
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 18 March 2021
Sec. Obesity
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.601160
This article is part of the Research Topic Non-Alcoholic Steatohepatitis (NASH) – Metabolic Contributors and Therapeutic Targets View all 25 articles
 Andrea M. Mueller1Robert Kleemann2,3Eveline Gart2,4Wim van Duyvenvoorde2Lars Verschuren5Martien Caspers5Aswin Menke2Natascha Krömmelbein1Kanita Salic2Yvonne Burmeister1Bernd Seilheimer1Martine C. Morrison2,4*
Andrea M. Mueller1Robert Kleemann2,3Eveline Gart2,4Wim van Duyvenvoorde2Lars Verschuren5Martien Caspers5Aswin Menke2Natascha Krömmelbein1Kanita Salic2Yvonne Burmeister1Bernd Seilheimer1Martine C. Morrison2,4*Background: Non-alcoholic fatty liver disease (NAFLD) is a complex multifactorial disorder that is characterised by dysfunctional lipid metabolism and cholesterol homeostasis, and a related chronic inflammatory response. NAFLD has become the most common cause of chronic liver disease in many countries, and its prevalence continues to rise in parallel with increasing rates of obesity. Here, we evaluated the putative NAFLD-attenuating effects of a multicomponent medicine consisting of 24 natural ingredients: Hepar compositum (HC-24).
Methods: Ldlr-/-.Leiden mice were fed a high-fat diet (HFD) with a macronutrient composition and cholesterol content comparable to human diets for 24 weeks to induce obesity-associated metabolic dysfunction, including hepatic steatosis and inflammation. HC-24 or vehicle control was administered intraperitoneally 3 times/week (1.5 ml/kg) for the last 18 weeks of the study. Histological analyses of liver and adipose tissue were combined with extensive hepatic transcriptomics analysis. Transcriptomics results were further substantiated with ELISA, immunohistochemical and liver lipid analyses.
Results: HFD feeding induced obesity and metabolic dysfunction including adipose tissue inflammation and increased gut permeability. In the liver, HFD-feeding resulted in a disturbance of cholesterol homeostasis and an associated inflammatory response. HC-24 did not affect body weight, metabolic risk factors, adipose tissue inflammation or gut permeability. While HC-24 did not alter total liver steatosis, there was a pronounced reduction in lobular inflammation in HC-24-treated animals, which was associated with modulation of genes and proteins involved in inflammation (e.g., neutrophil chemokine Cxcl1) and cholesterol homeostasis (i.e., predicted effect on ‘cholesterol’ as an upstream regulator, based on gene expression changes associated with cholesterol handling). These effects were confirmed by CXCL1 ELISA, immunohistochemical staining of neutrophils and biochemical analysis of hepatic free cholesterol content. Intrahepatic free cholesterol levels were found to correlate significantly with the number of inflammatory aggregates in the liver, thereby providing a potential rationale for the observed anti-inflammatory effects of HC-24.
Conclusions: Free cholesterol accumulates in the liver of Ldlr-/-.Leiden mice under physiologically translational dietary conditions, and this is associated with the development of hepatic inflammation. The multicomponent medicine HC-24 reduces accumulation of free cholesterol and has molecular and cellular anti-inflammatory effects in the liver.
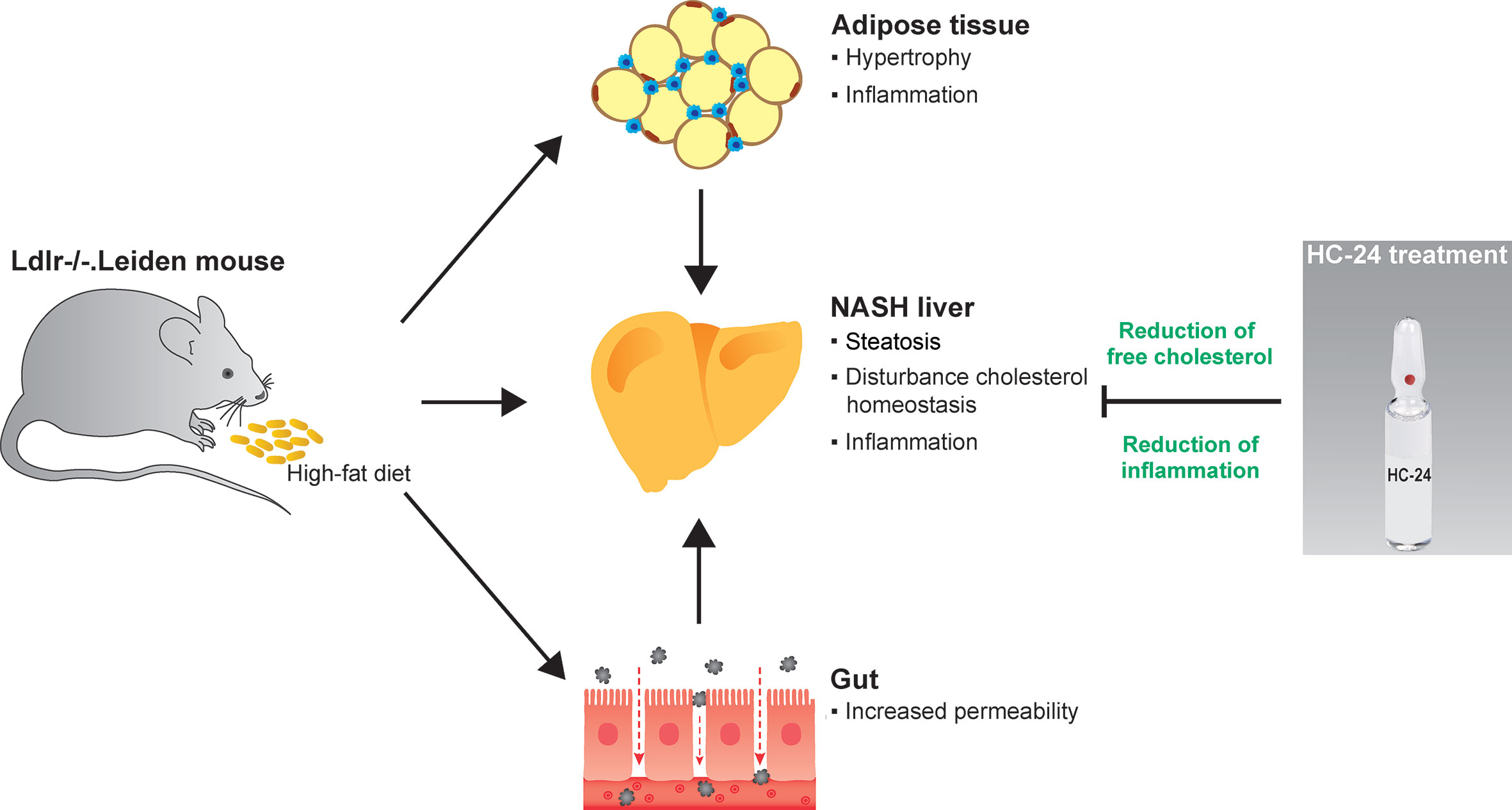
Graphical Abstract High-fat diet feeding in Ldlr-/-. Leiden mice induces obesity and associated metabolic dysfunction in adipose tissue (hypertrophy and inflammation), gut (increased permeability) and liver (non-alcoholic steatohepatitis; NASH). This NASH development in the liver is characterised by steatosis with disturbed cholesterol homeostasis and an associated inflammatory response, i.e. the infiltration of immune cells such as macrophages and neutrophils. Treatment with the multicomponent medicinal product HC-24 reduces hepatic accumulation of the cytotoxic lipid species free cholesterol and has anti-inflammatory effects in the liver – specifically reducing the infiltration of neutrophils into this tissue.
Non-alcoholic fatty liver disease (NAFLD) encompasses a spectrum of chronic liver disease which ranges from simple steatosis to non-alcoholic steatohepatitis (NASH), the progressive form of the disease which is characterised by hepatic inflammation and fibrosis in addition to steatosis. With a global prevalence of 25%, NAFLD is set to become the most common form of chronic liver disease worldwide (1). It is estimated that this prevalence will continue to increase, along with increasing rates of obesity and type 2 diabetes (2). As a result, NAFLD is projected to become the leading indication for liver transplantation within the next decade (3), especially since there is currently no approved pharmacological therapy for NAFLD. Therefore, there is an immense unmet need for efficacious therapeutics for NAFLD.
It is well accepted that the pathogenesis of NAFLD is multifactorial and also involves metabolic dysfunction in extrahepatic tissues such as the adipose tissue and the gut (4). In the liver, many molecular pathways contribute to the development and progression of the disease (5). An important factor that is thought to drive NAFLD progression from simple steatosis to steatohepatitis is cholesterol (6). It is becoming increasingly clear that accumulation of cholesterol in the liver (especially in its free, unesterified form) is a potent inducer of hepatic inflammation during NAFLD development (7, 8). In NAFLD patients, free cholesterol levels in the liver are associated with disease severity (9, 10), and many rodent models for NASH require dietary cholesterol supplementation to induce liver inflammation [reviewed in (11)]. It remains unclear, however, whether the disturbance of cholesterol homeostasis and the associated inflammatory response observed in humans is replicated in rodent models for NAFLD that use more human-like dietary conditions (i.e., diets with a macronutrient composition and a natural cholesterol content similar to human diets).
Given the multifactorial nature of the disease, it is conceivable that a successful therapy for NAFLD will need a multitarget therapy that targets several aspects of pathogenesis. Hepar compositum (HC-24) is a multicomponent medicinal product consisting of 24 ingredients including plant extracts, bioactive metabolites and animal-derived extracts (full composition shown in Supplemental Table 1). It has a long tradition of use as a supportive treatment for hepatic disorders of various origins (12), and there is anecdotal evidence for an effect of HC-24 on cholesterol metabolism (13), which possibly allows modulation of hepatic inflammatory responses. Many components of HC-24 have reported metabolic or anti-inflammatory effects. For instance, Silybum marianum (milk thistle) and its main constituent silymarin (rich in flavolignans) (14) have been shown to reduce plasma and liver markers of inflammation in high-fat diet (HFD)-fed mice (15). Similarly, Taraxacum officinale (dandelion) extracts have been shown to reduce LPS-induced inflammation in endothelial cells (16) and macrophages (17) and have cholesterol-modulating effects in rodents (18, 19) and type 2 diabetic patients (20). Likewise, Avena sativa (oat) is well-known for its anti-inflammatory and its hypocholesterolaemic effects (21).
Since both cholesterol metabolism and the inflammatory response are important interconnected physiological processes that are dysregulated during NAFLD development (8, 22), we questioned whether HC-24 might have beneficial effects in the treatment of NAFLD/NASH. To address this, we investigated the potential metabolic and anti-inflammatory effects of HC-24 in a translational diet-induced mouse model of NAFLD, the Ldlr-/-.Leiden mouse. In response to a HFD with a macronutrient composition and cholesterol content that is comparable to human diets (23–25), Ldlr-/-.Leiden mice develop histopathological characteristics of human NAFLD in the context of an obese phenotype with dyslipidaemia, hypertriglyceridaemia and insulin resistance (26–28) as is typical for a large proportion of NAFLD patients (29–31). In addition, many molecular processes that underlie the pathogenesis in NAFLD patients (both on the level of liver transcriptomics and of plasma metabolomics) are also reflected in the Ldlr-/-.Leiden mouse (32–34). Use of the aforementioned HFD with a translational cholesterol content enabled us to study the potential build-up of cholesterol in livers under physiologically relevant dietary conditions and to investigate the effects of HC-24 on the development of NAFLD.
The animal experiment was performed in the AAALAC-accredited animal facility at TNO Metabolic Health Research (Leiden, the Netherlands) in accordance with the rules and regulations set forward by the Netherlands Law on Animal Experiments with ethical approval by an independent Animal Welfare Body (IvD TNO; approval number 3682/TNO-210). Animals were group-housed in makrolon cages (2–4 mice/cage) in animal rooms with 50-60% relative humidity, temperature ~21°C and a 7 am to 7 pm light cycle with ad libitum access to food and water. Male Ldlr-/-.Leiden mice (age 13–16 weeks) were obtained from the breeding colony at TNO Metabolic Health Research (Leiden, the Netherlands). All animals were kept on a standard rodent maintenance diet until the start of the study (9 kcal% fat, 33 kcal% protein, 58 kcal% carbohydrate; R/M-H, Ssniff Spezialdiäten, Soest, Germany). At the start of the study, one group of mice (chow, n=10) was kept on this maintenance diet as a reference group. The rest of the mice (n=30) were switched to an energy-dense high-fat diet (HFD; 45 kcal% fat from lard, 20 kcal% protein, 35 kcal% carbohydrate; D12451, Research diets, New Brunswick, NJ, USA) to induce obesity, metabolic dysfunction and NAFLD. This diet is characterised by a macronutrient composition that mimics human diets (23–25), and the natural cholesterol content of this diet (approx. 0.02% w/w) was not further increased by addition of supraphysiological amounts of cholesterol. After 6 weeks of HFD feeding, these mice were matched into 2 groups based on body weight, blood glucose and plasma cholesterol levels. From that point onwards, the HFD+vehicle control group was treated with vehicle (saline), and the HFD+HC-24 group was treated with HC-24 (Heel GmbH, Baden-Baden, Germany; full composition shown in Supplemental Table 1). Both treatments were provided by intraperitoneal injection 3x weekly (Monday, Wednesday and Friday) at a dose of 1.5 ml/kg body weight. Body weight and food intake were monitored throughout the study. Body composition (lean mass and fat mass, expressed as % of body weight) was determined in the last week of the study (t=24 weeks) by EchoMRI (EchoMRI-LLC, Houston, TX, USA). Fasting (5 h) blood samples for EDTA plasma isolation were collected via the tail vein. In vivo gut permeability was assessed in week 23 using an FD4 gut permeability assay as described previously (28). In short, mice were fasted for 5 h after which a baseline blood sample was taken via the tail vein. Then, FITC-labeled dextran (three to five kDa FD4; Sigma Aldrich, St. Louis, MO, USA) was administered by oral gavage (900 mg/kg) and 4 h later a second blood sample was collected to determine the plasma FD4 concentration using a fluorometer (FLUOstar Galaxy, BMG labtech, Offenburg, Germany). The fluorescence reading was corrected for autofluorescence using the baseline blood sample. There was no drop out of mice during the study. All mice were terminated (unfasted) by gradual-fill CO2 asphyxiation after 24 weeks of chow/HFD feeding. A terminal blood sample for EDTA plasma was collected by cardiac puncture. The epididymal and mesenteric white adipose tissue depots were isolated and weighed and then fixed in formalin and embedded in paraffin for histological analyses. Livers were isolated and weighed, the medial lobe was fixed in formalin and embedded in paraffin for histological analyses, the left lobe was snap-frozen in liquid N2 and stored at -80°C for biochemical analyses.
Blood glucose was determined during blood sampling using a hand-held glucometer (Freestyle Freedom Light, Abbott Laboratories, Lake Bluff, IL, USA). Plasma insulin was determined by ELISA (Chrystal Chem Inc., Downers Grove, IL, USA). Plasma lipids (total cholesterol and triglycerides) were assayed in freshly prepared EDTA-plasma using commercially available enzymatic assays (CHOD-PAP and GPO-PAP respectively; Roche Diagnostics, Almere, the Netherlands). Plasma serum amyloid A (SAA) was measured by ELISA (Invitrogen/Thermo Fisher Waltham, MA, USA). Plasma alanine aminotransferase (ALT) was assessed using a spectrophotometric activity assay (Reflotron-plus, Roche Diagnostics). All these analyses were performed according to the manufacturers’ instructions.
Paraffin-embedded cross-sections (5 μm) of the epididymal and the mesenteric white adipose tissue were stained with hematoxylin-phloxine-saffron and digitised using a slide scanner (Aperio AT2, Leica Biosystems, Amsterdam, the Netherlands). Adipose tissue morphometry (average adipocyte size and adipocyte size distribution) and inflammation (number of crown-like structures; CLS per 1000 adipocytes) were analysed as described previously (35), using the automated image analysis software Adiposoft for morphometry analyses (36) and counting the number of CLS in the same fields used for morphometry analyses. Formalin-fixed and paraffin-embedded cross-sections of the medial liver lobe (3 μm) were stained with haematoxylin and eosin and scored blindly by a board-certified pathologist using an adapted grading method for human NASH (37, 38) as described previously (39). Briefly, two cross-sections/mouse were examined and the level of microvesicular steatosis, macrovesicular steatosis and hepatocellular hypertrophy were determined relative to the liver area analysed (expressed as a percentage). Hepatic inflammation was assessed by counting the number of inflammatory foci per field at a 100× magnification in five non-overlapping fields per specimen, expressed as the number of foci per mm2. Analysis of hepatic neutrophil counts was performed by immunohistochemical staining for GR-1 (with FITC-conjugated rat anti-mouse monoclonal antibody 1:1,000, eBioscience/Thermo Fisher; followed by rabbit anti-FITC recombinant monoclonal antibody 1:1,000, Invitrogen/Thermo Fisher; and BrightVision Poly-HRP goat anti-rabbit antibody 1:1, VWR International B.V., Amsterdam, the Netherlands) after enzymatic antigen retrieval using 0.025% pepsin (Sigma Aldrich) in 0.01 M HCl for 15 min at 37°C. Sections were stained with ImmPACT NovaRED HRP substrate (Vector Laboratories, Burlingame, CA, USA) and counterstained with haematoxylin. Neutrophils were quantified by counting the number of GR-1-positive inflammatory foci per field at 100× magnification in five non-overlapping fields per specimen, expressed as the number of positive foci per mm2. Analysis of hepatic macrophages was performed by immunohistochemical staining for F4/80 (rat anti-mouse monoclonal antibody 1:500, eBioscience; followed by biotinylated goat anti-rat secondary antibody 1:300, Abcam, Cambridge, UK) after heat-mediated antigen retrieval in sodium citrate buffer pH 6 in a PT link system (both DAKO/Agilent, Amstelveen, the Netherlands) and endogenous peroxidase block with 0.3% H2O2 in methanol. Sections were stained using Vectastain ABC-HRP Kit and DAB peroxidase substrate (both Vector Laboratories) and counterstained with haematoxylin. Macrophages were quantified by counting the number of F4/80-positive crown-like structures per field at 100× magnification in five non-overlapping fields per specimen, expressed as the number of CLS per mm2 as described previously (33).
RNA was isolated from snap-frozen liver tissue samples (left lobe) from all mice using RNA-Bee Total-RNA Isolation Kit (Bio-Connect, Huissen, the Netherlands). RNA concentration was determined spectrophotometrically using Nanodrop 1000 (Isogen Life Science, De Meern, the Netherlands) and RNA quality was assessed using a 2100 Bioanalyzer (Agilent Technologies, Amstelveen, the Netherlands). Then, next-generation RNA sequencing was performed on livers from all mice. For this, RNA was used to generate strand-specific cDNA libraries for Next Generation Sequencing according to the manufacturer’s protocol by GenomeScan B.V. (Leiden, the Netherlands; using oligo-dT magnetic beads, mRNA fragmentation, NEBNext Ultra Directional RNA Library Prep Kit from Illumina, NEB #E7420S/L resulting in 300–500 bp amplified libraries/sample). Libraries were multiplexed, clustered, and sequenced on a NextSeq500 system (Illumina, San Diego, CA, USA, using 1.6 pM of cDNA/sample and NextSeq control software version 2.0.2) with a single-read 75 cycles sequencing protocol, 12 million reads per sample and indexing. Image analysis, base calling, and quality check was performed with the Illumina data analysis pipeline RTA v2.4.11 and Bcl2fastq v2.17. Three mice were excluded as biological outliers (described below under statistical analysis), and one mouse (from the HFD + vehicle group) was excluded as a technical outlier (based on principal component analysis of the sequencing data), resulting in: n=10 chow, n=13 HFD+vehicle, n=13 HFD+HC-24 for further statistical analysis of the sequencing data. The gene expression data is publicly available via the NCBI Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/under accession number GSE163652). The number of differentially expressed genes (DEG) between groups was determined using an established statistical analysis procedure [DEseq2 pipeline (40)]. These DEG were determined using a statistical cut-off of p<0.01 for the following two comparisons: HFD+vehicle vs chow; and HFD+HC-24 vs HFD+vehicle. Next, these DEG were used as an input for an upstream regulator analysis through Ingenuity Pathway Analysis suite (IPA; www.ingenuity.com, accessed 2017). This analysis integrates the expression of a multitude of genes downstream from predefined upstream regulators (e.g., transcription factors, signalling proteins, metabolites) thus allowing summation of multiple (small) gene expression changes to provide information on the predicted activation state of such an upstream regulator. A negative Z-score <-2 indicates a predicted reduction in activity based on the direction of gene expression changes of target genes. A positive Z-score >2 indicates activation of the upstream regulator.
Liver homogenates were prepared from the sinister lobe in ice-cold lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 5mM CaCl2, 1% Triton X-100, pH 7.4) supplemented with complete mini protease inhibitor cocktail (Roche). Homogenates were centrifuged and the supernatant was used for CXCL1 determination by ELISA (Mouse CXCL1/KC Quantikine ELISA, R&D systems, Abingdon, UK) according to the manufacturer’s instructions. CXCL1 levels were expressed per mg liver protein, as determined by bicinchoninic acid (BCA) assay (Pierce/Thermo Fisher) in the same homogenates used for CXCL1 analysis.
Intrahepatic levels of triglycerides, cholesteryl esters and free cholesterol were determined in freshly prepared liver homogenates by high-performance thin-layer chromatography (HPTLC). Lipids were extracted from liver homogenates using methanol and chloroform as described previously (41, 42). After extraction, lipids were separated by high-performance thin-layer chromatography on silica gel 60 plates (0.20 mm). Lipid spots were stained with colour reagent (5 g of MnCl2∙4H2O, 32 ml of 95–97% H2SO4 added to 960 ml of CH3OH/H2O) and triglycerides, cholesteryl esters and free cholesterol were quantified using a Chemidoc Touch imaging system and Image Lab software (both Bio Rad Laboratories, Veenendaal, the Netherlands). Liver lipids were expressed as µg lipid per mg liver tissue.
Whole-body cholesterol balance (i.e., determination of net production or net excretion of cholesterol) was assessed by combining dietary cholesterol intake data with faecal cholesterol excretion data. In week 16 and week 20 of the study, faeces were collected from each cage over a period of 3 or 4 days respectively. Food intake was measured over the same period and dietary cholesterol intake was determined. Faecal neutral sterol and faecal bile acid levels were assessed to determine faecal excretion of cholesterol as described previously (43, 44). Faecal samples were first lyophilized and weighed. For extraction of bile acids, a 5 mg aliquot of faeces was incubated in 1 ml alkaline methanol (3:1 v/v) for 2 h at 80˚C, using nor-hyodeoxycholate as an internal standard. Samples were then diluted in distilled water, mixed and centrifuged. The supernatant was applied to a prepared Sep-Pak C18 solid-phase extraction cartridge (Waters Corporation, Wexford, Ireland) and bile acids were eluted with 100% methanol. Bile acids were derivatized by incubation with trifluoroacetic anhydride and 1,1,1,3,3,3-hexafluoro-2-propanol for 1 h at 60˚C after which they were separated using a 25 m × 0.25 mm capillary gas chromatography column (CP-Sil 5B, Agilent, Santa Clara, CA, USA; temperature programmed from 230°C to 280°C) in a Scion 436-GC gas chromatography system (Scion Instruments, Livingstone, UK) equipped with a flame ionization detector (kept at 300°C). Bile acid derivatives were introduced by split injection (split ratio, 20:1; injector temp 300°C). Quantitation of bile acids (cholic acid, deoxycholic acid, litocholic acid, α-muricholic acid, β-muricholic acid, ω-muricholic acid, and hyodeoxycholic acid) was based on the area ratio of the individual bile acid to the internal standard. Levels of α-muricholic acid were below the lower limit of quantitation and are therefore not reported. For extraction of neutral sterols, a 10 mg aliquot of faeces was incubated in 1 ml alkaline methanol as described for bile acid extraction, using 5α-cholestane as an internal standard. Next, neutral sterols were extracted three times with petroleum ether. The combined petroleum ether layers were then evaporated and the neutral sterols were silylated with DMF Sil-prep after which they were separated by GC using the same column and protocol as for the bile acids. Quantitation of neutral sterols (coprostanol, cholesterol, cholestanol, and lathosterol) was based on the area ratio of the individual neutral sterol to the internal standard. The data from the food intake measurements and the sterol and bile acid excretion analyses was used to calculate net cholesterol production as follows: cholesterol excretion (i.e., bile acid + neutral sterol excretion) – dietary cholesterol intake, expressed as µmol/mouse/day.
With the exception of the transcriptomics analysis, for which the statistical analysis has been described above, statistical analyses were performed using SPSS Statistics 24.0 (IBM, Armonk, NY, USA). Based on the robust regression and outlier removal test [ROUT (45)] with Q set to 1%, 3 mice (1 in the HFD + vehicle group and 2 in the HFD + HC-24 group) were identified as biological outliers and excluded from statistical analysis. Normal distribution of variables was analysed with the Shapiro-Wilk test, assuming normality at p>0.05. To test equality of variances, Levene’s test of homogeneity of variances was used, assuming equal variances at p>0.05. For normally distributed variables with equal variances, differences between groups were analysed by one-way analysis of variance (ANOVA) followed by one-sided Dunnet’s post-hoc tests. For normally distributed variables with unequal variances, differences between groups were analysed by analysis of variance (Brown-Forsythe) and one-sided Dunnett’s T3 post-hoc tests. The non-parametric Kruskall-Wallis test followed by post-hoc with one-sided Mann-Whitney U tests was used to determine differences between groups for variables that were not normally distributed. Correlation analyses were performed by Spearman’s rank correlation analysis. P-values <0.05 were considered statistically significant. Data are represented as means ± SD.
Ldlr-/-.Leiden mice were fed an energy-dense high-fat diet for 24 weeks to induce obesity, metabolic dysfunction and NAFLD. Relative to a chow-fed reference group, HFD feeding resulted in increased body weight and adiposity (reduced lean mass % and increased fat mass %; Table 1). HFD feeding did not affect blood glucose but did result in strongly increased plasma insulin levels (Table 1), indicative of reduced insulin sensitivity. Plasma lipids, both total cholesterol and triglycerides (Table 1), were strongly increased in HFD-fed mice. In addition, HFD feeding resulted in an increase in the systemic inflammation marker serum amyloid A (SAA; Table 1). Treatment with HC-24 from week 6 of HFD feeding until the end of the study did not affect the development of obesity and adiposity, food intake, hyperinsulinaemia, dyslipidaemia, or systemic inflammation (Table 1).
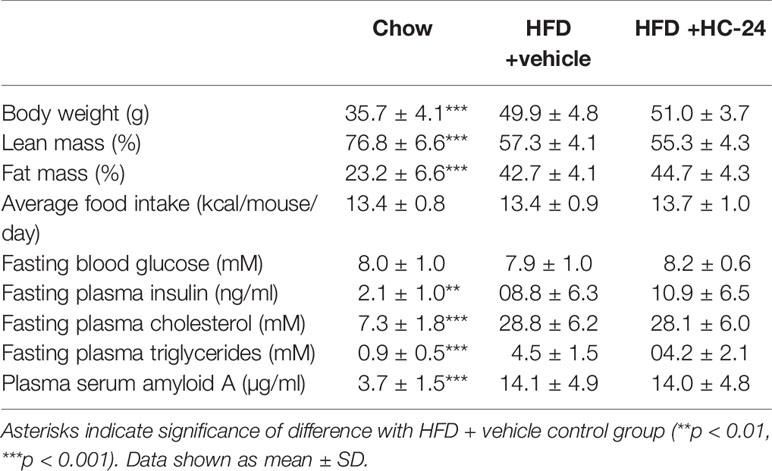
Table 1 Metabolic risk factors at t=24 weeks.
In line with the observed effects on body weight and fat mass, HFD feeding tended to increase the weight of the epididymal white adipose (eWAT) depot and significantly increased the weight of the mesenteric white adipose (mWAT) depot (Figures 1A, D). This HFD-induced increase in adipose depot mass was accompanied by an increase in adipocyte hypertrophy in both depots, as shown by an increase in the average adipocyte size and a shift in the distribution of adipocyte sizes towards a reduction in the percentage of smaller adipocytes and an increase in the percentage of large adipocytes (Figures 1B, C, E, F). This expansion and hypertrophy of both eWAT and mWAT was associated with adipose tissue inflammation as shown by the increased number of crown-like structures (Figures 1G, H). HC-24 treatment did not affect adipose mass, hypertrophy or inflammation in either of the depots studied. Since increased gut permeability is thought to contribute to the progression of NAFLD, we investigated gut permeability with an FD4 test in week 23 of the study. HFD feeding significantly increased the passage of FD4 from the intestinal lumen into the circulation (Figure 1I), indicative of increased gut permeability. This was not affected by HC-24 (Figure 1I).
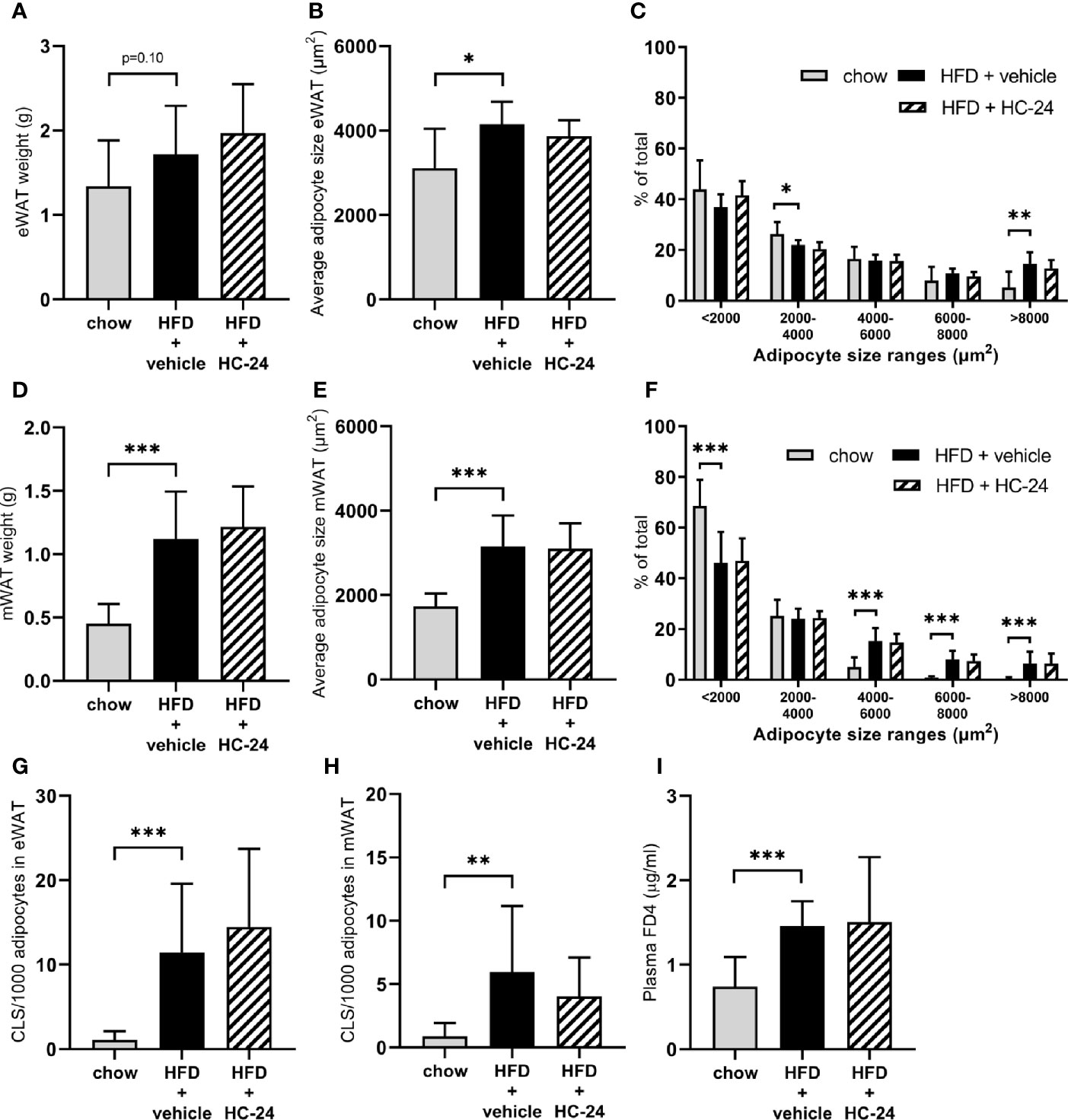
Figure 1 HC-24 does not affect adipose tissue mass, adipocyte hypertrophy or inflammation of epididymal and mesenteric white adipose tissue depots and has no effect on gut permeability in Ldlr-/-.Leiden mice fed a translational HFD for 24 weeks. (A) eWAT depot weight. (B) Average adipocyte size in eWAT. (C) Size distribution of adipocytes in eWAT. (D) mWAT depot weight. (E) Average adipocyte size in mWAT. (F) Size distribution of adipocytes in mWAT. (G) Number of CLS per 1,000 adipocytes in eWAT. (H) Number of CLS per 1000 adipocytes in mWAT. (I) In vivo functional gut permeability analysis using the Fluorescein isothiocyanate–labeled dextran (FD4) assay in week 23 of the study. eWAT, epididymal white adipose tissue; mWAT, mesenteric white adipose tissue; CLS, crown-like structure. Data are mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 vs. HFD + vehicle.
At the end of the 24-week study, liver weight was significantly increased in HFD-fed animals relative to chow (Figure 2A). This increase in liver weight was accompanied by an increase in plasma alanine aminotransferase (ALT; Figure 2B). Neither liver weight nor ALT were affected by HC-24 treatment. Histopathological analysis of NASH (representative images for all groups shown in Figure 2C) showed that HFD feeding resulted in pronounced hepatic steatosis (Figure 2D), which was roughly half in the macrovesicular form and half in the microvesicular form (Figures 2E, F). HC-24 treatment did not affect hepatic steatosis (Figure 2D) and did not affect the distribution of steatosis over the macrovesicular and the microvesicular form (Figures 2E, F). In line with the observations on hepatic steatosis, hepatocellular hypertrophy was increased by HFD-feeding, and was not affected by treatment with HC-24 (Figure 2G). However, quantitative analysis of the number of inflammatory aggregates in the liver showed a significant induction of hepatic inflammation in HFD-fed animals which was strongly and significantly reduced by treatment with HC-24 (Figure 2H). Altogether, this histopathological analysis of NASH shows a pronounced increase in steatosis and lobular inflammation in animals fed a HFD with a translational macronutrient composition and cholesterol content, and demonstrates a specific effect of HC-24 on the inflammatory component of the disease.
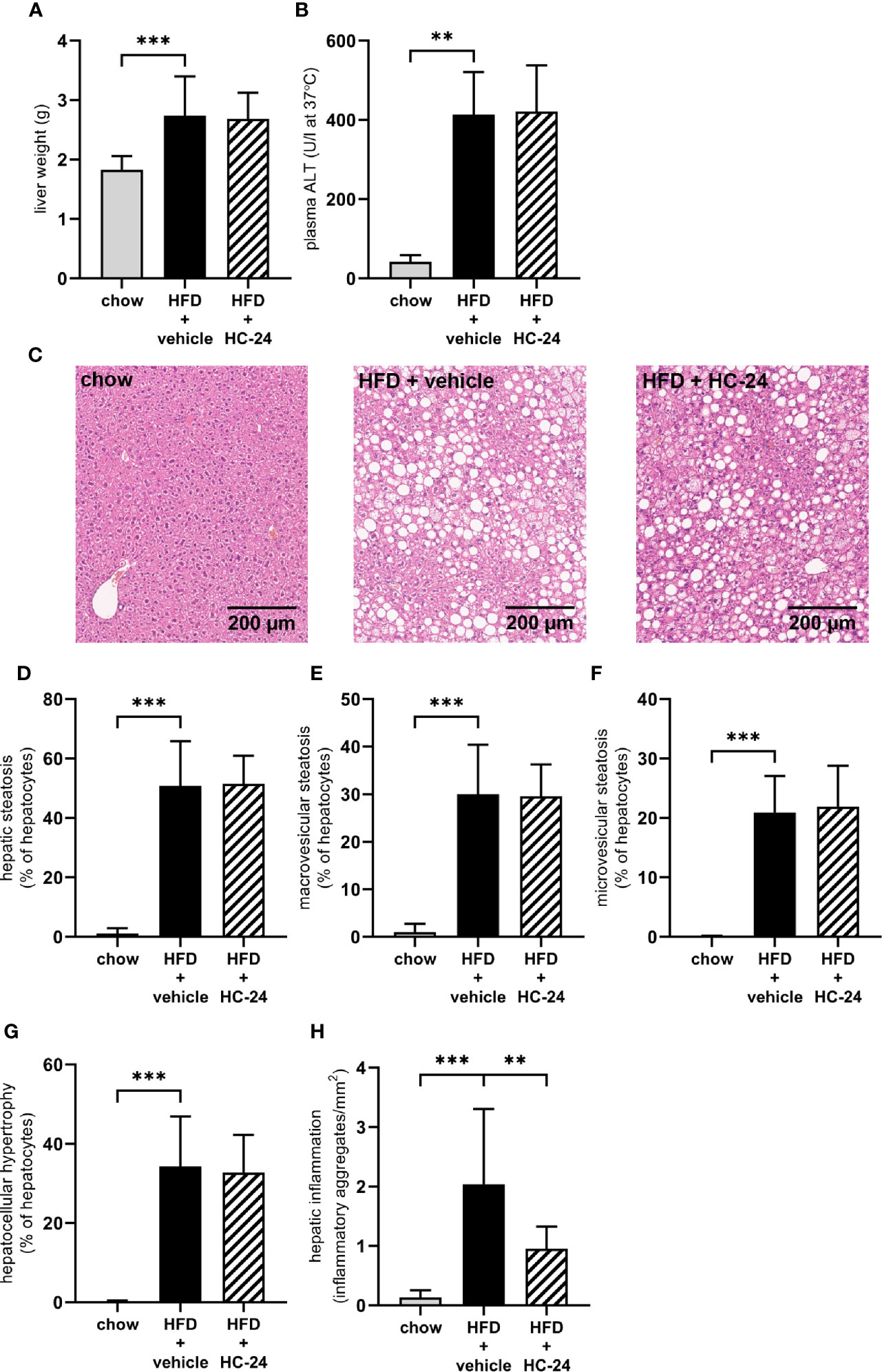
Figure 2 HC-24 treatment does not affect hepatic steatosis but does strongly reduce hepatic inflammation in Ldlr-/-.Leiden mice fed a translational HFD for 24 weeks. (A) Liver weight. (B) Plasma ALT measured in week 24 of the study. (C) Representative photomicrographs of Haemotoxylin and Eosin-stained cross sections of the medial lobe of the liver of chow, HFD + vehicle control and HFD + HC-24 groups. (D) Total steatosis. (E) Macrovesicular steatosis. (F) Microvesicular steatosis. (G) Hepatocellular hypertrophy. (H) Hepatic inflammation. HFD, high-fat diet; ALT, alanine aminotransferase. Data shown are mean ± SD. **p < 0.01, ***p < 0.001 compared with HFD + vehicle.
To investigate the molecular processes that may underlie the observed anti-inflammatory effects of HC-24 in the liver, we performed a hepatic gene expression analysis using genome-wide RNA sequencing. This gene expression profiling was followed by an upstream regulator analysis which predicts the activation state of key regulatory factors (e.g., cytokines, transcription factors, metabolites) based on the expression pattern of genes downstream from this factor. As expected and in line with histopathologically observed changes in the liver, HFD feeding resulted in gene expression changes related among others to lipid metabolism and inflammation (modulated upstream regulators shown in Supplemental Table 2). The metabolite “cholesterol” was identified as a significantly modulated upstream factor (i.e., there was a predicted HFD-dependent increase in hepatic cholesterol based on gene expression patterns downstream from cholesterol). HC-24 treatment significantly affected the expression of distinct genes in the liver (full list shown in Supplemental Table 3). In line with the observed anti-inflammatory effect of HC-24 on the histological level, HC-24 modulated several inflammation-related genes, such as Socs3 and Gzma and the neutrophil-attracting chemokine Cxcl1. Results from the upstream regulator analysis revealed further inflammation-modifying effects of HC-24, showing significant overlap with genes downstream of TGF-β, IL-17A, and IL-1β (Supplemental Table 4). Notably, this analysis also demonstrated significant inactivation of the upstream regulator cholesterol, suggesting reduced levels of cholesterol in the liver in HC-24 treated animals.
To investigate whether the anti-inflammatory effect of HC-24 was indeed linked to an effect on neutrophil infiltration in the liver as suggested by the gene expression data, we measured CXCL1 protein levels in the liver (Figure 3A). In line with the observed modulation of Cxcl1 gene expression, CXCL1 levels were low in chow-fed mice and significantly increased in HFD+vehicle controls. HC-24 treatment significantly lowered the levels of CXCL1 in liver tissue. Immunohistochemical staining of the neutrophil marker GR-1 followed by quantification of the number of GR-1-positive cell clusters in the liver (Figure 3B, with representative images shown in Figure 3C) further corroborated this effect: GR-1-positive cell aggregates were almost completely absent in chow controls, and their counts were significantly increased by HFD-feeding. Treatment with HC-24 significantly lowered the number of GR-1-positive cell aggregates in the liver, in line with the observed reduction in Cxcl1 gene and protein expression. To explore whether HC-24 affects neutrophil infiltration exclusively or has a broader effect on immune cell infiltration in general, we analysed the transcriptome dataset using previously described immune cell-type specific gene sets that allow assessment of the presence of particular leukocytes in a tissue (46, 47). This analysis revealed that while there was an increased presence of B-cells and T-cells in HFD-fed mice relative to chow controls, this was not affected by HC-24 treatment (B-cells: HFD + vehicle vs. chow: −log(p-value) = 2.4, HFD + HC-24 vs HFD + vehicle: non-significant; T-cells: HFD + vehicle vs. chow: −log(p-value) = 2.3, HFD + HC-24 vs. HFD + vehicle: non-significant). In addition, we performed an immunohistochemical analysis of the macrophage marker F4/80, followed by quantification of F4/80-positive crown-like structures in liver (Figure 3D, with representative images shown in Figure 3E) to assess potential effects of HC-24 on this immune cell subset. Similar to what was observed for the B- and T-cell population, F4/80-positive crown like structures were significantly increased by HFD relative to chow, and this was not affected by treatment with HC-24.
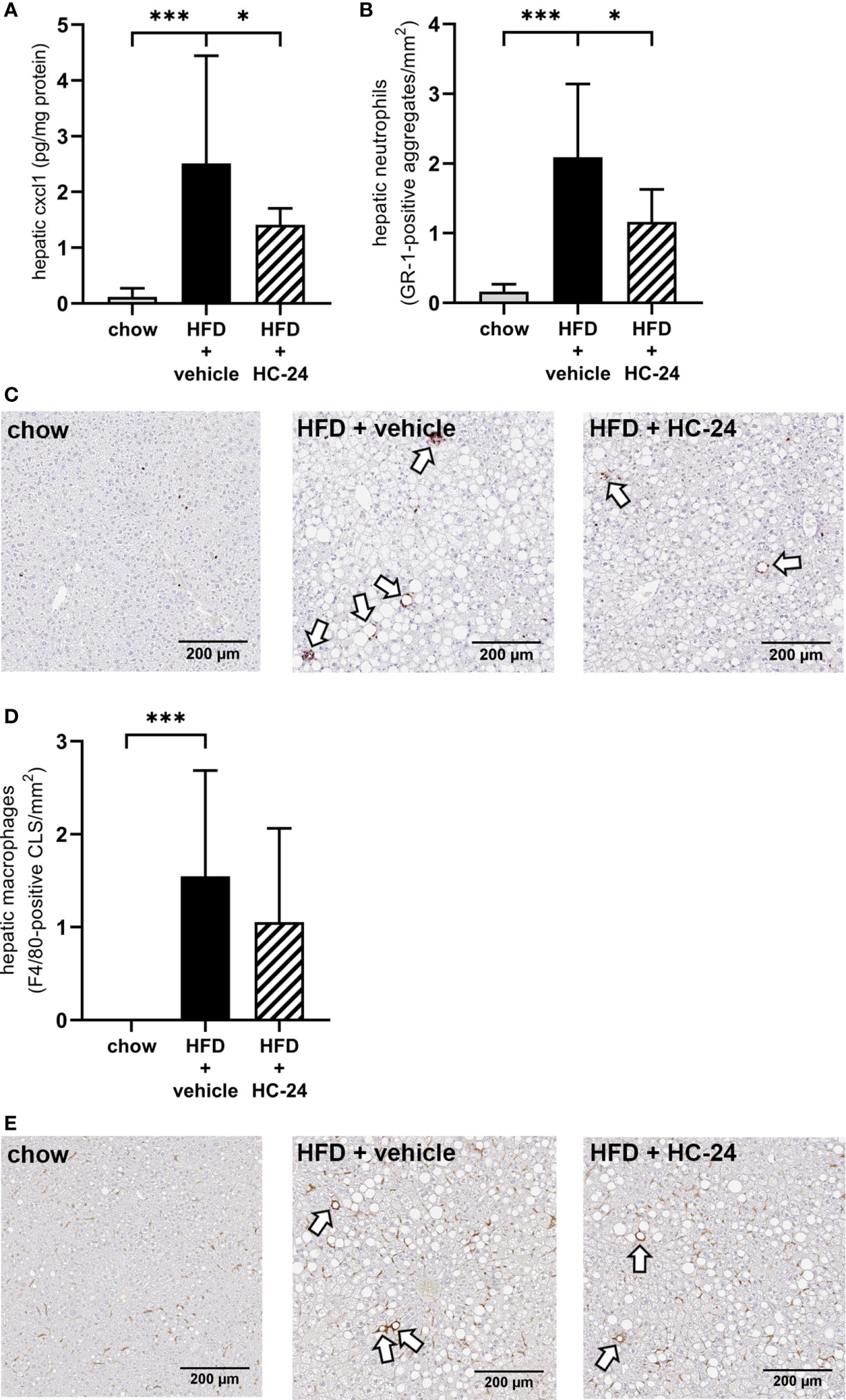
Figure 3 HC-24 treatment reduces neutrophil infiltration in Ldlr-/-.Leiden mice fed a translational HFD for 24 weeks. (A) Concentration of the neutrophil chemokine CXCL1 in liver tissue of chow, HFD + vehicle control and HFD + HC-24 groups. (B) Quantification of the immunohistochemical staining for the neutrophil marker GR-1, showing the number of GR-1-positive aggregates for each group. (C) Representative photomicrographs of immunohistochemical staining for the neutrophil marker GR-1 in cross sections of the medial lobe of the liver. Arrows indicate neutrophilic aggregates. (D) Quantification of the immunohistochemical staining for the macrophage marker F4/80, showing the number of F4/80-positive crown-like structures for each group. (E) Representative photomicrographs of immunohistochemical staining for the macrophage marker F4/80 in cross sections of the medial lobe of the liver. Arrows indicate F4/80-positive crown-like structures. HFD, high-fat diet; CLS, crown-like structure. Data shown are mean ± SD. *p < 0.05, ***p < 0.001 compared with HFD + vehicle.
Since free cholesterol is a potent inducer of hepatic inflammation and appeared to be critically affected by HFD feeding, we next investigated whether the predicted effect on cholesterol accumulation in the liver (based on observed gene signalling patterns downstream from cholesterol) may underlie the observed anti-inflammatory effects. For this, we first performed a biochemical analysis of intrahepatic lipids to confirm the predicted effect on hepatic cholesterol. This analysis showed that – like in NAFLD patients – triglycerides are the main lipid species that accumulate in the liver of HFD-fed mice (Figure 4A). In line with what was observed histologically, treatment with HC-24 did not affect the bulk storage of lipids as triglycerides (Figure 4A). In addition to triglycerides, cholesterol – both in the esterified form and in the free, unesterified form – also built up during NAFLD development, as can be observed in HFD-fed mice (Figures 4B, C). Treatment with HC-24 significantly reduced the build-up of free cholesterol (Figure 4C). To further substantiate this effect on hepatic cholesterol accumulation and to investigate how HC-24 affected hepatic free cholesterol levels, we investigated whether HC-24 had an effect on whole-body cholesterol balance. For this we measured faecal cholesterol excretion (in the form of neutral sterols and bile acids) and combined this with the dietary intake of cholesterol to calculate net excretion/production of cholesterol (Figures 4D, E and Supplemental Table 5). This analysis revealed that in HFD-fed Ldlr-/-.Leiden mice, there is a net production of cholesterol in the body (i.e., more cholesterol is excreted via the faeces than is consumed via the diet). Treatment with HC-24 significantly lowered the net production of cholesterol, in line with the effects on intrahepatic cholesterol accumulation. Furthermore, the levels of hepatic free cholesterol showed a significant positive correlation with hepatic inflammation (number of inflammatory cell foci; Spearman r = 0.78, p<0.001; Figure 4F), providing a rationale for the observed simultaneous effects of HC-24 on cholesterol homeostasis and liver inflammation.
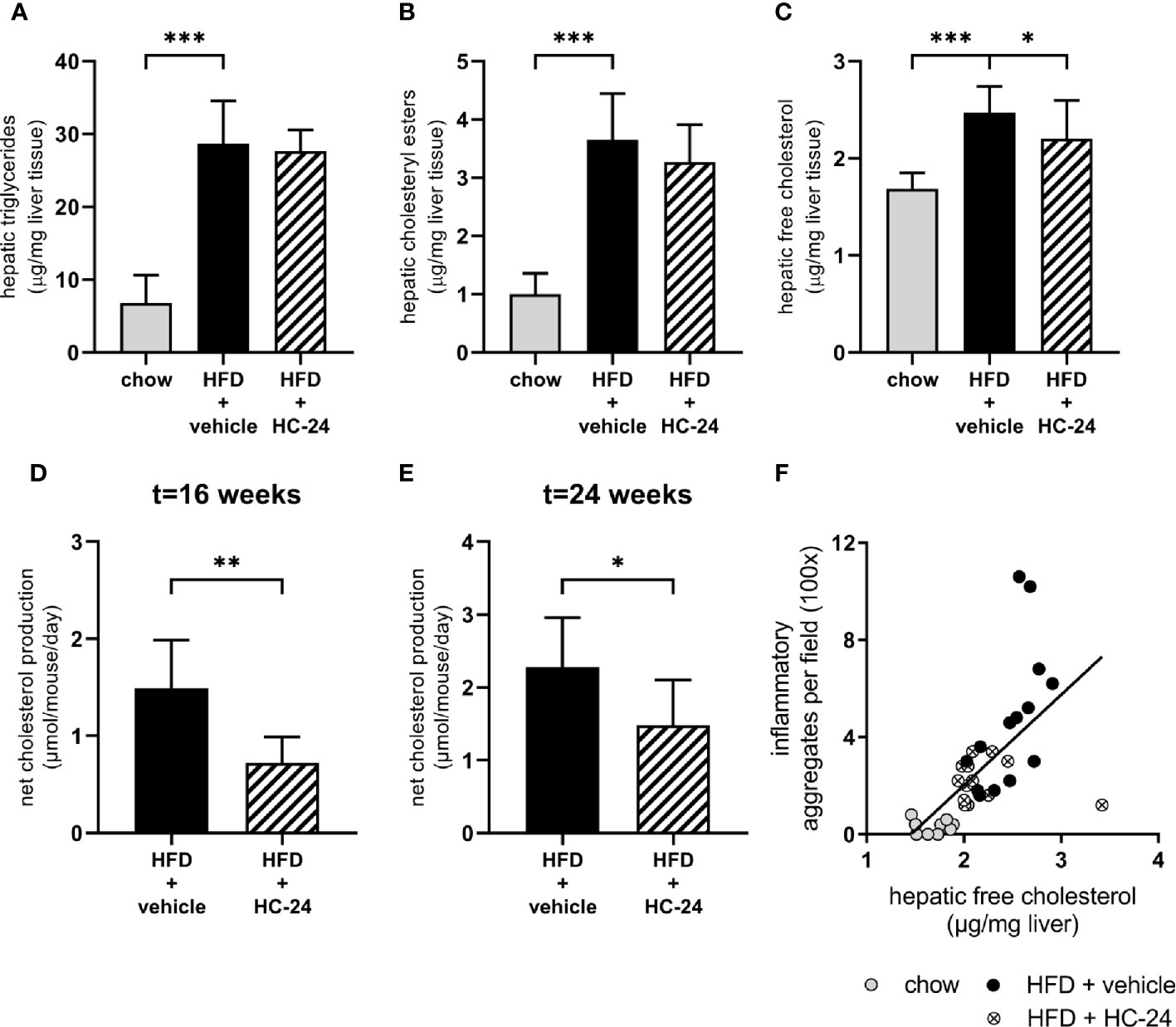
Figure 4 HC-24 treatment reduces free cholesterol accumulation in the liver of Ldlr-/-.Leiden mice fed a translational HFD for 24 weeks. (A) Hepatic triglyceride content. (B) Hepatic cholesteryl ester content. (C) Hepatic free cholesterol content. (D) Net whole-body cholesterol production in week 16 of the study. (E) Net whole-body cholesterol production in week 24 of the study. (F) Pearson correlation between free cholesterol in the liver and the histologically determined number of inflammatory aggregates in the liver. HFD, high-fat diet. Data shown are mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 compared with HFD + vehicle.
This study shows that accumulation of free cholesterol in the liver, under translational dietary conditions, is associated with hepatic inflammation and can be modulated by a multicomponent medicinal product HC-24, resulting in reduced inflammatory aggregates with fewer neutrophils, a cell type that is characteristic for human NASH.
Many conventional rodent models for NAFLD rely on supplementation of diets with high (supraphysiological) levels of cholesterol to induce liver inflammation (48). While the accumulation of cholesterol per se is not irrelevant for NAFLD pathogenesis, since hepatic free cholesterol is also increased and associated with disease severity in NAFLD patients (9, 10), such models strongly emphasise the role of dietary cholesterol in NAFLD. Furthermore, they imply that an increased cholesterol uptake and a greater hepatic influx of cholesterol the portal blood are pathogenically relevant processes for humans that are causal to the observed accumulation of free cholesterol and liver inflammation. Although there is some associative evidence from an observational study that showed increased dietary cholesterol intake in NASH patients relative to controls (49), this does not demonstrate a causal link between cholesterol intake and hepatic inflammation in humans as such. Indeed, a controlled human intervention study by Tannock et al. (50) showed no evidence for an inflammation-inducing effect of dietary cholesterol in insulin-resistant subjects (i.e., the patient group that is at increased risk for NAFLD/NASH). Despite a strongly increased dietary cholesterol intake, there was no increase in plasma SAA or CRP levels, the latter being the most sensitive liver-specific inflammation marker available. Hence, the available evidence does not support the concept that dietary cholesterol, at relevant doses achieved in human diets, is an inducer of liver inflammation in humans. Rodent models that rely strongly on supraphysiological cholesterol supplementation most likely specifically allow investigation of direct hepatotoxic effects of dietary cholesterol. In the current study, we used a more comprehensive model for NAFLD that makes use of more translational dietary conditions and may better reflect the multifactorial and multi-organ nature of the disease (e.g., with an obese and insulin-resistant phenotype, with the involvement of adipose tissue hypertrophy and inflammation and increased gut permeability). We show here that hepatic cholesterol homeostasis is also deregulated under these more translational experimental conditions with realistic dietary cholesterol concentrations, leading to free cholesterol accumulation which contributes to the hepatic inflammatory response.
Given the multifactorial aetiology of NAFLD that involves deregulation of a multitude of pathways as drivers of disease progression, it seems appropriate to use a multitarget approach to combat the disease. Such an approach is increasingly considered the new paradigm for drug development for NAFLD, with numerous combination therapies currently under investigation (51–53). Here we used a multicomponent therapy with natural ingredients (HC-24) to attenuate NAFLD development, and studied its effects on several organs considered to be involved in the pathogenesis, i.e., adipose tissue (54, 55) and gut (permeability) (56, 57) in addition to the liver. We show here that HC-24 did not affect adipose tissue mass (measured as total body fat percentage as well as by the weight of several individual adipose tissue depots), adipocyte hypertrophy or adipose tissue inflammation in two visceral white adipose tissue depots (the epididymal and the mesenteric depots), and that disease-associated increased gut permeability was also not affected by HC-24 treatment – together indicating that the NASH-attenuating effect of HC-24 is most likely localised to the liver specifically rather than (partly) attributable to indirect effects via the adipose tissue or the gut.
In the liver, we observed a pronounced reduction in lobular inflammation in animals treated with HC-24. This reduction in inflammatory aggregates was associated with a decrease in the accumulation of free cholesterol in the liver which corresponded with the observed reduction in whole-body cholesterol production. A subsequent correlation analysis revealed that the number of inflammatory aggregates in the liver was strongly and significantly correlated with hepatic free cholesterol content, thus providing a potential explanation for the observed beneficial effects of HC-24 in NAFLD. These findings are in line with an earlier rodent study that showed a positive correlation between hepatic free cholesterol concentrations and hepatic NFκB activation, both of which were reduced after treatment with an anthocyanin-rich plant extract (58). An orchestrated adaptation of cholesterol biosynthesis and metabolism pathways and connected inflammatory pathways in both the liver and the adipose tissue was also observed in another study in response to a high-fat diet with a natural cholesterol content (59).
Besides these effects on transcriptional control mechanisms, another proposed mechanism by which cholesterol induces inflammation is by induction of physical cellular damage [for instance through formation of cholesterol crystals (60)] – which can result in the release of damage-associated molecular patterns (DAMPs) that function as a chemotactic signal for neutrophils (61) and may thus underlie the observed effect on neutrophil infiltration. Additionally, cholesterol loading in macrophages has been shown to induce expression of IL-8 – a potent neutrophil chemoattractant (62) – and cholesterol enrichment in neutrophil membranes has been found to enhance neutrophil adhesion and arrest, thereby increasing their potential for extravasation (63). In line with this, we found that the neutrophil chemokine CXCL1 was increased in livers of HFD control mice. This corresponds with observations in human NASH livers, which show a strong upregulation of CXCL1 that is not observed in livers from patients with simple steatosis (64, 65). Of note, other HFD-fed mouse models of NASH do not show this upregulation of CXCL1 (65, 66). A recent report by Hwang et al. (65) showed that overexpression of Cxcl1 in HFD-fed mice was sufficient to induce NASH (recapitulating the pathological features of human NASH), thus further underlining the importance of this chemokine. Treatment with HC-24 significantly reduced CXCL1 expression in the liver — both on the gene expression level and on the protein level — and reduced infiltration of neutrophils into the liver. Although the infiltration of neutrophils is recognised as a defining characteristic of human NASH (67), their precise role in the pathogenesis of NASH has not been completely elucidated. However, their ability to release a potent cocktail of reactive oxygen species and proteases is thought to make them a potential cause of extensive tissue damage as demonstrated in other liver diseases (68, 69). This notion is further substantiated by the above-mentioned study on CXCL1-induced NASH (65), which provides several lines of evidence that neutrophils promote NASH development through production of ROS that activate several stress kinases (ASK1, p38/JNK-CASP3 pathway) in the liver, resulting in liver injury, inflammation and fibrosis. Conversely, depletion of neutrophils in HFD-fed mice reduces development of NASH (70). Since hepatic inflammation in the current study showed a clear correlation with hepatic free cholesterol levels, it is likely that the observed anti-inflammatory effects of HC-24 are (at least in part) secondary to the improvement of cholesterol homeostasis observed in HC-24-treated animals.
Overall, our results show that free cholesterol accumulates in the liver of Ldlr-/-.Leiden mice under physiologically translational dietary conditions and that this build-up of free cholesterol is associated with the development of hepatic inflammation. Treatment with the multicomponent medicinal product HC-24 reduces this free cholesterol accumulation and has molecular and cellular anti-inflammatory effects in the liver.
The data has been uploaded to the GEO database, under accession number GSE163652.
The animal study was reviewed and approved by Animal Welfare Body (IvD) TNO the Netherlands (approval number 3682/TNO-210).
RK, YB, BS, and MM contributed to conception and design of the study. EG, WD, and AMe collected the data. LV, MC, KS, and MM analyzed the data. AMu, RK, NK, YB, BS, and MM interpreted the data. AMu, RK, and MM wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was performed within the public-private partnership (PPP) ProLiver, a collaboration project that is co-funded by a PPP Allowance made available by Health~Holland, Top Sector Life Sciences & Health, to stimulate public-private partnerships. Heel GmbH was one of the private partners that contributed funding to the PPP ProLiver. The work described here was also supported by the TNO Research Programs Food and Nutrition and Biomedical Health.
AMu and BS are employees of Heel GmbH. NK and YB are former employees of Heel GmbH. Heel GmbH was involved in the design of the study and the preparation of the manuscript. Heel GmbH was not involved in data acquisition or data analysis. The publication of this study was a requirement of the funding received from Health~Holland, Top Sector Life Sciences & Health.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Simone van der Drift-Droog, Nikki van Trigt, Jessica Snabel, Marijke Voskuilen, and Ivana Bobeldijk-Pastorova for their excellent technical and organizational assistance during the study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.601160/full#supplementary-material
1. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol (2018) 15(1):11–20. doi: 10.1038/nrgastro.2017.109
2. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology (2018) 67(1):123–33. doi: 10.1002/hep.29466
3. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the united states. Gastroenterology (2015) 148(3):547–55. doi: 10.1053/j.gastro.2014.11.039
4. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med (2018) 24(7):908–22. doi: 10.1038/s41591-018-0104-9
5. Wegermann K, Diehl AM, Moylan CA. Disease pathways and molecular mechanisms of nonalcoholic steatohepatitis. Clin Liver Dis (Hoboken) (2018) 11(4):87–91. doi: 10.1002/cld.709
6. Parthasarathy G, Revelo X, Malhi H. Pathogenesis of nonalcoholic steatohepatitis: An overview. Hepatol Commun (2020) 4(4):478–92. doi: 10.1002/hep4.1479
7. Musso G, Gambino R, Cassader M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog Lipid Res (2013) 52(1):175–91. doi: 10.1016/j.plipres.2012.11.002
8. Ioannou GN. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol Metab (2016) 27(2):84–95. doi: 10.1016/j.tem.2015.11.008
9. Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology (2007) 46(4):1081–90. doi: 10.1002/hep.21763
10. Caballero F, Fernandez A, De Lacy AM, Fernandez-Checa JC, Caballeria J, Garcia-Ruiz C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol (2009) 50(4):789–96. doi: 10.1016/j.jhep.2008.12.016
11. Farrell G, Schattenberg JM, Leclercq I, Yeh MM, Goldin R, Teoh N, et al. Mouse models of nonalcoholic steatohepatitis: Toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology (2019) 69(5):2241–57. doi: 10.1002/hep.30333
12. Borner M, Weiser M. Biological therapy of liver disorders - results of drug monitoring with 801 patients. Biol Ther (1995) XIII(2):64–7.
13. Vovk AD, Solyanik IV. Experience of using of antihomotoxic preparations hepar compositum and engystol in complex treatment patients suffering with viral hepatitis. Biol Ther (Ukraine) (2003) 1):16–9.
14. Tajmohammadi A, Razavi BM, Hosseinzadeh H. Silybum marianum (milk thistle) and its main constituent, silymarin, as a potential therapeutic plant in metabolic syndrome: A review. Phytother Res (2018) 32(10):1933–49. doi: 10.1002/ptr.6153
15. Feng B, Meng R, Huang B, Shen S, Bi Y, Zhu D. Silymarin alleviates hepatic oxidative stress and protects against metabolic disorders in high-fat diet-fed mice. Free Radic Res (2016) 50(3):314–27. doi: 10.3109/10715762.2015.1116689
16. Jeon D, Kim SJ, Kim HS. Anti-inflammatory evaluation of the methanolic extract of taraxacum officinale in LPS-stimulated human umbilical vein endothelial cells. BMC Complement Altern Med (2017) 17(1). doi: 10.1186/s12906-017-2022-7
17. Park CM, Cho CW, Song YS. TOP 1 and 2, polysaccharides from taraxacum officinale, inhibit NFκB-mediated inflammation and accelerate Nrf2-induced antioxidative potential through the modulation of PI3K-akt signaling pathway in RAW 264.7 cells. Food Chem Toxicol (2014) 66:56–64. doi: 10.1016/j.fct.2014.01.019
18. Choi UK, Lee OH, Yim JH, Cho CW, Rhee YK, Lim SI, et al. Hypolipidemic and antioxidant effects of dandelion (taraxacum officinale) root and leaf on cholesterol-fed rabbits. Int J Mol Sci (2010) 11(1):67–78. doi: 10.3390/ijms11010067
19. Majewski M, Lis B, Juśkiewicz J, Ognik K, Borkowska-Sztachańska M, Jedrejek D, et al. Phenolic fractions from dandelion leaves and petals as modulators of the antioxidant status and lipid profile in an in vivo study. Antioxid (Basel) (2020) 9(2):131. doi: 10.3390/antiox9020131
20. Chatterji S, Fogel D. Study of the effect of the herbal composition SR2004 on hemoglobin A1c, fasting blood glucose, and lipids in patients with type 2 diabetes mellitus. Integr Med Res (2018) 7(3):248–56. doi: 10.1016/j.imr.2018.04.002
21. Sang S, Chu Y. Whole grain oats, more than just a fiber: Role of unique phytochemicals. Mol Nutr Food Res (2017) 61(7). doi: 10.1002/mnfr.201600715
22. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol (2018) 15(6):349–64. doi: 10.1038/s41575-018-0009-6
23. Linseisen J, Welch AA, Ocké M, Amiano P, Agnoli C, Ferrari P, et al. Dietary fat intake in the european prospective investigation into cancer and nutrition: Results from the 24-h dietary recalls. Eur J Clin Nutr (2009) 63 Suppl 4:S61–80. doi: 10.1038/ejcn.2009.75
24. Harika RK, Eilander A, Alssema M, Osendarp SJ, Zock PL. Intake of fatty acids in general populations worldwide does not meet dietary recommendations to prevent coronary heart disease: A systematic review of data from 40 countries. Ann Nutr Metab (2013) 63(3):229–38. doi: 10.1159/000355437
25. Liang W, Lindeman JH, Menke AL, Koonen DP, Morrison M, Havekes LM, et al. Metabolically induced liver inflammation leads to NASH and differs from LPS- or IL-1beta-induced chronic inflammation. Lab Invest (2014) 94:491–502. doi: 10.1038/labinvest.2014.11
26. Morrison MC, Mulder P, Salic K, Verheij J, Liang W, van Duyvenvoorde W, et al. Intervention with a caspase-1 inhibitor reduces obesity-associated hyperinsulinemia, non-alcoholic steatohepatitis and hepatic fibrosis in LDLR-/-.leiden mice. Int J Obes (Lond) (2016) 40(9):1416–23. doi: 10.1038/ijo.2016.74
27. Salic K, Gart E, Seidel F, Verschuren L, Caspers M, van Duyvenvoorde W, et al. Combined treatment with L-carnitine and nicotinamide riboside improves hepatic metabolism and attenuates obesity and liver steatosis. Int J Mol Sci (2019) 20(18):4359. doi: 10.3390/ijms20184359
28. Gart E, Souto Lima E, Schuren F, Schuren F, de Ruiter CGF, Attema J, Verschuren L, et al. Diet-independent correlations between bacteria and dysfunction of gut, adipose tissue, and liver: A comprehensive microbiota analysis in feces and mucosa of the ileum and colon in obese mice with NAFLD. Int J Mol Sci (2018) 20(1):1. doi: 10.3390/ijms20010001
29. Anderson N, Borlak J. Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol Rev (2008) 60(3):311–57. doi: 10.1124/pr.108.00001
30. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol (2013) 10(11):686–90. doi: 10.1038/nrgastro.2013.171
31. Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic steatohepatitis: A review. JAMA (2020) 323(12):1175–83. doi: 10.1001/jama.2020.2298
32. van Koppen A, Verschuren L, van den Hoek AM, Verheij J, Morrison MC, Li K, et al. Uncovering a predictive molecular signature for the onset of NASH-related fibrosis in a translational NASH mouse model. Cell Mol Gastroenterol Hepatol (2017) 5(1):83–98. doi: 10.1016/j.jcmgh.2017.10.001
33. Morrison MC, Verschuren L, Salic K, Verheij J, Menke A, Wielinga PY, et al. Obeticholic acid modulates serum metabolites and gene signatures characteristic of human NASH and attenuates inflammation and fibrosis progression in ldlr-/-.leiden mice. Hepatol Commun (2018) 2(12):1513–32. doi: 10.1002/hep4.1270
34. Morrison MC, Kleemann R, van Koppen A, Hanemaaijer R, Verschuren L. Key inflammatory processes in human NASH are reflected in ldlr(-/-).leiden mice: A translational gene profiling study. Front Physiol (2018) 9:132. doi: 10.3389/fphys.2018.00132
35. Jacobs SAH, Gart E, Vreeken D, Franx BAA, Wekking L, Verweij VGM, et al. Sex-specific differences in fat storage, development of non-alcoholic fatty liver disease and brain structure in juvenile HFD-induced obese ldlr-/-.leiden mice. Nutrients (2019) 11(8):1861. doi: 10.3390/nu11081861
36. Galarraga M, Campión J, Muñoz-Barrutia A, Boqué N, Moreno H, Martínez JA, et al. Adiposoft: Automated software for the analysis of white adipose tissue cellularity in histological sections. J Lipid Res (2012) 53(12):2791–6. doi: 10.1194/jlr.D023788
37. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology (2005) 41(6):1313–21. doi: 10.1002/hep.20701
38. Liang W, Menke AL, Driessen A, Koek GH, Lindeman JH, Stoop R, et al. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PloS One (2014) 9(12):e115922. doi: 10.1371/journal.pone.0115922
39. Schoemaker MH, Kleemann R, Morrison MC, Verheij J, Salic K, van Tol EAF, et al. A casein hydrolysate based formulation attenuates obesity and associated non-alcoholic fatty liver disease and atherosclerosis in LDLr-/-.leiden mice. PloS One (2017) 12(7):e0180648. doi: 10.1371/journal.pone.0180648
40. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol (2014) 15(12):550–014-0550-8. doi: 10.1186/s13059-014-0550-8
41. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol (1959) 37(8):911–7. doi: 10.1139/o59-099
42. Havekes LM, de Wit EC, Princen HM. Cellular free cholesterol in hep G2 cells is only partially available for down-regulation of low-density-lipoprotein receptor activity. Biochem J (1987) 247(3):739–46. doi: 10.1042/bj2470739
43. Post SM, de Crom R, van Haperen R, van Tol A, Princen HM. Increased fecal bile acid excretion in transgenic mice with elevated expression of human phospholipid transfer protein. Arterioscler Thromb Vasc Biol (2003) 23(5):892–7. doi: 10.1161/01.ATV.0000067702.22390.20
44. Salic K, Kleemann R, Wilkins-Port C, McNulty J, Verschuren L, Palmer M. Apical sodium-dependent bile acid transporter inhibition with volixibat improves metabolic aspects and components of non-alcoholic steatohepatitis in ldlr-/-.leiden mice. PloS One (2019) 14(6):e0218459. doi: 10.1371/journal.pone.0218459
45. Motulsky HJ, Brown RE. Detecting outliers when fitting data with nonlinear regression - a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinf (2006) 7. doi: 10.1186/1471-2105-7-123
46. Palmer C, Diehn M, Alizadeh AA, Brown PO. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics (2006) 7. doi: 10.1186/1471-2164-7-115
47. van den Hoek AM, Verschuren L, Worms N, van Nieuwkoop A, de Ruiter C, Attema J, et al. A translational mouse model for NASH with advanced fibrosis and atherosclerosis expressing key pathways of human pathology. Cells (2020) 9(9):2014. doi: 10.3390/cells9092014
48. Santhekadur PK, Kumar DP, Sanyal AJ. Preclinical models of non-alcoholic fatty liver disease. J Hepatol (2018) 68(2):230–7. doi: 10.1016/j.jhep.2017.10.031
49. Musso G, Gambino R, De Michieli F, Cassader M, Rizzetto M, Durazzo M, et al. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology (2003) 37(4):909–16. doi: 10.1053/jhep.2003.50132
50. Tannock LR, O’Brien KD, Knopp RH, Retzlaff B, Fish B, Wener MH, et al. Cholesterol feeding increases C-reactive protein and serum amyloid A levels in lean insulin-sensitive subjects. Circulation (2005) 111(23):3058–62. doi: 10.1161/CIRCULATIONAHA.104.506188
51. Corey KE, Chalasani N. Should combination therapy be the paradigm for future nonalcoholic steatohepatitis clinical trials? Hepatology (2011) 54(5):1503–5. doi: 10.1002/hep.24696
52. Younossi ZM, Loomba R, Rinella ME, Bugianesi E, Marchesini G, Neuschwander-Tetri BA, et al. Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology (2018) 68(1):361–71. doi: 10.1002/hep.29724
53. Johnston MP, Patel J, Byrne CD. Multi-drug approaches to NASH: What’s in the development pipeline? Expert Opin Investig Drugs (2020) 29(2):143–50. doi: 10.1080/13543784.2020.1668926
54. du Plessis J, van Pelt J, Korf H, Mathieu C, van der Schueren B, Lannoo M, et al. Association of adipose tissue inflammation with histologic severity of nonalcoholic fatty liver disease. Gastroenterology (2015) 149(3):635–48. doi: 10.1053/j.gastro.2015.05.044
55. Mulder P, Morrison MC, Wielinga PY, van Duyvenvoorde W, Kooistra T, Kleemann R. Surgical removal of inflamed epididymal white adipose tissue attenuates the development of non-alcoholic steatohepatitis in obesity. Int J Obes (Lond) (2016) 40(4):675–84. doi: 10.1038/ijo.2015.226
56. Volynets V, Küper MA, Strahl S, Maier IB, Spruss A, Wagnerberger S, et al. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig Dis Sci (2012) 57(7):1932–41. doi: 10.1007/s10620-012-2112-9
57. Xu L, Kitade H, Ni Y, Ota T. Roles of chemokines and chemokine receptors in obesity-associated insulin resistance and nonalcoholic fatty liver disease. Biomolecules (2015) 5(3):1563–79. doi: 10.3390/biom5031563
58. Morrison MC, Liang W, Mulder P, Verschuren L, Pieterman E, Toet K, et al. Mirtoselect, an anthocyanin-rich bilberry extract, attenuates non-alcoholic steatohepatitis and associated fibrosis in ApoE *3Leiden mice. J Hepatol (2015) 62:1180–6. doi: 10.1016/j.jhep.2014.12.011
59. Liang W, Tonini G, Mulder P, Kelder T, van Erk M, van den Hoek AM, et al. Coordinated and interactive expression of genes of lipid metabolism and inflammation in adipose tissue and liver during metabolic overload. PloS One (2013) 8(9):e75290. doi: 10.1371/journal.pone.0075290
60. Ioannou GN, Haigh WG, Thorning D, Savard C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J Lipid Res (2013) 54(5):1326–34. doi: 10.1194/jlr.M034876
61. Xu R, Huang H, Zhang Z, Wang FS. The role of neutrophils in the development of liver diseases. Cell Mol Immunol (2014) 11(3):224–31. doi: 10.1038/cmi.2014.2
62. Wang N, Tabas I, Winchester R, Ravalli S, Rabbani LE, Tall A. Interleukin 8 is induced by cholesterol loading of macrophages and expressed by macrophage foam cells in human atheroma. J Biol Chem (1996) 271(15):8837–42. doi: 10.1074/jbc.271.15.8837
63. Oh H, Mohler ER, Tian A, Baumgart T, Diamond SL. Membrane cholesterol is a biomechanical regulator of neutrophil adhesion. Arterioscler Thromb Vasc Biol (2009) 29(9):1290–7. doi: 10.1161/ATVBAHA.109.189571
64. Bertola A, Bonnafous S, Anty R, Patouraux S, Saint-Paul MC, Iannelli A, et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PloS One (2010) 5(10):e13577. doi: 10.1371/journal.pone.0013577
65. Hwang S, He Y, Xiang X, Seo W, Kim SJ, Ma J, et al. Interleukin-22 ameliorates neutrophil-driven nonalcoholic steatohepatitis through multiple targets. Hepatology (2020) 72(2):412–29. doi: 10.1002/hep.31031
66. Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, et al. Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology (2016) 151(3):513–25. doi: 10.1053/j.gastro.2016.05.051
67. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am J Gastroenterol (1999) 94(9):2467–74. doi: 10.1111/j.1572-0241.1999.01377.x
68. Jaeschke H. Neutrophil-mediated tissue injury in alcoholic hepatitis. Alcohol (2002) 27(1):23–7. doi: 10.1016/S0741-8329(02)00200-8
69. Jaeschke H. Mechanisms of liver injury. II. mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol (2006) 290(6):G1083–8. doi: 10.1152/ajpgi.00568.2005
Keywords: non-alcoholic fatty liver disease, obesity, cholesterol, inflammation, diet-induced, liver, neutrophils, hepar compositum
Citation: Mueller AM, Kleemann R, Gart E, van Duyvenvoorde W, Verschuren L, Caspers M, Menke A, Krömmelbein N, Salic K, Burmeister Y, Seilheimer B and Morrison MC (2021) Cholesterol Accumulation as a Driver of Hepatic Inflammation Under Translational Dietary Conditions Can Be Attenuated by a Multicomponent Medicine. Front. Endocrinol. 12:601160. doi: 10.3389/fendo.2021.601160
Received: 31 August 2020; Accepted: 04 February 2021;
Published: 18 March 2021.
Edited by:
Karine Clément, Sorbonne Universités, FranceReviewed by:
Giuseppe Danilo Norata, University of Milan, ItalyCopyright © 2021 Mueller, Kleemann, Gart, van Duyvenvoorde, Verschuren, Caspers, Menke, Krömmelbein, Salic, Burmeister, Seilheimer and Morrison. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martine C. Morrison, martine.morrison@tno.nl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.