity of Caspase 1, the predominant IL-1 processing protease. Caspase 1 activity is tightly controlled by cytosolic PRR-constituted inflammasome complex. Inflammasomes form the main class of cytosolic PPRs that are activated by diverse exogenous signals including anthrax lethal toxin (NLRP1), bacterial flagellin (NLRC4), double stranded DNA (AIM2), toxin-induced modifications of Rho-GTPase (Pyrin). Unlike other inflammasomes, the nucleotide-binding domain (NOD)-, Leucine-rich repeat (LRR)- and pyrin domain containing protein 3 (NLRP3) inflammasome is not only activated by microbial and environmental molecules but also by several metabolic products including ATP, cholesterol crystals and β amyloid fibers. In this regard, NLRP3 is unique because it is able to sense a wide range of threats. NLRP3 is therefore a central PAMPs and DAMPs sensor whose erratic activation leads to numerous NLRP3-driven diseases.
The NLRP3 inflammasome was first identified in the cryopyrin-associated periodic syndrome (CAPS) and was later recognized to be involved in many other inflammatory/metabolic diseases including gout, atherosclerosis, type 2 diabetes, non-alcoholic fatty liver diseases (NAFLD), colitis, and neurodegenerative diseases such as Alzheimer and Parkinson diseases. The NLRP3 inflammasome is not only expressed by leucocytes (macrophages, dendritic cells, neutrophils) but also by hepatocytes, neurons, endothelial cells, cardiomyocytes, and pancreatic beta cells (3).
The NLRP3 inflammasome is a supramolecular organizing center (SMOC) which consists of a sensor (NLRP3), an adaptor (Apoptosis-associated speck-like protein containing a Caspase recruitment domain (ASC) encoded by PYCARD), and an effector (Caspase 1) (4). NLPR3 contains an amino terminal pyrin domain (PYD) involved in protein–protein interaction, a central oligomerization domain (NOD, nucleotide-binding and oligomerization domain, NACHT) with an ATPase activity involved in the self-association and function of NLRP3 and a carboxy terminal leucin-rich repeat (LRR) domain inducing the autoinhibition of NLRP3 by folding back onto the NACHT domain (4). Apart from an Nter PYD domain, ASC also includes a Cter caspase recruitment domain (CARD) that plays a role of adaptor platform for the pro-Caspase 1 protein through a CARD–CARD domain interaction. Caspase 1 structure also includes a central catalytic domain (p20) and a Cter small catalytic subunit (p10) (4).
Upon stimulation, NLRP3 oligomerizes through homotypic interactions between NACHT domains of two NLRP3 proteins and the subsequent recruitment of ASC through PYD-PYD interactions (Figure 1). Then, helical ASC filaments nucleate and associate to form macromolecular ASC specks (5–7) (Figure 1). Finally, assembled ASC recruits pro-caspase 1 in a CARD-dependent manner that enables the proximity-driven self-cleavage of pro-caspase 1 to generate p33 (comprising the CARD and the p20 domains) and p10, which remains bound to ASC and becomes proteolytically active (Figure 1). Further processing then triggers the release of the p20 and p20–p10 complex from ASC. The p20–p10 complex is unstable in the cells, thus terminating its protease activity (8). Beyond the classical representation of NLRP3 inflammasome assembly, it has recently been demonstrated that the NIMA-related kinase 7 (NEK7) oligomerizes with the LRR domain of NLRP3 into a complex by bridging the gaps between adjacent NLRP3 subunits to mediate NLRP3 oligomerization that is essential for ASC speck formation and caspase 1 activation (9, 10) (Figure 1). Strikingly, NEK7 is specific to NLRP3 and does not interact with other inflammasomes such as NLRC4 (11). Regulation of NEK7-NLRP3 assembly is induced by ATP-driven potassium efflux (12) but also in a K+-efflux independent manner (13) and by reactive oxygen species (ROS) production (9). Activated-Caspase 1 is then able to process pro-IL-1β and pro-IL-18 into mature and functional IL-1β and IL-18 (Figure 1).
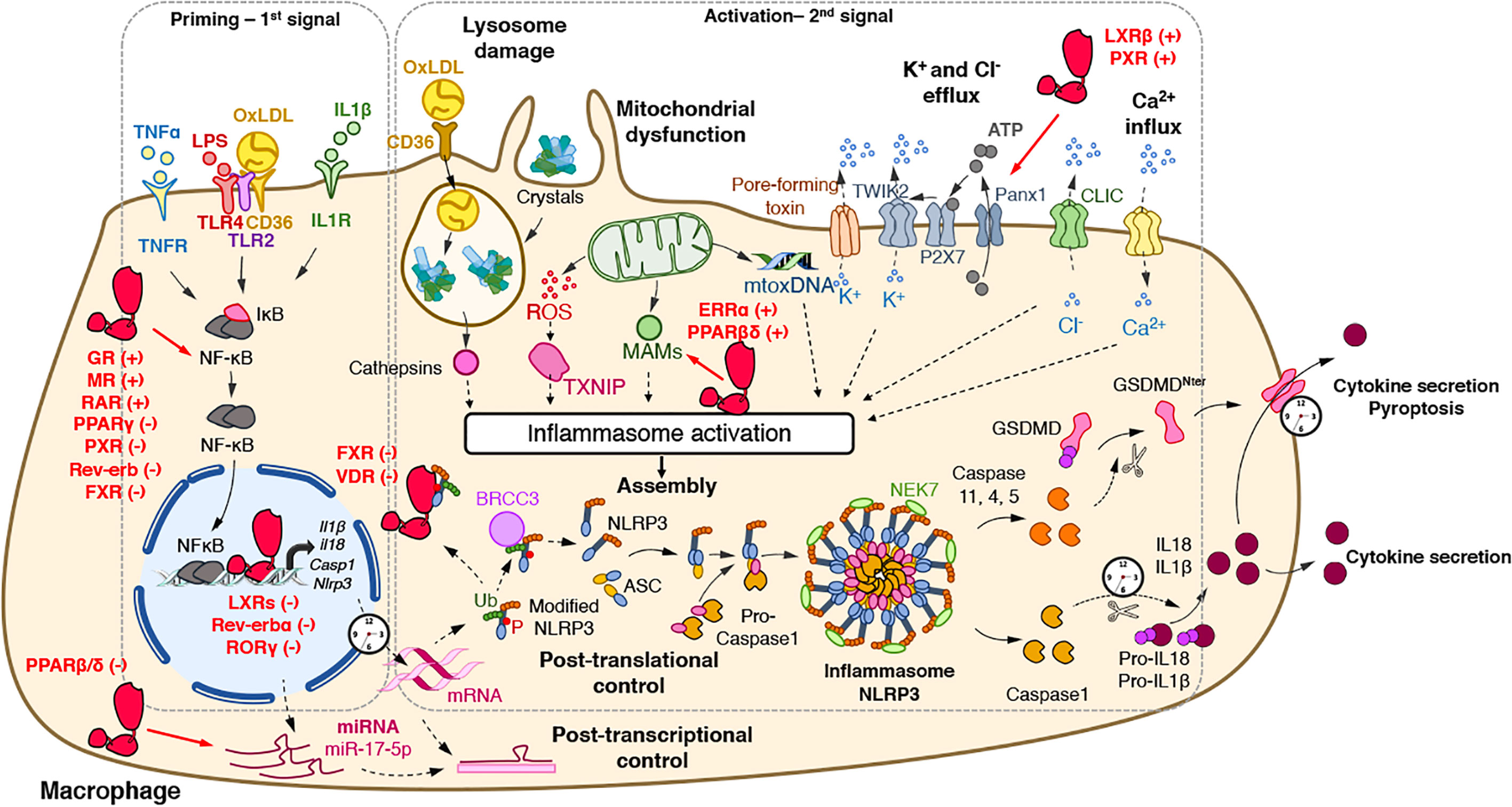
Figure 1 Regulatory activities of nuclear receptors on the NLRP3 inflammasome priming and activation steps. The priming (first step) of the NLRP3 inflammasome requires the binding and activation of PRRs (TLRs,…) by PAMPs such as LPS, cytokines or ox-LDL, resulting in the transcription of the NLRP3 inflammasome components. Its activation (second step) is the result of recognition of PAMPs (such as the bacterial pore-forming toxin nigericin) or DAMPs which are released by damaged or dying cells (such as ATP) following injury or metabolic imbalance (such as mtROS), or accumulate in tissues (such as crystals). These lead to lysosomal damage, mitochondrial damages (exposition of cardiolipin, mtDNA) which ultimately modify ion (K+, Ca2+) fluxes. Upon this two-step process, the NLRP3 inflammasome assembles, caspase 1 is activated, Gasdermin-D and pro-IL-1β and pro-IL-18 are cleaved, leading to mature cytokines secretion and cell death by pyroptosis. The activity of nuclear receptors on each step is indicated when appropriate. ASC, apoptosis-associated speck-like protein containing a CARD domain; ATP, adenosine triphosphate; BRCC3, Lys-63-specific deubiquitinase BRCC36; casp, caspase; CLIC, chloride intracellular channels; DAMPs, damage-associated molecular patterns; GSDMD, gasdermin-D; IL, interleukin; IL1R, interleukin-1 receptor; LPS, lipopolysaccharide; MAM, Mitochondria-associated ER membranes; mtoxDNA, mitochondrial oxidized DNA; NFκB, nuclear factor-kappa B; NLRP3, nucleotide-binding, LRR and PYD domains-containing protein 3; Ox-LDL, oxidized low-density lipoproteins; P, Phosphate; PAMPs, pathogen-associated molecular patterns; Panx1, Pannexin-1; PRRs, Pattern Recognition Receptors; ROS, reactive oxygen species; P2X7, purinergic receptor P2X7; TLR, Toll-like receptor; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; TWIK2, two-pore domain weak inwardly rectifying K+ channel; TXNIP, Thioredoxin-interacting protein Ub, ubiquitin. (+): activates; (−): inhibits.
In addition to the regulation of pro-inflammatory cytokine maturation, the NLRP3 inflammasome is also involved in the control of pyroptosis, defined as a rapid and inflammatory form of programmed cell death. Pyroptosis actually results from the cleavage of Gasdermin D (GSDMD) by inflammatory caspases including caspases 1, 4, 5, or 11 (14–16) (Figure 1). GSDMD possesses an Nter cell death domain (GSDMDNTerm), a central short region, which links to a Cter auto-inhibition domain. Caspase 1 cleaves pro-GSDMD, thereby removing the auto-inhibition domain, thus alleviating the inhibition on the cell death domain (Figure 1). GSDMDNTerm then binds to phosphatidylinositol phosphate and phosphatidylserine in the inner leaflet of the cell membrane, oligomerizes, and inserts into the plasma membrane, thus forming a pore of 16 symmetrical protomers that kill the cell (17).
The activation of the NLRP3 inflammasome, as most inflammasomes, is tightly controlled by a two-step process (4) (Figure 1). A priming step is required to increase gene and protein expression of its components in order to sense stimuli and become activated. Once the cytoplasmic levels of NLRP3 mRNA reach an activating threshold, inflammasome assembly can be triggered by a secondary signal. This activation step initiates the NLRP3 SMOC assembly that promotes Caspase 1 autocatalytic activity and its subsequent maturation.
The priming step has two main purposes: the transcriptional induction of the inflammasome complex components NLRP3, Caspase 1, IL-1β, and IL-18 and the induction of post-translational modifications of NLRP3 (Figure 1). The first one can be induced through the recognition of various PAMPs and DAMPs by PRRs such as TLRs and NLRs including NOD1 and NOD2 or cytokine receptors (e.g. TNFR) whose activation promotes nuclear factor κB (NF-κB) transcriptional factor activation and the subsequent induction of Nlrp3 and Il1β gene transcription (Figure 1).
In addition to classical TLR ligands, disruption of metabolic homeostasis has also been involved in the NLRP3 inflammasome priming. For instance, NLRP3 mediates trained immunity following western diet feeding (18), suggesting that a lipid-rich diet potentializes the NLRP3-mediated response to pro-inflammatory stimuli. Accordingly, oxidized LDL (oxLDL), but also islet amyloid polypeptide (IAPP) and Alzheimer Disease beta amyloid peptides (Aβ1–42), induce Nlrp3 and Il1β gene expression, and thus the priming of this pathway in a CD36-TLR2-TLR4 heterotrimer-dependent manner in bone marrow-derived macrophages (BMDM) (19, 20) (Figure 1). Finally, cholesterol crystal-induced release of Neutrophils Extracellular Traps (NETs) from neutrophils is also able to prime NLRP3 in macrophages through the activation of several TLRs (21).
In addition to transcriptional regulation, the stability of mRNA of inflammasome components such as NLRP3, Casp1, and Casp8 can be controlled at the post-transcriptional level by miRNA [see (22) for review]. For instance, miR-223-3p negatively regulates the NLRP3 inflammasome by targeting the 3′-untranslated region (UTR)-binding sites of NLRP3 mRNA in myeloid cells (23). In addition, miRNAs can also target the mRNA of upstream regulators of the NLRP3 inflammasome including TXNIP, TRAF6, and SOD2 (11). As an example, miR-17-5p decreases TXNIP mRNA stability and NLRP3 activation in insulin producing cells and in the brain, thus inhibiting NLRP3 pathway activation (24, 25) (Figure 1). Accordingly, altered expression of several miRNAs is associated with the development of numerous NLRP3-driven diseases such as rheumatoid arthritis (26), multiple sclerosis (27), and systemic lupus erythematosus (28, 29).
While this transcriptional priming allows the production of NLRP3 pathway components, additional mechanisms are necessary to maintain NLRP3 in an inactive but poised configuration to rapidly respond to an activation signal. The second function of priming is then the induction of rapid transcription-independent mechanisms that regulate NLRP3 stability in order to rapidly progress from this poised state to an active one. Such non-transcriptional mechanisms are mainly classical post-translational modifications including ubiquitination, phosphorylation and SUMOylation (Figure 1) [see for review (30)]. For instance, ubiquitination of NLRP3 by FBXL12, TRIM1, ARIH2 or the dopamine-induced E3 ligase MARCH7 promotes the proteasomal degradation of NLRP3 in resting macrophages (30), whereas deubiquitylation of NLRP3 LRR domain on K63 by BRCC3 triggers ASC oligomerization and inflammasome activation (31, 32) (Figure 1).
The NLRP3 inflammasome is unique as it can assemble in response to a wide range of stimuli with various chemical properties. These include exogenous molecules of various origins such as environmental particulates (silica crystals) or pathogens. In addition, many endogenous molecules that abnormally accumulate are able to activate the NLRP3 inflammasome. This abnormal accumulation usually reflects tissue damage or metabolic dysfunction, which are thus sensed by NLRP3. For instance, under physiological conditions, LDLs normally circulate in blood. When LDL level abnormally increases in the context of dyslipidemia and when the vascular endothelium is damaged, LDLs infiltrate into the vascular wall, are eventually oxidized and trigger macrophage recruitment as seen in atherogenesis. CD36-mediated uptake of oxLDLs by macrophages contributes to the formation of intracellular cholesterol crystals and leads to the subsequent activation of NLRP3 (19) (Figure 1). Likewise, while normal extracellular ATP levels are harmless, tissue damage or cell death increases extracellular ATP levels acting as NLRP3-activating DAMPs. NLRP3 activation is often due to cellular stress resulting in lysosomal destabilization, ion flux imbalance, and redox potential alteration.
Crystals (cholesterol, urea, hydroxyapatite crystals) or fibrillar protein aggregates (β-amyloid, IAPP) can be phagocytosed by immune cells and then traffic toward lysosomes. These crystals often lead to lysosomal damage resulting in the release of proteases such as cathepsins (Figure 1). Although lysosomal disruption appears as a critical step for NLRP3 activation (33), downstream mechanisms between lysosome alteration and activation of the NLRP3 inflammasome still need to be unequivocally identified. Lysosome-released cathepsin B was considered for long as an essential trigger of NLRP3 activation (33). Nevertheless, the use of a broad spectrum of cathepsin inhibitors and individual knock-out experiments of several cathepsins confirmed that NLRP3 activation however, relies on several cathepsins that may exert redundant activities (34, 35). Importantly, Leu-Leu-OMe-induced lysosomal damage enhances K+ and Ca2+ efflux that may account for lysosomal damage-controlled NLRP3 activation (36).
Ion fluxes are important regulators of NLRP3 inflammasome activation. Changes in ion homeostasis such as increased intracellular Ca2+ levels as well as decreased intracellular K+ and Cl− levels also appear to play a pivotal role in NLRP3 activation (Figure 1). Lower extracellular concentrations of K+ compared to intracellular K+ concentrations are sufficient to induce K+ efflux and to promote NLRP3 activation while high levels of extracellular K+ prevent its activation in THP1 cells and BMDM (37, 38). In addition, nigericin, a K+ ionophore, as well as the ATP-mediated activation of P2X purinoceptor 7 (P2rx7), a ligand-gated ion channel, promotes K+ efflux-dependent IL-1β maturation (39–41) (Figure 1). Interestingly, P2rx7 does not directly control K+ efflux, but instead, promotes Ca2+ and Na2+ influx after ATP stimulation and coordinates with the K+ channel two-pore domain weak inwardly rectifying K+ channel (TWIK2), which mediates K+ efflux (42) (Figure 1).
Interestingly, K+ efflux must be associated with Ca2+ influx to promote mitochondrial-mediated ROS production (43), where Ca2+ influx appears critical for NLRP3 activation (44, 45) (Figure 1). At the molecular level, CHOP, a transcription factor activated during ER stress, promotes Ca2+ release from the ER, thus stimulating the calcium-sensing receptor (CASR) and promoting NLRP3 assembly (44). K+ efflux also controls Ca2+ release from the ER demonstrating the interconnection between the different activating signals (43, 44).
In addition to K+ efflux and Ca2+ influx, Cl- flux has also been demonstrated to activate NLRP3 (Figure 1). Indeed, while low extracellular Cl− enhances ATP-induced IL-1β secretion, high extracellular Cl− concentration or Cl− channel blockers inhibit NLRP3 activation (46, 47). Two recent reports demonstrated that chloride intracellular channels (CLICs), especially CLIC1 and CLIC4 mediate NLRP3 activation by promoting Cl− efflux downstream nigericin-induced K+ efflux and mitochondrial ROS production, which promotes CLIC translocation to the plasma membrane (46, 47) (Figure 1). Interestingly, K+ seems to drive NLRP3 oligomerization, probably in a NEK7-dependent manner (12, 48), while Cl− efflux is prone to induce ASC polymerization (48). Finally, although ion fluxes were shown to control NLRP3 assembly and activation, the link between ion fluxes and the inflammasome activation remains to be identified.
Since ROS scavengers attenuate NLRP3 activation, the generation of ROS was considered a common cellular response critical for NLRP3 activation (49). Although the source of NLRP3-activating ROS was controversial, the inhibition of the lysosomal NADPH oxidase did not alter NLRP3 activation in mouse and human cells, thus suggesting an alternative source of NLRP3-activating ROS, likely the mitochondria (33, 50, 51). After stimulation with various NLRP3 activators, mitochondrial ROS (mtROS) altogether with Ca2+, contribute to the rapid release of mtDNA into the cytosol (52) where it is eventually oxidized (53). Oxidized mtDNA then specifically interacts with NLRP3 and activates the inflammasome (53) (Figure 1). In addition, mtROS promotes Thioredoxin-interacting protein (TXNIP)-NLRP3 interaction involved in NLRP3 expression (54) (Figure 1).
Notably, NLRP3 is mainly localized at the membrane surface of ER in unstimulated cells (49). However, in the presence of MSU, nigericin or alum, mtROS production leads to the rapid relocation of NLRP3 and cardiolipin at the mitochondria outer membrane and promotes K+ efflux (49). Then, the ASC adaptor accumulates at Mitochondria-associated ER membranes (MAMs) where the NLRP3-ACS complex is formed (49). In addition, NLRP3 may also interact with mitochondrial antiviral-signaling protein (MAVS), which is another mitochondrial outer MAM (55–57). In this context, mitofusin 2 can also be found in the outer mitochondrial membrane, the ER and MAM. Mitofusin 2 plays an important role in NLRP3 activation during RNA viral infections since it interacts with MAVS to support the relocation of NLRP3 to the mitochondria (58) (Figure 1).
In addition to the classical/canonical NLRP3 inflammasome activation, an alternative NLRP3 activation process has been identified in which LPS alone is sufficient to induce inflammasome activation without the involvement of another second activator (59). This signaling pathway relies on a cascade involving TLR4, TIR domain-containing adapter molecule 1 (TRIF), RIPK1, FADD and caspase 8 that finally promotes NLRP3 activation. Interestingly, in addition to LPS, the pro-atherogenic apolipoprotein ApoC3 is able to trigger TLR2 and TLR4 heterodimerization and promotes the alternative activation of NLRP3 (60), thus mirroring the effect of oxLDL in the canonical activation of NLRP3. Strikingly, the alternative inflammasome activation is characterized by its independency on K+ efflux and the absence of pyroptosome formation and pyroptosis. Then, this pathway is likely involved in the control of cytokine secretion without affecting cell viability.
In addition to caspase 1, cytosolic gram negative bacteria-derived LPS may also be sensed independently of TLR4 signaling by human caspases 4 and 5, and mouse caspase 11, to induce the non-canonical NLRP3 inflammasome (61, 62). In this pathway, Caspase-4/5/11 promote pyroptosis by processing pro-GSDMD and pannexin-1, a protein channel that releases ATP from the cell. This extracellular ATP then activates P2xr7 to promote K+ efflux and NLRP3 activation (63, 64).
In addition to the above-described regulators, priming and activation processes are also controlled by nuclear receptors (NRs), a subclass of transcription factors. Although numerous studies have reported this alternative activation pathway, such regulatory processes are rarely mentioned. We provide here the first review of the literature describing how these lipid-regulated receptors control both priming and activation processes in the context of different NLRP3-driven diseases. We will also describe in which pathophysiological contexts this regulation has been reported and how the pharmacological modulation of these NRs prevents the progression of NLRP3-driven diseases. Finally, we will discuss also the role of NLRP3 in NR regulation.
Discovered in the mid-80s, NRs represent a superfamily of structurally conserved ligand-dependent transcription factors that regulate gene expression (65–67). The nuclear receptor superfamily can be sub-divided into four classes based on their ligand- and DNA-binding properties and on the nature of their partner (68). NRs usually work as homo- or heterodimers, which bind to a specific response element composed of two AGGTCA half-sites separated by one to four nucleotides in the promoter of target genes (Figure 2). These half-sites are organized either as a palindromic sequence or a direct repeat. The first class, mostly classical steroid hormone receptors, is probably the best characterized and consists of nuclear hormone receptors such as Androgen Receptor (AR), Glucocorticoid receptor (GR), Estrogen receptors (ERs: ERα, ERβ), Mineralocorticoid Receptor (MR), Progesterone Receptor (PR). These NRs work as homodimers and are recruited to a palindromic arrangement of core recognition motifs. The second class consists of so-called adopted receptors that were initially identified as orphan receptors meaning without known ligands, but subsequent studies characterized naturally occurring ligands and determined their physiological roles. Its members encompass eicosanoid and fatty acid receptors Peroxisome Proliferator-Activated Receptors (PPARs: PPARα, PPARβ/δ, PPARγ), the oxysterol receptors Liver X Receptors (LXRs: LXRα and LXRβ), Thyroid hormone Receptors (TRs: TRα, TRβ), the Retinoic Acid Receptors (RARs: RARα, RARβ, RARγ), the Vitamin D3 Receptors (VDR), the xenobiotic receptor Pregnane X Peceptor (PXR). NRs from class II heterodimerize with one of the Retinoid X Receptors (RXRs: RXRα, RXRβ or RXRγ) and are recruited to a response element organized in two-half sites in tandem repeat (69–71). The third NR class is composed of adopted receptors such as RXRs, the heme receptors Rev-erb (Rev-erbα and Rev-erbβ), fatty acid receptor Human Nuclear Factor 4 (HNF4α, HNF4γ) and orphan receptors such as Chicken Ovalbumin Upstream Promoter Transcription Factor (COUP-TFI, COUP-TFII). The NRs from this third class act as monomers or homodimers bound on direct repeat response elements. Finally, the fourth class is made of orphan nuclear receptors such as Estrogen Related Receptors (ERRα, ERRβ, ERRγ), Retinoid-related Orphan Receptors (RORα, RORβ, RORγ), Nurr1, NOR1, Nurr77 and the steroidogenic factor 1 SF-1. Therefore, NRs represent a crucial superfamily of transcriptional factors whose transcriptional activity may be modulated by specific natural or synthetic ligands, identifying NRs as promising therapeutical targets in numerous diseases, and especially in NLRP3-driven diseases as described below.
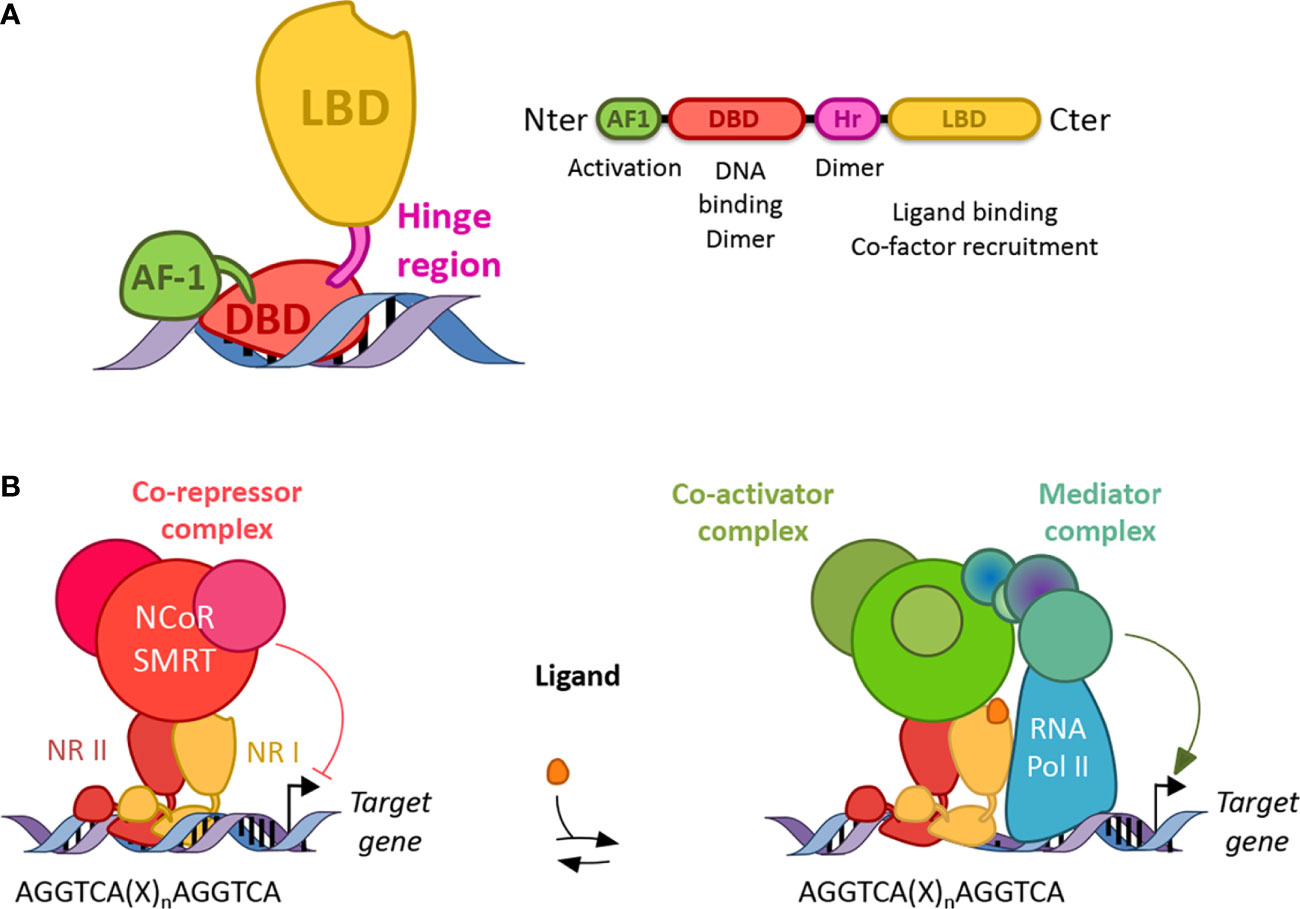
Figure 2 Structure and function of nuclear receptors. (A) Canonical structures of nuclear receptors. Nuclear receptors are composed of a N terminal activation function domain whose activity is independent of ligand binding, a DNA binding domain (DBD), a hinge region (Hr) and a ligand binding domain (LBD). Their respective activity is mentioned accordingly. Dimer: dimerization (B) Nuclear receptors work as homo or heterodimers which bind a response element present in the promoter of their target genes. Response elements are composed of two AGGTCA half-sites separated by one to four nucleotides (X). In the absence of ligand, NRs (except class I) preferentially bind co-repressor and inhibit gene transcription. In the presence of a ligand, co-repressors are degraded by the proteasome and co-activators are recruited, which then allows the binding of a mediator complex and the ARN polymerase II.
NRs consist of modular domains, including a variable amino N-terminal activation domain (AF-1), a highly conserved DNA-binding domain (DBD), a conserved hinge region linking the DBD with the conserved ligand-binding domain (LBD) (72) (Figure 2A). The DBD mediates the specific recruitment of NR monomers, homodimers, heterodimers to their DNA response element and is involved in the dimerization of NRs with their partner altogether with the hinge region and the LBD. In addition, the LBD mediates ligand-dependent interactions with transcriptional co-activators such as p300/CBP or co-repressors such as NCoR or SMRT (Figure 2). These interactions are controlled, at the structural level, by ligand-dependent conformational changes in the last α-helix 12 (αH12) of the LBD known as AF2 (73). In the absence of a ligand, co-repressors are preferentially bound to NRs, especially those of class II, while ligand binding induces a conformational change of the αH12 helix which then triggers the release of co-repressors, allowing co-activator binding (Figure 2B). If several NRs, especially those of the class II including PPARs, LXRs, RARs are then able to bind target genes in the absence of a ligand and recruit co-repressors to actively repress gene expression, class I steroid hormone receptors are usually sequestered into the cytoplasm in the absence of ligands and are translocated into the nucleus to bind their target genes in the presence of a ligand. Finally, the Rev-erb subfamily, Rev-erbα and Rev-erbβ, lacks the αH12, which then prevents the recruitment of co-activators (74, 75). Instead, although Rev-erbs are able to recruit co-repressors and actively repress gene expression in the absence of a ligand, ligand binding enhances co-repressor recruitment and the transcriptional activity of Rev-erbs to further inhibit the expression of their target genes (74, 75). Their transcriptional activity might be regulated by post-translational modifications including phosphorylation, ubiquitination, and SUMOylation (76–85).
NRs are involved in the control of numerous physiological activities including metabolism (86, 87), reproduction (88, 89), cell cycle (90), vasculature (91, 92), brain activity (93, 94), circadian rhythm (95–97) and immunity (98–103). NRs have then been widely implicated in the control of inflammatory processes and the control of immune cell activity (99). In macrophages, many NRs display anti-inflammatory activities by quenching the NF-κB dimer into the cytoplasm (99). For instance, GR inhibits the expression of TNFα and COX2. In addition, iNOS expression is inhibited by both PPARγ and GR, while TLR4 expression is dampened by Rev-erbα and PPARγ (78, 104). In the same manner, IL-6 expression is reduced by both GR and Rev-erbα (99, 105). Interestingly, LXRα is able to induce TLR4 in human macrophages only, emphasizing the species-specificity of such regulatory pathway and also induces its own negative regulatory loop by enhancing Rev-erbα expression to avoid TLR4 lasting expression (106). In addition, nuclear receptors such as PPARγ (107), LXRα (101, 108, 109), Nurr77 (110) and Rev-erbα (104, 111) also control the skewing of pro-inflammatory macrophages toward anti-inflammatory macrophages. Finally, nuclear receptors including LXR (112), GR (113), and Rev-erb (105, 114) regulate macrophage recruitment by controlling the production of adhesion molecules or the secretion of chemokines such as Monocyte Chemoattractant Protein 1 (MCP1).
Many NRs have been shown to interact with the NF-κB complex and to inhibit this pathway either by directly interacting with the NF-κB complex in the cytoplasm, a mechanism known as quenching (98, 99), or by preventing the polyubiquitination of the IKK complex, which then promotes NF-κB inhibition (98). However, although these regulatory processes are known, only few studies demonstrate the direct link between NR-controlled NF-κB pathway and NLRP3 priming. For instance, dexamethasone, a GR synthetic ligand, and cortisol treatments in human THP1 macrophages and in BMDMs induce the expression of NLRP3 mRNA and proteins in a GR-dependent manner but not those of Casp1 and Il1β (115) (Figure 1). Nevertheless, glucocorticoids enhance the secretion of mature IL-1β by these cells (115), thus demonstrating the ability of glucocorticoids to set up an active NLRP3 inflammasome pathway. Although the molecular mechanisms involved therein were not investigated in this report, this regulatory effect may be due, at least partially, to the activation of the NF-κB pathway. Indeed, dexamethasone as well as chronic stress, which triggers the production of cortisol, induce the NF-κB pathway in hippocampal neuroinflammation and depression-like behavior (116). In addition to GR, PXR agonists altogether with PXR overexpression induce NLRP3 and NLRP2 mRNA levels in endothelial HUVEC cells (117) (Figure 1). Interestingly, oxLDL has been shown to induce NLRP3 expression in a LOX1- and NF-κB-dependent manner (118). LOX1 is the main endothelial oxLDL receptor, whose stimulation by oxLDL induces NF-κB pathway (119), a mechanism reminiscent to the CD36-dependent one in macrophages (19). Interestingly, statins inhibit the activated NF-κB pathway and NLRP3 inflammasome by oxLDL in vascular endothelial cells through a PXR-dependent mechanism as well (118). Intriguingly, PXR blocks NF-κB binding in oxLDL-primed HUVEC, thus suggesting that PXR activation inhibits NLRP3 activation (118). Furthermore, epleronone-mediated inhibition of MR suppresses the expression of NLRP3 and Caspase 1 both in the liver and epididymal white adipose tissue (eWAT) (120) (Figure 1). However, whether these epleronone-mediated effects on NLRP3 pathway are dependent on MR remain to be confirmed. For instance, it is unknown whether MR-response elements are present in the promoter of inflammasome component coding genes. Accordingly, MR knock-down impairs aldosterone regulatory effect on IL-1β expression in LPS-stimulated BMDM, but this effect was likely due to an inhibition of NF-κB phosphorylation instead of a direct effect on NLRP3 gene expression (120). It is noteworthy that increased MR expression is associated with an increase in NLRP3 expression and altered microglia phenotype in hippocampus from spontaneously hypertensive rats (121). However, the actual functional impact of MR in this process needs further investigation to prove the implication of MR in this context. Furthermore, NRs such as RXRs and RAR are activated by retinoic acids including 9-cis-retinoic acid (9-cis-RA) and all-trans-retinoic acid (ATRA). Interestingly, human LPS-primed macrophages treated with ATRA exhibit elevated NLRP3 RNA and protein levels associated with an increase in caspase 1 and pro-IL-1β maturation. At the molecular level, ATRA alone induces NLRP3 expression and enhances LPS-induced NRLP3 and IL-1β mRNA levels by upregulating the phosphorylation of IκB, ERK, and p38 (122). Therefore, stimulation of GR, MR, PXR, and RAR induces NLRP3 priming. Besides, PPARγ also controls the NF-κB-dependent NLRP3 priming in different contexts including astrocytes and retinal ischemia/reperfusion (123, 124). Here, IL4-activated PPARγ inhibits NLRP3 protein levels in an NF-κB-dependent manner in High Mobility Group Box-1 (HMGB-1)-stimulated astrocytes (124), while treatment with pioglitazone, a PPARγ agonist, ameliorates retinal ischemia/reperfusion-mediated inflammatory response by suppressing NLRP3 activation in an NF-κB-dependent manner (123) (Figure 1). Furthermore, GW4004-mediated activation of FXR also inhibits the expression of TLR4 and Myd88 in ileum (125). The gene expression of the NLRP3 inflammasome pathway components was altered accordingly, although the direct impact of GW4004 on NF-κB activation was not reported in this context (125). Finally, in addition to PPARγ, Rev-erbα may also inhibit NLRP3 priming, at least partially, via the inhibition of p65 expression in mouse RAW264.7 macrophage cell line (126). Accordingly, modulation of Rev-erb activity revealed that Rev-erbs may inhibit p65 and IκB phosphorylation in RAW264.7 cells thus inhibiting NF-κB activity (127). Together these data indicate that PPARγ and Rev-erbα may inhibit NK-κB-dependent NLRP3 priming (Figure 1). Overall, as many NRs including the Constitutive Androstane Receptor (CAR) (128, 129) and PPARα (130, 131), have been demonstrated to control the NF-κB pathway (103), it could be anticipated that they may also be involved in NF-κB-dependent NLRP3 priming processes, although this still needs to be proven.
NRs are also able to directly control NLRP3 transcription. For instance, Rev-erbα, a transcriptional repressor, is directly recruited to four distinct Rev-erb Response Elements (RevRE) into the Nlrp3 gene promoter and actively inhibits Nlrp3 expression in both human and mouse primary macrophages (132) (Figure 1). Furthermore, the deletion of RORγ or the use of a RORγ inverse agonist decreases NLRP3 mRNA and protein levels, which is associated with a reduction of IL-1β secretion in LPS-primed BMDM (133). ROR and Rev-erb share the same consensus sequence allowing them to bind the same RORE/RevRE response elements (Figure 1). Accordingly, RORγ was found to be recruited to the same Rev-erbα sites in the Nlrp3 promoter (132, 133). Finally, although poorly investigated, NRs also control Nlrp3 mRNA post-transcriptional stability through the regulation of miRNA. Indeed, the PPARβ/δ agonist, GW0742, significantly reduces the number of activated pro-inflammatory microglial cells after hypoxia–ischemia in neonatal rat brain (134). This effect is mainly due to a decrease in TXNIP, NLRP3, IL-6 and TNFα (134). At the molecular level, the PPARβ/δ antagonist GSK3787 and the miR-17-5p inhibitor abolish GW0742 effect, thus demonstrating the dependency of GW0742 on the PPARβ/δ-miR-17-5p axis (134) (Figure 1). However, the identification of the precise mechanisms by which PPARβ/δ controls the regulation of miR-17-5-p still needs further investigations. It is not excluded either that other NRs may regulate miRNA expression implicated in the post-transcriptional regulation of Nlrp3 mRNA stability.
In addition to NLRP3 priming, nuclear receptors are also able to control NLRP3 activation, ie the second step of NLRP3 regulation. For instance, deletion of Rev-erbα increases nigericin- and ATP-induced ASC speck formation in mouse primary macrophages, thus suggesting that Rev-erbα prevents NLRP3 inflammasome assembly and its activation (132). However, the underlying mechanisms still need to be uncovered, and it cannot be excluded that this effect on NLRP3 activation reflects only the increase of Nlrp3 gene expression triggered after Rev-erbα deficiency. However, because Rev-erbα regulates mitochondrial function and autophagy processes in skeletal muscle (135), we may speculate that the inhibition of NLRP3 assembly by Rev-erbα could be mediated by a decrease in ROS production and an enhancement of mitochondrial function.
Interestingly, the bile acid receptor FXR is also able to physically interact with NLRP3 and Caspase1 thus inhibiting NLRP3 activity (136) (Figure 1). In addition, bile acids behave as DAMPs and inhibit the priming and activation of the NRLP3 inflammasome in the context of cholestatic and septic mice (136). At the molecular level, bile acids induce a prolonged Ca2+ influx and activate NLRP3 synergistically with ATP administration (136). It is noteworthy that these effects are independent of ROS production and K+ efflux (136). In this context, FXR deletion increases endotoxemia sensitivity while FXR overexpression increases mice resistance to endotoxemia, thus suggesting an FXR-independent effect of bile acids action in sepsis (136). Such FXR-independent effect of bile acid on NLRP3 inflammasome may be mediated by the membrane receptor Takeda G coupled Receptor 5 (TGR5), another bile acid receptor. Indeed, treatment of BMDM with bile acids suppresses LPS/Nigericin-mediated NLRP3 activation in a TGR5-cAMP-PKA dependent by inducing NLRP3 ubiquitination and phosphorylation (137–140).
Furthermore, vitamin D enhances VDR-mediated inhibition of NLRP3 activation (141). Indeed, vitamin D3 (VitD3) inhibits NLRP3 activation in LPS-primed mouse peritoneal macrophages in the presence of nigericin, MSU or alum (142). In addition, vitD3 dampens ASC speck formation by preventing the NLRP3/NEK7 interaction (142). Interestingly, vitD3 also promotes NLRP3 ubiquitination. Indeed, the LBD of VDR is able to physically interact with the NACHT-LRR domain of NLRP3 thus inhibiting the association of NLRP3 with BRCC3 and preventing NLRP3 deubiquitination (141) (Figure 1). Particularly, VDR has been shown to prevent NLRP3 modification on K63 and its subsequent activation (141). Finally, vitD3 also increases VDR-controlled UCP2 expression thus inhibiting ROS accumulation in LPS-primed peritoneal macrophages (141). Altogether, VDR inhibits NLRP3 inflammasome by favoring NLRP3 ubiquitination, preventing NLRP3 assembly and reducing ROS-mediated NLRP3 activation.
LXRs have also been shown to modulate the NLRP3 pathway. In colon cancer cells for instance, LXRβ activates NLRP3 inflammasome by inducing Pannexin1-dependent ATP release and autocrine P2x7R activation, which in turn leads to anti-tumoral effect of LXR agonists (143) (Figure 1). By contrast, LXRs have also been shown to inhibit Casp1, IL-1β and IL-18 expression through a direct DNA-dependent mechanism in human and mouse primary macrophages (109). In addition, LXRs enhance expression of IL-18BP, the decoy receptor of IL-18, through an indirect IRF8-dependent mechanism (101, 108, 109). In this study, LXRs did not appear to control Nlrp3 mRNA levels in macrophages. Instead, they inhibit the expression of other inflammasome components such as pro-casp1, pro-IL18 and pro-IL1β and they induce the expression of inhibitory factor including IL18BP. On the contrary, LXRα was recently shown to decrease NLRP3 mRNA and protein levels in renal cell carcinomas metastasis in vivo and in vitro, thus resulting in the reduction of pro-IL1β and pro-caspase1 protein levels and the inhibition of IL1β secretion (144). Finally, lysosomal acid lipase (LIPA)-mediated 25- and 27-hydroxycholesterol (OHC) production, two LXR natural agonists, decreases efferocytosis and metabolic inflammation by activating LXR and by inhibiting NLRP3 in THP1 human macrophages (145). However, the interdependency of each pathway needs further investigations as results from Viaud et al. suggest that 25-OHC dampens inflammasome function independently from LXR activation (145) (Figure 1). Instead, it may be due to reduced MAM-dependent mitochondrial repurposing leading to NLRP3 inhibition (145). Therefore, it seems that LXR activity on the NLRP3 inflammasome depends on the cellular and tissular context, underlying cell-specific and context-specific mechanisms that still need to be explained. Interestingly, ERRα and PPARβ/δ increase Mitofusin 2 expression (146, 147). Although the link between the regulation of MAM and NLRP3 has not been established yet, we may anticipate that both NR may be involved in NLRP3 activation.
Epleronone is an antagonist of MR while aldosterone is a MR activator. Interestingly, epleronone-mediated inhibition of MR inhibits IL-1β secretion from eWAT (120). At the molecular level, epleronone treatment prevents ROS production and ATP- or nigericin-induced IL-1β secretion in LPS-primed BMDM, thus suggesting an effect on NLRP3 activation (120). Accordingly, aldosterone induced renal tubular cell injury by activating NLRP3 in a mtROS-dependent manner. Aldosterone-induced IL-1β and IL-18 maturation was then inhibited by NLRP3 knock-down or epleronone-mediated MR inhibition. Epleronone abolishes aldosterone-induced NLRP3, ASC, Casp1, and IL-18 maturation in mouse kidney, but the mechanism is still uncovered (148).
PXR activation with xenobiotics also induces Caspase1 maturation and IL-1β secretion in human THP1 and mouse primary macrophages (149). At the molecular level, PXR promotes rapid ATP release thus acting as an activation signal 2 (149). In this context, SRC kinase (SFK) promotes Pannexin1 phosphorylation thus triggering rapid ATP release (149) (Figure 1). It is, however, uncertain whether PXR controls Pannexin1 and SFK at the genomic or non-genomic levels (149). However, since the release of ATP occurs only 15 seconds after PXR agonist stimulation, this effect is unlikely transcriptional but instead it may be due to post-translational modification, advocating for a non-genomic effect of PXR in the regulation of NLRP3 activation step.
Finally, glycolysis and metabolic intermediates were shown to impact NLRP3 activation and ROS production (150). Interestingly, ATRA treatment induces hexokinase 2 expression in human LPS-primed monocyte-derived macrophages, thus shifting the metabolism of macrophages toward glycolysis and activating the NLRP3 inflammasome (122). Imbalance of metabolic homeostasis then appears to be directly linked to the NLRP3 inflammasome activity and the innate immune system thus emphasizing the importance of metabolic sensors in the control of inflammatory pathway. As exemplified here, NRs play an important role in such regulatory processes by bridging metabolism sensing and immunity.
Until now, we have reviewed the regulatory effect of NRs on NLRP3. Interestingly, the NLRP3 pathway can also control the activity of NRs. For instance, the NLRP3/Caspase1 complex is able to cleave GR, thus impairing glucocorticoid activity in acute lymphoblastic leukemia (ALL) patients (151). Two cleavage sites of caspase 1, LLID and IKQE, have been identified in GR. Accordingly, increase in caspase 1 induced GR cleavage, decreased GR transcriptional activity and promoted glucocorticoid resistance (151). Interestingly, the comparison of NLRP3 and Caspase 1 expression between glucocorticoid sensitive and resistant primary leukemia cells isolated from 444 patients shows that high expression of Caspase 1 and NLRP3 is associated with an increase in glucocorticoid resistance (151). It is noteworthy that the higher expression of NRLP3 and Caspase 1 observed in glucocorticoid-resistant cells is likely due to lower somatic methylation of their respective promoter (151). Conversely, inhibition of Caspase 1 restores glucocorticoid sensitivity. Similar mechanisms were observed for AR (152).
Finally, the 17-oxo-DHA is a bioactive electrophilic α,β-unsaturated keto-derivative of the ω3 fatty acid docosahexaenoic acid (DHA) that is endogenously generated by COX2 in activated macrophages (153). The nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor that binds antioxidant response element (ARE) to control antioxidant and detoxifying enzyme transcription including heme oxygenase 1 (HO-1) and glutathione S-transferase (GST). 17-oxo-DHA displays anti-inflammatory and cytoprotective activities by inducing Nrf2-dependent anti-oxidant response and by suppressing NF-κB-dependent inflammatory reactions. Interestingly, 17-oxo-DHA inhibits nigericin-induced ASC speck formation in human THP-1 macrophage cell line. In the context of cigarette smoke-driven chronic obstructive pulmonary disease (COPD), the 17-oxo-DHA compound prevents inflammasome-dependent GR degradation in human peripheral blood mononuclear cells (PBMCs) (153). Although the underlying mechanisms are uncovered, we may speculate that 17-oxo-DHA controls Caspase 1 activity.
NLRP3 inflammasome upregulation is involved in numerous inflammatory diseases including joint, intestinal, respiratory, brain, hepatic, kidney, sexual organ and cardiometabolic diseases. Strikingly, NRs were widely involved in the regulation of these diseases through the control of NLRP3 (Table 1). It is then not surprising that the modulation of NR activity by specific agonists or antagonists regulates NLRP3 priming or activation and improves or worsens such diseases depending on the context.
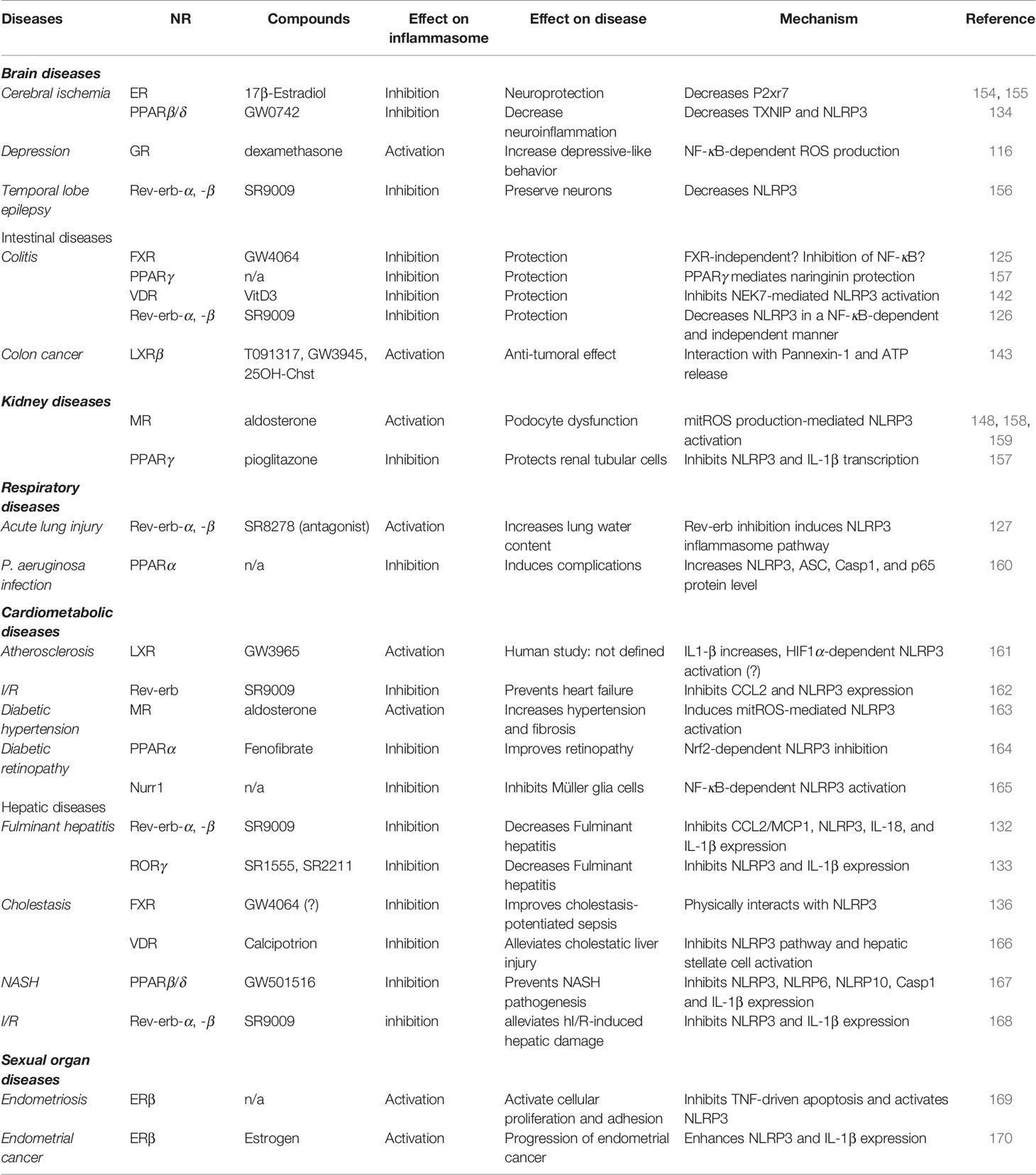
Table 1 Activity of NRs in NLRP3-driven diseases.
Cerebral ischemia is a particular condition promoting neuroinflammation (154, 155). The 17β-Estradiol (E2), an ER agonist, display neuroprotective effect in the context of global cerebral ischemia, a well-known condition in which NLRP3 pathway components are induced (154). In this context, E2 inhibits the expression of NLRP3 inflammasome components and NLRP3 activation by decreasing P2xr7 expression in protein and proline-glutamic acid and leucine-rich protein 1 (PELP1)-dependent manner (154) (Table 1). Accordingly, nicotine attenuates ERβ action on inflammasome activity and exacerbates ischemic brain damage (155). Indeed, nicotine inhibits ERβ protein levels in hippocampus and cortex while it increases ASC, IL1-β and Caspase 1 protein levels in brain of female rats (155). However, further investigations are needed to demonstrate whether nicotine regulatory effects on the NLRP3 pathway are mediated by ERβ and NLRP3 instead of a direct activation of the non-canonical or alternative pathway. In addition to ERs, a PPARβ/δ agonist significantly reduces neuroinflammation after hypoxia–ischemia by inhibiting the expression of TXNIP and NLRP3 (134). Furthermore, temporal lobe epilepsy (TLE) is characterized by spontaneous recurrent seizures leading to neuroinflammation features such as astrocytosis associated with microglia activation and inflammatory cytokine production (156) (Table 1). In the context of human and mouse TLE, the Rev-erb ligand, SR9009, prevents neuroinflammation by inhibiting NLRP3 mRNA and protein levels, reducing astrocytes and microglial activation and decreasing apoptosis, which then preserves neurons and provides neuroprotection (156). Finally, glucocorticoids induce NLRP3 in an NF-κB-dependent manner in hippocampal microglial cells, which mediates chronic stress-induced depressive-like behavior in rats (116) (Table 1). Altogether, these data demonstrate that NRs such as ERs, Rev-erbs, and GR play a regulatory role on NLRP3-induced brain disease such as cerebral ischemia, epilepsy and depressiveness. As such, the modulation of their activity with ligands may dampen the severity and the progression of such diseases.
Colitis is an inflammatory disease of the colon whose causes are still uncertain. We may differentiate acute ulcerative colitis from chronic Crohn’s disease. Strikingly, NLRP3 inflammasome is induced in dextran sulfate sodium (DSS)-induced colitis mouse model. Numerous NRs have then been shown to control DSS-induced colitis severity by modulating NRLP3 inflammasome pathway. For instance, the FXR agonist GW4064 exerts mild effect on colitis reduction by decreasing NLRP3 expression in LPS-induced ileum injury (125) (Table 1). However, GW4064 rapidly dampens both canonical and non-canonical NLRP3 activation in an FXR-independent manner, thus questioning the underlying mechanism involved in this fast response (171). Nevertheless, it is not excluded that FXR mediates GW4064 effect after a prolonged exposure to the agonist in this context (172). In obese patients, VDR polymorphisms were associated with increased inflammasome component expression, pro-inflammatory cytokine secretion and gut permeability, or dysbiosis, raising circulating LPS (173). Additionally, VitD3-activated VDR and SR9009-activated Rev-erbs also protect from DSS-induced colitis (126, 142), which then emphasizes the use of such NR-targeted approaches to control inflammatory bowel diseases (Table 1). It is noteworthy that numerous compounds derived from Chinese medicine are able to control the inflammasome pathway. For instance, Berberine, isolated from Rhizoma Coptidis, has been used for centuries in Chinese medicine to treat gastrointestinal disorders. Intriguingly, Berberine inhibits NLRP3 activation in DSS-induced colitis in a Rev-erbα-dependent manner (174). Naringin is a flavonoid extracted from grapefruit, sour orange and citrus seed that display anti-inflammatory properties (175). Interestingly, PPARγ mediates the anti-inflammatory effects of Naringin on DSS-induced ulcerative colitis (175). Whether thiazolidinediones, a PPARγ agonist class, prevent colitis progression as well remains to be determined (Table 1). Finally, LXRβ activates NLRP3 inflammasome in colon cancer cells leading to anti-tumoral effect of LXR agonists (143) (Table 1).
Podocytes are important glomerular cell types playing a key role in blood filtration by the kidney. Aldosterone, a MR agonist, drives NLRP3-dependent podocyte dysfunction in vivo and in vitro by inducing oxidative stress (158) (Table 1). Remarkably, eplerenone, that inhibits MR, protects podocytes from aldosterone-induced injury (158). However, the dependency of MR in this context still needs to be addressed. In addition to podocytes, aldosterone also induces renal tubular cell injury. These cells play a pivotal role in the absorption of glucose, amino acids and ions by the renal tubule. In this context, aldosterone promotes mtROS production and subsequent NLRP3 activation (159). Strikingly, aldosterone induces NLRP3, IL1β, IL18 and CASP1 expression in human immortalized normal kidney cells isolated from proximal tubules (HK-2 cells) in a dose- and time-dependent manner, thus inducing a phenotypic switch from HK-2 to fibroblast/pericyte cells in a MR and NLRP3-dependent manner (159). Accordingly, eplerenone abolishes these aldosterone-mediated effects. In addition, NLRP3 deletion in mice attenuates aldosterone-induced renal injury by protecting cells from apoptosis/pyroptosis and by preventing this phenotypic switch (159). Finally, aldosterone also induces tubulointerstitial fibrosis leading to kidney failure (148). As above, eplerenone abolishes aldosterone-induced macrophage infiltration, tubulointerstitial fibrosis in a MCP1- and ICAM1-dependent manner (148). Precisely, macrophage inflammasome was required to induce renal fibrosis and kidney dysfunction after aldosterone administration, whereas renal cells were involved in MCP1 expression, showing a cell-specific aldosterone action in renal failure. However, the dependency on MR in renal fibrosis is still elusive (148). Finally, PPARγ activation with pioglitazone inhibits MSU-induced NLRP3 and IL-1β mRNA and protein levels in HK-2 cells (157) (Table 1). Intriguingly, MSU and LPS were able to induce PPARγ expression in HK-2 cells after a short exposure, but not a long exposure, thus suggesting that PPARγ sets up a negative feedback loop to inhibit NLRP3 activation (157).
Acute lung injury is a severe IL-1β-associated complication that occurs after pulmonary inflammation and increases the mortality rate in patients. In mice, the Rev-erb antagonist SR8278 exacerbates LPS-induced lung permeability, which increases lung water contents (127) (Table 1). In this context, SR8278 increases macrophage recruitment in the lung and enhances IL-1β production in bronchioalveolar lavage fluid (127). In addition, PPARα ablation in mice increases NLRP3, ASC, Caspase 1 and p65 protein levels in the lung after infection with Pseudomonas aeruginosa (PA), which then promotes lung complications and subsequently worsens the pathophysiology of PA lung diseases (160) (Table 1).
Cardiometabolic diseases include hypertension, diabetes, non-alcoholic fatty liver diseases, vascular dysfunction, and heart failure. They share common inflammatory features including NLRP3 inflammasome activation. We may distinguish atherosclerosis, heart ischemia–reperfusion, obesity, type 2 diabetes, and diabetic retinopathy. Atherosclerosis is a lipid-driven inflammatory disease of the vascular wall during which infiltrating LDLs are eventually modified, triggering their uptake by macrophages. Oxidized LDLs are indeed internalized and promote both priming and cholesterol crystals-mediated activation of NLRP3 in a CD36-dependent manner (19, 20). Accordingly, ablation of the NLRP3 inflammasome pathway decreases atherosclerosis progression (176, 177). Numerous NRs have been shown to be involved in atherosclerosis development including PPARs, Rev-erbα, LXRs, and Nur77 (3, 86, 108, 178). Interestingly, an LXR agonist has lately been shown to increase IL-1β protein levels in an HIF1α-dependent manner in human atherosclerotic lesions. It is however unknown whether it relies on an LXR-dependent mechanism (161) (Table 1). However, as HIF1α induces NLRP3 inflammasome activation (179–181), such regulatory mechanism may then account for LXR-dependent activation of IL1-β production in hypoxic atherosclerotic lesions.
Diabetes and hypertension are common coexisting diseases that accelerate micro and macrovascular complication occurrence. Different groups evidenced that aldosterone-activated MR increases hypertension and fibrosis through mtROS-mediated NLRP3 activation (163). In a model of obese diabetic db/db mice, spironolactone-mediated MR inhibition ablates inflammasome activation in mesenteric arteries (163) (Table 1). In addition, spironolactone treatment ameliorates glucose homeostasis without affecting body mass and mesenteric artery KCl-induced contraction. However, spironolactone ameliorates acetylcholine-activated vasorelaxation in phenylephrine-contracted mesenteric artery ex vivo (163). Accordingly, the NLRP3 inhibitor MCC950 mimics spironolactone effect in this vasoreactivity model, then suggesting that NLRP3 controls vasoreactivity in a MR-dependent manner (163). Diabetic retinopathy is a common neurovascular complication of diabetes that represents the most frequent cause of vision loss and blindness worldwide. In early non-proliferative stages, hyperglycemia causes glucotoxicity and damages retinal small vessels. As the disease progresses, alteration of small vessels triggers hypoxia and the development of small, fragile neovessels that can bleed, clot, and alter the retina. Because of cell death, diabetic retinopathy may also be considered as a chronic low-grade inflammatory disease in which the NLRP3 inflammasome is activated (54, 182). Strikingly, treatment with the PPARα ligand fenofibrate (FF) ameliorates diabetic retinopathy by inducing Nrf2 signaling and inhibiting NLR3 inflammasome (164) (Table 1). FF inhibits Nrf2 expression in mouse retinal Müller glial cells and attenuates gliosis in diabetic retina (164). However, it is uncertain whether FF effect is mediated by PPARα activation. Finally, Nurr1 deficiency promotes high glucose-induced Müller glial cell activation by inducing NF-κB and the NLRP3 inflammasome axis (165) (Table 1).
Post-ischemia reperfusion (I/R), after a heart ischemic episode, triggers a profound inflammatory response called reperfusion injury, which provokes adverse cardiac remodeling and heart failure. Consistently, MCC950-mediated NLRP3 inhibition lowers infarct size and areas at risk (183). Remarkably, administration of SR9009 Rev-erb agonist, one day after myocardial I/R, prevents heart failure by targeting cardiomyocyte inflammasome in a Rev-erb-dependent manner (162) (Table 1). In addition, Rev-erb activation inhibits CCL2 secretion and leucocyte recruitment at ischemic sites, thus lowering cardiac inflammation that would prevent cardiac remodeling (162).
Cholestasis is a common liver complication in patients with extrahepatic infection or sepsis and consists in bile acid accumulation in liver and serum. Intriguingly, on the one hand, BAs behave as DAMPs which activate both priming and activation of NLRP3, while on the other hand, the BA receptor FXR inhibits NLRP3 activation by physically interacting with NLRP3 (136) (Table 1). However, because the GW4064 compound may modulate NLRP3 activity in an FXR-independent manner (171), we may anticipate that BA effects on NLRP3 in cholestatic mice may also occur in an FXR-independent manner, thus explaining this apparent discrepancy. However, as FXR expression is down-regulated in endotoxic mice, FXR synthetic ligands display a poor effect on cholestasis (136), thus advocating for the identification of an alternative therapeutic strategy such as promoting the increase of FXR expression. Finally, the VDR agonist calcipotriol is also able to alleviate cholestatic liver injury and fibrosis by inhibiting the NLRP3 inflammasome pathway involved in inflammation, and hepatic stellate cells activation likely responsible of fibrosis (166) (Table 1).
Non-alcoholic fatty liver diseases (NAFLD) are common chronic liver diseases, ranging from hepatic steatosis to non-alcoholic steatohepatitis (NASH), which is characterized by lipid accumulation, inflammation, and fibrosis (184). NASH may eventually progress to irreversible cirrhosis and hepatocarcinoma (184). Remarkably, inhibition of the NLRP3 inflammasome pathway reduces liver inflammation and fibrosis in an experimental mouse NASH model (185). Interestingly, the dual PPARα and PPARβ/δ agonist GFT505/Elafibranor displays hepatoprotective effects in different rodent models of NASH by reducing fibrosis and cytokine secretion including IL-1β (167). Consistently, administration of the PPARβ/δ agonist GW501516 inhibits Caspase 1 and IL-1β hepatic mRNA levels in mice fed a high fat diet (HFD) and co-treated with LPS (186) (Table 1). In human hepatic hepG2 cell line, palmitic acid and LPS co-treatment induces the expression of NLRP3, NLRP6 and NLRP10 as well as Caspase 1 and IL-1β (186). Consistently with in vivo data, GW501516 prevents palmitate/LPS-induced inflammasome component gene expression (186). Intriguingly, although GW501516 accordingly impairs Caspase 1 maturation, it does not control IL-1β secretion (186).
Fulminant hepatitis (FH) is a life-threatening condition characterized by fast evolving hepatic dysfunction associated with tissue necrosis, inflammation and hepatic encephalopathy (187). Albeit numerous factors including fungi intoxication, viral infection, and metabolic diseases trigger FH, the main cause of FH nowadays is drug overdose with acetaminophen as the main one (187). Acetaminophen accumulation induces P450-mediated overproduction of toxic metabolites leading to oxidative stress, mitochondrial membrane potential loss and hepatocellular death. Tissue necrosis is then responsible of the release of DAMPs such as ATP and subsequent NLRP3 inflammasome activation (188, 189). Strikingly, Rev-erbα-deficiency aggravates FH in a mouse model of LPS-galactosamine (GalN)-induced liver injury. This occurred in an NLRP3-dependent manner by alleviating its inhibitory effect on Caspase 1 activity and on IL-1β expression and secretion (132) (Table 1). As Rev-erbα also impairs CCL2/MCP1 chemokine expression, ablation of Rev-erbα worsened neutrophils and monocytes infiltration in LPS/GalN-challenged mice, thus contributing to increased liver injury (132). Consistently, pre-treatment with the Rev-erb agonist SR9009 prevents LPS/GalN-induced FH pathogenesis by inhibiting the NLRP3 inflammasome pathway and CCL2 expression, thereby delaying death and improving the survival rate from 10% in the control to 70% in the SR9009-treated mice (132). Finally, the RORγ inverse agonists SR1555 and SR2211 reduce the expression and secretion of IL-1β in LPS/GalN-induced FH and exert a hepatoprotective effect that improves the survival rate of treated FH mice (133) (Table 1). However, whether RORγ mediates SR1555 and SR2211 effect on NLRP3 pathway and FH protection still needs to be proven. Nevertheless, RORγ deletion in LPS-primed BMDM inhibits NLRP3 and IL-1β secretion, which is consistent with a RORγ-inhibiting effect of SR1555 and SR2211 on these processes (133).
Rev-erb-α has also been highlighted lately in the context of hepatic ischemia–reperfusion (hI/R). hI/R is a complex phenomenon during which hepatocyte damage hits when blood supply returns into the ischemic liver after a liver transplantation, hepatectomy, and ischemic shock (190). Inflammatory responses play an important role in hI/R injury during which activated Kupffer cells release ROS and pro-inflammatory cytokines including IL-1β. Consistently, NLRP3 deficiency protects against liver I/R injury in mice (191). Accordingly, Rev-erbα deletion sensitizes mice to hI/R and is accompanied by exacerbated NLRP3 activation and pro-inflammatory cytokine secretion (168). On the contrary, SR9009 treatment alleviates hI/R-induced hepatic damage by inhibiting IL-1β expression (168). In conclusion, Rev-erbs, RORγ, VDR, PPARβ/δ, and FXR then exhibit hepatoprotective effects in acute liver inflammatory diseases by dampening the NLRP3 inflammasome activity.
Endometriosis is a sexual organ disease originating from abnormal deposition of endometrial cells that grow outside from the uterine cavity. It affects 6–10% of reproductive-aged women. Endometriosis causes pelvic pain in 50% of cases and fertility problem in 40–50% of cases (169). Endometriosis is likely due to high production levels of 17β-estradiol that could play a role in the proliferation of endometriotic tissues (169) (Table 1). Compared to ERα, ERβ expression is significantly higher in endometriotic tissue than in normal uterine endometrium in human. In addition, the role and the specific expression of ERα in endometriotic tissues are controversial (169). Interestingly similar patterns were observed in the mouse (169). Interestingly, NLRP3−/− mice exhibit smaller ectopic lesions compared to wild type mice, thus suggesting that NLRP3 induces endometriosis (169). Strikingly, ERβ inhibits TNFα-driven apoptosis and activates NLRP3 in endometriotic tissues (169). Accordingly, ERβ then increases IL-1β secretion, which enhances cellular adhesion and proliferation (169). Consistently, NLRP3 inflammasome activation has been shown to promote the progression of human endometrial cancer in an ERβ-dependent manner (170) (Table 1). At the molecular level, ERβ interacts with the NLRP3 inflammasome in the cytoplasm (169). However, the exact regulatory mechanism still needs to be investigated.
Our ability to anticipate environmental changes imposed by the rotation of the Earth is controlled by the circadian clock, which properly gates many, if not all, physiological processes to the most appropriate time window (192). Among these physiological pathways, immune functions vary according to the time of day (3, 193), a process described as circadian immunity, in which innate immune cells such as macrophages harbor an intrinsic clockwork that drives circadian transcription of genes involved in the response to bacterial challenge (105, 194, 195). Pioneer studies have demonstrated that important features of the immune system such as trafficking and abundance of blood leucocytes, their recruitment to tissue, their ability to respond to pathogens and to secrete immune molecules vary in a circadian manner (196, 197). At the molecular level, the biological clock is a complex network of transcription factors and interlocked transcriptional feedback loops that orchestrate cellular circadian rhythms. Among the core clock components, the ligand-activated nuclear receptors Rev-erbs and RORs participate in the circadian control of the immune system (104, 106), whereas pharmacological activation of Rev-erbα and ROR modulates the expression and release of key pro-inflammatory cytokines (105, 133). It is noteworthy that Rev-erb nuclear receptors, altogether with RORα are the only core clock components whose activity may be directly modulated by a synthetic compound, thus representing an interesting therapeutical approach to directly modulate immune circadian behavior (198).
Over the past 100 years of global industrialization, mankind underwent some important changes in its lifestyle including its food habits, the ease of travel, the increase in shift work and social demands, and erratic exposure to artificial light from luminescent screens, which have dramatically altered circadian rhythms. It is now well-recognized that disruption of the intrinsic molecular clock impedes a proper immune response (199) and has severe repercussions on health. Indeed, numerous clinical studies have demonstrated that disruption of circadian rhythms in human represents an additional risk factor for neurological, metabolic and chronic inflammatory disorders (200–202) such as asthma, rheumatoid arthritis, atherosclerosis, type 2 diabetes or Alzheimer Disease (193, 202). Most of the clock-driven diseases demonstrate a chronic inflammatory component, either infiltration of macrophages in the vascular wall due to an accumulation of non-infectious DAMPs such as cholesterol crystal causing atherosclerosis, hydroxyapatite in joints leading to rheumatoid arthritis or the deposit of β-amyloid fibers, which activates microglial cells in Alzheimer disease (197).
Remarkably, clock disruption alters NLRP3 circadian oscillations in a mouse model of jetlag or in genetic and pharmacological models of clock alteration, thus modulating the progression of inflammatory diseases (3) including colitis (126), myocardial infarction/ischemia–reperfusion injury (162), lung injury (127) and fulminant hepatitis (132, 133). At the molecular level, NLRP3 expression altogether with IL-1β and IL-18 mRNA levels oscillate in a daily manner under the control of Rev-erbα in vivo and in vitro (132). Indeed, Rev-erbα ablation abolishes circadian oscillations in Nlrp3 gene expression in peritoneal macrophages and in serum shock-synchronized human and mouse primary macrophages, with functional repercussions on IL-1β and IL-18 oscillatory secretion (132). In vitro, Rev-erbα deletion promotes elevated expression of Nlrp3, Il1β and Il18, which is accompanied by an increase in IL-1β and IL-18 secretion (132). By contrast, activation of Rev-erbs with heme, their natural ligand, or with synthetic ligands reduces the secretion of these cytokines by inhibiting the expression of NLRP3 inflammasome component genes (132). Strikingly, the susceptibility to fulminant hepatitis and hepatic ischemia reperfusion injury is time-of-day dependent, upon the control of the molecular clock with Rev-erbα as an important regulator of the inflammasome (132, 168). Remarkably, pharmacological activation of both Rev-erbs and ROR reduces liver injury and improves the survival time and rate in a NLRP3-dependent manner in treated mice (132, 133). Consistently, time of cardiac ischemia/reperfusion and subsequent SR9009 treatment affect heart function recovery, the best response being obtained when Rev-erb expression is at its highest, ie when NLRP3 expression is at its lowest (162). Interestingly, the NF-κB-driven long non-coding RNA Lnc-UC has lately been shown to be induced by the core clock component Bmal1, thereby generating circadian expression of Lnc-UC (203). Then, Lnc-UC physically interacts with Cbx1 protein to reduce its gene silencing activity via H3K9me3, thereby enhancing Rev-erbα expression in an epigenetic manner (203). Then, by inducing Rev-erbα expression, Lnc-UC ablates NF-κB signaling and NLRP3 inflammasome signaling in macrophages (203). Consistently, Lnc-UC deletion disrupts clock gene expression, sensitizes mice to DSS-induced colitis and disrupts the diurnal rhythmicity in disease severity (203). Additionally, Rev-erbα-mediated effect of Berberine on DSS-induced colitis shows better effect when administered at ZT10 (late resting phase) compared to ZT2 (early resting phase), thus acknowledging the rationale to target core clock components in the control of NLRP3-driven diseases (174). Such circadian effect of drug efficiency might be explained by the lower severity of colitis at ZT10, which coincides with the maximum expression of Rev-erbα. In conclusion, circadian pharmacological effects of compounds on different diseases likely result from diurnal rhythms of both disease severity and daily oscillations of the drug target expression. Altogether, these observations advocate for chronotherapeutic practice on NLRP3-driven diseases.
NLRP3 inflammasome deregulation drives numerous diseases. Inhibition of NLRP3 using MCC950 demonstrates beneficial effects in fulminant hepatitis and in myocardial ischemia reperfusion (3). However, MCC950 displays hepatotoxic properties advocating for the development of alternative NLRP3 inhibitory strategies (3). Here, we provide the first extensive review showing the close links between nuclear receptors and the NLRP3 inflammasome pathway. Indeed, NRs are able to either activate the NLRP3 inflammasome or inhibit both priming and activation steps of the NLRP3 inflammasome pathways, acting at different levels, which offers numerous possibilities to modulate NLRP3-driven disorders. Indeed, the activity of NRs can be modulated by a plethora of synthetic, but also natural ligands. As such, NRs should be considered as sensors of environment changes including metabolic alterations, hormonal signal, pollutions and circadian rhythmicity. As NRs are able to control similar processes, we may consider that the entire NR family integrate these different environmental modifications, that may occur simultaneously, to deliver the best response. We may then anticipate that depending on their environment, NRs cooperate to appropriately modulate the NLRP3 inflammasome. NRs would then allow the adaptation of the innate immune system and the NLRP3 inflammasome to adjust its response from cytokine secretion to pyroptosis-induced cell death. Finally, nuclear receptors, including Rev-erb and ROR, control the circadian expression of NLRP3. As such, NLRP3 protein amounts are not equal across the day, thereby emphasizing the necessity of a chronotherapeutic approach. In the case of clock disruption as observed in shift workers or in elderlies for instance, targeting clock components to re-entrain the molecular clock and sustain circadian amplitude of NLRP3 expression may also be considered as an alternative or an additional approach.
BP and HD wrote and edited the manuscript and the figures. All authors contributed to the article and approved the submitted version.
Some of our work included in this review manuscript was supported by the Fondation pour la Recherche Médicale (FRM, EQU202003010310), INSERM, the ANR-Labex-EGID (EGID, ANR-10-LABX-46), the Fondation Francophone pour la recherche sur le diabète (FFRD) together with the Fédération Française des Diabétiques (AFD) AstraZeneca, Eli Lilly, Merck Sharp & Dohme (MSD), Novo Nordisk & Sanofi; the Région Hauts-de-France/FEDER (Chronoregeneration), Association Française contre les Myopathies (AFM), Fondation de France, Société Francophone du Diabète (SFD)-SERVIER and the ANR. This project is cofounded by the European Union under the European Region Development Fund (ERDF) and by the Hauts de France Region Council (contract_20000007), the MEL (contract-2020-ESR-02), and the French State (contract n°2019-R3-CTRL_IPL_Phase3). This project is cofounded by the European Union under the European Region Development Fund (ERDF) and by the Hauts de France Region Council (contract_20002842), the MEL (contract-2020-ESR-06), and the French State (contract n°2020-R3-CTRL_IPL_Phase4). This project is co-funded by the Agence Nationale pour la Recherche (ANR) (ANR-19-CE15-0033-01). The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Professor Steve Lancel as well as members of Hélène Duez Team for helpful discussions.
1. Evavold CL, Kagan JC. Inflammasomes: Threat-Assessment Organelles of the Innate Immune System. Immunity (2019) 51:609–24. doi: 10.1016/j.immuni.2019.08.005
2. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
3. Pourcet B, Duez H. Circadian Control of Inflammasome Pathways: Implications for Circadian Medicine. Front Immunol (2020) 11:1630. doi: 10.3389/fimmu.2020.01630
4. Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
5. Schmidt FI, Lu A, Chen JW, Ruan J, Tang C, Wu H, et al. A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assemblyA VHH defines mechanism of inflammasome assembly. J Exp Med (2016) 213:771–90. doi: 10.1084/jem.20151790
6. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell (2014) 156:1193–206. doi: 10.1016/j.cell.2014.02.008
7. Cai X, Chen J, Xu H, Liu S, Jiang Q-X, Halfmann R, et al. Prion-like Polymerization Underlies Signal Transduction in Antiviral Immune Defense and Inflammasome Activation. Cell (2014) 156:1207–22. doi: 10.1016/j.cell.2014.01.063
8. Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity Caspase-1 protease activity is self-limiting. J Exp Med (2018) 215:827–40. doi: 10.1084/jem.20172222
9. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol (2016) 17:250–8. doi: 10.1038/ni.3333
10. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature (2019) 570:338–43. doi: 10.1038/s41586-019-1295-z
11. Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, et al. (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J Biol Chem (2016) 291:103–9. doi: 10.1074/jbc.c115.700492
12. He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature (2016) 530:354–7. doi: 10.1038/nature16959
13. Groß CJ, Mishra R, Schneider KS, Médard G, Wettmarshausen J, Dittlein DC, et al. K+ Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity (2016) 45:761–73. doi: 10.1016/j.immuni.2016.08.010
14. Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, Dubyak GR. Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. J Immunol (2016) 197:1353–67. doi: 10.4049/jimmunol.1600699
15. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
16. He W, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res (2015) 25:1285–98. doi: 10.1038/cr.2015.139
17. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535:111–6. doi: 10.1038/nature18590
18. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell (2018) 172:162–175.e14. doi: 10.1016/j.cell.2017.12.013
19. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol (2013) 14:812–20. doi: 10.1038/ni.2639
20. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol (2010) 11:155–61. doi: 10.1038/ni.1836
21. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science (2015) 349:316–20. doi: 10.1126/science.aaa8064
22. Boxberger N, Hecker M, Zettl UK. Dysregulation of Inflammasome Priming and Activation by MicroRNAs in Human Immune-Mediated Diseases. J Immunol (2019) 202:2177–87. doi: 10.4049/jimmunol.1801416
23. Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, Hornung V. NLRP3 inflammasome activity is negatively controlled by miR-223. J Immunol (2012) 189:4175–81. doi: 10.4049/jimmunol.1201516
24. Chen D, Dixon BJ, Doycheva DM, Li B, Zhang Y, Hu Q, et al. IRE1α inhibition decreased TXNIP/NLRP3 inflammasome activation through miR-17-5p after neonatal hypoxic-ischemic brain injury in rats. J Neuroinflamm (2018) 15:32–18. doi: 10.1186/s12974-018-1077-9
25. Lerner AG, Upton J-P, Praveen PVK, Ghosh R, Nakagawa Y, Igbaria A, et al. IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab (2012) 16:250–64. doi: 10.1016/j.cmet.2012.07.007
26. Shibuya H, Nakasa T, Adachi N, Nagata Y, Ishikawa M, Deie M, et al. Overexpression of microRNA-223 in rheumatoid arthritis synovium controls osteoclast differentiation. Mod Rheumatol (2013) 23:674–85. doi: 10.1007/s10165-012-0710-1
27. Meira M, Sievers C, Hoffmann F, Rasenack M, Kuhle J, Derfuss T, et al. Unraveling Natalizumab Effects on Deregulated miR-17 Expression in CD4+ T Cells of Patients with Relapsing-Remitting Multiple Sclerosis. J Immunol Res (2014) 2014:1–11. doi: 10.1155/2014/897249
28. Kaga H, Komatsuda A, Omokawa A, Ito M, Teshima K, Tagawa H, et al. Downregulated expression of miR-155, miR-17, and miR-181b, and upregulated expression of activation-induced cytidine deaminase and interferon-α in PBMCs from patients with SLE. Mod Rheumatol (2015) 25:865–70. doi: 10.3109/14397595.2015.1030102
29. Sarhan RA, Aboelenein HRA, Sourour SKN, Fawzy IO, Salah S, Abdelaziz AI. Targeting E2F1 and c-Myc expression by microRNA-17-5p represses interferon-stimulated gene MxA in peripheral blood mononuclear cells of pediatric systemic lupus erythematosus patients. Discovery Med (2015) 19:419–25.
30. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci (2019) 20:3328. doi: 10.3390/ijms20133328
31. Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell (2013) 49:331–8. doi: 10.1016/j.molcel.2012.11.009
32. Lopez-Castejon G, Luheshi NM, Compan V, High S, Whitehead RC, Flitsch S, et al. Deubiquitinases Regulate the Activity of Caspase-1 and Interleukin-1β Secretion via Assembly of the Inflammasome. J Biol Chem (2013) 288:2721–33. doi: 10.1074/jbc.m112.422238
33. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9:847–56. doi: 10.1038/ni.1631
34. Orlowski GM, Sharma S, Colbert JD, Bogyo M, Robertson SA, Kataoka H, et al. Frontline Science: Multiple cathepsins promote inflammasome-independent, particle-induced cell death during NLRP3-dependent IL-1β activation. J Leukoc Biol (2017) 102:7–17. doi: 10.1189/jlb.3hi0316-152r
35. Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple Cathepsins Promote Pro–IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J Immunol (2015) 195:1685–97. doi: 10.4049/jimmunol.1500509
36. Katsnelson MA, Lozada-Soto KM, Russo HM, Miller BA, Dubyak GR. NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: roles for K+ efflux and Ca2+ influx. Am J Physiol-cell Ph (2016) 311:C83–C100. doi: 10.1152/ajpcell.00298.2015
37. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
38. Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ (2007) 14:1583–9. doi: 10.1038/sj.cdd.4402195
39. Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The Cytolytic P2Z Receptor for Extracellular ATP Identified as a P2X Receptor (P2X7). Science (1996) 272:735–8. doi: 10.1126/science.272.5262.735
40. Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J (1995) 14:1607–14. doi: 10.1002/j.1460-2075.1995.tb07149.x
41. Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem (1994) 269:15195–203. doi: 10.1016/S0021-9258(17)36591-2
42. Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity (2018) 49:56–65.e4. doi: 10.1016/j.immuni.2018.04.032
43. Yaron JR, Gangaraju S, Rao MY, Kong X, Zhang L, Su F, et al. K+ regulates Ca2+ to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis (2015) 6:e1954–4. doi: 10.1038/cddis.2015.277
44. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci (2012) 109:11282–7. doi: 10.1073/pnas.1117765109
45. Lee G-S, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature (2012) 492:123–7. doi: 10.1038/nature11588
46. Domingo-Fernández R, Coll RC, Kearney J, Breit S, O’Neill LAJ. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1β transcription and activate the NLRP3 inflammasome. J Biol Chem (2017) 292:12077–87. doi: 10.1074/jbc.m117.797126
47. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun (2017) 8:202. doi: 10.1038/s41467-017-00227-x
48. Green JP, Yu S, Martín-Sánchez F, Pelegrin P, Lopez-Castejon G, Lawrence CB, et al. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc Natl Acad Sci (2018) 115:201812744. doi: 10.1073/pnas.1812744115
49. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi: 10.1038/nature09663
50. Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun (2013) 4:1611–11. doi: 10.1038/ncomms2608
51. van Bruggen R, Köker MY, Jansen M, van Houdt M, Roos D, Kuijpers TW, et al. Human NLRP3 inflammasome activation is Nox1-4 independent. Blood (2010) 115:5398–400. doi: 10.1182/blood-2009-10-250803
52. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12:222–30. doi: 10.1038/ni.1980
53. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
54. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11:136–40. doi: 10.1038/ni.1831
55. Franchi L, Eigenbrod T, Muñoz-Planillo R, Ozkurede U, Kim Y-G, Chakrabarti A, et al. Cytosolic Double-Stranded RNA Activates the NLRP3 Inflammasome via MAVS-Induced Membrane Permeabilization and K+ Efflux. J Immunol (2014) 193:4214–22. doi: 10.4049/jimmunol.1400582
56. Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, et al. The Mitochondrial Antiviral Protein MAVS Associates with NLRP3 and Regulates Its Inflammasome Activity. J Immunol (2013) 191:4358–66. doi: 10.4049/jimmunol.1301170
57. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The Adaptor MAVS Promotes NLRP3 Mitochondrial Localization and Inflammasome Activation. Cell (2013) 153:348–61. doi: 10.1016/j.cell.2013.02.054
58. Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci (2013) 110:17963–8. doi: 10.1073/pnas.1312571110
59. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
60. Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol (2020) 21:30–41. doi: 10.1038/s41590-019-0548-1
61. Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479:117–21. doi: 10.1038/nature10558
62. Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol (2015) 45:2918–26. doi: 10.1002/eji.201545655
63. Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J (2006) 25:5071–82. doi: 10.1038/sj.emboj.7601378
64. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity (2015) 43:923–32. doi: 10.1016/j.immuni.2015.10.009
65. Green S, Walter P, Kumar V, Krust A, Bornert J-M, Argos P, et al. Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature (1986) 320:134–9. doi: 10.1038/320134a0
66. Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature (1985) 318:635–41. doi: 10.1038/318635a0
67. Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell (2014) 157:255–66. doi: 10.1016/j.cell.2014.03.012
68. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell (1995) 83:835–9. doi: 10.1016/0092-8674(95)90199-x
69. Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear Receptors and Lipid Physiology: Opening the X-Files. Science (2001) 294:1866–70. doi: 10.1126/science.294.5548.1866
70. Yu VC, Delsert C, Andersen B, Holloway JM, Devary OV, Näär AM, et al. RXRβ: A coregulator that enhances binding of retinoic acid, thyroid hormone, and vitamin D receptors to their cognate response elements. Cell (1991) 67:1251–66. doi: 10.1016/0092-8674(91)90301-e
71. Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling. Nature (1992) 355:446–9. doi: 10.1038/355446a0
72. Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, et al. Structure of the intact PPAR-gamma-RXR-alpha nuclear receptor complex on DNA. Nature (2008) 456(7220):350–6. doi: 10.1038/nature07413
73. Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev (2006) 20:1405–28. doi: 10.1101/gad.1424806
74. Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science (2011) 331:1315–9. doi: 10.1126/science.1198125
75. Yin L, Lazar MA. The Orphan Nuclear Receptor Rev-erbα Recruits the N-CoR/Histone Deacetylase 3 Corepressor to Regulate the Circadian Bmal1 Gene. Mol Endocrinol (2005) 19:1452–9. doi: 10.1210/me.2005-0057
76. Pourcet B, Pineda-Torra I, Derudas B, Staels B, Glineur C. SUMOylation of human peroxisome proliferator-activated receptor alpha inhibits its trans-activity through the recruitment of the nuclear corepressor NCoR. J Biol Chem (2010) 285:5983–92. doi: 10.1074/jbc.m109.078311
77. Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, et al. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell (2007) 25:57–70. doi: 10.1016/j.molcel.2006.11.022
78. Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature (2005) 437:759–63. doi: 10.1038/nature03988
79. Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, et al. A Nurr1/CoREST Pathway in Microglia and Astrocytes Protects Dopaminergic Neurons from Inflammation-Induced Death. Cell (2009) 137:47–59. doi: 10.1016/j.cell.2009.01.038
80. Blanquart C, Mansouri R, Fruchart JC, Staels B, Glineur C. Different ways to regulate the PPARalpha stability. Biochem Biophys Res Commun (2004) 319:663–70. doi: 10.1016/j.bbrc.2004.05.035
81. Gage MC, Becares N, Louie R, Waddington KE, Zhang Y, Tittanegro TH, et al. Disrupting LXRα phosphorylation promotes FoxM1 expression and modulates atherosclerosis by inducing macrophage proliferation. Proc Natl Acad Sci USA (2018) 115:E6556–65. doi: 10.1073/pnas.1721245115
82. Becares N, Gage MC, Voisin M, Shrestha E, Martin-Gutierrez L, Liang N, et al. Impaired LXRα Phosphorylation Attenuates Progression of Fatty Liver Disease. Cell Rep (2019) 26:984–995.e6. doi: 10.1016/j.celrep.2018.12.094
83. Torra IP, Ismaili N, Feig JE, Xu CF, Cavasotto C, Pancratov R, et al. Phosphorylation of liver X receptor alpha selectively regulates target gene expression in macrophages. Mol Cell Biol (2008) 28:2626–36. doi: 10.1128/mcb.01575-07
84. Blanquart C, Barbier O, Fruchart JC, Staels B, Glineur C. Peroxisome proliferator-activated receptor alpha (PPARalpha ) turnover by the ubiquitin-proteasome system controls the ligand-induced expression level of its target genes. J Biol Chem (2002) 277:37254–9. doi: 10.1074/jbc.M110598200
85. Berrabah W, Aumercier P, Lefebvre P, Staels B. Control of nuclear receptor activities in metabolism by post-translational modifications. FEBS Lett (2011) 585:1640–50. doi: 10.1016/j.febslet.2011.03.066
86. Pourcet B, Fruchart JC, Staels B, Glineur C. Selective PPAR modulators, dual and pan PPAR agonists: multimodal drugs for the treatment of type 2 diabetes and atherosclerosis. Expert Opin Emerg Drugs (2006) 11:379–401. doi: 10.1517/14728214.11.3.379
87. Barros RPA, Gustafsson J-Å. Estrogen Receptors and the Metabolic Network. Cell Metab (2011) 14:289–99. doi: 10.1016/j.cmet.2011.08.005
88. Maqdasy S, Trousson A, Tauveron I, Volle DH, Baron S, Lobaccaro J-MA. Once and for all, LXRα and LXRβ are gatekeepers of the endocrine system. Mol Aspects Med (2016) 49:31–46. doi: 10.1016/j.mam.2016.04.001
89. Chaudhuri G. Nuclear Receptors and Female Reproduction: A Tale of 3 Scientists, Jensen, Gustafsson, and O’Malley. Reprod Sci (2008) 15:110–20. doi: 10.1177/1933719108314516
90. Teboul M, Guillaumond F, Gréchez-Cassiau A, Delaunay F. Minireview: The Nuclear Hormone Receptor Family Round the Clock. Mol Endocrinol (2008) 22:2573–82. doi: 10.1210/me.2007-0521
91. Bishop-Bailey D. Nuclear Receptors in Vascular Biology. Curr Atheroscler Rep (2015) 17:27. doi: 10.1007/s11883-015-0507-8
92. Bishop-Bailey D. PPARs and angiogenesis. Biochem Soc Trans (2011) 39:1601–5. doi: 10.1042/bst20110643
93. Xu Y, O’Malley BW, Elmquist JK. Brain nuclear receptors and body weight regulation. J Clin Invest (2017) 127:1172–80. doi: 10.1172/jci88891
94. Förthmann B, Aletta JM, Lee Y, Terranova C, Birkaya B, Stachowiak EK, et al. Coalition of Nuclear Receptors in the Nervous System. J Cell Physiol (2015) 230:2875–80. doi: 10.1002/jcp.25036
95. Duez H, Staels B. Nuclear receptors linking circadian rhythms and cardiometabolic control. Arterioscler Thromb Vasc Biol (2010) 30:1529–34. doi: 10.1161/atvbaha.110.209098
96. Gerhart-Hines Z, Lazar MA. Rev-erbα and the circadian transcriptional regulation of metabolism. Diabetes Obes Metab (2015) 17:12–6. doi: 10.1111/dom.12510
97. Zhao X, Cho H, Yu RT, Atkins AR, Downes M, Evans RM. Nuclear receptors rock around the clock. EMBO Rep (2014) 15:518–28. doi: 10.1002/embr.201338271
98. Jin HS, Kim TS, Jo E-K. Emerging roles of orphan nuclear receptors in regulation of innate immunity. Arch Pharm Res (2016) 39:1491–502. doi: 10.1007/s12272-016-0841-6
99. Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol (2006) 6:44–55. doi: 10.1038/nri1748
100. Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr Rev (2018) 39:760–802. doi: 10.1210/er.2018-00064
101. Pourcet B, Pineda-Torra I. Transcriptional regulation of macrophage arginase 1 expression and its role in atherosclerosis. Trends Cardiovasc Med (2013) 23:143–52. doi: 10.1016/j.tcm.2012.10.003
102. Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol (2010) 10:365–76. doi: 10.1038/nri2748
103. Huang W, Glass CK. Nuclear Receptors and Inflammation Control: Molecular Mechanisms and Pathophysiological Relevance. Arterioscler Thromb Vasc Biol (2010) 30:1542–9. doi: 10.1161/atvbaha.109.191189
104. Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature (2013) 498:511–5. doi: 10.1038/nature12209
105. Gibbs JE, Blaikley J, Beesley S, Matthews L, Simpson KD, Boyce SH, et al. The nuclear receptor REV-ERBα mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc Natl Acad Sci USA (2012) 109:582–7. doi: 10.1073/pnas.1106750109
106. Fontaine C, Rigamonti E, Pourcet B, Duez H, Duhem C, Fruchart JC, et al. The nuclear receptor Rev-erbalpha is a liver X receptor (LXR) target gene driving a negative feedback loop on select LXR-induced pathways in human macrophages. Mol Endocrinol (2008) 22:1797–811. doi: 10.1210/me.2007-0439
107. Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab (2007) 6:137–43. doi: 10.1016/j.cmet.2007.06.010
108. Pourcet B, Feig JE, Vengrenyuk Y, Hobbs AJ, Kepka-Lenhart D, Garabedian MJ, et al. LXRalpha regulates macrophage arginase 1 through PU.1 and interferon regulatory factor 8. Circ Res (2011) 109:492–501. doi: 10.1161/circresaha.111.241810
109. Pourcet B, Gage MC, León TE, Waddington KE, Pello OM, Steffensen KR, et al. The nuclear receptor LXR modulates interleukin-18 levels in macrophages through multiple mechanisms. Sci Rep (2016) 6:25481. doi: 10.1038/srep25481
110. Hamers AA, Vos M, Rassam F, Marinkovic G, Kurakula K, van Gorp PJ, et al. Bone marrow-specific deficiency of nuclear receptor Nur77 enhances atherosclerosis. Circ Res (2012) 110:428–38. doi: 10.1161/circresaha.111.260760
111. Ma H, Zhong W, Jiang Y, Fontaine C, Li S, Fu J, et al. Increased atherosclerotic lesions in LDL receptor deficient mice with hematopoietic nuclear receptor Rev-erbalpha knock- down. J Am Heart Assoc (2013) 2:e000235. doi: 10.1161/jaha.113.000235
112. Kim S-M, Lee S-A, Kim B-Y, Bae S-S, Eo S-K, Kim K. 27-Hydroxycholesterol induces recruitment of monocytic cells by enhancing CCL2 production. Biochem Bioph Res Co (2013) 442:159–64. doi: 10.1016/j.bbrc.2013.11.052
113. Wenzel I, Roth J, Sorg C. Identification of a novel surface molecule, RM3/1, that contributes to the adhesion of glucocorticoid-induced human monocytes to endothelial cells. Eur J Immunol (1996) 26:2758–63. doi: 10.1002/eji.1830261131
114. Stujanna EN, Murakoshi N, Tajiri K, Xu D, Kimura T, Qin R, et al. Rev-erb agonist improves adverse cardiac remodeling and survival in myocardial infarction through an anti-inflammatory mechanism. PloS One (2017) 12:e0189330. doi: 10.1371/journal.pone.0189330
115. Busillo JM, Azzam KM, Cidlowski JA. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem (2011) 286:38703–13. doi: 10.1074/jbc.m111.275370
116. Feng X, Zhao Y, Yang T, Song M, Wang C, Yao Y, et al. Glucocorticoid-Driven NLRP3 Inflammasome Activation in Hippocampal Microglia Mediates Chronic Stress-Induced Depressive-Like Behaviors. Front Mol Neurosci (2019) 12:210:210. doi: 10.3389/fnmol.2019.00210
117. Wang S, Lei T, Zhang K, Zhao W, Fang L, Lai B, et al. Xenobiotic pregnane X receptor (PXR) regulates innate immunity via activation of NLRP3 inflammasome in vascular endothelial cells. J Biol Chem (2014) 289:30075–81. doi: 10.1074/jbc.m114.578781
118. Wang S, Xie X, Lei T, Zhang K, Lai B, Zhang Z, et al. Statins Attenuate Activation of the NLRP3 Inflammasome by Oxidized-LDL or TNF-α in Vascular Endothelial Cells through a PXR-dependent mechanism. Mol Pharmacol (2017) 92(3):256–64. doi: 10.1124/mol.116.108100
119. Hu C, Dandapat A, Sun L, Chen J, Marwali MR, Romeo F, et al. LOX-1 deletion decreases collagen accumulation in atherosclerotic plaque in low-density lipoprotein receptor knockout mice fed a high-cholesterol diet. Cardiovasc Res (2008) 79:287–93. doi: 10.1093/cvr/cvn110
120. Wada T, Ishikawa A, Watanabe E, Nakamura Y, Aruga Y, Hasegawa H, et al. Eplerenone prevented obesity-induced inflammasome activation and glucose intolerance. J Endocrinol (2017) 235:179–91. doi: 10.1530/joe-17-0351
121. Brocca ME, Pietranera L, Meyer M, Lima A, Roig P, de Kloet ER, et al. Mineralocorticoid receptor associates with pro-inflammatory bias in the hippocampus of spontaneously hypertensive rats. J Neuroendocrinol (2017) 29:100. doi: 10.1111/jne.12489
122. Alatshan A, Kovács GE, Aladdin A, Czimmerer Z, Tar K, Benkő S. All-Trans Retinoic Acid Enhances both the Signaling for Priming and the Glycolysis for Activation of NLRP3 Inflammasome in Human Macrophage. Cells (2020) 9:1591. doi: 10.3390/cells9071591
123. Zhang YL, Wang RB, Li WY, Xia FZ, Liu L. Pioglitazone ameliorates retinal ischemia/reperfusion injury via suppressing NLRP3 inflammasome activities. Int J Ophthalmol (2017) 10:1812–8. doi: 10.18240/ijo.2017.12.04
124. Yao X, Jiang Q, Ding W, Yue P, Wang J, Zhao K, et al. Interleukin 4 inhibits high mobility group box-1 protein-mediated NLRP3 inflammasome formation by activating peroxisome proliferator-activated receptor-γ in astrocytes. Biochem Biophys Res Commun (2019) 509:624–31. doi: 10.1016/j.bbrc.2018.11.145
125. Liu H-M, Liao J-F, Lee T-Y. Farnesoid X receptor agonist GW4064 ameliorates lipopolysaccharide-induced ileocolitis through TLR4/MyD88 pathway related mitochondrial dysfunction in mice. Biochem Biophys Res Commun (2017) 490:841–8. doi: 10.1016/j.bbrc.2017.06.129
126. Wang S, Lin Y, Yuan X, Li F, Guo L, Wu B. REV-ERBα integrates colon clock with experimental colitis through regulation of NF-κB/NLRP3 axis. NatCommun (2018) 9:4246. doi: 10.1038/s41467-018-06568-5
127. Yu D, Fang X, Xu Y, Xiao H, Huang T, Zhang Y, et al. Rev-erbα can regulate the NF-κB/NALP3 pathway to modulate lipopolysaccharide-induced acute lung injury and inflammation. Int Immunopharmacol (2019) 73:312–20. doi: 10.1016/j.intimp.2019.04.035
128. Gabbia D, Pozzo L, Zigiotto G, Roverso M, Sacchi D, Pozza AD, et al. Dexamethasone counteracts hepatic inflammation and oxidative stress in cholestatic rats via CAR activation. PloS One (2018) 13:e0204336. doi: 10.1371/journal.pone.0204336
129. Assenat E, Gerbal-Chaloin S, Larrey D, Saric J, Fabre J, Maurel P, et al. Interleukin 1β inhibits CAR-induced expression of hepatic genes involved in drug and bilirubin clearance. Hepatology (2004) 40:951–60. doi: 10.1002/hep.20387
130. Delerive P, Bosscher KD, Berghe WV, Fruchart JC, Haegeman G, Staels B. DNA binding-independent induction of IkappaBalpha gene transcription by PPARalpha. Mol Endocrinol (2002) 16:1029–39. doi: 10.1210/mend.16.5.0826
131. Delerive P, Bosscher KD, Besnard S, Berghe WV, Peters JM, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem (1999) 274:32048–54. doi: 10.1074/jbc.274.45.32048
132. Pourcet B, Zecchin M, Ferri L, Beauchamp J, Sitaula S, Billon C, et al. Nuclear Receptor Subfamily 1 Group D Member 1 Regulates Circadian Activity of NLRP3 Inflammasome to Reduce the Severity of Fulminant Hepatitis in Mice. Gastroenterology (2018) 154:1449–1464.e20. doi: 10.1053/j.gastro.2017.12.019
133. Billon C, Murray MH, Avdagic A, Burris TP. RORγ regulates the NLRP3 inflammasome. J Biol Chem (2019) 294:10–9. doi: 10.1074/jbc.ac118.002127
134. Gamdzyk M, Doycheva DM, Kang R, Tang H, Travis ZD, Tang J, et al. GW0742 activates miR-17-5p and inhibits TXNIP/NLRP3-mediated inflammation after hypoxic-ischaemic injury in rats and in PC12 cells. J Cell Mol Med (2020) 20:163. doi: 10.1111/jcmm.15698
135. Woldt E, Sebti Y, Solt LA, Duhem C, Lancel S, Eeckhoute J, et al. Rev-erb-alpha modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat Med (2013) 19:1039–46. doi: 10.1038/nm.3213
136. Hao H, Cao L, Jiang C, Che Y, Zhang S, Takahashi S, et al. Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab (2017) 25:856–867.e5. doi: 10.1016/j.cmet.2017.03.007
137. Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity (2016) 45:802–16. doi: 10.1016/j.immuni.2016.09.008
138. Iracheta-Vellve A, Calenda CD, Petrasek J, Ambade A, Kodys K, Adorini L, et al. FXR and TGR5 Agonists Ameliorate Liver Injury, Steatosis, and Inflammation After Binge or Prolonged Alcohol Feeding in Mice. Hepatology Commun (2018) 2:1379–91. doi: 10.1002/hep4.1256
139. Chen Y, Le TH, Du Q, Zhao Z, Liu Y, Zou J, et al. Genistein protects against DSS-induced colitis by inhibiting NLRP3 inflammasome via TGR5-cAMP signaling. Int Immunopharmacol (2019) 71:144–54. doi: 10.1016/j.intimp.2019.01.021
140. Hu X, Yan J, Huang L, Araujo C, Peng J, Gao L, et al. INT-777 attenuates NLRP3-ASC inflammasome-mediated neuroinflammation via TGR5/cAMP/PKA signaling pathway after subarachnoid hemorrhage in rats. Brain Behav Immun (2020) 91:587–600. doi: 10.1016/j.bbi.2020.09.016
141. Rao Z, Chen X, Wu J, Xiao M, Zhang J, Wang B, et al. Vitamin D Receptor Inhibits NLRP3 Activation by Impeding Its BRCC3-Mediated Deubiquitination. Front Immunol (2019) 10:2783:2783. doi: 10.3389/fimmu.2019.02783
142. Cao R, Ma Y, Li S, Shen D, Yang S, Wang X, et al. 1,25(OH)2 D3 alleviates DSS-induced ulcerative colitis via inhibiting NLRP3 inflammasome activation. J Leukoc Biol (2020) 34:12. doi: 10.1002/jlb.3ma0320-406rr
143. Derangère V, Chevriaux A, Courtaut F, Bruchard M, Berger H, Chalmin F, et al. Liver X receptor β activation induces pyroptosis of human and murine colon cancer cells. Cell Death Differ (2014) 21:1914–24. doi: 10.1038/cdd.2014.117
144. Wang K, Xu T, Ruan H, Xiao H, Liu J, Song Z, et al. LXRα promotes cell metastasis by regulating the NLRP3 inflammasome in renal cell carcinoma. Cell Death Dis (2019) 10:159–14. doi: 10.1038/s41419-019-1345-3
145. Viaud M, Ivanov S, Vujic N, Duta-Mare M, Aira L-E, Barouillet T, et al. Lysosomal Cholesterol Hydrolysis Couples Efferocytosis to Anti-Inflammatory Oxysterol Production. Circ Res (2018) 122:1369–84. doi: 10.1161/circresaha.117.312333
146. Liesa M, Borda-d’Água B, Medina-Gómez G, Lelliott CJ, Paz JC, Rojo M, et al. Mitochondrial Fusion Is Increased by the Nuclear Coactivator PGC-1β. PloS One (2008) 3:e3613. doi: 10.1371/journal.pone.0003613
147. Li Y, Yin R, Liu J, Wang P, Wu S, Luo J, et al. Peroxisome proliferator-activated receptor δ regulates mitofusin 2 expression in the heart. J Mol Cell Cardiol (2009) 46:876–82. doi: 10.1016/j.yjmcc.2009.02.020
148. Kadoya H, Satoh M, Sasaki T, Taniguchi S, Takahashi M, Kashihara N. Excess aldosterone is a critical danger signal for inflammasome activation in the development of renal fibrosis in mice. FASEB J (2015) 29:3899–910. doi: 10.1096/fj.15-271734
149. Hudson G, Flannigan KL, Venu VKP, Alston L, Sandall CF, Macdonald JA, et al. Pregnane X Receptor Activation Triggers Rapid ATP Release in Primed Macrophages That Mediates NLRP3 Inflammasome Activation. J Pharmacol Exp Ther (2019) 370:44–53. doi: 10.1124/jpet.118.255679
150. Hughes MM, O’Neill LAJ. Metabolic regulation of NLRP3. Immunol Rev (2018) 281:88–98. doi: 10.1111/imr.12608
151. Paugh SW, Bonten EJ, Savic D, Ramsey LB, Thierfelder WE, Gurung P, et al. NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat Genet (2015) 47:607–14. doi: 10.1038/ng.3283
152. Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, et al. Caspase Cleavage of Gene Products Associated with Triplet Expansion Disorders Generates Truncated Fragments Containing the Polyglutamine Tract. J Biol Chem (1998) 273:9158–67. doi: 10.1074/jbc.273.15.9158
153. Cipollina C, Vincenzo SD, Siena L, Sano CD, Gjomarkaj M, Pace E. 17-oxo-DHA displays additive anti-inflammatory effects with fluticasone propionate and inhibits the NLRP3 inflammasome. Sci Rep (2016) 6:37625–12. doi: 10.1038/srep37625
154. Thakkar R, Wang R, Sareddy G, Wang J, Thiruvaiyaru D, Vadlamudi R, et al. NLRP3 Inflammasome Activation in the Brain after Global Cerebral Ischemia and Regulation by 17β-Estradiol. Oxid Med Cell Longevity (2016) 2016:8309031–17. doi: 10.1155/2016/8309031
155. d’Adesky ND, Vaccari JP de R, Bhattacharya P, Schatz M, Perez-Pinzon MA, Bramlett HM, et al. Nicotine Alters Estrogen Receptor-Beta-Regulated Inflammasome Activity and Exacerbates Ischemic Brain Damage in Female Rats. Int J Mol Sci (2018) 19:1330. doi: 10.3390/ijms19051330
156. Yue J, He J, Wei Y, Shen K, Wu K, Yang X, et al. Decreased expression of Rev-Erbα in the epileptic foci of temporal lobe epilepsy and activation of Rev-Erbα have anti-inflammatory and neuroprotective effects in the pilocarpine model. J Neuroinflamm (2020) 17:43. doi: 10.1186/s12974-020-1718-7
157. Hong W, Hu S, Zou J, Xiao J, Zhang X, Fu C, et al. Peroxisome proliferator-activated receptor γ prevents the production of NOD-like receptor family, pyrin domain containing 3 inflammasome and interleukin 1β in HK-2 renal tubular epithelial cells stimulated by monosodium urate crystals. Mol Med Rep (2015) 12:6221–6. doi: 10.3892/mmr.2015.4145
158. Bai M, Chen Y, Zhao M, Zhang Y, He JC-J, Huang S, et al. NLRP3 inflammasome activation contributes to aldosterone-induced podocyte injury. Am J Physiol Renal Physiol (2017) 312:F556–64. doi: 10.1152/ajprenal.00332.2016
159. Ding W, Guo H, Xu C, Wang B, Zhang M, Ding F. Mitochondrial reactive oxygen species-mediated NLRP3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget (2016) 7:17479–91. doi: 10.18632/oncotarget.8243
160. Gugliandolo E, Fusco R, Ginestra G, D’amico R, Bisignano C, Mandalari G, et al. Involvement of TLR4 and PPAR-α Receptors in Host Response and NLRP3 Inflammasome Activation, Against Pulmonary Infection With Pseudomonas Aeruginosa. Shock (2019) 51:221–7. doi: 10.1097/shk.0000000000001137
161. Ménégaut L, Thomas C, Jalil A, Julla JB, Magnani C, Ceroi A, et al. Interplay between Liver X Receptor and Hypoxia Inducible Factor 1α Potentiates Interleukin-1β Production in Human Macrophages. Cell Rep (2020) 31:107665. doi: 10.1016/j.celrep.2020.107665
162. Reitz CJ, Alibhai FJ, Khatua TN, Rasouli M, Bridle BW, Burris TP, et al. SR9009 administered for one day after myocardial ischemia-reperfusion prevents heart failure in mice by targeting the cardiac inflammasome. Commun Biol (2019) 2:353. doi: 10.1038/s42003-019-0595-z
163. Ferreira NS, Bruder-Nascimento T, Pereira CA, Zanotto CZ, Prado DS, Silva JF, et al. NLRP3 Inflammasome and Mineralocorticoid Receptors Are Associated with Vascular Dysfunction in Type 2 Diabetes Mellitus. Cells (2019) 8:1595. doi: 10.3390/cells8121595
164. Liu Q, Zhang F, Zhang X, Cheng R, Ma J-X, Yi J, et al. Fenofibrate ameliorates diabetic retinopathy by modulating Nrf2 signaling and NLRP3 inflammasome activation. Mol Cell Biochem (2018) 445:105–15. doi: 10.1007/s11010-017-3256-x
165. Li W, Liu X, Tu Y, Ding D, Yi Q, Sun X, et al. Dysfunctional Nurr1 promotes high glucose-induced Müller cell activation by up-regulating the NF-κB/NLRP3 inflammasome axis. Neuropeptides (2020) 82:102057. doi: 10.1016/j.npep.2020.102057
166. Wang X, Wang G, Qu J, Yuan Z, Pan R, Li K. Calcipotriol Inhibits NLRP3 Signal Through YAP1 Activation to Alleviate Cholestatic Liver Injury and Fibrosis. Front Pharmacol (2020) 11:200:200. doi: 10.3389/fphar.2020.00200
167. Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology (2013) 58:1941–52. doi: 10.1002/hep.26461
168. Lin Y, Lin L, Gao L, Wang S, Wu B. Rev-erbα regulates hepatic ischemia-reperfusion injury in mice. Biochem Bioph Res Co (2020) 529:916–21. doi: 10.1016/j.bbrc.2020.06.152
169. Han SJ, Jung SY, Wu S-P, Hawkins SM, Park MJ, Kyo S, et al. Estrogen Receptor β Modulates Apoptosis Complexes and the Inflammasome to Drive the Pathogenesis of Endometriosis. Cell (2015) 163:960–74. doi: 10.1016/j.cell.2015.10.034
170. Liu S-G, Wu X-X, Hua T, Xin X-Y, Feng D-L, Chi S-Q, et al. NLRP3 inflammasome activation by estrogen promotes the progression of human endometrial cancer. OncoTargets Ther (2019) 12:6927–36. doi: 10.2147/ott.s218240
171. Xie S, Guo C, Chi Z, Huang B, Wu Y, Wang D, et al. A rapid administration of GW4064 inhibits the NLRP3 inflammasome activation independent of farnesoid X receptor agonism. FEBS Lett (2017) 591:2836–47. doi: 10.1002/1873-3468.12782
172. Wang Y, Chen W, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor κB in hepatic inflammatory response. Hepatology (2008) 48:1632–43. doi: 10.1002/hep.22519
173. Al-Daghri NM, Guerini FR, Al-Attas OS, Alokail MS, Alkharfy KM, Draz HM, et al. Vitamin D receptor gene polymorphisms are associated with obesity and inflammosome activity. PloS One (2014) 9:e102141. doi: 10.1371/journal.pone.0102141
174. Zhou Z, Lin Y, Gao L, Yang Z, Wang S, Wu B. Circadian pharmacological effects of berberine on chronic colitis in mice: Role of the clock component Rev-erbα. Biochem Pharmacol (2020) 172:113773. doi: 10.1016/j.bcp.2019.113773
175. Cao H, Liu J, Shen P, Cai J, Han Y, Zhu K, et al. Protective Effect of Naringin on DSS-Induced Ulcerative Colitis in Mice. J Agric Food Chem (2018) 66:13133–40. doi: 10.1021/acs.jafc.8b03942
176. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464:1357–61. doi: 10.1038/nature08938
177. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slütter B, et al. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler Thromb Vasc Biol (2017) 37:1457–61. doi: 10.1161/atvbaha.117.309575
178. Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, et al. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res (2012) 110:416–27. doi: 10.1161/circresaha.111.253377
179. Jiang Q, Geng X, Warren J, Cosky EEP, Kaura S, Stone C, et al. Hypoxia Inducible Factor-1α (HIF-1α) Mediates NLRP3 Inflammasome-Dependent-Pyroptotic and Apoptotic Cell Death Following Ischemic Stroke. Neuroscience (2020) 448:126–39. doi: 10.1016/j.neuroscience.2020.09.036
180. Huang J-J, Xia J, Huang L-L, Li Y-C. HIF-1α promotes NLRP3 inflammasome activation in bleomycin-induced acute lung injury. Mol Med Rep (2019) 20:3424–32. doi: 10.3892/mmr.2019.10575
181. Gupta N, Sahu A, Prabhakar A, Chatterjee T, Tyagi T, Kumari B, et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc Natl Acad Sci (2017) 114:4763–8. doi: 10.1073/pnas.1620458114
182. Loukovaara S, Piippo N, Kinnunen K, Hytti M, Kaarniranta K, Kauppinen A. NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol (2017) 95:803–8. doi: 10.1111/aos.13427
183. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21:248–55. doi: 10.1038/nm.3806
184. Haas JT, Francque S, Staels B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu Rev Physiol (2015) 78:1–25. doi: 10.1146/annurev-physiol-021115-105331
185. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Rooyen DMV, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatology (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
186. Lee HJ, Yeon JE, Ko EJ, Yoon EL, Suh SJ, Kang K, et al. Peroxisome proliferator-activated receptor-delta agonist ameliorated inflammasome activation in nonalcoholic fatty liver disease. World J Gastroenterol (2015) 21:12787–99. doi: 10.3748/wjg.v21.i45.12787
187. Cardoso FS, Marcelino P, Bagulho L, Karvellas CJ. Acute liver failure: An up-to-date approach. J Crit Care (2017) 39:25–30. doi: 10.1016/j.jcrc.2017.01.003
188. Guo S, Yang C, Diao B, Huang X, Jin M, Chen L, et al. The NLRP3 Inflammasome and IL-1beta Accelerate Immunologically Mediated Pathology in Experimental Viral Fulminant Hepatitis. PloS Pathog (2015) 11:e1005155. doi: 10.1371/journal.ppat.1005155
189. Seo MJ, Hong JM, Kim SJ, Lee SM. Genipin protects d-galactosamine and lipopolysaccharide-induced hepatic injury through suppression of the necroptosis-mediated inflammasome signaling. Eur J Pharmacol (2017) 812:128–37. doi: 10.1016/j.ejphar.2017.07.024
190. Jiménez-Castro MB, Cornide-Petronio ME, Gracia-Sancho J, Peralta C. Inflammasome-Mediated Inflammation in Liver Ischemia-Reperfusion Injury. Cells (2019) 8:1131. doi: 10.3390/cells8101131
191. Zhu P, Duan L, Chen J, Xiong A, Xu Q, Zhang H, et al. Gene Silencing of NALP3 Protects Against Liver Ischemia–Reperfusion Injury in Mice. Hum Gene Ther (2011) 22:853–64. doi: 10.1089/hum.2010.145
193. Curtis AM, Bellet MM, Sassone-Corsi P, O’Neill LA. Circadian clock proteins and immunity. Immunity (2014) 40:178–86. doi: 10.1016/j.immuni.2014.02.002
194. Bellet MM, Deriu E, Liu JZ, Grimaldi B, Blaschitz C, Zeller M, et al. Circadian clock regulates the host response to Salmonella. Proc Natl Acad Sci U.S.A. (2013) 110:9897–902. doi: 10.1073/pnas.1120636110
195. Keller M, Mazuch J, Abraham U, Eom GD, Herzog ED, Volk HD, et al. A circadian clock in macrophages controls inflammatory immune responses. Proc Natl Acad Sci USA (2009) 106:21407–12. doi: 10.1073/pnas.0906361106
196. Early JO, Curtis AM. Immunometabolism: Is it under the eye of the clock? Semin Immunol (2016) 25(5):478–90. doi: 10.1016/j.smim.2016.10.006
197. Man K, Loudon A, Chawla A. Immunity around the clock. Science (2016) 354:999–1003. doi: 10.1126/science.aah4966
198. Kojetin DJ, Burris TP. REV-ERB and ROR nuclear receptors as drug targets. Nat Rev Drug Discovery (2014) 13:197–216. doi: 10.1038/nrd4100
199. Castanon-Cervantes O, Wu M, Ehlen JC, Paul K, Gamble KL, Johnson RL, et al. Dysregulation of inflammatory responses by chronic circadian disruption. J Immunol (2010) 185:5796–805. doi: 10.4049/jimmunol.1001026
200. Cuesta M, Boudreau P, Dubeau-Laramee G, Cermakian N, Boivin DB. Simulated Night Shift Disrupts Circadian Rhythms of Immune Functions in Humans. J Immunol (2016) 196:2466–75. doi: 10.4049/jimmunol.1502422
201. Morris CJ, Purvis TE, Hu K, Scheer FA. Circadian misalignment increases cardiovascular disease risk factors in humans. Proc Natl Acad Sci U.S.A. (2016) 113:E1402–11. doi: 10.1073/pnas.1516953113
202. Videnovic A, Lazar AS, Barker RA, Overeem S. The clocks that time us’–circadian rhythms in neurodegenerative disorders. Nat Rev Neurol (2014) 10:683–93. doi: 10.1038/nrneurol.2014.206
Keywords: nuclear receptors, inflammasome, inflammatory disease, circadian rhythm, NLRP3, therapeutic strategy, inflammation and innate immunity
Citation: Duez H and Pourcet B (2021) Nuclear Receptors in the Control of the NLRP3 Inflammasome Pathway. Front. Endocrinol. 12:630536. doi: 10.3389/fendo.2021.630536
Received: 17 November 2020; Accepted: 04 January 2021;
Published: 25 February 2021.
Edited by:
Nicolas Venteclef, Sorbonne Universités, FranceReviewed by:
Wendong Huang, Beckman Research Institute, City of Hope, United StatesCopyright © 2021 Duez and Pourcet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benoit Pourcet, YmVub2l0LnBvdXJjZXRAcGFzdGV1ci1saWxsZS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.