 Yunting Zhou1†
Yunting Zhou1† Huiying Wang1†Junming Zhou2
Huiying Wang1†Junming Zhou2 Shanhu Qiu3,4
Shanhu Qiu3,4 Tingting Cai1Huiqin Li1Ziyang Shen1Yun Hu1Bo Ding1Menghui Luo1
Tingting Cai1Huiqin Li1Ziyang Shen1Yun Hu1Bo Ding1Menghui Luo1 Rong Huang1Rengna Yan1Wei Xu5Cong He6
Rong Huang1Rengna Yan1Wei Xu5Cong He6 Yumin Zhang3
Yumin Zhang3 Fengfei Li1Zilin Sun3Jianhua Ma1*
Fengfei Li1Zilin Sun3Jianhua Ma1*- 1Department of Endocrinology, Nanjing First Hospital, Nanjing Medical University, Nanjing, China
- 2Department of Cadre Gastroenterology, Jinling Hospital, Medical School of Nanjing University, Nanjing, China
- 3Department of Endocrinology, Zhongda Hospital, Institute of Diabetes, School of Medicine, Southeast University, Nanjing, China
- 4Department of Endocrinology, Shenzhen People’s Hospital, The Second Clinical Medical College of Jinan University, The First Affiliated Hospital of Southern University of Science and Technology, Shenzhen, China
- 5Department of Endocrinology, Xuzhou Central Hospital, Xuzhou Institute of Medical Sciences, Xuzhou Clinical School of Nanjing Medical University, Xuzhou, China
- 6State Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing, China
Vitamin A (VA), which is stored in several forms in most tissues, is required to maintain metabolite homeostasis and other processes, including the visual cycle, energy balance, epithelial cell integrity, and infection resistance. In recent years, VA molecules, also known as retinoids, have been extensively explored and used in the treatment of skin disorders and immune-related tumors. To date, several observational and interventional studies have explored the relationship between VA status and the pathogenesis of diabetes. In particular, VA micronutrients have been shown to regulate pancreatic development, β-cell function, pancreatic innate immune responses, and pancreatic stellate cells phenotypes through multiple mechanisms. However, there are still many problems to be proven or resolved. In this review, we summarize and discuss recent and available evidence on VA biological metabolism in the pancreas. Analysis of the effects of VA on metabolism in the pancreas will contribute to our understanding of the supportive physiological roles of VA in pancreas protection.
Introduction
The prevalence of diabetes mellitus (DM) is increasing rapidly worldwide. DM is a multifactorial disease that is typically linked to genetic information, life style and environmental stimulus (1). Nutrition metabolism, particularly most micronutrients in the organism, is also altered, either as part of the cause or effect, during the development of DM.
Vitamin A (VA), an essential nutrient that is only obtained from the diet, contributes significantly to the global health crisis affecting resource-constrained countries (2). Recent studies on the pancreas have demonstrated that VA and its receptors are directly associated with glucose metabolism (3–7). However, our understanding of the role of VA in the pathophysiological mechanisms of pre-DM and DM is still evolving. Thus, in this review, we thoroughly reviewed and summarized data regarding the influence and mechanisms of VA on endocrine function in the developing pancreas and adult pancreas.
VA Storage in the Pancreas
VA is a term including a variety of unsaturated organic compounds, such as retinol, retinal, and retinoic acid. The predominant VA in serum is retinol, which is derived from the carotenoid, β-carotene, or from pro-VA. In addition to VA in the circulatory system, hepatic stellate cells (HSCs) account for 80% of VA storage in the body and are responsible for VA metabolic responses in target tissues (8, 9). HSCs wrap its extended tentacles around the small blood vessels formed by hepatic sinusoidal endothelial cells and exhibit a remarkable capacity for regulation of cellular contraction and blood flow (10, 11). Available evidence indicates that hepatic endothelial cells can maintain the resting state of HSCs by producing nitric oxide (12, 13). Vascular disorder caused by liver injury in which nitric oxide synthase (endothelial Nitric Oxide Synthase, eNOS) activity is weakened, can effectively promote HSC activation with concomitant disappearance of the VA-storing lipid droplets (14). Activated HSCs in turn exacerbate endothelial dysfunction, the formation of this vicious circle promotes the development of liver fibrosis (15, 16). Therefore, the interaction between hepatic endothelial cells and HSCs may affect the storage, transport, and usage of VA.
Most retinoids are stored in the liver; however, this is not the only organ involved in retinoid storage. In cells, retinol can be bound to intracellular retinol binding proteins (CRBPs), among which CRBP1 is the most abundant and widely distributed (17). Several studies have shown that specific transport proteins for retinol (RBP4) in the serum (18) and cells (19) are distributed peripherally in a circular pattern within the pancreatic islets, and their anatomical locations resemble those of α cells. The presence of these retinoid-specific transport proteins in pancreatic islets suggests that retinoids and their related proteins may be involved in the metabolism of islets, supporting the endocrine functions of islets through various mechanisms (20). VA metabolic and signaling systems in cells were shown in Figure 1.
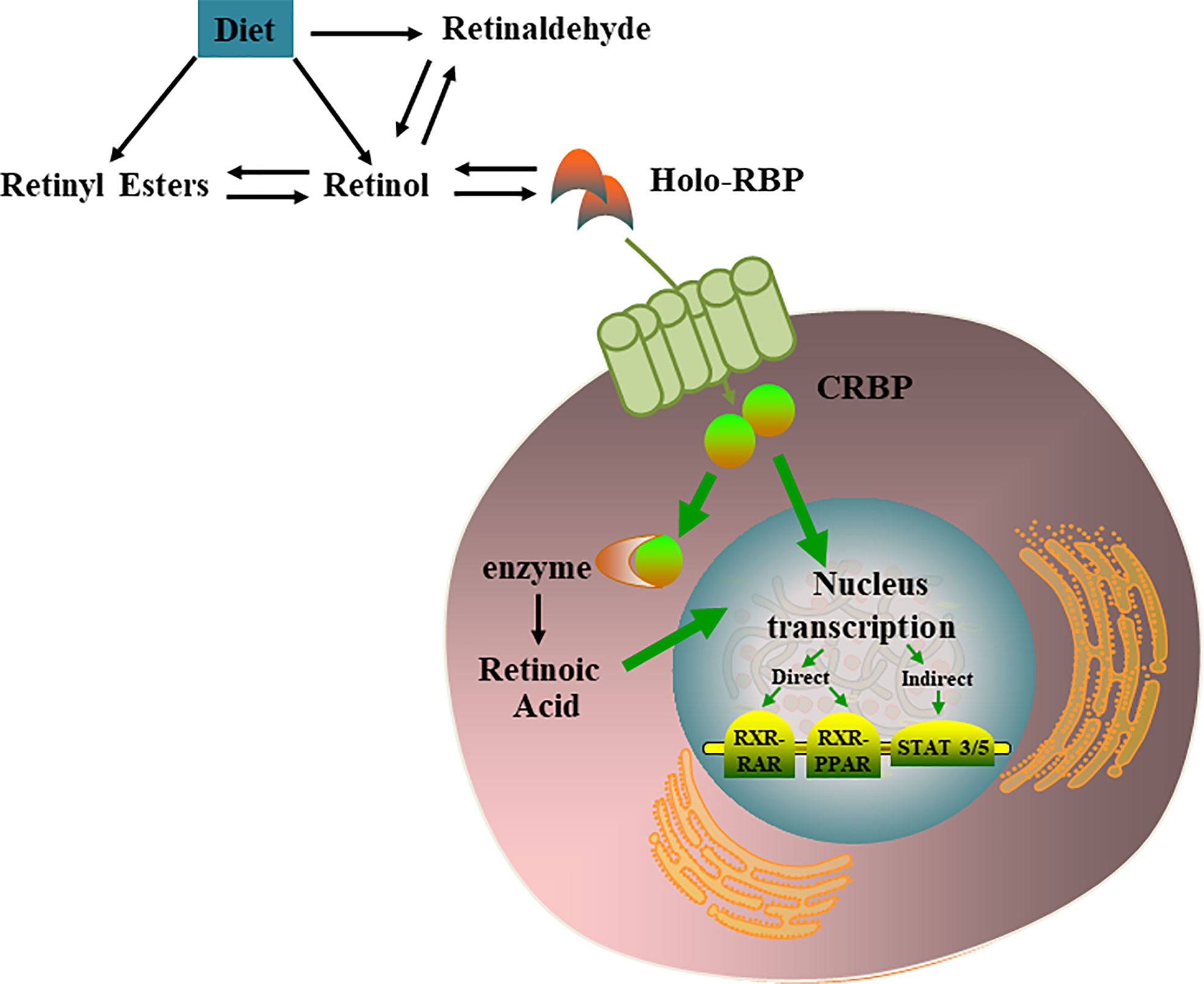
Figure 1 VA metabolic and signaling systems in cells. Retinol, retinal, and retinoic acid are three derivatives of VA. Each molecule has a cis and trans configuration, and the most active form is retinol. Retinol has 6 biologically active isoforms: all-trans, 11-cis, 13-cis, 9,13-di-cis, 9-cis, and 11,13-di-cis, with all-trans being the predominant form [21]. In cells, retinol can be converted to RA which regulates multiple nucleus transcription by activating the RXR-RAR, RXR-PPAR, STAT 3/5.
Pancreatic stellate cells (PSCs) exhibit VA-specific blue fluorescence (22) and are the only cell type enriched in droplets containing retinoid in human, rat, and mouse pancreas tissues (23–25). Under physiological conditions, quiescent PSCs are abundant in droplets of retinoids composed of retinyl palmitate. However, the specific roles of retinoids in quiescent PSCs have not yet been fully established. The results from in vitro studies have shown that all-trans retinoic acid (AT-RA) can promote the quiescent phenotype in cultured PSCs by inhibiting the activation of α-smooth muscle actin (α-SMA) and decreasing the expression of collagen synthesis (26–28). Zhou et al. (29) found that prolonged VA deficiency (VAD) alters the phenotype of resting islet stellate cells (ISCs, the subset of PSCs) compared with that of myofibroblast-like cells with increased α-SMA expression. Moreover, reintroduction of dietary VA to VA-deficient mice restores endocrine hormone profiles and induced ISCs/PSCs to become “re-quiescent,” similar to the results observed following induction of the VA-sufficient (VAS)-controlled ISCs/PSCs phenotype. However, Trasino et al. (5) detected a decrease in the CRBP1-positive PSC population in VA-deficient mice, but did not observe increased expression of α-SMA in PSCs. There is still no thorough experimental evidence supporting for the relationship between retinoid loss and PSC activation. Nevertheless, these seemingly contradictory studies have suggested that intracellular retinoid storage in the pancreas may be a key indicator for maintaining pancreatic function by accelerating or preventing PSC activation in vivo.
VA Status in Patients With Diabetes
In the context of type 1 diabetes patients (T1D), Basu et al. and Krempf et al. (30, 31) found that serum VA concentrations were significantly decreased in patients with impaired glucose tolerance (IGT) compared with those in normal individuals. In another study, serum VA levels were also decreased in young patients with T1D (32). Moreover, serum VA levels have been shown to be elevated in patients with T2D or pre-T2D, such as those with obesity and IGT (31–34). As a member of the lipocalin family of proteins, RBP4 functions together with transthyretin to transport retinol from the liver to peripheral tissues by binding to specific cell receptors (35). High serum RBP4 levels have been found to be positively associated with T2D and obesity in many human studies (36, 37). A recent meta-analysis also showed that increased RBP4 is a modest independent risk factor for women with gestational diabetes (GDM), similar to the results of case-control studies (38–41).
The Protective Effects of VA on the Pancreas
Effects of VA on Pancreas Development
By controlling cell specification and differentiation, VA-derived RA signals, such as the retinoid receptors, retinoic acid receptors (RAR) and retinoid X receptors (RXR), are essential for pancreatic β-cell development in the underlying endoderm (7, 42–44). RA signals imitate the directional indicator signal of the lateral mesoderm by regulating the expression of a series of growth factors and participate in the differentiation of uncommitted progenitor cells toward a pancreatic fate (45, 46). More importantly, RA can promote the formation of pancreatic duodenal homeobox-1 (Pdx1) foregut endoderm, which co-expresses pancreas transcription factor 1α (Ptf1α), a transcription factor indicative of pancreatic commitment (47). At the expense of the exocrine dorsal pancreas, Notch signaling controls early pancreatic differentiation through neurogenin 3 (Ngn3) repression, whereas RA promotes endocrine correlation with specific inhibition of Notch signaling activities (48). In vitro, RA also has important roles in chemical introduction protocols for induction of embryonic stem cells to differentiate into insulin-producing cells (47, 49, 50). Programming of ectodermal explants from Xenopus blastulae with a mixture containing RA is sufficient to drive pancreatic gene expression. The proportion of pancreatic tissue formed in such programmed explants is related to the RA concentration (51). In addition to the specific differentiation-promoting effects of RA on endocrine cells induced by stem/progenitor cell, RA can also reprogram cells to another cell type with or without reversion to pluripotent stem cells (52–54). Centroacinar cells were transdifferentiated into functional β-cells by regeneration after treatment with RA (53).
The interactions of mesenchymal-epithelial cells are necessary for proper maturation of tissues (55–57). Studies have indicated that the pancreatic mesenchyme not only influences the expansion of early pancreatic progenitors but also regulates the proliferation of terminally differentiated endocrine cells during the final phase of gestation (57). Moreover, PSCs are important mesenchymal supporting cells that can maintain the normal basement membrane to stabilize the cell cytoskeleton and structure, thereby protecting the normal function of parenchymal cells (58). Chen et al. (59) found that human fetal PSCs lost intracellular retinoid-storing lipid droplets and expressed specific activated stellate markers, α-SMA, and extracellular matrix (ECM) proteins as the cultures going on in vitro (e.g., collagen I, collagen IV, and fibronectin). The crosstalk between multiple integrins (β1, α3 and α5) and collagen I is essential for the cell adhesion, migration, proliferation, and growth factor production in human fetal PSCs, suggesting that human fetal PSCs may effectively regulate the ECM microenvironment required for pancreatic development. These findings initially elucidated the role of PSCs in pancreas specification induced by RA.
Involvement of VA in Glucose Homeostasis
In a study of insulin secretion, Chertow et al. (60) found that VA-deficient puppies born from mothers with mild VAD exhibited hyperglycemia and reduced glucose-stimulated insulin secretion. Dietary VA administered in the form of RA restored euglycemia and normalized islet insulin secretion. Both dietary VAD and decreased endogenous production of RA by genetic intervention blocked RA signals in mice, leading to reductions in fasting blood glucose levels and hepatic gluconeogenesis (61, 62). Mice lacking the RA-synthesizing enzyme aldehyde dehydrogenase-1 (ALDH-1) showed lower expression levels of the key gluconeogenic enzymes, glucose-6-phosphatase and phosphoenolpyruvate carboxykinase, the latter of which is an RA-inducible target gene containing a specific RA-receptor binding site as an RA response element (61). Other findings indicated an additional mechanism through which VA affects islet function by governing islets size distribution was correlated with the α-SMA-positive ISC pool in a mouse model of dietary VAD (29).
In studies of insulin responsiveness, RBP4 has attracted much research interest in the last decade owing to its effects on insulin resistance. Basic mechanistic studies have shown that elevated serum levels of RBP4 can induce the expression of phosphoenolpyruvate carboxykinase, the key gluconeogenesis-related enzyme expressed in the liver, and further increase circulating blood glucose levels via increased hepatic glucose production (63, 64). Other studies focusing on target organs have shown that overexpression of adipocyte-specific RBP4 increases the levels of pro-inflammatory markers and lipases associated with lipolysis, thereby promoting insulin resistance (65). Retinol-RBP4 complex is recognized by stimulated by retinoic acid 6 (STRA6), which transports retinol from the binding protein into cells (66, 67). STRA6 can effectively weaken the insulin response because STRA6-mediated retinol transport induces receptor phosphorylation, which in turn activates the Janus kinases 2/signal transducer and activator of signal transducers and activators of transcription 3/5 (STAT3/5) activation cascade, which contributes to the expression of the STAT target gene suppressor of cytokine signaling (66, 68). Additionally, mice lacking ALDH-1 are protected from high-fat diet-induced insulin resistance, potentially because retinaldehyde can increase the expression of mitochondrial uncoupling protein 1 to drive uncoupled respiration and adaptive thermogenesis in white adipose tissue, thereby promoting the development of the brown fat phenotype, increasing energy expenditure, and suppressing body weight increases. This may also be a compensatory protection mechanism for the body. RA is the ligand of peroxisome proliferator-activated receptor δ (PPARδ) and classical RAR. RA supplementation in obese mice leads to the upregulation of PPARδ and consequent ectopic lipid deposition. Therefore, PPARδ affects lipid and glucose homeostasis, thereby enhancing the expression of insulin signaling-related genes and reducing insulin intolerance (69). Furthermore, as retinoic transcription nuclear receptors, RARβ2 agonists also dramatically reduce lipid peroxidation and oxidative stress in the pancreas of both obese and diabetic mice. This suggests that RARβ2 agonists may be useful drugs for T2D therapy and for the treatment of hepatic steatosis, which may contribute to insulin sensitivity (70).
Effects of VA on Pancreatic Innate Immune Responses
VA and its derivatives regulate adaptive and innate immune responses through different mechanisms (71, 72). High VA levels can block the Th1 response and promote the Th2 response (73). According to studies on the effects of RA on monocytes/macrophages (74–77), RA not only suppresses the secretion of cytokines produced by Th1-type cells but also increases the secretion of cytokines produced by Th2-type cells (78, 79). Dalmas et al. (80) found that dendritic cells are endogenous RA producers in pancreatic islets. Dendritic cells in islets showed reduced ALDH activity in macrophages of interleukin (IL)-33–treated VA-deficient mice compared with mice fed a chow diet, indicating that IL-33–induced enhancement of β-cell function required VA and its conversion to RA. A similar study showed that VA exerted autoimmune protective effects in part by inhibiting CD4+CD8+ interferon (IFN)-γ-producing T cells, but had no effect on the IL-17–producing T-cell population (73, 81–83). Zunino et al. (84) demonstrated that intervention with VA dietary supplements protected against the development of T1D in mice by efficiently inhibiting the infiltration of T cells into the islets, thereby precluding the progression of insulitis and diabetes. A study by Van et al. (85) reported that ATRA-treated mice had fewer pancreatic islets and a reduced incidence of pre-insulitis, even after cell transfer with CD4+CD25+ cells, whereas mice from control group developed severe destructive insulitis. Overall, VA may have applications in the treatment of autoimmune inflammatory phenotypes to reduce the formation of autoimmune diseases, such as T1D (78, 85–88).
GDM and T2D exhibit various features associated with metabolic syndrome (89), such as obesity and low-grade inflammation (90–92). Immunologic-metabolic crosstalk also plays a role in the regulation of metabolic imbalances, which affect the immune system and obesity-associated inflammation (93). Few studies have focused on the effects of VA on the immune system in GDM and T2D. However, these data based on immunology-VA crosstalk provided us with insights into the metabolic imbalances driving GDM and T2D pathogenesis.
Effects of VA on the PSC Phenotype
PSC activation is thought to be a key cellular event for pancreatic fibrosis in the pathological processes of serious pancreatic diseases (94). The effects of VA and its analogs on PSC activation have been reported in several studies. A treatment medium containing retinoids from activated PSCs causes phenotypic reversal to the quiescent phenotype (26–28, 95, 96). Transition of quiescent PSCs to an activated myofibroblastic phenotype is marked by profound cytoskeletal changes and elevated actomyosin contractility (97, 98). Chronopoulos et al. (27) found that ATRA impairs the capacity of PSCs to remodel the ECM to promote cancer invasion. ATRA-treated PSCs showed a marked decrease in the overall traction force generation during the early and late stages of the spreading phase and had a severely reduced ability to deform the collagen matrigel matrix, thereby confirming that ATRA treatment inhibits force generation in PSCs. Thus, ATRA treatment affected the ability of PSCs to sense extracellular mechanical cues and induces cytoskeletal changes consistent with a resting-like phenotype. Zhou et al. (29) found CRBP1 knockdown restored the polygonal appearance of quiescent ISCs, and reduced the expression of activation-related proteins, such as α-SMA and collagen synthesis, thereby producing a resting-state phenotype. Maintaining ISCs being quiescent state enhanced glucose-induced insulin release and basal insulin secretion. Thus, regulation of VA metabolism-related molecules is required to maintain a quiescent ISC population and block islet fibrosis and exocrine pancreatitis. She et al. (99) found that overexpression of sterol regulatory element-binding protein-1c in activated HSCs, which have many biological features in common with PSCs, induces a drastic reversal of the cell phenotype to quiescent HSCs. Resting HSCs contain sufficient triglycerides (100); therefore, they can be used as a source of fatty acids for esterification of retinol.
Interestingly, our group previously reviewed that PSCs share similar biological phenotypes with “universal” pancreatic stem/progenitor cells; for example, PSCs share localization, stem cell markers, signaling pathways, and multi-potential differentiation abilities with pancreatic stem/progenitor cells (101) and have therefore been proposed as a new cell type of potential adult pancreatic stem/progenitor cells. However, further studies are still needed to determine whether and how RA signals suppress the capacity of the molecule to mediate the differentiation of PSCs into pancreatic endocrine cells. The effects of VA metabolism on pancreas were shown in Figure 2.
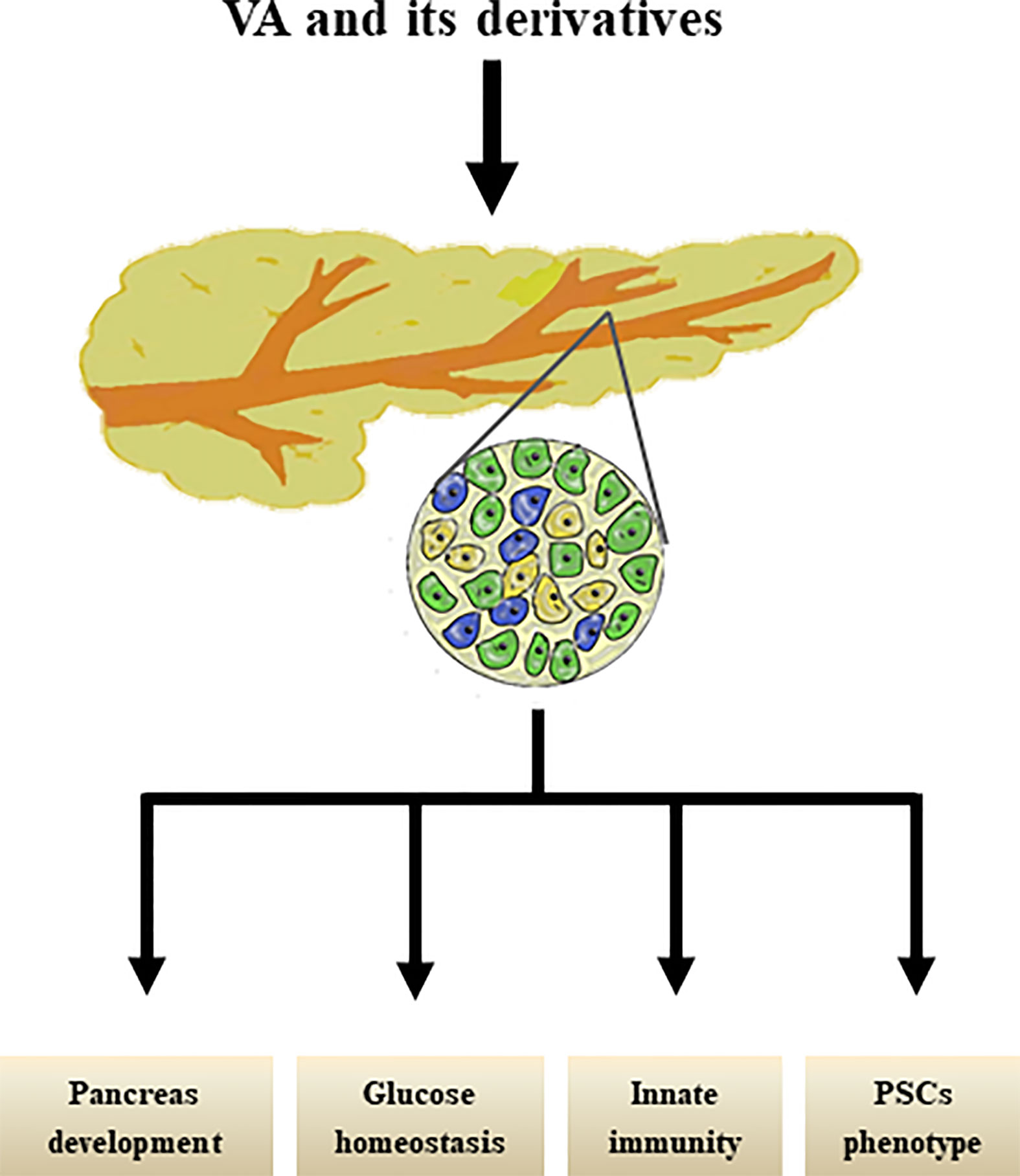
Figure 2 Effects of VA and its derivatives on pancreas. VA and its derivatives are reported to promote pancreas development, maintain glucose homestasis, regulate pancreatic innate immunity, and transform PSCs phenotype.
Conclusion
Based on current evidence, VA status is relevant in the pathogenesis of human DM and in the physiological processes of pancreatic development, β-cell function, pancreatic innate immune responses, and PSC phenotype. Further studies are needed to elucidate all of the physiological functions of RA, retinol, and their metabolites and to identify the mechanisms mediating the unique effects of VA on target cells and gene production.
Data Availability Statement
All data sets generated for this study are included in the manuscript.
Author Contributions
YtZ and HW conceived and wrote the manuscript. JZ, TC, ZyS, YH, HL, and BD collected articles. RY, RH, and ML analyzed the data. WX, CH, and FL reviewed articles. YtZ and SQ drew the figures. YmZ modified the manuscript. ZS and JM directed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Nature Science Foundation of China (NSFC-81870563).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Roden M, Shulman GI. The integrative biology of type 2 diabetes. Nature (2019) 576(7785):51–60. doi: 10.1038/s41586-019-1797-8
2. WHO. Global prevalence of vitamin A deficiency in populations at risk 1995–2005.pdf Vol. 2009. Geneva: WHO Library Cataloguing-inPublication Data (2009).
3. Trasino SE, Gudas LJ. Vitamin A: a missing link in diabetes? Diabetes Manag (Lond) (2015) 5(5):359–67. doi: 10.2217/dmt.15.30
4. Yoneda A, Sakai-Sawada K, Niitsu Y, Tamura Y. Vitamin A and insulin are required for the maintenance of hepatic stellate cell quiescence. Exp Cell Res (2016) 341(1):8–17. doi: 10.1016/j.yexcr.2016.01.012
5. Trasino SE, Benoit YD, Gudas LJ. Vitamin A deficiency causes hyperglycemia and loss of pancreatic beta-cell mass. J Biol Chem (2015) 290(3):1456–73. doi: 10.1074/jbc.M114.616763
6. Brossaud J, Pallet V, Corcuff JB. Vitamin A, endocrine tissues and hormones: interplay and interactions. Endocr Connect (2017) 6(7):R121–30. doi: 10.1530/EC-17-0101
7. Brun PJ, Grijalva A, Rausch R, Watson E, Yuen JJ, Das BC, et al. Retinoic acid receptor signaling is required to maintain glucose-stimulated insulin secretion and beta-cell mass. FASEB J (2015) 29(2):671–83. doi: 10.1096/fj.14-256743
8. Blaner WS, Hendriks HF, Brouwer A, de Leeuw AM, Knook DL, Goodman DS. Retinoids, retinoid-binding proteins, and retinyl palmitate hydrolase distributions in different types of rat liver cells. J Lipid Res (1985) 26(10):1241–51. doi: 10.1016/S0022-2275(20)34272-3
9. Hendriks HF, Verhoofstad WA, Brouwer A, de Leeuw AM, Knook DL. Perisinusoidal fat-storing cells are the main vitamin A storage sites in rat liver. Exp Cell Res (1985) 160(1):138–49. doi: 10.1016/0014-4827(85)90243-5
10. Iwakiri Y, Shah V, Rockey DC. Vascular pathobiology in chronic liver disease and cirrhosis - current status and future directions. J Hepatol (2014) 61(4):912–24. doi: 10.1016/j.jhep.2014.05.047
11. Rockey DC. Hepatic blood flow regulation by stellate cells in normal and injured liver. Semin Liver Dis (2001) 21(3):337–49. doi: 10.1055/s-2001-17551
12. Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology (2008) 48(3):920–30. doi: 10.1002/hep.22351
13. Langer DA, Das A, Semela D, Kang-Decker N, Hendrickson H, Bronk SF, et al. Nitric oxide promotes caspase-independent hepatic stellate cell apoptosis through the generation of reactive oxygen species. Hepatology (2008) 47(6):1983–93. doi: 10.1002/hep.22285
14. Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature (2014) 505(7481):97–102. doi: 10.1038/nature12681
15. Das A, Shergill U, Thakur L, Sinha S, Urrutia R, Mukhopadhyay D, et al. Ephrin B2/EphB4 pathway in hepatic stellate cells stimulates Erk-dependent VEGF production and sinusoidal endothelial cell recruitment. Am J Physiol Gastrointest Liver Physiol (2010) 298(6):G908–15. doi: 10.1152/ajpgi.00510.2009
16. Shimada H, Staten NR, Rajagopalan LE. TGF-beta1 mediated activation of Rho kinase induces TGF-beta2 and endothelin-1 expression in human hepatic stellate cells. J Hepatol (2011) 54(3):521–8. doi: 10.1016/j.jhep.2010.07.026
17. Brun PJ, Wongsiriroj N, Blaner WS. Retinoids in the pancreas. Hepatobiliary Surg Nutr (2016) 5(1):1–14. doi: 10.3978/j.issn.2304-3881.2015.09.03
18. Li Y, Wongsiriroj N, Blaner WS. The multifaceted nature of retinoid transport and metabolism. Hepatobiliary Surg Nutr (2014) 3(3):126–39. doi: 10.3978/j.issn.2304-3881.2014.05.04
19. Napoli JL. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim Biophys Acta (2012) 1821(1):152–67. doi: 10.1016/j.bbalip.2011.05.004
20. Kato M, Kato K, Blaner WS, Chertow BS, Goodman DS. Plasma and cellular retinoid-binding proteins and transthyretin (prealbumin) are all localized in the islets of Langerhans in the rat. Proc Natl Acad Sci U S A (1985) 82(8):2488–92. doi: 10.1073/pnas.82.8.2488
21. Rhee EJ, Plutzky J. Retinoid metabolism and diabetes mellitus. Diabetes Metab J (2012) 36(3):167–80. doi: 10.4093/dmj.2012.36.3.167
22. Watari N, Hotta Y, Mabuchi Y. Morphological studies on a vitamin A-storing cell and its complex with macrophage observed in mouse pancreatic tissues following excess vitamin A administration. Okajimas Folia Anat Jpn (1982) 58(4-6):837–58. doi: 10.2535/ofaj1936.58.4-6_837
23. Ikejiri N. The vitamin A-storing cells in the human and rat pancreas. Kurume Med J (1990) 37(2):67–81. doi: 10.2739/kurumemedj.37.67
24. Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut (1998) 43(1):128–33. doi: 10.1136/gut.43.1.128
25. Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology (1998) 115(2):421–32. doi: 10.1016/S0016-5085(98)70209-4
26. McCarroll JA, Phillips PA, Santucci N, Pirola RC, Wilson JS, Apte MV. Vitamin A inhibits pancreatic stellate cell activation: implications for treatment of pancreatic fibrosis. Gut (2006) 55(1):79–89. doi: 10.1136/gut.2005.064543
27. Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D, et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun (2016) 7:12630. doi: 10.1038/ncomms12630
28. Sarper M, Cortes E, Lieberthal TJ, Del Rio Hernandez A. ATRA modulates mechanical activation of TGF-beta by pancreatic stellate cells. Sci Rep (2016) 6:27639. doi: 10.1038/srep27639
29. Zhou Y, Zhou J, Sun B, Xu W, Zhong M, Li Y, et al. Vitamin A deficiency causes islet dysfunction by inducing islet stellate cell activation via cellular retinol binding protein 1. Int J Biol Sci (2020) 16(6):947–56. doi: 10.7150/ijbs.37861
30. Basu TK, Tze WJ, Leichter J. Serum vitamin A and retinol-binding protein in patients with insulin-dependent diabetes mellitus. Am J Clin Nutr (1989) 50(2):329–31. doi: 10.1093/ajcn/50.2.329
31. Krempf M, Ranganathan S, Ritz P, Morin M, Charbonnel B. Plasma vitamin A and E in type 1 (insulin-dependent) and type 2 (non-insulin-dependent) adult diabetic patients. Int J Vitam Nutr Res (1991) 61(1):38–42. doi: 10.1007/BF00266349
32. Tavridou A, Unwin NC, Laker MF, White M, Alberti KG. Serum concentrations of vitamins A and E in impaired glucose tolerance. Clin Chim Acta (1997) 266(2):129–40. doi: 10.1016/S0009-8981(97)00123-X
33. Basualdo CG, Wein EE, Basu TK. Vitamin A (retinol) status of first nation adults with non-insulin-dependent diabetes mellitus. J Am Coll Nutr (1997) 16(1):39–45. doi: 10.1080/07315724.1997.10718647
34. Kim M, Jee SH, Kim M, Yoo HJ, Kang M, Kim J, et al. Serum vitamin A-related metabolite levels are associated with incidence of type 2 diabetes. Diabetes Metab (2017) 43(3):287–91. doi: 10.1016/j.diabet.2016.09.012
35. Flower DR. The lipocalin protein family: structure and function. Biochem J (1996) 318( Pt 1):1–14. doi: 10.1042/bj3180001
36. Olsen T, Blomhoff R. Retinol, Retinoic Acid, and Retinol-Binding Protein 4 are Differentially Associated with Cardiovascular Disease, Type 2 Diabetes, and Obesity: An Overview of Human Studies. Adv Nutr (2020) 11(3):644–66. doi: 10.1093/advances/nmz131
37. Fan J, Yin S, Lin D, Liu Y, Chen N, Bai X, et al. Association of Serum Retinol-Binding Protein 4 Levels and the Risk of Incident Type 2 Diabetes in Subjects With Prediabetes. Diabetes Care (2019) 42(8):1574–81. doi: 10.2337/dc19-0265
38. Hu S, Liu Q, Huang X, Tan H. Serum level and polymorphisms of retinol-binding protein-4 and risk for gestational diabetes mellitus: a meta-analysis. BMC Pregnancy Childbirth (2016) 16:52. doi: 10.1186/s12884-016-0838-7
39. Jin C, Lin L, Han N, Zhao Z, Liu Z, Luo S, et al. Plasma retinol-binding protein 4 in the first and second trimester and risk of gestational diabetes mellitus in Chinese women: a nested case-control study. Nutr Metab (Lond) (2020) 17:1. doi: 10.1186/s12986-019-0425-9
40. Zhaoxia L, Mengkai D, Qin F, Danqing C. Significance of RBP4 in patients with gestational diabetes mellitus: a case-control study of Han Chinese women. Gynecol Endocrinol (2014) 30(2):161–4. doi: 10.3109/09513590.2013.871515
41. Chen Y, Lv P, Du M, Liang Z, Zhou M, Chen D. Increased retinol-free RBP4 contributes to insulin resistance in gestational diabetes mellitus. Arch Gynecol Obstet (2017) 296(1):53–61. doi: 10.1007/s00404-017-4378-9
42. Guo T, Hebrok M. Stem cells to pancreatic beta-cells: new sources for diabetes cell therapy. Endocr Rev (2009) 30(3):214–27. doi: 10.1210/er.2009-0004
43. Jun HS. In Vivo Regeneration of Insulin-Producing beta-Cells. Islets Langerhans (2010) 654:627–40. doi: 10.1007/978-90-481-3271-3_27
44. Perez RJ, Benoit YD, Gudas LJ. Deletion of retinoic acid receptor beta (RARbeta) impairs pancreatic endocrine differentiation. Exp Cell Res (2013) 319(14):2196–204. doi: 10.1016/j.yexcr.2013.05.032
45. Al Tanoury Z, Piskunov A, Rochette-Egly C. Vitamin A and retinoid signaling: genomic and nongenomic effects. J Lipid Res (2013) 54(7):1761–75. doi: 10.1194/jlr.R030833
46. Kedishvili NY. Enzymology of retinoic acid biosynthesis and degradation. J Lipid Res (2013) 54(7):1744–60. doi: 10.1194/jlr.R037028
47. Micallef SJ, Janes ME, Knezevic K, Davis RP, Elefanty AG, Stanley EG. Retinoic acid induces Pdx1-positive endoderm in differentiating mouse embryonic stem cells. Diabetes (2005) 54(2):301–5. doi: 10.2337/diabetes.54.2.301
48. Chen Y, Pan FC, Brandes N, Afelik S, Solter M, Pieler T. Retinoic acid signaling is essential for pancreas development and promotes endocrine at the expense of exocrine cell differentiation in Xenopus. Dev Biol (2004) 271(1):144–60. doi: 10.1016/j.ydbio.2004.03.030
49. Martin M, Gallego-Llamas J, Ribes V, Kedinger M, Niederreither K, Chambon P, et al. Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Dev Biol (2005) 284(2):399–411. doi: 10.1016/j.ydbio.2005.05.035
50. Ostrom M, Loffler KA, Edfalk S, Selander L, Dahl U, Ricordi C, et al. Retinoic acid promotes the generation of pancreatic endocrine progenitor cells and their further differentiation into beta-cells. PloS One (2008) 3(7):e2841. doi: 10.1371/journal.pone.0002841
51. Gere-Becker MB, Pommerenke C, Lingner T, Pieler T. Retinoic acid-induced expression of Hnf1b and Fzd4 is required for pancreas development in Xenopus laevis. Development (2018) 145(12). doi: 10.1242/dev.161372
52. Obinata A, Osakabe K, Yamaguchi M, Morimoto R, Akimoto Y. Tgm2/Gh, Gbx1 and TGF-beta are involved in retinoic acid-induced transdifferentiation from epidermis to mucosal epithelium. Int J Dev Biol (2011) 55(10-12):933–43. doi: 10.1387/ijdb.113326ao
53. Huang W, Beer RL, Delaspre F, Wang G, Edelman HE, Park H, et al. Sox9b is a mediator of retinoic acid signaling restricting endocrine progenitor differentiation. Dev Biol (2016) 418(1):28–39. doi: 10.1016/j.ydbio.2016.08.019
54. Lorberbaum DS, Kishore S, Rosselot C, Sarbaugh D, Brooks EP, Aragon E, et al. Retinoic acid signaling within pancreatic endocrine progenitors regulates mouse and human beta cell specification. Development (2020) 147(12). doi: 10.1242/dev.189977
55. Cunha GR. Mesenchymal-epithelial interactions: past, present, and future. Differentiation (2008) 76(6):578–86. doi: 10.1111/j.1432-0436.2008.00290.x
56. Le Guen L, Marchal S, Faure S, de Santa Barbara P. Mesenchymal-epithelial interactions during digestive tract development and epithelial stem cell regeneration. Cell Mol Life Sci (2015) 72(20):3883–96. doi: 10.1007/s00018-015-1975-2
57. Cozzitorto C, Mueller L, Ruzittu S, Mah N, Willnow D, Darrigrand JF, et al. A Specialized Niche in the Pancreatic Microenvironment Promotes Endocrine Differentiation. Dev Cell (2020) 55(2). doi: 10.1016/j.devcel.2020.08.003
58. Means AL. Pancreatic stellate cells: small cells with a big role in tissue homeostasis. Lab Invest (2013) 93(1):4–7. doi: 10.1038/labinvest.2012.161
59. Chen B, Li J, Fellows GF, Sun Z, Wang R. Maintaining human fetal pancreatic stellate cell function and proliferation require beta1 integrin and collagen I matrix interactions. Oncotarget (2015) 6(16):14045–59. doi: 10.18632/oncotarget.4338
60. Chertow BS, Blaner WS, Baranetsky NG, Sivitz WI, Cordle MB, Thompson D, et al. Effects of vitamin A deficiency and repletion on rat insulin secretion in vivo and in vitro from isolated islets. J Clin Invest (1987) 79(1):163–9. doi: 10.1172/JCI112778
61. Shin DJ, Odom DP, Scribner KB, Ghoshal S, McGrane MM. Retinoid regulation of the phosphoenolpyruvate carboxykinase gene in liver. Mol Cell Endocrinol (2002) 195(1-2):39–54. doi: 10.1016/S0303-7207(02)00215-0
62. Kiefer FW, Orasanu G, Nallamshetty S, Brown JD, Wang H, Luger P, et al. Retinaldehyde dehydrogenase 1 coordinates hepatic gluconeogenesis and lipid metabolism. Endocrinology (2012) 153(7):3089–99. doi: 10.1210/en.2011-2104
63. Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature (2005) 436(7049):356–62. doi: 10.1038/nature03711
64. Wolf G. Serum retinol-binding protein: a link between obesity, insulin resistance, and type 2 diabetes. Nutr Rev (2007) 65(5):251–6. doi: 10.1111/j.1753-4887.2007.tb00302.x
65. Lee SA, Yuen JJ, Jiang H, Kahn BB, Blaner WS. Adipocyte-specific overexpression of retinol-binding protein 4 causes hepatic steatosis in mice. Hepatology (2016) 64(5):1534–46. doi: 10.1002/hep.28659
66. Noy N. Vitamin A in regulation of insulin responsiveness: mini review. Proc Nutr Soc (2016) 75(2):212–5. doi: 10.1017/S0029665115004322
67. Chen G. Roles of Vitamin A Metabolism in the Development of Hepatic Insulin Resistance. ISRN Hepatol (2013) 2013:534972. doi: 10.1155/2013/534972
68. Berry DC, Jacobs H, Marwarha G, Gely-Pernot A, O’Byrne SM, DeSantis D, et al. The STRA6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J Biol Chem (2013) 288(34):24528–39. doi: 10.1074/jbc.M113.484014
69. Wolf G. Retinoic acid activation of peroxisome proliferation-activated receptor delta represses obesity and insulin resistance. Nutr Rev (2010) 68(1):67–70. doi: 10.1111/j.1753-4887.2009.00261.x
70. Trasino SE, Tang XH, Jessurun J, Gudas LJ. Retinoic acid receptor beta2 agonists restore glycaemic control in diabetes and reduce steatosis. Diabetes Obes Metab (2016) 18(2):142–51. doi: 10.1111/dom.12590
71. Surman SL, Penkert RR, Sealy RE, Jones BG, Marion TN, Vogel P, et al. Consequences of Vitamin A Deficiency: Immunoglobulin Dysregulation, Squamous Cell Metaplasia, Infectious Disease, and Death. Int J Mol Sci (2020) 21(15). doi: 10.3390/ijms21155570
72. Cantorna MT, Snyder L, Arora J. Vitamin A and vitamin D regulate the microbial complexity, barrier function, and the mucosal immune responses to ensure intestinal homeostasis. Crit Rev Biochem Mol Biol (2019) 54(2):184–92. doi: 10.1080/10409238.2019.1611734
73. Schambach F, Schupp M, Lazar MA, Reiner SL. Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur J Immunol (2007) 37(9):2396–9. doi: 10.1002/eji.200737621
74. Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci U S A (1980) 77(5):2936–40. doi: 10.1073/pnas.77.5.2936
75. Geissmann F, Revy P, Brousse N, Lepelletier Y, Folli C, Durandy A, et al. Retinoids regulate survival and antigen presentation by immature dendritic cells. J Exp Med (2003) 198(4):623–34. doi: 10.1084/jem.20030390
76. Jiang YJ, Xu TR, Lu B, Mymin D, Kroeger EA, Dembinski T, et al. Cyclooxygenase expression is elevated in retinoic acid-differentiated U937 cells. Biochim Biophys Acta (2003) 1633(1):51–60. doi: 10.1016/S1388-1981(03)00072-6
77. Mohty M, Morbelli S, Isnardon D, Sainty D, Arnoulet C, Gaugler B, et al. All-trans retinoic acid skews monocyte differentiation into interleukin-12-secreting dendritic-like cells. Br J Haematol (2003) 122(5):829–36. doi: 10.1046/j.1365-2141.2003.04489.x
78. Kinoshita K, Yoo BS, Nozaki Y, Sugiyama M, Ikoma S, Ohno M, et al. Retinoic acid reduces autoimmune renal injury and increases survival in NZB/W F1 mice. J Immunol (2003) 170(11):5793–8. doi: 10.4049/jimmunol.170.11.5793
79. Xu J, Storer PD, Chavis JA, Racke MK, Drew PD. Agonists for the peroxisome proliferator-activated receptor-alpha and the retinoid X receptor inhibit inflammatory responses of microglia. J Neurosci Res (2005) 81(3):403–11. doi: 10.1002/jnr.20518
80. Dalmas E, Lehmann FM, Dror E, Wueest S, Thienel C, Borsigova M, et al. Interleukin-33-Activated Islet-Resident Innate Lymphoid Cells Promote Insulin Secretion through Myeloid Cell Retinoic Acid Production. Immunity (2017) 47(5):928–42.e7. doi: 10.1016/j.immuni.2017.10.015
81. Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood (2008) 111(3):1013–20. doi: 10.1182/blood-2007-06-096438
82. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (2007) 317(5835):256–60. doi: 10.1126/science.1145697
83. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med (2007) 204(8):1775–85. doi: 10.1084/jem.20070602
84. Zunino SJ, Storms DH, Stephensen CB. Diets rich in polyphenols and vitamin A inhibit the development of type I autoimmune diabetes in nonobese diabetic mice. J Nutr (2007) 137(5):1216–21. doi: 10.1093/jn/137.5.1216
85. Van YH, Lee WH, Ortiz S, Lee MH, Qin HJ, Liu CP. All-trans retinoic acid inhibits type 1 diabetes by T regulatory (Treg)-dependent suppression of interferon-gamma-producing T-cells without affecting Th17 cells. Diabetes (2009) 58(1):146–55. doi: 10.2337/db08-1154
86. Driscoll HK, Chertow BS, Jelic TM, Baltaro RJ, Chandor SB, Walker EM, et al. Vitamin A status affects the development of diabetes and insulitis in BB rats. Metabolism (1996) 45(2):248–53. doi: 10.1016/S0026-0495(96)90062-1
87. Miyagawa N, Homma T, Kagechika H, Shudo K, Nagai H. Effect of synthetic retinoid, TAC-101, on experimental autoimmune disease. Pharmacology (2003) 67(1):21–31. doi: 10.1159/000066783
88. Osanai M, Nishikiori N, Murata M, Chiba H, Kojima T, Sawada N. Cellular retinoic acid bioavailability determines epithelial integrity: Role of retinoic acid receptor alpha agonists in colitis. Mol Pharmacol (2007) 71(1):250–8. doi: 10.1124/mol.106.029579
89. Noussitou P, Monbaron D, Vial Y, Gaillard RC, Ruiz J. Gestational diabetes mellitus and the risk of metabolic syndrome: a population-based study in Lausanne, Switzerland. Diabetes Metab (2005) 31(4 Pt 1):361–9. doi: 10.1016/S1262-3636(07)70205-7
90. Couch SC, Philipson EH, Bendel RB, Pujda LM, Lammi-Keefe CJ. Elevated lipoprotein lipids and gestational hormones in women with diet-treated gestational diabetes mellitus compared to healthy pregnant controls. J Diabetes Complications (1998) 12(1):1–9. doi: 10.1016/S1056-8727(97)00007-X
91. Koukkou E, Watts GF, Lowy C. Serum lipid, lipoprotein and apolipoprotein changes in gestational diabetes mellitus: a cross-sectional and prospective study. J Clin Pathol (1996) 49(8):634–7. doi: 10.1136/jcp.49.8.634
92. Bo S, Signorile A, Menato G, Gambino R, Bardelli C, Gallo ML, et al. C-reactive protein and tumor necrosis factor-alpha in gestational hyperglycemia. J Endocrinol Invest (2005) 28(9):779–86. doi: 10.1007/BF03347566
93. Prasad M, Chen EW, Toh SA, Gascoigne NRJ. Autoimmune responses and inflammation in type 2 diabetes. J Leukoc Biol (2020) 107(5):739–48. doi: 10.1002/JLB.3MR0220-243R
94. Apte MV, Pirola RC, Wilson JS. Pancreatic stellate cells: a starring role in normal and diseased pancreas. Front Physiol (2012) 3:344. doi: 10.3389/fphys.2012.00344
95. Froeling FE, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology (2011) 141(4):1486–97, 1497 e1-14. doi: 10.1053/j.gastro.2011.06.047
96. Jaster R, Hilgendorf I, Fitzner B, Brock P, Sparmann G, Emmrich J, et al. Regulation of pancreatic stellate cell function in vitro: biological and molecular effects of all-trans retinoic acid. Biochem Pharmacol (2003) 66(4):633–41. doi: 10.1016/S0006-2952(03)00390-3
97. Wilson JS, Pirola RC, Apte MV. Stars and stripes in pancreatic cancer: role of stellate cells and stroma in cancer progression. Front Physiol (2014) 5:52. doi: 10.3389/fphys.2014.00052
98. McCarroll JA, Naim S, Sharbeen G, Russia N, Lee J, Kavallaris M, et al. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front Physiol (2014) 5:141. doi: 10.3389/fphys.2014.00141
99. She H, Xiong S, Hazra S, Tsukamoto H. Adipogenic transcriptional regulation of hepatic stellate cells. J Biol Chem (2005) 280(6):4959–67. doi: 10.1074/jbc.M410078200
100. Yamada M, Blaner WS, Soprano DR, Dixon JL, Kjeldbye HM, Goodman DS. Biochemical characteristics of isolated rat liver stellate cells. Hepatology (1987) 7(6):1224–9. doi: 10.1002/hep.1840070609
Keywords: vitamin A, diabetes, pancreas, development, function, immune response, pancreatic stellate cells
Citation: Zhou Y, Wang H, Zhou J, Qiu S, Cai T, Li H, Shen Z, Hu Y, Ding B, Luo M, Huang R, Yan R, Xu W, He C, Zhang Y, Li F, Sun Z and Ma J (2021) Vitamin A and Its Multi-Effects on Pancreas: Recent Advances and Prospects. Front. Endocrinol. 12:620941. doi: 10.3389/fendo.2021.620941
Received: 24 October 2020; Accepted: 04 January 2021;
Published: 18 February 2021.
Edited by:
Vinod Tiwari, Indian Institute of Technology (BHU), IndiaReviewed by:
William Blaner, Columbia University, United StatesYoshihiro Mezaki, The Jikei University School of Medicine, Japan
Copyright © 2021 Zhou, Wang, Zhou, Qiu, Cai, Li, Shen, Hu, Ding, Luo, Huang, Yan, Xu, He, Zhang, Li, Sun and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianhua Ma, bWFqaWFuaHVhMTk2NTAzQDEyNi5jb20=
†These authors have contributed equally to this work