 Eugenia Bertoldo*
Eugenia Bertoldo* Giovanni Adami
Giovanni Adami Maurizio Rossini
Maurizio Rossini Alessandro Giollo
Alessandro Giollo Giovanni Orsolini
Giovanni Orsolini Ombretta Viapiana
Ombretta Viapiana Davide Gatti
Davide Gatti Angelo Fassio
Angelo Fassio- Rheumatology Unit, Department of Medicine, University of Verona, Verona, Italy
The relationship between endocrine hormones and the spectrum of rheumatic conditions has long been discussed in the literature, focusing primarily on sexual hormones, such as estrogens, androgens, prolactin (PRL). Estrogens are indeed involved in the pathogenesis of the main inflammatory arthritis thanks to their effects on the immune system, both stimulatory and inhibitory. The PRL system has been discovered in synovial tissue of rheumatoid arthritis (RA) and psoriatic arthritis (PsA), patients and has been propose as a new potential therapeutic target. Besides sexual hormones, in the last years scientific interest about the crosstalk of immune system with other class of hormones has grown. Hormones acting on the bone tissue (i.e. parathyroid hormone, vitamin D) and modulators of the Wnt pathway (i.e. Dickkopf-1) have been demonstrated to play active role in inflammatory arthritis course, defining a new field of research named osteoimmunology. PTH, which is one of the main determinants of Dkkopf-1, plays a crucial role in bone erosions in RA and a correlation between PTH, Trabecular Bone Score (TBS) and disease activity has been found in ankylosing spondylitis (AS). In PSA is under studying the interaction among IL-17 and bone metabolism. The purpose of this review is to discuss and summarize the recent data about the interaction between endocrine hormone and immune system in the main rheumatic disorders, covering in particular the role of bone-related hormones and cytokines. We will describe this relationship from a biochemical, diagnostic and therapeutic perspective, with a particular focus on RA, PsA and AS.
Introduction
Hormones are involved in various aspects of the immune response and rheumatic diseases. To date, there is a considerable body of evidence on the relationship between sex hormones and autoimmunity. In recent years, the scientific interest in the crosstalk between hormones and cytokines acting on bone metabolism has also grown, even in rheumatology. Emblematic examples are arthritic disorders such as RA, PsA, and AS.
Bone tissue is nowadays considered an ‘osteo-immune’ system and a principal actor in the pathogenesis of many rheumatic diseases; for those reasons, in the last decade, the term “osteoimmunology” has been increasingly used (1, 2).
In the following paragraphs, we will discuss and review the main concepts and the latest findings on the interplay between hormones, cytokines, and bone in the main arthritic conditions from a biochemical, diagnostic, and therapeutic perspective without attempting to be comprehensive (Figure 1).
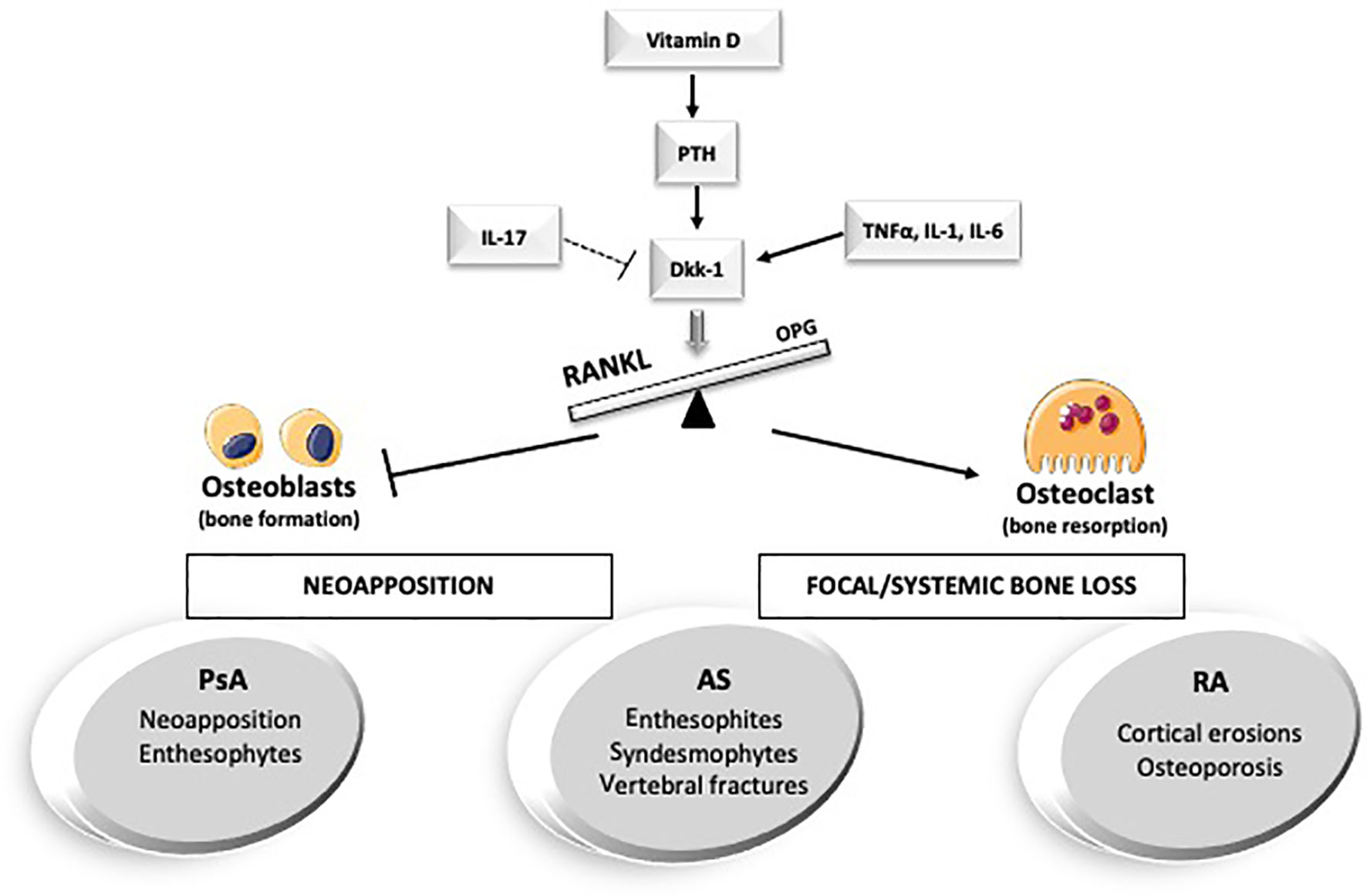
Figure 1 Hormones and cytokines involved in bone metabolism in the main arthritic disorders. AS, anskylosing spondilits; Dkk-1, Dickopf-1; IL-, interleukin-; PsA, Psoriatic Arthritis; PTH, parathyroid hormone; RA, Rheumatoid Arthritis; TNFalfa, Tumor necrosis factor alpha.
Bone remodeling is principally enacted by three types of cells: osteoblasts, osteoclasts, and osteocytes. One of the primary regulatory pathways of bone turnover is the Wnt/beta-catenin signaling (the canonical Wnt pathway) (3). Wnt signaling induces the commitment of the mesenchymal stem cell toward the osteoblast line (osteoblastogenesis) and favors their maturation and survival. In addition, in certain circumstances, it can also reduce osteoclastogenesis and bone resorption by promoting the osteoprotegerin (OPG) expression from the osteoblasts themselves (4). Dickkopf-related protein 1 (Dkk-1) is a secretory glycoprotein mainly expressed by osteoblasts and bone marrow stromal cells in the late phase of osteoblast differentiation. Dkk-1 is a potent inhibitor of the Wnt canonical pathway (5). Its role has also been investigated in various pathological conditions: low Dkk-1 and sclerostin serum levels have been described in diffuse idiopathic hyperostosis (6, 7), while its excessive overexpression seems to correlate with osteolytic lesions in multiple myeloma (8), with cortical erosions, low bone formation, and secondary osteoporosis in rheumatoid arthritis (9, 10).
Scloeristin is another inhibitor of Wnt signaling, and it is secreted mostly by osteocytes (11). Among the factors that influence Dkk-1 and sclerostin, we find several hormones, such as estrogens, androgens, parathyroid hormone (PTH) and vitamin D (12).
PTH is a polypeptide secreted by the parathyroid glands in response to decreases in plasma calcium, other regulators of PTH are 1,25- dihydroxyvitamin D, serum phosphate levels, and the phosphaturic hormone fibroblast growth factor-23 (FGF23). PTH acts via its own G protein-coupled receptors (GPCR) (Figure 2), a transmembrane protein expressed in different organs (13, 14). Parathyroid hormone 1 receptor (PTH1R) is expressed in bone and kidney and regulates calcium ion homeostasis through activation of adenylate cyclase and phospholipase C and the parathyroid hormone 2 receptor (PTH2R) that is expressed primarily in the central nervous system, pancreas, testis, and placenta.
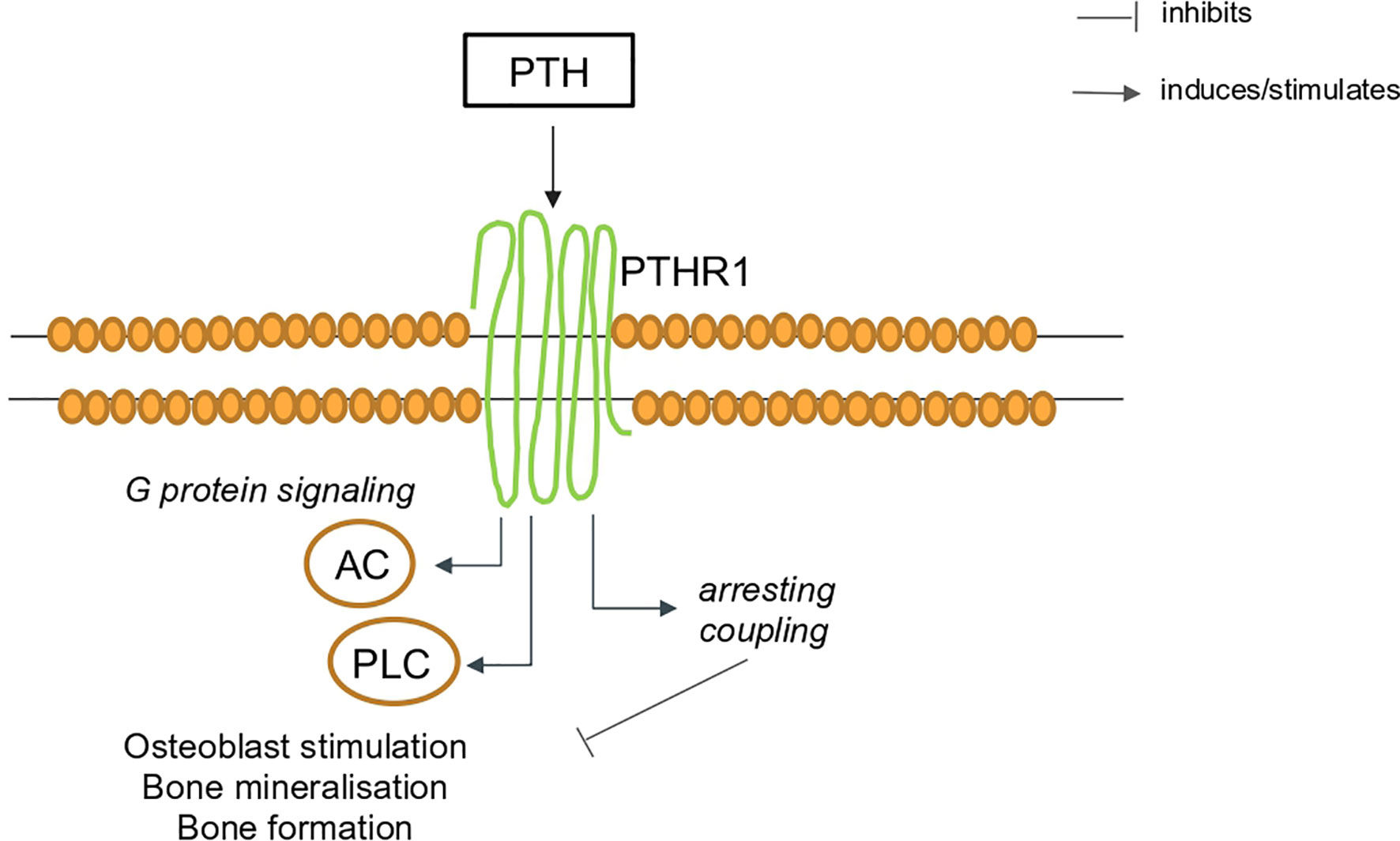
Figure 2 Schematic representation of PTH signaling via the PTH1R in bone. AC, adenylate cyclase; PTH, parathyroid hormone; PTH1R, parathyroid hormone 1 receptor, PLC, phospholipase C.
Vitamin D hormone, once metabolically converted in its active metabolite, 1,25-dihydroxyvitamin D [1,25(OH)2D], plays an essential role in calcium homeostasis and bone metabolism. Vitamin D, by acting on different pathways, can also modulate both the innate and adaptive systems thanks to its ubiquitously distributed cellular receptor (vitamin D receptor or VDR), which alters the transcription rates of target genes responsible for the biological responses.
Another key molecule involved in bone turnover and the immune system is the receptor activator of nuclear factor-kB ligand (RANKL). Its stimulation on the macrophage/dendritic progenitors leads to osteoclast differentiation (15), and its expression has been observed not only in the bone marrow but also in lymphocytes and in the lymphoid tissues (16), where it regulates the development of immune tolerance (7).
In recent years, lymphocytes, particularly T cells, have been found to play an important role in the bone health regulation (8). Lymphocytes play a dual role in the modulation of bone remodeling: on one side, resting T cells, via INFγ, inhibit in vitro osteoclast differentiation; on the other, activated T cells partake in the osteoclasts development (17).
Ankylosing Spondylitis
AS is a chronic rheumatic disease characterized by inflammation and extensive remodeling of the spine and joints. Eventually, it can lead to the development of spinal syndesmophytes and extra-articular enthesophytes (18). AS belongs to the spondyloarthritis (SpA) spectrum, such as PsA and axial spondyloarthritis (axSpA). These diseases are characterized by pathologic bone formation involving primarily the entheses complex and, at the same time, by cortical bone erosions (19).
Moreover, AS shows a dichotomous relationship with bone metabolism: the pathological neoformation coexists with an increased risk of fracture and impaired bone mineral density (BMD) (20, 21) (Figure 1).
The Wnt pathway seems to be one of the key players involved in the dual relationship with systemic bone metabolism found in AS. Unsurprisingly, the role of Dkk-1 in this disease has been the object of several investigations (22, 23). Rossini M. et al. (24) observed decreased serum levels of both Dkk-1 and sclerostin, suggesting a link between excessive Wnt exposure and the new focal bone formation. The Authors also reported a negative association between Dkk-1, spinal BMD, and vertebral fractures.
One metanalysis published in 2018 concluded that Dkk-1 serum levels in AS patients seem to be comparable to healthy controls (25). However, when the subanalysis considered only the studies characterized by high degrees of structural involvement (modified Stoke AS Spine Score >30), or increased levels of serum C reactive protein (CRP), Dkk-1 was found to be significantly reduced (25).
In addition, in AS patients, the incidence of osteoporosis is varying from 18.7 to 62%, according to studies (26). The risk of fracture is increased compared (27), despite normal or only slightly reduced BMD values (28, 29).
Boussoualim K et al. evaluated AS patients through the measurement of the Trabecular Bone Score (TBS) (30), an algorithm that leads a better assessment of vertebral bone quality (31), and documented an association between lower TBS values and disease activity and, an inverse association between TBS and parathyroid hormone (PTH) levels. Signaling through the PTH1R has been suggested as one of the main pathways that regulate Dkk-1 in AS patients and other inflammatory conditions (32).
In our opinion, it should be emphasized that the contradictory data on Dkk-1 in AS could be, at least in part, related to the variability of PTH and of vitamin D, one of its main determinants, whose metabolism could be impaired in inflammatory conditions (33).
Another relevant category of hormones implicated in rheumatic condition are sex hormons and nowadays gender medicine is an increasing topical issue.
The male-to-female ratio of SpA ranges approximately from 1:1 in patients with non radiographic-axSpA and 2:1 in patients with AS (34) and has been demonstrated that the proportion of female patients is significantly lower among the patients who progressed from nr-axSpA to AS (35). Furthermore male patients with AS have more severe radiographic damage than female (36).
Even if underlying biological differences between men and women with AS are still unknowns, estrogens are known to modulate T cell differentiation, type 2 cytokine production (37) and, in animal model, the inhibition of the differentiation of T helper17 cells (38).
Gooren et al. reported in 22 male AS patients reduced levels of testicular testosterone reserve, elevated levels of LH, inversion of estradiol testosterone ratio and slightly increased estradiol. Levels of estrogens (17β-estradiol) have been founded lower in active AS patients disease than in those in remission state (39). To explore this latter evidence, Jeong and colleagues have demonstrated how estrogens can suppress the development of arthritis in SpA mouse model, probably because estrogens inhibit Wnt signaling (40).
Rheumatoid Arthritis
RA is a chronic inflammatory disorder that leads to severe joint damage and disability (41). The most studied type of bone involvement in RA is the focal cortical bone loss (erosions). However, we can indeed label RA as a “bone disease”: cortical erosions, systemic bone loss (osteoporosis), and periarticular bone loss contribute to the disease burden (Figure 1). Local and systemic inflammation suppresses both bone formation and erosion healing (42, 43). Focal bone impairment is the result of the interaction between the dysregulated inflammation of the synovial membrane (synovitis) with the surrounding bone microenvironment. Eventually, this induces an excessive differentiation and activation of the osteoclast line and the development of cortical erosions (44).
At the systemic level, pro-inflammatory cytokines such as TNF-alfa, interleukin1(IL-1), and interleukin 6 (IL-6) play a crucial role in systemic bone impairment throughout, mainly via the RANK/RANKL/OPG system (45).
Among the hormones involved in the pathogenesis of RA, data suggest that PTH could play a significant role in bone erosions.
Indeed, in RA patients has been documented an association between joint erosions and higher PTH levels (10), probably in a vitamin D-independent way. T lymphocytes are hypothesized to promote PTH-induced osteoclastogenesis by increasing the medullary stromal cell responsiveness to PTH itself (46). As shown in both healthy and in subjects with primary hyperparathyroidism (47), persistent PTH signaling increases the RANKL/RANK pathway activity (48), negatively correlates with sclerostin and positively with Dkk-1.
As seen in AS (49), in mastocytosis with bone involvement (50), and in other conditions, also in RA PTH is suspected to be one of the main determinants of Dkk-1 serum levels (32). Furthermore, in RA patients, Dkk-1 and PTH serum levels are significantly higher, despite therapy with glucocorticoid (GC), tumor necrosis factor-alfa inhibitors (TNFi) or bisphosphonates (BPs) (32).
Different studies have remarked that levels of Dkk-1 in RA patients correlate inversely with BMD, in particular at cortical bone sites (32) and a significant association between low BMD and focal bony erosions has been described (51, 52). RA patients are generally characterized by increased diffuse bone loss together with a higher risk for hip and vertebral fractures (53).
Simon and colleagues analyzed in patients affected by RA the quantity and quality of intra-articular bone of the metacarpal heads (54), which are one of the typical erosion sites (55). In their study, a significant trabecular and cortical intra‐articular bone loss was observed, similar to the impairment that characterizes the bone microstructure after the menopause (54). These microstructural features in RA patients correlate with disease activity, with serum levels of pro‐inflammatory cytokines and, as already mentioned, with serum levels of Dkk-1 and PTH, as a consequence of a common pathological mechanism of both inflammatory and metabolic nature (32, 56, 57). For these reasons, we speculated that osteoporosis might be a significant and independent determinant of bone erosions in RA (10). Similar intra-articular microstructural alterations can also be secondary to aging and/or estrogen deficiency, and have also been found in extra-articular bone, such as the radius (58). Moreover, erosions and low mineral density share many risk factors: anti-citrullinated protein antibodies (ACPAs) (59, 60), disease activity, cigarette smoking, alcohol consumption, hypovitaminosis D, corticosteroid use, and aging (44). In line with this hypothesis, some osteoporosis treatments seem to prevent erosions in RA. Denosumab (Dmab), a monoclonal antibody that blocks RANKL, has been demonstrated to stop the structural progression in RA (61). Furthermore, it determines the increase in bone mass, especially in trabecular bone areas, regardless of GC use (62).
The keystone that allows Dmab to perform this protective effect is the inhibition of the osteoclast, a multinucleated cell that contributes not only to the development of erosions in the subchondral bone tissue but also to the destruction of the mineralized cartilage (61).
In our opinion, overlooking the bone metabolic status in osteoporotic RA patients might be one of the key determinants in those patients whose erosions continue to progress despite adequate treatment with DMARDs (63). To date, the meta-analyses on effects of DMARDs on radiographic progression of RA patients lack of BMD data, and this might represent an important bias (63).
Teriparatide (TPTD), a PTH analog, is an effective drug used in severe osteoporosis, with a completely different action than Dmab (64). TPTD boosts bone formation biomarkers already from the first month of treatment (64). Interestingly, long-term stimulation with PTH analogue seems to increase serum Dkk-1 in women postmenopausal osteoporosis (65). And this observation is consistent with the demonstrated relationship between serum levels of PTH, serum levels of Dkk1 and bone erosions in RA patients (32). These data suggest that PTH might contribute to determinate local DKK1 over-expression. Hence, high serum PTH levels may enhance local bone resorption and hinder bone repair by promoting DKK1 expression (32). Besides the prevention of erosions, there is still controversy on the possibility of their repairment over time. Apparently, this might occur in some cases (66).
In different settings, TPTD has been suggested as a possible treatment to enhance fracture healing. Furthermore, in a TNF-transgenic murine model of RA, combination therapy with TPTD and TNFi was associated with erosions repair (67). Unfortunately, in a recent trial on RA subjects, this endpoint was not achieved (68, 69). To explain this finding, one should remember that the bone microenvironment might be permanently altered in the setting of a longstanding inflammation. Therefore, this might cause a permanent impairment in the number and differentiation potential of the osteoblast progenitors (70), on which TPTD is supposed to exert its function. Furthermore, the chronically high serum PTH levels are associated with increased bone porosity (71) and decreased cortical thickness, contributing to the explanation for these disappointing results (68).
Ebina and colleagues compared the effects on joint erosions of the three different classes of therapies (BPs, Dmab, and TPDT) in biologic-naïve RA patients. Switching from BPs to Dmab was found to be more effective than continuing BPs or switching to TPTD in the prevention of structural progression (72).
As already mentioned, pro-inflammatory cytokines activate the osteoclast line via the RANK/RANKL/OPG axis, and are directly involved in bone complications (45). Pro-inflammatory cytokines are currently the main targets of the most widely used drugs for RA, i.e. TNFi and anti-IL6 receptor (IL-6R) monoclonal antibodies.
TNFα contributes substantially to RA pathogenesis, and it is involved in many pathways (73). Interestingly, some studies have demonstrated its relationship with bone turnover cytokines (9, 74, 75). TNF-alfa increases Dkk-1 levels in synovial fibroblasts both in vitro and in vivo, and it is correlated with the presence (9, 74) and the progression (75) of bone erosions.
On the other hand, in RA patients, TNFα inhibition demonstrated bone metabolic effects by reducing Dkk-1 and sclerostin serum levels (76, 77).
In RA, also IL-6 is strongly involved in bone loss through the inhibition of the Wnt canonical pathway (78). A recent study investigated the short-term effects of an anti-IL-6 treatment (tocilizumab) on bone turnover markers (BMTs) in RA patients, comparing it with TNFi and GCs (methyl-prednisone) (79). The strong and prompt influence of TNFis on bone turnover markers seen in previous studies (76) was not observed with the IL-6R blockade. Indeed, no significant change was observed either for sclerostin or Dkk-1 in the tocilizumab arm, while the arm receiving TNFis showed a decrease in markers of bone resorption and an increase in the markers of bone formation (80). To explain this difference, the Authors hypothesized a slower influence of tocilizumab on the Wnt pathway.
As seen in other arthritis, sex hormones, especially estrogens, are closely involved in RA. RA is more prevalent in women with a female-to-male sex ratio of 4:1 (81), even if the reason is still partially unclear. Estrogens can have both stimulatory and inhibitory effects on the immune system, as described by Straub (82), and estrogen exposure has been associated with increased risk of RA due to pro-inflammatory action of these hormones unbalanced with the anti-inflammatory one of androgens (82). However it must be noted that two conditions characterized by a low level of estrogens such as menopause and the use of anti-estrogen agents have been associated with an increased risk of developing RA (respectively HR, 2.1; (95% CI, 1.5–3.1) and OR, 2.4 (95% CI, 1.9–3.0) and OR, 1.9 (95% CI, 1.6–2.1) depending by dose and time of anti-estrogens exposure) (83–85). Furthermore, long duration of pharmacological estrogen exposure under oral contraceptives (OCs), seems to protect from the development of the disease, with a cumulative positive dose effect (86–88). Also the pregnancy condition has been described as protective against RA, probably thanks to a balance between progesterone to the high level of estrogens (89–91). On the contrary the post-partum and the lactating period, characterized respectively by a decline of estrogens and by the release of prolactin (PRL), has been consistently associated with an increased risk of RA (92). Captivatingly, in a small study, levels of 17β-estradiol and other hormones (progesterone, aldosterone and growth hormone) were founded higher in synovial fluid (SF) of RA patient in comparison to patients with osteoarthritis (93), suggesting a influencing roles also at local sites.
PRL is a sex hormones, mainly secreted by pituitary gland and with pleiotropic functions, among which the capacity to enhance or inhibit pro-inflammatory cytokine production (94). PRL can be locally produced by macrophages, T cells and synovial fibroblasts and its receptor (PRLR) is expressed in synovial macrophages and lymphocytes (94). For those reasons, PRL have long been thought to play an important role in RA disease (95, 96) even if there are contradictory results about its serum levels, that have been founded raised in RA patients compared to healthy controls in some studies (97–100). Levels of PRL has been dosed also in SF of RA patients without finding significant differences with patients with osteoarthritis (93). Nevertheless PRL and its receptors were suggested by Tang and colleagues to be engaged in RA thanks to a local crosstalk, via auto- or paracrine ways, between the immune and endocrine systems. The local presence of PRL system in synovial tissue of RA (and PsA, as we will see later) patients has been propose as a new potential therapeutic target (94).
Psoriatic Arthritis
PsA is a chronic, systemic inflammatory disease that affects peripheral joints, the axial skeleton, and it is associated with psoriasis of the skin and nails (101).
The bone involvement in PsA patients differs from the one seen in RA, presenting erosive damage associated with exuberant bone formation, especially in entheseal sites (102) (Figure 1). The different bone involvement in RA and PsA may be explained by a diverse interplay of the involved mediators and cytokines.
PsA is a strongly IL-17-driven disease (103). Among its functions, IL-17 is a potent osteoclastogenetic factor (104), particularly at inflamed sites undergoing mechanical stress, such as the entheses.
Concerning the etiopathogenesis of enthesopathy, it is well known that local trauma and inflammation play a pivotal role in the T cells activation, especially the gamma-delta subset (105). Cytokines such as IL-23 and IL-17 stimulate resident cells (chondrocytes, osteoblasts, and, to a lesser extent, osteoclasts) to secrete metalloproteinase and to overexpress the RANK-RANK-ligand, leading to both erosion and bone formation (106). However, entheses are poor in osteoclasts, and this consideration might suggest one explanation for a focal unbalance between bone formation and erosions. Furthermore, the mechanical stress in the synovial-entheseal complex might contribute to promote bone formation (19).
The relationship between anti IL-17 therapy and bone metabolism in PsA has been studied in a small longitudinal study in which Dkk-1 and sclerostin levels increased after treatment with secukinumab in a cohort of PsA patients, suggesting a possible drug-induced inhibition of local bone over-proliferation (107).
Conversely, one meta-analysis reported benefits in terms of BMD under TNFi treatment, thanks to the suppression of systemic inflammation (108). Over the past years, the relationship between inflammation and the consequent role of TNFi in the structural progression has been greatly debated both in AS (109) and PsA (110).
According to older RCTs, TNFi treatments failed to control the radiographic progression, despite the achievement of clinical improvement (111). For this reason, a few years ago, the so-called “TNF brake” hypothesis was proposed: early in the pathogenesis, TNFα might upregulate Dkk-1 expression. However, when the inflammatory lesions are established and mature, the bone microenvironment undergoes some other changes, activating bone formation pathways with the consequent expression of bone growth factors.
In this setting, TNFα has been hypothesized to act as a brake slowing down the new bone formation through the Dkk-1 upregulation. This model was postulated to explain why the TNFi failed to prevent radiological progression (i.e. syndesmophytes) (109). Nowadays, some new data suggest the efficacy of TNFis on radiological damage if started in a timely fashion. The suppression of local inflammation from the very beginning is thought to prevent the activation of bone formation pathways (112, 113).
Finally, in all inflammatory arthritis, the occurrence of physiological aging and senile osteoporosis can alter the function of bone mechanoreceptors, whose dysfunction can contribute to bone impairment through the impairment of different pathways that eventually converge on Wnt signaling (114).
As seen for RA, PRL has been studied also in PsA. As mention above for RA, PRL is locally expressed in the synovial tissue also of PsA patients and PRL mRNA expression positively correlates with disease activity (94).
The PRL-PRLR binding activates various signaling pathways among which the Janus kinase/signal transducer and activator of transcription (JAK-STAT) one, once the more recently studied therapeutic target both for RA and PSA.
Not many studies have faced the role of sex hormones in PsA, a disease with sex ratio of 1:1 and a bimodal distribution in female sex, with peaks of incidence during late adolescence and the perimenopausal period (115). Literature shows how psoriasis often improves during pregnancy and reappear in the post-partum, suggesting a direct link between estrogen and progesterone and disease severity (115). but less is known about PsA.
Other Rheumatic Conditions
The strong link between hormones, bone turnover, and rheumatic diseases has been described not only in the chronic arthritides but also in other rheumatic conditions, such as Polymyalgia rheumatica (PMR), Crystal-Induced Arthritides, and even connective tissue diseases (i.e. Systemic Erythematous Lupus, Sjogren Syndrome and Systemic Sclerosis) (116–119).
PMR is a chronic inflammatory disease affecting older adults that causes pain, stiffness, and inflammation of the shoulder and pelvic girdles, mainly treated with GCs (120). Data on the changes of bone metabolism induced by GCs and on the profile of bone markers its fine regulators (i.e. Dkk-1, sclerostin) in PMR are scarce.
In a very recent study (116), we showed an increase in Dkk-1 serum levels in also treatment-naive PMR patients. In this study, we also observed a significant decrease of Dkk-1, together with C-terminal telopeptide of type-1 collagen (CTX, a bone resorption marker) and in N-propeptide of type I collagen (PINP), after one month of GCs therapy. A similar trend was documented for sclerostin.
Systemic sclerosis (SSc) is a connective tissue disease characterized by tissue fibrosis and microvascular involvement. Even if difficult to assess due to the heterogeneity of the cohorts, it seems that SSc patients have an increased risk of developing osteoporosis (121). The most interesting aspect of bone involvement is that the modulators of bone metabolism are partially involved in SSc pathogenesis. The Wnt system plays a significant role in the development of fibrosis (122), through the endothelial-to-mesenchymal transition (117). For this reason, data about bone turnover markers are often conflicting, especially on Dkk-1. For instance, one study found a correlation between elevated Dkk-1 and low TBS (123), while in another, a similar correlation has been found with modified Rodnan Skin Score (mRSS) but not with BMD (124).
Furthermore, in SSc patients, increased serum RANKL levels have been observed (125), and TRIAL, a ligand of OPG with vascular protection properties (126), was found to be higher in SSc compared to the general population, suggesting a possible link between microvascular damage and bone loss.
Systemic Lupus Erythematosus (SLE) is an autoimmune connective tissue disease with a complex pathophysiology and a spectrum of clinical manifestations involving potentially every organ and system of the body (127).
As seen above in RA, bone tissue could be affected both at the systemic level, with low BMD and fragility fractures, and at the focal site, with joint erosions (118). Furthermore, the bone loss could be secondary to the disease itself and/or to steroid treatment (128), and its pathophysiology is extremely heterogeneous: its takes into account systemic inflammation, impairment of vitamin D-PTH-calcium system both for limited sun exposure and altered renal function, impairment of sex hormones (i.e. dehydroandrostenedione) (129). A two to threefold increased fracture risk has been reported in several large cohorts (130).
Decreased osteocalcin serum levels (a marker of bone formation) and increased levels of CTX in untreated premenopausal SLE patients have been observed, with a correlation between osteocalcin and disease activity (119).
A dysregulation in the Wnt/beta-catenin signaling has been observed in B and T cells involved in SLE pathogenesis and among bone mediators, Dkk-1 has been suggested as a potential biomarker for bone erosions (118) and as an independent biomarker for lupus nephritis (131).
Conclusions
Over the last years, rheumatology, endocrinology and immunology have intertwined especially with the domain of bone metabolism. This new field of research is providing new data that are contributing to the development of the evolving pathophysiological models of the rheumatic diseases. RA could be labled as “bone disease” in which have been described the connection between pro‐inflammatory cytokines, PTH, Dkk-1, bone erosions and bone loss. Some osteoporotic treatment, i.e. denosumab, demonstrates to stop the structural progression (61). In PsA, IL-23 and IL-17 interact with bone system through chondrocytes, osteoblasts, and osteoclasts and via RANKL/RANK signaling, leading to both erosion and bone formation. And in AS, the Wnt pathway seems to be one of the key players involved in the relationship with bone metabolism, characterized by pathological neoformation and impaired bone mineral density (20, 21). Hopefully, a deeper understanding of the relationship among bone turnover, hormones and the different rheumatic conditions’ phenotypes will be able to improve the clinical and therapeutic management of our patients.
Transparency declaration
The lead author (the manuscript’s guarantor) affirms that the manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned (and, if relevant, registered) have been explained.
Author Contributions
EB conceived and wrote the article. MR, GO, OV, DG, and AF revised the article critically for important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
This research received no external funding.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Tsukasaki M, Takayanagi H. Osteoimmunology: Evolving Concepts in Bone-Immune Interactions in Health and Disease. Nat Rev Immunol (2019) 19:626–42. doi: 10.1038/s41577-019-0178-8
3. Lerner UH, Ohlsson C. The WNT System: Background and its Role in Bone. J Intern Med (2015) 277:630–49. doi: 10.1111/joim.12368
4. Glass DA, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, et al. Canonical Wnt Signaling in Differentiated Osteoblasts Controls Osteoclast Differentiation. Dev Cell (2005) 8:751–64. doi: 10.1016/j.devcel.2005.02.017
5. Huang Y, Liu L, Liu A. Dickkopf-1: Current Knowledge and Related Diseases. Life Sci (2018) 209:249–54. doi: 10.1016/j.lfs.2018.08.019
6. Senolt L, Hulejova H, Krystufkova O, Forejtova S, Andres Cerezo L, Gatterova J, et al. Low Circulating Dickkopf-1 and its Link With Severity of Spinal Involvement in Diffuse Idiopathic Skeletal Hyperostosis. Ann Rheum Dis (2012) 71:71–4. doi: 10.1136/annrheumdis-2011-200357
7. Fassio A, Adami G, Idolazzi L, Giollo A, Viapiana O, Bosco E, et al. Diffuse Idiopathic Skeletal Hyperostosis (DISH) in Type 2 Diabetes: A New Imaging Possibility and a New Biomarker. Calcif Tissue Int (2020) 108(2):231–9. doi: 10.1007/s00223-020-00768-2
8. Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, et al. The Role of the Wnt-signaling Antagonist DKK1 in the Development of Osteolytic Lesions in Multiple Myeloma. N Engl J Med (2003) 349:2483–94. doi: 10.1056/NEJMoa030847
9. Wang S-Y, Liu Y-Y, Ye H, Guo J-P, Li R, Liu X, et al. Circulating Dickkopf-1 is Correlated With Bone Erosion and Inflammation in Rheumatoid Arthritis. J Rheumatol (2011) 38:821–7. doi: 10.3899/jrheum.100089
10. Rossini M, Bagnato G, Frediani B, Iagnocco A, LA Montagna G, Minisola G, et al. Relationship of Focal Erosions, Bone Mineral Density, and Parathyroid Hormone in Rheumatoid Arthritis. J Rheumatol (2011) 38:997–1002. doi: 10.3899/jrheum.100829
11. Weivoda MM, Youssef SJ, Oursler MJ. Sclerostin Expression and Functions Beyond the Osteocyte. Bone (2017) 96:45–50. doi: 10.1016/j.bone.2016.11.024
12. Delgado-Calle J, Sato AY, Bellido T. Role and Mechanism of Action of Sclerostin in Bone. Bone (2017) 96:29–37. doi: 10.1016/j.bone.2016.10.007
13. Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, et al. Cloning and Characterization of an Extracellular Ca 2+ -Sensing Receptor From Bovine Parathyroid. Nature (1993) 366:575–80. doi: 10.1038/366575a0
14. Thompson MD, Siminovitch KA, Cole DEC. G Protein-Coupled Receptor Pharmacogenetics. Methods Mol Biol Clifton NJ (2008) 448:139–85. doi: 10.1007/978-1-59745-205-2_8
15. Zaidi M. Skeletal Remodeling in Health and Disease. Nat Med (2007) 13:791–801. doi: 10.1038/nm1593
16. Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, et al. A Homologue of the TNF Receptor and its Ligand Enhance T-cell Growth and Dendritic-Cell Function. Nature (1997) 390:175–9. doi: 10.1038/36593
17. Zhang W, Dang K, Huai Y, Qian A. Osteoimmunology: The Regulatory Roles of T Lymphocytes in Osteoporosis. Front Endocrinol (2020) 11:465. doi: 10.3389/fendo.2020.00465
18. Sieper J, Poddubnyy D. Axial Spondyloarthritis. Lancet (2017) 390:73–84. doi: 10.1016/S0140-6736(16)31591-4
19. Schett G. Bone Formation in Psoriatic Arthritis: A Report From the GRAPPA 2013 Annual Meeting. J Rheumatol (2014) 41:1218–9. doi: 10.3899/jrheum.140173
20. Carette S, Graham D, Little H, Rubenstein J, Rosen P. The Natural Disease Course of Ankylosing Spondylitis. Arthritis Rheum (1983) 26:186–90. doi: 10.1002/art.1780260210
21. Klingberg E, Lorentzon M, Göthlin J, Mellström D, Geijer M, Ohlsson C, et al. Bone Microarchitecture in Ankylosing Spondylitis and the Association With Bone Mineral Density, Fractures, and Syndesmophytes. Arthritis Res Ther (2013) 15:R179. doi: 10.1186/ar4368
22. Haynes KR, Pettit AR, Duan R, Tseng H-W, Glant TT, Brown MA, et al. Excessive Bone Formation in a Mouse Model of Ankylosing Spondylitis is Associated With Decreases in Wnt Pathway Inhibitors. Arthritis Res Ther (2012) 14:R253. doi: 10.1186/ar4096
23. Daoussis D, Liossis S-NC, Solomou EE, Tsanaktsi A, Bounia K, Karampetsou M, et al. Evidence That Dkk-1 is Dysfunctional in Ankylosing Spondylitis. Arthritis Rheum (2010) 62:150–8. doi: 10.1002/art.27231
24. Rossini M, Viapiana O, Idolazzi L, Ghellere F, Fracassi E, Troplini S, et al. Higher Level of Dickkopf-1 is Associated With Low Bone Mineral Density and Higher Prevalence of Vertebral Fractures in Patients With Ankylosing Spondylitis. Calcif Tissue Int (2016) 98:438–45. doi: 10.1007/s00223-015-0093-3
25. Wu M, Chen M, Ma Y, Yang J, Han R, Yuan Y, et al. Dickkopf-1 in Ankylosing Spondylitis: Review and Meta-Analysis. Clin Chim Acta (2018) 481:177–83. doi: 10.1016/j.cca.2018.03.010
26. El Maghraoui A. Osteoporosis and Ankylosing Spondylitis. Joint Bone Spine (2004) 71:291–5. doi: 10.1016/j.jbspin.2003.06.002
27. Westerveld LA, Verlaan JJ, Oner FC. Spinal Fractures in Patients With Ankylosing Spinal Disorders: A Systematic Review of the Literature on Treatment, Neurological Status and Complications. Eur Spine J (2009) 18:145–56. doi: 10.1007/s00586-008-0764-0
28. Mitra D, Elvins DM, Speden DJ, Collins AJ. The Prevalence of Vertebral Fractures in Mild Ankylosing Spondylitis and Their Relationship to Bone Mineral Density. Rheumatology (2000) 39:85–9. doi: 10.1093/rheumatology/39.1.85
29. van der Weijden MAC, van der Horst-Bruinsma IE, van Denderen JC, Dijkmans BAC, Heymans MW, Lems WF. High Frequency of Vertebral Fractures in Early Spondylarthropathies. Osteoporos Int (2012) 23:1683–90. doi: 10.1007/s00198-011-1766-z
30. Boussoualim K, Amouzougan A, Pallot-Prades B, Denarié D, Collet P, Marotte H, et al. Evaluation of Bone Quality With Trabecular Bone Score in Active Spondyloarthritis. Joint Bone Spine (2018) 85:727–31. doi: 10.1016/j.jbspin.2018.02.006
31. Pothuaud L, Carceller P, Hans D. Correlations Between Grey-Level Variations in 2D Projection Images (TBS) and 3D Microarchitecture: Applications in the Study of Human Trabecular Bone Microarchitecture. Bone (2008) 42:775–87. doi: 10.1016/j.bone.2007.11.018
32. Rossini M, Viapiana O, Adami S, Fracassi E, Idolazzi L, Dartizio C, et al. In Patients With Rheumatoid Arthritis, Dickkopf-1 Serum Levels are Correlated With Parathyroid Hormone, Bone Erosions and Bone Mineral Density. Clin Exp Rheumatol (2015) 33:77–83.
33. Lin Z, Li W. The Roles of Vitamin D and Its Analogs in Inflammatory Diseases. Curr Top Med Chem (2016) 16:1242–61. doi: 10.2174/1568026615666150915111557
34. Rudwaleit M, Haibel H, Baraliakos X, Listing J, Märker-Hermann E, Zeidler H, et al. The Early Disease Stage in Axial Spondylarthritis: Results From the German Spondyloarthritis Inception Cohort. Arthritis Rheum (2009) 60:717–27. doi: 10.1002/art.24483
35. Jeong H, Yoon JY, Park E-J, Hwang J, Kim H, Ahn JK, et al. Clinical Characteristics of Nonradiographic Axial Spondyloarthritis in Korea: A Comparison With Ankylosing Spondylitis. Int J Rheum Dis (2015) 18:661–8. doi: 10.1111/1756-185X.12458
36. Lee W, Reveille JD, Davis JC, Learch TJ, Ward MM, Weisman MH. Are There Gender Differences in Severity of Ankylosing Spondylitis? Results From the PSOAS Cohort. Ann Rheum Dis (2007) 66:633–8. doi: 10.1136/ard.2006.060293
37. Nalbandian G, Kovats S. Estrogen, Immunity & Autoimmune Disease. Curr Med Chem - Immunol Endocr Metab Agents (2005) 5:85–91. doi: 10.2174/1568013053005418
38. Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen Deficiency Induces the Differentiation of IL-17 Secreting Th17 Cells: A New Candidate in the Pathogenesis of Osteoporosis. PloS One (2012) 7(9):e44552. doi: 10.1371/journal.pone.0044552
39. Jimenez-Balderas FJ, Tapia-Serrano R, Madero-Cervera JI, Murrieta S, Mintz G. Ovarian Function Studies in Active Ankylosing Spondylitis in Women. Clinical Response to Estrogen Therapy. J Rheumatol (1990) 17:497–502.
40. Jeong H, Bae E-K, Kim H, Eun YH, Kim IY, Kim H, et al. Estrogen Attenuates the Spondyloarthritis Manifestations of the SKG Arthritis Model. Arthritis Res Ther (2017) 19(1):198. doi: 10.1186/s13075-017-1407-9
41. Pincus T, Callahan LF, Sale WG, Brooks AL, Payne LE, Vaughn WK. Severe Functional Declines, Work Disability, and Increased Mortality in Seventy-Five Rheumatoid Arthritis Patients Studied Over Nine Years. Arthritis Rheum (1984) 27:864–72. doi: 10.1002/art.1780270805
42. Walsh N, Reinwald S, Manning C, Condon K, Iwata K, Burr D, et al. Osteoblast Function is Compromised At Sites of Focal Bone Erosion in Inflammatory Arthritis. Rheumatol Publ (2009) 24(9):1572–85. doi: 10.1359/jbmr.090320
43. Matzelle MM, Gallant MA, Condon KW, Walsh NC, Manning CA, Stein GS, et al. Resolution of Inflammation Induces Osteoblast Function and Regulates the Wnt Signaling Pathway. Arthritis Rheum (2012) 64:1540–50. doi: 10.1002/art.33504
44. Schett G, Gravallese E. Bone Erosion in Rheumatoid Arthritis: Mechanisms, Diagnosis and Treatment. Nat Rev Rheumatol (2012) 8:656–64. doi: 10.1038/nrrheum.2012.153
45. Rossini M, Adami G, Viapiana O, Idolazzi L, Orsolini G, Fassio A, et al. Osteoporosis: An Independent Determinant of Bone Erosions in Rheumatoid Arthritis? J Bone Miner Res Off J Am Soc Bone Miner Res (2017) 32:2142–3. doi: 10.1002/jbmr.3209
46. Gao Y, Wu X, Terauchi M, Li J-Y, Grassi F, Galley S, et al. T CELLS Potentiate PTH Induced CORTICAL Bone LOSS Through CD40L Signaling. Cell Metab (2008) 8:132–45. doi: 10.1016/j.cmet.2008.07.001
47. Viapiana O, Fracassi E, Troplini S, Idolazzi L, Rossini M, Adami S, et al. Sclerostin and DKK1 in Primary Hyperparathyroidism. Calcif Tissue Int (2013) 92:324–9. doi: 10.1007/s00223-012-9665-7
48. Ma YL, Cain RL, Halladay DL, Yang X, Zeng Q, Miles RR, et al. Catabolic Effects of Continuous Human PTH (1–38) In Vivo is Associated With Sustained Stimulation of RANKL and Inhibition of Osteoprotegerin and Gene-Associated Bone Formation. Endocrinology (2001) 142:4047–54. doi: 10.1210/endo.142.9.8356
49. Orsolini G, Adami G, Rossini M, Ghellere F, Caimmi C, Fassio A, et al. Parathyroid Hormone is a Determinant of Serum Dickkopf-1 Levels in Ankylosing Spondylitis. Clin Rheumatol (2018) 37:3093–8. doi: 10.1007/s10067-018-4205-0
50. Rossini M, Adami S, Zanotti R, Viapiana O, Idolazzi L, Biondan M, et al. Serum Levels of Bone Cytokines in Indolent Systemic Mastocytosis Associated With Osteopenia or Osteoporosis. J Allergy Clin Immunol (2014) 133:933–5. doi: 10.1016/j.jaci.2013.12.007
51. Jensen T, Hansen M, Jensen KE, Pødenphant J, Hansen TM, Hyldstrup L. Comparison of Dual X-ray Absorptiometry (DXA), Digital X-ray Radiogrammetry (DXR), and Conventional Radiographs in the Evaluation of Osteoporosis and Bone Erosions in Patients With Rheumatoid Arthritis. Scand J Rheumatol (2005) 34:27–33. doi: 10.1080/03009740510017986
52. Forslind K, Keller C, Svensson B, Hafström I, Group BS. Reduced Bone Mineral Density in Early Rheumatoid Arthritis is Associated With Radiological Joint Damage At Baseline and After 2 Years in Women. J Rheumatol (2003) 30:2590–6.
53. Cooper C, Coupland C, Mitchell M. Rheumatoid Arthritis, Corticosteroid Therapy and Hip Fracture. Ann Rheum Dis (1995) 54:49–52. doi: 10.1136/ard.54.1.49
54. Simon D, Kleyer A, Stemmler F, Simon C, Berlin A, Hueber AJ, et al. Age- and Sex-Dependent Changes of Intra-articular Cortical and Trabecular Bone Structure and the Effects of Rheumatoid Arthritis. J Bone Miner Res Off J Am Soc Bone Miner Res (2017) 32:722–30. doi: 10.1002/jbmr.3025
55. Scott DL, Wolfe F, Huizinga TW. Rheumatoid Arthritis. Lancet (2010) 376:1094–108. doi: 10.1016/S0140-6736(10)60826-4
56. Zhu TY, Griffith JF, Qin L, Hung VW, Fong T-N, Au S-K, et al. Alterations of Bone Density, Microstructure, and Strength of the Distal Radius in Male Patients With Rheumatoid Arthritis: A Case-Control Study With HR-Pqct. J Bone Miner Res Off J Am Soc Bone Miner Res (2014) 29:2118–29. doi: 10.1002/jbmr.2221
57. Sambrook PN. The Skeleton in Rheumatoid Arthritis: Common Mechanisms for Bone Erosion and Osteoporosis? J Rheumatol (2000) 27:2541–2.
58. Zhu TY, Griffith JF, Qin L, Hung VWY, Fong T-N, Kwok AW, et al. Bone Density and Microarchitecture: Relationship Between Hand, Peripheral, and Axial Skeletal Sites Assessed by HR-pQCT and DXA in Rheumatoid Arthritis. Calcif Tissue Int (2012) 91:343–55. doi: 10.1007/s00223-012-9644-z
59. Kleyer A, Finzel S, Rech J, Manger B, Krieter M, Faustini F, et al. Bone Loss Before the Clinical Onset of Rheumatoid Arthritis in Subjects With Anticitrullinated Protein Antibodies. Ann Rheum Dis (2014) 73:854–60. doi: 10.1136/annrheumdis-2012-202958
60. Llorente I, Merino L, Ortiz AM, Escolano E, González-Ortega S, García-Vicuña R, et al. Anti-Citrullinated Protein Antibodies are Associated With Decreased Bone Mineral Density: Baseline Data From a Register of Early Arthritis Patients. Rheumatol Int (2017) 37:799–806. doi: 10.1007/s00296-017-3674-9
61. Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, Sharp JT, et al. Denosumab Treatment Effects on Structural Damage, Bone Mineral Density, and Bone Turnover in Rheumatoid Arthritis: A Twelve-Month, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase II Clinical Trial. Arthritis Rheum (2008) 58:1299–309. doi: 10.1002/art.23417
62. Takeuchi T, Tanaka Y, Ishiguro N, Yamanaka H, Yoneda T, Ohira T, et al. Effect of Denosumab on Japanese Patients With Rheumatoid Arthritis: A Dose-Response Study of AMG 162 (Denosumab) in Patients With RheumatoId Arthritis on Methotrexate to Validate Inhibitory Effect on Bone Erosion (DRIVE)-a 12-Month, Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase II Clinical Trial. Ann Rheum Dis (2016) 75:983–90. doi: 10.1136/annrheumdis-2015-208052
63. Rossini M, Adami G, Viapiana O, Idolazzi L, Gatti D. Denosumab, Cortical Bone and Bone Erosions in Rheumatoid Arthritis. Ann Rheum Dis (2016) 75:e70. doi: 10.1136/annrheumdis-2016-210022
64. Compston J. Recombinant Parathyroid Hormone in the Management of Osteoporosis. Calcif Tissue Int (2005) 77:65–71. doi: 10.1007/s00223-005-0012-0
65. Gatti D, Viapiana O, Idolazzi L, Fracassi E, Rossini M, Adami S. The Waning of Teriparatide Effect on Bone Formation Markers in Postmenopausal Osteoporosis Is Associated With Increasing Serum Levels of DKK1. J Clin Endocrinol Metab (2011) 96:1555–9. doi: 10.1210/jc.2010-2552
66. Sharp JT, Van Der Heijde D, Boers M, Boonen A, Bruynesteyn K, Emery P, et al. Repair of Erosions in Rheumatoid Arthritis Does Occur. Results From 2 Studies by the OMERACT Subcommittee on Healing of Erosions. J Rheumatol (2003) 30:1102–7.
67. Redlich K, Görtz B, Hayer S, Zwerina J, Doerr N, Kostenuik P, et al. Repair of Local Bone Erosions and Reversal of Systemic Bone Loss Upon Therapy With Anti-Tumor Necrosis Factor in Combination With Osteoprotegerin or Parathyroid Hormone in Tumor Necrosis Factor-Mediated Arthritis. Am J Pathol (2004) 164:543–55. doi: 10.1016/S0002-9440(10)63144-6
68. Solomon DH, Kay J, Duryea J, Lu B, Bolster MB, Yood RA, et al. Effects of Teriparatide on Joint Erosions in Rheumatoid Arthritis: A Randomized Controlled Trial. Arthritis Rheumatol Hoboken NJ (2017) 69:1741–50. doi: 10.1002/art.40156
69. Duryea J, Gravallese EM, Wortman JR, Xu C, Lu B, Kay J, et al. Healing of Erosions in Rheumatoid Arthritis Remains Elusive: Results With 24 Months of the Anabolic Agent Teriparatide. Scand J Rheumatol (2020) 50(1):11–4. doi: 10.1080/03009742.2020.1772362
70. Lepperdinger G. Inflammation and Mesenchymal Stem Cell Aging. Curr Opin Immunol (2011) 23:518–24. doi: 10.1016/j.coi.2011.05.007
71. Zanchetta JR, Bogado CE, Ferretti JL, Wang O, Wilson MG, Sato M, et al. Effects of Teriparatide [Recombinant Human Parathyroid Hormone (1-34)] on Cortical Bone in Postmenopausal Women With Osteoporosis. J Bone Miner Res Off J Am Soc Bone Miner Res (2003) 18:539–43. doi: 10.1359/jbmr.2003.18.3.539
72. Ebina K, Hirao M, Hashimoto J, Matsuoka H, Iwahashi T, Chijimatsu R, et al. Impact of Switching Oral Bisphosphonates to Denosumab or Daily Teriparatide on the Progression of Radiographic Joint Destruction in Patients With Biologic-Naïve Rheumatoid Arthritis. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA (2018) 29:1627–36. doi: 10.1007/s00198-018-4492-y
73. Firestein GS. Evolving Concepts of Rheumatoid Arthritis. Nature (2003) 423:356–61. doi: 10.1038/nature01661
74. Garnero P, Tabassi NC-B, Voorzanger-Rousselot N. Circulating Dickkopf-1 and Radiological Progression in Patients With Early Rheumatoid Arthritis Treated With Etanercept. J Rheumatol (2008) 35:2313–5. doi: 10.3899/jrheum.080356
75. de Rooy DPC, Yeremenko NG, Wilson AG, Knevel R, Lindqvist E, Saxne T, et al. Genetic Studies on Components of the Wnt Signalling Pathway and the Severity of Joint Destruction in Rheumatoid Arthritis. Ann Rheum Dis (2013) 72:769–75. doi: 10.1136/annrheumdis-2012-202184
76. Fassio A, Adami G, Gatti D, Orsolini G, Giollo A, Idolazzi L, et al. Inhibition of Tumor Necrosis Factor-Alpha (TNF-Alpha) in Patients With Early Rheumatoid Arthritis Results in Acute Changes of Bone Modulators. Int Immunopharmacol (2019) 67:487–9. doi: 10.1016/j.intimp.2018.12.050
77. Orsolini G, Adami G, Viapiana O, Idolazzi L, Fassio A, Vitiello M, et al. Ab0313 Short-Term Effects of Tnf Inhibitors on Bone Turnover Markers and Bone Mineral Density in Rheumatoid Arthritis. Ann Rheum Dis (2016) 75:1007–8. doi: 10.1136/annrheumdis-2016-eular.4582
78. Malysheva K, de Rooij K, Löwik CWGM, Baeten DL, Rose-John S, Stoika R, et al. Interleukin 6/Wnt Interactions in Rheumatoid Arthritis: Interleukin 6 Inhibits Wnt Signaling in Synovial Fibroblasts and Osteoblasts. Croat Med J (2016) 57:89–98. doi: 10.3325/cmj.2016.57.89
79. Fassio A, Adami G, Giollo A, Viapiana O, Malavolta N, Saviola G, et al. Acute Effects of Glucocorticoid Treatment, Tnfα or IL-6R Blockade on Bone Turnover Markers and Wnt Inhibitors in Early Rheumatoid Arthritis: A Pilot Study. Calcif Tissue Int (2020) 106:371–7. doi: 10.1007/s00223-019-00649-3
80. Zerbini C a. F, Clark P, Mendez-Sanchez L, Pereira RMR, Messina OD, Uña CR, et al. Biologic Therapies and Bone Loss in Rheumatoid Arthritis. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA (2017) 28:429–46. doi: 10.1007/s00198-016-3769-2
81. Neogi T, Aletaha D, Silman AJ, Naden RL, Felson DT, Aggarwal R, et al. The 2010 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Rheumatoid Arthritis: Phase 2 Methodological Report. Arthritis Rheum (2010) 62:2582–91. doi: 10.1002/art.27580
82. Straub RH. The Complex Role of Estrogens in Inflammation. Endocr Rev (2007) 28:521–74. doi: 10.1210/er.2007-0001
83. Bengtsson C, Malspeis S, Orellana C, Sparks JA, Costenbader KH, Karlson EW. Menopausal Factors are Associated With Seronegative RA in Large Prospective Cohorts: Results From the Nurses’ Health Studies. Arthritis Care Res (2017) 69:1676–84. doi: 10.1002/acr.23194
84. Chen JY, Ballou SP. The Effect of Antiestrogen Agents on Risk of Autoimmune Disorders in Patients With Breast Cancer. J Rheumatol (2015) 42:55–9. doi: 10.3899/jrheum.140367
85. Alpízar-Rodríguez D, Finckh A. Environmental Factors and Hormones in the Development of Rheumatoid Arthritis. Semin Immunopathol (2017) 39:461–8. doi: 10.1007/s00281-017-0624-2
86. Berglin E, Kokkonen H, Einarsdottir E, Agren A, Rantapää Dahlqvist S. Influence of Female Hormonal Factors, in Relation to Autoantibodies and Genetic Markers, on the Development of Rheumatoid Arthritis in Northern Sweden: A Case-Control Study. Scand J Rheumatol (2010) 39:454–60. doi: 10.3109/03009741003742763
87. Pedersen M, Jacobsen S, Klarlund M, Pedersen BV, Wiik A, Wohlfahrt J, et al. Environmental Risk Factors Differ Between Rheumatoid Arthritis With and Without Auto-Antibodies Against Cyclic Citrullinated Peptides. Arthritis Res Ther (2006) 8:R133. doi: 10.1186/ar2022
88. Chen Q, Jin Z, Xiang C, Cai Q, Shi W, He J. Absence of Protective Effect of Oral Contraceptive Use on the Development of Rheumatoid Arthritis: A Meta-Analysis of Observational Studies. Int J Rheum Dis (2014) 17:725–37. doi: 10.1111/1756-185X.12413
89. Silman A, Kay A, Brennan P. Timing of Pregnancy in Relation to the Onset of Rheumatoid Arthritis. Arthritis Rheum (1992) 35:152–5. doi: 10.1002/art.1780350205
90. Guthrie KA, Dugowson CE, Voigt LF, Koepsell TD, Nelson JL. Does Pregnancy Provide Vaccine-Like Protection Against Rheumatoid Arthritis? Arthritis Rheum (2010) 62:1842–8. doi: 10.1002/art.27459
91. Peschken CA, Robinson DB, Hitchon CA, Smolik I, Hart D, Bernstein CN, et al. Pregnancy and the Risk of Rheumatoid Arthritis in a Highly Predisposed North American Native Population. J Rheumatol (2012) 39:2253–60. doi: 10.3899/jrheum.120269
92. Wallenius M, Skomsvoll JF, Irgens LM, Salvesen KÅ, Koldingsnes W, Mikkelsen K, et al. Postpartum Onset of Rheumatoid Arthritis and Other Chronic Arthritides: Results From a Patient Register Linked to a Medical Birth Registry. Ann Rheum Dis (2010) 69:332–6. doi: 10.1136/ard.2009.115964
93. Rovensky J, Kvetnansky R, Radikova Z, Imrich R, Greguska O, Vigas M, et al. Hormone Concentrations in Synovial Fluid of Patients With Rheumatoid Arthritis. Clin Exp Rheumatol (2005) 23:292–6.
94. Tang MW, Garcia S, Gerlag DM, Tak PP, Reedquist KA. Insight Into the Endocrine System and the Immune System: A Review of the Inflammatory Role of Prolactin in Rheumatoid Arthritis and Psoriatic Arthritis. Front Immunol (2017) 8:720. doi: 10.3389/fimmu.2017.00720
95. McMurray RW. Estrogen, Prolactin, and Autoimmunity: Actions and Interactions. Int Immunopharmacol (2001) 1:995–1008. doi: 10.1016/s1567-5769(01)00045-5
96. Clapp C, Adán N, Ledesma-Colunga MG, Solís-Gutiérrez M, Triebel J. Martínez De La Escalera G. The Role of the Prolactin/Vasoinhibin Axis in Rheumatoid Arthritis: An Integrative Overview. Cell Mol Life Sci CMLS (2016) 73:2929–48. doi: 10.1007/s00018-016-2187-0
97. Ram S, Blumberg D, Newton P, Anderson NR, Gama R. Raised Serum Prolactin in Rheumatoid Arthritis: Genuine or Laboratory Artefact? Rheumatol Oxf Engl (2004) 43:1272–4. doi: 10.1093/rheumatology/keh307
98. Mateo L, Nolla JM, Bonnin MR, Navarro MA, Roig-Escofet D. High Serum Prolactin Levels in Men With Rheumatoid Arthritis. J Rheumatol (1998) 25:2077–82.
99. Orbach H, Zandman-Goddard G, Amital H, Barak V, Szekanecz Z, Szucs G, et al. Novel Biomarkers in Autoimmune Diseases. Ann N Y Acad Sci (2007) 1109:385–400. doi: 10.1196/annals.1398.044
100. Tang MW, Reedquist KA, Garcia S, Fernandez BM, Codullo V, Vieira-Sousa E, et al. The Prolactin Receptor is Expressed in Rheumatoid Arthritis and Psoriatic Arthritis Synovial Tissue and Contributes to Macrophage Activation. Rheumatol Oxf Engl (2016) 55:2248–59. doi: 10.1093/rheumatology/kew316
101. Gladman D, Antoni C, Mease P, Clegg D, Nash P. Psoriatic Arthritis: Epidemiology, Clinical Features, Course, and Outcome. Ann Rheum Dis (2005) 64:ii14–7. doi: 10.1136/ard.2004.032482
102. Heiland GR, Appel H, Poddubnyy D, Zwerina J, Hueber A, Haibel H, et al. High Level of Functional Dickkopf-1 Predicts Protection From Syndesmophyte Formation in Patients With Ankylosing Spondylitis. Ann Rheum Dis (2012) 71:572–4. doi: 10.1136/annrheumdis-2011-200216
103. Raychaudhuri SP. Role of IL-17 in Psoriasis and Psoriatic Arthritis. Clin Rev Allergy Immunol (2013) 44:183–93. doi: 10.1007/s12016-012-8307-1
104. Zrioual S, Toh M-L, Tournadre A, Zhou Y, Cazalis M-A, Pachot A, et al. Il-17RA and IL-17RC Receptors are Essential for IL-17A-induced Elr+ CXC Chemokine Expression in Synoviocytes and are Overexpressed in Rheumatoid Blood. J Immunol Baltim Md 1950 (2008) 180:655–63. doi: 10.4049/jimmunol.180.1.655
105. Nguyen CT, Maverakis E, Eberl M, Adamopoulos IE. γδ T Cells in Rheumatic Diseases: From Fundamental Mechanisms to Autoimmunity. Semin Immunopathol (2019) 41:595–605. doi: 10.1007/s00281-019-00752-5
106. Maeda S, Hayami Y, Naniwa T, Ueda R. The Th17/IL-23 Axis and Natural Immunity in Psoriatic Arthritis. Int J Rheumatol (2012) 2012:539683. doi: 10.1155/2012/539683
107. Fassio A, Gatti D, Rossini M, Idolazzi L, Giollo A, Adami G, et al. Secukinumab Produces a Quick Increase in WNT Signalling Antagonists in Patients With Psoriatic Arthritis. Clin Exp Rheumatol (2019) 37:133–6.
108. Simon D, Kleyer A, Bayat S, Tascilar K, Kampylafka E, Meinderink T, et al. Effect of Disease-Modifying Anti-Rheumatic Drugs on Bone Structure and Strength in Psoriatic Arthritis Patients. Arthritis Res Ther (2019) 21:162. doi: 10.1186/s13075-019-1938-3
109. Maksymowych WP, Chiowchanwisawakit P, Clare T, Pedersen SJ, Østergaard M, Lambert RGW. Inflammatory Lesions of the Spine on Magnetic Resonance Imaging Predict the Development of New Syndesmophytes in Ankylosing Spondylitis: Evidence of a Relationship Between Inflammation and New Bone Formation. Arthritis Rheum (2009) 60:93–102. doi: 10.1002/art.24132
110. Poggenborg RP, Wiell C, Bøyesen P, Boonen A, Bird P, Pedersen SJ, et al. No Overall Damage Progression Despite Persistent Inflammation in Adalimumab-Treated Psoriatic Arthritis Patients: Results From an Investigator-Initiated 48-Week Comparative Magnetic Resonance Imaging, Computed Tomography and Radiography Trial. Rheumatology (2014) 53:746–56. doi: 10.1093/rheumatology/ket426
111. van der Heijde D, Salonen D, Weissman BN, Landewé R, Maksymowych WP, Kupper H, et al. Assessment of Radiographic Progression in the Spines of Patients With Ankylosing Spondylitis Treated With Adalimumab for Up to 2 Years. Arthritis Res Ther (2009) 11:R127. doi: 10.1186/ar2794
112. Landewé R, Ritchlin CT, Aletaha D, Zhang Y, Ganz F, Hojnik M, et al. Inhibition of Radiographic Progression in Psoriatic Arthritis by Adalimumab Independent of the Control of Clinical Disease Activity. Rheumatol Oxf Engl (2019) 58:1025–33. doi: 10.1093/rheumatology/key417
113. Molnar C, Scherer A, Baraliakos X, Hooge M, Micheroli R, Exer P, et al. TNF Blockers Inhibit Spinal Radiographic Progression in Ankylosing Spondylitis by Reducing Disease Activity: Results From the Swiss Clinical Quality Management Cohort. Ann Rheum Dis (2018) 77:63–9. doi: 10.1136/annrheumdis-2017-211544
114. Carina V, Della Bella E, Costa V, Bellavia D, Veronesi F, Cepollaro S, et al. Bone’s Response to Mechanical Loading in Aging and Osteoporosis: Molecular Mechanisms. Calcif Tissue Int (2020) 107:301–18. doi: 10.1007/s00223-020-00724-0
115. Zachary C, Fackler N, Juhasz M, Pham C, Mesinkovska NA. Catamenial Dermatoses Associated With Autoimmune, Inflammatory, and Systemic Diseases: A Systematic Review. Int J Womens Dermatol (2019) 5:361–7. doi: 10.1016/j.ijwd.2019.09.008
116. Fassio A, Adami G, Idolazzi L, Giollo A, Viapiana O, Vantaggiato E, et al. Wnt Inhibitors and Bone Turnover Markers in Patients With Polymyalgia Rheumatica and Acute Effects of Glucocorticoid Treatment. Front Med (2020) 7:551. doi: 10.3389/fmed.2020.00551
117. Piera-Velazquez S, Mendoza FA, Jimenez SA. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. J Clin Med (2016) 5(4):45. doi: 10.3390/jcm5040045
118. Long L, Liu Y, Wang S, Zhao Y, Guo J, Yu P, et al. Dickkopf-1 as Potential Biomarker to Evaluate Bone Erosion in Systemic Lupus Erythematosus. J Clin Immunol (2010) 30:669–75. doi: 10.1007/s10875-010-9436-z
119. Sarkissian A, Sivaraman V, Bout-Tabaku S, Ardoin SP, Moore-Clingenpeel M, Mruk V, et al. Bone Turnover Markers in Relation to Vitamin D Status and Disease Activity in Adults With Systemic Lupus Erythematosus. Lupus (2019) 28:156–62. doi: 10.1177/0961203318815593
120. Buttgereit F, Dejaco C, Matteson EL, Dasgupta B. Polymyalgia Rheumatica and Giant Cell Arteritis: A Systematic Review. JAMA (2016) 315:2442. doi: 10.1001/jama.2016.5444
121. Caimmi C, Caramaschi P, Barausse G, Orsolini G, Idolazzi L, Gatti D, et al. Bone Metabolism in a Large Cohort of Patients With Systemic Sclerosis. Calcif Tissue Int (2016) 99:23–9. doi: 10.1007/s00223-016-0119-5
122. Piersma B, Bank RA, Boersema M. Signaling in Fibrosis: Tgf-β, WNT, and YAP/TAZ Converge. Front Med (2015) 2:59. doi: 10.3389/fmed.2015.00059
123. Ruaro B, Casabella A, Paolino S, Pizzorni C, Ghio M, Seriolo C, et al. Dickkopf-1 (Dkk-1) Serum Levels in Systemic Sclerosis and Rheumatoid Arthritis Patients: Correlation With the Trabecular Bone Score (Tbs). Clin Rheumatol (2018) 37:3057–62. doi: 10.1007/s10067-018-4322-9
124. Taylan A, Birlik M, Kenar G, Toprak B, Gundogdu B, Gurler O, et al. Osteoprotegrin Interacts With Biomarkers and Cytokines That Have Roles in Osteoporosis, Skin Fibrosis, and Vasculopathy in Systemic Sclerosis: A Potential Multifaceted Relationship Between OPG/RANKL/TRAIL and Wnt Inhibitors. Mod Rheumatol (2019) 29:619–24. doi: 10.1080/14397595.2018.1500736
125. Dovio A, Data V, Carignola R, Calzolari G, Vitetta R, Ventura M, et al. Circulating Osteoprotegerin and Soluble Rank Ligand in Systemic Sclerosis. J Rheumatol (2008) 35:2206–13. doi: 10.3899/jrheum.080192
126. Harper E, Rochfort KD, Forde H, Davenport C, Smith D, Cummins PM. TRAIL Attenuates RANKL-mediated Osteoblastic Signalling in Vascular Cell Mono-Culture and Co-Culture Models. PloS One (2017) 12(11):e0188192. doi: 10.1371/journal.pone.0188192
127. Tsokos GC. Systemic Lupus Erythematosus. N Engl J Med (2011) 365:2110–21. doi: 10.1056/NEJMra1100359
128. Bultink IEM, Lems WF. Lupus and Fractures. Curr Opin Rheumatol (2016) 28:426–32. doi: 10.1097/BOR.0000000000000290
129. Park S-G, Hwang S, Kim J-S, Park K-C, Kwon Y, Kim K-C. The Association Between Dehydroepiandrosterone Sulfate (Dhea-S) and Bone Mineral Density in Korean Men and Women. J Bone Metab (2017) 24:31–6. doi: 10.11005/jbm.2017.24.1.31
130. Tedeschi SK, Kim SC, Guan H, Grossman JM, Costenbader KH. Comparative Fracture Risks Among US Medicaid Enrollees With and Without Systemic Lupus Erythematosus. Arthritis Rheumatol Hoboken NJ (2019) 71:1141–6. doi: 10.1002/art.40818
Keywords: bone metabolism, hormones, bone turnover markers, rheumatic disorders, parathyroid hormone
Citation: Bertoldo E, Adami G, Rossini M, Giollo A, Orsolini G, Viapiana O, Gatti D and Fassio A (2021) The Emerging Roles of Endocrine Hormones in Different Arthritic Disorders. Front. Endocrinol. 12:620920. doi: 10.3389/fendo.2021.620920
Received: 24 October 2020; Accepted: 20 April 2021;
Published: 21 May 2021.
Edited by:
Zhong Zheng, University of California, Los Angeles, United StatesReviewed by:
Sergio Minucci, Università della Campania Luigi Vanvitelli, ItalyMiles Douglas Thompson, Rady Children’s Hospital-San Diego, University of California, San Diego, United States
Copyright © 2021 Bertoldo, Adami, Rossini, Giollo, Orsolini, Viapiana, Gatti and Fassio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eugenia Bertoldo, ZXVnZW5pYS5iZXJ0b2xkb0BnbWFpbC5jb20=