
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 19 March 2021
Sec. Thyroid Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.620117
This article is part of the Research Topic Inborn Errors of Synthesis and Sensitivity to Thyroid Hormone View all 11 articles
 Cao-Xu Zhang1†Jun-Xiu Zhang2†Liu Yang1Chang-Run Zhang1Feng Cheng3
Cao-Xu Zhang1†Jun-Xiu Zhang2†Liu Yang1Chang-Run Zhang1Feng Cheng3 Rui-Jia Zhang1Ya Fang1Zheng Wang1
Rui-Jia Zhang1Ya Fang1Zheng Wang1 Feng-Yao Wu1Pei-Zhang Li1
Feng-Yao Wu1Pei-Zhang Li1 Jun Liang4*Rui Li1*
Jun Liang4*Rui Li1* Huai-Dong Song1*
Huai-Dong Song1*Background and Objectives: Defects in the human sodium/iodide symporter (SLC5A5) gene have been reported to be one of the causes of congenital hypothyroidism (CH). We aimed to identify SLC5A5 mutations in Chinese patients with CH and to evaluate the function of the mutation.
Methods: Two hundred and seventy-three patients with primary CH were screened for mutations in SLC5A5 using next-generation sequencing. We investigated the expression and cellular localization of the novel compound heterozygous mutation in SLC5A5. The functional activity of the mutants was further examined in vitro.
Results: In 273 patients with CH, two previously undescribed pathogenic mutations p.Gly51AlafsTer45 (G51fs) and p.Gly421Arg (G421R) in a compound heterozygous state in SLC5A5 were identified in a pediatric patient. G51fs was located in the first intercellular loop connecting transmembrane segment I and II, whereas G421R was in the transmembrane segment (TMS) XI. G51fs and G421R resulted in a truncated NIS and reduced protein expression, respectively. In vitro experiments further showed that the normal function of iodine transport of sodium-iodide symporter (NIS) mutants was markedly impaired.
Conclusion: The undescribed compound heterozygous mutation of SLC5A5 was discovered in a Chinese CH patient. The mutation led to significantly reduced NIS expression and impaired iodide transport function accompanied by the impaired location of the NIS on the plasma membrane. Our study thus provides further insights into the roles of SLC5A5 in CH pathogenesis.
Congenital hypothyroidism (CH), which is defined by inadequate thyroid hormone production in newborn infants, is a common neonatal endocrine disorder with the incidence at about 1:2,000–4,000 worldwide (1). Thyroid hormones are critical for neurodevelopment; severe congenital hypothyroidism can lead to growth retardation and permanent intellectual disability (1, 2).
The human SLC5A5 (GenBank reference sequence: NM_000453.3) gene, encoding the sodium-iodide symporter (NIS), a 643 amino acid protein, is located on chromosome 19 and consists of 15 exons (3). The NIS protein is composed of 13 transmembrane segments (TMS), an extracellular amino terminus, an intracellular carboxy terminus and three asparagine-linked glycosylation sites. Previous research showed that glycosylation was not critical for NIS targeting to the plasma membrane or normal function (4).
NIS mediates the active transport of iodide from the bloodstream into the tissues, particularly in the thyroid grand, by coupling the inward translocation of Na+ down its electrochemical gradient to the inward movement of I- against its electrochemical gradient. The driving force for this process is the electrochemical sodium gradient generated by Na+/K+ ATPase (2, 5–9). Sufficient dietary iodine intake is essential for the production of the thyroid hormones. In thyroid tissue, iodide uptake is typically the first step in thyroid hormone synthesis. The SLC5A5 is responsible for the transport of iodide mainly in the thyroid gland (2, 10). The mutation of SLC5A5 is a cause of thyroid dyshormonogenesis, which leads to CH in patients. Currently, several iodine transport defect-related mutations of SLC5A5 have been identified, including G18R, V59E, G93R, R124H, Q267E, V270E, C272X, D331N, Y348D, T354P, G395R, R516X, G543E, and S547R, which have provided valuable structural evidence about the symporter and helped us to understand the pathogenesis of this disease (11–24).
The reported mutations of SLC5A5 almost all affect its normal functional activity, leading to thyroid dyshormonogenesis (5, 10, 25). Our study, therefore, aimed to identify additional mutations in SLC5A5 from the Chinese patients with CH and analyzed the function of the mutations in vitro, to evaluate the roles of SLC5A5 in the pathogenesis of CH in the Chinese patients.
Herein, we report a novel compound heterozygous missense mutation G51fs/G421R of SLC5A5 gene in a pediatric patient diagnosed as congenital hypothyroidism from newborn screening. Our results showed that G51fs and G421R markedly affected the function of iodine intake accompanied by impairment of the location of NIS on the plasma membrane. Our study provided further insights into the roles of SLC5A5 in CH pathogenesis.
All subjects were recruited from the Chinese Han population in Jiangsu province, Fujian province, Anhui province, and Shanghai through collaboration with multiple hospitals in China. A total of 273 CH patients (141 females and 132 males) identified through newborn screening were enrolled in this study. The diagnosis of permanent CH was confirmed in infants based on the following criteria: CH patients with elevated TSH (thyrotropin) levels, with or without T4 (thyroxine) or FT4 (free thyroxine) levels less than the normal range, and the restoration of normal thyroid parameters after receiving replacement therapy with L-thyroxine; however, after stopping treatment, a rise in TSH and a drop in FT4 were observed again (26–28). Written consent was obtained from the parents of the CH patients, and the study was approved by the Ethics Committee of Shanghai Ninth People’s Hospital affiliated with the Shanghai Jiao Tong University School of Medicine.
The sequencing methods and analysis pipelines used were previously reported (28). Genomic DNA was extracted from the whole blood of the probands and family members if available according to standard extraction procedures. All the exons and exon-intron boundaries of SLC5A5 were amplified by performing multiplex polymerase chain reaction (PCR) using a 48×48Access Array™ microfluidic platform (Fluidigm) according to the manufacturer’s protocol. The primers were designed using iPLEX Assay Design software (Sequenom). The HiSeq3000 platform (Illumina, San Diego, CA) was used to perform deep sequencing of these amplicon libraries.
The target sequences were amplified and deep sequenced in duplicate for each sample to avoid base pair variants caused by multiplex PCR. Descriptions of the SLC5A5 mutations were based on NM_000453.3. We analyzed the raw sequence data in fastq format and obtained the quality scores by following the method indicated by previous studies (28). We filtered out the variants with frequencies >1% in the dbSNP 135 and ESP6500 v2 databases and focused on the functional (protein altering) variants (removal of intergenic and 3’/5’UTR (untranslated region) variants, non-splice-related intronic variants, and synonymous variants) identified in duplicate samples (https://doi.org/10.6084/m9.figshare.13134824.v1). Then, the two remaining variants were selected for validation by Sanger sequencing.
The ORF (open reading frame, nucleotide 1 to 1932) of human wild-type (WT) SLC5A5 was amplified by PCR from genomic DNA using primers containing the EcoRI and XbaI restriction sites. The amplified PCR fragment was inserted into a p3xFLAG-CMV-10 expression vector and the FLAG tag was inserted upstream to the NIS coding region. Similarly, a WT NIS-EGFP fusion protein was expressed in a pEGFP-N2 expression vector (TransGen Biotech) using EcoRI and BamHI restriction sites, and the GFP was tagged downstream to the NIS coding region. The missense mutations were generated by site-directed mutagenesis using the Fast Mutagenesis System kit (TransGen Biotech). Due to premature termination of NIS, p.Gly51AlafsTer45 (G51fs) containing the EcoRI and BamHI restriction sites was cloned into the pEGFP-N2 vector to generate the fusion plasmid. All the plasmid constructs were verified by Sanger sequencing. The primers used are listed in Supplemental Table 3.
The human embryonic kidney 293T cell line was cultured in Dulbecco’s modified Eagle’s medium (DMEM)/high-glucose medium (Gibco) supplemented with 10% fetal bovine serum (FBS; Sigma Aldrich) at 37 °C in a humidified atmosphere containing 5% CO2. Transfections were performed on cells by Lipofectamine 2000 Transfection Reagent (Invitrogen) following the manufacturer’s guides. Cells were plated in a 20 mm glass bottom cell culture dish (NEST) and transiently transfected with 1 µg plasmid DNA to detect the cell localization of the WT or mutants of SLC5A5-pEGFP-N2 plasmids. Iodide uptake assays were performed on the 293T cells. The cells were cultured in 12-well plates, transfected with 1 µg WT or mutant 3xFLAG-NIS plasmids (cotransfection with 0.5 µg WT and 0.5 µg mutant plasmids to simulate the heterozygous state), and the treated cells were then kept in a humidified incubator at 37 °C with 5% CO2 for 48 h.
RNA extraction was performed as described previously (27). RNA was prepared using Trizol reagent (Invitrogen, H10522) and the total RNA (1 μg) was reverse transcribed using the cDNA synthesis kit (Takara). Quantitative PCR (qPCR) was performed using TB Green® Premix Ex Taq™ II (Tli RNaseH Plus), ROX plus (Takara), and the ABI QuantStudio 12K Flex Real-time PCR System (Life Technologies). The primers used are described in Supplemental Table 3.
The iodide uptake assay was performed as described previously (29). In brief, 293T cells were seeded in 12-well plates, which contained sterilized cover slips (WHB Biotech), incubated at 37°C for 24 h, and transiently transfected alone or co-transfected with WT or mutant pEGFP-N2 plasmids. 48 h after transfection, the 293T cells were washed once in serum-free DMEM medium and incubated for 1 h in 1 ml serum-free medium containing 131I at 5 KBq/ml as the only source of iodide. For the inhibition of NIS-mediated uptake, NaClO4 (final concentration 1 mM) was included in parallel incubations (30). The cells were washed briefly in Hank’s Balanced Salt Solution (HBSS) buffer and then incubated with 1 ml HBSS for 5 min. The cells were solubilized by the addition of 1 ml 1 M NaOH, and the radioactivity was measured using a γ counter (GC1200, Anhui, China). DNA was determined by the Qubit Fluorometer using ssDNA Assay Kit (Q10212, Thermofisher). Each value represents the mean ± SD of picomoles 131I per microgram of DNA of three independent experiments done in triplicate.
Representative Western blot analysis of whole cell lysates from transiently transfected 293T cells was probed with anti-FLAG antibodies (AE063, ABclonal) and anti-GAPDH (AM4300, Thermo Fisher Scientific) antibodies. Proteins were visualized by the Odyssey CLx Infra-Red Imaging system.
The 293T cells were seeded in 12-well plates, which contained sterilized cover slips (WHB Biotech), incubated at 37 °C for 24 h, and transiently transfected with WT or mutant pEGFP-N2 plasmids. One day later, the cells were washed twice with PBS (phosphate buffer saline), fixed with 4% PBS-buffered formaldehyde (PFA) for 30 min at room temperature, washed twice with PBS, and then incubated with fluorescent probe Dil (Beyotime Biotech) targeted to the cell membrane for 5 min at 37°C. After washing with PBS, the nuclei were stained with DAPI (Beyotime Biotech) at room temperature for 5 min. Cover slips were mounted, and the plates were examined using a confocal microscope (Nikon A1 Microsystems).
The sequence of SLC5A5 was analyzed using the SWISS-MODEL (31). The 3D structure of the Vibrio parahaemolyticus sodium/galactose symporter (vSGLT) was chosen as the modeling template. Theoretical modeling of the protein structure was performed using PyMOL 2.4 (https://pymol.org/2/).
Statistical analysis was performed using Prism 8.0 software (GraphPad Software) from more than three independent experiments. Data were presented as the mean ± SD. Comparisons between two groups were analyzed using unpaired two-tailed Student’s t test (*p < 0.05, **p < 0.01, and ***p < 0.001). Differences were considered statistically significant at values of p < 0.05.
In our study, mutations in SLC5A5 for congenital hypothyroidism were screened for in the cohort of 273 CH patients. Only one patient was found to carry the mutations in SLC5A5 with no other mutations in the reported candidate genes. The proband was a full-term girl, born from healthy non-consanguineous Chinese parents. Newborn screening found an abnormally high TSH level (specific value unknown). At 18 days after birth, diagnostic confirmation of congenital hypothyroidism was achieved by measuring the serum TSH at >100 uIU/ml (normal range 0.34–5.6 uIU/ml), free T4 0.45 ng/dl (normal range 0.58-1.64ng/dL), and free T3 0.85 pg/ml (free triiodothyronine normal range 2.5–3.9pg/ml) (Table 1). Her height was 54.5 cm, and her weight was 3.3 kg.
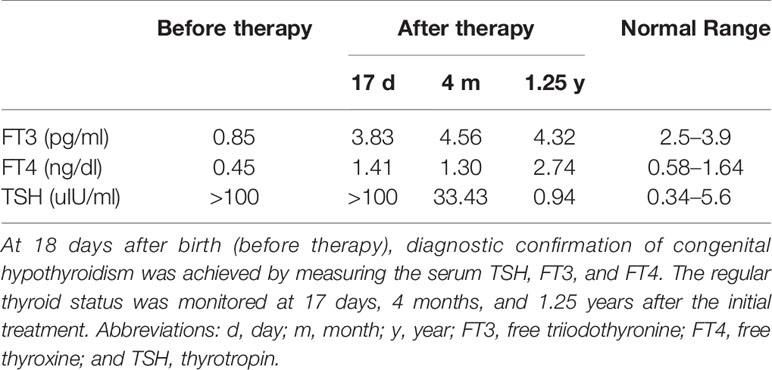
Table 1 Thyroid function tests during follow-up before and after thyroid hormone replacement therapy.
Thyroid hormone supplementation was started immediately after diagnosis, with a daily dose of 28 μg levothyroxine. However, 17 days after initial treatment, thyroid function was still abnormal (TSH level >100 uIU/ml; FT4:1.41 ng/dl; FT3:3.83 pg/ml) (Table 1), and the local pediatrician adjusted the daily dose of levothyroxine to 40 μg. Four months after treatment, her height was 75 cm, and weight was 10 kg. Her thyroid function was slightly improved but still abnormal (TSH:33.43 uIU/ml; FT4:1.3 ng/dL; FT3:4.56 pg/ml) (Table 1). At the age of 1 years and 3 months, the proband was recruited to our study. Her height was 81.5 cm, and her weight was 11 kg.
The TSH level returned to the normal range during thyroid hormone replacement therapy (TSH:0.94 uIU/ml; FT4:2.74 ng/dL; FT3:4.32 pg/ml) (Table 1). Due to poor compliance, the results of thyroid scintigraphy and the concentration of serum thyroglobulin and autoantibody were unavailable. Normal growth was observed during the first years of life. Of note, the patient’s hypothyroidism did not appear to affect her intellectual development. Thyroid ultrasonography showed a normal size and well-located gland. No other members of the family had a known history of hypothyroidism. The proband’s parents were euthyroid with no autoantibodies and normal thyroid volume by ultrasound.
Further molecular analysis of the whole blood of the proband and family members revealed the presence of an undescribed compound heterozygous mutation G51fs/G421R. Based on the current secondary structure model for NIS, G51fs is located in the first intercellular loop connecting transmembrane segment I and II, whereas G421R is in the TMS XI (Figure 1). Sequencing of the patient’s SLC5A5 gene revealed a G deletion at nucleotide +152 in exon 1, resulting in a frameshift mutation and, thus, premature termination of NIS and a loss of about 85% of the amino acids of the protein (Figure 2A, Supplemental Table 1). The patient also had a novel heterozygous G>A transversion at nucleotide +1261 in exon 11, which leads to the substitution of Gly by Arg at residue 421 (Figure 2A).
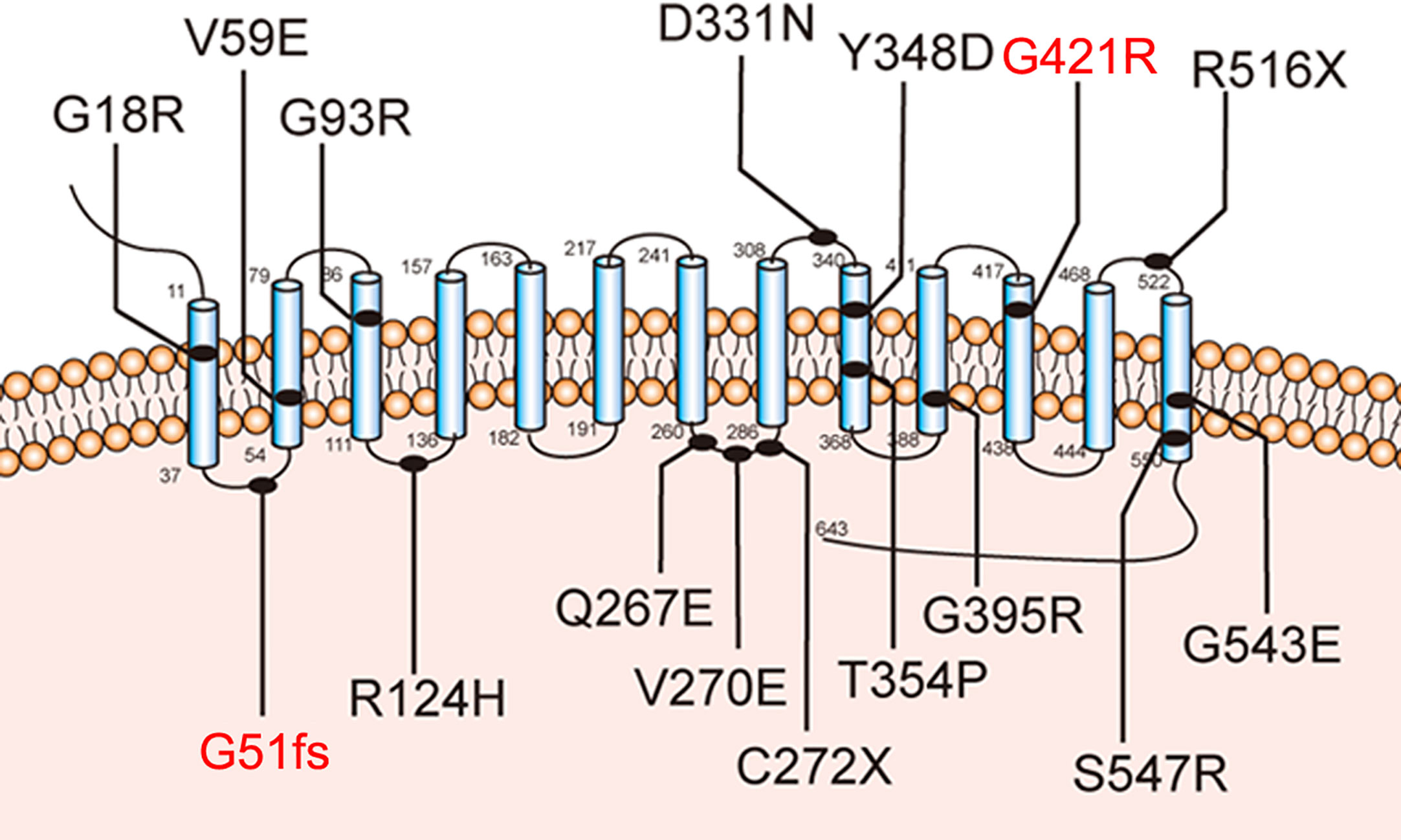
Figure 1 Sodium-iodide symporter (NIS) secondary structure model. Blue cylinders represent the 13 transmembrane segments (TMS). The amino-terminus faces the extracellular space and the carboxy-terminus is in the cytosol. The novel pathogenic variants p.G51fs and p.G421R are highlighted with blue lines.
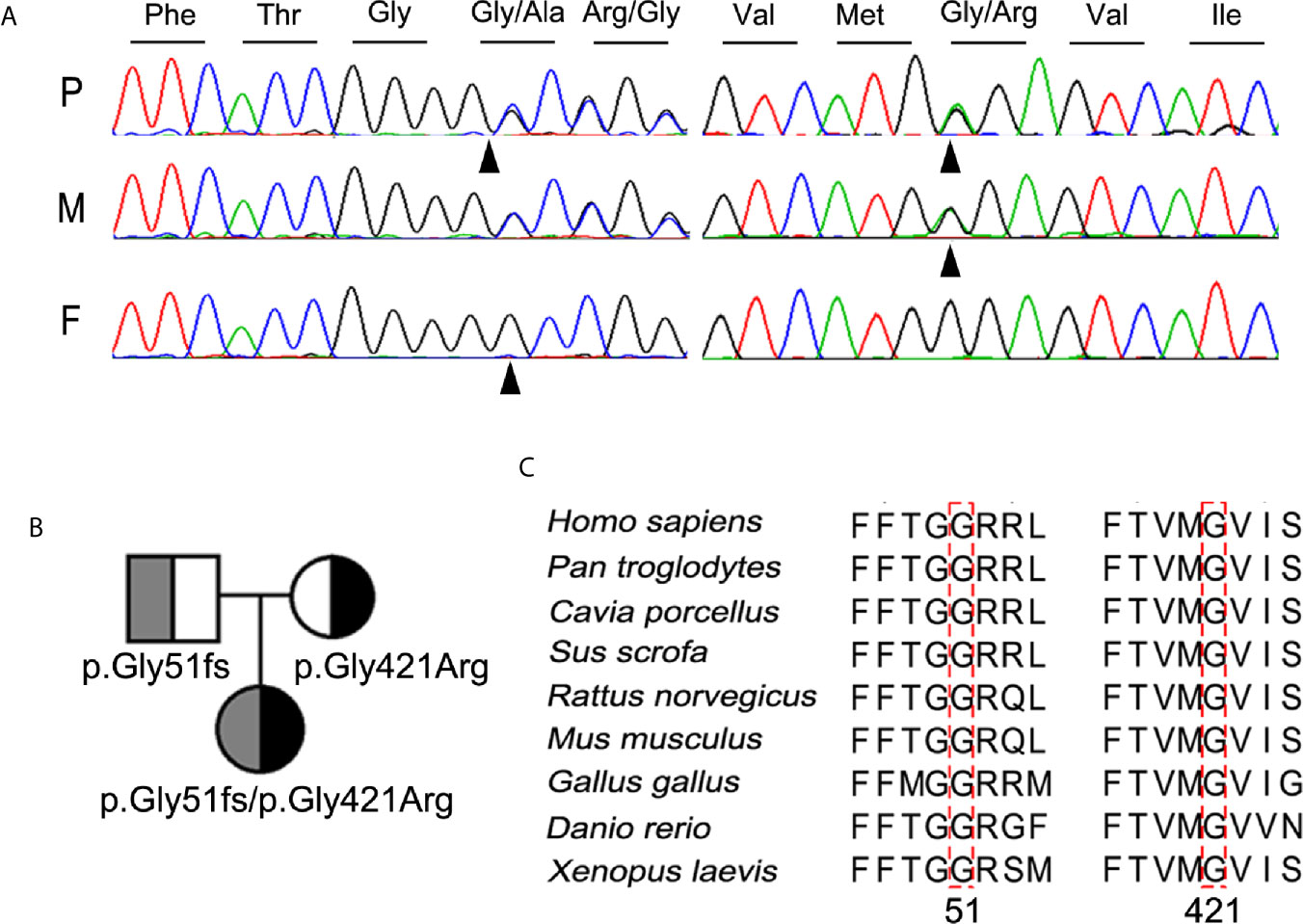
Figure 2 Identification and characterization of the compound heterozygous NIS mutation p.G51fs/p.G421R. (A) Partial sequence chromatograms cover the region of the variants in exon 1 (c.152del, p.G51fs) and 11 (c.1261G>A, p.G421R) of SLC5A5 gene. The nucleotide and corresponding amino acid sequences using the three-letter code are indicated. The proband was compound heterozygous for the mutant alleles p.G51fs and p.G421R NIS. Arrowheads denote the mutated sites. P, proband; F, father; M, mother. (B) Pedigree analysis of the members of the proband’s family. The mutated alleles for p.G51fs and p.G421R NIS are noted below. (C) Amino acid sequences of NIS from other metazoan species were aligned with those of humans by PSI/TM-Coffee (http://tcoffee.crg.cat/apps/tcoffee/index.html). The mutated amino acids in all NIS homologs are indicated by dotted boxes.
The mutations contribute to a compound heterozygous mutation G51fs/G421R in NIS (Figure 2B). On the other hand, analysis of the euthyroid parents of the proband showed that the father was heterozygous for G51fs NIS, and the mother was heterozygous for G421R NIS, suggesting recessive inheritance of the mutation (Figures 2A, B). This information is summarized in a pedigree diagram (Figure 2B). Multiple amino acid sequence alignment of NIS orthologs from different species revealed that two glycine residues at position 51 and 421 were highly conserved (Figure 2C).
We next used the Vibrio parahaemolyticus sodium/galactose symporter (vSGLT) as a template to model the 3D structure of G421R. The substitution of the neutral side chain of Gly by the basic amino acid arginine residue at position 421 changed the net surface positive charges and distorted its surface structure (Figure 3A). We observed that the interatomic distance between G421 and T354 was changed from 15.0 Å to 8.7 Å (Figure 3B). Substituting gly with a bulkier arginine destabilized the helix by causing steric hindrance between the arginine side chain and the nearby amino acid side chains. We further utilized bioinformatic tools to predict the effects of G421R. G421R was categorized by PolyPhen-2 as being “Probably Damaging” with a score of 0.999, and by SIFT as “Damaging” with a score of 0.001 (Supplemental Table 2).
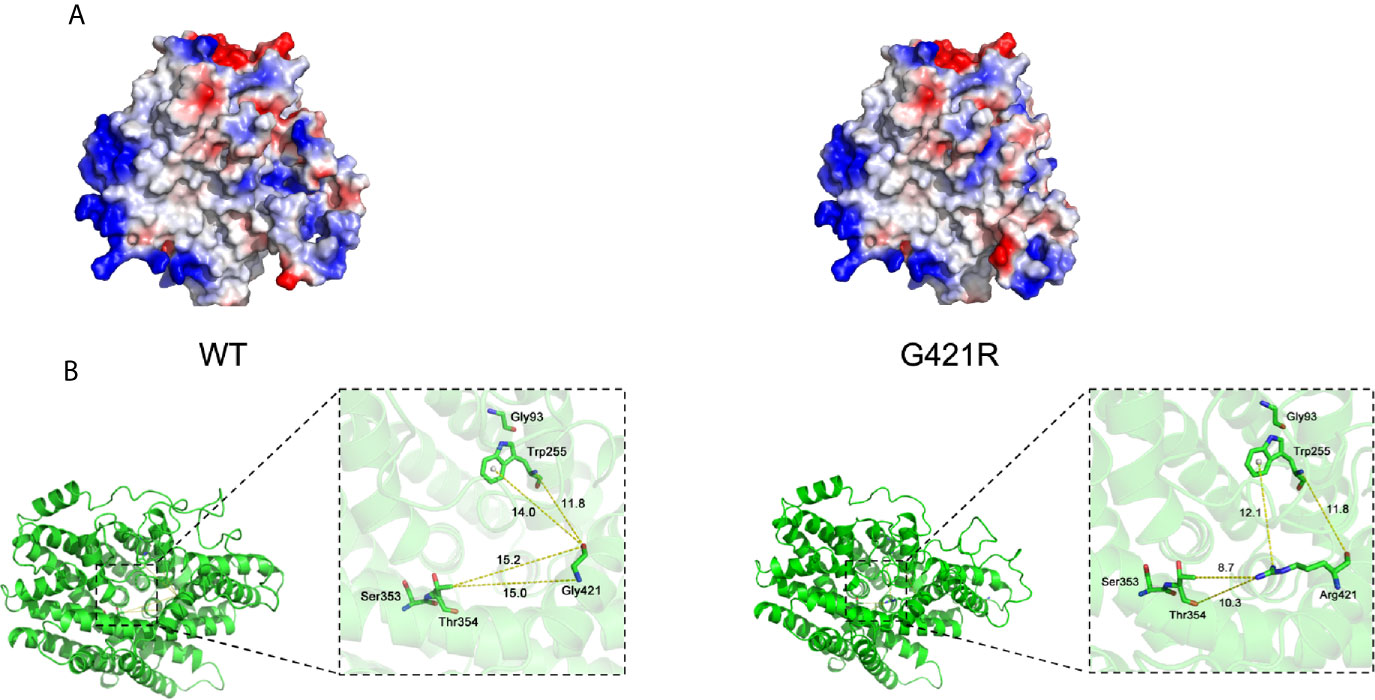
Figure 3 Electrostatic surface and inter-atomic distances of WT NIS and G421R. (A) Local solid surface and electrostatic potentials are greatly changed by G421R mutation. Positive and negative potentials are shown in blue and red, respectively. (B) The cartoon representation of the crystal structure of NIS based on vSGLT. Those previously reported residues, which are particularly close to the mutation site, are shown in the magnified regions. Dotted lines connect atoms for which inter-atomic distances are given in angstroms. The specific values are shown in the figure. The left part of figure represents WT NIS, and the right G421R.
To assess the effect of mutations on the activity of iodide transport, we expressed wild-type or mutant 3xFLAG-NIS plasmids in 293T cells. A p3xFLAG-CMV-10 empty vector was employed as a negative control, and a reported D331N mutation construct was used as the positive control. Quantitative PCR analysis was performed to ensure effective transcription (Supplemental Figure 1A). Western Blot analysis was done to investigate the effect of G51fs and G421R on protein expression. No band was detected in 293T cells transfected with empty vector (Supplementary Figure 1B), while truncated NIS with a molecular weight of 13 kDa (monomer) and 26 kDa (dimer) were observed after G51fs transfection (Figure 4A). Moreover, protein expression was decreased in 293T cells transfected with the G421R mutant plasmid compared to those transfected with wild-type (immature unglycosylated NIS ~ 55kDa, mature glycosylated NIS~ 100 kDa) (Figure 4A).
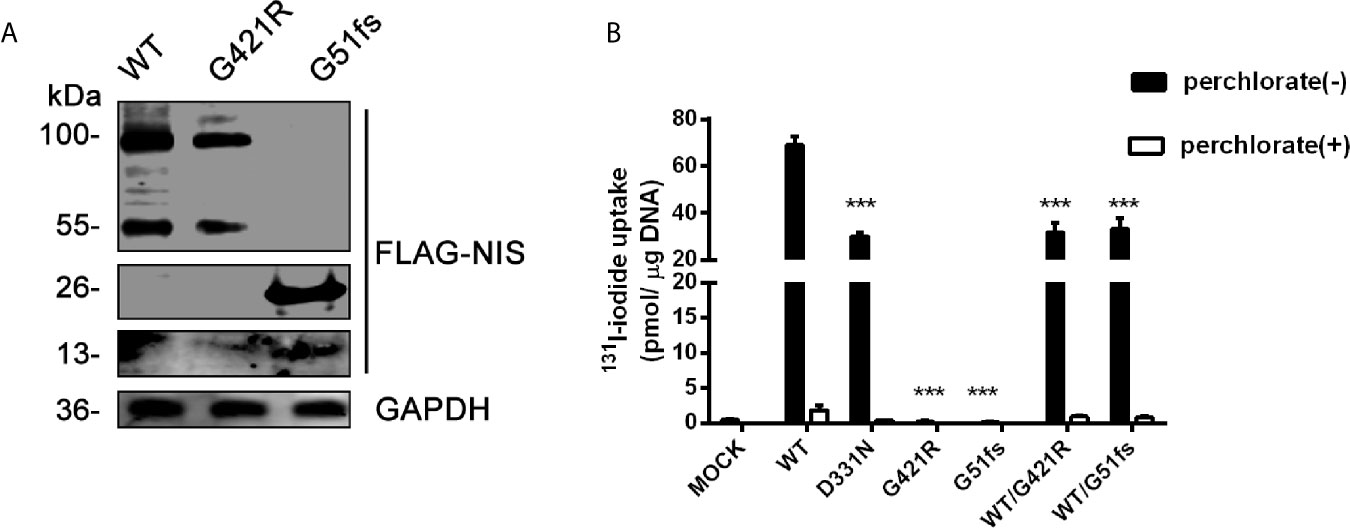
Figure 4 (A) Representative Western blot analysis of whole-cell lysates from transiently transfected 293T cells probed with anti-FLAG and GAPDH antibodies. The proteins of WT (wild-type), G421R and G51fs were visualized by Odyssey CLx Infra-Red Imaging system. The molecular weight band was on the right (WT, G421R unglycosylated~ 55 kDa, mature glycosylated~ 100 kDa; G51fs monomer~13 kDa, dimer~ 26 kDa). (B) Iodide transport activity for of G421R and G51fs NIS mutants. Mock (p3xFLAG-CMV-10 plasmids), or expressing WT (wild-type) NIS, D331N, G421R and G51fs NIS plasmids were transfected alone or co-transfected into 293T cells. The cells were incubated with with131I at 5 KBq/mL in the absence (black bars) or presence (white bars) of 1 mM perchlorate and the intracellular iodide accumulation was detected using a γ counter. The values are representative of more than three independent experiments and represent the mean ± SD of picomoles 131I per microgram of DNA; each experiment was performed in triplicate. ***p < 0.001.
The iodide transport activity assay demonstrated that 293T cells transfected with WT NIS showed a marked accumulation of radio-iodide in the cells, while addition of the competitive inhibitor perchlorate significantly inhibited NIS-mediated iodide transport (Figure 4B). Consistent with the previous study, cells carrying the expression vector encoding D331 NIS exhibited partially reduced, although still detectable, 131I uptake, compared with the iodide uptake levels in wild-type NIS-expressing cells. G421R and G51fs NIS-expressing cells showed almost no iodide transport activity, with similar radio-iodide levels to cells transfected with a p3xFLAG-CMV-10 empty vector (Figure 4B). In addition, substituting of G421 with other amino acids, aspartic acid and phenylalanine), showed the same results (Supplemental Figure 2). Further, co-transfection of WT and mutant plasmids to mimic the heterozygous state, no dominant negative effect was observed (Figure 4B). Our data showed that G421R and G51fs mutations markedly impaired iodide transport activity of the cells, which was consistent with the impaired membrane patterns.
The sodium/iodide symporter has been shown by immunofluorescence analysis to be located at the basolateral side of thyroid follicular cells and to mediate active iodide trapping. To perform its physiological function, this transporter must be properly localized at the plasma membrane. To assess the effect of SLC5A5 mutations on the membrane localization, we expressed wild-type and G421R or G51fs mutants of the SLC5A5-pEGFP-N2 plasmid in 293T cells and examined the cellular localization using a confocal fluorescence microscope. A pEGFP-N2 empty vector was employed as a negative control. As we can see from Figure 5, NIS was clearly present at the cell membrane in WT cells, while in D331N transfected cells, normal plasma membrane trafficking of NIS was partially affected. In cells transfected with G51fs and G421R mutants, the NIS expressions on the cell membranes were decreased in varying degrees and appeared to localize in the cytoplasm (Figure 5), suggesting impaired plasma membrane localization of the mutated NIS proteins.
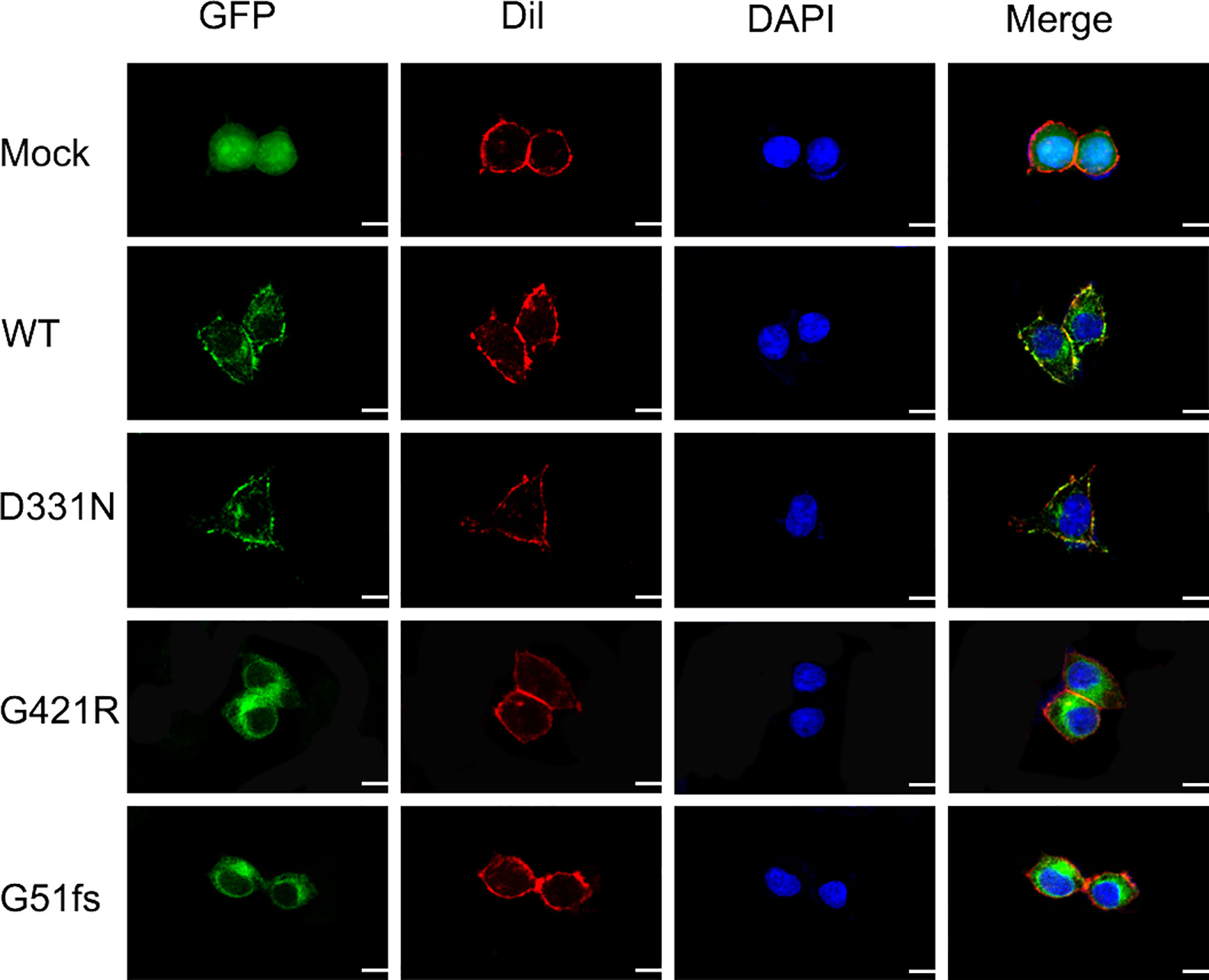
Figure 5 Characterization of cellular localization of G421R and G51fs NIS mutants in 293T cells. pEGFP-N2 empty vector was employed as a negative control (Mock). Human WT NIS, D331N, G421R, and G51fs mutants were cloned into pEGFP-N2 plasmids (green). The cell membrane was labeled with a fluorescent probe Dil (red). Mock, WT NIS, D331N, G421R, and G51fs plasmids were transfected into 293T cells 24 h after seeding. Mutant D331N NIS showed slightly decreased membrane fluorescence, compared with the WT. However, G51fs and G421R mutants displayed severely reduced cell membrane localization in 293T cells. Nuclei were stained with DAPI (blue). Scale: 10 μm.
In the present study, the undescribed compound heterozygous mutation of G51fs/G421R in SLC5A5 was discovered in a Chinese pediatric patient with CH. The mutations significantly reduced the NIS expression and impaired iodide transport function accompanied by impairing the location of NIS on the plasma membrane. Our study, thus, provided further insights into the roles of SLC5A5 in CH pathogenesis.
Clinical heterogeneity in patients with different SLC5A5 mutations was reported (10). In our patient, hypothyroidism was discovered by neonatal screening. After the diagnosis of CH, the patient was treated with L-thyroxine replacement immediately. However, in many other cases, the onset age of hypothyroidism was variable, ranging from the neonatal period to childhood, and even adulthood (32). Due to delayed treatments, patients presented irreversible intellectual disabilities. Research stated that less than 10% of iodine transport defect patients detected by neonatal screening showed clinical features of congenital hypothyroidism.
In addition, the occurrence of goiters varies between individuals. Because the formation of goiters depends on the level and the duration of TSH overstimulation, as well as on the iodide availability. The patients carrying the same described mutation did not develop goiters. Researchers speculated that early diagnosis and treatment with hormone replacement therapy during the neonatal period might prevent the development of goiters (33). However, even if these patients without goiters at birth are treated and maintain euthyroid, diffuse goiters will still occur after, which indicates that other factors besides TSH stimulation could also cause goiters. Notably, patients carrying the mutation G395R developed goiters has not been reported so far. The majority of these patients were severely hypothyroid, which was detected during the neonatal period (16). The molecular studies of G395R mutation indicated that G395R NIS was synthesized and properly targeted to the plasma membrane but intrinsically inactive (2, 34). It was highly possible that a “milder” pathogenic variant causing a partial loss-of-function might exist and could allow for a lower risk of goiters. Hence, regular long-term follow-up is necessary, and more comprehensive research is required to explore the mechanisms in detail.
In our study, we screened for mutations in SLC5A5 in a cohort of the 273 CH patients. The frequency of SLC5A5 mutation in the Chinese patients with CH was about 0.37% (1/273) in our study. However, according to the allele frequency of SLC5A5 in the Chinese Millionome Database (CMBD), we speculated that the frequency of SLC5A5 deficiency in CH in Chinese population was about 0.036%, which is significantly lower than what was found in our study. The relatively small cohort size may be a plausible reason for this discrepancy. The G51fs/G421R compound heterozygous mutation in SLC5A5 in a pediatric patient diagnosed with congenital hypothyroidism has not been reported before. Analysis of the proband’s family showed that the father and mother were heterozygous for G51fs and G421R NIS, respectively (Figures 2A, B).
Along with the clinical manifestations of the proband, we proposed that the two novel mutation sites were crucial for normal functional activity of NIS. As expected, cells transfected with the G51fs NIS construct showed no iodide uptake and impaired membrane location due to the loss of most of the functional domains (Figures 4B, 5). Additionally, our studies revealed that the G421R mutation located in the TMS XI was associated with diminished iodide uptake activity (Figure 4B). The mutated NIS proteins could not correctly target the plasma membrane (Figure 5). In vitro experiments in 293T cells transiently co-transfected with WT and mutant plasmids, mimicking the parental heterozygous state, indicated that the mutated allele did not interrupt the activity of WT NIS (Figure 4B). This observation was in accordance with the clinical observation that the parents who showed the same mutant allele in the heterozygous state (Figure 2B) were euthyroid.
Multiple lines of computational evidence supported a deleterious effect on NIS due to G421R missense mutation (Supplemental Table 2). As previously described, the structure of vSGLT, comprised of a central group of seven helices (TMS II-IV, TMS VII-IX, and TMS XI), can form a large cavity participating in the transport of galactose (35). We speculated that the arginine residue may occupy a comparably larger conformational space than glycine in the cavity, thereby, blocking the entry of ions. Previous studies on T354P mutation located in TMS IX revealed that the hydroxyl group at the β-carbon of the residue at position 354 was essential for its function in Na+ binding and translocation (23, 24). In addition, the architecture of the Na+ binding site was conserved among other transport proteins, such as bacterial leucine transporter(LeuT) and vSGLT (36, 37). We found that the interatomic distance between G421 and T354 was changed from 15.0 Å to 8.7 Å. Replacement of the hydrophobic neutral glycine residue with the hydrophilic and positively charged amino acid changed the net surface positive charges and distorted the structure of the surface, which may disturb the required rigidity of the protein and affect the trafficking of NIS (Figure 3). To perform its physiological function of maintaining an active iodide concentration from the blood using the positive sodium gradient, NIS is required to be properly localized at the plasma membrane (30).
In summary, we identified an undescribed compound heterozygous mutation of G51fs/G421R in SLC5A5 from 273 Chinese patients with CH. These variants were associated with a significant reduction in NIS protein expression and subsequently impaired the membrane localization, resulting in a dramatic reduction in iodide transport and thyroid hormone biosynthesis. Our data demonstrated that these mutations were the direct cause of the observed CH phenotype. This study advanced our understanding of the possible mechanisms of NIS in CH pathogenesis.
The data presented in the study are deposited in the FigShare repository (https://doi.org/10.6084/m9.figshare.13134824.v1).
The studies involving human participants were reviewed and approved by the Ethics Committee of Shanghai Ninth People’s Hospital affiliated to Shanghai Jiao Tong University School of Medicine. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s) and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
C-XZ: writing-original draft and data curation. J-XZ: supervision. LY: project administration. C-RZ: methodology. FC: funding acquisition. R-JZ: resources. YF: resources. ZW: software. F-YW: visualization. P-ZL: validation. JL: funding acquisition. RL: funding acquisition, review, and editing. H-DS: funding acquisition, conceptualization, and investigation. All authors contributed to the article and approved the submitted version.
This work was supported by the Chinese National Key Research Program (2017YFC1001801) and the National Natural Science Foundation of China (82070816, 81770786, 81661168016, 81870537, 81670717 and 81870540), and the Shanghai Municipal Education Commission-Gao feng Clinical Medicine Grant Support (20161318).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank all patients and their families for the participation in our study and for allowing us to publish their cases.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.620117/full#supplementary-material
1. Wassner AJ. Congenital Hypothyroidism. Clinics Perinatol (2018) 45:1–18. doi: 10.1016/j.clp.2017.10.004
2. Ravera S, Reyna-Neyra A, Ferrandino G, Amzel LM, Carrasco N. The Sodium/Iodide Symporter (NIS): Molecular Physiology and Preclinical and Clinical Applications. Annu Rev Physiol (2017) 79:261–89. doi: 10.1146/annurev-physiol-022516-034125
3. Smanik PA, Liu Q, Furminger TL, Ryu K, Xing S, Mazzaferri EL, et al. Cloning of the human sodium lodide symporter. Biochem Biophys Res Commun (1996) 226:339–45. doi: 10.1006/bbrc.1996.1358
4. Levy O, De la Vieja A, Ginter CS, Riedel C, Dai G, Carrasco N. N-linked glycosylation of the thyroid Na+/I- symporter (NIS). Implications for its secondary structure model. J Biol Chem (1998) 273:22657–63. doi: 10.1074/jbc.273.35.22657
5. Nilsson M. Molecular and cellular mechanisms of transepithelial iodide transport in the thyroid. BioFactors (Oxford England) (1999) 10:277–85. doi: 10.1002/biof.5520100228
6. Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature (1996) 379:458–60. doi: 10.1038/379458a0
7. Levy O, Dai G, Riedel C, Ginter CS, Paul EM, Lebowitz AN, et al. Characterization of the thyroid Na+/I- symporter with an anti-COOH terminus antibody. Proc Natl Acad Sci U States America (1997) 94:5568–73. doi: 10.1073/pnas.94.11.5568
8. Dohán O, De la Vieja A, Paroder V, Riedel C, Artani M, Reed M, et al. The sodium/iodide Symporter (NIS): characterization, regulation, and medical significance. Endocr Rev (2003) 24:48–77. doi: 10.1210/er.2001-0029
9. Portulano C, Paroder-Belenitsky M, Carrasco N. The Na+/I- symporter (NIS): mechanism and medical impact. Endocr Rev (2014) 35:106–49. doi: 10.1210/er.2012-1036
10. Spitzweg C, Morris JC. Genetics and phenomics of hypothyroidism and goiter due to NIS mutations. Mol Cell Endocrinol (2010) 322:56–63. doi: 10.1016/j.mce.2010.02.007
11. Martin M, Bernal Barquero CE, Geysels RC, Papendieck P, Peyret V, Masini-Repiso AM, et al. Novel Sodium/Iodide Symporter Compound Heterozygous Pathogenic Variants Causing Dyshormonogenic Congenital Hypothyroidism. Thyroid (2019) 29:1023–6. doi: 10.1089/thy.2019.0046
12. Watanabe Y, Ebrhim RS, Abdullah MA, Weiss RE. A Novel Missense Mutation in the SLC5A5 Gene in a Sudanese Family with Congenital Hypothyroidism. Thyroid (2018) 28:1068–70. doi: 10.1089/thy.2018.0137
13. Fujiwara H, Tatsumi K, Tanaka S, Kimura M, Nose O, Amino N. A novel V59E missense mutation in the sodium iodide symporter gene in a family with iodide transport defect. Thyroid (2000) 10:471–4. doi: 10.1089/thy.2000.10.471
14. Stoupa A, Al Hage Chehade G, Kariyawasam D, Tohier C, Bole-Feysot C, Nitschke P, et al. First case of fetal goitrous hypothyroidism due to SLC5A5/NIS mutations. Eur J Endocrinol (2020) 183:K1–k5. doi: 10.1530/EJE-20-0255
15. Kosugi S, Inoue S, Matsuda A, Jhiang SM. Novel, missense and loss-of-function mutations in the sodium/iodide symporter gene causing iodide transport defect in three Japanese patients. J Clin Endocrinol Metab (1998) 83:3373–6. doi: 10.1210/jcem.83.9.5243
16. Kosugi S, Bhayana S, Dean HJ. A novel mutation in the sodium/iodide symporter gene in the largest family with iodide transport defect. J Clin Endocrinol Metab (1999) 84:3248–53. doi: 10.1210/jc.84.9.3248
17. Pohlenz J, Medeiros-Neto G, Gross JL, Silveiro SP, Knobel M, Refetoff S. Hypothyroidism in a Brazilian kindred due to iodide trapping defect caused by a homozygous mutation in the sodium/iodide symporter gene. Biochem Biophys Res Commun (1997) 240:488–91. doi: 10.1006/bbrc.1997.7594
18. Reed-Tsur MD, De la Vieja A, Ginter CS, Carrasco N. Molecular characterization of V59E NIS, a Na+/I- symporter mutant that causes congenital I- transport defect. Endocrinology (2008) 149:3077–84. doi: 10.1210/en.2008-0027
19. Paroder-Belenitsky M, Maestas MJ, Dohán O, Nicola JP, Reyna-Neyra A, Follenzi A, et al. Mechanism of anion selectivity and stoichiometry of the Na+/I- symporter (NIS). Proc Natl Acad Sci U States America (2011) 108:17933–8. doi: 10.1073/pnas.1108278108
20. Paroder V, Nicola JP, Ginter CS, Carrasco N. The iodide-transport-defect-causing mutation R124H: a δ-amino group at position 124 is critical for maturation and trafficking of the Na+/I- symporter. J Cell Sci (2013) 126:3305–13. doi: 10.1242/jcs.120246
21. De La Vieja A, Ginter CS, Carrasco N. The Q267E mutation in the sodium/iodide symporter (NIS) causes congenital iodide transport defect (ITD) by decreasing the NIS turnover number. J Cell Sci (2004) 117:677–87. doi: 10.1242/jcs.00898
22. Nicola JP, Reyna-Neyra A, Saenger P, Rodriguez-Buritica DF, Gamez Godoy JD, Muzumdar R, et al. Sodium/Iodide Symporter Mutant V270E Causes Stunted Growth but No Cognitive Deficiency. J Clin Endocrinol Metab (2015) 100:E1353–61. doi: 10.1210/jc.2015-1824
23. Levy O, Ginter CS, De la Vieja A, Levy D, Carrasco N. Identification of a structural requirement for thyroid Na+/I- symporter (NIS) function from analysis of a mutation that causes human congenital hypothyroidism. FEBS Lett (1998) 429:36–40. doi: 10.1016/S0014-5793(98)00522-5
24. De la Vieja A, Reed MD, Ginter CS, Carrasco N. Amino acid residues in transmembrane segment IX of the Na+/I- symporter play a role in its Na+ dependence and are critical for transport activity. J Biol Chem (2007) 282:25290–8. doi: 10.1074/jbc.M700147200
25. Targovnik HM, Citterio CE, Rivolta CM. Iodide handling disorders (NIS, TPO, TG, IYD). Best Pract Res Clin Endocrinol Metab (2017) 31:195–212. doi: 10.1016/j.beem.2017.03.006
26. Fang Y, Sun F, Zhang RJ, Zhang CR, Yan CY, Zhou Z, et al. Mutation screening of the TSHR gene in 220 Chinese patients with congenital hypothyroidism. Clinica chimica acta; Int J Clin Chem (2019) 497:147–52. doi: 10.1016/j.cca.2019.07.031
27. Zhang RJ, Sun F, Chen F, Fang Y, Yan CY, Zhang CR, et al. The TPO mutation screening and genotype-phenotype analysis in 230 Chinese patients with congenital hypothyroidism. Mol Cell Endocrinol (2020) 506:110761. doi: 10.1016/j.mce.2020.110761
28. Sun F, Zhang JX, Yang CY, Gao GQ, Zhu WB, Han B, et al. The genetic characteristics of congenital hypothyroidism in China by comprehensive screening of 21 candidate genes. Eur J Endocrinol (2018) 178:623–33. doi: 10.1530/EJE-17-1017
29. Taylor JP, Metcalfe RA, Watson PF, Weetman AP, Trembath RC. Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: implications for thyroid dysfunction in Pendred syndrome. J Clin Endocrinol Metab (2002) 87:1778–84. doi: 10.1210/jcem.87.4.8435
30. Tonacchera M, Agretti P, de Marco G, Elisei R, Perri A, Ambrogini E, et al. Congenital hypothyroidism due to a new deletion in the sodium/iodide symporter protein. Clin Endocrinol (2003) 59:500–6. doi: 10.1046/j.1365-2265.2003.01877.x
31. Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinf (Oxford England) (2006) 22:195–201. doi: 10.1093/bioinformatics/bti770
32. Szinnai G, Kosugi S, Derrien C, Lucidarme N, David V, Czernichow P, et al. Extending the clinical heterogeneity of iodide transport defect (ITD): a novel mutation R124H of the sodium/iodide symporter gene and review of genotype-phenotype correlations in ITD. J Clin Endocrinol Metab (2006) 91:1199–204. doi: 10.1210/jc.2005-1832
33. Nicola JP, Nazar M, Serrano-Nascimento C, Goulart-Silva F, Sobrero G, Testa G, et al. Iodide transport defect: functional characterization of a novel mutation in the Na+/I- symporter 5’-untranslated region in a patient with congenital hypothyroidism. J Clin Endocrinol Metab (2011) 96:E1100–7. doi: 10.1210/jc.2011-0349
34. Dohán O, Gavrielides MV, Ginter C, Amzel LM, Carrasco N. Na(+)/I(-) symporter activity requires a small and uncharged amino acid residue at position 395. Mol Endocrinol (Baltimore Md) (2002) 16:1893–902. doi: 10.1210/me.2002-0071
35. Faham S, Watanabe A, Besserer GM, Cascio D, Specht A, Hirayama BA, et al. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science (2008) 321:810–4. doi: 10.1126/science.1160406
36. Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl–dependent neurotransmitter transporters. Nature (2005) 437:215–23. doi: 10.1038/nature03978
Keywords: iodine transport, mutation, SLC5A5, next-generation sequencing, congenital hypothyroidism
Citation: Zhang C-X, Zhang J-X, Yang L, Zhang C-R, Cheng F, Zhang R-J, Fang Y, Wang Z, Wu F-Y, Li P-Z, Liang J, Li R and Song H-D (2021) Novel Compound Heterozygous Pathogenic Mutations of SLC5A5 in a Chinese Patient With Congenital Hypothyroidism. Front. Endocrinol. 12:620117. doi: 10.3389/fendo.2021.620117
Received: 22 October 2020; Accepted: 05 February 2021;
Published: 19 March 2021.
Edited by:
Juan Pablo Nicola, National University of Cordoba, ArgentinaReviewed by:
Antonio De La Vieja, Instituto de Salud Carlos III (ISCIII), SpainCopyright © 2021 Zhang, Zhang, Yang, Zhang, Cheng, Zhang, Fang, Wang, Wu, Li, Liang, Li and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huai-Dong Song, aHVhaWRvbmdfczE5NjZAMTYzLmNvbQ==; Rui Li, cnVpX2xpMjAyMEAxNjMuY29t; Jun Liang, bXdsajUyMUAxNjMuY29t
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.