 Panyisha Wu
Panyisha Wu Moya Zhang4
Moya Zhang4 Nicholas J. G. Webster
Nicholas J. G. Webster
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 26 February 2021
Sec. Obesity
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.613213
This article is part of the Research Topic Non-Alcoholic Steatohepatitis (NASH) – Metabolic Contributors and Therapeutic Targets View all 25 articles
Alternative RNA splicing is a process by which introns are removed and exons are assembled to construct different RNA transcript isoforms from a single pre-mRNA. Previous studies have demonstrated an association between dysregulation of RNA splicing and a number of clinical syndromes, but the generality to common disease has not been established. Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease affecting one-third of adults worldwide, increasing the risk of cirrhosis and hepatocellular carcinoma (HCC). In this review we focus on the change in alternative RNA splicing in fatty liver disease and the role for splicing regulation in disease progression.
Eukaryotic protein-coding genes are usually split into exons and intervening introns that are removed by the process of RNA splicing (1). Alternative splicing is the process that selectively removes introns or exons, or parts thereof, to generate multiple messenger RNAs (mRNAs) from a single precursor mRNA (pre-mRNA) (2). First reported in 1980, RNA alternative splicing events have been found in the majority of eukaryotic genes. Indeed, it is estimated that more than 95% of the genes in the human cell undergo alternative splicing and produce isoforms of different or even opposing biological roles (3). Therefore, alternative RNA splicing plays a critical role in defining the transcriptome and fine-tuning the proteome of the cell.
Recent studies have shown that changes in RNA splicing and RNA binding protein expression occur during the maturation of the liver (4, 5), are associated with aging and sexual dimorphism in the liver (6), and have also been documented in hepatocellular carcinoma (7–16), but little is known about changes in RNA splicing in early liver disease (17). Non-alcoholic fatty liver disease (NAFLD) is the leading cause of liver disease in western countries, affecting almost 25% of the adult population in the world (18). It is defined as the fat accumulation in the liver after the exclusion of secondary causes (19). NAFLD can progress from simple steatosis to non-alcoholic steatohepatitis (NASH), cirrhosis and even hepatocellular carcinoma (HCC) (20). Not surprisingly, NAFLD is strongly associated with the metabolic syndrome (21, 22) and the prevalence of NAFLD is rising due to the global obesity epidemic (23). Yet the risk of NAFLD is affected by both environmental and genetic factors (24). Many studies have assessed gene expression changes in NAFLD patients and identified transcripts that are associated with specific metabolic comorbidities in patients with NAFLD and NASH (25–28). Changes in splicing factor expression have also been observed which would suggest alterations in RNA splicing (29, 30). Furthermore, some studies have demonstrated alternative splicing of obesity-related genes in the liver and other metabolic tissues, suggesting that splicing variants may play a role in NAFLD development (31–35). Thus, the evidence to date points to a potential association between liver disease and RNA splicing that deserves further investigation. In this article, we review prior evidence and discuss more recent reports of alternative RNA splicing events and their significance in the process of fatty liver disease.
The human genome, as well as many other eukaryotic genomes, contains stretches of intronic sequence between sparser and relatively smaller, exonic sequences. The process of removing the introns from the pre-mRNA is called RNA splicing and has been extensively reviewed (2, 36–38). Of interest here, these introns can be spliced out in the pre-mRNA at different locations and efficiencies to form different arrangements of exons in the final mRNA transcripts through the use of alternative splice sites (2). This process allows for greater RNA and protein diversity than would be predicted by the number of genes (3, 39). Briefly, a complex RNA-based molecular machine called the spliceosome recognizes the splice sites and performs the intron excision and exon joining (40). The spliceosome is formed from five main subunits of small nuclear ribonucleoprotein particles (snRNPs) (41, 42). In canonical splicing, the U1 snRNP recognizes a GU-containing hexamer sequence at the 5′ site of the intron, while the U2 subunit in combination with an auxiliary factor U2AF recognizes the branch-point sequences, a pyrimidine-rich sequence, and the AG-splice site at the 3′ site of the intron. Once these molecules are bound to the RNA, they recruit the U4/U5/U6 snRNPs to form the spliceosome which then rearranges to form an active splicing complex. This complex cleaves the intron at the 5′ and 3′ ends then ligates the exons together by a trans-esterification mechanism (7, 43). While a U1 and U2 interaction across the intron (intron definition) has been demonstrated in vitro, it is only effective when the introns are relatively short (less than 250 nucleotides). For the majority of long introns in vivo, this U1–U2 splice site recognition process is thought to occur across exons, in a process called exon definition (43).
Recognition of 5′ splice sites is mediated by base pairing of the U1 snRNA, a component of the U1 snRNP, with the sequences surrounding the splice site, whereas the 3′ splice site is recognized in a less sequence specific manner by U2AF binding to the polypyrimidine tract at the 3′ splice site allowing the U2snRNA to base-pair with the branch-point sequence. As such, any divergence of the splice site sequence from the consensus will weaken recognition by the snRNPs and use of the splice site by the spliceosome. This allows for selection of alternate exons, or the use of alternative splice sites in exons. Recognition of these weak splice sites is modulated through the binding of RNA-binding proteins (RBPs) to adjacent short regulatory sequences. These sequences can either repress or enhance splicing at the adjacent site. Many RBPs are involved in alternative splicing, including the serine/arginine-rich (SR) proteins and the heterogeneous nuclear ribonucleoprotein (hnRNPs) families, that together target the spliceosome to the appropriate splice site (7). As a general, but not hard-and-fast rule, SR proteins are typically involved with exonic splicing enhancers (ESEs) and help stabilize the snRNPs at splice sites (44, 45), but hnRNPs interact with exonic splicing silencers (ESSs) and prevent the binding of SR proteins or snRNPs. The hnRNPs thus mainly play a role in exon skipping, in which the exon and its neighboring introns are spliced out of the pre-mRNA (7, 44).
Extracellular factors have also been shown to contribute to the regulation of alternative splicing (46). Extracellular stimuli can affect the phosphorylation of splicing factors, either promoting or inhibiting binding to the RNA (47). Kinases and phosphatases that perform post-translational modifications of splicing factors have also been identified (7, 46). These different mechanisms expand the versatility of alternative splicing, allowing the body to produce a wide variety of proteins and molecules based off of a relatively limited genome.
To understand the factors involved in the development of NAFLD, transcriptome profiling of human livers has been performed by microarray and RNAseq, and changes in the expression of many transcription factors have been documented, such as FOXO1, SREBP1, IRF1, IRF3, C/EBPβ, SMAD3, SMAD7, PPARα and PPARβ (48–51); however, most of the studies did not investigate changes in RNA splicing factors or spliceosome components. In recent years, in recognition of the increasingly important role for RNA splicing, a number of studies (10, 29, 48, 52) have investigated whether the components of the alternative splicing machinery may be altered in NAFLD and NASH that might underlie changes in splicing variants (Table 1).
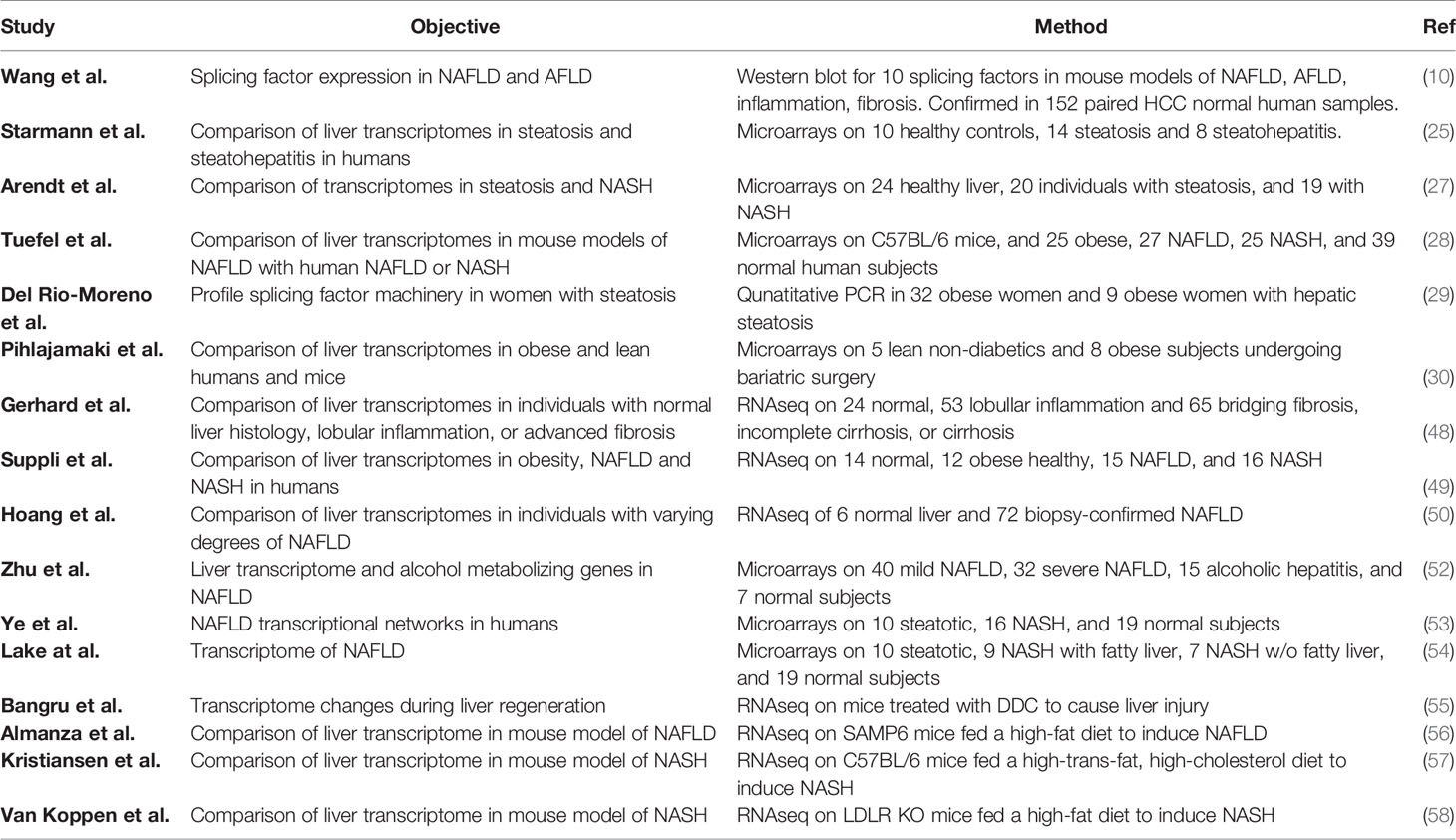
Table 1 Studies reporting alterations of RNA splicing components in NAFLD or NASH.
The association of splicing factors with obesity and NAFLD was initially investigated by microarray analysis (53, 59, 60), which allowed quantification of gene expression but not RNA splicing. NAFLD is strongly associated with obesity, and several obesity-linked genes have been shown to be regulated by alternative splicing (61–64); therefore, Pihlajamaki et al. identified differentially expressed genes in the liver and muscle biopsies from obese patients by high-density oligonucleotide arrays (30). Five lean control subjects and eight obese subjects were included for the liver cohort. Though none of them had abnormalities in glucose metabolism at the start of the study, six of the obese subjects were diagnosed with type 2 diabetes mellitus (T2D) during the study. When they analyzed the pathways affected by the alterations in expression, they found the top two pathways were mRNA processing and RNA splicing pathways. Forty-six of the 199 analyzed RNA splicing genes were downregulated in obese livers, and the expression of 13 genes decreased in both liver and muscle, including SFRS10 (TRA2b), SFRS7 (9G8), SF3A1, SFRS2 (SC35), SFPQ, HNRPA1, HNRPK. The decreased expression of SFRS10, SFRS7, SF3A1, SFPQ and HNRPK in obese liver was further confirmed in a mouse model of diet-induced obesity. This study demonstrated that RNA splicing factor expression inversely correlated with hepatic fat accumulation and hyperinsulinemia and that alterations in RNA splicing factor expression may contribute to obesity-related phenotypes.
Starmann et al. (25) profiled healthy controls (n = 10) and patients with simple steatosis (n = 14) or steatohepatitis (n = 8) by microarray. They found 4,963 genes altered in the steatohepatitis patients versus healthy liver or 2,542 genes altered versus the simple steatosis patients. Inspection of these differentially expressed genes for known splicing factors showed that 136 splicing factors were altered compared to healthy liver and 41 compared to simple steatosis. Among the genes altered were eight HNRNPs (A2B1, H1/2/3, L, F, D & U), RBFOX2, eight RBMs (12B, 14, 22, 7, 10, 20, 4 & 6), four SF3 genes (B6, B5, A3, & B2), SLU7, SFPQ, SRSF11, MBNL3, and TRA2A.
Zhu et al. (52) examined hepatic gene expression in 72 patients with mild NAFLD (fibrosis stage 0–1, n = 40) and severe NAFLD (fibrosis stages 3–4, n = 32), alcoholic hepatitis (AH, n = 15), or healthy liver (n = 7) by microarray. The mild and severe NAFLD and AH clustered together but were distinct from the healthy controls. Although not addressed in the paper, reanalysis of their dataset showed that the expression of 92 splicing factor genes was altered in subjects with mild NAFLD versus healthy controls. Among these altered genes were ESRP1/2, MBNL1/2/3, SLU7, nine SF3 genes and ten SRSF proteins.
Ye et al. (53) used weighted gene co-expression network analysis on a NASH-NAFLD microarray dataset generated by Lake et al. (54) and found that modules involved in RNA processing with enrichment for genes involved in RNA binding, mRNA processing and the spliceosome in the NASH and NAFLD groups.
More recently in 2018, Gerhard et al. (48) performed RNAseq of liver samples from individuals with normal histology (n = 24), lobular inflammation (n = 53), or advanced fibrosis, defined by bridging fibrosis, incomplete cirrhosis, or cirrhosis (n = 65). They reported differential expression of 3,820 and 2,980 genes in the lobular inflammation and advanced fibrosis groups compared to normal histology. In addition to genes involved in inflammation, extracellular matrix, cytokine and PI-3K signaling, and focal adhesion, 35 splicing factors were altered in the lobular inflammation group, including ESRP1, RBM4/20/24, SF3B5, HNRNPU, CELF3/4/5, ELAVL2/4, NOVA1/2, and RBFOX1/3, and 20 were altered in the advanced fibrosis group including CELF3/4/5, ELAVL2/4, RBM20/24, NOVA1/2, and RBFOX1/3.
To specifically address whether the splicing machinery is altered in steatosis, Del Rio-Moreno et al. (29) profiled the expression of spliceosome components and splicing factors in liver samples obtained from 41 obese women with (n = 32) and without (n = 9) hepatic steatosis by qPCR. The patients with steatosis were further classified into mild, moderate, or severe steatosis by liver echography. It should be noted that all the patients in this study presented with steatosis at an early stage of NAFLD without any evidence of NASH or cirrhosis. The expression of 17 splicing machinery components and 28 splicing factors was determined. It was found that the expression of 16 of these 45 genes was clearly different between patients with and without hepatic steatosis, including eight spliceosome components (RNU6ATAC, RNU6, SF3B1, RNU2, RNU4ATAC, RBM22, U2AF1, U2AF2) and eight splicing factors (PTBP1, SRRM1, SND1, KHDRSB1, SRSF2, SRSF10, ESRP2, TIA1). In patients with steatosis the expression of RNU6ATAC, RNU6, SF3B1, RNU2, RNU4ATAC, TIA1 was downregulated, but the expression of the other 10 genes was elevated significantly. When the patients with steatosis were grouped according to similar expression patterns of spliceosome components and splicing factors, patients in Cluster A (characterized by lower SRSF4 and TRA2B) showed increased blood glucose and haptoglobin levels, whereas patients in Cluster B (higher RBM45 and TRA2A) had higher plasma triglycerides, GGT, and lower alkaline phosphatase levels, and Cluster C (higher SND1 and RAVER1) exhibited elevated insulin, ALT, and AST levels. Moreover, Cluster C presented a worse response to bariatric surgery, compared with Clusters A+B, exhibiting less normalization of plasma GGT, glucose, triglycerides, alkaline phosphatase, and HDL levels. The differences of these three molecular NAFLD phenotypes suggest that the dysregulation of specific splicing machinery components is associated with distinct clinical/metabolic alterations. To test whether a causal relationship could exist, the authors used RNAi knockdown in HepG2 cells. Knockdown of specific splicing factors (PTBP1, SRSF4, RBM22, RBM45, SND1, RAVER1) significantly lowered lipid accumulation in HepG2 hepatoma cells after lipid loading with oleic acid. Although expression of the selected splicing factors was not altered by oleic acid treatment, the expression of some splicing machinery components was modulated by other metabolic factors. For example, elevated glucose decreased SND1, and leptin decreased RBM22, but insulin-like growth factor 1 (IGF-1) increased RAVER1, and palmitic acid increased PTBP1 and RBM22. Thus, this study provided evidence that not only could the overexpression of SRSF4, RBM45, SND1, and RAVER1 that is seen in the three molecular clusters enhance the development of NAFLD, but also that the metabolic milieu could contribute to the altered RNA splicing.
In 2019, Suppli et al. (49) also published an RNAseq study on liver samples from healthy normal (n = 14), obese individuals (n = 12), and NAFLD (n = 15) and NASH (n = 16) patients. They found that genes involved in RNA metabolism were enriched in samples from NASH patients compared to NAFLD. Although many genes (8,244) were differentially expressed in NAFLD and NASH compared to healthy liver, the authors did not provide the lists of differentially expressed genes and did not investigate whether RNA splicing genes were altered. A reanalysis of these data, however, showed that many RNA splicing factor genes were altered in NAFLD (174) and NASH (204).
That same year, Hoang et al. (50) published a study of 72 patients with varying degrees of biopsy-confirmed NAFLD compared to six healthy controls by RNAseq. Patients’ samples were assessed by NAFLD activity score (NAS) or fibrosis stage. The authors used ordinal regression to identify genes that significantly changed with severity of disease either by NAS or fibrosis stage. At a false discovery rate of 1%, they identified 2,970 genes associated with NAS and 1,656 genes associated with fibrosis stage. Integration of these genes with protein-interaction networks demonstrated that genes involved in immune signaling, extracellular matrix organization, and cell cycle were enriched. They also identified genes enriched in RNA metabolism associated with both NAS and fibrosis stage. Inspection of the list of significant genes shows that 88 splicing factors are associated with NAS and 52 with fibrosis stage, suggesting a change in the splicing machinery with disease progression.
Similar to the human data, there is evidence that splicing factor expression is altered in different liver disease stages in six mouse models of non-alcoholic and alcoholic fatty liver disease (AFLD). In a recent study, Wang et al. (10) evaluated the expression of 10 splicing factors (PSF, NONO, SRSF1, SRSF3, SRSF6, SRSF7, hnRNPA2B1, hnRNPH, La, and SF1) in mouse livers by western blot. A significant decrease of SRSF3 and increases of NONO, SRSF6, hnRNPA2B1 and hnRNPH protein were detected in livers of HFD-induced NAFLD mice. The AFLD mouse model also showed increased expression of PSF, p47, SRSF7 and La. To model the effect of disease progression, male mice were injected with LPS and CCl4 to induce liver inflammation and fibrosis, respectively. As a result, the level of p47, SRSF3, SRSF6 and La was upregulated in the LPS-induced inflammatory livers, while SRSF6, SRSF7, and SF1 levels were elevated in fibrotic livers. Furthermore, they confirmed alteration in many of these splicing factors in RNA from 152 paired human HCC and normal samples.
Consistent with the known developmental changes in RNA splicing during liver maturation, liver injury caused by 0.1% 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) ingestion causes hepatocyte regeneration and a switch to a neo-natal or fetal splicing program (55). This is accompanied by reductions in splicing factor ESRP2, hnRNPH, hnRNPC, and CELF1 protein expression and increases in the MBNL1, PTBP1, hnRNPA1, and SRSF1 protein expression. As a consequence, livers showed alterations in RNA splicing, with exon skipping/retention being the predominant mode. Gene ontology analysis indicated that mRNA processing and spliceosome regulation of splicing were enriched with these alternatively spliced genes. Downregulation of ESRP2 was sufficient to phenocopy DDC treatment (see below).
Not all studies have found changes in the splicing machinery. A number of earlier studies did not report alterations in splicing factor expression. A study carried by Teufel et al. (28) compared gene expression in liver samples from patients at different stages of NAFLD with mouse models of NAFLD. Liver tissues of 25 NASH patients, 27 NAFLD patients, 15 healthy obese patients, and 39 controls were collected, in addition to liver tissues from nine NAFLD mouse models [mice on a high-fat diet (with or without fructose), mice on a Western-type diet, mice on a methionine- and choline-deficient diet, mice on a high-fat diet given streptozotocin, and mice with disruption of Pten in hepatocytes]. They found that, although there was very little overlap of gene expression profiles in NAFLD liver tissues between human and mouse, at the pathway level the gene expression patterns in livers of mice with NAFLD due to high-fat diet feeding closely matched the human liver profiles, especially pathways associated with lipid metabolism. Analysis of the human samples uncovered 65 and 177 differentially expressed genes in the NAFLD and NASH groups, but only 12 genes in the healthy obese group. Although splicing factor expression was not significantly altered in the human samples, Hnrnpab, Hnrnpl, Rbm4, Rbm4b, Rbm42, Srsf2, and Srsf5 were altered in the mouse MCD and HFD NASH models.
Arendt et al. (27) profiled liver expression in 20 patients with simple steatosis, 19 with NASH and 24 healthy controls by microarray and identified 556 genes altered in the NASH group, while 530 genes were altered in the simple steatosis group and only 22 genes were different between the steatosis and NASH groups. While they were able to show enrichment of genes for fibrosis, inflammation, oxidative stress and lipid metabolism, the dataset did not show enrichment of genes involved in RNA splicing.
Almanza et al. (56) performed transcriptional profiling by RNAseq in SAMP6 mice fed a high-fat diet to induce NAFLD. The authors identified a pre-fibrotic and pre-malignant gene signature in the 350 differentially expressed genes in this mouse model, but splicing factors were not found to be altered.
Kristiansen et al. (57) used a high-cholesterol (2%), high trans-fat (44%) diet to induce NASH in C57BL/six mice compared to obese db/db mice. Although they reported 868 genes induced and 510 genes repressed in their NASH model, the data were not provided so it was not possible to determine whether any splicing factors were among the genes altered.
Similarly, Van Koppen et al. (58) performed RNAseq on livers from LDL-R KO mice fed a high-fat diet to induce NASH. While they reported fibrosis, inflammation, and lipid metabolism signatures, they did not provide the data, so it was again not possible to determine if any splicing factors were altered.
Given the dysregulation of the RNA splicing machinery that has been observed in human and mouse fatty liver disease, a number of studies have looked at the role of individual splicing factors in the liver using mouse genetics (Table 2).
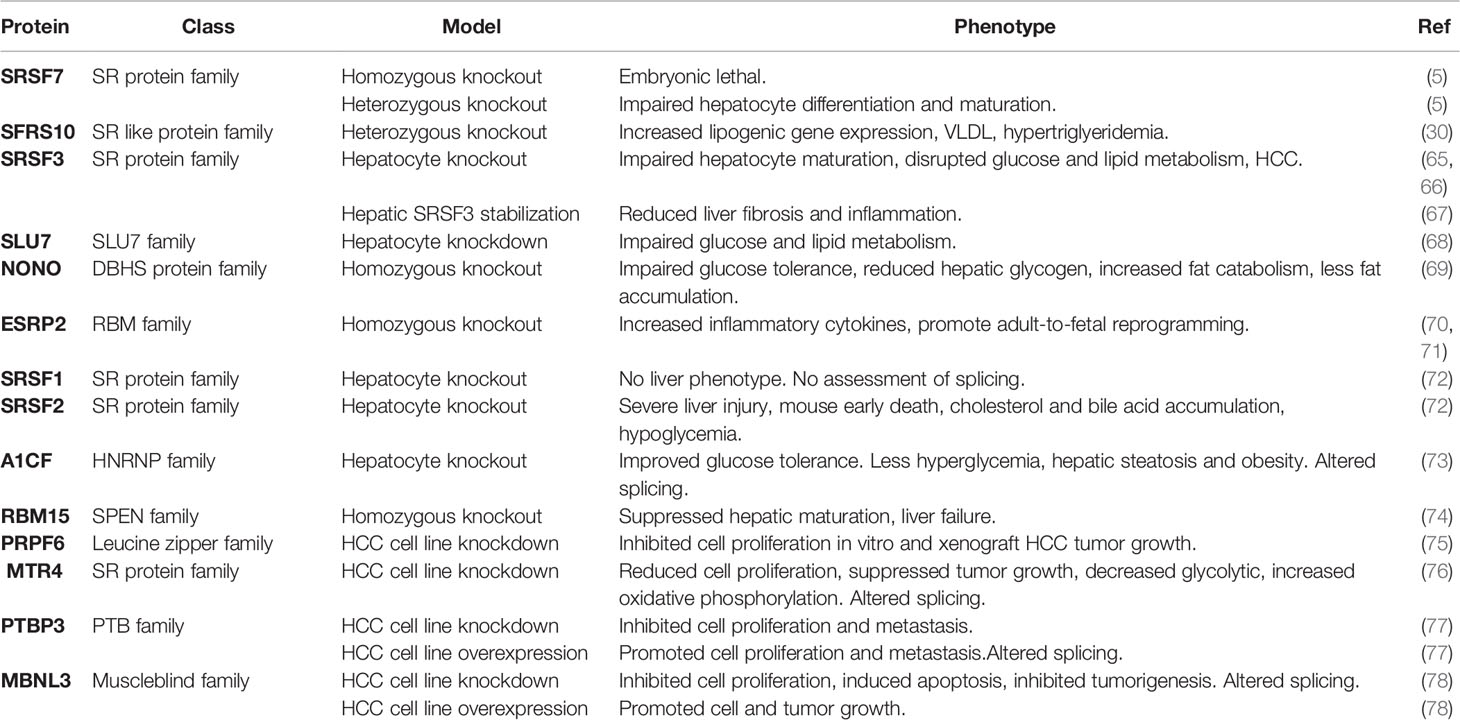
Table 2 Genetic manipulation of splicing factors implicated in liver disease.
SFRS10 (also known as TRA2b) belongs to SR-like protein family (79) and was identified as being reduced in the liver and muscle of obese individuals in the paper by Pihlajamaki et al. (30). Like many other SR proteins, the level of SFRS10 protein expression is regulated by a negative feedback loop. SFRS10 binds to its own exon 2 and promotes exon inclusion, generating an mRNA isoform that is subject to non-sense-mediated decay and unable to be translated into protein. When SFRS10 levels decline, exon 2 is skipped and the mRNA isoform encoding full length SFRS10 is produced (80). SiRNA-mediated SFRS10 knockdown increased the expression of several lipogenic genes and increased lipogenesis in vitro. Despite the negative feedback regulation, SFRS10 heterozygous mice showed increased expression of lipogenic gene expression in the liver, increased VLDL secretion and hypertriglyceridemia in vivo (30).
SRSF3 (also known as SRp20) is the smallest member of the SR protein family and was previously identified as a regulator of inclusion of exon 11 in the insulin receptor mRNA, which modulates the affinity of the receptor for IGF2. Sen et al. (65) showed that genetic loss of SRSF3 in hepatocytes impaired hepatocyte maturation and disrupted glucose and lipid metabolism. Furthermore, the SRSF3 knockout mice all developed HCC with aging (66). SRSF3 was found to be downregulated in human HCC samples, and recent studies showed SRSF3 was also reduced in NAFLD, NASH and cirrhosis liver samples in both human and mouse, and as a consequence, changes in the splicing of known SRSF3 target genes were observed (FN1, MYO1B, INSR, SLK) (10, 81). The increase of Slk spliced isoform and decrease of Dgkd and Insr spliced isoforms have also been detected in mouse livers with inflammation and fibrosis (10). Although SRSF3 protein levels were decreased in fatty liver disease, the levels of its mRNA and the ratio of SRSF3 mRNA isoforms did not change. The level of SRSF3 protein in the liver was determined by proteosomal degradation in the cytoplasm and was controlled by covalent attachment of NEDD8 on lysine 11 (67). Preventing SRSF3 degradation by inhibiting neddylation prevented NAFLD progression in mice, consistent with the fact that the inhibition of neddylation pathway was shown to reverse liver fibrosis in vivo (67). Thus, destabilization of a splicing factor under lipid overload is able to trigger liver disease progression. SRSF3 is also implicated in hepatitis B virus pathology as the HBx protein sequesters SRSF3 in the cytoplasm in a 14-3-3β complex, which has been shown to enhance Ras/FOXO4 signaling through increased expression of CCDC50S (82).
SLU7 is a splicing factor that ensures the correct selection of the 3′ splice site (83), and its expression was downregulated in patients with cirrhosis (84). Elizalde et al. (68) analyzed the splice events that occurred in HCC cells with downregulated SLU7 and found the most influenced category of genes was RNA post-translational modification. Knockdown of SLU7 in human liver cells and mouse liver impaired glucose and lipid metabolism, and the knockdown mice were unresponsive to normal feeding and fasting. SLU7 was more recently shown to be essential for maintaining genome integrity by suppressing the inclusion of exon 4 in the SRSF3 mRNA. This transcript is normally subject to non-sense-mediated decay but if translated gives rise to a truncated form of SRSF3 and causes intron retention in sororin mRNA and defects in sister chromatid cohesion and hence, DNA damage (85). In contrast to cirrhosis, SLU7 was shown to be elevated in alcoholic steatohepatitis in humans, and SLU7 knockdown prevented oxidative stress and liver damage in alcohol-treated mice. So, these studies suggest that both overexpression and loss of SLU7 are detrimental to liver function.
The RNA binding protein NONO (non-POU domain-containing octamer binding) belongs to the Drosophila Behavior Human Splicing family and binds primarily to introns within pre-mRNAs (86). NONO is an RNA binding protein that forms a heterodimer with SFPQ, a splicing factor that has been shown to be decreased in obesity. Using RNA-immunoprecipitation and sequencing (RIP-seq) NONO bind sites were found to be enriched in metabolic and circadian genes especially after feeding. NONO regulated glucose-responsive genes, including Gck and Glut2, in the liver post-transcriptionally. NONO-deficient mice had impaired glucose tolerance and reduced hepatic glycogen, were lean and stored less fat, and exhibited increased fat catabolism during the light phase (69). Viral overexpression of NONO improved the glucose tolerance in the NONO-deficient mice. Supporting a role in liver disease, NONO was also found to be elevated in livers from mice on high-fat diet (10). NONO-SFPQ is also the target for the lncRNA Morrbid and modulates Nras splicing (87). The NONO and SFPQ genes are frequently co-expressed in HCC where they promote the inclusion of exon12a in the bridging integrator 1 (BIN1) gene causing the expression of the BIN1-L isoform that binds and stabilizes PLK1 (88).
Epithelial splicing regulatory protein 2 (ESRP2) belongs to the RBM family of RNA-binding proteins and was originally identified as an epithelium-specific splicing regulator (89). A more recent study showed ESRP2 controlled the neonatal-to-adult shift of alternative splicing in the liver (70). Furthermore, Hyun et al. (71) found ESRP2 was suppressed in severe alcoholic hepatitis (ASH) in both humans and mouse models. They further showed that the release of inflammatory cytokines (TNF-α, IL-1β) by excessive alcohol ingestion reprogramed adult hepatocytes into fetal-like cells by suppressing ESRP2. Indeed, depleting ESRP2 exacerbated alcohol-induced steatohepatitis in mouse models. As inflammatory cytokines are involved in liver injury in many disease settings (90–92), ESRP2 suppression and adult-to-fetal reprogramming were also observed in a carbon tetrachloride (CCl4)-induced liver fibrosis model (71). Downregulation of ESRP2 activates a neonatal splicing program and causes exon skipping in the Yap1 and Tead1 genes, rewiring Hippo signaling and supporting progenitor cell proliferation upon liver injury (55). Altered ESRP2 expression has also been found in human HCC samples (93), so it is reasonable to expect that ESRP2-mediated splicing plays a role in liver disease as inflammation is a marker of progression from NAFLD to NASH (94).
SRSF2 (also known as SC35) also belongs to the SR protein family and binds exonic splicing enhancers (95, 96). Cheng et al. (72) reported that hepatocyte-specific deletion of SRSF2 caused severe liver injury and early death in mice. RNA-Seq analysis identified SRSF2-regulated cell death and stress-related alternative splicing events, including Becn1, Mfge8, Trp53inp, and Trp53inp2. Furthermore, inactivation of SRSF2 caused hepatic metabolic disorders by controlling expression of transcription factors responsible for energy homeostasis and bile acid metabolism, including PPARα, C/EBPα, SREBF1c, and NR1I3, that led to cholesterol and bile acid accumulation in the SRSF2-KO mice. Loss of SRSF2 also decreased expression of metabolic genes such Ebp, Baat, Slc27a5, resulting in hypoglycemia indicating an essential role of SRSF2 in hepatic metabolism. Interestingly, the few Srsf2 knockout mice that did not die from liver failure showed impaired hepatocyte maturation, activation of hepatocyte progenitor cells, and eventually developed HCC (97). Deletion of a related SR protein SRSF1 (SF2/ASF) did not result in this phenotype, so the effects are specific and not related to global effects on RNA splicing. Overexpression of SRSF2 has been observed in HCC and knockdown of SRSF2 in human hepatoma cells prevents tumor growth (98).
SRSF7 (also known as 9G8) is closely related to SRSF3 (99). A study performed by Peng et al. (100) profiled mouse liver transcriptomes during liver development, and SRSF7 expression was shown to decrease during liver maturation. This finding was further supported by Jam et al. who analyzed hepatocytes from juvenile and adult mice (101) and found SRSF7 was expressed more highly in juvenile hepatocytes. Depletion of SRSF7 led to premature maturation, whereas forced expression of SRSF7 suppressed cellular senescence in vitro. SRSF7 depletion also impaired cellular anabolism and increased glycolysis consistent with a more fetal-like state. SRSF7 knockout mice also exhibited suppression of juvenility-associated genes in hepatocytes, including Igf2, which functions as an enhancer of body growth (5). Thus, SRSF7 is essential for hepatocyte differentiation and maturation.
A1CF (also known as APOBEC1 complementation factor) belongs to the HNRNP family of RNA binding proteins. It was originally identified as an essential component of the ApoB editing complex but recent reports have shown that it is dispensable for RNA editing (102–104). Mice lacking A1cf expression in the liver exhibit improved glucose tolerance and are protected from fructose-induced hyperglycemia, hepatic steatosis, and obesity. The mice have altered RNA splicing of 84 genes including Gk and Khk, and PAR-CLIP studies indicated that A1CF binds to a UGGG sequence and competes with HNRNPH to regulate splicing of various RNA transcripts (73).
RNA binding motif protein 15 (RBM15) belongs to the SPEN family and determines cell fate of many tissues (105). RBM15 binds to RNA to regulate post-transcriptional modifications (106) such as alternative RNA splicing, polyadenylation, and protein translation. RBM15 is not only essential for megakaryocyte differentiation (107), but also indispensable for liver development (74). Hu et al. (74) found RBM15 was expressed in the liver during its differentiation, and depletion of RBM15 specifically suppressed hepatic maturation and caused liver failure but did not affect hepatocyte proliferation and apoptosis. More studies are needed to understand the role of RBM15 in hepatic maturation and liver diseases.
Pre-mRNA processing factor 6 is a splicing factor involved in spliceosome formation. Depletion of Rpr6 inhibits cell proliferation and HCC tumor growth potentially by upregulating the expression of the androgen receptor splice variant 7 (AR-V7) (75).
MTR4 is an RNA helicase that is present in the TRMAP complex that targets incorrectly processed transcripts for degradation by the nuclear exosome (108, 109). Knockdown of MTR4 expression in HCC cells causes changes in alternative splicing predominantly through exon skipping (76). The authors demonstrated that the glycolytic enzymes Glut1 and Pkm2 are two MTR4 targets, and knockdown of MTR4 increases splicing of the Glut1b and Pkm1 isoforms, causing a metabolic switch from glycolysis to oxidative phosphorylation. Mechanistically, MTR4 acts by recruiting the poly-pyrimidine tract binding protein PTBP1 to 3′ splice sites.
The polypyrimidine tract binding protein 3 (PTBP3) is overexpressed in HCC and regulates alternative splicing at the 3′ end of the lncRNA NEAT1. This lncRNA controls p53 and CCND1 signaling and hence proliferation (77).
The muscle blind protein 3 (MBNL3) is expressed highly in fetal liver and is re-expressed in HCC. Transcriptomic analysis of SMMC-7721 HCC cells with Mbnl3 knockdown revealed 527 MBNL3-dependent alternative splicing events (78). The authors showed that MBNL3 induces exon 4 inclusion in the lncRNA PXN-AS1 that is transcribed from the anti-sense strand of the paxillin (PXN) gene. These anti-sense transcripts have different effects on PXN mRNA translation, with the short isoform inhibiting translation but the long isoform preventing mRNA degradation by miR-24. Paxillin is a focal adhesion protein that promotes tumor cell proliferation, and the authors demonstrated that the oncogenic effects of MBNL3 overexpression are mediated by changes in PXN translation.
Alternative splicing of mRNA can alter the sequence of the encoded protein. This can alter the biochemical properties of the protein, the intracellular localization, the stability, the ability to be regulated by post-translational modifications, or interactions with other proteins (110). In extreme cases, such as the Bcl-X gene, alternative splicing can generate isoforms with antagonistic activity (111). Changes in splicing factor expression can alter the splicing of hundreds of target genes that could potentially be responsible for the observed phenotype. So, in this section, we will look at individual splice variants in target genes and their possible role in fatty liver disease.
Kruppel-like factor 6 (KLF6), a member of the Kruppel-like family, was identified as an activator of several genes involved in the development of liver fibrosis (112), and its expression is increased during progression to fibrosis in a rat NASH model (113). Genetic association studies have also shown that a SNP located in the first intron is associated with NAFLD. The functional SNP creates a novel binding site for a splicing factor SRp40 that alters the splicing of the KLF6 pre-mRNA allowing production of a shorter isoform. While the KLF6 full length isoform was increased in NAFLD patients with more advanced disease, the alternative spliced isoform of KLF6 enhanced by the SNP was associated with reduced fibrosis in NAFLD. Miele et al. (114) found that the shorter KLF6 isoform was anti-fibrogenic and could abrogate the induction of α-smooth muscle actin and type 1 collagen mRNAs in vitro by full length KLF6. Moreover, another study (115) revealed that the alternatively spliced variant of KLF6 lowered the hepatic insulin resistance and blood glucose by reducing glucokinase expression.
The peroxisome proliferator-activated receptor family (PPARs) consists of three genes: PPARα, PPARβ/δ and PPARγ, with a highly conserved structure (116). The PPARs regulate genes involved in multiple processes, such as fatty acid uptake and oxidation, lipid metabolism and inflammation (117). PPARα and PPARβ/δ play a role in lipid catabolism, while PPARγ regulates lipid anabolism and is essential for induction of adipogenesis (118). PPARγ has multiple alternatively spliced transcripts but two major protein isoforms: PPARγ1 and PPARγ2. Many transcripts encode PPARγ1, but only the PPARG-201 transcript, which initiates from a downstream promoter, encodes the PPARγ2 protein (119). Though PPARγ1 expression is very low in normal liver, PPARγ2 expression is significantly elevated in terms of both mRNA and protein levels in the livers of obese mice compared to the wide type mice. Furthermore, the increased PPARγ2 expression was positively correlated with liver steatosis in obese patients and with insulin resistance in mice (120–122). Given PPARγ2’s role in induction of adipogenic genes, the elevated expression in the liver is consistent with the induction of steatosis in obesity. PPARγ2 activation induced lipogenic genes (including ADRP, SREBP-1, and FAS) and promoted de novo lipogenesis resulting in lipid accumulation in hepatocytes (123–125).
The insulin receptor (INSR) is a tyrosine kinase receptor that mediates both the metabolic and mitogenic effects of insulin (126). The association between INSR and NAFLD has been widely investigated since Marchesini et al. first demonstrated that NAFLD patients had reduced insulin sensitivity and impaired hepatic glucose production (127). INSR has two isoforms due to alternative splicing of exon 11: INSR-A and INSR-B (128, 129). INSR-A lacks exon 11 and binds both insulin and IGF-2 with high affinity, but INSR-B that contains the additional 12 amino acids encoded by exon 11 only binds insulin with high affinity (130). The expression of these two different isoforms is regulated at both mRNA transcription and post-transcription levels (128, 129). In many cancers, INSR is overexpressed and the A:B ratio increased (129). Kumar et al. (81) have reported that the splicing of the INSR is altered in patients with NAFLD, NASH, and cirrhosis and in mice on high-fat diet or NASH diets, with increased expression of INSR-A. In pre-clinical studies, Lopez-Pastor et al. reported that AAV expression of INSR-A or INSR-B significantly reduced the NAFLD activity score (NAS) and improved insulin secretion, but did not affect body weight or glucose tolerance in mice on a high fat diet (131). Interestingly, INSR-A improved insulin sensitivity and increased glucose uptake into liver and muscle. Similar studies in a liver Insr knockout mouse showed that INSR-A was more effective at ameliorating glucose intolerance (132). The two receptors are known to signal differentially and INSR-A and INSR-B showed different effects on gene expression as has been reported in pancreatic beta cells (133). Thus, altered INSR splicing in liver could potentially alter NAFLD progression.
LPIN1 is a member of the lipin gene family that dephosphorylates phosphatidic acid to diacylglycerol in the penultimate step in triglyceride metabolism (134). LPIN1 gene encodes two mRNA isoforms, lipin-1α, lipin-1β by alternative mRNA splicing (135). Lipin-1β includes exon 6 compared with lipin-1α (35). The two isoforms of lipin-1 differ in expression pattern, subcellular localization, and function (35). During the adipocyte differentiation, lipin-1α decreases, and lipin-1β increases. In mature adipocytes, lipin-1α localizes in nucleus but lipin-1β is primarily cytoplasmic. Unlike lipin-1α, lipin-1β expression leads to induction of lipogenic genes (35). The expression of lipin-1β mRNA but not lipin-1α increased in the livers of NASH mice induced by choline-deficient diet (136). Pihlajamaki et al. (30) reported that SFRS10 regulates lipin-1 mRNA splicing by binding to exon 6 causing its inclusion. SFRS10 levels are reduced in the livers of obese mice and obese humans and, although overall expression of lipin-1 did not change, the lipin-1β/α ratio increased. Yin et al. (137) demonstrated that lipin-1β/α is also increased in alcoholic fatty liver disease (AFLD) in mice. They also showed that alcohol reduces SIRT1 expression in mouse liver which in turn reduces SRSF10 expression. Genetic deletion of SIRT1 in hepatocytes causes hepatic steatosis suggesting this may be causative in AFLD (138). The downregulation of SIRT1 was also observed in obese human subjects, and LPIN1 splicing was altered, but paradoxically, SRSF10 levels were not changed (139). More evidence is needed, therefore, to fully establish the SIRT1–SFRS10–LIPIN-1 axis in fatty liver in humans.
Tissue factor (TF) is produced by the liver and is required for blood coagulation (140). An alternatively spliced form of TF is found in the plasma of patients with chronic liver disease including liver fibrosis, cirrhosis and HCC. This isoform lacks exon 5 leading to premature termination of translation in exon 6, the last exon (141). The resulting protein lacks the transmembrane domain that anchors TF in the plasma membrane as TF acts as an angiogenic factor by stimulating integrin signaling, triggering proliferation and tumor cell metastasis.
The prenyldiphosphate synthase subunit 2 (PDSS2) is a key factor in coenzyme Q10 synthesis. Six splicing variants are produced, but only full length PDSS2 is catalytically active, the other five variants showing loss of function. Loss of PDSS2 function causes a shift from mitochondrial respiration to aerobic glycolysis and increased proliferation of HCC cells that can only be restored by the full length Pdss2 isoform (142). Knockdown of PDSS2 in MIHA immortalized liver cells caused chromosomal instability and transformation.
It is generally accepted that alternative RNA splicing plays an important role in fine tuning gene expression and cellular function in various tissues and contexts. There is also evidence that changes in RNA splicing may be involved in the pathogenesis of obesity in various tissues (143). In the liver, changes in RNA splicing have been documented during development and maturation of hepatocytes, but studies showing a causal relationship are rare. In 2017 (17), we reviewed the status of the field and the published studies of alternative splicing in the liver. Since then, a number of large human studies have been published so we have updated our review to include these and other new studies that further support the concept that alternative splicing is an early feature of liver disease. While RNA splicing changes have been documented in HCC (8–16) and reviewed elsewhere (144–146), most studies in early liver disease, NAFLD or NASH, have focused on total mRNA changes rather than changes in individual mRNA isoforms and alternative splicing (48–50). These studies, however, can be informative as changes in the expression of splicing machinery components can be indicative of changes in RNA splicing. The idea that these changes may be causative for, rather than the result of, liver disease is supported by accumulating evidence from genetic manipulation of individual splicing factors or isoform variants in mice that contribute to liver disease. It is worth noting that most of the altered mRNAs or splicing factors are associated with lipid metabolism which is not unexpected given the common steatotic phenotype. Beyond that, gluconeogenesis and fibrosis are also disease processes that may be influenced by alternative splicing, which may contribute to disease progression, inflammation and fibrosis. Given the potential role of individual mRNA isoforms in liver disease, further investigation into the extent of splicing dysregulation in liver disease, the splicing factor target networks involved, and the function of the alternatively spliced isoforms is clearly required. Additionally, as the transcriptome profiles between humans and mice with NAFLD have been shown to differ, it would be important to compare alternative splicing patterns in human liver and mouse models of liver disease. This is particularly important as studying hepatic-specific KO mice and splice variants is a powerful tool for functional evaluation of alternative splicing variants in vivo. Functional assessment is essential as it is not possible a priori to predict the effect of a splice variant on protein activity. Unlike transcriptome profiling where an increase in gene expression generally leads to an increase in protein expression and activity, this is not the case for alternative splicing. Indeed, there are many examples of splice variants having antagonistic activities. Gene-to-gene mapping for transcriptome studies is possible but mapping alternative splicing events is more problematic. There is currently no widely accepted method to name splicing events, and the output from most software programs is not compatible with cross-comparisons. The development of standardized formats and nomenclature in the splicing field will be required to enable the use of animal models to predict human disease.
The recent studies provide a strong rationale for the development and testing of novel therapeutic strategies targeting specific isoforms based on an understanding of alternative splice variants in NAFLD. Small molecule splicing inhibitors have been developed to interfere with the activity of the spliceosome and are being tested in cancer models (147). Yet these general inhibitors are unlikely to be useful in other diseases due to broad effects to inhibit all splicing. A more promising approach is the development of splice-switching anti-sense oligonucleotides (ASO) for diseases related to RNA mis-splicing, thus minimizing toxic side effects (148, 149). Although anti-sense oligonucleotides (ASO) delivery is a challenge, recent advances in the modifications have improved the stability, affinity (150, 151) and specificity (152). For example, the BCL-X gene has two major isoforms: antiapoptotic BCL-XL and proapoptotic BCL-XS. ASO can induce a shift from BCL-XL to Bcl-XS in human hepatic stellate cells, which are the major producers of fibrotic ECM, showing that BCL-X ASO is a potential therapy for liver fibrosis (153). Other ASOs have been proved to protect mice from NAFLD through aiming at gene silencing (154, 155). Alternative therapeutic approaches include the development of modified U1 snRNAs that target specific splicing mutations (156). These have been used successfully in preclinical studies in familial dysautonomia (157), hemophilia B (158), and spinal muscular atrophy (159). Development of such targeted therapeutics for liver disease will require further studies on alternative splicing in NAFLD and the role of individual splice variants.
PW wrote the first draft. MZ helped revise the manuscript. NW edited and finalized the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded partly by a Veterans Affairs Merit Review awards (I01BX000130 and I01BX004848) and Senior Research Career Scientist Award (IK6BX005224), and partly by NIH grants (CA196853, AI25860, and HL141999) to NW.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Licatalosi DD, Darnell RB. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet (2010) 11(1):75–87. doi: 10.1038/nrg2673
2. Anczukow O, Krainer AR. Splicing-factor alterations in cancers. RNA (2016) 22(9):1285–301. doi: 10.1261/rna.057919.116
3. Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet (2008) 40(12):1413–5. doi: 10.1038/ng.259
4. Gunewardena SS, Yoo B, Peng L, Lu H, Zhong X, Klaassen CD, et al. Deciphering the Developmental Dynamics of the Mouse Liver Transcriptome. PloS One (2015) 10(10):e0141220. doi: 10.1371/journal.pone.0141220
5. Kadota Y, Jam FA, Yukiue H, Terakado I, Morimune T, Tano A, et al. Srsf7 Establishes the Juvenile Transcriptome through Age-Dependent Alternative Splicing in Mice. iScience (2020) 23(3):100929. doi: 10.1016/j.isci.2020.100929
6. Chaturvedi P, Neelamraju Y, Arif W, Kalsotra A, Janga SC. Uncovering RNA binding proteins associated with age and gender during liver maturation. Sci Rep (2015) 5:9512. doi: 10.1038/srep09512
7. Berasain C, Goni S, Castillo J, Latasa MU, Prieto J, Avila MA. Impairment of pre-mRNA splicing in liver disease: mechanisms and consequences. World J Gastroenterol (2010) 16(25):3091–102. doi: 10.3748/wjg.v16.i25.3091
8. Li S, Hu Z, Zhao Y, Huang S, He X. Transcriptome-Wide Analysis Reveals the Landscape of Aberrant Alternative Splicing Events in Liver Cancer. Hepatology (2019) 69(1):359–75. doi: 10.1002/hep.30158
9. Chen QF, Li W, Wu P, Shen L, Huang ZL. Alternative splicing events are prognostic in hepatocellular carcinoma. Aging (Albany NY) (2019) 11(13):4720–35. doi: 10.18632/aging.102085
10. Wang H, Lekbaby B, Fares N, Augustin J, Attout T, Schnuriger A, et al. Alteration of splicing factors’ expression during liver disease progression: impact on hepatocellular carcinoma outcome. Hepatol Int (2019) 13(4):454–67. doi: 10.1007/s12072-019-09950-7
11. Wang J, Wang X, Bhat A, Chen Y, Xu K, Mo YY, et al. Comprehensive Network Analysis Reveals Alternative Splicing-Related lncRNAs in Hepatocellular Carcinoma. Front Genet (2020) 11:659. doi: 10.3389/fgene.2020.00659
12. Xiong Y, Yang G, Wang K, Riaz M, Xu J, Lv Z, et al. Genome-Wide Transcriptional Analysis Reveals Alternative Splicing Event Profiles in Hepatocellular Carcinoma and Their Prognostic Significance. Front Genet (2020) 11:879. doi: 10.3389/fgene.2020.00879
13. Lee SE, Alcedo KP, Kim HJ, Snider NT. Alternative Splicing in Hepatocellular Carcinoma. Cell Mol Gastroenterol Hepatol (2020) 10(4):699–712. doi: 10.1016/j.jcmgh.2020.04.018
14. Jin YJ, Byun S, Han S, Chamberlin J, Kim D, Kim MJ, et al. Differential alternative splicing regulation among hepatocellular carcinoma with different risk factors. BMC Med Genomics (2019) 12(Suppl 8):175. doi: 10.1186/s12920-019-0635-z
15. Yang L, He Y, Zhang Z, Wang W. Systematic analysis and prediction model construction of alternative splicing events in hepatocellular carcinoma: a study on the basis of large-scale spliceseq data from The Cancer Genome Atlas. PeerJ (2019) 7:e8245. doi: 10.7717/peerj.8245
16. Wu HY, Peng ZG, He RQ, Luo B, Ma J, Hu XH, et al. Prognostic index of aberrant mRNA splicing profiling acts as a predictive indicator for hepatocellular carcinoma based on TCGA SpliceSeq data. Int J Oncol (2019) 55(2):425–38. doi: 10.3892/ijo.2019.4834
17. Webster NJG. Alternative RNA Splicing in the Pathogenesis of Liver Disease. Front Endocrinol (Lausanne) (2017) 8:133. doi: 10.3389/fendo.2017.00133
18. Marchisello S, Di Pino A, Scicali R, Urbano F, Piro S, Purrello F, et al. Pathophysiological, Molecular and Therapeutic Issues of Nonalcoholic Fatty Liver Disease: An Overview. Int J Mol Sci (2019) 20(8):1948. doi: 10.3390/ijms20081948
19. Munteanu MA, Nagy GA, Mircea PA. Current Management of NAFLD. Clujul Med (2016) 89(1):19–23. doi: 10.15386/cjmed-539
20. Asrih M, Jornayvaz FR. Metabolic syndrome and nonalcoholic fatty liver disease: Is insulin resistance the link? Mol Cell Endocrinol (2015) 418:55–65. doi: 10.1016/j.mce.2015.02.018
21. Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology (2006) 43(2 Suppl 1):S99–S112. doi: 10.1002/hep.20973
22. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology (2016) 64(1):73–84. doi: 10.1002/hep.28431
23. Sherif ZA, Saeed A, Ghavimi S, Nouraie SM, Laiyemo AO, Brim H, et al. Global Epidemiology of Nonalcoholic Fatty Liver Disease and Perspectives on US Minority Populations. Dig Dis Sci (2016) 61(5):1214–25. doi: 10.1007/s10620-016-4143-0
24. Koutsari C, Lazaridis KN. Emerging genes associated with the progression of nonalcoholic fatty liver disease. Hepatology (2010) 52(3):807–10. doi: 10.1002/hep.23869
25. Starmann J, Falth M, Spindelbock W, Lanz KL, Lackner C, Zatloukal K, et al. Gene expression profiling unravels cancer-related hepatic molecular signatures in steatohepatitis but not in steatosis. PloS One (2012) 7(10):e46584. doi: 10.1371/journal.pone.0046584
26. Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology (2014) 59(2):471–82. doi: 10.1002/hep.26661
27. Arendt BM, Comelli EM, Ma DW, Lou W, Teterina A, Kim T, et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology (2015) 61(5):1565–78. doi: 10.1002/hep.27695
28. Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, et al. Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues From Patients. Gastroenterology (2016) 151(3):513–25 e0. doi: 10.1053/j.gastro.2016.05.051
29. Del Rio-Moreno M, Alors-Perez E, Gonzalez-Rubio S, Ferrin G, Reyes O, Rodriguez-Peralvarez M, et al. Dysregulation of the Splicing Machinery Is Associated to the Development of Nonalcoholic Fatty Liver Disease. J Clin Endocrinol Metab (2019) 104(8):3389–402. doi: 10.1210/jc.2019-00021
30. Pihlajamaki J, Lerin C, Itkonen P, Boes T, Floss T, Schroeder J, et al. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab (2011) 14(2):208–18. doi: 10.1016/j.cmet.2011.06.007
31. Moller DE, Yokota A, Caro JF, Flier JS. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol Endocrinol (1989) 3(8):1263–9. doi: 10.1210/mend-3-8-1263
32. Huang Z, Bodkin NL, Ortmeyer HK, Zenilman ME, Webster NJ, Hansen BC, et al. Altered insulin receptor messenger ribonucleic acid splicing in liver is associated with deterioration of glucose tolerance in the spontaneously obese and diabetic rhesus monkey: analysis of controversy between monkey and human studies. J Clin Endocrinol Metab (1996) 81(4):1552–6. doi: 10.1210/jcem.81.4.8636366
33. Kaminska D, Hamalainen M, Cederberg H, Kakela P, Venesmaa S, Miettinen P, et al. Adipose tissue INSR splicing in humans associates with fasting insulin level and is regulated by weight loss. Diabetologia (2014) 57(2):347–51. doi: 10.1007/s00125-013-3097-4
34. Kulseth MA, Berge KE, Bogsrud MP, Leren TP. Analysis of LDLR mRNA in patients with familial hypercholesterolemia revealed a novel mutation in intron 14, which activates a cryptic splice site. J Hum Genet (2010) 55(10):676–80. doi: 10.1038/jhg.2010.87
35. Peterfy M, Phan J, Reue K. Alternatively spliced lipin isoforms exhibit distinct expression pattern, subcellular localization, and role in adipogenesis. J Biol Chem (2005) 280(38):32883–9. doi: 10.1074/jbc.M503885200
36. Lee Y, Rio DC. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu Rev Biochem (2015) 84:291–323. doi: 10.1146/annurev-biochem-060614-034316
37. Escobar-Hoyos L, Knorr K, Abdel-Wahab O. Aberrant RNA Splicing in Cancer. Annu Rev Cancer Biol (2019) 3(1):167–85. doi: 10.1146/annurev-cancerbio-030617-050407
38. Montes M, Sanford BL, Comiskey DF, Chandler DS. RNA Splicing and Disease: Animal Models to Therapies. Trends Genet (2019) 35(1):68–87. doi: 10.1016/j.tig.2018.10.002
39. Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature (2008) 456(7221):470–6. doi: 10.1038/nature07509
40. Shi Y. The Spliceosome: A Protein-Directed Metalloribozyme. J Mol Biol (2017) 429(17):2640–53. doi: 10.1016/j.jmb.2017.07.010
41. Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell (2009) 136(4):701–18. doi: 10.1016/j.cell.2009.02.009
42. Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem (2003) 72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720
43. Srebrow A, Kornblihtt AR. The connection between splicing and cancer. J Cell Sci (2006) 119(Pt 13):2635–41. doi: 10.1242/jcs.03053
44. Graveley BR. Sorting out the complexity of SR protein functions. RNA (2000) 6(9):1197–211. doi: 10.1017/S1355838200000960
45. Sahebi M, Hanafi MM, van Wijnen AJ, Azizi P, Abiri R, Ashkani S, et al. Towards understanding pre-mRNA splicing mechanisms and the role of SR proteins. Gene (2016) 587(2):107–19. doi: 10.1016/j.gene.2016.04.057
46. Shin C, Manley JL. Cell signalling and the control of pre-mRNA splicing. Nat Rev Mol Cell Biol (2004) 5(9):727–38. doi: 10.1038/nrm1467
47. Stamm S. Regulation of alternative splicing by reversible protein phosphorylation. J Biol Chem (2008) 283(3):1223–7. doi: 10.1074/jbc.R700034200
48. Gerhard GS, Legendre C, Still CD, Chu X, Petrick A, DiStefano JK. Transcriptomic Profiling of Obesity-Related Nonalcoholic Steatohepatitis Reveals a Core Set of Fibrosis-Specific Genes. J Endocr Soc (2018) 2(7):710–26. doi: 10.1210/js.2018-00122
49. Suppli MP, Rigbolt KTG, Veidal SS, Heeboll S, Eriksen PL, Demant M, et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am J Physiol Gastrointest Liver Physiol (2019) 316(4):G462–G72. doi: 10.1152/ajpgi.00358.2018
50. Hoang SA, Oseini A, Feaver RE, Cole BK, Asgharpour A, Vincent R, et al. Gene Expression Predicts Histological Severity and Reveals Distinct Molecular Profiles of Nonalcoholic Fatty Liver Disease. Sci Rep (2019) 9(1):12541. doi: 10.1038/s41598-019-48746-5
51. Steensels S, Qiao J, Ersoy BA. Transcriptional Regulation in Non-Alcoholic Fatty Liver Disease. Metabolites (2020) 10(7):283. doi: 10.3390/metabo10070283
52. Zhu R, Baker SS, Moylan CA, Abdelmalek MF, Guy CD, Zamboni F, et al. Systematic transcriptome analysis reveals elevated expression of alcohol-metabolizing genes in NAFLD livers. J Pathol (2016) 238(4):531–42. doi: 10.1002/path.4650
53. Ye H, Liu W. Transcriptional networks implicated in human nonalcoholic fatty liver disease. Mol Genet Genomics (2015) 290(5):1793–804. doi: 10.1007/s00438-015-1037-3
54. Lake AD, Novak P, Fisher CD, Jackson JP, Hardwick RN, Billheimer DD, et al. Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos (2011) 39(10):1954–60. doi: 10.1124/dmd.111.040592
55. Bangru S, Arif W, Seimetz J, Bhate A, Chen J, Rashan EH, et al. Alternative splicing rewires Hippo signaling pathway in hepatocytes to promote liver regeneration. Nat Struct Mol Biol (2018) 25(10):928–39. doi: 10.1038/s41594-018-0129-2
56. Almanza D, Gharaee-Kermani M, Zhilin-Roth A, Rodriguez-Nieves JA, Colaneri C, Riley T, et al. Nonalcoholic Fatty Liver Disease Demonstrates a Pre-fibrotic and Premalignant Molecular Signature. Dig Dis Sci (2019) 64(5):1257–69. doi: 10.1007/s10620-018-5398-4
57. Kristiansen MN, Veidal SS, Rigbolt KT, Tolbol KS, Roth JD, Jelsing J, et al. Obese diet-induced mouse models of nonalcoholic steatohepatitis-tracking disease by liver biopsy. World J Hepatol (2016) 8(16):673–84. doi: 10.4254/wjh.v8.i16.673
58. van Koppen A, Verschuren L, van den Hoek AM, Verheij J, Morrison MC, Li K, et al. Uncovering a Predictive Molecular Signature for the Onset of NASH-Related Fibrosis in a Translational NASH Mouse Model. Cell Mol Gastroenterol Hepatol (2018) 5(1):83–98. doi: 10.1016/j.jcmgh.2017.10.001
59. Lopez-Vicario C, Gonzalez-Periz A, Rius B, Moran-Salvador E, Garcia-Alonso V, Lozano JJ, et al. Molecular interplay between Delta5/Delta6 desaturases and long-chain fatty acids in the pathogenesis of non-alcoholic steatohepatitis. Gut (2014) 63(2):344–55. doi: 10.1136/gutjnl-2012-303179
60. Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology (2013) 145(5):1076–87. doi: 10.1053/j.gastro.2013.07.047
61. Kishore S, Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science (2006) 311(5758):230–2. doi: 10.1126/science.1118265
62. Lefai E, Roques M, Vega N, Laville M, Vidal H. Expression of the splice variants of the p85alpha regulatory subunit of phosphoinositide 3-kinase in muscle and adipose tissue of healthy subjects and type 2 diabetic patients. Biochem J (2001) 360(Pt 1):117–26. doi: 10.1042/bj3600117
63. Patel NA, Kaneko S, Apostolatos HS, Bae SS, Watson JE, Davidowitz K, et al. Molecular and genetic studies imply Akt-mediated signaling promotes protein kinase CbetaII alternative splicing via phosphorylation of serine/arginine-rich splicing factor SRp40. J Biol Chem (2005) 280(14):14302–9. doi: 10.1074/jbc.M411485200
64. Ghosh N, Patel N, Jiang K, Watson JE, Cheng J, Chalfant CE, et al. Ceramide-activated protein phosphatase involvement in insulin resistance via Akt, serine/arginine-rich protein 40, and ribonucleic acid splicing in L6 skeletal muscle cells. Endocrinology (2007) 148(3):1359–66. doi: 10.1210/en.2006-0750
65. Sen S, Jumaa H, Webster NJ. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat Commun (2013) 4:1336. doi: 10.1038/ncomms2342
66. Sen S, Langiewicz M, Jumaa H, Webster NJ. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology (2015) 61(1):171–83. doi: 10.1002/hep.27380
67. Zubiete-Franco I, Fernandez-Tussy P, Barbier-Torres L, Simon J, Fernandez-Ramos D, Lopitz-Otsoa F, et al. Deregulated neddylation in liver fibrosis. Hepatology (2017) 65(2):694–709. doi: 10.1002/hep.28933
68. Elizalde M, Urtasun R, Azkona M, Latasa MU, Goni S, Garcia-Irigoyen O, et al. Splicing regulator SLU7 is essential for maintaining liver homeostasis. J Clin Invest (2014) 124(7):2909–20. doi: 10.1172/JCI74382
69. Benegiamo G, Mure LS, Erikson G, Le HD, Moriggi E, Brown SA, et al. The RNA-Binding Protein NONO Coordinates Hepatic Adaptation to Feeding. Cell Metab (2018) 27(2):404–18. doi: 10.1016/j.cmet.2017.12.010
70. Bhate A, Parker DJ, Bebee TW, Ahn J, Arif W, Rashan EH, et al. ESRP2 controls an adult splicing programme in hepatocytes to support postnatal liver maturation. Nat Commun (2015) 6(1):8768. doi: 10.1038/ncomms9768
71. Hyun J, Sun Z, Ahmadi AR, Bangru S, Chembazhi UV, Du K, et al. Epithelial splicing regulatory protein 2-mediated alternative splicing reprograms hepatocytes in severe alcoholic hepatitis. J Clin Invest (2020) 130(4):2129–45. doi: 10.1172/JCI132691
72. Cheng Y, Luo C, Wu W, Xie Z, Fu X, Feng Y. Liver-Specific Deletion of SRSF2 Caused Acute Liver Failure and Early Death in Mice. Mol Cell Biol (2016) 36(11):1628–38. doi: 10.1128/MCB.01071-15
73. Nikolaou KC, Vatandaslar H, Meyer C, Schmid MW, Tuschl T, Stoffel M. The RNA-Binding Protein A1CF Regulates Hepatic Fructose and Glycerol Metabolism via Alternative RNA Splicing. Cell Rep (2019) 29(2):283–300 e8. doi: 10.1016/j.celrep.2019.08.100
74. Hu L, Li H, Chi Z, He J. Loss of the RNA-binding protein Rbm15 disrupts liver maturation in zebrafish. J Biol Chem (2020) 295(33):11466–72. doi: 10.1074/jbc.RA120.014080
75. Song H, Sun N, Lin L, Wei S, Zeng K, Liu W, et al. Splicing factor PRPF6 upregulates oncogenic androgen receptor signaling pathway in hepatocellular carcinoma. Cancer Sci (2020) 111(10):3665–78. doi: 10.1111/cas.14595
76. Yu L, Kim J, Jiang L, Feng B, Ying Y, Ji KY, et al. MTR4 drives liver tumorigenesis by promoting cancer metabolic switch through alternative splicing. Nat Commun (2020) 11(1):708. doi: 10.1038/s41467-020-14437-3
77. Yang X, Qu S, Wang L, Zhang H, Yang Z, Wang J, et al. PTBP3 splicing factor promotes hepatocellular carcinoma by destroying the splicing balance of NEAT1 and pre-miR-612. Oncogene (2018) 37(50):6399–413. doi: 10.1038/s41388-018-0416-8
78. Yuan JH, Liu XN, Wang TT, Pan W, Tao QF, Zhou WP, et al. The MBNL3 splicing factor promotes hepatocellular carcinoma by increasing PXN expression through the alternative splicing of lncRNA-PXN-AS1. Nat Cell Biol (2017) 19(7):820–32. doi: 10.1038/ncb3538
79. Nayler O, Cap C, Stamm S. Human transformer-2-beta gene (SFRS10): complete nucleotide sequence, chromosomal localization, and generation of a tissue-specific isoform. Genomics (1998) 53(2):191–202. doi: 10.1006/geno.1998.5471
80. Stoilov P, Daoud R, Nayler O, Stamm S. Human tra2-beta1 autoregulates its protein concentration by influencing alternative splicing of its pre-mRNA. Hum Mol Genet (2004) 13(5):509–24. doi: 10.1093/hmg/ddh051
81. Kumar D, Das M, Sauceda C, Ellies LG, Kuo K, Parwal P, et al. Degradation of splicing factor SRSF3 contributes to progressive liver disease. J Clin Invest (2019) 129(10):4477–91. doi: 10.1172/JCI127374
82. Wang H, Zhang CZ, Lu SX, Zhang MF, Liu LL, Luo RZ, et al. A Coiled-Coil Domain Containing 50 Splice Variant Is Modulated by Serine/Arginine-Rich Splicing Factor 3 and Promotes Hepatocellular Carcinoma in Mice by the Ras Signaling Pathway. Hepatology (2019) 69(1):179–95. doi: 10.1002/hep.30147
83. Reed R. Mechanisms of fidelity in pre-mRNA splicing. Curr Opin Cell Biol (2000) 12(3):340–5. doi: 10.1016/S0955-0674(00)00097-1
84. Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science (2010) 330(6009):1340–4. doi: 10.1126/science.1193494
85. Jimenez M, Urtasun R, Elizalde M, Azkona M, Latasa MU, Uriarte I, et al. Splicing events in the control of genome integrity: role of SLU7 and truncated SRSF3 proteins. Nucleic Acids Res (2019) 47(7):3450–66. doi: 10.1093/nar/gkz014
86. Knott GJ, Bond CS, Fox AH, The DBHS proteins SFPQ. NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res (2016) 44(9):3989–4004. doi: 10.1093/nar/gkw271
87. Fefilova A, Melnikov P, Prikazchikova T, Abakumova T, Kurochkin I, Mazin PV, et al. Murine Long Noncoding RNA Morrbid Contributes in the Regulation of NRAS Splicing in Hepatocytes In Vitro. Int J Mol Sci (2020) 21(16):5605. doi: 10.3390/ijms21165605
88. Hu Z, Dong L, Li S, Li Z, Qiao Y, Li Y, et al. Splicing Regulator p54(nrb) /Non-POU Domain-Containing Octamer-Binding Protein Enhances Carcinogenesis Through Oncogenic Isoform Switch of MYC Box-Dependent Interacting Protein 1 in Hepatocellular Carcinoma. Hepatology (2020) 72(2):548–68. doi: 10.1002/hep.31062
89. Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell (2009) 33(5):591–601. doi: 10.1016/j.molcel.2009.01.025
90. Liu Y, Wen PH, Zhang XX, Dai Y, He Q. Breviscapine ameliorates CCl4induced liver injury in mice through inhibiting inflammatory apoptotic response and ROS generation. Int J Mol Med (2018) 42(2):755–68. doi: 10.3892/ijmm.2018.3651
91. Aguilar-Bravo B, Rodrigo-Torres D, Arino S, Coll M, Pose E, Blaya D, et al. Ductular Reaction Cells Display an Inflammatory Profile and Recruit Neutrophils in Alcoholic Hepatitis. Hepatology (2019) 69(5):2180–95. doi: 10.1002/hep.30472
92. Hwang W, Lee J. Pathophysiologic Implications of Cytokines Secretion during Liver Transplantation Surgery. Int J Med Sci (2018) 15(14):1737–45. doi: 10.7150/ijms.28382
93. Tremblay MP, Armero VE, Allaire A, Boudreault S, Martenon-Brodeur C, Durand M, et al. Global profiling of alternative RNA splicing events provides insights into molecular differences between various types of hepatocellular carcinoma. BMC Genomics (2016) 17:683. doi: 10.1186/s12864-016-3029-z
94. Machado MV, Cortez-Pinto H. Non-alcoholic fatty liver disease: what the clinician needs to know. World J Gastroenterol (2014) 20(36):12956–80. doi: 10.3748/wjg.v20.i36.12956
95. Howard JM, Sanford JR. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip Rev RNA (2015) 6(1):93–110. doi: 10.1002/wrna.1260
96. Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J (2009) 417(1):15–27. doi: 10.1042/BJ20081501
97. Zhang C, Shen L, Yuan W, Liu Y, Guo R, Luo Y, et al. Loss of SRSF2 triggers hepatic progenitor cell activation and tumor development in mice. Commun Biol (2020) 3(1):210. doi: 10.1038/s42003-020-0893-5
98. Luo C, Cheng Y, Liu Y, Chen L, Liu L, Wei N, et al. SRSF2 Regulates Alternative Splicing to Drive Hepatocellular Carcinoma Development. Cancer Res (2017) 77(5):1168–78. doi: 10.1158/0008-5472.CAN-16-1919
99. Cavaloc Y, Bourgeois CF, Kister L, Stevenin J. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA (1999) 5(3):468–83. doi: 10.1017/S1355838299981967
100. Peng L, Yoo B, Gunewardena SS, Lu H, Klaassen CD, Zhong XB. RNA sequencing reveals dynamic changes of mRNA abundance of cytochromes P450 and their alternative transcripts during mouse liver development. Drug Metab Dispos (2012) 40(6):1198–209. doi: 10.1124/dmd.112.045088
101. Jam FA, Kadota Y, Mendsaikhan A, Tooyama I, Mori M. Identification of juvenility-associated genes in the mouse hepatocytes and cardiomyocytes. Sci Rep (2018) 8(1):3132. doi: 10.1038/s41598-018-21445-3
102. Fossat N, Tourle K, Radziewic T, Barratt K, Liebhold D, Studdert JB, et al. C to U RNA editing mediated by APOBEC1 requires RNA-binding protein RBM47. EMBO Rep (2014) 15(8):903–10. doi: 10.15252/embr.201438450
103. Blanc V, Xie Y, Kennedy S, Riordan JD, Rubin DC, Madison BB, et al. Apobec1 complementation factor (A1CF) and RBM47 interact in tissue-specific regulation of C to U RNA editing in mouse intestine and liver. RNA (2019) 25(1):70–81. doi: 10.1261/rna.068395.118
104. Snyder EM, McCarty C, Mehalow A, Svenson KL, Murray SA, Korstanje R, et al. APOBEC1 complementation factor (A1CF) is dispensable for C-to-U RNA editing in vivo. RNA (2017) 23(4):457–65. doi: 10.1261/rna.058818.116
105. Zhang L, Tran NT, Su H, Wang R, Lu Y, Tang H, et al. Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife (2015) 4:e07938. doi: 10.7554/eLife.07938
106. Tran NT, Su H, Khodadadi-Jamayran A, Lin S, Zhang L, Zhou D, et al. The AS-RBM15 lncRNA enhances RBM15 protein translation during megakaryocyte differentiation. EMBO Rep (2016) 17: (6):887–900. doi: 10.15252/embr.201541970
107. Niu C, Zhang J, Breslin P, Onciu M, Ma Z, Morris SW. c-Myc is a target of RNA-binding motif protein 15 in the regulation of adult hematopoietic stem cell and megakaryocyte development. Blood (2009) 114(10):2087–96. doi: 10.1182/blood-2009-01-197921
108. Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, et al. Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell (2011) 43(4):624–37. doi: 10.1016/j.molcel.2011.06.028
109. Nag A, Steitz JA. Tri-snRNP-associated proteins interact with subunits of the TRAMP and nuclear exosome complexes, linking RNA decay and pre-mRNA splicing. RNA Biol (2012) 9(3):334–42. doi: 10.4161/rna.19431
110. Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, Falaleeva M, et al. Function of alternative splicing. Gene (2013) 514(1):1–30. doi: 10.1016/j.gene.2012.07.083
111. Stevens M, Oltean S. Modulation of the Apoptosis Gene Bcl-x Function Through Alternative Splicing. Front Genet (2019) 10:804. doi: 10.3389/fgene.2019.00804
112. Ratziu V, Lalazar A, Wong L, Dang Q, Collins C, Shaulian E, et al. Zf9, a Kruppel-like transcription factor up-regulated in vivo during early hepatic fibrosis. Proc Natl Acad Sci U.S.A. (1998) 95(16):9500–5. doi: 10.1073/pnas.95.16.9500
113. Stärkel P, Sempoux C, Leclercq I, Herin M, Deby C, Desager J-P, et al. Oxidative stress, KLF6 and transforming growth factor-β up-regulation differentiate non-alcoholic steatohepatitis progressing to fibrosis from uncomplicated steatosis in rats. J Hepatol (2003) 39(4):538–46. doi: 10.1016/S0168-8278(03)00360-X
114. Miele L, Beale G, Patman G, Nobili V, Leathart J, Grieco A, et al. The Kruppel-Like Factor 6 Genotype Is Associated With Fibrosis in Nonalcoholic Fatty Liver Disease. Gastroenterology (2008) 135(1):282–91.e1. doi: 10.1053/j.gastro.2008.04.004
115. Bechmann LP, Gastaldelli A, Vetter D, Patman GL, Pascoe L, Hannivoort RA, et al. Glucokinase links Kruppel-like factor 6 to the regulation of hepatic insulin sensitivity in nonalcoholic fatty liver disease. Hepatology (2012) 55(4):1083–93. doi: 10.1002/hep.24793
116. Keller H, Dreyer C, Medin J, Mahfoudi A, Ozato K, Wahli W. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc Natl Acad Sci U S A (1993) 90(6):2160–4. doi: 10.1073/pnas.90.6.2160
117. Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab (2012) 23(7):351–63. doi: 10.1016/j.tem.2012.05.001
118. Clemot M, Senos Demarco R, Jones DL. Lipid Mediated Regulation of Adult Stem Cell Behavior. Front Cell Dev Biol (2020) 8:115. doi: 10.3389/fcell.2020.00115
119. Fajas L, Fruchart J-C, Auwerx J. PPARγ3 mRNA: a distinct PPARγ mRNA subtype transcribed from an independent promoter. FEBS Lett (1998) 438(1-2):55–60. doi: 10.1016/S0014-5793(98)01273-3
120. Rahimian R, Masih-Khan E, Lo M, van Breemen C, McManus BM, Dube GP. Hepatic over-expression of peroxisome proliferator activated receptor gamma2 in the ob/ob mouse model of non-insulin dependent diabetes mellitus. Mol Cell Biochem (2001) 224(1-2):29–37. doi: 10.1023/A:1011927113563
121. Memon RA, Tecott LH, Nonogaki K, Beigneux A, Moser AH, Grunfeld C, et al. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology (2000) 141(11):4021–31. doi: 10.1210/endo.141.11.7771
122. Pettinelli P, Videla LA. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J Clin Endocrinol Metab (2011) 96(5):1424–30. doi: 10.1210/jc.2010-2129
123. Schadinger SE, Bucher NL, Schreiber BM, Farmer SR. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am J Physiol Endocrinol Metab (2005) 288(6):E1195–205. doi: 10.1152/ajpendo.00513.2004
124. Zhang YL, Hernandez-Ono A, Siri P, Weisberg S, Conlon D, Graham MJ, et al. Aberrant hepatic expression of PPARgamma2 stimulates hepatic lipogenesis in a mouse model of obesity, insulin resistance, dyslipidemia, and hepatic steatosis. J Biol Chem (2006) 281(49):37603–15. doi: 10.1074/jbc.M604709200
125. Lee YK, Park JE, Lee M, Hardwick JP. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Res (2018) 2(4):209–15. doi: 10.1016/j.livres.2018.12.001
126. Tatulian SA. Structural Dynamics of Insulin Receptor and Transmembrane Signaling. Biochemistry (2015) 54(36):5523–32. doi: 10.1021/acs.biochem.5b00805
127. Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med (1999) 107(5):450–5. doi: 10.1016/S0002-9343(99)00271-5
128. Belfiore A, Malaguarnera R, Vella V, Lawrence MC, Sciacca L, Frasca F, et al. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr Rev (2017) 38(5):379–431. doi: 10.1210/er.2017-00073
129. Vella V, Milluzzo A, Scalisi N, Vigneri P, Sciacca L. Insulin Receptor Isoforms in Cancer. Int J Mol Sci (2018) 19(11):3615. doi: 10.3390/ijms19113615
130. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev (2009) 30(6):586–623. doi: 10.1210/er.2008-0047
131. Lopez-Pastor AR, Gomez-Hernandez A, Diaz-Castroverde S, Gonzalez-Aseguinolaza G, Gonzalez-Rodriguez A, Garcia G, et al. Liver-specific insulin receptor isoform A expression enhances hepatic glucose uptake and ameliorates liver steatosis in a mouse model of diet-induced obesity. Dis Model Mech (2019) 12(2):1–11. doi: 10.1242/dmm.036186
132. Diaz-Castroverde S, Gomez-Hernandez A, Fernandez S, Garcia-Gomez G, Di Scala M, Gonzalez-Aseguinolaza G, et al. Insulin receptor isoform A ameliorates long-term glucose intolerance in diabetic mice. Dis Model Mech (2016) 9(11):1271–81. doi: 10.1242/dmm.025288
133. Leibiger B, Leibiger IB, Moede T, Kemper S, Kulkarni RN, Kahn CR, et al. Selective Insulin Signaling through A and B Insulin Receptors Regulates Transcription of Insulin and Glucokinase Genes in Pancreatic β Cells. Mol Cell (2001) 7(3):559–70. doi: 10.1016/S1097-2765(01)00203-9
134. Csaki LS, Reue K. Lipins: multifunctional lipid metabolism proteins. Annu Rev Nutr (2010) 30:257–72. doi: 10.1146/annurev.nutr.012809.104729
135. Khalil MB, Blais A, Figeys D, Yao Z. Lipin — The bridge between hepatic glycerolipid biosynthesis and lipoprotein metabolism. Biochim Biophys Acta (BBA) - Mol Cell Biol Lipids (2010) 1801(12):1249–59. doi: 10.1016/j.bbalip.2010.07.008
136. Arai T, Tanaka M, Goda N. HIF-1-dependent lipin1 induction prevents excessive lipid accumulation in choline-deficient diet-induced fatty liver. Sci Rep (2018) 8(1):14230. doi: 10.1038/s41598-018-32586-w
137. Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, et al. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology (2014) 146(3):801–11. doi: 10.1053/j.gastro.2013.11.008
138. Wang RH, Li C, Deng CX. Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int J Biol Sci (2010) 6(7):682–90. doi: 10.7150/ijbs.6.682
139. Kurylowicz A, Owczarz M, Polosak J, Jonas MI, Lisik W, Jonas M, et al. SIRT1 and SIRT7 expression in adipose tissues of obese and normal-weight individuals is regulated by microRNAs but not by methylation status. Int J Obes (2016) 40(11):1635–42. doi: 10.1038/ijo.2016.131
140. Caversaccio NI, Reina Caro MD, Prince R, Muller M, Lewis CS, Bogdanov VY, et al. Alternatively spliced tissue factor levels are elevated in the plasma of patients with chronic liver diseases. Eur J Gastroenterol Hepatol (2018) 30(12):1470–5. doi: 10.1097/MEG.0000000000001236
141. Bogdanov VY, Balasubramanian V, Hathcock J, Vele O, Lieb M, Nemerson Y. Alternatively spliced human tissue factor: a circulating, soluble, thrombogenic protein. Nat Med (2003) 9(4):458–62. doi: 10.1038/nm841
142. Li Y, Lin S, Li L, Tang Z, Hu Y, Ban X, et al. PDSS2 Deficiency Induces Hepatocarcinogenesis by Decreasing Mitochondrial Respiration and Reprogramming Glucose Metabolism. Cancer Res (2018) 78(16):4471–81. doi: 10.1158/0008-5472.CAN-17-2172
143. Wong CM, Xu L, Yau MY. Alternative mRNA Splicing in the Pathogenesis of Obesity. Int J Mol Sci (2018) 19(2):632. doi: 10.3390/ijms19020632
144. Sen S. Aberrant pre-mRNA splicing regulation in the development of hepatocellular carcinoma. Hepatoma Res (2018) 4(7):37–44. doi: 10.20517/2394-5079.2018.39
145. Jimenez M, Arechederra M, Avila MA, Berasain C. Splicing alterations contributing to cancer hallmarks in the liver: central role of dedifferentiation and genome instability. Transl Gastroenterol Hepatol (2018) 3:84. doi: 10.21037/tgh.2018.10.11
146. Liu L, Xie S, Zhang C, Zhu F. Aberrant regulation of alternative pre-mRNA splicing in hepatocellular carcinoma. Crit Rev Eukaryot Gene Expr (2014) 24(2):133–49. doi: 10.1615/CritRevEukaryotGeneExpr.2014007702
147. Effenberger KA, Urabe VK, Jurica MS. Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip Rev RNA (2017) 8(2):e1381. doi: 10.1002/wrna.1381
148. Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res (2016) 44(14):6549–63. doi: 10.1093/nar/gkw533
149. Urbanski LM, Leclair N, Anczukow O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip Rev RNA (2018) 9(4):e1476. doi: 10.1002/wrna.1476
150. Juliano RL. The delivery of therapeutic oligonucleotides. Nucleic Acids Res (2016) 44(14):6518–48. doi: 10.1093/nar/gkw236
151. Hagedorn PH, Hansen BR, Koch T, Lindow M. Managing the sequence-specificity of antisense oligonucleotides in drug discovery. Nucleic Acids Res (2017) 45(5):2262–82. doi: 10.1093/nar/gkx056
152. Shen X, Corey DR. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res (2018) 46(4):1584–600. doi: 10.1093/nar/gkx1239
153. Wu L, Mao C, Ming X. Modulation of Bcl-x Alternative Splicing Induces Apoptosis of Human Hepatic Stellate Cells. BioMed Res Int (2016) 2016:7478650. doi: 10.1155/2016/7478650
154. Cansby E, Nunez-Duran E, Magnusson E, Amrutkar M, Booten SL, Kulkarni NM, et al. Targeted Delivery of Stk25 Antisense Oligonucleotides to Hepatocytes Protects Mice Against Nonalcoholic Fatty Liver Disease. Cell Mol Gastroenterol Hepatol (2019) 7(3):597–618. doi: 10.1016/j.jcmgh.2018.12.004
155. Linden D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andreasson AC, et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol Metab (2019) 22:49–61. doi: 10.1016/j.molmet.2019.01.013
156. Rogalska ME, Tajnik M, Licastro D, Bussani E, Camparini L, Mattioli C, et al. Therapeutic activity of modified U1 core spliceosomal particles. Nat Commun (2016) 7:11168. doi: 10.1038/ncomms11168
157. Donadon I, Pinotti M, Rajkowska K, Pianigiani G, Barbon E, Morini E, et al. Exon-specific U1 snRNAs improve ELP1 exon 20 definition and rescue ELP1 protein expression in a familial dysautonomia mouse model. Hum Mol Genet (2018) 27(14):2466–76. doi: 10.1093/hmg/ddy151
158. Tajnik M, Rogalska ME, Bussani E, Barbon E, Balestra D, Pinotti M, et al. Molecular Basis and Therapeutic Strategies to Rescue Factor IX Variants That Affect Splicing and Protein Function. PloS Genet (2016) 12(5):e1006082. doi: 10.1371/journal.pgen.1006082
Keywords: RNA splicing, splicing factor, NAFLD, NASH, cirrhosis
Citation: Wu P, Zhang M and Webster NJG (2021) Alternative RNA Splicing in Fatty Liver Disease. Front. Endocrinol. 12:613213. doi: 10.3389/fendo.2021.613213
Received: 01 October 2020; Accepted: 13 January 2021;
Published: 26 February 2021.
Edited by:
Karine Clément, Sorbonne Universités, FranceReviewed by:
Ann K. Daly, Newcastle University, United KingdomCopyright © 2021 Wu, Zhang and Webster. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas J. G. Webster, bndlYnN0ZXJAdWNzZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.