
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 11 March 2021
Sec. Cancer Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.627819
This article is part of the Research Topic Organoid Models and Cancer Endocrinology View all 4 articles
 Krijn K. Dijkstra1,2†José G. van den Berg3
Krijn K. Dijkstra1,2†José G. van den Berg3 Fleur Weeber1,2Joris van de Haar1,2Arno Velds4Sovann Kaing1,2
Fleur Weeber1,2Joris van de Haar1,2Arno Velds4Sovann Kaing1,2 Dennis D. G. C. Peters5
Dennis D. G. C. Peters5 Ferry A. L. M. Eskens6Derk-Jan A. de Groot7
Ferry A. L. M. Eskens6Derk-Jan A. de Groot7 Margot E. T. Tesselaar8
Margot E. T. Tesselaar8 Emile E. Voest1,2*
Emile E. Voest1,2*Gastroenteropancreatic neuroendocrine carcinoma (GEP-NEC) is a poorly understood disease with limited treatment options. A better understanding of this disease would greatly benefit from the availability of representative preclinical models. Here, we present the potential of tumor organoids, three-dimensional cultures of tumor cells, to model GEP-NEC. We established three GEP-NEC organoid lines, originating from the stomach and colon, and characterized them using DNA sequencing and immunohistochemistry. Organoids largely resembled the original tumor in expression of synaptophysin, chromogranin and Ki-67. Models derived from tumors containing both neuroendocrine and non-neuroendocrine components were at risk of overgrowth by non-neuroendocrine tumor cells. Organoids were derived from patients treated with cisplatin and everolimus and for the three patients studied, organoid chemosensitivity paralleled clinical response. We demonstrate the feasibility of establishing NEC organoid lines and their potential applications. Organoid culture has the potential to greatly extend the repertoire of preclinical models for GEP-NEC, supporting drug development for this difficult-to-treat tumor type.
NEC are poorly differentiated, fast-growing tumors that arise in various tissues, with a median survival of 5 months for metastatic disease (1, 2). Most extrapulmonary NEC arise in the gastroenteropancreatic tract (GEP-NEC) (3, 4). Patients with GEP-NEC often present with advanced disease at diagnosis (4, 5) and treatment options are limited. First-line treatment for patients with metastatic GEP-NEC consists of combination platinum-based chemotherapy. Response rate is relatively high (42%–67%), but patients relapse rapidly (5). No standard second-line therapy has been established.
Due to its low incidence (3, 4), GEP-NEC is a relatively understudied disease. Genetic characterization is limited to small studies and targeted sequencing of a few cancer genes (6–8). Because of its low incidence, accrual into clinical trials and development of new treatments is slow. There is an unmet need for better preclinical models to increase understanding of this disease and accelerate drug development. However, only few GEP-NEC cell lines have been established, the majority of which are derived from colorectal NEC (9).
Tumor organoids are three-dimensional cultures of tumor cells that retain histomorphologic and genetic features of the original tumor and can be established with high success rate from various tumors (10). Organoid models have been established from four patients with metastatic neuroendocrine prostate cancer (11) two colorectal NEC (12) and a NEC originating from the ampulla of Vater (13). In addition, recently an organoid biobank of neuroendocrine neoplasms has been established (14). Encouraged by these reports, we set out to systematically evaluate the feasibility of establishing GEP-NEC organoid lines, as well as their potential applications. Here, we established three organoid lines of NEC originating from the stomach and colon, describe their histomorphological, immunohistochemical, and genetic properties and drug response. Organoid models could greatly expand the repertoire of pre—clinical models available for the study of GEP-NEC.
Tissue was obtained from patients with metastatic or unresectable neuroendocrine carcinoma of extrapulmonary origin, being treated with everolimus and cisplatin in a phase II clinical trial (clinicaltrials.gov identifier NCT02695459). The study was approved by the Medical Ethical Committee of the Netherlands Cancer Institute – Antoni van Leeuwenhoek and written informed consent was obtained from all patients. Patients were treated in cycles of 75 mg/m2 intravenous cisplatinum on day 1 and 7.5 mg everolimus daily orally on days 1–21. Prior to treatment, a 18G core needle biopsy of the tumor was taken and processed for organoid culture within 24 h.
Organoids were established based on the protocol for colorectal cancer (CRC) organoids (15). Tumor tissue was dissociated into small pieces using razor blades or needles and enzymatically digested using 1.5 mg/ml collagenase type II (Sigma-Aldrich), 10 µg/ml hyaluronidase type IV (Sigma-Aldrich), 10 µM Y-27632 (Sigma-Aldrich) and 1:500 Primocin (Invivogen) at 37°C for 30 min. After washing, tumor cells were embedded in 10 mg/ml lactate dehydrogenase elevating virus (LDEV)-free reduced growth factor basement membrane extract (Gibco) and plated in small drops on a six-well culture plate. Geltrex drops were solidified for 20 min at 37°C before overlaying them with NEC organoid medium. NEC organoid medium consisted of Advanced DMEM/F12 (GIBCO) supplemented with 2 mM Ultraglutamine I (Lonza), 10 mM HEPES (GIBCO), and 100/100 U/ml Pencillin/Streptomycin (GIBCO), 50% Wnt-3a-conditioned medium (CM), 10% Noggin-CM, 20% R-spondin1-CM, 1x B27 supplement without vitamin A (GIBCO), 1.25 mM N-Acetylcysteine (Sigma-Aldrich), 10 mM nicotinamide (Sigma-Aldrich), 50 ng/ml human recombinant EGF (Peprotech), 100 ng/ml IGF-1 (Peprotech), 500 nM A83-01 (Tocris), 10 µM SB202190 (Cayman Chemicals), 10 µM Y-27632, 10 nM gastrin (Sigma-Aldrich) and 10 nM prostaglandin E2 (Cayman Chemicals). Until cryopreservation, organoids were cultured in an incubator at 37°C, under normoxic conditions with 5% CO2, in the presence of 1:500 Primocin. Medium was refreshed every 3–4 days and organoids were passaged every 1–2 weeks at a 1:2 to 1:5 split ratio. For passaging, medium was removed, organoids were resuspended in 1 ml/well dispase type II and incubated for 5 min at 37°C. Organoids were collected in 10 ml PBS and 100 µl of 0.5 M EDTA was added for every 1 ml of dispase used. After washing, organoids were resuspended in 1–4 ml accutase (Stem Cell Technologies) and incubated for 5–10 min at 37°C. Organoids were washed in PBS and replated in Geltrex drops. Organoids were cryopreserved in Recovery Cell Culture Recovery Freezing Medium (Gibco) or 10% DMSO/FCS at −80°C or in liquid nitrogen.
Some lines were cultured in tissue-specific medium rather than the generic medium described above (Supplementary Table 1). Medium formulation was based on previously published reports (pancreas (16), ovarium (17), stomach (18), colorectal (15)).
Organoids were authenticated by single nucleotide polymorphism (SNP) array, short tandem repeat (STR) profiling or human leukocyte antigen (HLA) haplotyping. For SNParray, organoids and matching blood were typed for 26 single nucleotide polymorphisms (SNPs) using a Taqman-based SNParray (Hartwig Medical Foundation, Amsterdam). For STR profiling, samples were matches using a PowerPlex 16 HS System (Promega, DC2101). HLA-A, -B, -DRB1 low-intermediate resolution typing was performed at the dept. of Sanquin HLA diagnostic services by means of the Sequence Specific Oligonucleotide probes (SSOP) typing method using the Luminex microbead technology which involved polymerase chain reaction (PCR) amplification of the HLA class I and II regions (Lifecodes HLA typing kits, Genprobe, San Diego CA-USA). All samples were regularly tested for Mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza).
Organoids were isolated from Geltrex by incubating in 2 mg/ml dispase for 5 min at 37°C, followed by washing in 10 ml 5 mM EDTA/PBS. In some cases, dispase treatment resulted in organoid dissociation. Therefore we resupended organoids in ice-cold PBS to isolate organoids from Geltrex in some cases. Organoids were pelleted and fixed in 4% paraformaldehyde for 30 min at room temperature, or for 24 h at 4°C. After fixation, organoids were washed in PBS and embedded in paraffin blocks. 3 µm paraffin sections were cut and routine hematoxylin and eosin (H&E) staining was performed.
Immunohistochemistry of the formalin-fixed, paraffin-embedded (FFPE) tumor samples was performed on a BenchMark or Discovery Ultra autostainer (Ventana Medical Systems). Briefly, paraffin sections were cut at 3 µm, heated at 75°C for 28 min and deparaffinized in the instrument with EZ prep solution (Ventana Medical Systems). Heat-induced antigen retrieval was carried out using Cell Conditioning 1 (CC1, Ventana Medical Systems) for 32 min at 95°C (Synaptophysin, Chromogranin, MLH1, MSH2, MSH6, CD56), 64 min at 95°C (Ki-67) or 72 min at 95°C (PMS2). The following primary antibodies were used: chromogranin was detected using clone LK2H10 (Ready-to-Use, 32 min at 37°C, Roche), Synaptophysin using clone 27G12 (Leica) 1/50 dilution for 32 min at 37°C, Ki-67 using clone MIB1 (1/100 dilution, 1 hour at 37°C, Agilent/DAKO), MLH1 using clone ES05 (1/20 dilution, 32 min at 37°C, Agilent/DAKO), MSH2 using clone G219-1129 (Ready-To-Use, 12 min at 37°C, Roche/Ventana), MSH6 using clone EP49 (1/50 dilution, 32 min at 37°C, Epitomics), PMS2 using clone A16-4 (Ready-To-Use, 32 min at 37°C, Roche/Ventana), and CD56 using Clone MRQ-42 (Ready-To-Use, 4 min at 37°C, Ventana Medical Systems).
To reduce background staining, after incubation of the primary antibodies chromogranin A, synaptophysin and Ki67, a rabbit anti-mouse IgG1, IgG2a, IgG3 (Clone M204-3, Abcam, ab1334698, 1/500, 32 min at 37°C) was added, after which bound antibody for chromogranin A, synaptophysin, Ki67 and CD56 was visualized using Anti-Rabbit HQ (Ready to Use, 12 min at 37°C, Ventana Medical Systems), Anti-HQ HRP (Ready to Use, 12 min at 37°C, Ventana Medical Systems) and ChromoMap DAB (Ventana Medical Systems). For negative controls, stainings were performed without the primary antibody. For MLH1 and PMS2 signal amplification was applied using the Optiview Amplification Kit (4 min, Ventana Medical Systems). For MLH1, MSH2, MSH6 and PMS2 bound antibody was visualized using the OptiView DAB Detection Kit (Ventana Medical Systems). Slides were counterstained with Hematoxylin and Bluing Reagent (Ventana Medical Systems).
Organoids were isolated from Geltrex 4 days after passaging by incubating in 2 mg/ml dispase type II for 5 min at 37°C, followed by washing in 5 mM EDTA/PBS. Organoids were counted and resuspended in 10 mg/ml Geltrex at 50 organoids/µl. Five-microliter drops were plated in pre-warmed clear-bottom white-walled flat-bottom 96-well culture plates (Greiner). Geltrex drops were allowed to solidify at 37°C for 15 min before overlaying with 200 µl/well of drug-containing NEC organoid medium. Five-step ten-fold serial dilutions were made of cisplatin (50 µg/ml to 5 ng/ml, PBS, Accord Health Care, Netherlands) and everolimus (25 µM to 250 fM, DMSO, Selleck Chemicals). For untreated controls, the volume of DMSO corresponding to the highest drug concentration was added to NEC organoid medium. After 6 days culture in the presence or absence of drugs, viability was evaluated using CellTiter-Glo 3D (Promega) on an Infinite 200 Pro plate reader (Tecan Life Sciences). Medium was replaced by 50 µl Ad-DF+++ mixed with 50 µl CellTiter-Glo reagent, incubated in the dark for 5 min on an orbital shaker (room temperature), followed by 25 min incubation without shaking. Baseline measurements were taken at the time of plating (day 0).
Growth rate (GR) corrected viability was calculated as described before (19)
where Vd is the viability of drug-treated organoids at day 6, V0 is median viability of baseline measurement (day 0), and Vctrl is the viability of control-treated organoids at day 6 (based on CellTiter Glo). Data was analyzed using GraphPad Prism V7.03. Dose response curves were fit on data normalized to control-treated samples using Prism based with a hill slope of −1, and top constrained to 100. IC50 or GR0 values are absolute and interpolated from dose response curves.
DNA was isolated from organoids and patient-matched blood using a DNEasy blood and tissue kit (Qiagen). At least 200 ng of DNA was used for whole-exome sequencing after exome capture (Integrated DNA Technologies probe set) and sequenced on an Illumina NextSeq DNA analyzer using 150 base pair paired-end reads. Reads were aligned to a reference genome (GRCh38) with BWA-MEM (20), tagged, if duplicate, by Picard MarkDuplicates, and base quality score normalized using GATK BaseRecalibrator (GATK version 4.1) (21). Hereafter somatic and germline single nucleotide variants (SNVs) were called by GATK MuTect 2 and HaplotypeCaller, respectively. Copy number, cellularity and ploidy were estimated, from the paired tumor-normal samples, and visualized using Sequenza (22).
Somatic mutations were annotated using SnpEff (23) and SnpSift (24) was used to remove silent and non-coding mutations. Specifically, retained mutations comprised those annotated as high or moderate impact, and/or having an effect of the following categories: ‘coding_sequence_variant’, ‘disruptive_inframe_deletion’, ‘disruptive_inframe_insertion’, ‘exon_variant’, ‘feature_ablation’, ‘frameshift_variant’, ‘gene_variant’, ‘inframe_deletion’, ‘inframe_insertion’, ‘missense_variant’, ‘protein_protein_contact’, ‘rare_amino_acid_variant’, ‘stop_gained’, ‘stop_lost’, ‘structural_interaction_variant’, ‘transcript_variant’. Mutated genes were filtered for cancer genes, using the MSK-IMPACT cancer gene panel (25).
In addition, DNA was used for low-coverage whole genome sequencing for copy number analysis on an Illumina HiSeq2500 DNA analyzer (High Output Mode) using 65 base pair single reads. Reads were aligned to GRCh38 using BWA-MEM (20). Per sample, per bin of 20 kb, mapping quality 37 read counts were rated against mappability, predicted by a likewise mapping of commensurate GRCh38 reference derived sequences, and log2 transformed. Sample counts were corrected per bin for local GC effects using a non-linear fit of mappabilities over 0.8 on autosomes. Reference values were scaled according to the slope of a linear fit of reference mappabilities after GC correction. Ratios of corrected sample counts and reference values left out bins with mappability below 0.2 or overlapping ENCODE blacklisted regions (26). Resulting copy number data were analyzed using Nexus v10.0.
IC50 or GR0 values were compared to NEC-01013 as a reference using a one-way ANOVA with Dunnett’s multiple comparison test. Other statistics are based on two-sided Student’s t test. P-values < 0.05 are considered statistically significant. The number of asterisks in figures indicates the level of significance: * P < 0.05; ** P < 0.01; *** P < 0.001.
We generated GEP-NEC organoid lines using a procedure similar to that established for CRC organoids (15). We initially based our culture medium on that used for CRC organoids, with the addition of IGF-1 based on high expression of phosphorylated mTOR in NEC (27). We used this medium to establish organoids from core needle biopsies from 22 patients with metastatic or inoperable extrapulmonary NEC. From this initial cohort, organoid lines were established from three patients: NEC-01010, derived from a liver metastasis originating from the ampulla of Vater; NEC-01013, derived from a liver metastasis of a gastric NEC; and NEC-02002, derived via a colonic biopsy.
As we received NEC from various tissues (Supplementary Figure 1), in a second phase we attempted organoid culture using a modified protocol in which the culture medium was adjusted (where possible) for each sample based on its tissue of origin (Supplementary Table 1). This resulted in the establishment of two additional organoid lines (NEC-02006, from a liver metastasis of a NEC originating from the colon; and NEC-02007, a gastric NEC). Organoid establishment rates were not significantly different for samples cultured in generic NEC medium (three out of 24 successful) versus tissue-specific medium (two out of 7, Fisher’s exact test: p = 0.31). Overall establishment rate (combining both protocols) was 16% (5 out of 31).
Organoids grew as solid, slightly irregular spheres (Figure 1A) and could be passaged with a biweekly split ratio of 1:2 to 1:5. After 5–10 weeks of expansion, organoids were frozen, from which they recovered well (only one sample, which we classified as a failed culture, did not survive cryopreservation). Organoids were cultured until passages 17–22, or 7–10 months in culture. We did not observe any decrease in proliferation at higher passage, and cultures were terminated for practical reasons rather than because of ceased growth.
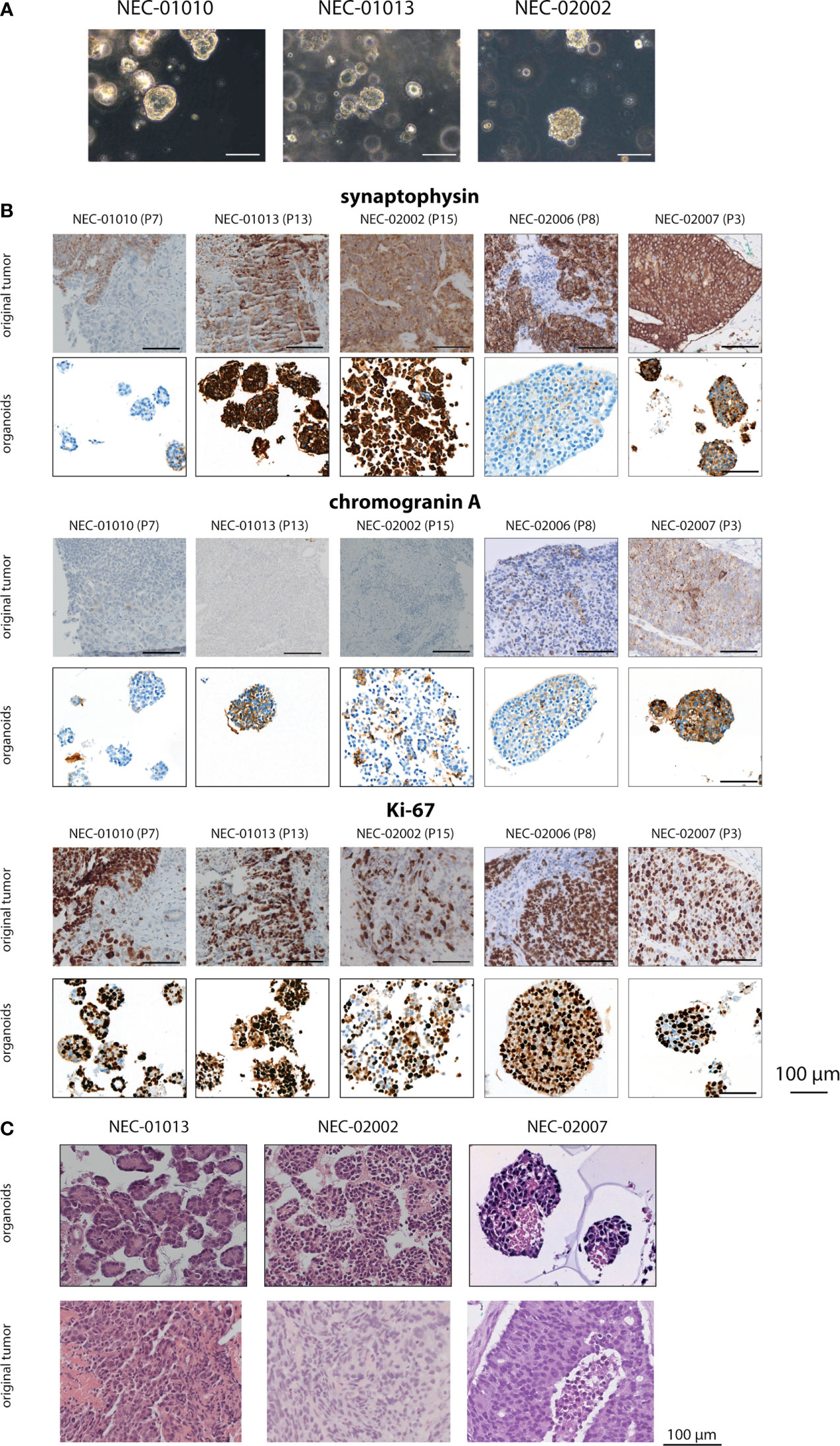
Figure 1 Histomorphology of GEP-NEC organoid lines and matching original tumor. (A) Phase-contrast photomicrographs of GEP-NEC organoid lines. Scale bar = 100 µm. (B) Immunostainings of synaptophysin, chromogranin and Ki-67 for organoids and original tumor. P7 indicates passage 7. (C) Hematoxylin and eosin (H&E) stained slides of GEP-NEC organoid lines and original tumors. NEC-01013: passage (P) 12; NEC-02002: P20; NEC-02007: P3.
To confirm the neuroendocrine nature of the established organoid lines, we evaluated the expression of the neuroendocrine markers synaptophysin and chromogranin (Figure 1B, Supplementary Figure 2A and Table 1).

Table 1 Expression of Ki-67 and neuroendocrine markers in original tumor and organoids1.
Based on these markers, organoid lines NEC-01013, NEC-02002, and NEC-02007 could be reliably classified as NEC organoids. The original tumor and corresponding organoid lines expressed synaptophysin in all three cases. NEC-01013 and NEC-02002 were negative for chromogranin (both in the original tumor and the matching organoid lines), but NEC-02007 showed chromogranin expression in organoids as well as the original tumor. The percentage of cells expressing Ki-67 was comparable between organoids and original tumor (Table 1).
In contrast, expression of neuroendocrine makers was discordant between organoids and original tumor for lines NEC-01010 and NEC-02006. The original tumor from NEC-01010 was negative for chromogranin and showed heterogeneous synaptophysin expression (50% overall, varying from absent to strong, both in solid and in glandular parts of the tumor (Supplementary Figure 2B). However, organoids were negative for both chromogranin and synaptophysin. Even though 30% of organoids expressed CD56, the original tumor showed only limited CD56 expression, which did not track closely with expression of synaptophysin (Supplementary Figure 3). NEC-02006 organoids (a liver metastasis of a NEC originating from the colon) were negative for all NEC markers tested (synaptophysin, chromogranin A and CD56; Figure 1B and Supplementary Figure 3). Based on the absence of NEC marker expression in NEC-01010 and NEC-02006, we cannot confidently classify these lines as NEC organoids.
Histomorphologically, NEC organoid lines NEC-01013, NEC-02002 and NEC-02007 grew as irregular nests, with large, hyperchromatic nuclei and limited cytoplasm (Figure 1C). If only organoids that can reliably be classified as NEC are considered, organoid establishment rate is 10% (3 out of 31).
We characterized organoid lines NEC-01013 and NEC-02002 in more detail. We performed whole exome sequencing (WES) to detect mutations, and low-coverage whole genome sequencing (WGS) for copy number analysis. NEC-01013 and NEC-02002 contained 49 and 532 mutations, respectively (Table 2). The high number of mutations in NEC-02002 was paralleled by a relatively high proportion of indels. This genomic profile is characteristic of mismatch repair (MMR) deficiency, and NEC-02002 organoids as well as the original tumor indeed showed loss of expression of the MMR proteins PMS2 and MLH1 (Figure 2). Loss of RB1 expression and mutations in TP53 are frequently found in NEC (6–8, 28). TP53 mutations were present in both NEC-01013 and NEC-02002 (Table 2). NEC-01013 also contained an RB1 mutation. An RB1 mutation was also identified in NEC-02002 (Supplementary Table 2), but this was an intron variant with low allele frequency and therefore unlikely to be a strong driver. Other mutated cancer genes included those involved in Wnt signaling (APC, AMER1), mitogen signaling (KRAS, BRAF), epigenetic modification (ARID1A, ARID2, DNMT3A, NCOA3) and DNA repair (RAD50, BRIP1, ERCC5, RECQL4).

Table 2 Mutations in NEC organoids identified by whole exome sequencing.4
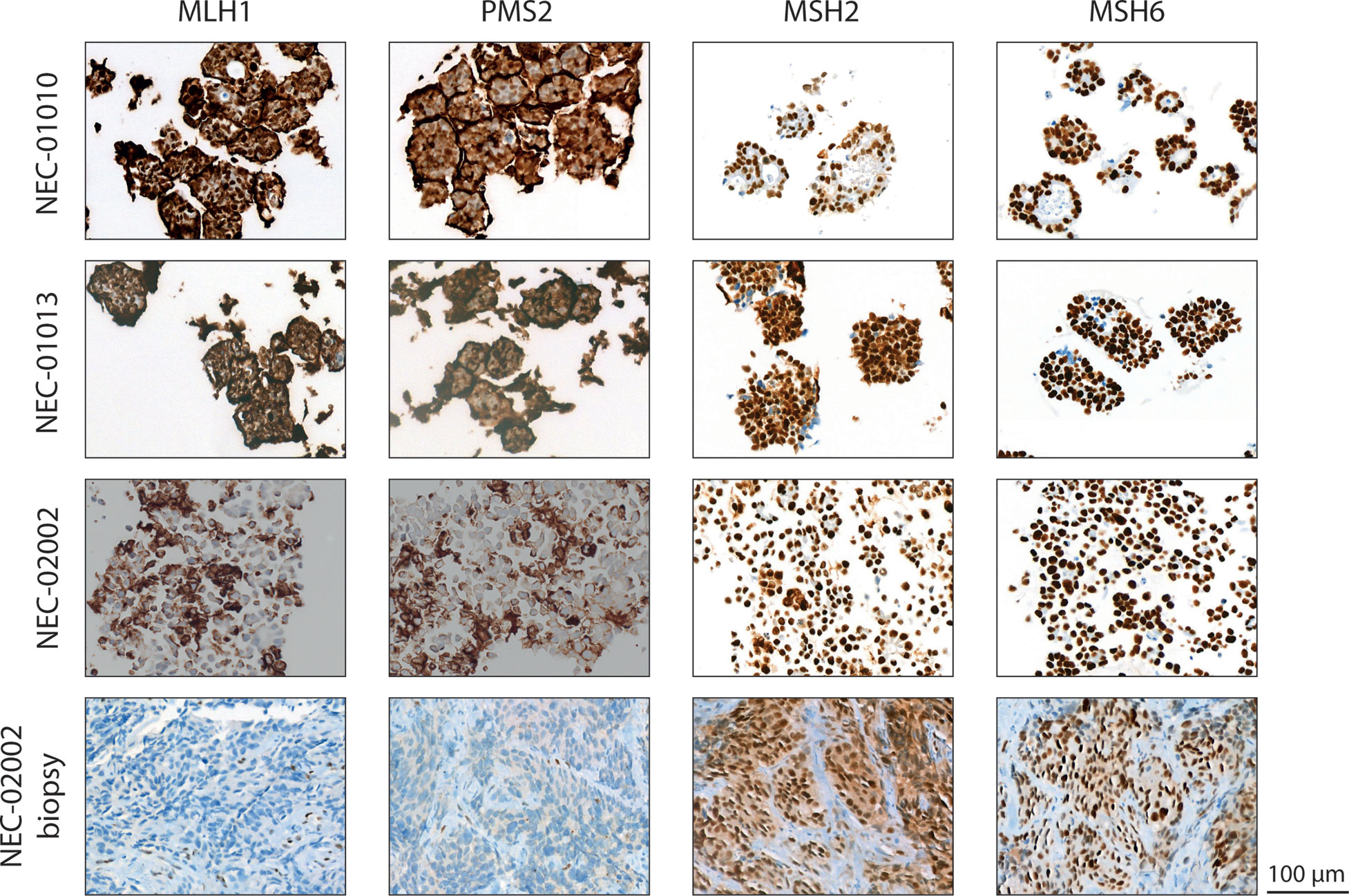
Figure 2 Mismatch repair status of GEP-NEC organoid lines. Staining of slides from NEC-01010 (P7 and P11), NEC-01013 (P13), and NEC-02002 organoids (P15), as well as the original tumor of NEC-02002, for mismatch repair proteins MLH1, PMS2, MSH2, and MSH6. Note 100% nuclear expression of MHS2 and MSH6 but absence of nuclear staining of MLH1 and PMS2 in both organoids and original tumor of NEC-02002. The brown halo on the cell border is a specific staining caused by residual Geltrex in organoid slides. Scale bar = 100 µm.
NEC organoids showed variable degrees of chromosomal abnormalities (Figure 3A and Table 3). NEC-01013 showed a relatively normal genome. In contrast, NEC-02002 showed a highly disorganized genome, with 87% of the genome showing abnormal copy number. We used Sequenza to determine allele-specific copy number based on SNPs identified by WES. This revealed whole-chromosomal losses resulting in loss of heterozygosity (LOH) (Figure 3B), further underscoring the level of genomic disorganization of NEC-02002.
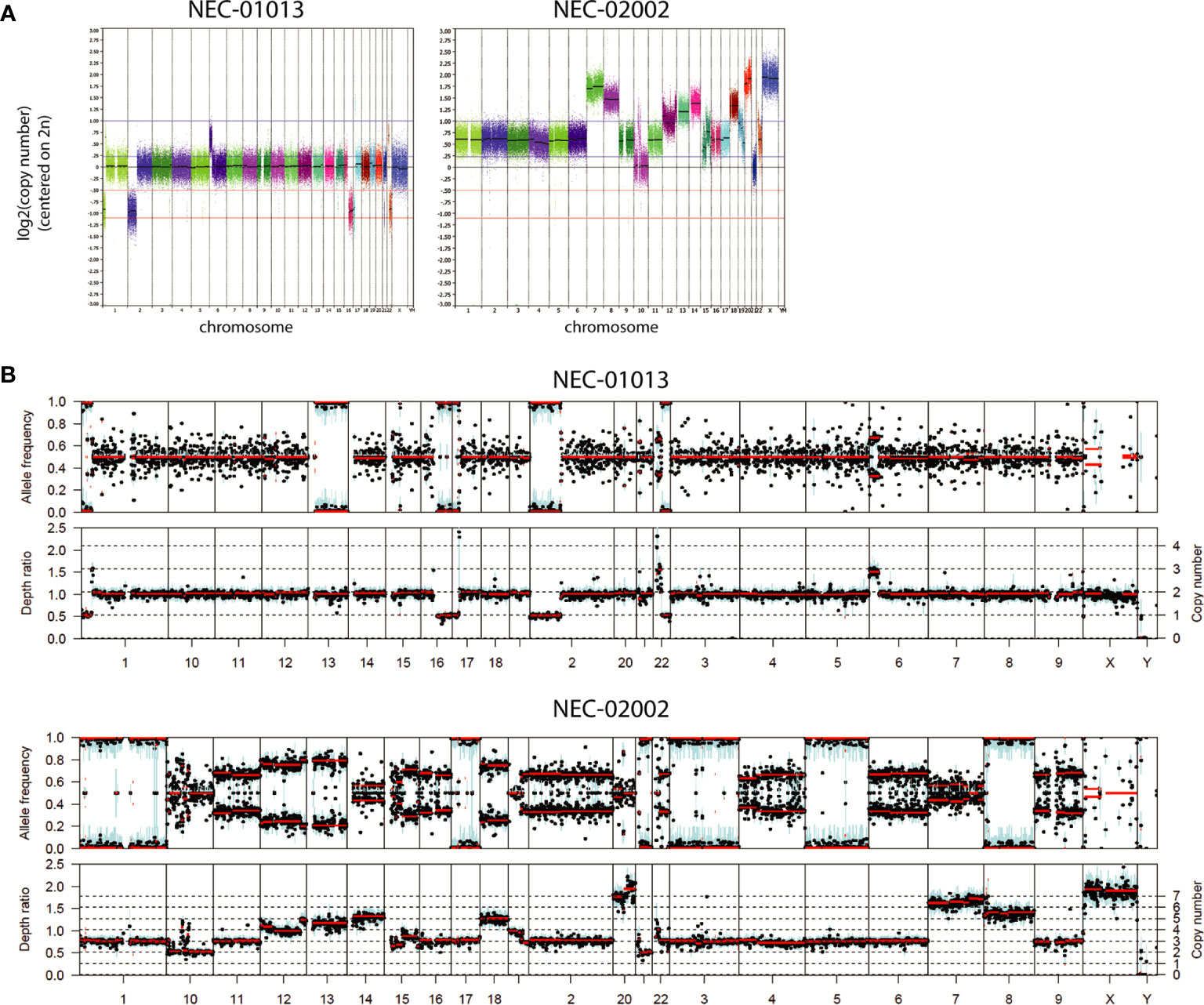
Figure 3 Copy number profiles of GEP-NEC organoids. (A) Copy number profiles of GEP-NEC organoids are generated based on low-coverage whole genome sequencing and represented on a log2 scale. NEC-01013: passage (P) 11; NEC-02002: P5. (B) Copy number profiles were based on SNPs identified by whole exome sequencing and analyzed by Sequenza. Allele frequencies are given as ratios and used to infer copy number. NEC-01013: P11; NEC-02002: P15.
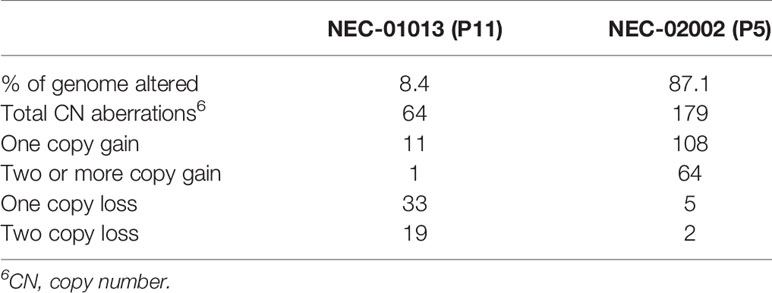
Table 3 Copy number alterations in NEC organoids.
Organoids were derived from patients treated with cisplatin and the mTOR inhibitor everolimus. We therefore determined the response of NEC organoids to these drugs and compared this to patient response for NEC-01010, NEC-01013 and NEC-02002. NEC-01013 was the only patient who showed an objective partial response (PR) after three cycles of chemotherapy. Patient NEC-01010 showed stable disease (SD) after three treatment cycles, but a mixed response at the next evaluation, with some lesions progressing and others regressing (the lesion from which the organoids were derived had not changed in size, but had become more hypodense). Patient NEC-02002 showed limited progression of the liver metastases that still fell within the boundaries of SD at the first two evaluations, and then progressed.
Because the clinical effect of everolimus is often limited to disease stabilization rather than tumor regression (29), we considered two different metrics to determine organoid drug sensitivity. First, we generated dose response curves and calculated IC50 (Figures 4A, C). We also evaluated the effect of drug treatment on organoid growth rate (Figures 4B, D). The growth rate metric (GR) compares the amount of tumor cells between the start and end of the drug assay (6 days), with a value of –1 indicating complete tumor cell death, a value of 0 complete growth inhibition, and a value of 1 no effect of the drug (19).
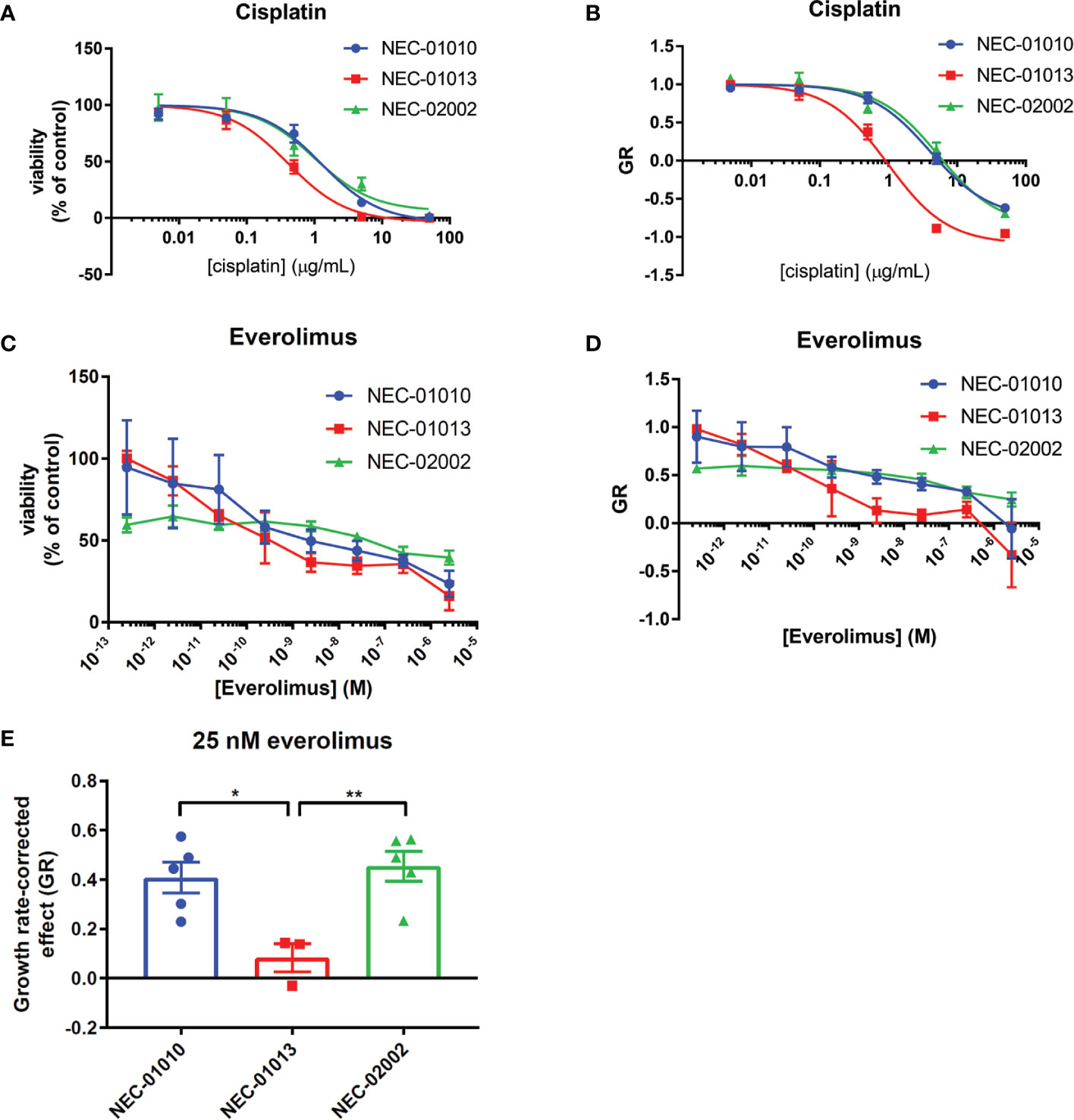
Figure 4 Drug response of GEP-NEC organoids. (A, B) Viability (A) or growth-rate corrected viability (B) of cisplatin-treated GEP-NEC organoids. n = 3–4. NEC-01010: passage (P) 6-14; NEC-01013: P10-19; NEC-02002: P6-16. (C, D) Viability (C) or growth-rate corrected viability (D) of everolimus-treated GEP-NEC organoids. n = 3–5. NEC-01010: P6-16; NEC-01013: P10-20; NEC-02002: P6-17. (E) GR at 25 nM everolimus (corresponding to Cmax). n = 3–5. Error bars represent s.e.m. Student’s t test. *P < 0.05; **P < 0.01.
NEC-01013 (the only patient with a partial response) was more sensitive to cisplatin than NEC-01010 and NEC-02002, showing statistically significant differences in GR0 (Figures 4A, B, and Table 4). Everolimus did not lead to complete tumor cell death, even at concentrations far exceeding the maximum plasma concentration (Cmax = 25 nM) (30) (Figure 4C). However, based on GR, everolimus effectively inhibited growth of NEC-01013, with values approaching zero in the nanomolar range (Figure 4D). The effect of everolimus at 25 nM (Cmax) was significantly stronger in NEC-01013 compared to NEC-01010 and NEC-02002 (Figure 4E). In summary, in this limited set of patient derived-organoids, organoids from the responding tumor showed higher sensitivity to both cisplatin and everolimus than organoids from the patients without an objective response.
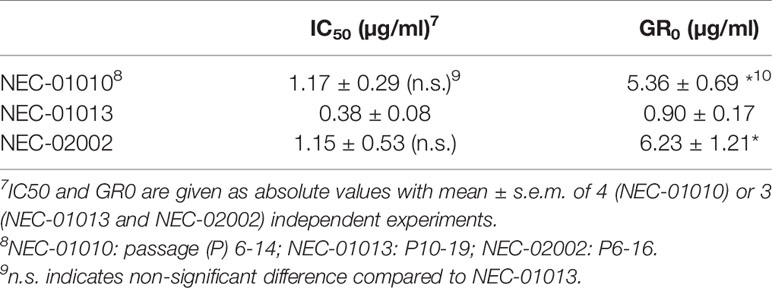
Table 4 Cytotoxicity of cisplatin on NEC organoid lines.
Here, we describe the establishment and characterization of three novel GEP-NEC organoid lines. Organoid culture may expand the current scientific toolbox to study this rare disease.
Organoids could be classified as neuroendocrine based on the expression of neuroendocrine markers (synaptophysin, chromogranin and CD56). Two organoid lines could not be classified as NEC because they lacked expression of these markers. The tumors from which these organoids were derived were heterogeneous, containing both synaptophysin-positive and -negative tumor cells. Absence of synaptophysin-positive organoids could indicate selective outgrowth of non-neuroendocrine tumor cells or loss of synaptophysin expression during organoid culture. Optimization of media formulation to promote selective outgrowth of neuroendocrine cells could increase the purity of NEC organoid cultures.
Organoid sensitivity to cisplatin and everolimus mirrored patient response in this limited set. Such an association is reassuring, and others have seen correlations between patient response and organoid drug sensitivity (31–33). The predictive value of organoid drug response differs between treatment regimens (32) and studies with larger cohorts are required to determine the validity of organoid response to cisplatin and everolimus as a predictor of clinical response.
Our current culture success rate is 16% (or 10% if only considering organoids confidently classified as NEC), which is too low for clinical implementation at the level of individual patients. This establishment rate is lower than for CRC biopsies (71%) (34) but in the same range as for non-small lung cell cancer (35, 36) or advanced prostate cancer (37). Higher success rates may be achieved by optimizing the culture medium formulation, which may differ depending on the tissue of origin. In our preliminary analysis, establishment rate was not significantly higher for samples grown in tissue-specific rather than generic NEC medium, but higher numbers are needed to draw definite conclusions. Recently, an organoid biobank of neuroendocrine neoplasms has been established, with a considerably higher establishment rate (14). The difference in culture success rate may be attributable to different medium composition. In this study, we received biopsies, which limits the number of different culture conditions that can be tested and may also have contributed to the low culture success rate. Obtaining more tissue, for example by surgical resection of a tumor, will help to further optimize culture conditions.
Two of the NEC models (NEC-01013 and NEC-02007) were derived from metastases and one (NEC-02002) from a primary tumor. Establishment of larger numbers of organoid models from both primary tumors and metastases could serve to evaluate how biological behavior and drug sensitivity changes throughout NEC progression.
NEC organoid models may also be valuable beyond approaches for individual patients. Tumor organoids can help prioritize drug candidates based on genetic or other biological features. Mutations were detected in several DNA repair genes, including those involved in homologous recombination (RAD50, BRIP1, RECQL4). Organoids could be used to evaluate whether drugs targeting DNA repair mechanisms (e.g. PARP inhibitors) are effective in GEP-NEC. We also noted very extensive chromosomal abnormalities in NEC-02002. A high number of copy number alterations has been described previously in GEP-NEC (38). Targeting aneuploidy and/or chromosomal instability (CIN) may present other therapeutic options, such as therapies aimed at increasing proteotoxic stress, or increasing CIN by combining spindle checkpoint inhibitors with taxanes (39) We also identified a BRAFV600E mutation in the colonic NEC-02002. BRAF mutations have been found in 5%–59% of colorectal NEC (40) and two recent case reports describe three colorectal NEC patients that responded to combined BRAF/MEK inhibition (41, 42). NEC organoids could be used for future drug discovery studies.
In conclusion, we have generated three NEC organoid lines that may serve as preclinical models to further the understanding of the biology of NEC. Our approach may also be of value for other rare diseases.
Requests for sequencing and other data, and for sharing of organoid lines and other resources should be directed to the corresponding author, EV (ZS52b2VzdEBua2kubmw=) and will be evaluated by the Institutional Review Board of the Netherlands Cancer Institute (SVJCQG5raS5ubA==). The informed consent signed by patients participating in this study limits the deposition of DNA sequencing data in publicly available repositories. Producer cell lines for Wnt-3a, R-spondinand Noggin-CM were obtained under a material transfer agreement (MTA). Requests for these lines should be directed to the relevant institutions [Hubrecht Institute for Wnt-3a and Noggin, and Calvin Kuo (Stanford) for R-spondin producer lines].
The studies involving human participants were reviewed and approved by the Medical Ethical Committee of the Netherlands Cancer Institute – Antoni van Leeuwenhoek. The patients/participants provided their written informed consent to participate in this study.
KD: study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript, statistical analysis. JB: study concept and design, acquisition of pathology data, analysis and interpretation of data, critical revision of manuscript. FW: study concept and design. JH and AV: analysis of sequencing data. SK: processing of tumor tissue. DP: immunohistochemistry. FE and D-JG: patient inclusion and clinical aspects. MT: study concept and design, patient inclusion and clinical aspects, critical revision of manuscript, obtained funding, study supervision. EV: study concept and design, obtained funding, critical revision of manuscript, study supervision. All authors contributed to the article and approved the submitted version.
This work was supported by the NWO Gravitation Program (grant number NWO 2012-2022) (to EV on behalf of CancerGenomics.nl), and Ammodo foundation (to MT).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Judith Westra, Sandra Visser, Louisa Hoes, Luuk Schipper, and Daphne van der Velden for help with acquisition of human samples. We acknowledge Ingrid Hofland and Annegien Broeks and other members of the NKI-AVL Core Facility Molecular Pathology & Biobank (CFMPB) for supplying NKI-AVL Biobank material and lab support. We thank Ron Kerkhoven, Marja Nieuwland, Roel Kluin, Charlaine Steenis, and Wim Brugman for DNA sequencing and analysis. We thank Loes van Velthuysen for providing immunohistochemistry slides.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.627819/full#supplementary-material
1. Sorbye H, Welin S, Langer SW, Vestermark LW, Holt N, Osterlund P, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): The NORDIC NEC study. Ann Oncol (2013) 24:152–60. doi: 10.1093/annonc/mds276
2. Surveillance Epidemiology, and End Results (SEER) Program. SEER*Stat Database: Incidence – SEER 9 Regs Research Data. (2013), November 2011 submission (1973-2010). Available at: www.seer.cancer.gov.
3. Korse CM, Taal BG, van Velthuysen M-LF, Visser O. Incidence and survival of neuroendocrine tumours in the Netherlands according to histological grade: experience of two decades of cancer registry. Eur J Cancer (2013) 49:1975–83. doi: 10.1016/j.ejca.2012.12.022
4. Van Der Zwan JM, Siesling S, Van Velthuysen L, Links T, Walenkamp A, Tesselaar M. Extra-Pulmonary Neuroendocrine Carcinomas: A Population-Based Study in the Netherlands. Neuroendocrinology (2018) 107:50–9. doi: 10.1159/000488987
5. Sorbye H, Strosberg J, Baudin E, Klimstra DS, Yao JC. Gastroenteropancreatic high-grade neuroendocrine carcinoma. Cancer (2014) 120:2814–23. doi: 10.1002/cncr.28721
6. Yachida S, Vakiani E, White CM, Zhong Y, Saunders T, Morgan R, et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol (2012) 36:173–84. doi: 10.1097/PAS.0b013e3182417d36
7. Jesinghaus M, Konukiewitz B, Keller G, Kloor M, Steiger K, Reiche M, et al. Colorectal mixed adenoneuroendocrine carcinomas and neuroendocrine carcinomas are genetically closely related to colorectal adenocarcinomas. Mod Pathol (2017) 30:610–9. doi: 10.1038/modpathol.2016.220
8. Takizawa N, Ohishi Y, Hirahashi M, Takahashi S, Nakamura K, Tanaka M, et al. Molecular characteristics of colorectal neuroendocrine carcinoma; Similarities with adenocarcinoma rather than neuroendocrine tumor. Hum Pathol (2015) 46:1890–900. doi: 10.1016/j.humpath.2015.08.006
9. Kawasaki K, Fujii M, Sato T. Gastroenteropancreatic neuroendocrine neoplasms: genes, therapies and models. Dis Model Mech (2018) 11:1–12. doi: 10.1242/dmm.029595
10. Weeber F, Ooft SN, Dijkstra KK, Voest EE. Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chem Biol (2017) 24:1092–100. doi: 10.1016/j.chembiol.2017.06.012
11. Puca L, Bareja R, Prandi D, Shaw R, Benelli M, Karthaus WR, et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat Commun (2018) 9:1–10. doi: 10.1038/s41467-018-04495-z
12. Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell (2016) 18:827–38. doi: 10.1016/j.stem.2016.04.003
13. Saito Y, Muramatsu T, Kanai Y, Ojima H, Sukeda A, Hiraoka N, et al. Establishment of Patient-Derived Organoids and Drug Screening for Biliary Tract Carcinoma. Cell Rep (2019) 27:1265–76.e4. doi: 10.1016/j.celrep.2019.03.088
14. Kawasaki K, Toshimitsu K, Matano M, Fujita M, Fujii M, Togasaki K, et al. An Organoid Biobank of Neuroendocrine Neoplasms Enables Genotype-Phenotype Mapping. Cell (2020) 183(5):1420–35.e2. doi: 10.1016/j.cell.2020.10.023
15. Van De Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell (2015) 161:933–45. doi: 10.1016/j.cell.2015.03.053
16. Boj SF, Il HC, LA B, Chio IIC, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell (2015) 160:324–38. doi: 10.1016/j.cell.2014.12.021
17. Kopper O, de Witte CJ, Lõhmussaar K, Valle-Inclan JE, Hami N, Kester L, et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat Med (2019) 25:838–49. doi: 10.1038/s41591-019-0422-6
18. Bartfeld S, Bayram T, Van De Wetering M, Huch M, Begthel H, Kujala P, et al. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology (2015) 148:126–36. doi: 10.1053/j.gastro.2014.09.042
19. Hafner M, Niepel M, Chung M, Sorger PK. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat Methods (2016) 13:521–7. doi: 10.1038/nmeth.3853
20. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv (2013) 1303.3997.
21. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr Protoc Bioinforma (2013) 43(1110):11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43
22. Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, et al. Sequenza: Allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol (2015) 26:64–70. doi: 10.1093/annonc/mdu479
23. Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) (2012) 6:80–92. doi: 10.4161/fly.19695
24. Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front Genet (2012) 3:1–9. doi: 10.3389/fgene.2012.00035
25. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagnostics (2015) 17:251–64. doi: 10.1016/j.jmoldx.2014.12.006
26. Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature (2012) 489:57–74. doi: 10.1038/nature11247
27. Shida T, Kishimoto T, Furuya M, Nikaido T, Koda K, Takano S, et al. Expression of an activated mammalian target of rapamycin (mTOR) in gastroenteropancreatic neuroendocrine tumors. Cancer Chemother Pharmacol (2010) 65:889–93. doi: 10.1007/s00280-009-1094-6
28. Rickman DS, Beltran H, Demichelis F, Rubin MA. Biology and evolution of poorly differentiated neuroendocrine tumors. Nat Med (2017) 23:664–73. doi: 10.1038/nm.4341
29. Cirkel GA, Weeber F, Bins S, Gadellaa-van Hooijdonk CGM, van Werkhoven E, Willems SM, et al. The time to progression ratio: a new individualized volumetric parameter for the early detection of clinical benefit of targeted therapies. Ann Oncol Off J Eur Soc Med Oncol (2016) 27:1638–43. doi: 10.1093/annonc/mdw223
30. de Melo AC, Grazziotin-Reisner R, Erlich F, Fontes Dias MS, Moralez G, Carneiro M, et al. A phase I study of mTOR inhibitor everolimus in association with cisplatin and radiotherapy for the treatment of locally advanced cervix cancer: PHOENIX I. Cancer Chemother Pharmacol (2016) 78:101–9. doi: 10.1007/s00280-016-3064-0
31. Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández-Mateos J, Khan K, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Sci (80- ) (2018) 359:920–6. doi: 10.1126/science.aao2774
32. Ooft SN, Weeber F, Dijkstra KK, McLean CM, Kaing S, van Werkhoven E, et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci Transl Med (2019) 11:1–9. doi: 10.1126/scitranslmed.aay2574
33. Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discovery (2018) 8:1112–29. doi: 10.1158/2159-8290.CD-18-0349
34. Weeber F, van de Wetering M, Hoogstraat M, Dijkstra KK, Krijgsman O, Kuilman T, et al. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc Natl Acad Sci U S A (2015) 112:13308–11. doi: 10.1073/pnas.1516689112
35. Sachs N, Papaspyropoulos A, Zomer-van Ommen DD, Heo I, Böttinger L, Klay D, et al. Long-term expanding human airway organoids for disease modeling. EMBO J (2019) 38:1–20. doi: 10.15252/embj.2018100300
36. Dijkstra KK, Monkhorst K, Schipper LJ, Hartemink KJ, Smit EF, Kaing S, et al. Challenges in Establishing Pure Lung Cancer Organoids Limit Their Utility for Personalized Medicine. Cell Rep (2020) 31. doi: 10.1016/j.celrep.2020.107588
37. Drost J, Karthaus WR, Gao D, Driehuis E, Sawyers CL, Chen Y, et al. Organoid culture systems for prostate epithelial and cancer tissue. Nat Protoc (2016) 11:347–58. doi: 10.1038/nprot.2016.006
38. Furlan D, Cerutti R, Uccella S, La Rosa S, Rigoli E, Genasetti A, et al. Different Molecular Profiles Characterize Well-Differentiated Endocrine Tumors and Poorly Differentiated Endocrine Carcinomas of the Gastroenteropancreatic Tract. Clin Cancer Res (2004) 10:947–57. doi: 10.1158/1078-0432.CCR-1068-3
39. van Jaarsveld RH, Kops GJPL. Difference Makers: Chromosomal Instability versus Aneuploidy in Cancer. Trends Cancer (2016) 2:561–71. doi: 10.1016/j.trecan.2016.09.003
40. Girardi DM, Silva ACB, Rêgo JFM, Coudry RA, Riechelmann RP. Unraveling molecular pathways of poorly differentiated neuroendocrine carcinomas of the gastroenteropancreatic system: A systematic review. Cancer Treat Rev (2017) 56:28–35. doi: 10.1016/j.ctrv.2017.04.002
41. Klempner SJ, Gershenhorn B, Tran P, Lee TK, Erlander MG, Gowen K, et al. BRAFV600E mutations in high-grade colorectal neuroendocrine tumors may predict responsiveness to BRAF-MEK combination therapy. Cancer Discovery (2016) 6:594–600. doi: 10.1158/2159-8290.CD-15-1192
Keywords: gastroenteropancreatic neuroendocrine carcinoma (GEP-NEC), extrapulmonary neuroendocrine carcinoma, disease modeling, pre-clinical models, organoids
Citation: Dijkstra KK, van den Berg JG, Weeber F, van de Haar J, Velds A, Kaing S, Peters DDGC, Eskens FALM, de Groot D-JA, Tesselaar MET and Voest EE (2021) Patient-Derived Organoid Models of Human Neuroendocrine Carcinoma. Front. Endocrinol. 12:627819. doi: 10.3389/fendo.2021.627819
Received: 10 November 2020; Accepted: 26 January 2021;
Published: 11 March 2021.
Edited by:
Priya Dedhia, The Ohio State University, United StatesReviewed by:
Ilaria Marinoni, University of Bern, SwitzerlandCopyright © 2021 Dijkstra, van den Berg, Weeber, van de Haar, Velds, Kaing, Peters, Eskens, de Groot, Tesselaar and Voest. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emile E. Voest, ZS52b2VzdEBua2kubmw=
†Present address: Krijn K. Dijkstra, Cancer Evolution and Genome Instability Laboratory, The Francis Crick Institute, London, United Kingdom
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.